

Abstract
A key feature of alcohol use disorder (AUD) is negative affect during withdrawal, which often contributes to relapse and is thought to be caused by altered brain function, especially in circuits that are important mediators of emotional behaviors. Both the agranular insular cortex (AIC) and the basolateral amygdala (BLA) regulate emotions and are sensitive to ethanol-induced changes in synaptic plasticity. The AIC and BLA are reciprocally connected; and the effects of chronic ethanol exposure on this circuit have yet to be explored. Here, we use a combination of optogenetics and electrophysiology to examine the pre- and postsynaptic changes that occur to AIC-BLA synapses following withdrawal from 7- or 10-days of chronic intermittent ethanol (CIE) exposure. While CIE/withdrawal did not alter presynaptic glutamate release probability from AIC inputs, withdrawal from 10, but not 7, days of CIE increased AMPA receptor-mediated postsynaptic function at these synapses. Additionally, NMDA receptor-mediated currents evoked by electrical stimulation of the external capsule, which contains AIC afferents, were also increased during withdrawal. Notably, a single subanesthetic dose of ketamine administered at the onset of withdrawal prevented the withdrawal-induced increases in both AMPAR and NMDAR postsynaptic function. Ketamine also prevented the withdrawal-induced increases in anxiety-like behavior measured using the elevated zero maze. Together, these findings suggest that chronic ethanol exposure increases postsynaptic function within the AIC-BLA circuit and that ketamine can prevent ethanol withdrawal-induced alterations in synaptic plasticity and negative affect.
1. Introduction
Alcohol use disorder (AUD) is characterized by prolonged and excessive alcohol consumption. This type of chronic alcohol exposure produces neuroadaptations in brain circuits that regulate of emotional states (Gilpin & Koob 2008, McCool 2011, Valenzuela 1997). In humans, withdrawal in alcohol-dependent patients significantly increases brain glutamate levels and significantly lowers brain GABA concentrations relative to healthy controls (Hermann et al. 2012, Tsai et al. 1998). Similarly, in rodent models of ethanol dependence, withdrawal increases glutamatergic synaptic function in the basolateral amygdala (BLA) which can contribute to increased anxiety-like behavior (Lack et al. 2007, McGinnis et al. 2019). The BLA serves as the primary input nuclei in the amygdala’s emotional-related neural circuitry (Sah et al. 2003), receiving highly processed sensory information from cortical afferents arriving via the external capsule (EC) (Rainnie et al. 1991). We have previously reported using electrical stimulation that EC glutamatergic inputs onto BLA principal neurons undergo predominantly postsynaptic alterations characterized by increased AMPA receptor function following withdrawal (WD) from chronic intermittent ethanol (CIE) (Christian et al. 2012, Morales et al. 2018). However, EC afferents onto BLA neurons potentially arise from many different cortical regions; and, it is unclear if CIE/WD-induced postsynaptic changes can be localized to individual circuits.
The agranular insula cortex (AIC), a subdivision of the polymodal association cortex located on the ventrolateral surface of the rostral cerebral cortex, is reciprocally connected with the BLA via the EC (Allen et al. 1991, Matyas et al. 2014, McDonald & Mascagni 1996). In addition to sensory inputs, the AIC integrates affective, anticipatory, and reward-related information arising from limbic regions (Maffei et al. 2012). Recent functional imaging studies in humans highlight the insula across many psychiatric and neurological disorders (Gogolla 2017). A large body of evidence indicates that the AIC helps mediate fear and anxiety as well as addiction-related behaviors. For example, pharmacological inactivation of the AIC decreases both operant responding for alcohol and alcohol consumption (Pushparaj & Le Foll 2015).
Optogenetic inhibition of glutamatergic AIC inputs into the nucleus accumbens (NAc) reduces quinine-resistant alcohol intake, suggesting this pathway sustains aversion-resistant alcohol intake (Seif et al. 2013). Additionally, chemogenetic silencing of these AIC-NAc projections decreases alcohol intake in rats trained to self-administer alcohol (Jaramillo et al. 2018). Despite the mounting evidence that that AIC circuits directly modulate AUD-related behaviors, very few studies have examined potential modulation of these circuits by ethanol itself. Notably, pharmacologically relevant concentrations of ethanol inhibit both AIC N-methyl-D-aspartate receptor (NMDAR)-mediated excitatory postsynaptic currents (EPSCs) and the induction of long-term depression (Shillinglaw et al. 2018). However, the effects of chronic ethanol exposure on downstream AIC projections, such as the AIC-BLA terminals, are yet to be examined.
Since AIC inputs arrive at the BLA via the EC and previous work shows that EC-BLA synapses undergo postsynaptic changes following withdrawal from chronic ethanol exposure, we hypothesized that AIC-BLA synapses would express a similar form of ethanol-induced plasticity. To test this hypothesis, we exposed rats to chronic, intermittent ethanol (CIE) using ethanol vapor chambers, a commonly used method for inducing dependence in rodent models of AUD (Gilpin et al. 2008). We have previously reported that varying durations of this type of ethanol exposure produce behavioral alterations indicative of a dependence-like phenotype including increased voluntary ethanol-self administration and increased anxiety-like behavior during withdrawal (Morales et al. 2015, Morales et al. 2018). Using a combination of electrophysiology and optically-evoked glutamatergic responses from AIC terminals in the BLA, we characterized the presynaptic and postsynaptic function of AIC-BLA synapses in CIE- and air-exposed animals.
2. Materials and Methods
2.1. Animals
Male Sprague-Dawley rats (100g) were purchased from Envigo (Indianapolis, IN) and given access to food and water ad libitum upon arrival. Rats were pair-housed in a humidity and temperature-controlled room and maintained on a reverse 12:12h light-dark cycle (lights off at 9 AM). At ∼5 weeks of age, rats underwent surgery (N=96); and rats that did not undergo surgery (N=32) were aged ∼8 weeks (250g) at arrival. All rats were aged ∼10 weeks (300g) at the time of behavioral measures and electrophysiology recordings. All animal care procedures were in accordance with the NIH Guide for the Care and Use of Laboratory Animals and were approved in advance by the Institutional Animal Care and Use Committee at Wake Forest University Health Sciences.
2.2. Stereotaxic Surgery
Rats were kept under continuous isoflurane anesthesia (3–5% induction, 1–3% maintenance, 1L/min oxygen) throughout the surgery as previously described (McGinnis et al. 2019). An adeno-associated viral vector containing Channelrhodopsin (AAV5-CamKIIα-hChR2(H134R)-EYFP; UNC Vector Core, Chapel Hill, NC) was bilaterally microinjected (1μL/side, 0.1μL/min over 10min) into the agranular insular cortex (AIC) using a Neurostar StereoDrive (Germany) with the following coordinates (in mm, relative to bregma): 2.76AP, ± 3.50ML, 5.10DV. Injectors were left in place for an additional 5min after injection. Rats were given 2mL of warmed sterile saline and 3mg/kg ketoprofen (I.P.; Patterson Veterinary, Devens, MA) at the end of the surgery and were individually housed until 1 week after surgery when sutures were removed and housing pairs were re-established. A total of 4 weeks was allowed for virus expression and transport of hChR2 to BLA terminal fields prior to experimentation. Injection sites were confirmed by visualizing EYFP in coronal slices of the AIC using fluorescence microscopy postmortem. Rats were excluded if there was unintended delivery or spread outside of the AIC.
2.3. Chronic Intermittent Ethanol (CIE) Vapor Exposure
Using standard procedures from our laboratory (Morales et al. 2018), pair-housed rats were exposed to CIE for 7 or 10 consecutive days in their home cages placed inside larger, custom-build Plexiglas chambers (Triad Plastics, Winston-Salem, NC). Ethanol vapor was pumped into the chambers beginning at 9 PM (start of the light cycle) at a constant rate of 16L/min and maintained at ∼25mg/L throughout the exposure for 12h/day. Control animals were identically housed but only exposed to room-air. All rats were weighed daily. To monitor and adjust ethanol vapor levels as necessary, tail blood samples were collected periodically throughout the CIE exposure; and, blood ethanol concentrations (BECs) were determined from plasma using an alcohol dehydrogenase/NADH enzymatic assay (Carolina Liquid Chemistries, Greensboro, NC). Average BECs in the CIE animals were 259.40 ± 9.26mg/dL. In some experiments, rats received either ketamine (10mg/kg in saline, I.P.; KetaVed, Patterson Veterinary, Devens, MA) or saline (vehicle) at the onset of withdrawal, 24h before behavioral testing and electrophysiology recording. All behavioral experiments and electrophysiology recordings were conducted 24h after the last ethanol or air exposure.
2.4. Elevated Zero Maze
Rats were tested on the elevated zero maze (EZM; Med Associates, Fairfax, VT) 24h after the last ethanol or air exposure. The circular EZM consists of two open sections that are dimly lit (∼40lux) and two enclosed sections. To assess anxiety-like behavior (avoidance of open spaces) and general locomotion, the rat center point, tail base, and nose position were tracked throughout the 5min test using a monochrome camera (Basler AG, Germany) and EthoVision XT video tracking software (Noldus; Leesburg, VA). The apparatus was cleaned with warm water and mild soap and then thoroughly dried between animals.
2.5. Electrophysiology
2.5.1. Slice Preparation.
Rats were deeply anesthetized with isoflurane and decapitated. Brains were quickly removed and incubated for 5min in ice-cold, modified artificial cerebral spinal fluid (aCSF, equilibrated with 95%O2 and 5%CO2) containing (in mM): 180 Sucrose, 30 NaCl, 4.5 KCl, 1 MgCl2·6 H2O, 26 NaHCO3, 1.2 NaH2PO4, 10 D-glucose, 0.10 ketamine. 400-micron thick coronal slices containing the BLA were collected using a VT1200/S vibrating blade microtome (Leica, Buffalo Grove, IL) and transferred semi-wet to a holding chamber containing ∼500mL of room temperature (∼25°C), oxygenated standard aCSF (in mM: 126 NaCl, 3 KCl, 1.25 NaH2PO4, 2 MgSO4·7 H2O, 26 NaHCO3, 10 D-glucose, and 2 CaCl2·2H2O) for at least 1h and up to 5h prior to recordings. Unless otherwise noted, all chemicals were obtained from Tocris (Ellisville, Missouri) or Sigma-Aldrich (St. Louis, MO).
2.5.2. Whole-Cell Patch-Clamp Recording.
Using standard methods for whole-cell voltage-clamp electrophysiology from our laboratory (Morales et al. 2018), we recorded synaptic responses from BLA slices in a submersion-type recording chamber that was continuously perfused with oxygenated, room temperature aCSF (2mL/min). Glutamatergic synaptic responses were isolated with the GABAA antagonist, picrotoxin (100μM). Recordings electrodes were filled with an intracellular solution containing (in mM): 145CsOH, 10EGTA, 5NaCl, 1MgCl2·6H2O, 10HEPES, 4Mg-ATP, 0.4Na-GTP, 0.4QX314, 1CaCl2·2H2O; osmolarity adjusted to ∼285Osm/L; pH adjusted to ∼7.3 using gluconic acid.
Optically-driven synaptic responses were evoked using a 473nm laser connected to a fiber optic cable (Thorlabs, Newton, NJ). The naked end of the cable was placed just above the external capsule along the lateral boundary of the BLA. Five-millisecond laser pulses were delivered to activate Channelrhodopsin expressed in the AIC terminals. Sweeps were recorded every 30sec; light stimulation intensities were submaximal and normalized across cells to elicit synaptic responses with amplitudes 100–200pA. In some recordings, 1μM tetrodotoxin (TTX; Tocris) and 20mM 4-aminopyridine (4-AP; Tocris) were included to isolate monosynaptic transmission (Cruikshank et al. 2010, Petreanu et al. 2009).
To measure NMDA-mediated EPSCs, extracellular Mg2+ concentrations were lowered to 0.2mM in standard aCSF (Ca2+-containing); and neurons were voltage-clamped at −60mV membrane potential in the presence of the AMPA receptor antagonist, 6,7-dinitroquinoxaline-2,3-dione (DNQX; 20μM) and picrotoxin. Electrically-evoked, NMDA-mediated EPSCs were measured every 30sec using brief (0.2msec) square-wave stimulations delivered to the external capsule using a platinum/iridium concentric bipolar stimulating electrode with an inner pole diameter of 12.5μm (FHC, Bowdoinham, ME). Increasing stimulus intensities (10μA to 200μA) produced graded, monosynaptic responses.
All electrophysiology data were acquired using a Axopatch 700B amplifier (Molecular Devices, Foster City, CA) and were low-pass filtered at 2kHz. pClamp 10 software (Molecular Devises, Foster City, CA) was used for later analysis. BLA principal neurons were included based on their electrophysiological characteristics of low access resistance (≤ 25MΩ) and high membrane capacitance (>100pF) (Washburn & Moises 1992). Cells in which capacitance or access resistance changed ≥20% during the recording or that did not meet principal neuron criteria were excluded from analysis.
2.5.3. Paired-Pulse Ratio.
Two 5msec light stimuli of equal intensity were delivered to the external capsule at an inter-stimulus interval of 50msec. This short interval is traditionally viewed as an indicator of presynaptic release probability (Fioravante & Regehr 2011). The paired-pulse ratio (PPR) was conservatively calculated using the evoked EPSC amplitudes as: ([Peak2 amplitude – Peak1 amplitude]/Peak1 amplitude). The average paired-pulse ratio was determined from a 5min, 11 sweep recording.
2.5.4. Strontium Substitution.
To measure optically- and electrically evoked asynchronous EPSCs from AIC synapses, we perfused slices at the beginning of the recordings with aCSF containing 2mM strontium chloride substituted for calcium chloride. Strontium flows through voltage-gated calcium channels but poorly substitutes for calcium during vesicle fusion and neurotransmitter release, producing an initial ‘synchronous’ release event similar to an electrically evoked response followed for several hundred milliseconds by asynchronous vesicle fusion events. The amplitude and interevent interval of these delayed, strontium-dependent asynchronous EPSCs (Sr-aEPSCs) have been extensively used as measures of pre- and postsynaptic function (respectively) at defined synapses (Choi & Lovinger 1997, Dodge et al. 1969, Morales et al. 2018, Xu-Friedman & Regehr 2000). As described previously (Christian et al. 2012, Morales et al. 2018), a bipolar stimulating electrode or an optical fiber was placed at the external capsule where an electrical or light stimulation was applied every 30sec. Semiautomated Sr-aEPSC analysis was conducted on responses during a 400msec window beginning 50msec post-stimulation. The median interevent intervals and amplitudes were extracted using the Mini Analysis Program (Synaptosoft, Fort Lee, NJ).
2.6. Statistics
All data are represented as mean±SEM throughout the text and figures. Primary statistical analyses were conducted using Prism 5 (GraphPad, La Jolla, CA). Differences between groups were analyzed using t-tests and one- or two-way ANOVA depending on the experimental design. A value of p<0.05 was considered statistically significant. When significant effects were defined using ANOVA tests, the indicated post hoc comparisons defined differences between experimental groups with reported P values adjusted to account for multiple comparisons.
3. Results
3.1. Neurons in the AIC make monosynaptic, glutamatergic synapses onto BLA principal neurons.
Previous studies have shown that the AIC and BLA are reciprocally connected (Matyas et al. 2014; McDonald & Mascagni 1996). Microinjection of Channelrhodopsin-eYFP into the dorsal and ventral aspects of the AIC (Fig. 1A) produced dense eYFP fluorescence in the BLA (Fig. 1B). Optically stimulating hChR2-expressing AIC terminals (Fig. 1C) produced EPSCs in BLA neurons that were abolished by the sodium channel blocker, TTX (1μM, Fig. 1D); addition of the potassium channel inhibitor 4-AP (20mM) rescued these light-evoked EPSCs. On average, the peak amplitude of EPSCs recorded in the presence of TTX and 4-AP were 84.8±1.9% (n=3) of the baseline peak amplitude. The AMPA receptor antagonist, DNQX (20μM) completely abolished these monosynaptic light-evoked ESPCs. Thus, AIC-evoked EPSCs recorded from BLA principal neurons are monosynaptic and glutamatergic.
Figure 1. The agranular insular cortex sends monosynaptic glutamatergic projections to basolateral amygdala principal neurons.
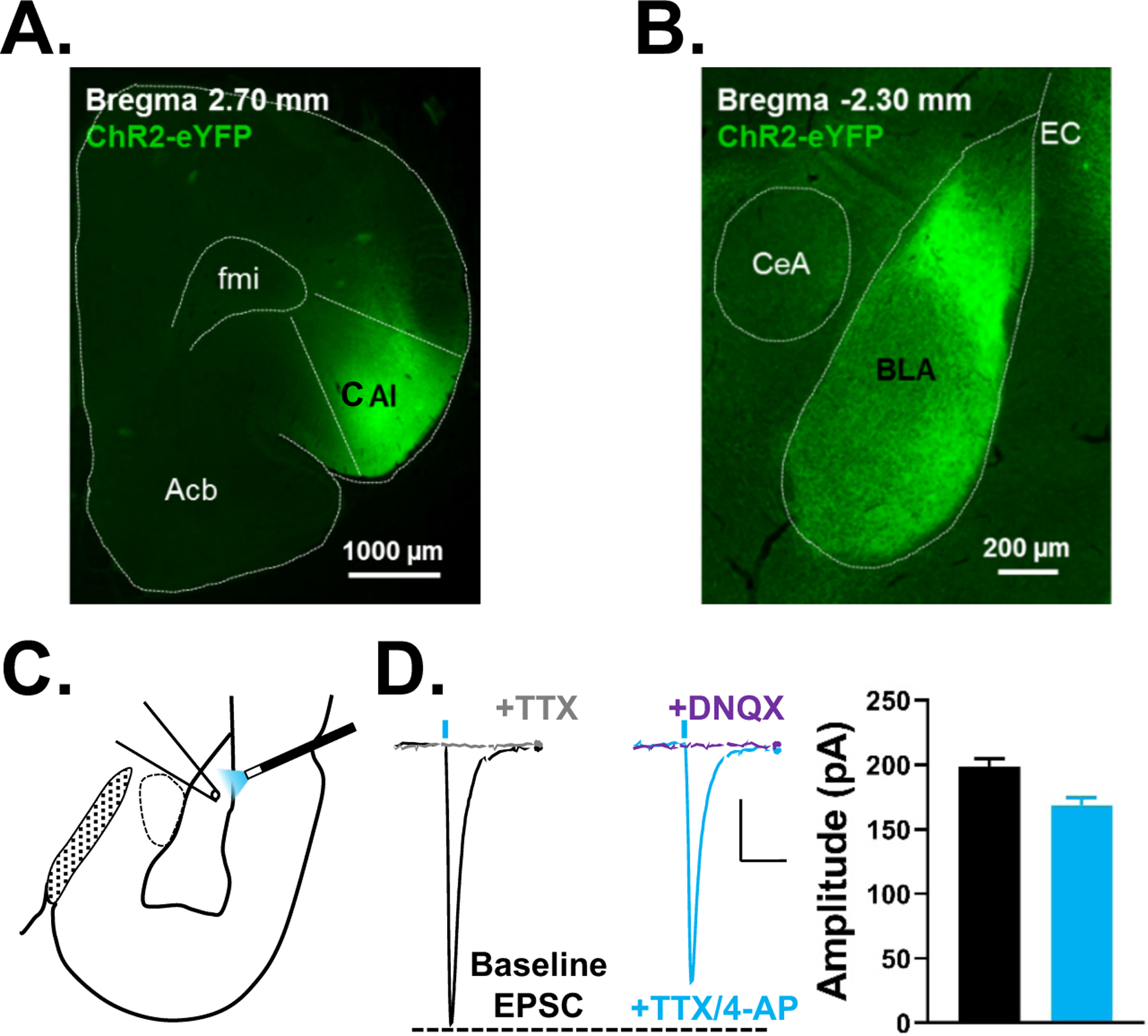
A, Representative fluorescent image of the agranular insular cortex (AIC) injection site and B, resulting terminal field in the basolateral amygdala (BLA) 4 weeks after injection of Channelrhodopsin. C, Schematic depicting the typical placement of the optical stimulating fiber and patch electrode for electrophysiology recordings in the BLA. The optical fiber delivered 470 nm blue light and was placed just above the lateral external capsule (EC) to activate the Channelrhodopsin-expressing inputs entering the BLA from the AIC. Recording electrodes were placed where the YFP-expressing terminals were most dense. D, Representative traces of optically-evoked EPSCs recorded from AIC-BLA synapses at baseline (with picrotoxin, 100μM; black trace), in the presence of tetrodotoxin (TTX, 1μM; gray trace), with 4-aminopyridine (4-AP, 20mM; cyan trace), and then 6,7-dinitroquinoxaline-2,3-dione (DNQX, 20μM; purple trace). Vertical blue dashes represent the approximate timing of optogenetic stimulation (5msec). Scale bars= 25pA × 50msec. Summary of response amplitudes for each condition are shown to the right (see text). Acb= nucleus accumbens, AIC= agranular insular cortex, BLA= basolateral amygdala, CeA= central nucleus of the amygdala, EC= external capsule, fmi= anterior forceps of corpus callosum.
3.2. AIC-BLA synapses do not express presynaptic changes following chronic ethanol exposure.
Using electrical stimulation, several studies from our laboratory have reported that external capsule glutamatergic synapses onto BLA principal neurons do not express changes in presynaptic function following CIE (Christian et al. 2012, Morales et al. 2018). To examine presynaptic function at AIC-BLA synapses, we exposed animals to 7 or 10 consecutive days of CIE (Fig. 2A) and recorded PPRs from BLA principal neurons 24h later by optically stimulating hChR2-expressing AIC terminals arriving via the external capsule. We found no difference in PPRs in AIR (PPR = −0.19±0.06, N=15) and 7d CIE/WD neurons (−0.17±0.08, N=15; unpaired t-test, t(28)=0.245, p=0.809). Similarly, a longer 10 day CIE exposure did not alter AIC-BLA PPRs (AIR = −0.19±0.05, N=15; 10d CIE/WD = −0.15±0.06, N=15; unpaired t-test, t(28)=0.406, p=0.688). Like electrically evoked EC responses (Christian et al. 2013, Christian et al. 2012, Diaz et al. 2011, Lack et al. 2009, Lack et al. 2007, Morales et al. 2015, Morales et al. 2018), optically-evoked responses from AIC terminals onto BLA principal neurons do not express CIE-dependent presynaptic alterations.
Figure 2. Withdrawal from 10, but not 7, days of chronic exposure enhances optically-evoked, Sr2+-dependent, asynchronous EPSC (Sr-aEPSC) amplitudes measured at agranular insular synapses onto basolateral amygdala principal neurons.
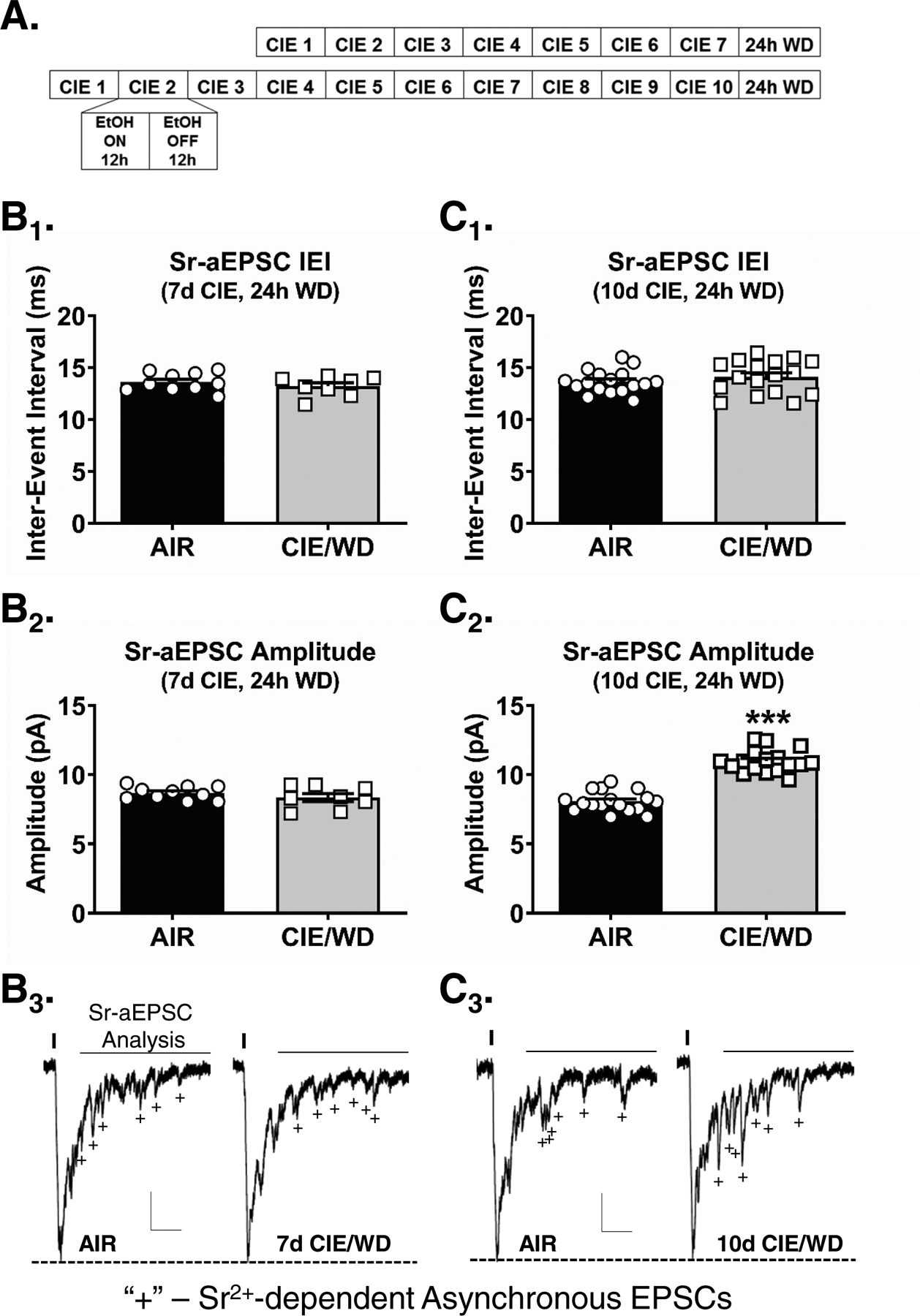
A, Experimental timeline depicting the 7- or 10- day chronic intermittent ethanol (CIE) exposures. Each day, rats were exposed to 12-hours of ethanol (EtOH) and 12-hours of air. All electrophysiology recordings were conducted 24-hours after the last ethanol exposure. Control animals were identically housed but only exposed to air. B, Following a 7 day CIE treatment, the interevent interval (B1) and amplitude (B2) of optically-stimulated Sr-aEPSCs evoked from AIC→BLA synapses were similar in AIR (N= 10) and WD neurons (N= 8). C, Following 10d CIE/WD, there was no change in the interevent interval of Sr-aEPSCs (C1) recorded from AIR (N= 17) and 10d CIE/WD (N= 17) animals. However, the optically-evoked Sr-aEPSC amplitudes were significantly larger in 10d CIE/WD animals as compared to AIR (C2, *** - p<0.0001, t-test). Representative Sr-aEPSC traces from 7- (B3) and 10-day CIE (C3) exposures. Vertical lines indicate the timing of 473nm laser activation. ‘+’ represent exemplar Sr-aEPSCs. Horizontal lines represent the ‘window’ for Sr-aEPSC analysis (see Methods). Scale bars= 30pA × 50msec. Unpaired t-test, ***p < 0.0001.
3.3. Withdrawal from 10, but not 7, days of CIE increases postsynaptic function at AIC – BLA synapses.
To measure the pre- and postsynaptic impact of the CIE exposure at these synapses, we measured the frequency and amplitude of light-evoked, strontium-dependent asynchronous EPSCs (Sr-aEPSCs) from AIC-BLA synapses following 7 or 10 days of CIE. We found no change in the Sr-aEPSC interevent intervals between AIR (Fig. 2B1, 13.61±0.28msec, N=10) and 7d CIE/WD neurons (13.20±0.34msec, N=8; unpaired t-test, t(16)=0.939, p=0.362). Surprisingly, 7dCIE/WD likewise did not alter Sr-aEPSC amplitude (Fig. 2B2; 8.36±0.29pA, N=8) relative to AIR-exposed neurons (8.68±0.15pA; unpaired t-test, t(16)=1.035, p=0.316). After a 10 day exposure, we again found no differences in the Sr-aEPSC intervals between AIR (Fig. 2C1; 13.63±0.27msec, N=17) and 10d CIE/WD neurons (14.11±0.39msec, N=17; unpaired t-test, t(32)=1.008, p=0.321). This confirms the absence of presynaptic effect of the CIE treatment on AIC synapses measure with paired light pulses. However, 10d CIE/WD significantly increased Sr-aEPSC amplitude (Fig. 2C2; 10.98±0.20pA) relative to AIR (8.06±0.18pA; unpaired t-test, t(32)=10.94, p<0.0001). These data strongly suggest that 10d CIE/WD increases the postsynaptic, but not the presynaptic, function at AIC-BLA synapses.
3.4. In vivo ketamine administration prevents the CIE/WD-induced increases postsynaptic function.
NMDA receptors play an integral role in activity-dependent long-term plasticity, typically expressed as postsynaptic changes in AMPA receptor function (Hunt & Castillo 2012). Recent work (Vranjkovic et al. 2018) showed that a single ketamine injection following long-term ethanol consumption increases the capacity for NMDA-dependent LTP in the bed nucleus of the stria terminalis (BNST). Given CIE-dependent increases in Sr-aEPSC amplitude mimic NMDA receptor-dependent postsynaptic plasticity, we administered ketamine (10mg/kg, I.P.) at the onset of withdrawal from 10d CIE or Air (Fig. 3A) and examined the effect on optically-evoked AIC-BLA Sr-aEPSCs. A two-way ANOVA analysis of median Sr-aEPSC interevent interval (Fig. 3B1) revealed no main effect of CIE (F(1,43)=2.857, p=0.098), no main effect of ketamine treatment (F(1,43)=0.215, p=0.645), and no interaction (F(1,43)=0.556, p=0.460). However, a two-way ANOVA on median Sr-aEPSC amplitudes (Fig. 3B2) revealed a significant interaction between CIE exposure and ketamine treatment (F(1,43)=22.53, p<0.0001) as well as significant main effects (CIE, F(1,43)=33.59, p<0.0001; ketamine, F(1,43)=28.19, p<0.0001). Bonferroni posttests indicated a significant increase in Sr-aEPSC amplitude in the vehicle-injected CIE/WD neurons (12.4±0.6pA, N=13) relative to saline-injected AIR-exposed controls (8.0±0.3pA, N=10, t=7.35, p<0.0001), ketamine-injected Air controls (8.0±0.3pA, N=13, t=8.13, p<0.0001), and ketamine-injected CIE/WD neurons (8.6±0.5pA, N=13, t=7.36, p<0.0001). There were no significant differences between ketamine-injected CIE/WD neurons and either the Air-Saline (t=0.333, p>0.9999) or Air-ketamine groups (t=0.753, p>0.9999). In vivo ketamine injection immediately following the last CIE exposure appeared to prevent CIE-dependent increases in AIC-BLA Sr-aEPSC amplitude.
Figure 3. Ketamine administration attenuates CIE-induced increases in AMPA receptor function measured from both optically-stimulated AIC-BLA synapses and electrically-stimulated EC – BLA synapses.

A, Experimental timeline depicting the 10-day CIE exposure followed by a ketamine injection (10mg/kg, I.P., N=16) at the onset of withdrawal, 24-hours prior to the electrophysiology recordings. B, Neither CIE/WD nor ketamine changed the interevent interval (B1) of optically-evoked Sr-aEPSCs measured from AIC-BLA synapses (see text). While CIE/WD+Saline (N=12) increased Sr-aEPSC amplitude (B2) compared to AIR+Saline (N= 12), ketamine administration blocked this effect. Ketamine administration did not alter optically-evoked Sr-aEPSC amplitude in neurons (N=13) from air-exposed animals. Two-way ANOVA interaction p<0.0001 (F(1,43)=22.5), *** - p<0.0001 with Bonferroni posttest CIE/WD + Saline versus cells from the other three treatment groups. C, Representative traces of optically-evoked responses from AIC-BLA synapses. Horizontal lines represent the ‘window’ for Sr-aEPSC analysis (see Methods). Scale bars = 30pA × 50msec. ‘+’ indicate examples of Sr-aEPSCs. D, Neither CIE/WD nor ketamine altered Sr-aEPSC interevent intervals (D1) recorded from electrically-stimulated EC-BLA synapses (see text). Electrically-evoked Sr-aEPSC amplitudes (D2) from EC-BLA inputs were significantly greater (Two-way ANOVA, interaction p=0.0018, F(1,48)=10.3) in CIE/WD+Saline neurons (N= 11) compared to neurons from Air+Saline (N=12, ## −− p=0.0028, Bonferroni posttest), Air + ketamine (N= 13, ## −− p=0.006), and CIE/WD + ketamine (N = 16, *** − p<0.0001).
Given the postsynaptic nature of the CIE facilitation at AIC-BLA synapses, we wanted to determine if ketamine inhibition of increased optically-evoked AIC-BLA Sr-aEPSC amplitudes could be generalizable to all EC-BLA synapses. Using electrical stimulation of the EC, a two-way ANOVA on median Sr-aEPSC frequency (Fig. 3D1) again revealed no interaction between the main factors of CIE exposure and ketamine treatment (F(1,49)=0.010, p=0.922) and no main effects (CIE, F(1,49)=3.091, p=0.085; ketamine, F(1,49)=0.132, p=0.718). Similar to the optical stimulation findings, a two-way ANOVA on electrically-evoked Sr-aEPSC amplitudes (Fig. 3D2) revealed a significant interaction (F(1,49)=10.90, p=0.002), a main effect of CIE exposure (F(1,49)=5.01, p=0.030), and a main effect of ketamine treatment (F(1,49)=15.42, p=0.001). Sr-aEPSC amplitudes in saline-injected CIE/WD animals (11.7±0.6pA, N=11) were significantly larger than saline-injected AIR-exposed controls (9.0±0.6pA, N=12; t=3.754, p=0.003), ketamine-injected Air-exposed neurons (8.7±0.2pA, N=13; t=4.260, p=0.001), and ketamine-injected CIE/WD neurons (8.2 ± 0.3pA, N=16; t=5.236, p<0.0001). Notably, Sr-aEPSC amplitudes recorded from neurons within the ‘CIE/WD+ketamine’ group were not significantly different from neurons recorded from either the saline-injected Air (t=1.223, p>0.9999) or ketamine-injected Air groups (t=0.788, p>0.9999). These data suggest that the in vivo ketamine-mediated attenuation of optically-evoked CIE/WD Sr-aESPC amplitudes at AIC-BLA synapses is generalizable to the population of EC-BLA synapses activated by electrical stimulation.
3.5. In Vivo ketamine administration prevents CIE/WD-induced increases in NMDA receptor function.
We previously reported that NMDA receptor function is upregulated by CIE (Floyd et al. 2003, Lack et al. 2007). Using electrical stimulations delivered to the external capsule, we recorded NMDAR-mediated EPSCs from BLA principal neurons across a range of stimulation intensities in AIR and 10d CIE/WD neurons from animals that received either ketamine (10mg/kg) or vehicle (saline). Mixed effects analysis found a significant interaction between stimulation intensity and treatment group (Fig. 4A&B; F(12, 156)=8.918, p<0.0001) and significant main effects of both stimulation intensity (F(4,156)=53.41, p<0.0001) and ketamine/CIE (F(3,47)=14.41, p<0.0001). Using a planned comparison between Air+saline controls and the other treatment groups, Dunnett’s multiple comparison test indicated that only the CIE+Saline group was significantly different from AIR controls at the 100μA (p=0.001), 150μA (p<0.0001), and 200μA (p<0.0001) stimulation intensities. These results demonstrate that NMDAR function at EC-BLA synapses is increased following 10 days of CIE and that in vivo ketamine administration can block the effects of CIE.
Figure 4. Ketamine administration prevents chronic ethanol-induced increase in NMDA receptor function at EC-BLA synapses.
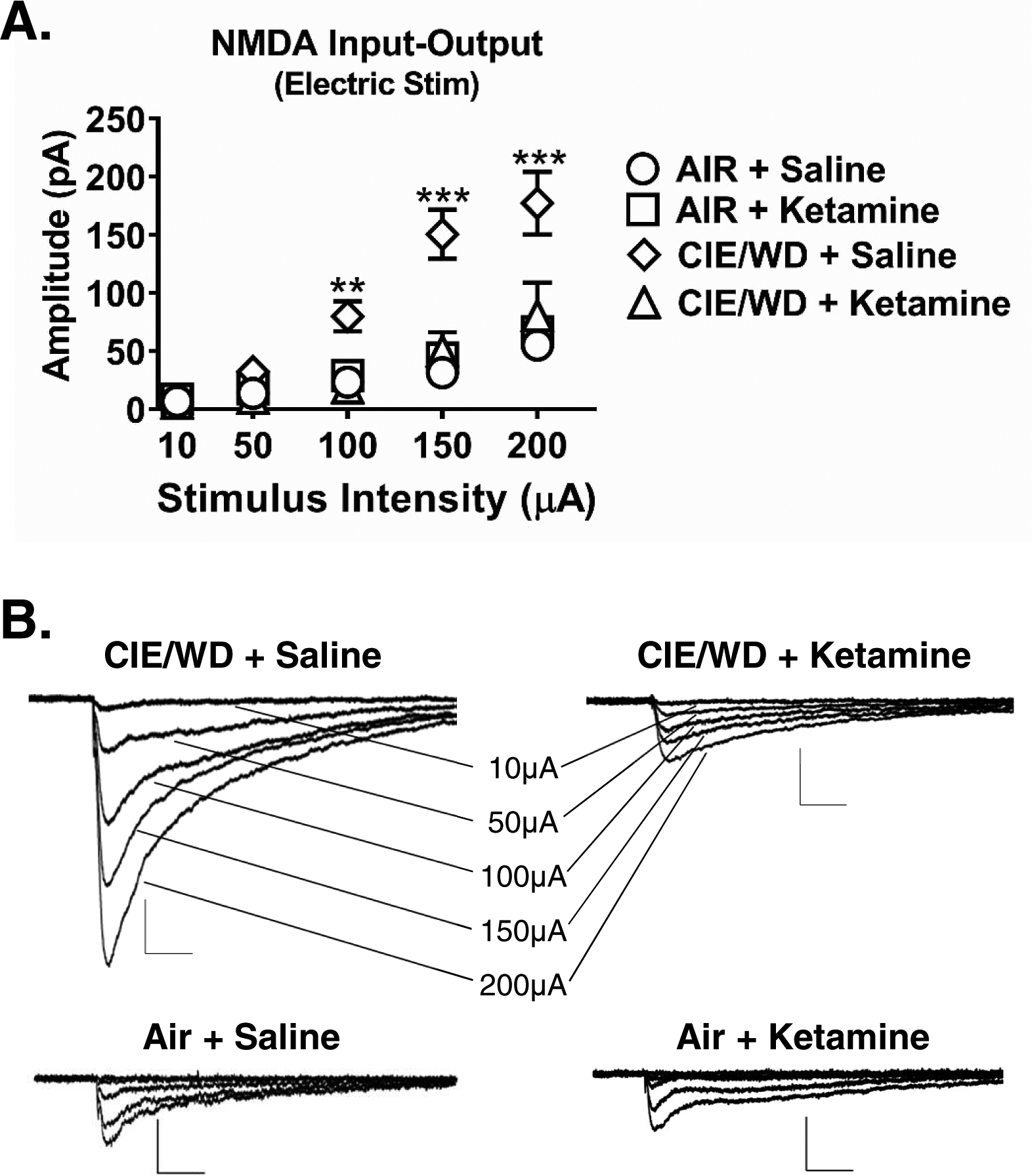
A, Significant increase in NMDAR synaptic function in CIE/WD + Saline (N= 18) group as compared to AIR + saline (N= 14), Air+ketamine (N= 10), and CIE/WD + ketamine (N= 9) groups across multiple stimulation intensities (Two-way ANOVA mixed effects analysis; interaction, F(12, 156)=8.92, p<0.0001; Dunnett’s posttest, ** − p=0.001, *** − p<0.0001). There were no significant differences between AIR+saline, Air+ketamine, and CIE/WD+ketamine (all p>0.05, see text for details). B, Representative traces of electrically-evoked NMDAR-mediated EPSCs recorded from CIE/WD + Saline, CIE/WD + Ketamine, Air + Saline, and Air + Ketamine EC – BLA synapses. Scale bars= 50pA × 100msec.
3.6. In vivo ketamine administration attenuates withdrawal-induced anxiety-like behavior.
The reversal of CIE/WD-dependent postsynaptic facilitation of NMDA and AMPA receptor function suggested that in vivo ketamine exposure may also alter withdrawal-related behaviors. To examine this, we delivered single 10mg/kg ketamine injection (I.P.) at the end of a 10 day CIE exposure (Fig. 3A) and measured anxiety-like behavior in the elevated zero maze 24h later (Fig. 5C). A two-way ANOVA of time spent in open areas on the EZM (Fig. 5A) found a significant interaction (F(1,32)=11.70, p=0.002) but no main effects of either ketamine treatment (F(1,32)=0.14, p=0.708) or CIE exposure (F(1,32)=0.86, p=0.359). Multiple comparison posttests indicated that saline-injected CIE/WD animals spent significantly less time in the open areas of the EZM (30.7±8.0sec, N=9) compared to both saline-injected AIR-exposed control animals (92.2±21.7sec, N=9, t=3.077, p=0.004) and ketamine-injected CIE/WD animals (73.7±14.0sec, N=9, t=2.152, p=0.039). Surprisingly, ketamine-injected AIR animals also spent significantly less time (38.6±8.2sec, N=9, t=2.686, p=0.011) in the open areas than AIR animals injected with saline. Air-ketamine and CIE-saline groups were not significantly different from each other (t=0.391, p=0.699). Measures of head-dip frequency (number explorations over the side of the open portions of the apparatus) and closed area → open area transitions closely paralleled these observations (not shown). A two-way ANOVA of total distance moved (Fig. 5B) found no main effect of CIE exposure (F(1,32)=1.29, p=0.265), no main effect of ketamine treatment (F(1,32)=0.017, p= 0.895), and no interaction (F(1,32)=2.83, p=0.103). These data suggest that a single ketamine injection delivered at the end of the last CIE exposure can ameliorate withdrawal-related anxiety-like behavior measured 24h later.
Figure 5. Ketamine administration prevents ethanol withdrawal-associated increases in anxiety-like behavior.
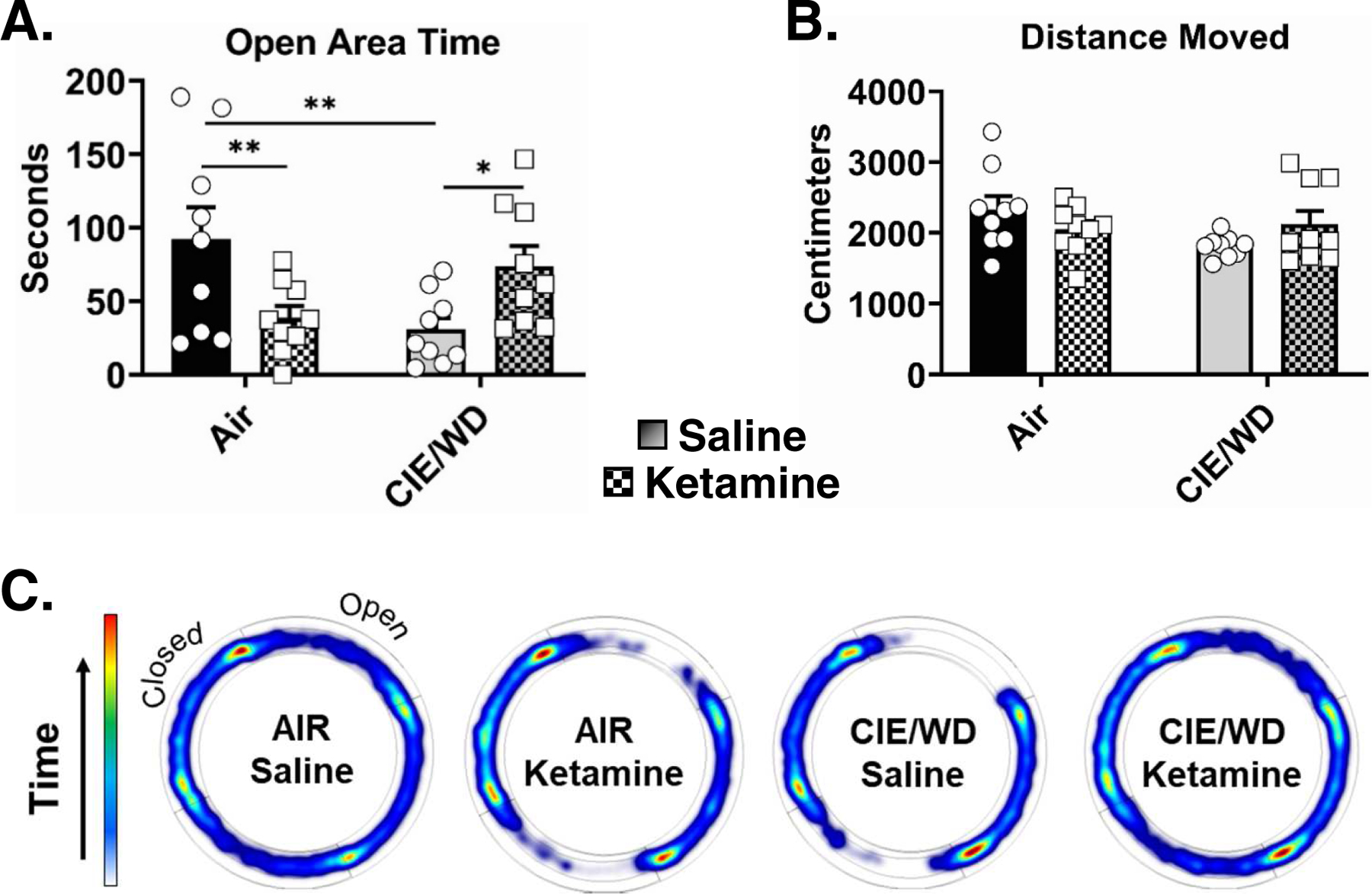
A., Time spent in the open areas of the EZM measured 24 hours after air or 10-days of CIE ±ketamine administration (10mg/kg, IP, same timing as Figure 3A). Animals in the CIE/WD+saline group (N= 9) spent significantly less time (Two-way ANOVA, interaction p=0.0017, F(1,32)=11.7) in the open areas compared to the AIR+saline (N= 9, Bonferonni posttest, ** − p=0.004) and CIE/WD+ketamine groups (N= 9, * − p=0.039). AIR+ketamine animals (N= 9) also spent significantly less time in the open compared to Air+saline (** −p=0.011). B, There were no significant differences in total distance moved between any of the treatment groups (Two-way ANOVA, interaction p=0.054, F(1,32)=4.0; exposure p=0.143, F(1,32)=0.15; treatment p=0.895, F(1,32)=0.90). C, EZM heat plots representing group data for times spent in the various parts of the apparatus.
4. Discussion
In this study we examined the effects of chronic ethanol exposure and withdrawal on glutamate transmission in the AIC-BLA circuit. Using a combination of optogenetics and electrophysiology, we demonstrate that AIC terminals make monosynaptic glutamatergic synapses onto BLA principal neurons (Fig. 1). Following withdrawal from 10 days CIE (but not 7 days), AIC-BLA synapses express increased postsynaptic function represented by increased AMAPR-mediated, strontium-dependent asynchronous EPSC amplitudes (Fig. 2). Neither 7 nor 10 days of CIE produced presynaptic changes at AIC-BLA synapses as reflected by either paired-pulse ratios or Sr-aEPSC interevent intervals. Importantly, a subanesthetic dose of ketamine delivered at the onset of withdrawal prevented increases in postsynaptic function at AIC-BLA synapses that was also generalizable to the population of EC synapses activated by electrical stimulation (Fig. 3). We also found that ketamine administration prevented CIE-induced changes in NMDA receptor synaptic function expressed in these EC-BLA synapses (Fig. 4). Finally, we found that in vivo ketamine attenuates the development of increased negative-affect in CIE/WD animals (Fig. 5). Surprisingly, we found that ketamine alone increased anxiety-like behavior in air-exposed control animals. Together, these results indicate that postsynaptic function of the AIC-BLA pathway is sensitive to chronic ethanol exposure and that acute ketamine is an effective modulator of both this form of BLA glutamatergic plasticity and anxiety-like behavior.
The effects of chronic ethanol on the AIC-BLA pathway had not been examined prior to this study. However, previous studies demonstrate that both the AIC and the BLA are independently altered by chronic ethanol exposure. In monkeys, chronic ethanol self-administration enhanced glutamatergic EPSCs within the AIC (Alexander et al. 2012). Human alcohol-dependent subjects exhibit greater insular cortical atrophy compared to the non-dependent controls (Chattopadhyay et al. 2011). In the amygdala, our laboratory has reported that chronic ethanol exposure increases postsynaptic NMDAR function measured from ‘local’ stimulation of BLA synapses (Lack et al. 2007) and increases synaptic responses mediated by both kainate- (Lack et al. 2009) and AMPA-type (Christian et al. 2012) glutamate receptors at EC-BLA inputs. Our current findings demonstrate that AIC-BLA synapses express post- but not presynaptic, plasticity paralleling this published work. Given that increased amygdala and insula reactivity are related to emotional processing (Stein et al. 2007), our findings help support the notion that chronic ethanol disrupts emotional-processing across a range of contributing neural circuits.
With electrical stimulation, we’ve previously shown that postsynaptic changes at the population of EC-BLA synapses occur following 7 days of CIE exposure in adolescent animals (∼P35). However, in the present study, 10 days CIE, but not 7 days, increased in postsynaptic function in AIC-BLA synapses using optogenetic activation of these inputs. While it is possible that the AIC-BLA synapses are require longer chronic ethanol exposures compared to the population of EC inputs in which they reside, a more parsimonious explanation for the relative delay in the development of CIE-dependent postsynaptic facilitation is the age of the animals during the ethanol exposure. Expression of channelrhodopsin in the AIC terminal fields requires several weeks; and, as a result, CIE exposure in the current study occurred when rats were ‘young adults’ (∼P80). Prior studies from our lab reporting that 7 days of CIE were sufficient to produce postsynaptic alterations at electrically evoked EC synapses (Christian et al. 2012, Morales et al. 2018) used animals that were several weeks younger (∼P45 at the time of the electrophysiology). Notably, in the current study, we used identical experimental approaches (electrical stimulation of EC synapses, Fig. 3D) in older animals and found that 10 days of CIE produced similar alterations in SR-aEPSC amplitude compared to optical stimulation of AIC→BLA synapses. These data strongly suggest that postsynaptic alterations at AIC→BLA synapses mimic the general population of synapses activated by EC electrical stimulation and potentially highlight the unique vulnerability of adolescent animals to CIE. There are numerous reports suggesting adolescent animals are more vulnerable to ethanol (reviewed in (McCool & McGinnis 2019, Spear 2015)). Our findings may suggest that age-dependent decreases in ethanol sensitivity are reflected even at the level of the synapse.
NMDA receptor-dependent synaptic plasticity, such as long-term potentiation (LTP), is believed to play a critical role in learning and memory. Strong evidence suggests that this postsynaptic potentiation depends on increased calcium influx through NMDARs, activation of calcium/calmodulin-dependent kinase II (CamKII), and the insertion of AMPARs into the postsynaptic membrane (Luscher & Malenka 2012). Notably, chronic ethanol exposure increases CamKII activation and phosphorylation which influence AMPA receptor postsynaptic function (Christian et al. 2012). Ethanol-induced plasticity may therefore occur through mechanisms similar to NMDAR-dependent LTP. In the present study, a subanesthetic dose of ketamine administered in vivo immediately after the last CIE exposure blocked the ethanol-induced increase in NMDAR function (Fig. 5), which we hypothesize may lead to ketamine attenuation of the ethanol-induced increase in postsynaptic AMPAR function (Fig. 4). We found that ketamine was also able to alleviate withdrawal-induced increases in anxiety-like behavior. These findings parallel a recent study that found 1) ketamine administered at the end of chronic ethanol drinking prevented abstinence-dependent anxiety and 2) restored the capacity for the expression of LTP occluded by chronic ethanol within the BNST which is normally occluded by chronic ethanol (Vranjkovic et al. 2018). Interestingly, Vranjkovic and colleagues also report that the timing of ketamine administration was critical. This appears consistent with our own findings that increased AMPA receptor synaptic function at EC synapses occurs during the first 24h of withdrawal from CIE (Christian et al. 2012). It should be noted that we used a low concentration of ketamine (100μM) during slice preparation since this greatly enhances the health of BLA principal neurons. Given that individual slices are transferred semi-wet from the slicing apparatus to the holding chamber, ketamine concentrations in that chamber would be very low (less than 50nM); and slices are extensively washed in aCSF prior to recording. Further, we used identical slice preparation procedures for both Air- and CIE-exposed brains. This strongly suggests that the brief in situ exposure to ketamine in ice-cold solutions during tissue preparation had minimal long-term impact on the significant differences noted between exposure groups for both Sr-aEPSC and NMDA-mediated EPSCs.
Notably, ketamine administration in air-exposed control animals significantly increased anxiety-like behavior in the EZM (Fig. 5) without altering glutamate function at AIC-BLA synapses or within the EC-BLA pathway (Fig. 3 & 4). First, this suggests that the anxiogenic effect of ketamine in control animals is not directly related to alterations in BLA glutamatergic transmission at these synapses. Secondly, other studies have also reported anxiogenic effects of single or repeated ketamine injections (Becker & Grecksch 2004, de Carvalho Cartagenes et al. 2019, Frohlich & Van Horn 2014). In one of these studies, the atypical antipsychotics clozapine and risperidone attenuated ketamine-induced alterations in a variety of anxiety-related behaviors (Becker & Grecksch 2004). This suggests that ketamine acting on serotonin and dopamine systems may underlie the anxiety-provoking effects of ketamine in air-exposed animals. Regardless, our findings are consistent with the notion that ketamine may be protective against chronic ethanol-induced changes in both glutamate synaptic function and negative affect.
In conclusion, we demonstrate that AIC-BLA synapses express increases in postsynaptic AMPA receptor function following 10 days of CIE. This paralleled and may be a result of increased NMDA receptor synaptic function at EC-BLA synapses. Importantly, a subanesthetic dose of ketamine, administered at the onset of ethanol withdrawal, blocks both neurophysiological effects. Acute ketamine administration was also able to attenuate withdrawal-dependent anxiety-like behavior. Future studies will focus on establishing a role of AIC-BLA circuit in anxiety-like behavior as well as understanding the mechanisms by which ketamine modulates these outcomes.
Highlights.
Agranular insula projections to basolateral amygdala are monosynaptic and glutamatergic
Insula synapses express postsynaptic facilitation after chronic ethanol exposure
A subanesthetic dose of ketamine reverses ethanol facilitation of insula synapses
Ketamine also reverses the effects of ethanol on anxiety-like behavior
Acknowledgements
This work was supported by the National Institutes of Health/National Institute on Alcohol Abuse and Alcoholism [T32 AA007565 and F31 AA025514 (MMM), R01 AA023999, R01 AA014445, R21 AA026572, and P50 AA026117 (BAM)].
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alexander GM, Graef JD, Hammarback JA, Nordskog BK, Burnett EJ, Daunais JB, Bennett AJ, Friedman DP, Suomi SJ, Godwin DW (2012) Disruptions in serotonergic regulation of cortical glutamate release in primate insular cortex in response to chronic ethanol and nursery rearing. Neuroscience 207: 167–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen GV, Saper CB, Hurley KM, Cechetto DF (1991) Organization of visceral and limbic connections in the insular cortex of the rat. J Comp Neurol. 311: 1–16 [DOI] [PubMed] [Google Scholar]
- Becker A, Grecksch G (2004) Ketamine-induced changes in rat behaviour: a possible animal model of schizophrenia. Test of predictive validity. Prog Neuropsychopharmacol Biol Psychiatry. 28: 1267–77 [DOI] [PubMed] [Google Scholar]
- Chattopadhyay S, Srivastava M, Srivastava AS, Srivastava A (2011) Structural changes in the insular cortex in alcohol dependence: A cross sectional study. Iran J Psychiatry. 6: 133–7 [PMC free article] [PubMed] [Google Scholar]
- Choi S, Lovinger DM (1997) Decreased frequency but not amplitude of quantal synaptic responses associated with expression of corticostriatal long-term depression. J Neurosci 17: 8613–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian DT, Alexander NJ, Diaz MR, McCool BA (2013) Thalamic glutamatergic afferents into the rat basolateral amygdala exhibit increased presynaptic glutamate function following withdrawal from chronic intermittent ethanol. Neuropharmacology 65: 134–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian DT, Alexander NJ, Diaz MR, Robinson S, McCool BA (2012) Chronic intermittent ethanol and withdrawal differentially modulate basolateral amygdala AMPA-type glutamate receptor function and trafficking. Neuropharmacology 62: 2429–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruikshank SJ, Urabe H, Nurmikko AV, Connors BW (2010) Pathway-specific feedforward circuits between thalamus and neocortex revealed by selective optical stimulation of axons. Neuron 65: 230–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Carvalho Cartagenes S, Fernandes LMP, Carvalheiro T, de Sousa TM, Gomes ARQ, Monteiro MC, de Oliveira Paraense RS, Crespo-Lopez ME, Lima RR, Fontes-Junior EA, Prediger RD, Maia CSF (2019) “Special K” drug on adolescent rats: oxidative damage and neurobehavioral impairments. Oxid Med Cell Longev. 2019: 5452727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz MR, Christian DT, Anderson NJ, McCool BA (2011) Chronic ethanol and withdrawal differentially modulate basolateral amygdala paracapsular and local GABAergic synapses. J Pharmacol Exp Ther 337: 162–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodge FA Jr., Miledi R, Rahamimoff R (1969) Strontium and quantal release of transmitter at the neuromuscular junction. J Physiol 200: 267–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fioravante D, Regehr WG (2011) Short-term forms of presynaptic plasticity. Curr Opin Neurobiol 21: 269–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floyd DW, Jung KY, McCool BA (2003) Chronic ethanol ingestion facilitates N-methyl-D-aspartate receptor function and expression in rat lateral/basolateral amygdala neurons. J Pharmacol Exp Ther 307: 1020–9 [DOI] [PubMed] [Google Scholar]
- Frohlich J, Van Horn JD (2014) Reviewing the ketamine model for schizophrenia. J Psychopharmacol. 28: 287–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilpin NW, Koob GF (2008) Neurobiology of alcohol dependence: focus on motivational mechanisms. Alcohol Res Health 31: 185–95. [PMC free article] [PubMed] [Google Scholar]
- Gilpin NW, Richardson HN, Cole M, Koob GF (2008) Vapor inhalation of alcohol in rats. Curr Protoc Neurosci Chapter 9: Unit 9 29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gogolla N (2017) The insular cortex. Curr Biol. 27: R580–R86 [DOI] [PubMed] [Google Scholar]
- Hermann D, Weber-Fahr W, Sartorius A, Hoerst M, Frischknecht U, Tunc-Skarka N, Perreau-Lenz S, Hansson AC, Krumm B, Kiefer F, Spanagel R, Mann K, Ende G, Sommer WH (2012) Translational magnetic resonance spectroscopy reveals excessive central glutamate levels during alcohol withdrawal in humans and rats. Biol Psychiatry. 71: 1015–21 [DOI] [PubMed] [Google Scholar]
- Hunt DL, Castillo PE (2012) Synaptic plasticity of NMDA receptors: mechanisms and functional implications. Curr Opin Neurobiol. 22: 496–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaramillo AA, Van Voorhies K, Randall PA, Besheer J (2018) Silencing the insular-striatal circuit decreases alcohol self-administration and increases sensitivity to alcohol. Behav Brain Res. 348: 74–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lack AK, Christian DT, Diaz MR, McCool BA (2009) Chronic ethanol and withdrawal effects on kainate receptor-mediated excitatory neurotransmission in the rat basolateral amygdala. Alcohol 43: 25–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lack AK, Diaz MR, Chappell A, DuBois DW, McCool BA (2007) Chronic ethanol and withdrawal differentially modulate pre- and postsynaptic function at glutamatergic synapses in rat basolateral amygdala. J Neurophysiol 98: 3185–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luscher C, Malenka RC (2012) NMDA receptor-dependent long-term potentiation and long-term depression (LTP/LTD). Cold Spring Harb Perspect Biol. 4: pii: a005710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maffei A, Haley M, Fontanini A (2012) Neural processing of gustatory information in insular circuits. Curr Opin Neurobiol. 22: 709–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matyas F, Lee J, Shin HS, Acsady L (2014) The fear circuit of the mouse forebrain: connections between the mediodorsal thalamus, frontal cortices and basolateral amygdala. Eur J Neurosci. 39: 1810–23 [DOI] [PubMed] [Google Scholar]
- McCool BA (2011) Ethanol modulation of synaptic plasticity. Neuropharmacology 61: 1097–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCool BA, McGinnis MM (2019) Adolescent Vulnerability to Alcohol Use Disorder: Neurophysiological Mechanisms from Preclinical Studies. Handb. Exp. Pharmacol 9: DOI: 10.1007/164_2019_296 [DOI] [PubMed] [Google Scholar]
- McDonald AJ, Mascagni F (1996) Cortico-cortical and cortico-amygdaloid projections of the rat occipital cortex: a Phaseolus vulgaris leucoagglutinin study. Neuroscience 71: 37–54 [DOI] [PubMed] [Google Scholar]
- McGinnis MM, Parrish BC, Chappell AM, Alexander NJ, McCool BA (2019) Chronic Ethanol Differentially Modulates Glutamate Release from Dorsal and Ventral Prefrontal Cortical Inputs onto Rat Basolateral Amygdala Principal Neurons. eNeuro 23: 0132–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales M, McGinnis MM, McCool BA (2015) Chronic ethanol exposure increases voluntary home cage intake in adult male, but not female, Long-Evans rats. Pharmacol Biochem Behav. 139: 67–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales M, McGinnis MM, Robinson SL, Chappell AM, McCool BA (2018) Chronic intermittent ethanol exposure modulation of glutamatergic neurotransmission in rat lateral/basolateral amygdala is duration-, input-, and sex-dependent. Neuroscience 371: 277–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petreanu L, Mao T, Sternson SM, Svoboda K (2009) The subcellular organization of neocortical excitatory connections. Nature 457: 1142–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pushparaj A, Le Foll B (2015) Involvement of the caudal granular insular cortex in alcohol self-administration in rats. Behav Brain Res. 293: 203–7 [DOI] [PubMed] [Google Scholar]
- Rainnie DG, Asprodini EK, Shinnick-Gallagher P (1991) Excitatory transmission in the basolateral amygdala. J Neurophysiol 66: 986–98. [DOI] [PubMed] [Google Scholar]
- Sah P, Faber ES, Lopez De Armentia M, Power J (2003) The amygdaloid complex: anatomy and physiology. Physiol Rev. 83: 803–34 [DOI] [PubMed] [Google Scholar]
- Seif T, Chang SJ, Simms JA, Gibb SL, Dadgar J, Chen BT, Harvey BK, Ron D, Messing RO, Bonci A, Hopf FW (2013) Cortical activation of accumbens hyperpolarization-active NMDARs mediates aversion-resistant alcohol intake. Nat Neurosci. 16: 1094–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shillinglaw JE, Morrisett RA, Mangieri RA (2018) Ethanol modulates glutamatergic transmission and NMDAR-mediated synaptic plasticity in the agranular insular cortex. Front Pharmacol. 9: 1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spear LP (2015) Adolescent alcohol exposure: Are there separable vulnerable periods within adolescence? Physiol Behav. 148: 122–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein MB, Simmons AN, Feinstein JS, Paulus MP (2007) Increased amygdala and insula activation during emotion processing in anxiety-prone subjects. Am J Psychiatry. 164: 318–27 [DOI] [PubMed] [Google Scholar]
- Tsai GE, Ragan P, Chang R, Chen S, Linnoila VM, Coyle JT (1998) Increased glutamatergic neurotransmission and oxidative stress after alcohol withdrawal. Am J Psychiatry. 155: 726–32 [DOI] [PubMed] [Google Scholar]
- Valenzuela CF (1997) Alcohol and neurotransmitter interactions. Alcohol Health Res World 21: 144–8. [PMC free article] [PubMed] [Google Scholar]
- Vranjkovic O, Winkler G, Winder DG (2018) Ketamine administration during a critical period after forced ethanol abstinence inhibits the development of time-dependent affective disturbances. Neuropsychopharmacology. 43: 1915–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washburn MS, Moises HC (1992) Electrophysiological and morphological properties of rat basolateral amygdaloid neurons in vitro. J Neurosci 12: 4066–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu-Friedman MA, Regehr WG (2000) Probing fundamental aspects of synaptic transmission with strontium. J Neurosci. 20: 4414–22. [DOI] [PMC free article] [PubMed] [Google Scholar]