

Abstract
The number and quality of oocytes, as well as the decline in both of these parameters with age, determines reproductive potential in women. However, the underlying mechanisms of this diminution are incompletely understood. Previously, we identified novel roles for CHTF18 (Chromosome Transmission Fidelity Factor 18), a component of the conserved Replication Factor C-like complex, in male fertility and gametogenesis. Currently, we reveal crucial roles for CHTF18 in female meiosis and oocyte development. Chtf18−/− female mice are subfertile and have fewer offspring beginning at 6 months of age. Consistent with age-dependent subfertility, Chtf18−/− ovaries contain fewer follicles at all stages of folliculogenesis than wild type ovaries, but the decreases are more significant at 3 and 6 months of age. By 6 months of age, both primordial and growing ovarian follicle pools are markedly reduced to near depletion. Chromosomal synapsis in Chtf18−/− oocytes is complete, but meiotic recombination is impaired resulting in persistent DNA double-strand breaks, fewer crossovers, and early homolog disjunction during meiosis I. Consistent with poor oocyte quality, the majority of Chtf18−/− oocytes fail to progress to metaphase II following meiotic resumption and a significant percentage of those that do progress are aneuploid. Collectively, our findings indicate critical functions for CHTF18 in ensuring both the quantity and quality of the mammalian oocyte pool.
Keywords: CHTF18; meiosis; ovarian reserve; oocyte quantity; oocyte quality; DNA double-strand break repair; female fertility, cohesion; cohesin aneuploidy
CHTF18 ensures both the quantity and quality of the mammalian oocyte pool and may play a role in delaying ovarian aging.
Introduction
Meiosis is a monumental developmental task accomplished through a series of carefully orchestrated steps including the critical processes of DNA double-strand break (DSB) repair and crossover formation. These processes must occur in a timely and accurate manner to ensure proper chromosome segregation and prevent aneuploidy. Although meiosis is tightly regulated, it is highly error-prone in females [1–3]. In females, meiosis begins during fetal development and arrests at the end of prophase I; it does not resume and complete until puberty through adulthood, as many as 50 years later. This extended time frame of the first meiotic division and abnormalities arising during meiosis that evade detection in females each contributes to the error-prone nature of female meiosis. As a result, errors occurring during meiosis I in the oocyte account for the majority of aneuploidy in humans [4, 5]. Female meiosis is especially susceptible to errors and aneuploidy with increasing age in women [6–9]. The decline in oocyte quality caused by errors that arise during meiosis I with advancing female age is also accompanied by decreasing oocyte number. Although ovarian reserve refers to the quantity of oocytes, from a clinical vantage point of reproductive potential and aging, ovarian reserve is viewed in relation to both the quantity and quality of remaining oocytes [10]. Diminution in it ultimately leads to infertility, miscarriage, and chromosomal anomalies in offspring. Although many chromosomal factors impact oocyte quality and number, DSB repair, crossover formation, and chromosome cohesion during meiosis are especially crucial. Meiotic recombination begins during the leptotene stage of meiotic prophase I with formation of DSBs, which are initiated by SPO11, a topoisomerase-like protein [11–13]. Repair of DSBs then ensues and progresses to completion in the late pachytene stage, when a small subset becomes crossovers that are cytologically seen as chiasmata at diakinesis and metaphase I. Cohesion between sister chromatids is necessary during meiosis for DSB repair, recombination, and to stabilize chiasmata [9, 14–17]; together cohesion and chiasmata prevent homologs from separating prematurely [18–20]. Defects in DSB repair during prophase I can lead to reduced recombination events and crossovers, resulting in chromosome missegregation and subsequent aneuploidy [9, 21, 22]. In addition, repair of DSBs during meiosis I is essential to preserve and protect the ovarian reserve. Unrepaired and accumulated DSBs can trigger the elimination of defective oocytes by quality control mechanisms [23–27]. Despite our expanding knowledge about the causes of errors inherent in female meiosis, the exact molecular mechanisms underlying advancing age are still incompletely understood. Therefore, physiologically relevant mammalian models are needed to elucidate the basis of defects leading to deterioration in both the quantity and quality of the ovarian reserve.
Proteins involved in DNA replication and repair have recognized functions in genome integrity and meiosis too [28–32]. Chromosome Transmission Fidelity Factor 18 (CHTF18) is a conserved subunit of the alternative Replication Factor C-Like complex, RLC-CHTF18. The canonical functions of RLC-CHTF18 are to aid in the establishment of sister chromatid cohesion and to load Proliferating Cell Nuclear Antigen (PCNA) onto DNA during replication [33–35]. CTF18, the yeast ortholog, is involved in stabilizing replication forks and likely couples sister chromatid cohesion with DNA replication, although the exact mechanism is unknown [36, 37]. In cultured human epithelial cells, CHTF18 is implicated in controlling fork progression and in facilitating both the acetylation of the cohesin subunit, SMC3 and sister chromatid cohesion [38]. Interestingly, CHTF18 is expressed throughout the murine germline, suggesting a role in mammalian germ cell development [39]. Previously, we reported novel functions for CHTF18 in male meiosis [28]. Chtf18−/− male mice are subfertile with defects in spermatogenesis and meiotic recombination. Given the essential functions of CHTF18 in male gametogenesis, we predicted vital roles for CHTF18 in female fertility and meiosis. Here, we reveal critical functions of CHTF18 in female meiotic DSB repair and in ensuring a normal number of crossovers in oocytes. Consistent with these functions, CHTF18 prevents the premature separation of homologs during prophase I and aneuploidy at metaphase II. Decreases in the ovarian follicle pools also become significantly worse as females age in the absence of CHTF18. Collectively, our findings suggest that CHTF18 plays essential roles in regulating both the quantity and quality of oocytes and may play a role in delaying ovarian aging.
Materials and methods
Mice
Derivation of Chtf18-null (Chtf18−/−) mice was previously described [28]. All experiments involving mice were approved by the Institutional Animal Care and Use Committee at Drexel University College of Medicine and according to the National Institutes of Health guidelines.
Fertility assessment
Six adult Chtf18−/− or three wild-type (Chtf18+/+) females were each bred with adult wild-type (Chtf18+/+) males (1 female/male pair per cage) over a 6-month period, and the numbers of litters and offspring per litter were recorded.
Ovarian follicle histology, morphometric, and apoptotic analyses
Ovaries from mice at postnatal day 7 and 21 and at 3 and 6 months of age were collected, processed, paraffin embedded, and follicles counted. Briefly, dissected ovaries were fixed in Bouin’s solution or 4% paraformaldehyde solution at 4 °C overnight, embedded in paraffin blocks, and sectioned. Every tenth section of ovary pairs was stained with hematoxylin and eosin (H&E) and used for morphometric analysis. Ovaries from mice at postnatal day 3 were harvested, fixed in Davidson’s solution at 4 °C for 5–6 h, then processed, paraffin-embedded, sectioned, and H&E stained at Rutgers University Research Pathology Services (Piscataway, NJ). Every fifth stained section was imaged with an EVOS FL Auto Imaging System and a 40× objective under brightfield for analysis. Ovarian follicles were quantified in sections of ovary pairs using counting techniques as described [40]. Follicles were categorized and scored as primordial, primary, secondary, and antral according to morphological criteria. Specifically, each primordial follicle was identified by an oocyte surrounded by a single layer of flattened granulosa cells; each primary follicle contained an oocyte surrounded by one layer of cuboidal granulosa cells; each antral follicle contained an oocyte and antral spaces surrounded by greater than one layer of granulosa cells (Supplementary Figure S1). Only follicles each containing an oocyte and discernable nucleus were counted to avoid repeated counting. A correction factor of 10 was multiplied to obtain a total count of follicles for ovaries in which every tenth section was analyzed, and a correction factor of 5 was used to obtain a total count of primordial follicles in postnatal day 3 ovaries in which every fifth section was analyzed. To evaluate apoptosis, the second, sixth, and eleventh sections of ovary pairs were immunostained with anti-cleaved Caspase-3 (1:100; CP3, BioCare Medical, Ref. # CP229A). Briefly, after quenching endogenous peroxidase, slides were blocked with horse serum and incubated with CP3 antibody overnight at 4 °C. Slides were washed and incubated with a biotinylated goat anti-rabbit IgG secondary antibody (Vectastain ABC Kit, Vector Labs, Ref. # PK-6200) for 45 min at 37 °C and then incubated in an avidin/biotin complex for 30 min at 37 °C. A DAB (3,3′-diaminobenzidine) HRP substrate kit (Vector SK4100) was used to detect the presence of CP3. Sections were then counterstained with hematoxylin and imaged with an EVOS FL Auto Imaging System under brightfield and a 20× objective to quantify CP3 positive cells. Histological sections of adult wild-type ovaries were used as a second control to verify a positive signal for cleaved caspase-3 (Supplementary Figure S2).
Meiotic chromosome spreads and immunofluorescence
Pairs of Chtf18+/− females were mated with Chtf18+/− males, and the presence of a copulation plug in the vaginal orifice the following morning was designated as embryonic day 0.5. Embryos were harvested at embryonic days 17.5 and 18.5 to evaluate the pachytene and diplotene stages of prophase I in fetal ovaries, respectively. Pairs of ovaries were dissected from female embryos and placed into 50-μL drops of hypotonic extraction buffer (30 mM Tris-HCl, 50 mM sucrose, 17 mM trisodium citrate dihydrate, 5 mM EDTA, 0.5 mM DTT, 0.1 mM PMSF, pH 8.2) for 30 min. Each pair of ovaries was then moved to a 20-μL drop of 100-mM sucrose, and the ovaries poked with a 27G½ inch needle to release oocytes. Another 30 μL was added to resuspend the cells and 20 μL of the suspension was placed on each of the two slides previously dipped in fresh 2% paraformaldehyde/0.15% Triton X-100. Slides were placed on a flat surface and allowed to slowly dry in a humidified chamber for 2 h, then washed twice in 0.4% Kodak Photo-Flo 200/1X PBS and once in 0.4% Kodak Photo-Flo 200/dH2O for 5 minutes each. Slides were then allowed to air dry and stored at −80 °C. Tail samples from each of the female embryos were collected for genotyping by PCR. For immunofluorescence, meiotic chromosome spreads were covered with 2–3 drops of signal enhancer solution (Life Technologies; ref 136933) followed by parafilm in a humidified chamber for 30 min. Slides were blocked in phosphate-buffered saline containing 0.2% bovine serum albumin, 0.2% fish gelatin, and 0.05% Tween-20 for 45 min at room temperature, and then incubated with primary antibodies in a humidified chamber overnight at 4 °C. The following primary antibodies were used: rabbit anti-SYCP3 antibody (Abcam, ab15093; 1:200), mouse anti-SYCP3 (Abcam, ab97672; 1:200), human CREST autoimmune serum (Immunovision, HCT-0100; 1:500), rabbit anti-RAD51 (Abcam, ab176458, 1:250), and mouse anti-MLH1 (BD Pharmingen, 550838; 1:50). After four 15-min washes in blocking solution, slides were incubated for 1 h at 37 °C with the appropriate Alexa-Fluor conjugated secondary antibody (Invitrogen) diluted 1:1000 in blocking solution. After an additional four 15-min washes in blocking solution, coverslips were mounted on slides with mounting medium and slides were imaged with fluorescence microscopy. Only foci that co-localized with SYCP3-positive chromosome axes were quantified.
Assessment of meiotic progression of oocytes
Oocytes were cultured and allowed to mature in vitro. Three–four week old female mice were each injected with 5 IU of PMSG (Pregnant Mare Serum Gonadotropin) (Calbiochem, 3672222) intraperitoneally and sacrificed 44–48 h later. Ovaries were excised and collected in minimal essential medium and then punctured with 27G½ inch needles to release antral follicles. The oocyte-cumulus cell complexes were then collected and transferred to fresh medium. Cumulus cells were mechanically removed from oocytes by pipetting, and cumulus-free oocytes were then incubated in Chatot Ziomek Bavister (CZB) medium without milrinone at 37 °C in 5% CO2 and air. The total number of oocytes per genotype was recorded, and oocytes were scored for the following stages of meiotic progression: presence of a germinal vesicle, germinal vesicle breakdown (GVBD), or extrusion of first polar body at collection time and after 18 h in culture. GVBD signifies the resumption of meiosis I, and the extrusion of a polar body signifies progression to metaphase II. We calculated the percentage progression of germinal vesicle-intact fully grown oocytes from prophase I arrest to metaphase II by calculating percentages of oocytes reaching each stage (i.e., number of oocytes with polar body extrusion [PBE] after 18 h in culture divided by the total number of GV-intact oocytes placed into culture minus the number of GV-intact oocytes after 18 h in culture for each genotype). Stages were assessed morphologically with brightfield microscopy using Nomarski optics.
Evaluation of aneuploidy in metaphase II eggs
An in situ chromosome counting assay was performed to score numbers of paired kinetochores in chromosomes of Chtf18−/− compared to wild-type eggs (as described [41]). Briefly, oocytes were obtained from the antral follicles of ovaries from 3-week-old PMSG-primed Chtf18−/− and wild-type females (as described above). Cultured metaphase II eggs were incubated in CZB containing 100-μM monastrol, a kinesin-5 inhibitor (Sigma-Aldrich, M8515), for 1 h and then fixed in 2% paraformaldehyde/0.1-mM Triton X-100. Fixed eggs were immunostained with CREST autoimmune serum (Immunovision, HCT - 0100; 1:30) to visualize kinetochores of sister chromatids and counterstained with 4′,6-diamidino-2-phenylindole (DAPI) to visualize the chromosomes. Images were obtained with an Olympus FV1000 laser scanning confocal microscope using a 100x oil immersion objective. Pairs of sister kinetochores were scored using Image J software (National Institutes of Health, Bethesda, MD) taking care to accurately count kinetochores present in more than one z-stack image.
Statistical analyses
The unpaired t-test with or without Welch’s correction, and the Mann-Whitney U test were used to calculate differences between groups using GraphPad Prism Software. A value of P < 0.05 was considered statistically significant.
Results
Chtf18 −/− females have age-dependent subfertility and marked attrition of ovarian follicle pools
Previously, we employed targeted deletion of Chtf18 and constructed a null allele to generate a classic knockout mouse model [28]. We demonstrated by immunohistochemistry, too, that the gene is deleted in both males and females [39]. Initially, we characterized the phenotype in males. Chtf18−/− males, though healthy, have impaired spermatogenesis and are subfertile. Meiotic recombination is also defective, and homologous chromosomes (homologs) separate during prophase I [28]. Given the importance of CHTF18 in male germ cell development, we investigated its role in female fertility, ovarian folliculogenesis, and meiosis. We chose to take an anatomic approach and began by evaluating fertility and ovaries of Chtf18−/− compared to wild-type females. Six Chtf18−/− or three adult wild-type (Chtf18+/+) females were bred with adult wild-type (Chtf18+/+) males over a 6-month period. Chtf18−/− females had similar total numbers of litters compared to wild-type females (mean number of 6 litters each) (Figure 1A). Although we observed no difference in litter size between wild-type and Chtf18−/− females at 5 months of age for the first five litters, we did observe a difference in size of the sixth and seventh litters. At an age of 6 months and older for each, wild-type females had a mean litter size of 11.5 offspring per litter, whereas Chtf18−/− females had a mean litter size of 6.8 offspring per litter (Figure 1B). These results are consistent with reduced fertility of Chtf18−/− females at advanced maternal age.
Figure 1.
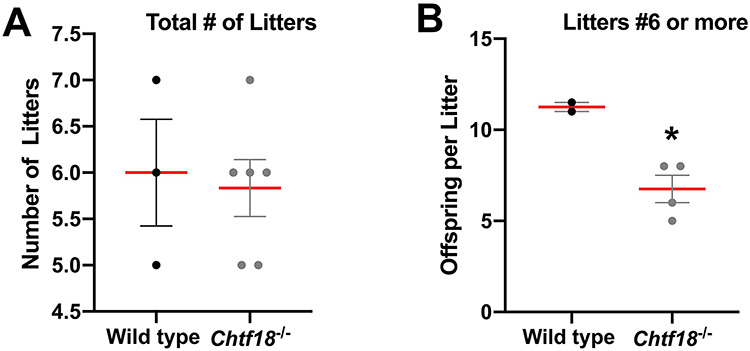
Chtf18 −/− females exhibit age-dependent subfertility. Adult wild-type and Chtf18−/− females were each bred with wild-type (Chtf18+/+) males over a 6-month period. Scatter plots represent the number of total litters (A) and the number of offspring per litter for 6 or more litters (B). Values are the mean ± SEM (n = 6 Chtf18−/− and 3 wild-type females). P = 0.017* (number of offspring per litter for 6 or more litters), unpaired t-test.
In female mice, primordial germ cells, called oogonia, develop in clusters or nests until they enter meiosis as oocytes at approximately embryonic day 13.5. Oocytes progress in meiotic prophase I and arrest at the diplotene stage. At birth, nests of oocytes separate into single oocytes that become surrounded by somatic cells to give rise to the finite pool of primordial follicles by postnatal day 7 [42–44]. Primary follicles develop from primordial follicles beginning at about 3 days postnatally and then grow into larger secondary follicles and, ultimately, into antral follicles. Although most antral follicles undergo atresia, some are recruited to grow under cyclic hormonal regulation beginning at puberty [45, 46]. To explore the role of CHTF18 in folliculogenesis and to begin to understand the physiological basis of decreased fertility in aged Chtf18−/− females, we examined ovaries morphologically. Paraffin-embedded fixed ovaries of adult Chtf18−/− and wild-type mice were sectioned and stained with H&E. Histologically, Chtf18−/− ovaries showed all stages of follicle development, but they appeared to contain fewer follicles than wild-type ovaries (Figure 2A–F). Additionally, a statistically significant greater percentage of Chtf18−/− follicles were abnormal and appeared to be degenerating compared to wild-type follicles (14.8 ± 2.5% in wild-type vs. 27.1 ± 3.5% in Chtf18−/−) (Figure 2C–G).
Figure 2.
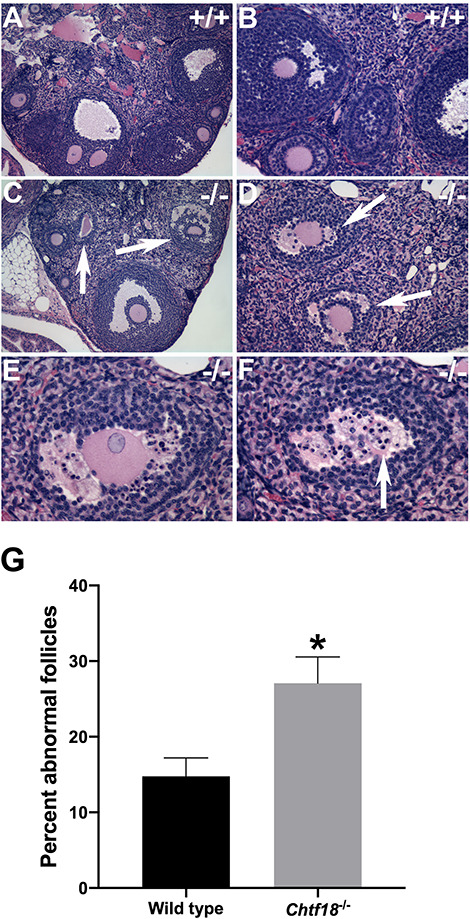
Chtf18 −/− ovaries contain fewer follicles and more follicles that appear to be degenerating. (A–F) Representative images of H&E-stained ovarian sections from adult wild-type (+/+) and Chtf18−/− (−/−) females. (C–G) Chtf18−/− ovaries contain fewer follicles and a greater percentage of abnormal follicles compared to wild-type ovaries (arrows in C, D, and F; shown in higher power view E and F). Original magnification ×100 (A and C), ×200 (B and D), ×400 (E and F). (G) Bar graph represents the percentage of abnormal follicles per ovary pair. Values are the mean ± SEM (n = 3 wild-type and 3 Chtf18−/− females). P = 0.0443*, unpaired t-test.
To better characterize the ovarian follicle defects observed in Chtf18−/− females, we performed morphometric analyses during the early and growing stages of follicle development. Follicle counting techniques were performed on paraffin-embedded, H&E-stained sections of Chtf18−/− and wild-type ovaries [40]. While primordial, primary, secondary, and antral follicles were all present, numbers of follicles at each of these stages were significantly decreased in Chtf18−/− females (Figure 3). Compared to wild-type ovaries, Chtf18−/− ovaries contained 25% fewer primordial follicles at postnatal day 7 (4086 ± 426.4 vs. 3059 ± 200.4, respectively) (Figure 3A) and 58% fewer primordial follicles at postnatal day 21 (2175 ± 298.4 vs. 916.3 ± 231.9) (Figure 3B). Notably, there were fewer primary, secondary, and antral follicles in Chtf18−/− ovaries at 3 months of age, especially primordial follicles (Figure 3C). The mean numbers in wild-type and Chtf18−/− ovary pairs, respectively, were 1427 ± 221.5 vs. 143.3 ± 18.56 primordial follicles; 310 ± 36.91 vs. 163.6 ± 28.22 primary follicles; 265.7 ± 29.68 vs. 142.9 ± 26.53 secondary follicles; and 40 ± 5.742 vs. 25 ± 6.692 antral follicles. Strikingly, by 6 months of age, there were less than half the number of follicles in each of these categories compared to wild-type ovaries (Figure 3D). The mean numbers in wild-type and Chtf18−/− ovary pairs, respectively, were 425 ± 106.9 vs. 95 ± 32.27 primordial follicles; 328 ± 43.91 vs. 117 ± 34.93 primary follicles; 247 ± 35.25 vs. 105 ± 28.22 secondary follicles; and 42 ± 10.62 vs. 15.3 ± 5.743 antral follicles. Although the initial primordial follicle pool at postnatal days 7 and 21 was decreased in Chtf18−/− compared to wild-type females, this pool was markedly decreased at 3 and 6 months of age (Figure 3A–D). We also scrutinized the decreases in the primordial follicle pools of wild-type and Chtf18−/− females at different time points. Although the decline in primordial follicles from postnatal day 7 to 6 months of age was marked in Chtf18−/− females, we were struck by the much greater attrition of primordial follicle numbers in Chtf18−/− ovaries (Figure 3E). Diminution of the growing ovarian follicles was also more significant in Chtf18−/− females at 3 and 6 months of age (Figure 3C and D). This finding is highlighted by the observation that in morphometrically analyzed ovaries, significantly more Chtf18−/− females lacked ovarian follicles. At 3 months of age, 9.5% of Chtf18−/− compared to 2.4% of wild-type females lacked antral follicles, whereas at 6 months of age, 23.3% of Chtf18−/− females lacked follicles at the primary, secondary, or antral stages of development (Supplementary Table S1). Collectively, these data indicate that follicles are almost depleted in Chtf18−/− females by 6 months of age.
Figure 3.
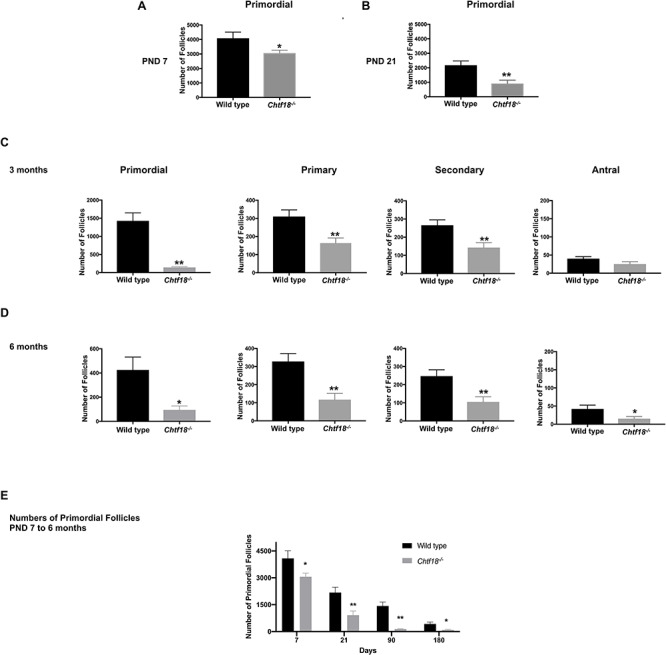
Ovarian follicle attrition is significant and pools become depleted by 6 months of age in Chtf18−/− females. Morphometric analyses were performed on sections of paraffin-embedded H&E-stained ovary pairs from Chtf18−/− and wild-type females. Values are the mean ± SEM by the unpaired t-test. (A and B) Numbers of primordial follicle numbers at postnatal day 7 (PND 7) (n = 9 mice/genotype; P = 0.0446*) and postnatal day 21 (PND 21) (n = 8 mice/genotype, P = 0.0049**). (C) Numbers of primordial, primary, secondary, and antral follicles at 3 months, n = 14 mice/genotype (primordial: P = 0.0045**; primary: P = 0.0041**; secondary: P = 0.0048**; and antral: P = 0.1008) and (D) at 6 months (n = 10 mice/genotype) (primordial: P = 0.0254*; primary: P = 0.0014**; secondary: P = 0.0056**; and antral: P = 0.0402*). (E) Bar graph represents the numbers of primordial follicles of wild-type vs. Chtf18−/− ovaries at postnatal days 7 and 21, and at 3 and 6 months of age. Values are the mean ± SEM with numbers of mice and P values stated in A–D.
Chtf18 −/− neonates have reduced ovarian reserve and more follicles with apoptotic cells
We questioned whether the dramatic reductions in the follicle pools of juvenile and adult Chtf18−/− females could be due to a smaller initial endowment of primordial follicles from which to recruit and/or increased apoptosis of the early ovarian reserve of follicles. A few days after birth, single oocytes resulting from the breakdown of germ cell cysts become enclosed in primordial follicles [44]. To evaluate the early primordial follicle pool, we examined histological sections of ovaries at postnatal day 3 and found that Chtf18−/− neonatal females had significantly fewer primordial follicles than wild-type females. Chtf18−/− ovaries contained a mean of 76.10 ± 15.61 primordial follicles per section compared to wild-type ovaries, (142.5 ± 11.59) (Figure 4A and B). To assess the possibility of increased cell death in neonatal ovaries, we scored apoptotic cells in histological sections with an antibody to cleaved caspase-3, an apoptotic cell marker. We found a more than 2-fold increase in the percentage of follicles with cleaved caspase-3 positive cells in Chtf18−/− neonatal ovaries (1.7 ± 0.17% in wild-type vs. 4.2 ± 0.84% in Chtf18−/−) (Figure 4C and D). Together, these results suggest that a diminished initial endowment and increased atresia of follicles contribute to the decreased early ovarian reserve and declining growing follicle pools of Chtf18−/− females.
Figure 4.
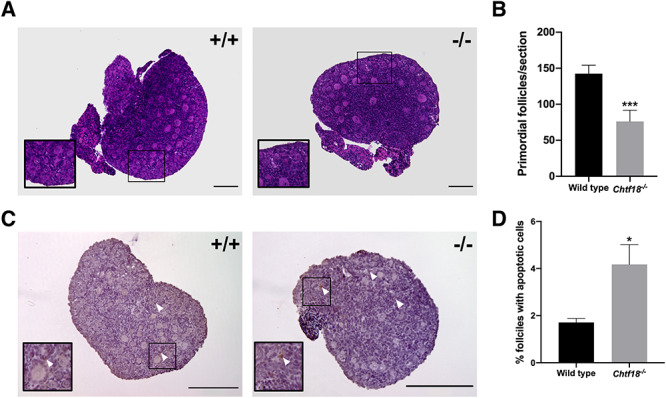
Chtf18 −/− ovaries have a decreased primordial follicle endowment and increased apoptotic cells at birth. (A) Representative images of paraffin-embedded H&E-stained sections of ovaries from wild-type and Chtf18−/− postnatal day 3 mice. Insets show primordial follicles. (B) Bar graph shows the total numbers of primordial follicles per section as the mean ± SEM; wild-type n = 13 sections (4 mice); Chtf18−/−n = 10 sections (3 mice); and P = 0.0005*** by the Mann-Whitney U test. (C) Representative images of paraffin-embedded sections of wild-type and Chtf18−/− ovaries at postnatal day 3 immunostained with cleaved caspase-3 antibody and counterstained with hematoxylin (apoptotic cells, brown). Arrowheads indicate apoptotic cells in follicles. (D) Bar graph represents the percent ovarian follicles with apoptotic cells as the mean ± SEM; wild-type n = 15 sections (5 mice); Chtf18−/−n = 12 sections (4 mice); and P = 0.0144* by the unpaired t-test. Scale bars, 200 μm.
Synapsis is complete but homologs separate prematurely in Chtf18−/− oocytes during prophase I
Given the Chtf18−/− ovarian follicle depletion we observed and a known role for CHTF18 in DSB repair during male meiosis [28], we suspected that defective meiotic recombination in Chtf18−/− females might cause ovarian follicle elimination. Therefore, we analyzed meiotic chromosome spreads prepared from fetal oocytes to evaluate the function of CHTF18 during female meiosis. In mammals, meiotic recombination commences with the formation of DSBs. During the zygotene stage of prophase I, pairing and synapsis of homologous chromosomes are facilitated by the tripartite synaptonemal complex. DSBs undergo repair by homologous recombination, and during the pachytene stage, a subset of these DSBs are resolved as crossovers (reviewed in [19, 21]). By the end of the pachytene stage, the 20 homologs have paired, consistent with complete chromosomal synapsis. Although the synaptonemal complex disassembles in the diplotene stage at the end of prophase I, homologs remain synapsed at centromeres and physically connected at sites of crossovers. Thus, during the pachytene and diplotene stages of prophase I, a count of 20 centromeres corresponds to complete synapsis.
To assess the pachytene and diplotene stages of prophase I, chromosome spreads of fetal oocytes from Chtf18−/− and wild-type females were prepared and immunostained at embryonic days 17.5 and 18.5, respectively. Anti-SYCP3 antibody and CREST autoimmune serum were used to label the central and axial/lateral elements of the synaptonemal complex and centromeres, respectively, and the numbers of CREST foci were quantified. Both Chtf18−/− and wild-type pachytene oocytes showed the expected 20 CREST foci per nucleus consistent with complete synapsis (Figure 5A). However, in diplotene Chtf18−/− oocytes, the numbers of CREST foci were significantly greater (25.04 ± 0.9322) than those of wild-type oocytes (20.39 ± 0.2221) (Figure 5B and C). Since synapsis was complete during pachynema, these findings indicate that homologs separated prematurely during the diplotene stage of prophase I in Chtf18−/− oocytes.
Figure 5.
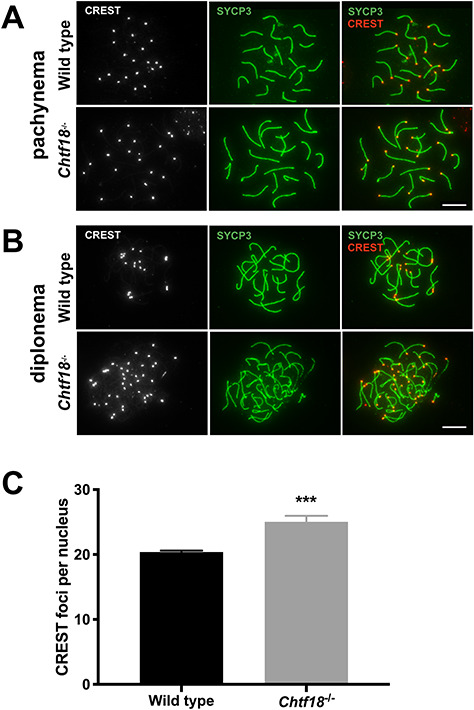
Premature homolog disjunction in Chtf18−/− oocytes during prophase I. Meiotic chromosome spreads from wild-type and Chtf18−/− oocytes were prepared and immunostained with SYCP3 antibody (synaptonemal complex, green) and CREST autoimmune serum (centromeres, gray or red). CREST foci were scored during the pachytene (A) and diplotene (B) stages of prophase I. Although Chtf18−/− and wild-type oocytes contained 20 CREST foci per nucleus during the pachytene stage (n = 10 embryos per genotype, P = 0.4), Chtf18−/− oocytes contained significantly greater foci per nucleus during the diplotene stage. Scale bars, 10 μm. (C) Bar graph depicting CREST foci per nucleus during diplonema. Values are the mean ± SEM (wild-type n = 8 embryos; Chtf18−/−n = 13 embryos; P = 0.0003***, unpaired t-test with Welch’s correction).
CHTF18 is necessary for DSB repair and meiotic progression
To investigate the cause of premature homolog disjunction and the possibility of impaired meiotic recombination, we evaluated RAD51, a single-stranded DNA binding protein that localizes to DSB sites [47, 48] and MLH1, a marker of crossovers [49, 50]. We performed immunofluorescence staining of chromosome spreads with a RAD51 antibody and found that the number of RAD51 foci was significantly greater during pachynema in Chtf18−/− oocytes (17.92 ± 1.775) compared to wild-type controls (4.28 ± 0.627) (Figure 6A and B). Since the number of RAD51 foci is known to decrease in pachynema as DSBs are repaired [51, 52], these results indicate the persistence of unrepaired DSBs in Chtf18−/− oocytes. We also scored crossovers, which are necessary to keep homologs physically joined until they segregate in anaphase I. In the process of repair during prophase I, DSBs are ultimately resolved as either crossovers or noncrossovers at the end of pachynema. Crossovers can be immunolocalized as MLH1 foci during the mid-to-late pachytene stages. MLH1 is a mismatch repair protein that marks the sites of crossovers and future chiasmata [49, 50]. To assess the completion of meiotic recombination and successful formation of crossovers, we performed immunofluorescence staining of chromosome spreads with an antibody to MLH1. Interestingly, we found significantly decreased numbers of MLH1 foci during pachynema in Chtf18−/− (20.74 ± 0.461) compared to wild-type oocytes (25.06 ± 0.473) (Figure 6C and D). In addition, whereas each homolog should have at least one crossover, the obligate crossover, some Chtf18−/− homologs completely lacked MLH1 foci. These results demonstrate that DSB repair and crossover formation are defective in Chtf18−/− oocytes and they indicate a role for CHTF18 in female meiotic recombination.
Figure 6.
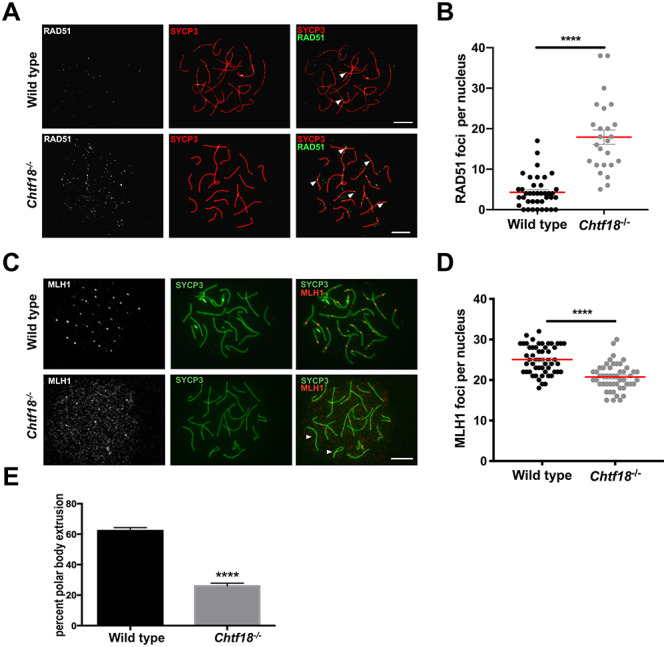
Absence of CHTF18 causes defective DSB repair and poor quality oocytes that fail to progress. (A) Meiotic chromosome spreads prepared from fetal oocytes of wild-type and Chtf18−/− embryos at E16.5 (early pachynema) were immunostained with SYCP3 antibody (synaptonemal complex, red) and RAD51 antibody (RAD51 foci, gray or green). Arrowheads indicate RAD51 foci. (B) Scatter plot depicting numbers of RAD51 foci per nucleus with the mean ± SEM; wild-type n = 39 nuclei (3 embryos); Chtf18−/−n = 25 nuclei (3 embryos); P < 0.0001**** by the unpaired t-test. (C) Meiotic chromosome spreads prepared from fetal oocytes of wild-type and Chtf18−/− embryos at E17.5 (pachynema) were immunostained with SYCP3 antibody (green) and MLH1 antibody (crossovers, gray or red). Arrowheads indicate homologs lacking MLH1 foci. Scale bars, 10 μm. (D) Scatter plot depicting numbers of MLH1 foci per nucleus (late pachytene) with the mean ± SEM; wild-type n = 54 nuclei (5 embryos); Chtf18−/−n = 50 nuclei (4 embryos); P < 0.0001**** by the unpaired t-test. (E) PBE was scored in wild-type and Chtf18−/− eggs at metaphase II. Values are percent progression as the mean ± SEM; wild-type n = 17 mice; Chtf18−/−n = 11; P < 0.0001**** by the unpaired t-test.
Because defective meiotic recombination could compromise oocyte quality and developmental competence, we next evaluated the ability of Chtf18−/− oocytes to progress following meiotic resumption. We cultured mouse oocytes in vitro and examined their progression from the germinal vesicle stage to the completion of meiosis I indicated by PBE at metaphase II. Given our low oocyte yield with hormonally primed 6 week and older females in this mouse strain, we used 3–4 week old females to improve oocyte yields [53, 54]. Although 62% of wild-type oocytes displayed a somewhat low rate of progression, in age-matched Chtf18−/− females, only 26% of oocytes reached metaphase II (Figure 6E). This marked impairment of meiotic progression strongly suggests that Chtf18−/− oocytes are poor in quality and indicates that CHTF18 is necessary for oocyte maturation following meiotic resumption.
Absence of CHTF18 causes aneuploidy and weakened centromeric cohesion in metaphase II eggs
Given our findings of early homolog disjunction, defective meiotic recombination, and impaired meiotic progression consistent with poor-quality oocytes and eggs, we suspected that Chtf18−/− eggs might be aneuploid. To assess aneuploidy, we analyzed numbers of sister chromatid pairs in metaphase II eggs. We utilized an in situ chromosome counting assay (previously described [41]) to score the numbers of paired kinetochores in chromosomes of Chtf18−/− compared to wild-type eggs. We analyzed a total of 88 wild-type and 79 Chtf18−/− eggs in 4 mice per genotype (Table 1).
Table 1.
Results of chromosome counting in Chtf18−/− and wild-type metaphase II eggs treated with monastrol.
| +/+ | −/− | |
|---|---|---|
| Euploid total | 87 | 66 |
| Aneuploid total | 1 | 12 |
| Aneuploid, ± single chromatid | 0 | 2 |
| Aneuploid, ± chromosome pair | 1 | 10 |
| Total eggs | 88 | 79 |
Chtf18 +/+, n = 4 mice; Chtf18−/−, n = 4 mice; a minimum of 17 eggs were analyzed per mouse. Values depict the numbers of metaphase II eggs per genotype.
Chromosomes were counted in metaphase II eggs treated with monastrol, a kinesin-5 inhibitor that collapses the spindle and disperses the chromosomes in situ [55]. Although most Chtf18−/− eggs were euploid and possessed the expected 20 pairs of sister kinetochores per egg (Figure 7A and Table 1), a significantly greater percentage of Chtf18−/− eggs were aneuploid compared to wild-type eggs. We found that 15.2% of Chtf18−/− eggs were aneuploid and showed the loss or gain of a single sister chromatid (Figure 7B–C and Table 1) or chromosome pair (Figure 7D and Table 1), whereas only 1.1% of wild-type eggs (a single egg of 88 total analyzed) showed the loss of a single pair of kinetochores (Table 1). Strikingly, while assessing metaphase II eggs for aneuploidy, we noted that kinetochores of sister chromatid pairs appeared to be farther apart in Chtf18−/− eggs. We measured distances between sister kinetochore pairs and found that the mean outer kinetochore distances were significantly greater in Chtf18−/− eggs (1.419 ± 0.0438 μm), compared to wild-type eggs (1.232 ± 0.0766 μm), indicating that centromeric cohesion is weakened in the absence of CHTF18 (Figure 7E–H). Together, these results indicate that CHTF18 is crucial for accurate chromosome segregation, and they suggest a role in preserving chromosome cohesion during female meiosis. Collectively, our findings also reveal that CHTF18 ensures oocyte quality and prevents aneuploidy in eggs.
Figure 7.
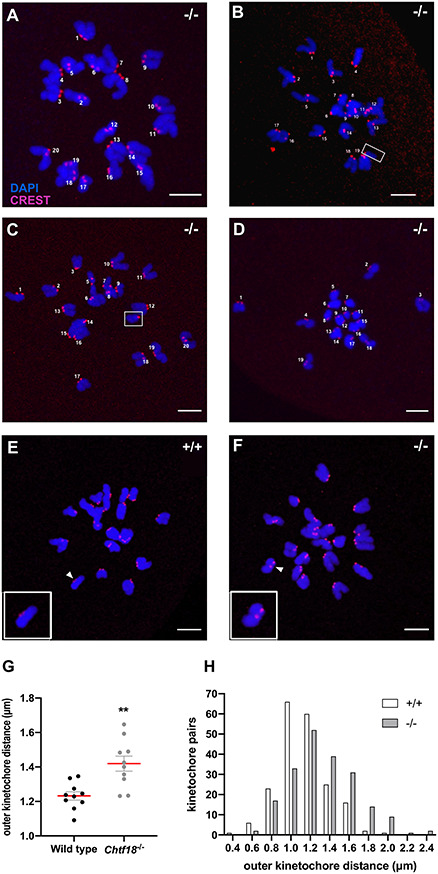
Chtf18 −/− eggs are aneuploid with weakened centromeric cohesion at metaphase II. Monastrol-treated metaphase II wild-type and Chtf18−/− eggs were immunostained with CREST autoimmune serum (kinetochores, red) and DAPI (DNA, blue), and pairs of sister chromatids were scored for aneuploidy. Representative images of the four types of Chtf18−/− (−/−) eggs observed are shown. (A) Euploid Chtf18−/− egg containing 20 pairs of sister chromatids. (B) Aneuploid Chtf18−/− egg containing 19 pairs of sister chromatids +1 single sister chromatid (shown in box). (C) Aneuploid Chtf18−/− egg containing 20 pairs of sister chromatids +1 extra single sister chromatid (shown in box). (D) Aneuploid Chtf18−/− egg lacking one chromosome pair (contains 19 pairs of sister chromatids). Scale bars, 5 μm. (E and F) Outer distances between kinetochore pairs were measured in images of monastrol-treated, metaphase II wild-type (+/+) and Chtf18−/− (−/−) eggs, and representative images are shown (eggs were stained as above). White arrowheads indicate kinetochore pairs magnified in the insets. Scale bars, 5 μm. (G) Scatter plot depicting the outer kinetochore distances for sister kinetochore pairs. Values are the mean ± SEM; wild-type, n = 10; Chtf18−/−, n = 10; and P = 0.0015** by the unpaired t-test. (H) Bar graph depicting the populations of kinetochore distances in wild-type and Chtf18−/− eggs. Wild-type, n = 2 mice; Chtf18−/−, n = 2 mice; 100 kinetochore pairs were assessed in 5 oocytes for each mouse. Distances between sister kinetochores that were not in the same plane were determined with the Pythagorean theorem.
Discussion
Errors in meiosis, including those of homologous recombination and chromosome segregation, are a common cause of aneuploidy in humans [3, 5, 22]. In particular, the frequency of these errors increases in women as they age and corresponds to decreased egg quality, a hallmark of ovarian aging. However, the molecular basis of the defects leading to these errors is incompletely understood. Thus, appropriate model organisms are needed to better elucidate the underlying causes of these defects. Here, we demonstrate that female mice lacking Chtf18 have age-dependent subfertility with oocytes that exhibit defective meiotic recombination, precocious homolog segregation, and aneuploidy. Ovarian follicle attrition is severe and significantly worse in postnatal Chtf18−/− compared to wild-type females, especially at advanced female age. These findings are consistent with the age-dependent subfertility exhibited in Chtf18−/− females. Thus, CHTF18 has vital functions in female fertility and meiosis and may play a role in delaying ovarian aging.
Previously, we revealed roles for CHTF18, the conserved DNA replication protein and subunit of CHTF18-RLC in male fertility and meiosis [28]. Absence of CHTF18 causes persistent DSBs and defective crossover formation in Chtf18−/− pachytene spermatocytes, leading to impaired spermatogenesis, oligospermia, and subfertility in males [28]. Here, we demonstrate that like Chtf18−/− males, females lacking Chtf18 are subfertile with impaired gametogenesis, as well as defective meiotic recombination and premature segregation of homologs during prophase I. Univalent chromosomes are present in Chtf18−/− oocytes during both meiosis I and II. Thus, although sexual dimorphism of meiotic phenotypes is common, it is not observed here, suggesting that CHTF18 has fundamentally important functions in mammalian germ cell development and meiosis.
It is well established that in humans, the majority of meiotic aneuploidy arises from errors in chromosome segregation during female meiosis I [4, 22, 56]. The rate of oocyte aneuploidy rises with increasing maternal age and has been demonstrated in both humans and mouse models [7, 8, 57–59]. Recent studies demonstrate that in aged oocytes, premature separation of bivalents occurs during MI in both mice and women [60, 61]. These studies suggest that early homolog separation during meiosis I is a key defect causing age-associated aneuploidy in oocytes. Consistent with these studies, we show that in Chtf18−/− oocytes, premature separation of homologs leads to aneuploidy. Chtf18−/− oocytes also possess fewer crossovers, and some homologs completely lack crossovers. In fact, decreased recombination events, namely reduced crossovers, have also been strongly associated with aneuploidy in humans [9, 22, 62].
Deterioration of chromosomal cohesion is another important cause of female age-related aneuploidy [63–65], and loss of cohesins, the multiprotein complexes that mediate cohesion, have been strongly implicated [9, 66]. Cohesion deterioration has also been shown to occur prior to chromosome missegregation and likely in conjunction the destabilization of chiasmata [16, 67, 68]. Removal of cohesin and resolution of chiasmata are both necessary for homolog disjunction during meiosis I [14, 15, 69, 70]. Thus, it is biologically plausible that in Chtf18−/− oocytes, impaired cohesion causes defective recombination and crossover formation, leading to premature separation of homologs during prophase I. In addition, our findings of increased distances between sister kinetochores in Chtf18−/− eggs during metaphase II suggest weakened centromeric cohesion. This possibility is consistent with prior studies, suggesting a role for CHTF18 in meiotic cohesion during male meiosis [28], as well as studies in yeast and human cells [33, 37, 38, 71, 72]. Collectively, the findings of early homolog separation in diplonema of prophase I and aneuploidy in metaphase II support a function for CHTF18 in chromosome segregation and cohesion during meiosis. Based on the known function of CHTF18 as a clamp loader of PCNA during mitosis [33–35], it is interesting to postulate that premature homolog disjunction in Chtf18−/− meiocytes might be due to improper loading or maintenance of cohesins during premeiotic S phase or meiosis, respectively. Future investigation will explore in depth a role for CHTF18 in meiotic chromosome cohesion.
Defective meiotic recombination in Chtf18−/− oocytes results in persistent DSBs. Previously, we showed that defective homologous recombination and crossover formation in Chtf18−/− spermatocytes leads to DSBs that persist well into the diplotene substage of prophase I [28]. Impaired crossover formation and subsequent failure of Chtf18−/− oocytes to progress following meiotic resumption are consistent with DSBs continuing beyond prophase I and into the late stages of meiosis. Although it is somewhat surprising that a DSB repair defect in Chtf18−/− oocytes does not lead to pachytene arrest and elimination of oocytes prior to diplonema, this finding is consistent with the male Chtf18−/− phenotype [28]. Unrepaired DSBs in poor-quality Chtf18−/− oocytes may activate the spindle assembly checkpoint (SAC) resulting in failure to mature following meiotic resumption. Recent studies support a role for the SAC in preventing meiotic progression of oocytes with excessive DNA damage [73–75]. Studies from several groups also suggest that the persistence of DSBs leads to ovarian follicle loss or depletion and ovarian aging in mice [23–25, 27, 29, 76]. A role for DSB repair in mouse and human oocyte survival has also been demonstrated [76, 77]. CHTF18 may influence the rate of elimination and/or survival of oocytes with excessive unrepaired DSBs. However, the reduced early ovarian reserve likely reflects fewer follicles available for activation, thus impacting the pool of growing follicles at 3 and 6 months, too. These effects are likely due, in part, to the absence of CHTF18 early in gametogenesis. CHTF18 is expressed throughout female and male germ cell development from embryonic day 13.5 through 15.5, as well as in somatic cells [39]. Consistent with findings of decreased early ovarian reserve in Chtf18−/− females, the number of early prospermatogonia, a mitotically proliferating population, is markedly decreased in Chtf18−/− neonatal seminiferous tubules compared to wild-type controls [28]. Persistent DSBs that arise during prophase I, as indicated by increased numbers of RAD51 foci in Chtf18−/− oocytes, may activate the CHK2 DNA damage checkpoint leading to postnatal oocyte elimination as reported in Chk2−/− and Mre11ALTD1/ALTD1 females [23, 27, 29]. Increased apoptosis observed in Chtf18−/− neonatal ovaries is consistent with early postnatal elimination. Although a certain threshold of DSBs may need to be exceeded to activate checkpoint elimination, our results are somewhat reminiscent of Mre11ALTD1/ALTD1 females that exhibit persistent DSBs and elimination of follicles postnatally [29]. However, unlike Mre11ALTD1/ALTD1 mice, Chtf18−/− females do not exhibit defects in homolog pairing, synapsis, or complete elimination of ovarian follicles postnatally. In fact, the deficit of primordial follicles in Chtf18−/− compared to wild-type females at P3, P7, and P21 is consistent with persistent DSB-induced follicle elimination [24]. In addition, we note that the loss of ovarian follicles becomes most significant in females of advanced age, and by 6 months, Chtf18−/− ovaries possess fewer than half the numbers of primary, secondary, and antral follicles compared to wild-type females. These findings are consistent with the age-dependent subfertility observed in Chtf18−/− females and resemble those of advancing female age, in which both the quantity and quality of oocytes in the follicle pool diminishes, especially when women reach the age of 35 and beyond [10, 78]. Interestingly, numbers of primordial follicles are already significantly decreased in Chtf18−/− at postnatal days 7 and 21 compared to wild-type ovaries. In addition, more Chtf18−/− females have ovaries completely lacking primary, secondary, or antral follicles at 3 and 6 months of age compared to wild-type females (Supplemental Table S1). Strikingly, by 6 months of age, 7 of 10 (23.3%) Chtf18−/− females completely lack ovarian follicles at the primary, secondary, or antral stages. Taken together, these data suggest that CHTF18 aids in maintaining the early and developing ovarian follicle pools and prevents severe attrition. The initial endowment of germ cells and primordial follicles is decreased early on and likely contributes to the shortfall seen in Chtf18−/− growing follicles. Female flies with a loss of function mutation in Cutlet, the Drosophila melanogaster ortholog of Chtf18, exhibit cessation of germline stem cell proliferation during the mitotic stages of amplification [79]. Accordingly, impaired establishment or maintenance of the early germ cell pools could contribute to the decreased numbers of follicles seen in Chtf18−/− ovaries. Early homolog disjunction, defective meiotic recombination, and ovarian follicle loss may also lead to poor oocyte quality and ultimately aneuploidy in Chtf18−/− females. Absence of CHTF18 in mitotic cells could also contribute to defects in Chtf18−/− mice. In fact, the partial embryonic lethality of Chtf18−/− mice seen in Chtf18+/− intercross matings [28] may be due to defects in somatic tissues during early fetal development. However, the loss of CHTF18 function in meiotic cells appears to have a more significant impact on the Chtf18−/− phenotype of surviving offspring.
Thus, CHTF18 has crucial functions in female meiosis and oocyte development and may play a role in delaying ovarian aging. In particular, our data suggest that CHTF18 maintains the early and developing ovarian follicle pools and prevents premature chromosome segregation and ultimately aneuploidy. The phenotype of age-dependent subfertility and reduced oocyte quantity and quality in Chtf18−/− females closely resembles that of women with diminished ovarian reserve. Therefore, Chtf18−/− mice are a physiologically relevant model to elucidate mechanisms of ovarian aging in women. Future studies will address the molecular basis of ovarian follicle defects and aneuploidy in Chtf18−/− female mice.
Supplementary Material
{kind=link}
Acknowledgments
We are grateful to Karen Schindler for technical assistance and valuable input. We also thank Dr Richard Huneke, Andrea McCurry, and all the staff of Drexel University Laboratory Animal Resources (ULAR), as well as graduate students in the Master of Laboratory Animal Science program at Drexel University for outstanding animal husbandry and care provided.
Footnotes
† Grant Support: This work was supported by the Pennsylvania Department of Health Tobacco Settlement Formula Funds, the DeWitt Pettit Award, the American Society for Reproductive Medicine Research Grant, and the National Institutes of Health (NIH) R01 GM106262 grant to K.M.B.
References
- 1. Warburton D. Human female meiosis: new insights into an error-prone process. Am J Hum Genet 1997; 61:1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hunt PA, Hassold TJ. Sex matters in meiosis. Science 2002; 296:2181–2183. [DOI] [PubMed] [Google Scholar]
- 3. Hassold T, Hall H, Hunt P. The origin of human aneuploidy: where we have been, where we are going. Hum Mol Genet 2007; 16:R203–R208. [DOI] [PubMed] [Google Scholar]
- 4. Hall HE, Surti U, Hoffner L, Shirley S, Feingold E, Hassold T. The origin of trisomy 22: evidence for acrocentric chromosome-specific patterns of nondisjunction. Am J Med Genet A 2007; 143A:2249–2255. [DOI] [PubMed] [Google Scholar]
- 5. Lamb NE, Sherman SL, Hassold TJ. Effect of meiotic recombination on the production of aneuploid gametes in humans. Cytogenet Genome Res 2005; 111:250–255. [DOI] [PubMed] [Google Scholar]
- 6. Sauer MV. The impact of age on reproductive potential: Lessons learned from oocyte donation. Maturitas 1998; 30:221–225. [DOI] [PubMed] [Google Scholar]
- 7. Fragouli E, Alfarawati S, Goodall NN, Sanchez-Garcia JF, Colls P, Wells D. The cytogenetics of polar bodies: Insights into female meiosis and the diagnosis of aneuploidy. Mol Hum Reprod 2011; 17:286–295. [DOI] [PubMed] [Google Scholar]
- 8. Kuliev A, Zlatopolsky Z, Kirillova I, Spivakova J, Cieslak JJ. Meiosis errors in over 20,000 oocytes studied in the practice of preimplantation aneuploidy testing. Reprod Biomed Online 2011; 22:2–8. [DOI] [PubMed] [Google Scholar]
- 9. Chiang T, Schultz RM, Lampson MA. Meiotic origins of maternal age-related aneuploidy. Biol Reprod 2012; 86:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Practice Committee of the American Society for Reproductive Medicine Testing and interpreting measures of ovarian reserve: a committee opinion. Fertil Steril 2015; 103:e9–e17. [DOI] [PubMed] [Google Scholar]
- 11. Baudat F, Manova K, Yuen JP, Jasin M, Keeney S. Chromosome synapsis defects and sexually dimorphic meiotic progression in mice lacking Spo11. Mol Cell 2000; 6:989–998. [DOI] [PubMed] [Google Scholar]
- 12. Keeney S, Giroux CN, Kleckner N. Meiosis-specific DNA double-strand breaks are catalyzed by Spo11, a member of a widely conserved protein family. Cell 1997; 88:375–384. [DOI] [PubMed] [Google Scholar]
- 13. Romanienko PJ, Camerini-Otero RD. The mouse Spo11 gene is required for meiotic chromosome synapsis. Mol Cell 2000; 6:975–987. [DOI] [PubMed] [Google Scholar]
- 14. Buonomo SB, Clyne RK, Fuchs J, Loidl J, Uhlmann F, Nasmyth K. Disjunction of homologous chromosomes in meiosis I depends on proteolytic cleavage of the meiotic cohesin Rec8 by separin. Cell 2000; 103:387–398. [DOI] [PubMed] [Google Scholar]
- 15. Lee J, Iwai T, Yokota T, Yamashita M. Temporally and spatially selective loss of Rec8 protein from meiotic chromosomes during mammalian meiosis. J Cell Sci 2003; 116:2781–2790. [DOI] [PubMed] [Google Scholar]
- 16. Revenkova E, Eijpe M, Heyting C, Hodges CA, Hunt PA, Liebe B, Scherthan H, Jessberger R. Cohesin SMC1 beta is required for meiotic chromosome dynamics, sister chromatid cohesion and DNA recombination. Nat Cell Biol 2004; 6:555–562. [DOI] [PubMed] [Google Scholar]
- 17. Xu H, Beasley MD, Warren WD, Horst GT, McKay MJ. Absence of mouse REC8 cohesin promotes synapsis of sister chromatids in meiosis. Dev Cell 2005; 8:949–961. [DOI] [PubMed] [Google Scholar]
- 18. Brooker AS, Berkowitz KM. The roles of cohesins in mitosis, meiosis, and human health and disease. Methods Mol Biol 2014; 1170:229–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Handel MA, Schimenti JC. Genetics of mammalian meiosis: regulation, dynamics and impact on fertility. Nat Rev Genet 2010; 11:124–136. [DOI] [PubMed] [Google Scholar]
- 20. Suja JA, Barbero JL. Cohesin complexes and sister chromatid cohesion in mammalian meiosis. Genome Dyn 2009; 5:94–116. [DOI] [PubMed] [Google Scholar]
- 21. Gray S, Cohen PE. Control of meiotic crossovers: from double-strand break formation to designation. Annu Rev Genet 2016; 50:175–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hassold T, Hunt P. To err (meiotically) is human: the genesis of human aneuploidy. Nat Rev Genet 2001; 2:280–291. [DOI] [PubMed] [Google Scholar]
- 23. Bolcun-Filas E, Rinaldi VD, White ME, Schimenti JC. Reversal of female infertility by Chk2 ablation reveals the oocyte DNA damage checkpoint pathway. Science 2014; 343:533–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Di Giacomo M, Barchi M, Baudat F, Edelmann W, Keeney S, Jasin M. Distinct DNA-damage-dependent and -independent responses drive the loss of oocytes in recombination-defective mouse mutants. Proc Natl Acad Sci U S A 2005; 102:737–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li XC, Schimenti JC. Mouse pachytene checkpoint 2 (trip13) is required for completing meiotic recombination but not synapsis. PLoS Genet 2007; 3:e130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Qiao H, Rao H, Yun Y, Sandhu S, Fong JH, Sapre M, Nguyen M, Tham A, Van BW, Chng TYH, Lee A, Hunter N. Impeding DNA break repair enables oocyte quality control. Mol Cell 2018; 72:211–221e213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rinaldi VD, Bolcun-Filas E, Kogo H, Kurahashi H, Schimenti JC. The DNA damage checkpoint eliminates mouse oocytes with chromosome synapsis failure. Mol Cell 2017; 67:1026–1036e1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Berkowitz KM, Sowash AR, Koenig LR, Urcuyo D, Khan F, Yang F, Wang PJ, Jongens TA, Kaestner KH. Disruption of CHTF18 causes defective meiotic recombination in male mice. PLoS Genet 2012; 8:e1002996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Inagaki A, Roset R, Petrini JH. Functions of the MRE11 complex in the development and maintenance of oocytes. Chromosoma 2016; 125:151–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lutzmann M, Grey C, Traver S, Ganier O, Maya-Mendoza A, Ranisavljevic N, Bernex F, Nishiyama A, Montel N, Gavois E, Forichon L, Massy B et al. MCM8- and MCM9-deficient mice reveal gametogenesis defects and genome instability due to impaired homologous recombination. Mol Cell 2012; 47:523–534. [DOI] [PubMed] [Google Scholar]
- 31. Lyndaker AM, Lim PX, Mleczko JM, Diggins CE, Holloway JK, Holmes RJ, Kan R, Schlafer DH, Freire R, Cohen PE, Weiss RS. Conditional inactivation of the DNA damage response gene Hus1 in mouse testis reveals separable roles for components of the RAD9-RAD1-HUS1 complex in meiotic chromosome maintenance. PLoS Genet 2013; 9:e1003320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. McNairn AJ, Rinaldi VD, Schimenti JC. Repair of meiotic DNA breaks and homolog pairing in mouse meiosis requires a minichromosome maintenance (MCM) paralog. Genetics 2017; 205:529–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Merkle CJ, Karnitz LM, Henry-Sanchez JT, Chen J. Cloning and characterization of hCTF18, hCTF8, and hDCC1. Human homologs of a Saccharomyces cerevisiae complex involved in sister chromatid cohesion establishment. J Biol Chem 2003; 278:30051–30056. [DOI] [PubMed] [Google Scholar]
- 34. Ohta S, Shiomi Y, Sugimoto K, Obuse C, Tsurimoto T. A proteomics approach to identify proliferating cell nuclear antigen (PCNA)-binding proteins in human cell lysates. Identification of the human CHL12/RFCs2-5 complex as a novel PCNA-binding protein. J Biol Chem 2002; 277:40362–40367. [DOI] [PubMed] [Google Scholar]
- 35. Shiomi Y, Shinozaki A, Sugimoto K, Usukura J, Obuse C, Tsurimoto T. The reconstituted human Chl12-RFC complex functions as a second PCNA loader. Genes Cells 2004; 9:279–290. [DOI] [PubMed] [Google Scholar]
- 36. Ansbach AB, Noguchi C, Klansek IW, Heidlebaugh M, Nakamura TM, Noguchi E. RFCCtf18 and the Swi1-Swi3 complex function in separate and redundant pathways required for the stabilization of replication forks to facilitate sister chromatid cohesion in Schizosaccharomyces pombe. Mol Biol Cell 2008; 19:595–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lengronne A, McIntyre J, Katou Y, Kanoh Y, Hopfner KP, Shirahige K, Uhlmann F. Establishment of sister chromatid cohesion at the S. cerevisiae replication fork. Mol Cell 2006; 23:787–799. [DOI] [PubMed] [Google Scholar]
- 38. Terret ME, Sherwood R, Rahman S, Qin J, Jallepalli PV. Cohesin acetylation speeds the replication fork. Nature 2009; 462:231–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Berkowitz KM, Kaestner KH, Jongens TA. Germline expression of mammalian CTF18, an evolutionarily conserved protein required for germ cell proliferation in the fly and sister chromatid cohesion in yeast. Mol Hum Reprod 2008; 14:143–150. [DOI] [PubMed] [Google Scholar]
- 40. Sharan SK, Pyle A, Coppola V, Babus J, Swaminathan S, Benedict J, Swing D, Martin BK, Tessarollo L, Evans JP, Flaws JA, Handel MA. BRCA2 deficiency in mice leads to meiotic impairment and infertility. Development 2004; 131:131–142. [DOI] [PubMed] [Google Scholar]
- 41. Duncan FE, Chiang T, Schultz RM, Lampson MA. Evidence that a defective spindle assembly checkpoint is not the primary cause of maternal age-associated aneuploidy in mouse eggs. Biol Reprod 2009; 81:768–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Choi Y, Rajkovic A. Genetics of early mammalian folliculogenesis. Cell Mol Life Sci 2006; 63:579–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pepling ME, Spradling AC. Female mouse germ cells form synchronously dividing cysts. Development 1998; 125:3323–3328. [DOI] [PubMed] [Google Scholar]
- 44. Pepling ME, Spradling AC. Mouse ovarian germ cell cysts undergo programmed breakdown to form primordial follicles. Dev Biol 2001; 234:339–351. [DOI] [PubMed] [Google Scholar]
- 45. McGee EA, Hsueh AJ. Initial and cyclic recruitment of ovarian follicles. Endocr Rev 2000; 21:200–214. [DOI] [PubMed] [Google Scholar]
- 46. Adhikari D, Liu K. Molecular mechanisms underlying the activation of mammalian primordial follicles. Endocr Rev 2009; 30:438–464. [DOI] [PubMed] [Google Scholar]
- 47. Shinohara A, Ogawa H, Ogawa T. Rad51 protein involved in repair and recombination in S. cerevisiae is a RecA-like protein. Cell 1992; 69:457–470. [DOI] [PubMed] [Google Scholar]
- 48. Shinohara A, Ogawa H, Matsuda Y, Ushio N, Ikeo K, Ogawa T. Cloning of human, mouse and fission yeast recombination genes homologous to RAD51 and recA. Nat Genet 1993; 4:239–243. [DOI] [PubMed] [Google Scholar]
- 49. Baker SM, Plug AW, Prolla TA, Bronner CE, Harris AC, Yao X, Christie DM, Monell C, Arnheim N, Bradley A, Ashley T, Liskay RM. Involvement of mouse Mlh1 in DNA mismatch repair and meiotic crossing over. Nat Genet 1996; 13:336–342. [DOI] [PubMed] [Google Scholar]
- 50. Edelmann W, Cohen PE, Kane M, Lau K, Morrow B, Bennett S, Umar A, Kunkel T, Cattoretti G, Chaganti R, Pollard JW, Kolodner RD et al. Meiotic pachytene arrest in MLH1-deficient mice. Cell 1996; 85:1125–1134. [DOI] [PubMed] [Google Scholar]
- 51. Ashley T, Plug AW, Xu J, Solari AJ, Reddy G, Golub EI, Ward DC. Dynamic changes in Rad51 distribution on chromatin during meiosis in male and female vertebrates. Chromosoma 1995; 104:19–28. [DOI] [PubMed] [Google Scholar]
- 52. Plug AW, Peters AH, Keegan KS, Hoekstra MF, Boer P, Ashley T. Changes in protein composition of meiotic nodules during mammalian meiosis. J Cell Sci 1998; 111:413–423. [DOI] [PubMed] [Google Scholar]
- 53. Gates AH, Bozarth JL. Ovulation in the PMSG-treated immature mouse: effect of dose, age, weight, puberty, season and strain (BALB/c, 129 and C129F1 hybrid). Biol Reprod 1978; 18:497–505. [DOI] [PubMed] [Google Scholar]
- 54. Hoogenkamp H, Lewing P. Superovulation in mice in relation to their age. Vet Q 1982; 4:47–4844. [DOI] [PubMed] [Google Scholar]
- 55. Mayer TU, Kapoor TM, Haggarty SJ, King RW, Schreiber SL, Mitchison TJ. Small molecule inhibitor of mitotic spindle bipolarity identified in a phenotype-based screen. Science 1999; 286:971–974. [DOI] [PubMed] [Google Scholar]
- 56. Handyside AH. Molecular origin of female meiotic aneuploidies. Biochim Biophys Acta 2012; 1822:1913–1920. [DOI] [PubMed] [Google Scholar]
- 57. Camlin NJ, McLaughlin EA, Holt JE. The use of C57Bl/6 x CBA F1 hybrid cross as a model for human age-related oocyte aneuploidy. Mol Reprod Dev 2017; 84:6–7. [DOI] [PubMed] [Google Scholar]
- 58. Merriman JA, Jennings PC, McLaughlin EA, Jones KT. Effect of aging on superovulation efficiency, aneuploidy rates, and sister chromatid cohesion in mice aged up to 15 months. Biol Reprod 2012; 86:49. [DOI] [PubMed] [Google Scholar]
- 59. Nagaoka SI, Hassold TJ, Hunt PA. Human aneuploidy: mechanisms and new insights into an age-old problem. Nat Rev Genet 2012; 13:493–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sakakibara Y, Hashimoto S, Nakaoka Y, Kouznetsova A, Hoog C, Kitajima TS. Bivalent separation into univalents precedes age-related meiosis I errors in oocytes. Nat Commun 2015; 6:7550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zielinska AP, Holubcova Z, Blayney M, Elder K, Schuh M. Sister kinetochore splitting and precocious disintegration of bivalents could explain the maternal age effect. Elife 2015; 4:e11389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wang S, Hassold T, Hunt P, White MA, Zickler D, Kleckner N, Zhang L. Inefficient crossover maturation underlies elevated aneuploidy in human female meiosis. Cell 2017; 168:977–989e917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Chiang T, Duncan FE, Schindler K, Schultz RM, Lampson MA. Evidence that weakened centromere cohesion is a leading cause of age-related aneuploidy in oocytes. Curr Biol 2010; 20:1522–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Chiang T, Schultz RM, Lampson MA. Age-dependent susceptibility of chromosome cohesion to premature separase activation in mouse oocytes. Biol Reprod 2011; 85:1279–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Duncan FE, Hornick JE, Lampson MA, Schultz RM, Shea LD, Woodruff TK. Chromosome cohesion decreases in human eggs with advanced maternal age. Aging Cell 2012; 11:1121–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Jessberger R. Age-related aneuploidy through cohesion exhaustion. EMBO Rep 2012; 13:539–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hodges CA, Revenkova E, Jessberger R, Hassold TJ, Hunt PA. SMC1beta-deficient female mice provide evidence that cohesins are a missing link in age-related nondisjunction. Nat Genet 2005; 37:1351–1355. [DOI] [PubMed] [Google Scholar]
- 68. Lister LM, Kouznetsova A, Hyslop LA, Kalleas D, Pace SL, Barel JC, Nathan A, Floros V, Adelfalk C, Watanabe Y, Jessberger R, Kirkwood TB et al. Age-related meiotic segregation errors in mammalian oocytes are preceded by depletion of cohesin and Sgo2. Curr Biol 2010; 20:1511–1521. [DOI] [PubMed] [Google Scholar]
- 69. Lee J, Okada K, Ogushi S, Miyano T, Miyake M, Yamashita M. Loss of Rec8 from chromosome arm and centromere region is required for homologous chromosome separation and sister chromatid separation, respectively, in mammalian meiosis. Cell Cycle 2006; 5:1448–1455. [DOI] [PubMed] [Google Scholar]
- 70. Kudo NR, Wassmann K, Anger M, Schuh M, Wirth KG, Xu H, Helmhart W, Kudo H, McKay M, Maro B, Ellenberg J, Boer P et al. Resolution of chiasmata in oocytes requires separase-mediated proteolysis. Cell 2006; 126:135–146. [DOI] [PubMed] [Google Scholar]
- 71. Bylund GO, Burgers PM. Replication protein A-directed unloading of PCNA by the Ctf18 cohesion establishment complex. Mol Cell Biol 2005; 25:5445–5455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Bermudez VP, Maniwa Y, Tappin I, Ozato K, Yokomori K, Hurwitz J. The alternative Ctf18-Dcc1-Ctf8-replication factor C complex required for sister chromatid cohesion loads proliferating cell nuclear antigen onto DNA. Proc Natl Acad Sci U S A 2003; 100:10237–10242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Collins JK, Lane SIR, Merriman JA, Jones KT. DNA damage induces a meiotic arrest in mouse oocytes mediated by the spindle assembly checkpoint. Nat Commun 2015; 6:8553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Greaney J, Wei Z, Homer H. Regulation of chromosome segregation in oocytes and the cellular basis for female meiotic errors. Hum Reprod Update 2018; 24:135–161.. [DOI] [PubMed] [Google Scholar]
- 75. Marangos P, Stevense M, Niaka K, Lagoudaki M, Nabti I, Jessberger R, Carroll J. DNA damage-induced metaphase I arrest is mediated by the spindle assembly checkpoint and maternal age. Nat Commun 2015; 6:8706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Titus S, Li F, Stobezki R, Akula K, Unsal E, Jeong K, Dickler M, Robson M, Moy F, Goswami S, Oktay K. Impairment of BRCA1-related DNA double-strand break repair leads to ovarian aging in mice and humans. Sci Transl Med 2013; 5:172ra121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Oktay K, Turan V, Titus S, Stobezki R, Liu L. BRCA mutations, DNA repair deficiency, and ovarian aging. Biol Reprod 2015; 93:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Hansen KR, Knowlton NS, Thyer AC, Charleston JS, Soules MR, Klein NA. A new model of reproductive aging: The decline in ovarian non-growing follicle number from birth to menopause. Hum Reprod 2008; 23:699–708. [DOI] [PubMed] [Google Scholar]
- 79. Jaffe AB, Jongens TA. Structure-specific abnormalities associated with mutations in a DNA replication accessory factor in drosophila. Dev Biol 2001; 230:161–176. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.