

Abstract
Adenosine to-Inosine (A-to-I) RNA editing is a conserved post-transcriptional modification that is critical for a variety of cellular processes. A-to-I editing is widespread in nearly all types of RNA, directly imparting significant global changes in cellular function and behavior. Dysfunctional RNA editing is also implicated in a number of diseases, and A-to-I editing activity is rapidly becoming an important biomarker for early detection of cancer, immune disorders, and neurodegeneration. While millions of sites have been identified, the biological function of the majority of these sites is unknown, and the regulatory mechanisms for controlling editing activity at individual sites is not well understood. Robust detection and mapping of A-to-I editing activity throughout the transcriptome is vital for understanding these properties and how editing affects cellular behavior. However, accurately identifying A-to-I editing sites is challenging because of inherent sampling errors present in RNA-seq. We recently developed Endonuclease V immunoprecipitation enrichment sequencing (EndoVIPER-seq) to directly address this challenge by enrichment of A-to-I edited RNAs prior to sequencing. This protocol outlines how to process cellular RNA, enrich for A-to-I edited transcripts with EndoVIPER pulldown, and prepare libraries suitable for generating RNA-seq data.
Keywords: A-to-I Editing, RNA Editing, RNA-seq, Epitranscriptomics
INTRODUCTION
RNA is modified extensively after transcription. Adenosine-to-inosine (A-to-I) RNA editing is one of the most common post-transcriptional modifications, and is catalyzed by adenosine deaminases acting on RNAs (ADARs) (Bass, 2002). A-to-I RNA editing alters the chemical structure and hydrogen bonding characteristics of the nucleobase, where inosine instead base pairs with cytidine to effectively recode this site as guanosine (Figure 1a, b). A-to-I editing is ubiquitous in nearly all types of RNA, and millions of sites across the transcriptome have been identified to date. Cellular homeostasis is reliant on this type of editing, and editing has been linked to regulating innate cellular immunity, embryogenesis, and stem cell differentiation pathways. Dysregulation of A-to-I RNA editing is also implicated in a number of disorders, including several cancer types and a variety of autoimmune and neurodegenerative diseases. Despite a growing body of research surrounding A-to-I RNA editing, the precise biological function of editing at individual sites is unknown, and the regulatory mechanisms that govern editing activity are still unclear. Accurately detecting, measuring, and mapping A-to-I editing at individual sites is crucial to addressing these gaps in understanding, and conventional methods utilize high-throughput RNA sequencing (RNA-seq). Due to the base pairing change imparted by editing, inosine locations can be directly inferred from A-G transitions in sequencing data (Oakes, Vadlamani, & Hundley, 2017). However, accurately detecting A-to-I editing with RNA-seq remains challenging. Despite the large number of known A-to-I editing sites across several databases (~5 million to date), these sites are relatively rare in the context of the entire transcriptome (Kiran & Baranov, 2010; Picardi, D’Erchia, Lo Giudice, & Pesole, 2016; Ramaswami & Li, 2013). Moreover, RNA-seq is prone to random sampling and high technical variability between replicates, making it difficult to consistently capture and detect these A-to-I sites. To address these limitations, we recently developed Endonuclease V immunoprecipitation enrichment (EndoVIPER) as a straightforward method to enrich edited transcripts prior to sequencing, increasing the efficiency and accuracy of detecting A-to-I editing in RNA-seq workflows (Knutson et. al., 2020). In particular, we employ an inosine-binding enzyme Endonuclease V (EndoV) to selectively capture A-to-I edited transcripts. EndoV normally requires Mg2+ as a cofactor to bind and cleave inosine-containing RNAs (Morita et al., 2013; Vik et al., 2013; Yao, Hatahet, Melamede, & Kow, 1994), but in the presence of Ca2+ only binding activity is maintained, enabling capture and enrichment of edited transcripts from cellular RNA. We combine EndoV with an optimized protocol using random fragmentation and glyoxal denaturation of RNA to maximize efficiency of recovery of edited transcripts (Figure 2).
Figure 1.
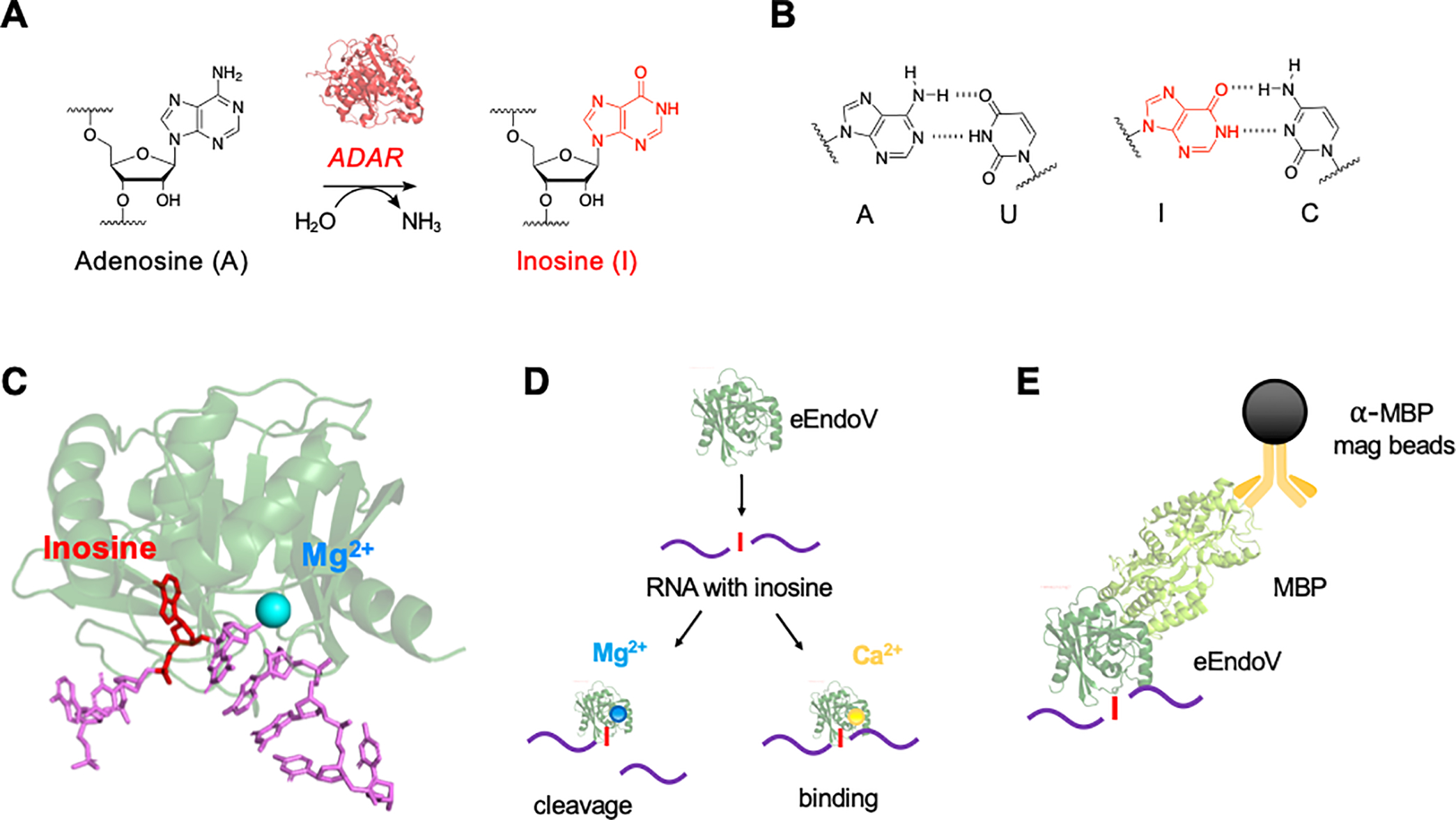
(A) A-to-I RNA editing is the result of a site selective deamination reaction catalyzed by ADAR enzymes. (B) A-to-I editing alters base pairing properties of the nucleobase, resulting in I:C hydrogen bonding. (C) Crystal structure of EndoV (PDB 2W35) bound to a single-stranded DNA substrate (purple) with inosine (red) and a Mg2+ ion (blue). (D) EndoV supplemented with Mg2+ results in cleavage of inosine containing transcripts, while Ca2+ promotes binding. (E) General concept of immunoprecipitation using E. coli EndoV (eEndoV) – maltose binding protein (MBP) fusion and anti-MBP magnetic beads.
Figure 2.
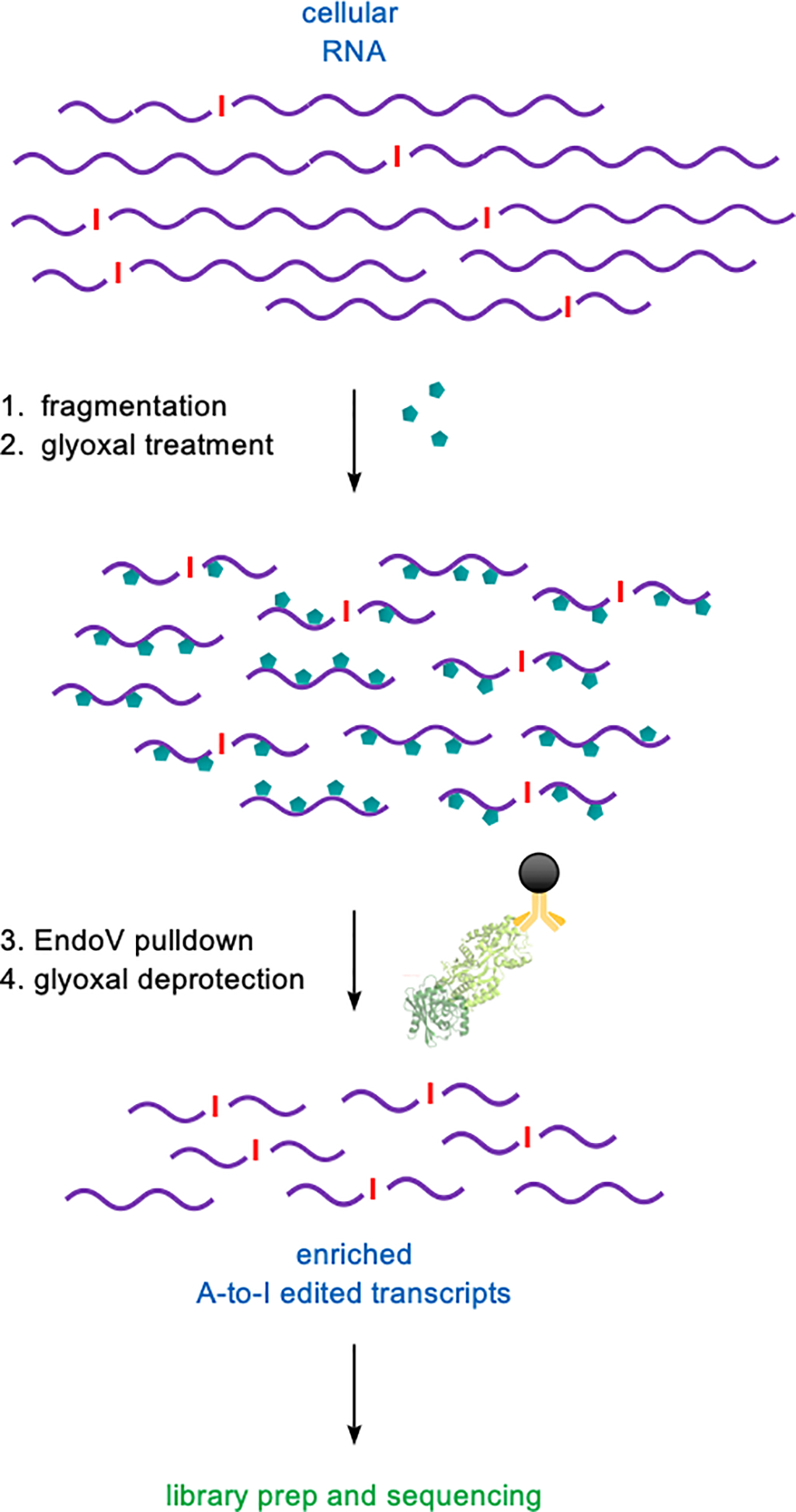
EndoVIPER-seq protocol workflow. mRNA starting material is 1) randomly hydrolyzed into 200–500 nt fragments followed by 2) glyoxal denaturation (turquoise hexagons). A-to-I edited transcripts are enriched with 3) EndoVIPER pulldown and 4) glyoxal adducts are removed prior to RNA-seq library preparation and sequencing.
As we describe in this chapter, Endonuclease V immunoprecipitation enrichment (EndoVIPER) is a straightforward approach toward isolating inosine-containing transcripts upstream of RNA-seq library preparation and sequencing. The first protocol outlines how to fragment and treat cellular RNA so that is it compatible with enrichment. The second protocol details the EndoVIPER enrichment of edited RNAs using our pulldown approach. We also include a third protocol that guides users through library preparation and culminates in the production of Illumina-capable libraries for acquiring RNA-seq data. Detailed procedures for basic quality control (Williams, Thomas, Wyman, & Holloway, 2014), read mapping (Ahn & Xiao, 2015), and A-to-I editing analysis (Picardi, D’Erchia, Montalvo, & Pesole, 2015) of RNA-seq datasets have been outlined in other protocols and are not included in this chapter.
STRATEGIC PLANNING
These protocols require experience with RNA handling. RNA is highly susceptible to hydrolysis and nuclease degradation, so it is essential that the starting RNA material is of good integrity and that all glassware, tubes, pipet tips, and work areas are clean and free of contaminants. It is advisable to read this protocol in its entirety to gain familiarity and ensure a smooth transition between key steps. While it is recommended to proceed directly to each step in a timely fashion, there are several stopping points noted in protocols where samples can be stably stored for long periods of time. This overall workflow is also optimized for use with mRNA, so if using total RNA isolated directly from cells, it is advisable to have experience enriching polyA+ mRNA from this type of sample.
BASIC PROTOCOL 1 - mRNA Fragmentation and Glyoxalation
In this first protocol, starting mRNA material is randomly hydrolyzed into ~200–500 nt fragments using high temperature and Mg2+. While we previously optimized this hydrolysis time to be roughly 1 minute, it is highly advisable that you follow this protocol and test several heating times to determine precise fragment size distribution generated using your own equipment and reagents. Hydrolysis time will also largely depend on the integrity of the starting mRNA material, and can be adjusted accordingly. After recovering fragmented mRNA material and verifying desired fragment size, mRNA is then covalently denatured (Figure 3) using glyoxal to ensure all fragments are single-stranded prior to immunoprecipitation. Practice proper precaution to prevent RNase contamination and further unwanted degradation of material. We routinely use RNaseZap™ (Thermo Fisher Scientific) to decontaminate gloves, instruments, and workspaces.
Figure 3.
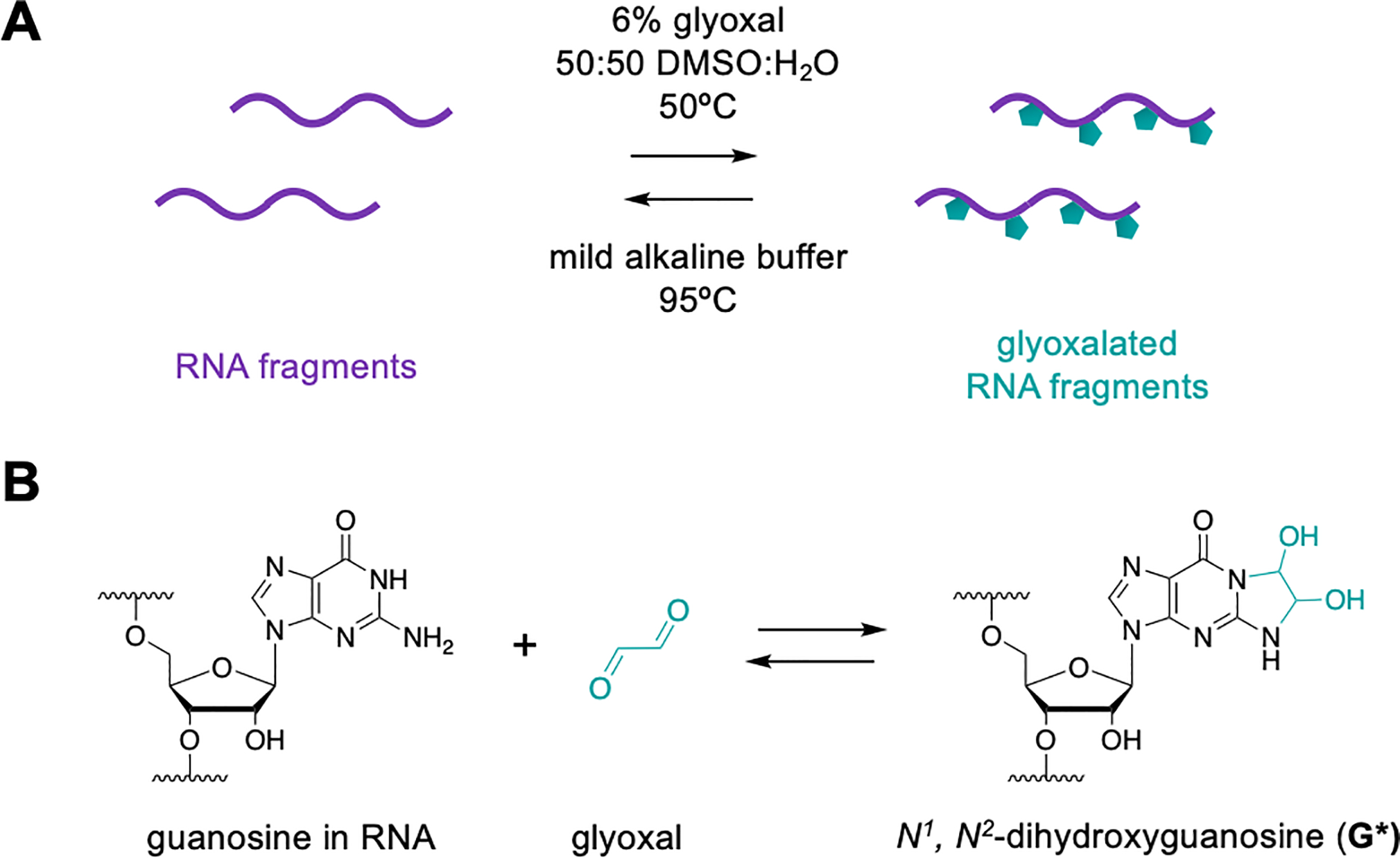
(A) Glyoxal is used to covalently and reversibly denature RNA fragments. (B) Glyoxal reacts with the Watson-Crick-Franklin face of nucleobases, strongly inhibiting secondary structure and maintaining RNA in single-stranded conformation for optimal EndoVIPER pulldown.
Materials:
mRNA material, (we have used Human Brain Poly A+ RNA from Takara Bio, cat. No 636102, but this can be isolated from desired biological source or purchased elsewhere)
RNaseZap™ (Thermo Fisher Scientific, cat. No. AM9780)
Nuclease-free water
NEBNext® Magnesium RNA Fragmentation Module (New England Biolabs, cat. No. E6150S)
Sodium acetate anhydrous, molecular biology grade, ≥99% (Sigma Aldrich, cat. No. S2889)
Acetic acid, glacial, ≥99% (Sigma Aldrich, cat. No. A6283)
Glycogen, RNA grade, 20mg/mL (Thermo Fisher Scientific, cat. No. R0551)
Ethanol, anhydrous, 200 Proof (Decon Labs, cat. No. 2701)
Glyoxal solution, 40 wt. % in H2O (Sigma Aldrich, cat. No. 128465)
Dimethyl sulfoxide (DMSO), molecular biology grade (Sigma Aldrich, cat. No. D8418)
Agilent 6000 RNA Pico Assay Kit (Agilent, cat. No. 5067-1514)
Nuclease-free filter pipet tips
Nuclease-free 0.25 mL PCR tubes
Nuclease-free 1.5 mL microcentrifuge tubes
NanoDrop 2000/2000c UV-Vis spectrophotometer
Thermal cycler
−20 °C freezer
−80 °C ultrafreezer
Refrigerated microcentrifuge
Agilent 2100 Bioanalyzer instrument
mRNA Fragmentation
-
1
After obtaining mRNA from desired origin, check the concentration of this material with a Nanodrop spectrophotometer.
A 260/280 nm absorbance ratio of approximate 2.0 is indicative of high purity for RNA. If mRNA was obtained from cells and/or purified from total RNA, a ratio lower than 2.0 may indicate the presence of contaminants and require additional purification before use. A high absorbance at ~230 nm may indicate chemical contaminants present.
-
2
Preheat a thermal cycler to 94 °C.
-
3
In separate 0.25 mL PCR tubes, combine 500 ng mRNA with 2 μL of the included 10× RNA Fragmentation Buffer and nuclease-free water to a total volume of 20 μL. Prepare 4 total samples.
-
4
Incubate tubes in the preheated thermal cycler at 94 °C for precisely 0 minutes, 1 minute, 2 minutes, and 3 minutes. At each stopping point, place tube on ice and add 2 μL of the 10× RNA Fragmentation Stop Solution. Mix by pipetting up and down.
The timing of this step is crucial. It is advisable to only work with one tube at a time to ensure that stop solution is added in a precise and timely manner.
-
5
Transfer reactions to a 1.5 mL microcentrifuge tube. Precipitate each sample by adding, in order, 2.2 μL 3 M sodium acetate pH 5.2, 2 μL 20 mg/mL glycogen, and 60 μL cold 100% ethanol (EtOH). Mix gently by pipetting up and down and incubate at −80 °C for at least an hour.
While glycogen is not completely necessary for this procedure, it makes locating the precipitated pellet significantly easier and does not interfere in downstream portions of the assay. Protocol can be stopped at this point and samples can be kept at −80 °C indefinitely until ready to proceed.
-
6
Remove samples from freezer and centrifuge at max speed (~17,000 x g) for 20 minutes in a refrigerated microcentrifuge. Carefully remove and discard supernatant, and add 1 mL of ice cold EtOH to the tube. Wash pellet by inverting tube several times, and repeat spin step.
RNA will appear as a small white/translucent pellet on the bottom of the tube. It is helpful to orient the “hinge” of the tube outward in the centrifuge rotor to make it easier to locate the pellet.
-
7
Carefully remove and discard supernatant. Leave caps on tubes open and allow pellets to air dry for ~5 minutes at room temperature. Reconstitute each pellet in 10 μL nuclease free water. Quantify concentration with Nanodrop. Samples should be ~ 50 ng/μL.
Do not overdry the pellet as this can render it more difficult to dissolve in water. When reconstituting, it is helpful to carefully add nuclease-free water on top of the pellet and let incubate undisturbed for 5–10 minutes. Afterwards, gently mix the solution by pipetting up and down to fully dissolve the material. Material can be stored frozen at −80 °C at this step indefinitely until ready to proceed to size distribution analysis.
-
8
Using the entire sample recovered from step 7, dilute fragmented mRNA samples to ~5 ng/μL in nuclease free water. Denature mRNA samples by heating to 70 °C for 2 minutes. Store on ice.
-
9
Determine size distribution using the Agilent 6000 RNA Pico Assay Kit and Agilent Bioanalyzer 2100 instrument according to the manufacturer’s specifications. Generated electropherograms should show an overall reduction in fragment size distributions with increasing exposure to 94 °C (Figure 4).
It is also possible to assess size distribution of mRNA samples using a denaturing formaldehyde agarose gel compared to ssRNA standards (Rio, 2015).
-
10
After a suitable fragmentation time that yields fragments having a centered distribution ~200 – 500 nt has been identified, it is advisable to maintain the scale of fragmentation reactions for all subsequent assays to ensure reproducibility.
Figure 4.
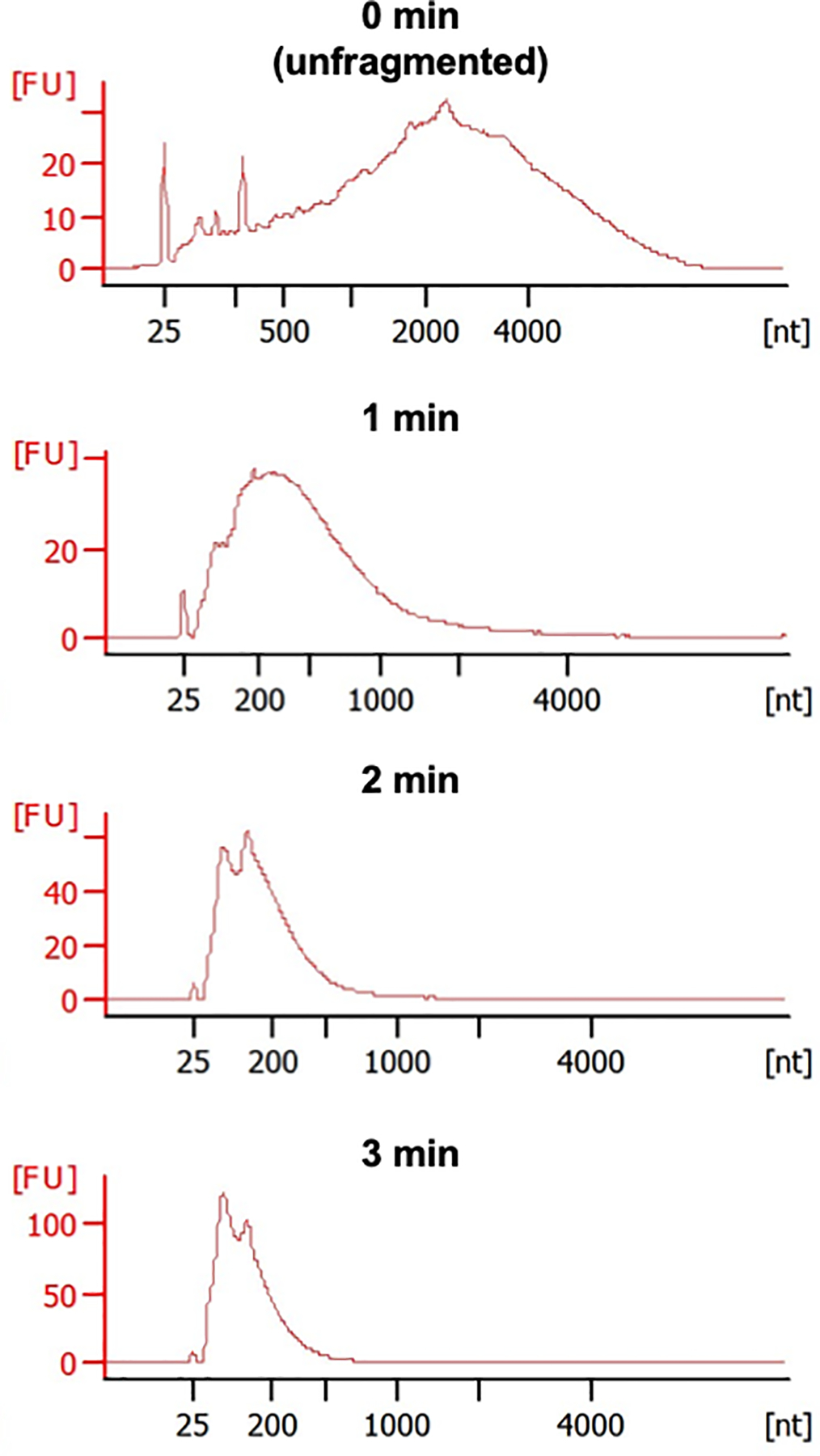
Representative Bioanalyzer electropherograms of mRNA samples after increasing fragmentation times at 94 °C (FU indicates fluorescent units). mRNA with good integrity should consist of very large fragments across a broad range of lengths. The overall size distribution should become smaller and more homogenous with increasing fragmentation. 1 minute incubation time yields fragments that center around 200–500 nt in length.
Glyoxal Denaturation
-
11
In separate 0.25 mL PCR tubes, combine 500 ng fragmented mRNA from earlier (~10 μL) with 14.5 μL 40% glyoxal solution, 50 μL DMSO and nuclease-free water up to 100 μL. Mix gently by pipetting up and down. React at 50 °C in a thermal cycler for 1 hour.
We have found that 1 hour incubation time is sufficient for this scale and proceeds essentially to completion. Additional reaction time beyond this will not increase performance downstream, but shouldn’t adversely affect the assay either. This reaction can be scaled up or down as needed, but if performing a large volume glyoxalation reaction, it is recommended that it be divided it into multiple PCR tubes (100 μL, each).
-
12
Transfer each reaction to a 1.5 mL microcentrifuge tube. Precipitate each sample by adding 11 μL 3 M sodium acetate pH 5.2, 2 μL 20 mg/mL glycogen, and 250 μL cold 100% ethanol (EtOH). Mix gently by pipetting up and down and incubate at −80 °C overnight.
Because of the high amount of DMSO in this reaction, it will not completely freeze at −80 °C. This is normal. It is recommended to let the samples precipitate overnight, and this is an excellent stopping point in the protocol. Samples can be kept at −80 °C indefinitely until ready to proceed.
-
13
Remove samples from freezer and centrifuge at max speed (~17,000 x g) for 20 minutes in a refrigerated microcentrifuge. Carefully remove and discard supernatant, and add 1 mL of ice cold EtOH to the tube. Wash pellet by inverting tube several times, and repeat spin step.
-
14
Carefully remove and discard supernatant. Leave caps on tubes open and allow pellets to air dry for ~5 minutes at room temperature. Reconstitute each pellet in 50 μL nuclease free water.
The pellet will be more difficult to dissolve in water after glyoxal treatment. When reconstituting, it is helpful to carefully add nuclease-free water on top of the pellet and let incubate undisturbed for 20–30 minutes. Afterwards, gently mix the solution by pipetting up and down repeatedly to fully dissolve the material. It is also common to see small particulates when mixing. Brief periods of heating at 95 °C (< 30–45 seconds at a time) can also be combined with pipet mixing to completely dissolve material.
-
15
If not directly proceeding to EndoVIPER pulldown, store fragmented and glyoxalated mRNA samples at −80 °C. Samples can be stored indefinitely at this stage.
BASIC PROTOCOL 2 - EndoVIPER Pulldown
In this second protocol, mRNA fragments containing inosine will be enriched through EndoV binding and immunoprecipitation (Figure 5). Fragmented and glyoxalated mRNA will be first incubated with a E. coli EndoV-MBP fusion protein in the presence of Ca2+ to form ribonucleoprotein complexes, followed by immobilization and solid-phase capture using anti-MBP magnetic beads. After washing away unbound RNA, enriched edited transcripts are then eluted from the beads. Lastly, we will deprotect glyoxal adducts and subsequently proceed to RNA-seq library preparation. Practice proper precaution to prevent RNase contamination and further unwanted degradation of material. We routinely use RNaseZap™ (Thermo Fisher Scientific) to decontaminate gloves, instruments, and workspaces.
Figure 5.
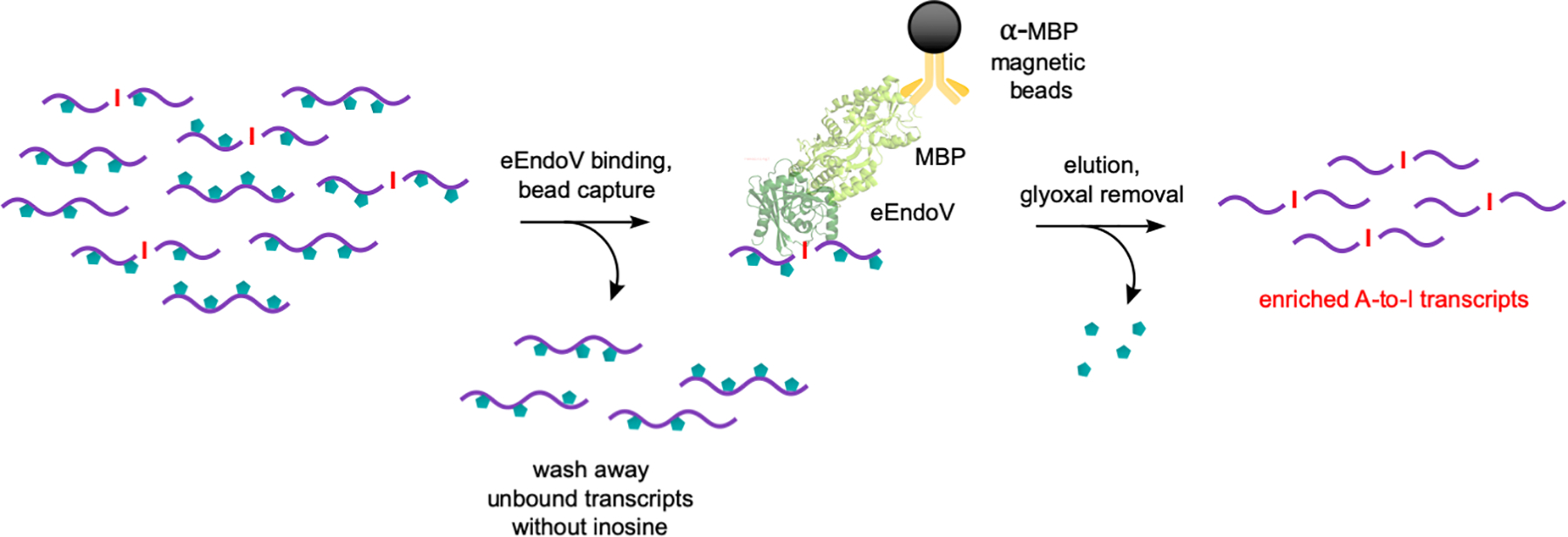
Key EndoVIPER pulldown steps for enriching A-to-I edited transcripts from fragmented and glyoxalated mRNA. Fragments containing inosine are first bound to an E. coli EndoV (eEndoV) and MBP fusion protein, followed by capture using anti-MBP magnetic beads. Unbound RNA is washed, and captured transcripts are eluted and glyoxal groups are removed.
Materials:
RNaseZap™ (Thermo Fisher Scientific, cat. No. AM9780)
Nuclease-free water
Calcium chloride dihydrate, molecular biology grade ≥99% (Sigma Aldrich, cat. No. C3306)
Tris hydrochloride ≥99.0% (Sigma Aldrich, cat. No. T5941)
Sodium chloride (Millipre Sigma, cat. No. SX0420-1)
Glycerol, molecular biology grade ≥99% (Sigma Aldrich, cat. No. G5516)
Endonuclease V recombinant protein (New England Biolabs, cat. No. M0305S)
Diluent C solution (New England Biolabs, cat. No. M0305S)
RNasin® Plus RNase Inhibitor (Promega, cat. No. N2611)
Anti-MBP Magnetic Beads (New England Biolabs, cat. No. E8037S)
Costar® Spin-X® Centrifuge Tube Filters, 0.22 μm pore, cellulose acetate membrane (Corning, cat. No. 8161)
Monarch® RNA Cleanup Kit, 10μg capacity (New England Biolabs, cat. No. T2030S)
Phosphate-Buffered Saline (PBS), 10X, pH 7.4, RNase-free (Thermo Fisher Scientific, cat. No. AM9624)
Sodium acetate anhydrous, molecular biology grade, ≥99% (Sigma Aldrich, cat. No. S2889)
Acetic acid, glacial, ≥99% (Sigma Aldrich, cat. No. A6283)
Glycogen, RNA grade, 20 mg/mL (Thermo Fisher Scientific, cat. No. R0551)
Ethanol, anhydrous, 200 Proof (Decon Labs, cat. No. 2701)
Agilent 6000 RNA Pico Assay Kit (Agilent, cat. No. 5067-1514)
Nuclease-free filter pipet tips
Nuclease-free 0.25 mL PCR tubes
Nuclease-free 1.5 mL microcentrifuge tubes
DynaMag™−2 Magnetic Stand (Thermo Fisher Scientific 12321D)
Rotary shaker incubator
Thermal cycler
−20 °C freezer
−80 °C ultrafreezer
Refrigerated microcentrifuge
Agilent 2100 Bioanalyzer instrument
EndoVIPER Pulldown
-
1
Prepare an EndoV dilution in “diluent C” (NEB). Mix 4.8 μL of the concentrated EndoV stock (~6 μM) with 35.2 μL Diluent C solution. Mix gently by pipetting up and down. Keep on ice.
EndoV concentrations vary between production lots obtained from NEB. While most batches are probably similar in final protein concentration values, it is advisable to contact their technical support department (https://www.neb.com/support/neb-technical-support) and request lot-specific information. EndoV stock solution and Diluent C are also both quite viscous, so care must be taken to ensure accurate pipetting.
-
2
In a 1.5 mL microcentrifuge tube, combine 50 μL of fragmented and glyoxalated mRNA with 3 μL RNasin® Plus, 35 μL EndoV dilution prepared above in step 1, 25 μL 10× EndoVIPER binding and wash buffer, and 137 μL nuclease-free water to bring the total volume up to 250 μL. Incubate at room temperature for 30 minutes.
This reaction can be scaled up or down depending on desired application. We have optimized this particular ratio of Endonuclease V to input RNA, but there is likely some flexibility in these amounts in the reaction. If changing these conditions for a particular assay, it is advisable to optimize these parameters for the desired outcome. RNase inhibitor is also added as a precaution to prevent further degradation of mRNA fragments. We have had good success with RNasin® Plus, however this can likely be substituted with alternative protein-based inhibitors. However, DO NOT use small molecule inhibitors (such as ribonucleoside vanadyl complexes) as these appear to also inhibit EndoV from binding inosine-containing transcripts.
-
3
While EndoV binding reactions are incubating, aliquot 300 μL of anti-MBP magnetic bead suspension for every pulldown reaction into a sterile microcentrifuge tube.
The magnetic beads are supplied as a suspension and will be settled at the bottom of the tube. Beads are stored at 4 °C when not in use. Make sure to allow beads to come to room temperature prior to use, and fully reconstitute this solution before aliquoting into new tubes by repeated inversion of stock. Beads can also be vortexed gently or rotated for 10–30 minutes to achieve good mixing. Bead suspensions should be promptly returned to 4 °C. DO NOT freeze beads at any time.
-
4
Place tubes with beads on magnetic stand and let sit for 1–5 minutes to allow magnetic beads to collect on the side of the tube. Carefully remove storage solution and discard. Remove tubes from stand and add 500 μL 1× EndoVIPER binding and wash buffer. Wash the beads by inverting the tube several times or by gently vortexing. Remove wash buffer with magnetic stand. Repeat washing step for a total of two times.
The added glycerol (7%) in the binding and wash buffer can require longer magnetic separation times. Allow tubes to incubate on stand for as long as needed to completely clarify solution. Bead loss will occur if separation time is insufficient.
-
5
After last wash, apply magnet and completely remove and discard supernatant. Add the entire binding reaction from step 2 to the beads and mix with gentle vortexing or inversion. Incubate at room temp for 2 hours with end-over end-rotation.
-
6
Remove supernatant and wash beads three times each with 500 μl 1× EndoVIPER binding and wash buffer.
-
7
After last wash, completely remove supernatant and add 200 μL of 1× EndoVIPER elution buffer. Resuspend beads by gently vortexing or pipetting and divide evenly into two PCR tubes and heat to 95 °C for 10 minutes.
It is advisable to use a thermal cycler for this step to enable precise control of temperature while also taking advantage of the heated lid to avoid evaporation. Our thermal cycler accommodates volumes up to 100 μL so we divide each elution into two tubes.
-
8
After elution step, apply magnet and collect supernatant. To remove any residual beads, apply collected supernatant directly onto a CoStar 0.2 μm spin filter tube. Centrifuge sample at 10,000 x g for 2 minutes. Collect flowthrough and set aside.
Pressure will have likely built up in tubes after heating, so quickly opening the tube cap can be used to relieve this pressure and preventing popping. Some condensation will have also likely appear near the top, so tubes can be quickly spun down to collect this material prior to magnetic separation.
-
9
Using the Monarch RNA Cleanup Kit, add 400 μL of the included RNA Cleanup Binding Buffer to the pooled 200 μL flowthrough sample collected in the previous step. Add 600 μL (1 volume) of 100 % Ethanol to the sample and mix by pipetting or flicking the tube. Do not vortex. Insert Monarch column into a collection tube, and apply 600 μL of the sample to the column. Centrifuge for 1 minute at 17,000 x g and discard flow-through. Load remaining 600 μL of sample and repeat spin.
-
10
Re-insert column into collection tube. Add 500 μL RNA Cleanup Wash Buffer and spin for 1 minute. Discard the flow-through. Repeat wash step once.
-
11
Transfer column to a new 1.5 mL microcentrifuge tube and add 30 μL nuclease-free water directly to the center of the column. Let incubate for 1 minute, and then centrifuge for 1 minute at 17,000× g to collect sample.
Final Glyoxal Deprotection
-
12
To remove any remaining glyoxal adducts, combine 30 μL of purified mRNA from the previous step with 10 μL 10× PBS, 50 μL DMSO, nuclease-free water up to 100 μL in a 0.25 mL PCR tube. Incubate in a thermal cycler at 65 °C for 2 hours.
-
13
Transfer reactions to a 1.5 mL microcentrifuge tube. Precipitate each sample by adding 100 μL nuclease-free water, 20 μL 3M sodium acetate pH 5.2, 2 μL 20 mg/mL glycogen, and 500 μL cold 100% ethanol (EtOH). Mix gently by pipetting up and down and incubate at −80 °C for at least an hour.
Protocol can be stopped at this point and samples can be kept at −80 °C indefinitely until ready to proceed.
-
14
Remove samples from freezer and centrifuge at max speed (~17,000 × g) for 20 minutes in a refrigerated microcentrifuge. Carefully remove and discard supernatant, and add 1mL of ice cold EtOH to the tube. Wash pellet by inverting the tube several times, and repeat spin step.
-
15
Carefully remove and discard supernatant. Leave caps on tubes open and allow pellets to air dry for ~5 minutes at room temperature. Reconstitute each pellet in 25 μL nuclease free water.
When reconstituting, it is helpful to carefully add nuclease-free water on top of the pellet and let incubate undisturbed for 10–20 minutes. Afterwards, gently mix the solution by pipetting up and down repeatedly to fully dissolve the material. It is also common to see small particulates when mixing. Brief periods of heating at 95 °C (< 30–45 seconds at a time) can also be combined with pipet mixing to completely dissolve material.
-
16
Determine approximate concentration and size distribution of enriched RNA using the Agilent 6000 RNA Pico Assay Kit and Agilent Bioanalyzer 2100 instrument according to the manufacturer’s specifications. Remaining material should be stored at −80 °C until ready for library preparation. At this stage, RNA material can also be used for other assays, including RT-qPCR.
This step is not necessary for proceeding to RNA-seq library prep, but is advisable to ensure that no appreciable degradation of mRNA has occurred. Generated electropherograms should show that pulldown samples are lower in concentration, but the overall size distribution should be similar (Figure 6). If fragments are significantly smaller after pulldown this is typically indicative of hydrolysis of nuclease-degradation of mRNA fragments. Based on concentration, you should expect to recover about 0.5–5% (0.1 ng/μL – 1 ng/μL, or 2.5 – 25 ng total) of your input mRNA (500 ng). However, this number is a rough estimate and can vary.
Figure 6.
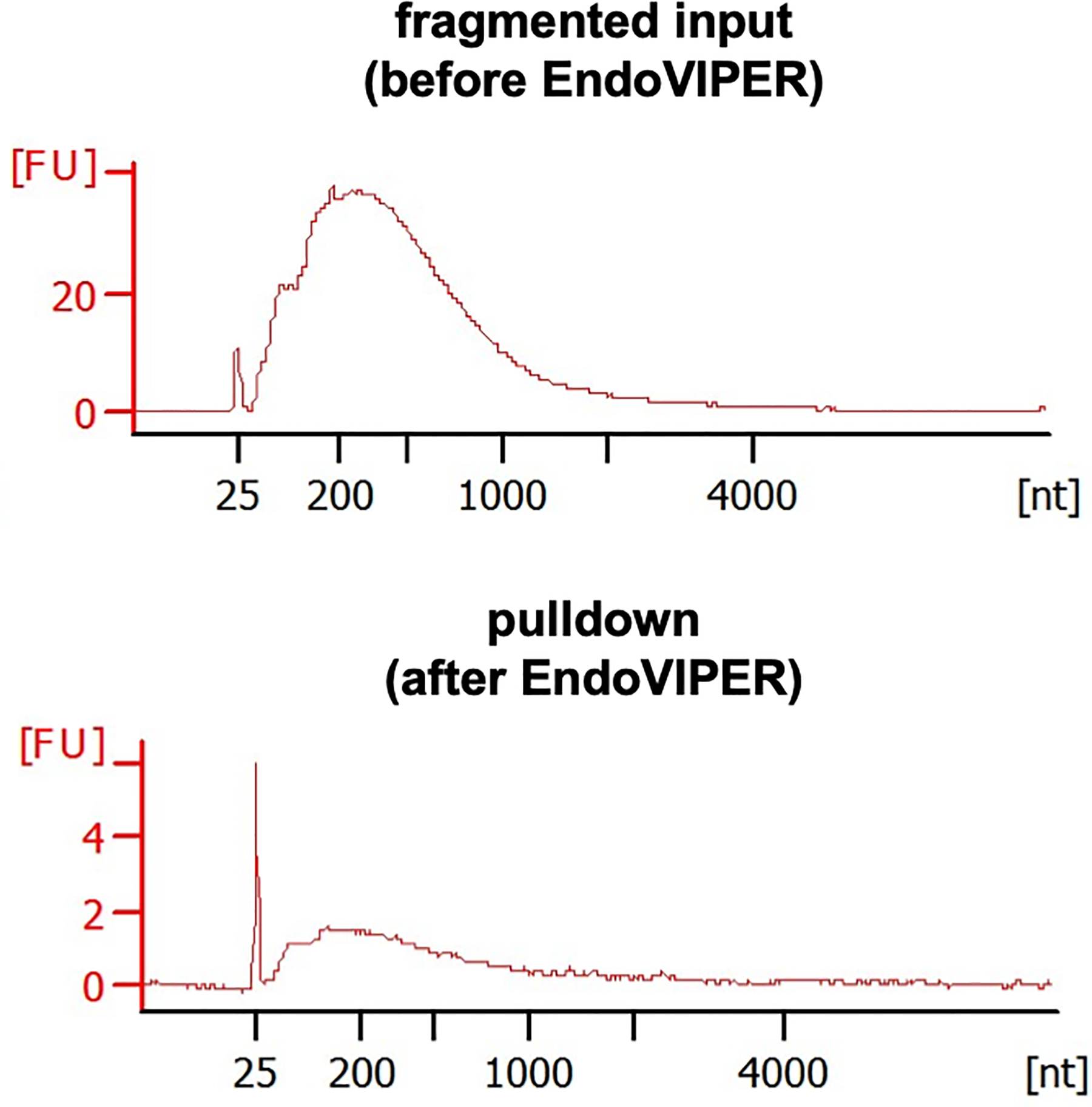
Representative Bioanalyzer electropherograms of fragmented mRNA samples before and EndoVIPER pulldown. Fluorescent units (FU) should be smaller from mRNA after pulldown, but should be roughly the same overall size distribution.
BASIC PROTOCOL 3 - RNA-seq Library Preparation
In this last protocol, enriched mRNA fragments containing inosine are reverse-transcribed and processed to generate DNA libraries for high throughput sequencing. Because we are starting with fragmented material, we use the SMARTer® Stranded Total RNA-Seq Kit v2 kit available from Takara Bio. This kit requires very low input RNA amounts (pg to ng amounts) and enables random priming of fragments. This workflow first generates cDNA from mRNA fragments using random hexamer primers (N6), and then using a provided template-switching oligonucleotide, libraries are amplified and barcoded using PCR to produce DNA libraries that are ready for sequencing with Illumina instrumentation (Figure 7a). After library preparation, each fragment will contain the necessary flow cell adaptor sequences (P7 and P5), barcode index sequences to discriminate between pooled libraries, and primer binding sites for forward and reverse reads (Figure 7b). Because of our insert size (~200 – 500 nt), we recommend using 150 bp paired-end sequencing to ensure adequate coverage of this region. Illumina next-generation sequencing (NGS) instruments can utilize these prepared libraries depending on the desired number of reads and coverage depth, including the iSeq®, MiniSeq®, MiSeq®, HiSeq®and NextSeq® platforms.
Figure 7.
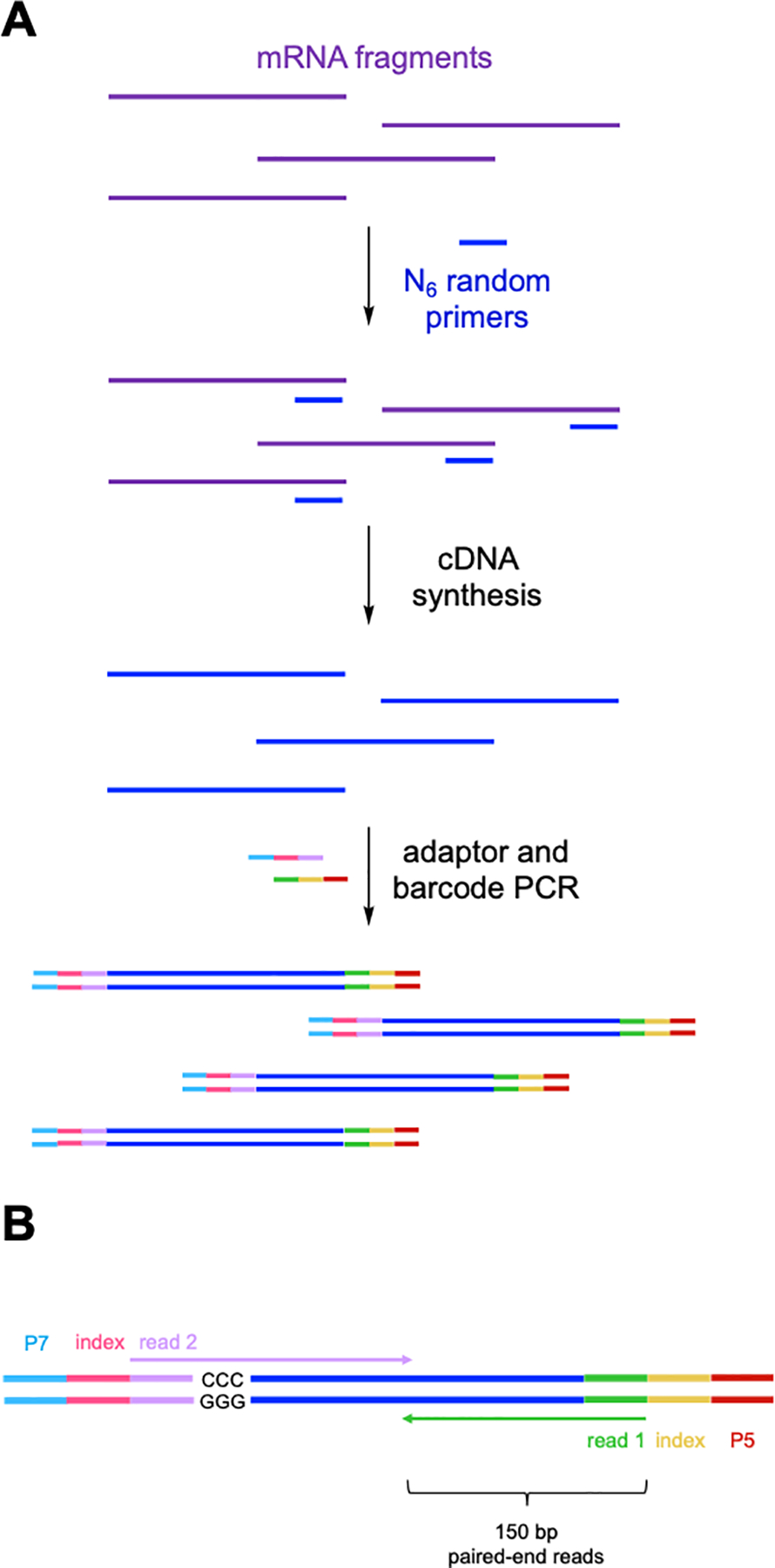
(A) Key steps in SMARTer® RNA-seq library preparation following EndoVIPER pulldown. Enriched mRNA fragments are first reverse-transcribed using random hexamer (N6) DNA primers. Resulting cDNA is then PCR amplified to attach (B) Illumina flow cell adaptors (P7 and P5), Illumina TruSeq® index barcodes, and read primer binding sites (reads 1 and 2). 150 bp paired-end sequencing is then used to ensure coverage of the insert space from both reads. The template switching oligo step produces “GGG/CCC” during cDNA synthesis. These must be removed from the beginning of read 2 during data analysis.
Materials:
SMARTer® Stranded Total RNA-seq Kit v2 - Pico Input Mammalian kit (Takara Bio, cat. No 634411)
Nuclease-free water
AMPure XP Magnetic Beads, 5 mL (Beckman Coulter, cat. No. A63880)
Ethanol, anhydrous, 200 Proof (Decon Labs, cat. No. 2701)
Ethanol, 80% solution in nuclease-free water
Nuclease-free filter pipet tips
Nuclease-free 0.25 mL PCR tubes
Nuclease-free 1.5 mL microcentrifuge tubes
DynaMag™−2 Magnetic Stand (Thermo Fisher Scientific, cat. No. 12321D)
Polarsafe™ Cooling Blocks for 0.25 mL PCR tubes (Thomas Scientific, cat. No. 1184J02)
Thermal cycler
−20 °C freezer
−80 °C ultrafreezer
Refrigerated microcentrifuge
First Strand cDNA Synthesis
-
1
In a 0.25 mL PCR tube for each sample, combine recovered mRNA with 1 μL of the SMART Pico Oligos Mix v2 and nuclease-free water to a total volume of 9 μL. Preheat a thermal cycler and incubate tubes at 72 °C for precisely 3 minutes, followed by immediate incubation on an ice-cold PCR chiller rack for 2 minutes.
The amount of input mRNA material is flexible with this library preparation kit, however it should be in the range of 250 pg–10 ng as recommended by the manufacturer. All libraries prepared across assays should be prepared using the same amount of input material to allow for effective comparison.
-
2
In a separate PCR tube for each sample, combine 4 μL 5× first-strand buffer, 4.5 μL SMART TSO Mix v2, 0.5 μL included RNase inhibitor, and 2 μL SMARTScribe reverse transcriptase included in the SMARTer® Stranded Total RNA-seq Kit. Mix gently by pipetting up and down.
The SMART TSO Mix v2 is very thick and can be hard to pipet. Ensure that the mixture is homogenous by gently pipetting up and down.
-
3
For each sample, combine the two mixtures from steps 1 and 2 into a single PCR tube. Mix the tube by briefly vortexing for a few seconds, then spin the tubes down to collect liquid to the bottom of the tube. Incubate the reaction in a preheated thermal cycler using the following conditions: 42 °C for 90 minutes, 70°C for 10 minutes, and infinite hold at 4 °C.
cDNA has been generated at this step and this is a safe stopping point in the protocol. Samples can be kept in the thermal cycler overnight at 4 °C until proceeding. If not moving directly to the next step, it is advisable to store the cDNA reaction at −20 °C for longer term storage (2 weeks).
PCR Addition of Illumina Adapters and Indexes
-
4
In a separate 0.25 mL PCR tube for each library, combine 2 μL nuclease-free water, 25 μL 2× SeqAmp CB PCR buffer, 1 μL SeqAmp DNA polymerase, and 1 μL 3’ index (i5) primer*. Mix gently by pipetting up and down and spin briefly.
*This step adds unique barcode indexes that are eventually used to identify pooled libraries after sequencing data is obtained. It is critical that you note which indexes are used to barcode each library. If fewer than 12 libraries are being prepared, a single 3’ index can be used for all samples at this step. If more than 12 libraries are being prepared, more than one unique 3’ index primer must be used across the samples.
-
5
Combine this PCR mixture with the cDNA transcription reaction from earlier. Mix by gently pipetting up and down.
-
6
Add 1 μL of a unique 5’ index (i7) primer* to each library separately. Mix by gently pipetting up and down.
*This step also adds a unique barcode indexes that are eventually used to identify pooled libraries after sequencing data is obtained. It is critical that you note which indexes are used to barcode each library so that data can be correctly demultiplexed.
-
7
Preheat a thermal cycler and perform the following program: 94 °C for 1 minute, followed by 5 cycles of 98 °C for 15 seconds, 55 °C for 15 seconds, and 68 °C for 30 seconds. Lastly, perform a final extension step at 68 °C for 2 minutes followed by infinite hold at 4 °C.
This is a safe stopping point in the protocol. Samples can be kept in the thermal cycler overnight at 4 °C until proceeding. If not moving directly to the next step, it is advisable to store the PCR reaction at −20 °C for longer term storage (2 weeks).
Purification of RNA-seq Library using AMPure Beads
-
8
Remove AMPure beads from refrigerator and allow to equilibrate to room temperature for at least 30 minutes.
-
9
Transfer PCR reactions generated from previous step to 1.5 mL microcentrifuge tubes. Add 40 μL beads directly to each PCR reaction. Mix by vortexing for 5 seconds or by pipetting up and down at least 10 times.
The magnetic beads are supplied as a suspension and will be settled at the bottom of the tube. Beads are stored at 4 °C when not in use. Make sure to allow beads to come to room temperature prior to use, and fully reconstitute this solution before adding beads to PCR reactions. DO NOT freeze beads at any time. The beads are also viscous, and it is important to thoroughly mix the suspension when added to PCR reactions to ensure adequate purification.
-
10
Incubate the PCR bead mixture for 8 minutes at room temperature to allow PCR product to bind the magnetic beads.
-
11
Place tubes with beads on magnetic stand and let sit for 5 minutes or until the solution is completely clear so as to allow magnetic beads to collect on the side of the tube. While tubes are still on the magnetic stand, carefully remove supernatant and discard.
-
12
While tubes are still on the magnetic stand, carefully add 200 μL of freshly prepared 80% EtOH. Let incubate on beads for 30 seconds to 1 minute, and carefully remove and discard using a pipette. Repeat washing step once.
It is important not to disturb the bead pellet when adding this solution. It is helpful to slowly add the liquid to the side of tube and not directly onto the beads.
-
13
Centrifuge tubes briefly at 2,000 × g to collect any residual EtOH at the bottom. Place tubes back on the magnetic stand for at least 30 seconds, and use a pipette to remove any remaining EtOH without disturbing the bead pellet.
-
14
Air dry the bead pellet by opening the cap to each tube and allowing to incubate at room temperature for 3–5 minutes or until the bead pellets appear dry.
A tiny crack will be visible in bead pellets when they are dry. It is critical that you do not overdry the pellet.
-
15
Immediately after the bead pellets are dry, carefully add 52 μL nuclease-free water to each tube to elute PCR product. Remove the tubes from the magnetic stand and wash the beads down to the bottom of the tube by repeatedly pipetting the liquid on the wall of the tube. Let beads hydrate at room temperature for 5 minutes to fully elute DNA.
-
16
Centrifuge tubes briefly at 2,000 × g to collect any liquid at the bottom of the tube. Place tubes back on the magnetic stand for at least 1 minute until solution is completely clear. Using a pipette, carefully remove supernatant and transfer to a new 1.5 mL microcentrifuge tube.
-
17
Begin purifying the PCR product a second time by adding 40 μL of fresh beads directly to each PCR reaction. Mix by vortexing for 5 seconds or by pipetting up and down at least 10 times.
-
18
Incubate the PCR bead mixture for 8 minutes at room temperature to allow PCR product to bind the magnetic beads.
-
19
Place tubes with beads on magnetic stand and let sit for 5 minutes or until the solution is completely clear so as to allow magnetic beads to collect on the side of the tube. While tubes are still on the magnetic stand, carefully remove supernatant and discard.
-
20
While tubes are still on the magnetic stand, carefully add 200 μL of freshly prepared 80% EtOH. Let incubate on beads for 30 seconds to 1 minute, and carefully remove and discard using a pipette. Repeat washing step once.
-
21
Centrifuge tubes briefly at 2,000 × g to collect any residual EtOH at the bottom of the tube. Place tubes back on the magnetic stand for at least 30 seconds, and use a pipette to remove any remaining EtOH without disturbing the bead pellet.
-
22
Air dry the bead pellet by opening the cap to each tube and allowing to incubate at room temperature for 3–5 minutes or until the bead pellets appear dry.
The beads will dry quicker during this second purification stage, so be careful not to overdry the beads.
-
23
Carefully add 22 μL nuclease-free water to each tube to elute PCR product. Remove the tubes from the magnetic stand and wash the beads down to the bottom of the tube by repeatedly pipetting the liquid on the wall of the tube. Let beads hydrate at room temperature for 5 minutes to fully elute DNA.
-
24
Centrifuge tubes briefly at 2,000 × g to collect any liquid at the bottom of the tube. Place tubes back on the magnetic stand for at least 1 minute until solution is completely clear. Using a pipette, carefully remove supernatant and transfer to a new 0.25 mL PCR tube.
This is a safe stopping point in the protocol. Samples can be kept overnight at 4 °C until proceeding. If not moving directly to the next step, it is advisable to store the sample at −20 °C for longer term storage (2 weeks).
Final Library PCR Amplification
-
25
In a separate 0.25 mL PCR tube for each library, combine 26 μL nuclease-free water, 50 μL 2× SeqAmp CB PCR buffer, 2 μL PCR2 primers v2, and 2 μL SeqAmp DNA polymerase. Mix gently by pipetting up and down and spin briefly.
Because all libraries have been barcoded at this point, every sample can use the same set of primers for the final amplification step.
-
26
Combine this PCR mixture with the bead purified supernatant (~20 μL) from earlier. Mix by gently pipetting up and down.
-
27
Preheat a thermal cycler and perform the following program: 94 °C for 1 minute, followed by 16 cycles of 98 °C for 15 sec, 55 °C for 15 seconds, and 68 °C for 30 seconds. Lastly, samples can be put at infinite hold at 4 °C.
This is a safe stopping point in the protocol. Samples can be kept in the thermal cycler overnight at 4 °C until proceeding. If not moving directly to the next step, it is advisable to store the PCR reaction at −20 °C for longer term storage (2 weeks).
Final Purification of RNA-seq Library using AMPure Beads
-
28
Remove AMPure beads from refrigerator and allow to equilibrate to room temperature for at least 30 minutes.
-
29
Transfer PCR reactions from previous step to 1.5 mL microcentrifuge tubes so that they can fit on the magnetic separator rack. Add 100 μL beads directly to each PCR reaction. Mix by vortexing for 5 seconds or by pipetting up and down at least 10 times.
-
30
Incubate the PCR bead mixture for 8 minutes at room temperature to allow PCR product to bind the magnetic beads.
-
31
Place tubes with beads on magnetic stand and let sit for 5–10 minutes or until the solution is completely clear so as to allow magnetic beads to collect on the side of the tube. While tubes are still on the magnetic stand, carefully remove supernatant and discard.
This step will likely take longer than the previous bead purification because of the higher volume. It is important to wait until the solution is completely clear so that bead loss is minimized.
-
32
While tubes are still on the magnetic stand, carefully add 200 μL of freshly prepared 80% EtOH. Let incubate on beads for 30 seconds to 1 minute, and carefully remove and discard using a pipette. Repeat washing step once.
-
33
Centrifuge tubes briefly at 2,000 ×g to collect any residual EtOH at the bottom of the tube. Place tubes back on the magnetic stand for at least 30 seconds, and use a pipette to remove any remaining EtOH without disturbing the bead pellet.
-
34
Air dry the bead pellet by opening the cap to each tube and allowing to incubate at room temperature for 3–5 minutes or until the bead pellets appear dry.
A tiny crack will be visible in bead pellets when they are dry. It is critical that you do not overdry the pellet.
-
35
Immediately after the bead pellets are dry, carefully add 20 μL of the Tris elution buffer included in the SMARTer® Stranded Total RNA-seq Kit to each pellet. Remove the tubes f rom the magnetic stand and wash the beads down to the bottom of the tube by repeatedly pipetting the liquid on the wall of the tube. Let beads hydrate at room temperature for 5 minutes to fully elute DNA.
Elution volume can be reduced to 10 μL if anticipated yield is low and a higher concentration is desired.
-
36
Centrifuge tubes briefly at 2,000 × g to collect any liquid at the bottom of the tube. Place tubes back on the magnetic stand for at least 1 minute until solution is completely clear. Using a pipette, carefully remove supernatant and transfer to a new 1.5 mL microcentrifuge tube. Proceed to sequencing immediately or store long term at −20°C (< 2 weeks).
REAGENTS AND SOLUTIONS:
Use distilled or deionized water that is nuclease-free for all solutions listed.
3 M Sodium Acetate, pH 5.2
24.6 g sodium acetate
Slowly add and dissolve sodium acetate powder in ~ 90 mL nuclease free water until fully in solution. Adjust pH to 5.2 with glacial acetic acid, and top off to a final volume of 100 mL with nuclease free water. Filter or autoclave to sterilize. Can be stored at room temperature for extended periods of time.
70% Ethanol
350 mL ethanol, anhydrous, 200 proof
Combine ethanol and nuclease free water to a total volume of 500 mL in a sterile glass bottle. Mix gently and place at −20 °C. Can be stored in freezer for extended periods of time.
80% Ethanol
80 mL ethanol, anhydrous, 200 proof
Combine ethanol and nuclease-free water to a total volume of 100 mL in a sterile glass bottle. Mix gently and store at room temperature for immediate use. Prepare fresh for each library preparation protocol and discard leftover amounts.
EndoVIPER Binding and Wash Buffer (10×)
29.2 g sodium chloride
14.9 g tris hydrochloride
735 mg calcium chloride dihydrate
Slowly add and dissolve sodium chloride, tris hydrochloride, and calcium chloride powder in ~ 450 mL nuclease-free water until fully in solution. Adjust pH to 7.4, and top off to a final volume of 500 mL with nuclease-free water. Filter to sterilize. Can be stored at room temperature for extended periods of time.
EndoVIPER Binding and Wash Buffer (1×)
5 mL 10× EndoVIPER binding and wash buffer
3.5 mL glycerol
Carefully measure glycerol and add to a 50 mL conical tube. Glycerol is very viscous, so it can be poured directly or made into a 50% solution with water to make it easier. Add 5mL 10X× EndoVIPER binding and wash buffer and bring volume up to 50 mL total volume with nuclease-free water. Mix by gentle inversion. Can be stored at room temperature but should be made fresh for each assay or within a few days time.
EndoVIPER Elution Buffer (1×)
9.5 mL formamide
100 mg SDS
First make a 10% SDS solution by dissolving 100 mg of the powder in a total volume of 1 mL nuclease-free water. Prepare elution buffer by combining 9.5 mL formamide with 20 μL 10% SDS and 480 μL nuclease-free water.
COMMENTARY
BACKGROUND INFORMATION
Despite A-to-I editing being one of most abundant RNA modifications in the transcriptome, accurately detecting and mapping editing sites using RNA-seq remains challenging. RNA-seq is limited by stochastic sampling and technical constraints in the number of obtainable reads, and actual inosine frequency is relatively low in the scope of cellular RNA. Moreover, transcript abundances and editing rates at individual sites can vary considerably (Paul & Bass, 1998; Tan et al., 2017; Yang et al., 2003). Because of this difficulty in characterizing A-to-I editing, there has been great interest in developing new methods to address these shortcomings. The characterization of several other RNA modifications, including N6-methyladenosine (Dominissini et al., 2012), 5-methylcytosine (Edelheit, Schwartz, Mumbach, Wurtzel, & Sorek, 2013), 5-hydroxymethylcytosine (Delatte et al., 2016) and pseudouridine (X. Li et al., 2015) have benefited significantly from enrichment-based approaches, allowing more sensitive detection and mapping in complex RNA samples. However, these techniques employ specific antibodies toward each respective base for immunoprecipitation, or employ site-selective chemical approaches to specifically tag and enrich modified nucleotides, and it has been challenging to apply similar strategies toward inosine in cellular RNA. Generating antibodies that are specific to inosine has been particularly challenging and has resulted in significant off-target affinity to other nucleobases, likely owing to the fact that inosine has few unique chemical features and structurally resembles several nucleotides (Inouye, Fuchs, Sela, & Littauer, 1973). Chemical labeling and pulldown of inosine has also proved challenging to implement. It has been known for several decades that Michael acceptors, in particular acrylonitrile, can react with N1 on inosine to form a stable adduct on the Watson-Crick-Franklin face (Yoshida & Ukita, 1965). We and others have applied this chemistry toward the design of inosine-enrichment reagents, however these compounds also reacted with pseudouridine and uridine, which overall limited their enrichment potential (Knutson, Ayele, & Heemstra, 2018; Y. Li, Göhl, Ke, Vanderwal, & Spitale, 2019). In search of improved methods to address this enrichment problem we were inspired by Endonuclease V (EndoV), an evolutionary conserved repair enzyme that recognizes and cleaves inosine in nucleic acids. In particular, prokaryotic EndoV appears to facilitate repair of inosine lesions in DNA that are the result of reactive oxygen species (Yao et al., 1994). Human and eukaryotic EndoV homologs have been implicated in enzymatic digestion and metabolism of inosine-containing transcripts (Morita et al., 2013; Vik et al., 2013). EndoV normally requires Mg2+ for strand-scission, but in order to study binding properties of the enzyme researchers have also replaced this metal cofactor with Ca2+, which facilitates binding but does not support cleavage of substrates (Morita et al., 2013; Vik et al., 2013; Yao et al., 1994). We leveraged this property to supplement Endo V with Ca2+ and enable recognition, capture, and immunoprecipitation and sequencing of A-to-I edited RNA transcripts from cellular RNA, which we term EndoVIPER-seq (Endonuclease V immunoprecipitation enrichment sequencing) (Knutson et. al., 2020). This new approach addresses many of the difficulties present in detecting A-to-I editing with conventional RNA-seq, and significantly increases the sensitivity and accuracy of studying editing in the transcriptome.
CRITICAL PARAMETERS
Designing and performing RNA-seq experiments with immunoprecipitation and/or enrichment is well documented and fairly straightforward to execute properly. However, attention must be paid to the overall quality of the starting materials used, and in particular the integrity of the mRNA and pulldown components (EndoV and anti-MBP beads) are both critical to the success of this workflow. In addition, RNase contamination is an especially important consideration, and its prevention is paramount to robustly generating usable data.
mRNA Starting Material
The chemical integrity of mRNA used in EndoVIPER is a critical component towards eventual success in this protocol and its overall size distribution must be known. It is advisable that mRNA be either purchased commercially, as this is the material that was used for optimizing our protocol. RNA material freshly isolated from cells and purified away from total RNA may also work, but performance may vary. While it is possible that total RNA is compatible with EndoVIPER, we do not know for certain if other non-coding RNAs may interfere with performance and this protocol deviation will require testing and optimization. In any case, with mRNA material in hand, it is highly advisable to assess integrity of this initial material by either Bioanalyzer capillary electrophoresis (Agilent) or formaldehyde agarose gel (Rio, 2015) methods. A representative mRNA sample with good composition is shown in the top panel of Figure 4, illustrating long fragments that constitute a wide range of sizes. If mRNA has signs of degradation, this material can still likely be used in EndoVIPER. However, this starting composition must be taken into account when fragmenting mRNA prior to glyoxalation and pulldown. A final fragment size ~200–500 nt was optimized for this protocol and should be sought in the first series of experiments. Other sizes may still be functional, but may not produce desirable results and could require further optimization.
RNase Contamination
This protocol requires many steps that come in direct contact with RNA. A number of solutions must be made, and liquid is transferred between different tubes and supports throughout the entire workflow. It is critical that all of these components be of high quality and free of any nuclease contamination. In reactions, protein-based RNase inhibitors (such as RNaisin plus) are included to potentially inhibit any contaminating enzymes. However, we have tried small-molecule based inhibitors (such as ribonucleoside-vanadyl complex) and these have resulted in inhibition of EndoV as well and are hence not advisable to use. Benchtop water purifiers (such as MilliQ) that are rated for nuclease removal can be used for EndoVIPER. Many vendors also offer pre-packaged nuclease-free water and a variety of pack sizes are available that are useful for making buffer stocks. Diethyl pyrocarbonate (DEPC) is also sometimes used to inactivate RNase enzymes. We have not directly tested the effect of this additive on EndoVIPER performance, so we cannot guarantee that its use will not adversely effect results. In addition, it is advisable to regularly clean the workspace being used. We typically use RNase Zap (Thermo Fisher Scientific) as a topical spray to use on both flat surfaces as well as regularly cleaning gloved hands.
Integrity of Protein Components
The other critical and potentially labile components in this workflow are the actual EndoV fusion protein and the anti-MBP magnetic beads. Both of these items are commercially available and are tested by the manufacturer (New England Biolabs) for integrity and performance, and we have not encountered any problems in using freshly purchased and obtained material. Despite this, it is generally advisable to regularly replace inventory of these items (every 3–−6 months) to ensure optimal performance. In addition, EndoV concentrations can vary from lot to lot, so it is wise to contact NEB technical support to obtain lot-specific characterization data. They will provide this information upon request. The magnetic beads are stable when refrigerated but DO NOT freeze (−20 °C or lower) them at any time. NEB specifically notes user to refrain from doing this, as it may damage the bead support or irreversibly denature the antibody.
TROUBLESHOOTING
Table 1 below describes potential common problems with the protocols, their causes, and potential solutions.
Table 1.
Troubleshooting commonly encountered problems.
| Problem | Possible Cause | Solution |
|---|---|---|
| Starting mRNA integrity poor | Obtained material has been degraded, or purification protocol suboptimal, RNase contamination of material | Verify starting material again by Bioanalzyer or formaldehyde gel. If necessary, purchase new material or freshly purify from cellular origin |
| mRNA size distribution after fragmentation smaller than desired (< 200 nt) | Excessive hydrolysis caused by extended incubation at 94 °C with Mg2+ fragmentation kit | Reduce incubation time at 94 °C. Compare several different incubation times followed by size distribution analysis. |
| mRNA pellet not visible after precipitation | Omission of glycogen | Ensure addition of glycogen co-precipitant when performing EtOH precipitation of RNA, particularly when trying to precipitate small RNAs at low concentration. |
| mRNA pellet will not dissolve after glyoxalation | Excessive glyoxalation or overdrying of pellet after precipitation | Reduce glyoxal treatment time or limit drying step after EtOH precipitation. Hydrate pellet and leave undisturbed for 30 minutes or longer. Brief heating (30–60 second intervals) at 95 °C can also be used to aid pellet dissolving. |
| mRNA size distribution smaller after EndoVIPER | RNase degradation or chemical hydrolysis of material | Check integrity of all components used in assay and remake or order new items if needed. Additional RNase inhibitor may also help. |
| No detectable mRNA after EndoVIPER | Degradation or hydrolysis of material, loss of material during precipitation or purification steps | See above notes. Check integrity of all components. Ensure use of glycogen and practice care during precipitation and purification steps. |
UNDERSTANDING RESULTS
Expected results for basic protocol 1 can be seen in Figure 4. mRNA material with excellent integrity can be seen in the top panel, and obtained data should ideally match this standard. It is advisable to perform this analysis with any new potential mRNA sample or prior to enrichment. If starting material is partially degraded, it can likely still be used but this starting composition must be taken into account with optimizing fragmentation time as it will likely require shorter hydrolysis times to achieve the desired size distribution (200–500 nt). Size distribution and quantification results of mRNA before and after EndoVIPER pulldown (basic protocol 2) are shown in Figure 6. Overall concentration should be markedly reduced after pulldown, but size distribution should be roughly identical compared to input material.
TIME CONSIDERATIONS
Basic protocol 1 (mRNA fragmentation optimization) can be performed in a few hours, but it is advisable to fully optimize this portion of the protocol before moving on to other procedures. RNA material after fragmentation can be left to precipitate overnight and purified material can be stored frozen in water (−80 °C) indefinitely. mRNA glyoxalation only takes ~1 hour, but it is advisable to precipitate this material overnight because of the added DMSO in the reaction. After purification this material can also be frozen in water and at stored at −80 °C indefinitely. EndoVIPER pulldown takes ~2.5–3 hours for binding and immobilization onto magnetic beads, washing, and elution, and ~0.5 hours for initial purification. Final deprotection takes about 2–2.5 hours and can then be ethanol precipitated overnight. When ready to proceed to library preparation, users should dedicate an entire day to complete the procedure and to minimize distractions and opportunities for errors.
SIGNIFICANCE STATEMENT:
A-to-I RNA editing is critical for cellular function. While millions of editing sites are present across the transcriptome, detecting these sites is challenging with RNA-seq, which is prone to sampling errors. EndoVIPER-seq addresses this limitation by enriching edited RNAs prior to sequencing.
ACKNOWLEDGEMENTS
This work was supported by the National Institutes of Health (R01GM116991 to J.M.H.). This study was supported in part by the Emory Integrated Genomics Core (EIGC), which is subsidized by the Emory University School of Medicine and is one of the Emory Integrated Core Facilities. In particular we would like to thank Lyra Griffiths for her helpful advice and technical expertise in next-generation sequencing. We would also like to thank Viren Patel for his help in computational analysis.
Footnotes
INTERNET RESOURCES
Product information pages for both EndoV and anti-MBP magnetic bead components:
https://www.neb.com/products/m0305-endonuclease-v
https://www.neb.com/products/e8037-anti-mbp-magnetic-beads#Product%20Information
Product information page for SMARTER RNA-seq library preparation kit:
Product information page for commercially available RNA:
https://www.takarabio.com/products/cdna-synthesis/purified-total-rna-and-mrna
LITERATURE CITED
- Ahn J, & Xiao X (2015). RASER: reads aligner for SNPs and editing sites of RNA. Bioinformatics, 31(24), 3906–3913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass BL (2002). RNA editing by adenosine deaminases that act on RNA. Annu Rev Biochem, 71, 817–846. doi: 10.1146/annurev.biochem.71.110601.135501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delatte B, Wang F, Ngoc LV, Collignon E, Bonvin E, Deplus R, … Awe S (2016). Transcriptome-wide distribution and function of RNA hydroxymethylcytosine. Science, 351(6270), 282–285. [DOI] [PubMed] [Google Scholar]
- Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, … Kupiec M (2012). Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature, 485(7397), 201. [DOI] [PubMed] [Google Scholar]
- Edelheit S, Schwartz S, Mumbach MR, Wurtzel O, & Sorek R (2013). Transcriptome-wide mapping of 5-methylcytidine RNA modifications in bacteria, archaea, and yeast reveals m5C within archaeal mRNAs. PLoS genetics, 9(6), e1003602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inouye H, Fuchs S, Sela M, & Littauer UZ (1973). Detection of inosine-containing transfer ribonucleic acid species by affinity chromatography on columns of anti-inosine antibodies. Journal of Biological Chemistry, 248(23), 8125–8129. [PubMed] [Google Scholar]
- Kiran A, & Baranov PV (2010). DARNED: a DAtabase of RNa EDiting in humans. Bioinformatics, 26(14), 1772–1776. [DOI] [PubMed] [Google Scholar]
- Knutson SD, Ayele TM, & Heemstra JM (2018). Chemical labeling and affinity capture of inosine-containing RNAs using acrylamidofluorescein. Bioconjugate Chemistry, 29(9), 2899–2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knutson SD, Arthur RA, Johnston HR, & Heemstra JM (2020). Selective enrichment of A-to-I edited transcripts from cellular RNA using Endonuclease V. Journal of the American Chemical Society, 142(11), 5241–5251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Zhu P, Ma S, Song J, Bai J, Sun F, & Yi C (2015). Chemical pulldown reveals dynamic pseudouridylation of the mammalian transcriptome. Nature Chemical Biology, 11(8), 592. [DOI] [PubMed] [Google Scholar]
- Li Y, Göhl M, Ke K, Vanderwal CD, & Spitale RC (2019). Identification of adenosine-to-inosine RNA editing with acrylonitrile reagents. Organic letters. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita Y, Shibutani T, Nakanishi N, Nishikura K, Iwai S, & Kuraoka I (2013). Human endonuclease V is a ribonuclease specific for inosine-containing RNA. Nature communications, 4, 2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakes E, Vadlamani P, & Hundley HA (2017). Methods for the detection of adenosine-to-inosine editing events in cellular RNA mRNA Processing (pp. 103–127): Springer. [DOI] [PubMed] [Google Scholar]
- Paul MS, & Bass BL (1998). Inosine exists in mRNA at tissue‐specific levels and is most abundant in brain mRNA. The EMBOJournal, 17(4), 1120–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picardi E, D’Erchia AM, Lo Giudice C, & Pesole G (2016). REDIportal: a comprehensive database of A-to-I RNA editing events in humans. Nucleic acids research, 45(D1), D750–D757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picardi E, D’Erchia AM, Montalvo A, & Pesole G (2015). Using REDItools to detect RNA editing events in NGS datasets. Current Protocols in Bioinformatics, 49(1), 12.12.11–12.12.15. [DOI] [PubMed] [Google Scholar]
- Ramaswami G, & Li JB (2013). RADAR: a rigorously annotated database of A-to-I RNA editing. Nucleic Acids Research, 42(D1), D109–D113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rio DC (2015). Denaturation and electrophoresis of RNA with formaldehyde. Cold Spring Harbor Protocols, 2015(2), pdb. prot080994. [DOI] [PubMed] [Google Scholar]
- Tan MH, Li Q, Shanmugam R, Piskol R, Kohler J, Young AN, … Ariyoshi K (2017). Dynamic landscape and regulation of RNA editing in mammals. Nature, 550(7675), 249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vik ES, Nawaz MS, Andersen PS, Fladeby C, Bjørås M, Dalhus B, & Alseth I (2013). Endonuclease V cleaves at inosines in RNA. Nature Communications, 4, 2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams AG, Thomas S, Wyman SK, & Holloway AK (2014). RNA‐seq data: challenges in and recommendations for experimental design and analysis. Current Protocols in Human Genetics, 83(1), 11.13.11–11.13.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JH, Luo X, Nie Y, Su Y, Zhao Q, Kabir K, … Rabinovici R (2003). Widespread inosine‐containing mRNA in lymphocytes regulated by ADAR1 in response to inflammation. Immunology, 109(1), 15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao M, Hatahet Z, Melamede RJ, & Kow YW (1994). Purification and characterization of a novel deoxyinosine-specific enzyme, deoxyinosine 3’endonuclease, from Escherichia coli. Journal of Biological Chemistry, 269(23), 16260–16268. [PubMed] [Google Scholar]
- Yoshida M, & Ukita T (1965). Selective modifications of inosine and Ø-uridine with acrylonitrile out of the other ribonucleosides. The Journal of Biochemistry, 57(6), 818–821. [DOI] [PubMed] [Google Scholar]