

Key Points
TRIM14 interacts with TBK1 to promote STAT3 activation and limit IFNAR signaling.
Loss of TRIM14 in macrophages leads to hyperinduction of Ifnb and related ISGs.
Trim14−/− macrophages restrict M. tuberculosis growth by hyperinducing iNOS.
Visual Abstract
Abstract
Tripartite motif-containing proteins (TRIMs) play a variety of recently described roles in innate immunity. Although many TRIMs regulate type I IFN expression following cytosolic nucleic acid sensing of viruses, their contribution to innate immune signaling and gene expression during bacterial infection remains largely unknown. Because Mycobacterium tuberculosis is an activator of cGAS-dependent cytosolic DNA sensing, we set out to investigate a role for TRIM proteins in regulating macrophage responses to M. tuberculosis. In this study, we demonstrate that TRIM14, a noncanonical TRIM that lacks an E3 ubiquitin ligase RING domain, is a critical negative regulator of the type I IFN response in Mus musculus macrophages. We show that TRIM14 interacts with both cGAS and TBK1 and that macrophages lacking TRIM14 dramatically hyperinduce IFN stimulated gene (ISG) expression following M. tuberculosis infection, cytosolic nucleic acid transfection, and IFN-β treatment. Consistent with a defect in resolution of the type I IFN response, Trim14 knockout macrophages have more phospho-Ser754 STAT3 relative to phospho-Ser727 and fail to upregulate the STAT3 target Socs3, which is required to turn off IFNAR signaling. These data support a model whereby TRIM14 acts as a scaffold between TBK1 and STAT3 to promote phosphorylation of STAT3 at Ser727 and resolve ISG expression. Remarkably, Trim14 knockout macrophages hyperinduce expression of antimicrobial genes like Nos2 and are significantly better than control cells at limiting M. tuberculosis replication. Collectively, these data reveal an unappreciated role for TRIM14 in resolving type I IFN responses and controlling M. tuberculosis infection.
Introduction
Mycobacterium tuberculosis, one of the world’s most successful pathogens, elicits a carefully orchestrated immune response that allows bacteria to survive and replicate in humans for decades. Infection of macrophages with M. tuberculosis sets off a number of pathogen sensing cascades, most notably those downstream of TLR2, which senses mycobacterial lipomannan (1, 2), and cGAS, which senses bacterial DNA in the host cytosol (3–5). cGAS-dependent DNA sensing during M. tuberculosis infection elicits two distinct and somewhat paradoxical responses: targeting of a population of bacilli for destruction in lysosomes via ubiquitin-mediated selective autophagy (6), and activation of a type I IFN gene expression program (7), which is inadequate at controlling bacterial pathogenesis in vivo. Because both selective autophagy and type I IFN have been repeatedly shown in animal and human studies to be important in dictating M. tuberculosis infection outcomes (5–7), there is a critical need to elucidate the molecular mechanisms that regulate their activation.
Many members of the tripartite motif-containing protein (TRIM) family of proteins have emerged as important regulators of a variety of innate immune responses (8–10). Defined on the basis of their tripartite domain architecture, TRIMs generally encode an RING domain with E3 ubiquitin ligase activity, a B-box zinc-binding domain with an RING-like fold (11), and a coil-coiled domain that mediates dimer/multimerization and protein–protein interactions (12). In addition to these, TRIMs have highly variable C-terminal domains. Since the initial discovery of TRIM5 as a potent HIV restriction factor (13), a variety of TRIMs have been shown to play critical roles in antiviral innate immunity through polyubiquitination of key molecules in DNA and RNA sensing cascades, including MDA5 by TRIM13, TRIM40, and TRIM65 (14–17); RIG-I by TRIM25 and TRIM40 (14, 15); and TBK1 by TRIM11 and TRIM23 (16). We are just beginning to appreciate the complex and dynamic network of factors, including TRIMs, that cells employ to regulate innate immune signaling and gene expression (17).
Recent work has shown that cytosolic nucleic acid sensing pathways are engaged during infection with a variety of intracellular bacterial pathogens in addition to M. tuberculosis, including Legionella pneumophila, Listeria monocytogenes, Francisella novicida, and Chlamydia trachomatis (18). Some of these pathogens, like M. tuberculosis and C. trachomatis, have been shown to activate cGAS via bacterial dsDNA (3, 19), whereas others like L. monocytogenes directly activate STING by secreting cyclic di-AMP (20). It is becoming increasingly clear that activation of nucleic acid sensing pathways can benefit intracellular bacterial pathogens, so the ability to engage with and manipulate regulatory molecules like TRIM proteins is likely to be a conserved bacterial adaptation. Consistent with this notion, Salmonella Typhimurium has been shown to secrete SopA, an effector molecule that targets TRIM56 and TRIM65 to stimulate innate immune signaling through RIG-I and MDA5 (21, 22). Likewise, TRIM8 has been shown to regulate inflammatory gene expression downstream of TLR3 and TLR4 during S. Typhimurium–induced septic shock (23). In addition, ablation of TRIM72 in alveolar macrophages enhances phagocytosis and clearance of Pseudomonas aeruginosa (24).
Realizing the potential for TRIM proteins in tipping the balance between pro- and antibacterial innate immune outcomes, we sought to study TRIMs during M. tuberculosis infection, specifically a noncanonical TRIM family member, TRIM14. Like most TRIMs, TRIM14 encodes a coiled-coil, a B-box, and a C-terminal PRY/SPRY domain, but curiously, it lacks an E3 ubiquitin ligase RING domain, likely rendering it unable to catalyze ubiquitination events. Consistent with it being a major player in antiviral innate immunity, TRIM14 has been shown to directly influence replication of several RNA viruses including influenza A via interaction with the viral NP protein (25), hepatitis B via interaction with HBx (26), and hepatitis C via interaction with NS5A (27). In the context of RNA sensing, TRIM14 has been shown to localize to mitochondria and interact with the antiviral signaling adapter MAVS (28). More recently, TRIM14 has been shown to promote cGAS stability by recruiting the deubiquitinase USP14 and preventing autophagosomal targeting of cGAS (29).
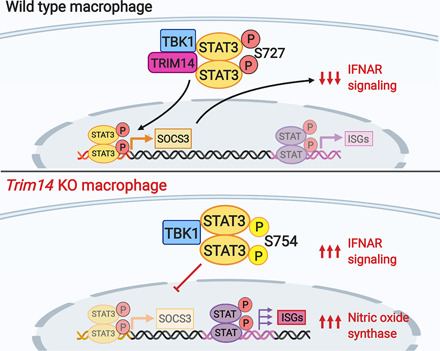
In this article, we demonstrate that TRIM14 is a crucial negative regulator of IFN-β and IFN stimulated gene (ISG) expression during macrophage infection with M. tuberculosis. TRIM14 was recruited to the M. tuberculosis phagosome and directly interacted with two proteins critical for cytosolic DNA sensing: the pattern recognition receptor cGAS and the kinase TBK1. Deletion of Trim14 led to dramatic hyperinduction of Ifnb and ISGs in response to M. tuberculosis and other cytosolic nucleic acid agonists. In Trim14 knockout (KO) macrophages we observed preferential phosphorylation of the transcription factor STAT3 at Ser754 and a lack of association of STAT3 with the chromatin loci of target genes like suppressor of cytokine signaling (Socs3), a negative regulator of IFN-α/β receptor (IFNAR) signaling. These data argue that TRIM14 acts as a negative regulator of type I IFN responses by bringing TBK1 and STAT3 together to promote phosphorylation of STAT3 at Ser727. Surprisingly, Trim14 KO macrophages were remarkably efficient at limiting M. tuberculosis replication by virtue of overexpressing inducible NO synthase (iNOS). Collectively, this work suggests that TRIM14 is a critical regulatory node of type I IFN induction and resolution in macrophages and highlights a previously unappreciated role for TRIM14 in cell-intrinsic anti–M. tuberculosis innate immunity.
Materials and Methods
Cell culture
RAW 264.7 macrophages, HEK293T, murine embryonic fibroblasts (MEF), and Lenti-X cells were cultured at 37°C with 5% CO2. Cell culture medium was composed of high glucose, sodium pyruvate, DMEM (Thermo Fisher Scientific) with 10% FBS (Sigma-Aldrich), 0.5% HEPES (Thermo Fisher Scientific).
Primary cell culture
Bone marrow–derived macrophages (BMDMs) were differentiated from bone marrow cells isolated by washing mouse femurs with 10 ml DMEM. Cells were then centrifuged for 5 min at 1000 rpm and resuspended in BMDM media (DMEM, 20% FBS, 1 mM sodium pyruvate, 10% MCSF conditioned media). Bone marrow cells were counted and plated at 5 × 106 in 15-cm non–tissue culture–treated dishes in 30 ml complete media and fed with an additional 15 ml of media on day 3. On day 7, cells were harvested with 1× PBS-EDTA.
Coimmunoprecipitations
A total of 1.8 × 106 HEK293T cells in a 10-cm plate were transfected with 1–10 μg of pDEST 3xFlag TRIM14, pDEST hemagglutinin (HA) cGAS, pDEST HA-STING, pDEST HA TBK1, pDEST HA IRF3, pDEST Flag STAT3, or pDEST HA TRIM14 using PolyJet In Vitro DNA Transfection Reagent. Cells were harvested in PBS plus 0.5 M EDTA 24 h posttransfection, and pellets were lysed on ice with lysis buffer (50 mM Tris HCl [pH 7.4], 150 mM NaCl, 1 mM EDTA, 0.075% NP-40) containing protease inhibitor (A32955; Pierce). Strep-Tactin Superflow Plus beads (Qiagen) were washed using buffer containing 5% 1 M Tris [pH 7.4], 3% NaCl, and 0.2% 0.5 M EDTA. One milliliter of the cleared lysate was added to the beads and inverted for 2 h at 4°C. Beads were then washed four times with wash buffer (50 mM Tris HCl [pH 7.4] 150 mM NaCl 0.5 M EDTA, 0.05% NP-40) and eluted using 1× Biotin. Whole-cell lysate inputs and elutions were boiled in 4× SDS loading buffer with 10% 2-ME. Proteins were run on SDS-PAGE gels (Bio-Rad Laboratories) and then transferred to nitrocellulose membrane (GE Healthcare). Membranes were blocked in TBS (Odyssey Blocking buffer; Li-COR) for 1 h and incubated with primary Ab overnight at 4°C. LI-COR secondary was used (IRDye CW 680 goat anti-rabbit, IRDye CW 680 goat anti-rat 680, IRDye CW800 goat anti-mouse [LI-COR]) and developed with the Odyssey Fc by LI-COR. Immunoprecipitation experiment were also performed as stated above but with Pierce Anti-HA agarose (26181; Thermo Fisher Scientific). Beads were eluted three times at room temperature for 15 min each using Influenza HA peptide (I2149; Sigma-Aldrich).
Immunoblot analysis
Protein samples were run on Any kDa Mini-PROTEAN TGX precast protein gels (Bio-Rad Laboratories) and transferred to 0.45 μm nitrocellulose membranes (GE Healthcare). Membranes were incubated in the primary Ab of interest overnight and washed three times with TBST. Membranes were then incubated in secondary Ab for 1 h and imaged using LI-COR Odyssey FC Imaging System. Primary Abs used in this study were mouse monoclonal α-FLAG M2 Ab (F3165; Sigma-Aldrich), α-HA high affinity rat mAb (3F10; Roche), α-strep (A00626; Genscript), α-phospho-Stat3 (Ser727) (no. 9134; Cell Signaling Technology), α-phospho-Stat3 (Ser754) (no. 98543; Cell Signaling Technology), α-Stat3 (124H6) Mouse mAb (no. 9139; Cell Signaling Technology), α-phospho-Stat1 (Tyr701) (58D6) Rabbit mAb no. 9167, α-TRIM14 G-15 (sc79761; Santa Cruz Biotechnology), α-TRIM14 (ARP34737; Aviva), and α-mouse monoclonal β-Actin (no. 6276; Abcam). Secondary Abs used in this study were IRDye CW 680 goat anti-rabbit, IRDye CW 680 goat anti-rat 680, and IRDye CW800 goat anti-mouse (LI-COR).
Construction of single guide RNA/Cas9 LentiCRISPR and viral transduction
Guide RNAs (gRNAs) targeting the first exon of Trim14 were designed using the Broad online tool (https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design). The top five hits were used to design five gRNA constructs that were cloned into Lenti CRISPR vector (Puromycin) at the BsmB1 site. Constructs were sequenced and verified. Plasmids were then transfected into Lenti-X cells with PAX2 and VSVG packing plasmids. Virus was collected 24 and 48 h posttransfection and stored at −80°C. RAW 264.7 cells stably expressing Cas9 were then transduced with virus and selected using puromycin for 72 h. Cells were then clonally selected using serial dilutions and clones were selected from wells calculated to contain a single cell. To verify the KO, genomic DNA was collected from cells, and the first exon of Trim14 was amplified using PCR. This reaction was sent for sequencing to verify the mutation.
Generation of short hairpin RNA–expressing stable cell lines
To generate knockdown (KD) RAW 264.7 macrophages, plasmids of scramble nontargeting short hairpin RNA (shRNA) constructs and Trim14 shRNA constructs targeted toward the 3′ untranslated region of Trim14 were transfected into Lenti-X cells with PAX2 and VSVG packing plasmids. Virus was collected 24 and 48 h posttransfection and stored at −80°C. RAW 264.7 cells were then transduced with virus and selected using hygromycin at 10 mg/ml (Invitrogen) to select for cells containing the shRNA plasmid.
Small interfering RNA KD
To knock down Socs3 expression in RAW 264.7 macrophages, we followed the same approach as described in (30) but with an Socs3-specific small interfering RNA (siRNA) (assay identifier 160219; Thermo Fisher Scientific).
Macrophage stimulation
RAW 264.7, CRISPR/Cas9 RAW 264.7, or shRNA RAW 264.7 macrophages were plated on 12-well tissue culture–treated plates at a density of 3 × 105 cells per well and allowed to grow overnight. Cells were then transfected with 1 μg/ml ISD or 1 μg/ml poly(I:C) with lipofectamine or treated with 200 U recombinant mouse IFN-β (Cat. no. 12400-1; PBL Assay Science).
M. tuberculosis infection
Low passaged laboratory stocks of each M. tuberculosis strain (Erdman wild-type [WT], Erdman luxBCADE, or Erdman mCherry) were thawed for each experiment to ensure virulence was preserved. M. tuberculosis was cultured in roller bottles at 37°C in Middlebrook 7H9 broth (BD Biosciences) supplemented with 10% OADC, 0.5% glycerol, and 0.1% Tween 80. All work with M. tuberculosis was performed under Biosafety Level 3 containment using procedures approved by the Texas A&M University Institutional Biosafety Committee. To prepare the inoculum, bacteria grown to log phase (OD 0.6–0.8) were spun at low speed (500 g) to remove clumps and then pelleted and washed with PBS twice. Resuspended bacteria were briefly sonicated and spun at low speed once again to further remove clumps. The bacteria were diluted in DMEM plus 10% horse serum and added to cells at a multiplicity of infection (MOI) of 10 for RNA and cytokine analysis and MOI of 1 for microscopy studies. Cells were spun with bacteria for 10 min at 1000 × g to synchronize infection, washed twice with PBS, and then incubated in fresh media. Prior to infection, BMDMs were seeded at 3 × 105 cells per well (12-well dish), and RAW 264.7 or CRISPR/Cas9 RAW 264.7 macrophages were plated on 12-well tissue culture–treated plates at a density of 3 × 105 cells per well and allowed to grow overnight. Where applicable, RNA was harvested from infected cells using 0.5 ml TRIzol reagent at each time point.
M. tuberculosis survival/replication
RAW 264.7 or CRISPR/Cas9 RAW 264.7 macrophages were plated on 12-well tissue culture–treated plates at a density of 2.5 × 105 cells per well. Luminescence was read for M. tuberculosis luxBCADE by lysing in 400 μl 0.5% Triton X-100 and splitting into two wells of a 96-well white plate and using the luminescence feature of the INFINITE 200 PRO by TECAN at 0, 24, 48, and 72 h postinfection. For CFU enumeration, cells were lysed in 500 μl PBS plus 0.1% Tween 80, and serial dilutions were plated on 7H11 plates. Colonies were counted after plates were incubated at 37°C for 2 wk. For iNOS inhibitor experiments, RAW 264.7 or CRISPR/Cas9 RAW 264.7 macrophages were plated on 12-well tissue culture–treated plates at a density of 2.5 × 105 cells per well overnight and treated with 1400W dihydrochloride (W4262; Sigma-Aldrich) at 0, 10, or 100 μM final concentration.
RNA isolation and real-time quantitative PCR analysis
To analyze transcripts, cells were harvested in TRIzol at the specified time points and RNA was isolated using Direct-zol RNA Miniprep kits (Zymo Research) with 1 h DNase treatment. cDNA was synthesized with iScript cDNA Synthesis Kit (Bio-Rad Laboratories). cDNA was diluted to 1:20 for each sample. A pool of cDNA from each treated or infected sample was used to make a 1:10 standard curve, with each standard sample diluted 1:5 to produce a linear curve. Real-time quantitative PCR (RT-qPCR) was performed using Power-Up SYBR Green Master Mix (Thermo Fisher Scientific) using a Quant Studio Flex 6 (Applied Biosystems). Samples were run in triplicate wells in a 96-well or 384-well plate. Averages of the raw values were normalized to average values for the same sample with the control gene, Bact (β-actin). To analyze fold induction, the average of the treated sample was divided by the untreated control sample, which was set at 1.
ELISAs
Supernatants were collected at 8 and 24 h and analyzed using ELISA for IFN-β. Briefly, Nanopore Maxisorp 96-well plates were coated with 50 μl of capture Ab diluted 1:500 (no. sc-57201; Santa Cruz Biotechnology) in 0.1 M carbonate buffer and were incubate 4°C overnight. Samples were then blocked using PBS plus 10% FBS for 2 h at 37°C. Samples were loaded at a 1:4 dilution for a total of 50 μl added to each well. Standard (12400-1; PBL Assay Science) was diluted 1:4 for serial dilutions and incubated at room temperature overnight. Samples were washed with PBS plus 0.05% Tween before each step. Detection Ab (32400-1; R&D Systems) was added at a 1:2000 dilution and incubated at room temperature overnight. After washing, secondary Ab (7074; Cell Signaling Technology) was added at a 1:2000 dilution and incubated for 3 h. Following washes, TMB substrate (SeraCare) was added and the reaction was stopped with 2 N H2SO4. ELISA was read at 450 nm.
Immunofluorescence microscopy
Glass coverslips were incubated in 100 μl poly-lysine at 37°C for 30 min. MEF cells were plated at a density of 2 × 104 on glass coverslips in 24-well plates and left to grow overnight. Cells were then transfected with 250 ng of the desired plasmid(s) using PolyJet. The next day, cells were treated with 1 μg ISD as described above. At the designated time points, cells were washed with PBS (Thermo Fisher Scientific) and then fixed in 4% paraformaldehyde for 10 min. Fixed cells were washed three times in PBS and permeabilized by incubating them in PBS containing 5% nonfat milk and 0.05% saponin (Calbiochem). Coverslips were placed in primary Ab for 1 h, then washed three times in PBS and placed in secondary Ab (Alexa Fluor 488, 594, or 647 for corresponding species [Invitrogen]).These were washed twice in PBS and twice in deionized water, followed by mounting onto a glass slide using ProLong Diamond antifade mountant (Invitrogen). Images were acquired on a Nikon A1-Confocal Microscope. DAPI was used for nuclear staining (Thermo Fisher Scientific).
Colocalization experiments with M. tuberculosis
RAW 264.7 macrophages were plated on glass coverslips at a density of 1.5 × 105 cells per well in 24-well plates. Cells were infected with mCherry-expressing M. tuberculosis at an MOI of 1 and fixed in 4% paraformaldehyde for 20 min. Coverslips were stained as above using α-TRIM14 (clone G-15, sc79761; 1:100; Santa Cruz Biotechnology), α-ubiquitin (clone FK2, 04-263; 1:500; Millipore), α-LC3 (L10382; 1:200; Thermo Fisher Scientific), α-p62 (A302-855A; 1:300; Bethyl Laboratories), and α-TAX1BP1 (A303-791A; 1:300; Bethyl Laboratories) primary Abs and Alexa Fluor 488 anti-goat and Alexa Fluor 647 anti-rabbit or anti-mouse (Invitrogen) secondary Abs. Colocalization with M. tuberculosis was visualized by fluorescence microscopy. A series of images were captured and analyzed by counting the number of bacteria that colocalized with the corresponding marker. At least 100 events were analyzed per coverslip, and each condition was performed with triplicate coverslips. Images were acquired on a Nikon A1-Confocal Microscope or an Olympus Fluoview FV3000 Confocal Laser Scanning Microscope.
STAT3 translocation
RAW 264.7 cells were plated on glass coverslips as described above and transfected with 100 μg ISD. At 6 h posttransfection, cells were fixed in 4% PFA for 10 min and washed twice with PBS. Cells were permeabilized for 20 min in PBS with 5% nonfat dry milk and 0.5% Triton X-100. Using the same buffer for Ab dilutions, coverslips were stained with α-STAT3 (no. 9139; Cell Signaling Technology) for 3 h, washed, stained with Alexa Fluor 488 and DAPI for 1 h, washed, and mounted. Cells were imaged on a Biotek Lionheart Automated Microscope, and STAT3 translocation was quantified using Gen5 software. Total and nuclear STAT3 was quantified in transfected and untreated cells, and DAPI signal was used as the primary mask to delineate the nuclear compartment.
Protein expression and purification
The cDNA encoding mouse Trim14 (residues 247–440) and mouse Irf3 (residues 184–419) were cloned into a modified pET28(a) vector containing an N-terminal Avi-His6-SUMO tag. Sequences of the plasmids were confirmed by DNA sequencing. The BL21 (DE3) cells were cotransformed with the pET28(a) plasmids coding for the target proteins and the pBirAcm plasmid coding for BirA and induced with 0.4 mM IPTG in the presence of 5 μg ml−1 biotin and cultured at 16°C overnight. The Biotin-labeled Avi-His6-SUMO proteins were purified using a nickel-NTA column followed by gel-filtration chromatography using a HiLoad 16/60 Superdex 75 column (GE Healthcare). Mouse and human TBK1 (residues 1–657) were cloned into the pAcGHLTc baculovirus transfer vector. The plasmid was transfected together with Baculo-Gold bright linearized baculovirus DNA (BD Biosciences) into sf9 insect cells to generate recombinant baculovirus. The original recombinant viruses were amplified for at least two rounds before the large-scale protein expression. The insect cells at a density of 2.5 × 106 cells/ml were infected by TBK1 recombinant baculovirus and cultured at 27°C and harvested 72 h postinfection. The cells were lysed in the buffer containing 150 mM NaCl, 0.2 M Tris HCl, 1% NP-40, 1 mM PMSF [pH 8]. The target protein in the supernatant was purified using nickel affinity chromatography followed by size-exclusion chromatography.
Surface plasmon resonance binding study
The binding studies between mouse TRIM14 and TBK1 were performed using a Biacore X100 surface plasmon resonance (SPR) instrument (GE Healthcare). Biotin-labeled SUMO-fusion TRIM14 was coupled on the sensor chip SA (GE Healthcare). Dilution series of TBK1 or IRF3 (1.25, 2.5, 5, 10, 20 μM) in 1× HBS-EP+ buffer (GE Healthcare) were injected over the sensor chip at a flow rate of 30 μl/min. The single-cycle kinetic/affinity protocol was used in all binding studies. All measurements were duplicated under the same conditions. The equilibrium Kd was determined by fitting the data to a steady-state 1:1 binding model using Biacore X100 Evaluation software version 2.0 (GE Healthcare).
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) was adapted from Abcam’s protocol. Briefly, one confluent 15-cm dish of CRISPR/Cas9 RAW 264.7 macrophages was cross-linked in formaldehyde to a final concentration of 0.75% and rotated for 10 min. Glycine was added to stop the cross-linking by shaking for 5 min at a concentration of 125 mM. Cells were rinsed with PBS twice and then scraped into 5 ml PBS and centrifuged at 1000 × g for 5 min at 4°C. Cellular pellets were resuspended in ChIP lysis buffer (50 mM HEPES-KOH [pH 7.5], 140 mM NaCl, 1 mM EDTA [pH 8], 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS protease inhibitors) (750 μl per 1 × 107 cells) and incubated for 10 min on ice. Cellular lysates were sonicated for 40 min (30 s on, 30 s off) on high in a Bioruptor UCD-200 (Diagenode). After sonication, cellular debris was pelleted by centrifugation for 10 min, 4°C, 8000 × g. Input samples were taken at this step and stored at −80°C until decross-linking. Approximately 25 μg of DNA diluted to 1:10 with RIPA buffer was used for overnight immunoprecipitation. Each ChIP had one sample for the specific Ab and one sample for Protein G beads only that were preblocked for 1 h with single-stranded herring sperm DNA (75 ng/μl) and BSA (0.1 μg/μl). The respective primary Ab was added to all samples except the beads-only sample at a concentration of 5 μg and rotated at 4°C overnight. Beads were washed three times with a final wash in high salt (500 mM NaCl). DNA was eluted with elution buffer and rotated for 15 min at 30°C. Centrifuge for 1 min at 2000 × g and transfer the supernatant into a fresh tube. Supernatant was incubated in NaCl, RNase A (10 mg/ml), and proteinase K (20 mg/ml) and incubated at 65°C for 1 h. DNA was purified using phenol/chloroform extraction. DNA levels were measured by RT-qPCR. Primers were designed by tiling each respective gene every 500 bp that were inputted into National Center for Biotechnology Information primer design.
mRNA sequencing
RNA was sequenced from four biological replicates for each condition; uninfected BMDMs, ESX-1–infected BMDMs, and M. tuberculosis–infected BMDMs. Raw reads were processed with expHTS to trim low-quality sequences and adapter contamination and to remove PCR duplicates. Trimmed reads for each sample were aligned to the GRCm38 GENCODE primary genome assembly using STAR v.2.5.2b aligner and the GENCODE v.M10 annotation (gtf file). Each of the four replicates were merged into a single BAM file for further analysis. Prior to analysis, genes with expression <2 counts per million reads were filtered, leaving 11,808 genes. Differential gene expression was conducted using a single factor ANOVA model in the limma-voom Bioconductor pipeline. Log2 fold change values with a p value <0.05 are represented in heatmaps where uninfected samples were the denominator and ESX-1 or M. tuberculosis–infected samples were the numerator in their respective datasets. Heatmaps were generated with GraphPad Prism Software.
Vesicular stomatitis virus infection
RAW 264.7 cells were seeded in 12-well plates at 8 × 105 16 h before infection. Cells were infected with vesicular stomatitis virus (VSV)-GFP virus at MOI of 1 in serum-free DMEM (SH30022.01; HyClone). After 1 h of incubation with media containing virus, supernatant was removed, and fresh DMEM plus 10% FBS was added to each well. At indicated times postinfection, cells were harvested with TRIzol and prepared for RNA isolation.
Statistics
Statistical analysis of data were performed using GraphPad Prism software (GraphPad). Two-tailed unpaired Student t tests were used for statistical analyses, and unless otherwise noted, all results are representative of at least three independent biological experiments and are reported as the mean ± SD (n = 3 per group).
Results
TRIM14 is upregulated during M. tuberculosis infection and localizes to the M. tuberculosis–containing phagosome
Having previously described a crucial role for the ESX-1 virulence-associated secretion system in eliciting cGAS-dependent cytosolic DNA sensing and type I IFN expression during M. tuberculosis infection (3, 6, 7), we first set out to better define gene expression differences in macrophages infected with a wild-type and a ΔESX-1 strain. Briefly, we infected BMDMs with wild-type M. tuberculosis (Erdman strain) and the Tn5370::Rv3877/EccD1 mutant (ΔESX-1) (31), which lacks a functional ESX-1 secretion system and has been shown to be defective in eliciting cGAS-dependent responses (7, 32). We performed RNA-sequencing (RNA-seq) at an established key innate immune time point of 4 h and an average log2 fold change of four biological replicates is shown (p < 0.05) (Supplemental Fig. 1A). Consistent with previous microarray and RNA-seq data (7, 33), we observed dramatic induction of proinflammatory cytokines (Il6, Il1b) and antimicrobial molecules like Nos2 in macrophages infected with both wild-type and ΔESX-1 M. tuberculosis (Supplemental Fig. 1A). We also observed downregulation of several protein-coding genes (Epha2, Gpr34, Rtn4rl1) and noncoding RNAs (Gm13391, Gm15564, Gm24270) (Supplemental Fig. 1B). To identify genes/pathways whose induction required ESX-1 secretion, we performed Ingenuity Pathway Analysis (Qiagen) and found “IFN signaling” and “Activation of IRF by Cytosolic PRRs” as the major pathways enriched for in an ESX-1–dependent manner (Supplemental Fig. 1C). This analysis is in agreement with earlier data demonstrating a requirement for ESX-1 phagosome permeabilization in activating type I IFN expression downstream of cGAS (7). We next used RT-qPCR to measure expression of a number of important innate immune transcripts, both in BMDMs to validate our RNA-seq results and in RAW 264.7 murine macrophage-like cells (Supplemental Fig. 1D) to justify our use of these genetically tractable cells moving forward. In both cell types, maximal ISG induction was ESX-1 dependent [consistent with ESX-1 secretion being required for perturbation of the M. tuberculosis–containing phagosome and release of M. tuberculosis–derived dsDNA into the cytosol, where it activates the cGAS/STING/IRF3 DNA sensing axis (3)], whereas expression of NF-κB genes was ESX-1 independent [consistent with M. tuberculosis surface lipoarabinomannan and lipomannan activating NF-κB via TLR2 signaling (34)] (Supplemental Fig. 1D). In analyzing the lists of ESX-1–dependent upregulated genes, we noticed that several belonged to the TRIM family, consistent with TRIMs themselves being ISGs (Fig. 1A) (35). Because so little is known about how TRIM proteins regulate antibacterial immunity, we set out to better understand how TRIMs influence cGAS-dependent innate immune outcomes during M. tuberculosis infection.
FIGURE 1.

TRIMs are players in the innate immune response to M. tuberculosis. (A) Heatmap of significant (p < 0.05) gene expression differences (log2 fold-change) in TRIM family genes in uninfected BMDMs versus BMDMs infected with WT M. tuberculosis. MOI = 10. (B) RT-qPCR of fold-change in Trim14 transcripts in BMDMs infected with WT M. tuberculosis or stimulated with ISD (n = 3 biological replicates). (C) RAW 264.7 cells infected with mCherry M. tuberculosis (Mtb, red) for 6 h and immunostained for TRIM14 (green); nuclei stained with DAPI (blue). (D) As in (C) with the addition of costaining for endogenous selective autophagy markers (purple). Quantification shows the proportion of TRIM14+ bacilli that are also positive for indicated selective autophagy marker. Statistical significance was determined using two-tailed Student t test. ***p < 0.001.
Based on its recent characterization as a regulator of cGAS stability (29), we elected to investigate the role of TRIM14 during M. tuberculosis infection. RT-qPCR analysis confirmed that Trim14 expression was modestly upregulated after M. tuberculosis infection of RAW 264.7 cells (Fig. 1B). Transfection of RAW 264.7 cells with dsDNA (ISD, IFN stimulatory DNA) (36) recapitulated this effect, albeit with slightly different kinetics (Fig. 1B). This result suggests that Trim14 upregulation during M. tuberculosis infection occurs downstream of cytosolic DNA sensing. To further implicate TRIM14 in M. tuberculosis infection of macrophages, we asked whether TRIM14 protein colocalized with the M. tuberculosis phagosome. Using immunofluorescence microscopy and an Ab against endogenous TRIM14, we detected TRIM14 at ∼30% of M. tuberculosis phagosomes (Fig. 1C) (6). We have previously shown that 30% is the typical percentage of M. tuberculosis–containing phagosomes that become LC3+ over the course of early macrophage infection. To confirm that the TRIM14+ population of M. tuberculosis phagosomes were indeed positive for other selective autophagy markers, we costained M. tuberculosis–infected macrophages for TRIM14 and for ubiquitin, the selective autophagy adapters p62 or TAX1BP1, or the autophagosome marker LC3 (Fig. 1D). As predicted, virtually all TRIM14+ M. tuberculosis phagosomes were also positive for these autophagosomal markers (Fig. 1D). Together, these data begin to suggest that TRIM14 is a player in the macrophage response to M. tuberculosis.
TRIM14 interacts with components of the DNA sensing pathway
Based on its recruitment to the M. tuberculosis phagosome, we hypothesized that TRIM14 may interact with one or more components of the cytosolic DNA sensing pathway (e.g., cGAS, TBK1), which we have previously observed at the M. tuberculosis phagosome (model in Fig. 2A and Ref. 3). To test this, we transfected epitope-tagged versions of mouse TRIM14 (3xFLAG-TRIM14) and major components of the DNA sensing pathway (mouse HA-cGAS, HA-STING, and HA-TBK1) into MEFs. Following 24 h of expression, cells were fixed and coimmunostained. Generally, 3xFLAG-TRIM14 showed a punctate pattern with some perinuclear enrichment. Consistent with a previous report (24), we observed that a population TRIM14 colocalized with a portion of the cytosolic fraction of cGAS (Fig. 2B), but little to no 3xFLAG-TRIM14 colocalized with HA-STING. Intriguingly, we also observed overlap between a fraction of the 3xFLAG-TRIM14 signal and that of HA-TBK1, which suggests that TRIM14 may interact with more than one component of the cytosolic DNA sensing pathway.
FIGURE 2.

TRIM14 interacts with both cGAS and TBK1 in the DNA sensing pathway. (A) Model of DNA sensing pathway during M. tuberculosis infection. Model created using BioRender software (B) Immunofluorescence microscopy of MEFs expressing 3xFLAG-TRIM14 with HA-cGAS, HA-STING, or HA-TBK1 costained with α-HA and α-FLAG Abs. Nuclei stained with DAPI (blue). (C) Immunoblot analysis of coimmunoprecipitation of 3xFLAG-TRIM14 coexpressed with HA-cGAS, HA-STING, HA-TBK1, or HA-IRF3 in HEK293T cells. Blot is representative of >3 independent biological replicates. (D) Diagram of mTRIM14, mTBK1, hTBK1, mIRF3 gene domains and truncations used in SPR studies. (E) Equilibrium binding study of mTRIM14 and mTBK1 by SPR. mIRF3 was used as a negative control. Dissociation constant (Kd = 24.3 μM) was derived by fitting of the equilibrium binding data to a one-site binding model. (F) As in (E) but with mTRIM14 and hTBK1. Dissociation constant (Kd = 42.6 μM) was derived by fitting of the equilibrium binding data to a one-site binding model.
To further characterize this novel association between TRIM14 and TBK1, we coexpressed mouse 3xFLAG-TRIM14 with mouse HA-cGAS, HA-STING, HA-TBK1, and HA-IRF3 in HEK293T cells, immunopurified each of the DNA sensing pathway components, and probed for TRIM14 by immunoblot. Consistent with our immunofluorescence microscopy data, we found that TRIM14 coimmunoprecipitated with both cGAS and TBK1 but not with STING or IRF3 (Fig. 2C).
Next, to determine whether these biochemical associations were direct interactions, we performed SPR experiments. Briefly, truncated versions of mouse TRIM14 (residues 247–440), human cGAS (residues 157–522), and mouse and human TBK1 (residues 11–657) were expressed using a baculovirus system. A portion of mouse IRF3 (residues 184–419) served as the negative control (Fig. 2D). Each of these protein truncations had previously been shown to stably express at high levels and remain soluble in insect cells (Supplemental Fig. 2A) (37–39). Equilibrium binding studies measured a binding affinity of 24.3 μM for binding between mTRIM14 and mTBK1 (Fig. 2E) and a slightly lower affinity of 42.6 μM for mouse TRIM14 and a portion of human TBK1 (Fig. 2F). Mouse TRIM14 and full-length hTBK1 also showed direct binding (Kd = 11 μM) (Supplemental Fig. 2D), as did human cGAS and mouse TRIM14 (Kd = 25.8 μM) (Supplemental Fig. 2B, 2C). No binding was detected between mTRIM14 and mIRF3 in any of the experiments (Fig. 2E, 2F, Supplemental Fig. 2C, 2D). Combined, these in vivo and in vitro biochemical data strongly indicate a direct interaction between TRIM14 and TBK1.
Loss of TRIM14 leads to type I IFN and ISG hyperinduction during M. tuberculosis infection
To investigate the contribution of TRIM14 to cytosolic DNA sensing outcomes during M. tuberculosis infection, we first tested how KD of Trim14 affects Ifnb gene expression. Trim14 KD macrophages were generated by transducing RAW 264.7 cells with lentiviral shRNA constructs designed to target the 3′ untranslated region of Trim14 or a control scramble shRNA (SCR). RT-qPCR analysis confirmed ∼50 and 70% KD of Trim14 using two different shRNA constructs (KD1 and KD2, respectively) (Supplemental Fig. 3A). Using these Trim14 KD and SCR control RAW 264.7 cells, we either infected with M. tuberculosis or transfected with ISD to directly engage cGAS and then measured Ifnb transcripts after 4 h. In both stimulations, we observed lower levels of Ifnb transcript induction in Trim14 KD cells compared with SCR control cells (Supplemental Fig. 3B, 3C). Because residual levels of TRIM14 protein in the KD cell lines could potentially complicate the interpretation of phenotypes, we next generated Trim14 KO using CRISPR-Cas9. Briefly, Trim14-specific gRNAs were designed to target Trim14 exon 1, and a GFP-specific gRNA was designed as a negative control. Two clonally expanded cell lines with distinct frameshift mutations, each of which introduced a stop codon early in exon 1, were identified and used for subsequent experiments (Fig. 3A). KO of Trim14 was confirmed by immunoblot using an Ab against the endogenous protein and by anti-TRIM14 immunofluorescence of control and Trim14 KO cells (Fig. 3B).
FIGURE 3.

Loss of TRIM14 leads to hyperinduction of Ifnb in response to M. tuberculosis and cytosolic DNA. (A) Sequencing chromatogram depicting mutations in Trim14 gRNA CRISPR-Cas9 RAW 264.7 macrophages compared with GFP gRNA control (WT). (B) Immunoblot analysis and immunofluorescence microscopy of Trim14 in WT versus Trim14 KO RAW 264.7 macrophages using an endogenous α-TRIM14 Ab. (C) RT-qPCR of Ifnb transcripts in WT and Trim14 KO RAW 264.7 macrophages infected with M. tuberculosis at specified times postinfection. (D) RT-qPCR of Ifnb transcripts in WT and Trim14 KO RAW 264.7 macrophages treated with ISD at specified times after treatment. (E) RT-qPCR of Ifnb transcripts in WT and Trim14 KO RAW 264.7 macrophages treated with poly(I:C) at specified times after treatment. (F) IFN stimulated response element reporter cells expressing luciferase with relative light units measured as a readout for IFN-β protein. (G) IFN-β protein ELISA of WT and Trim14 KO RAW 264.7 macrophages treated with ISD. All RT-qPCRs represent n = 3 biological replicates. Statistical significance was determined using two-tailed Student t test. ***p < 0.001.
To test how genetic ablation of Trim14 affects cytosolic DNA sensing, we infected Trim14 KO and control macrophages with wild-type M. tuberculosis and collected RNA over a 24-h time course of infection. Surprisingly, although we again found lower Ifnb expression at 4 h postinfection, we observed a dramatic hyperinduction of Ifnb in the absence of TRIM14 at later infection time points (Fig. 3C). To determine the contribution of cytosolic DNA sensing to this phenotype, we transfected Trim14 KO and control cells with ISD and again measured significantly higher induction of Ifnb in the absence of TRIM14 at 8 h posttransfection (Fig. 3D). We also observed Ifnb hyperinduction in Trim14 KO cells transfected with poly(I:C), which stimulates cytosolic dsRNA sensing via RIG-I and MAVS, with slightly faster kinetics (Fig. 3E). To verify that the transcriptional changes we observed translated to differences in protein levels, we used IFN stimulated response element luciferase reporter cells to analyze IFN-β protein secretion in supernatants from cells 8 and 24 h after M. tuberculosis infection. Using relative light units as a proxy for IFN-β, we indeed observed higher IFN-β levels in the absence of TRIM14 (Fig. 3F). We also found higher levels of IFN-β protein in the supernatants of Trim14 KO macrophages 8 h after ISD transfection using direct measurement via ELISA (Fig. 3G). Together, these data suggest that one major phenotype associated with Trim14 ablation in RAW 264.7 macrophages is hyperinduction of Ifnb downstream of cytosolic DNA or RNA sensing.
Having observed higher levels of both Ifnb transcript and protein in Trim14 KO RAW 264.7 macrophages, we predicted that these cells would also hyperinduce ISGs following treatment with innate immune agonists that stimulate IRF3 signaling (downstream of cGAS) or STAT signaling (downstream of IFNAR). Indeed, RT-qPCR analysis of RNA from a time course of either M. tuberculosis infection or ISD transfection showed hyperinduction of Ifit1, Isg15, and Irf7 (Fig. 4A, 4B). Likewise, high levels of these same ISGs were observed in cells transfected with 1 μg poly(I:C) (Fig. 4C) or treated directly with rIFN-β (Fig. 4D). Importantly, non-ISGs like Il1b, Tnfa, and Il6 were induced to equivalently high levels during M. tuberculosis infection in Trim14 KO and control cells (Supplemental Fig. 3D). Collectively, these results demonstrate that although TRIM14 is needed for the initial induction of Ifnb downstream of cGAS-dependent cytosolic DNA sensing, it also plays a crucial role in resolving the response at later time points, arguing for a model whereby TRIM14 regulates the type I IFN response in two distinct ways.
FIGURE 4.

Loss of TRIM14 leads to hyperinduction of ISGs in response to multiple innate immune agonists. (A) RT-qPCR of Ifit1, Isg15, and Irf7 transcripts in WT and Trim14 KO RAW 264.7 macrophages infected with M. tuberculosis at specified times postinfection. (B) RT-qPCR of Ifit1, Isg15, and Irf7 transcripts in WT and Trim14 KO RAW 264.7 macrophages transfected with 1 μg ISD. (C) As in (B) but with transfection of 1 μg poly(I:C). (D) As in (B) but with IFN-β treatment (200 IU). All RT-qPCRs represent n = 3 biological replicates. Statistical significance was determined using two-tailed Student t test. *p < 0.05, **p < 0.01, ***p < 0.001.
TRIM14 regulates STAT3 activation through TBK1
Because we detected in vivo and in vitro interactions between TRIM14 and TBK1, we hypothesized that TRIM14-dependent misregulation of TBK1 activity could drive hyperinduction of Ifnb and ISGs. TBK1 is a prolific innate immune serine/threonine kinase with many known targets (40, 41). One such target, STAT3 has been repeatedly implicated in the negative regulation of type I IFN responses (42, 43). Therefore, we sought to determine if the presence of TRIM14 and its interaction with TBK1 was required to control STAT3 activity in macrophages.
Previous studies have demonstrated that TBK1 can directly phosphorylate STAT3 at Ser727 and Ser754 upon cytosolic DNA sensing (44) (Fig. 5A). Phosphorylation at Ser754 inhibits STAT3’s ability to interact with target genes, whereas phosphorylation at Ser727 promotes STAT3 activity and enhances transcription of STAT3 targets (45). To determine whether TRIM14 influences STAT3 phosphorylation, we transfected control and Trim14 KO cells with ISD to activate TBK1 and analyzed the dynamics of STAT3 phosphorylation at Ser727 and Ser754 by immunoblot over a time course. We observed substantially more phospho-Ser754 STAT3 and significantly less phospho-Ser727 STAT3 in the absence of TRIM14 (Fig. 5B; replicate blot in Supplemental Fig. 3E). Importantly, loss of TRIM14 had no effect on JAK tyrosine kinase phosphorylation of STAT1 at Y701. These data suggest a role for TRIM14 in influencing TBK1’s preference to phosphorylate particular serine residues in the transactivation domain of STAT3. To further implicate TRIM14 in mediating STAT3 activation through TBK1, we performed cellular fractionations and used immunoblots to measure the amount of STAT3 in the nucleus following ISD transfection in Trim14 KO and control cells. Consistent with reduced activation of STAT3, we observed significantly less STAT3 accumulation in the nuclei of Trim14 KO cells (Fig. 5C). We also measured STAT3 nuclear translocation via immunofluorescence microscopy with automated image analysis and again saw that following ISD transfection, less STAT3 redistributed into the nucleus in Trim14 KO cells compared with controls (Fig. 5D).
FIGURE 5.

Loss of TRIM14 promotes inhibitory phosphorylation of STAT3 at Ser754. (A) Diagram of STAT3 phosphorylation sites. (B) Immunoblot of phospho-Ser754 STAT3, phospho-Ser727 STAT3, and phospho-Y701 STAT1 in WT and Trim14 KO RAW 264.7 macrophages at 1, 2, 4, 6, and 8 h after ISD transfection. ACTIN is shown as a loading control. (C) Cellular fractionation showing nuclear STAT3 in WT and Trim14 KO RAW 264.7 macrophages treated with ISD at specified times after treatment. Histone 3 shown as loading/nuclear control. (D) WT and Trim14 KO RAW 264.7 macrophages immunostained for STAT3 6 h after ISD transfection. Nuclear translocation calculated by automated image analysis of nuclear STAT3 relative to total cellular STAT3 (n > 800 cells per genotype/condition). (E) Coimmunoprecipitation and immunoblot analysis of HEK293T cells cotransfected with FLAG-STAT3 and HA-TRIM14. (F) Coimmunoprecipitation and immunoblot analysis of TBK1 and STAT3 in RAW 264.7 macrophages stably expressing 3xFLAG-TRIM14 over a time course of ISD treatment. A total of 1% total cell lysate loaded, 16% immunoprecipitation loaded. (G) RT-qPCR of Ifit1, Isg15, and Irf7 transcripts in control (Tbk1+/−Tnfr−/−) and Tbk1−/−Tnfr−/− BMDMs treated with IFN-β (200 IU). All immunoblots are representative of >3 independent biological replicates. Statistical significance was determined using two-tailed Student t test. **p < 0.01, ***p < 0.001.
We next predicted that TRIM14 might control TBK1’s ability to phosphorylate STAT3 by interacting with both factors, bringing them together in a complex or conformation that promotes phosphorylation at Ser727 while inhibiting phosphorylation at Ser754. Previous studies have demonstrated that TBK1 and STAT3 can coimmunoprecipitate (45), so we tested whether STAT3 and TRIM14 interact. Indeed, when coexpressed in HEK293T cells, we detected association between STAT3 and TRIM14 (Fig. 5E). We next assessed the ability of TRIM14 to interact with TBK1 and STAT3 in macrophages and whether these interactions might be impacted by activation of cytosolic DNA sensing. To this end, we transfected RAW 264.7 macrophages stably expressing 3xFLAG-TRIM14 with ISD, immunopurified 3xFLAG-TRIM14 at 2, 4, and 6 h posttransfection, and probed for STAT3 or TBK1 via immunoblot using Abs directed against endogenous proteins. Interestingly, we saw that the interaction between TRIM14 and TBK1 was independent of ISD, but ISD transfection enhanced the interaction between TRIM14 and STAT3 (Fig. 5F). We propose that TRIM14, through interactions with STAT3 and TBK1, influences TBK1’s ability to phosphorylate STAT3 at particular serine residues.
We reasoned that because loss of TRIM14 caused hyperinduction of ISGs via a TBK1- and STAT3-dependent mechanism, then loss of either TBK1 or STAT3 would phenocopy loss of TRIM14. Indeed, previous studies have shown that Stat3 KO MEFs and BMDMs hyperinduce ISGs following viral infection (42). To test whether loss of TBK1 could also lead to ISG hyperinduction, we generated BMDMs from Tbk1−/−/Tnfr−/− mice (44, 46) and treated them with rIFN-β. Because treatment with IFN-β directly engages IFNAR, it allows us to assess TBK1’s contribution to IFNAR signaling without the confounding factor of its role in cytosolic DNA sensing, which is activated by both ISD transfection and M. tuberculosis infection (47, 48). Remarkably, we measured dramatic hyperinduction of ISGs in Tbk1−/−/Tnfr−/− BMDMs over a 6 h time course after IFN-β treatment (Fig. 5G). This result argues that TBK1 plays a crucial, yet mostly unappreciated, role in downregulating the type I IFN response downstream of IFNAR signaling, and it supports a model whereby TRIM14 downregulates type I IFN gene expression via TBK1-dependent phosphorylation of STAT3.
TRIM14 is required for STAT3-dependent transcription of Socs3, a negative regulator of the type I IFN response
Because an uncontrolled type I IFN response is deleterious to the host, cells have evolved multiple mechanisms to dampen type I IFN gene expression following pathogen sensing or IFNAR activation. The hyperinduction of Ifnb and ISGs we observe in Trim14 KO macrophages is consistent with a loss of negative regulation; therefore, we hypothesized that expression of one or more negative regulators would be lower in Trim14 KO cells. One well-characterized mechanism of type I IFN downregulation is inhibition of JAK1-STAT signaling (49), and SOCS family proteins are ISGs that interfere with JAK1 kinase activity to limit STAT signaling and dampen type I IFN responses (43). Similarly, ubiquitin specific peptidase 18 (USP18) is an ISG that has been shown to block the interaction between JAK1 and the IFNAR2 subunit to inhibit type I IFN expression (50). To test whether Trim14 KO cells were defective in expressing these negative regulators, we measured Socs3, Socs1, and Usp18 transcripts in control and Trim14 KO RAW 264.7 cells after M. tuberculosis infection or ISD transfection. Similar to all the other ISGs we examined in these studies, Socs1 and Usp18 were hyperinduced in Trim14 KO macrophages. However, we observed a specific defect in Socs3 induction, suggesting that one or more Socs3 transcription factors were impacted by loss of TRIM14 (Fig. 6A, 6B).
FIGURE 6.

Trim14 KO macrophages fail to induce expression of the negative regulator of the type I IFN response, Socs3. (A) RT-qPCR of Socs3, Socs1, and Usp18 transcripts in WT and Trim14 KO RAW 264.7 macrophages infected with M. tuberculosis at specified times postinfection. (B) RT-qPCR of Socs3, Socs1, and Usp18 transcripts in WT and Trim14 KO RAW 264.7 macrophages treated with ISD at specified times after treatment. (C) Quantitative PCR (qPCR) primers designed to tile Socs3 gene for ChIP experiments. (D) ChIP qPCR of STAT3-associated genomic DNA from the Socs3 locus in WT and Trim14 KO RAW 264.7 macrophages transfected with 1 μg ISD. (E) RT-qPCR of Socs3 transcript in RAW 264.7 macrophages stably expressing siRNA to either control or Socs3. (F) RT-qPCR of Ifnb transcript in RAW 264.7 macrophages stably expressing siRNA to either control or Socs3 treated with ISD at specified times after treatment. (G) RT-qPCR of Ifnb transcript in RAW 264.7 macrophages stably expressing siRNA to either control or Socs3 infected with M. tuberculosis at specified times postinfection. Statistical significance was determined using two-tailed Student t test. *p < 0.05; **p < 0.01; ***p < 0.001.
Previous reports have shown that Socs3 is a major target gene of STAT3 (51, 52). Having measured increased phosphorylation at the “inhibitory” Ser754 residue of STAT3 in Trim14 KO macrophages, we hypothesized that a lack of Socs3 induction could be due to the inability of STAT3 to bind at the Socs3 promoter. To test this, we transfected control and Trim14 KO cells with ISD and performed ChIP with an Ab for total STAT3 at 0, 1, and 6 h after ISD transfection (Fig. 6C). Consistent with low Socs3 transcription, we detected significantly less recruitment of STAT3 to the Socs3 genomic locus at both 1 and 6 h after ISD transfection (Fig. 6D). We also detected lower STAT3 recruitment to other non-ISG target genes, including Bcl3 and Cxcl9 (Supplemental Fig. 3C), suggesting that loss of TRIM14 broadly impacts STAT3’s ability to translocate to the nucleus and/or associate with DNA. These data support a model whereby defective nuclear translocation of STAT3 and subsequent lack of Socs3 transcriptional activation result in ISG hyperinduction in the absence of TRIM14. Consistent with such a model, siRNA KD of Socs3 in RAW 264.7 macrophages resulted in Ifnb hyperinduction following both M. tuberculosis infection and ISD transfection (Fig. 6E–G).
Loss of TRIM14 impacts macrophages’ ability to control infection
Having demonstrated an important role for TRIM14 in regulating the type I IFN response, we next investigated how loss of TRIM14 impacts cell-intrinsic innate immune responses to infection. To test how loss of TRIM14 impacts survival and replication of M. tuberculosis, we infected control and Trim14 KO macrophages with M. tuberculosis expressing the luxBCADE operon from Vibrio harveyi (53) and quantified bacterial replication as a measure of luminescence over a 72 h time course (53, 54). Remarkably, we observed a dramatic inhibition of M. tuberculosis replication in Trim14 KO macrophages (Fig. 7A). Importantly, this lack of M. tuberculosis replication did not correspond to loss of cells, as infected monolayers remained completely intact 72 h postinfection (Supplemental Fig. 4A). We also observed a significant inhibition of M. tuberculosis replication in Trim14 KO cells using the enumeration of CFUs (Fig. 7B). To begin to identify the molecular mechanisms driving M. tuberculosis restriction in Trim14 KO macrophages, we measured the expression of several genes whose proteins have been purported to have antimicrobial activity against M. tuberculosis. RT-qPCR revealed hyperinduction of iNOS (Nos2) and guanylate binding proteins 1 and 5 (Gbp1 and Gbp5) in M. tuberculosis–infected Trim14 KO macrophages (Fig. 7C). We predicted that the overabundance of one or more of these factors might contribute to enhanced control of M. tuberculosis replication in the absence of TRIM14. Consistent with this idea, we found that chemical inhibition of iNOS in Trim14 KO cells was sufficient to abrogate the restriction of M. tuberculosis replication and return the level of M. tuberculosis growth to that of control cells by either relative light units (Fig. 7D) or CFUs (Fig. 7E). This remarkable result strongly implicates Nos2/iNOS hyperinduction as the mechanism of M. tuberculosis restriction in the absence of TRIM14.
FIGURE 7.

Trim14 KO macrophages restrict M. tuberculosis replication via hyperinduction of iNOS. (A) Fold replication of M. tuberculosis luxBCADE in WT and Trim14 KO RAW 264.7 macrophages. (B) CFUs in WT and Trim14 KO RAW 264.7 macrophages at specified times postinfection. (C) RT-qPCR of Nos2, Gbp1, and Gbp5 transcript levels in M. tuberculosis–infected WT and Trim14 KO RAW 264.7 macrophages. (D) Fold replication of M. tuberculosis luxBCADE in WT and Trim14 KO RAW 264.7 macrophages ± 1400W dihydrochloride (iNOS inhibitor) at indicated concentration. (E) CFUs in WT and Trim14 KO RAW 264.7 macrophages ± 1400W dihydrochloride. (F) Proposed model of TRIM14’s dual role in regulating cytosolic DNA sensing. TRIM14/cGAS interaction is required to inhibit proteasomal degradation of cGAS. TRIM14/TBK1 interaction is required to promote TBK1-dependent phosphorylation of STAT3 at Ser727 and activate transcription of negative regulators of the type I IFN response like Socs3. Model created using BioRender software. **p < 0.01; ***p < 0.001.
Because another recent report demonstrated that loss of TRIM14 leads to hyperreplication of VSV, an enveloped RNA virus, we infected Trim14 KO and control RAW 264.7 cells with VSV and followed viral replication and ISG expression by RT-qPCR over a 12 h time course. Although uptake of virus, as inferred by viral genome measurements, at the 1 h time point was very similar between the two genotypes, we observed a dramatic hyperreplication of VSV in Trim14 KO macrophages (Supplemental Fig. 4B) and significantly higher Ifnb and ISG expression (Supplemental Fig. 4D). Although hyperinduction of ISGs is seemingly at odds with hyperreplication of VSV, we consistently observed lower basal levels of Ifnb and ISGs in resting Trim14 KO macrophages (Supplemental Fig. 4C) [likely due to cGAS instability (29)]. This defect may give VSV the opportunity to begin replicating unimpeded in Trim14 KO cells before subsequent TRIM14-dependent negative regulation of type I IFN responses begins.
Discussion
To prevent chronic inflammation and damage to host cells and tissues, potent innate immune responses like type I IFN induction require tight control. In this study, we demonstrate a previously unappreciated role for TRIM14 in resolving Ifnb and ISG expression following a variety of cytosolic innate immune stimuli. By providing evidence that TRIM14 can directly interact with both cGAS and TBK1, our work uncovers a complex mechanism through which TRIM14 can both up and downregulate type I IFN responses in macrophages. Notably, we report that loss of TRIM14 has significant consequences on cell-intrinsic control of both bacterial and viral replication, with dramatic restriction of M. tuberculosis replication and uncontrolled replication of VSV in Trim14 KO macrophages (Fig. 7A, 7B, Supplemental Fig. 4). These results reveal a crucial role for TRIM14 in regulating macrophage innate immunity and point to TRIMs as potential targets for host-directed therapies to enhance a macrophage’s antimicrobial capacity.
Our data support a model whereby TRIM14 acts as a scaffold between TBK1 and STAT3, promoting TBK1-dependent phosphorylation of STAT3 at Ser727 and transcriptional activation of negative regulators of JAK/STAT signaling like SOCS3 (Fig. 7F). There is mounting evidence that a complex network of posttranslational modifications regulates STAT3’s ability to dimerize, translocate to the nucleus, and/or bind DNA (43). In addition to inhibitory and activating STAT3 phosphorylation at Ser754 and Ser727 (45), respectively, several other modifications are known to control STAT3 activity, including acetylation at lysine 685 and phosphorylation of tyrosine 705, both of which increase the protein’s ability to bind DNA and translocate to the nucleus (55, 56). We propose that in the context of DNA sensing, TBK1-dependent phosphorylation of STAT3 acts as a control point for ramping up or down the STAT3 transcriptional regulon and the presence of TRIM14 can tip this balance. It is curious that these two modifications (Ser727 and Ser754) have dramatic opposing effects on STAT3 activity, as both residues reside in the transactivation domain and are in close proximity to one another. Structural studies will be needed to shed light on how modulation of TBK1/STAT3 interactions by TRIM14 promote Ser727 phosphorylation over Ser754 phosphorylation. It is possible that the presence of TRIM14 makes one site more accessible either directly through interactions with STAT3 or indirectly by modulating interactions with other binding partners that influence serine availability.
The apparent reliance of Socs3 on STAT3 for its activation in our RAW 264.7 cells is also notable. In addition to being expressed by STAT3, depending on the cell type and context, Socs3 can be regulated by STAT1, and the Socs3 promoter also contains AP-1, Sp3, and NF-κB binding elements (57–59). The fact that these remaining transcription factors do not compensate for loss of STAT3 nuclear translocation in Trim14 KO macrophages (Fig. 5C) hints at the potential for cross-talk between STAT3 and other transcription factors. This notion is consistent with previous reports (60–62), but the extent to which the entire STAT3-transcriptional regulon is impacted by loss of TRIM14 remains unclear. Adding further complexity to this interplay is the fact that following STAT3-dependent expression of Socs3, SOCS3 can subsequently downregulate STAT3 via a negative feedback loop (63, 64). Additional experiments are needed to determine precisely how this loop is broken in Trim14 KO macrophages. As STAT3 and SOCS3 are important not only for controlling inflammatory responses during infection but also for regulating embryogenesis, cancer metastasis, and apoptosis, there is a critical need to understand how TRIM14 influences their activation (65, 66).
Another recent study also shows a requirement for TRIM14 in controlling VSV replication but reported that Trim14 KO cells had lower ISG expression compared with controls (29). The authors ascribed these phenotypes to TRIM14’s role in promoting cGAS stabilization and provide evidence that loss of TRIM14 allows for cGAS degradation via the E3 ligase USP14, which targets cGAS to p62-dependent selective autophagy. We also observed lower Ifnb in response to M. tuberculosis infection and ISD transfection at our earliest measurements (4 h for M. tuberculosis infection; 2, 4, and 6 h for ISD transfection) (Fig. 3C, 3D), but the phenotype of Trim14 KO macrophages dramatically shifts to hyperinduction at later time points. It is not entirely clear what accounts for the discrepancies in our data, although notably, our studies focus almost exclusively on the very early time points following viral infection or innate immune activation (1–12 h) as opposed to the 12–24 h examined by Chen et al. (29). At these later time points, cell death resulting from high viral titers may complicate the interpretation of transcript abundance. Additionally, it is possible that loss of TRIM14 results in different phenotypes depending on the cell type and/or the agonist that activates the type I IFN response. TRIM14 may act differently in the context of infection with a virus that is actively working to turn off type I IFN as opposed to M. tuberculosis, which primarily activates cytosolic DNA sensing.
Taking the conclusions of our study and these others into account (28, 67), it seems likely that TRIM14 plays multiples roles in type I IFN regulation whereby it interacts with cGAS and/or MAVS to promote type I IFN expression and with TBK1/STAT3 to dampen it. To provide insights into these complexities, future experiments will need to investigate the precise spatiotemporal distribution of TRIM14-containing complexes over the course of type I IFN induction in different cell types following treatment with different agonists. It will also be important to investigate how and when TRIM14 itself is posttranslationally modified. Recent work from Jia et al. (68) provides evidence for RNF125-mediated polyubiquitination and proteasomal degradation of a mitochondrially associated population of TRIM14 during viral infection. This posttranslational modification and others could be critical for controlling whether TRIM14 influences type I IFN responses at the level of cGAS, MAVS, and/or TBK1/STAT3.
Our finding that Trim14 KO macrophages are better at controlling M. tuberculosis replication is quite remarkable. As M. tuberculosis replicates very slowly (∼24 h doubling time), we propose that unlike VSV, whose replication can be influenced by basal ISGs in Trim14 KO cells, M. tuberculosis replication is restricted by hyperinduction of ISGs that dominate later during infection. It is unlikely that TRIM14’s contribution to cGAS stability accounts for M. tuberculosis restriction in Trim14 KO macrophages, as our previous work has shown that knocking out cGAS actually renders macrophages more permissive to M. tuberculosis infection, likely through loss of selective autophagy targeting downstream of cytosolic DNA sensing (3). Consistent with these data and our model, another group has reported that siRNA KD of STAT3 in human macrophages enhances NO synthesis and restricts M. tuberculosis replication (69). However, it is formally possible that TRIM14 contributes to M. tuberculosis restriction through more direct mechanisms as well. Curiously, in the context of Listeria monocytogenes infection of STAT1-deficient fibroblasts, overexpression of TRIM14 was protective, suggesting TRIM14 may have ISG-independent antibacterial functions (70). Future experiments designed to investigate what proteins TRIM14 interacts with on the M. tuberculosis phagosome and how loss of TRIM14 impacts maturation of the autophagosome will provide important insights into how TRIM14 controls M. tuberculosis replication and shed light on how we may be able to manipulate TRIM14 as an antituberculosis therapeutic.
Supplementary Material
Acknowledgments
We thank Monica Britton at the University of California, Davis, DNA Technologies and Expression Analysis Core Library for help with initial analysis of our RNA-seq data. We thank Dr. Malea Murphy and the Texas A&M Health Science Center Integrated Microscopy and Imaging Laboratory for assistance with immunofluorescence microscopy. We would also like to acknowledge the members of the Patrick and Watson laboratories for valuable discussions, feedback, and help editing this manuscript.
The online version of this article contains supplemental material.
- BMDM
- bone marrow–derived macrophage
- ChIP
- chromatin immunoprecipitation
- gRNA
- guide RNA
- HA
- hemagglutinin
- IFNAR
- IFN-α/β receptor
- iNOS
- inducible NO synthase
- ISG
- IFN stimulated gene
- KD
- knockdown
- KO
- knockout
- MEF
- murine embryonic fibroblast
- MOI
- multiplicity of infection
- RNA-seq
- RNA sequencing
- RT-qPCR
- real-time quantitative PCR
- SCR
- scramble shRNA
- shRNA
- short hairpin RNA
- siRNA
- small interfering RNA
- Socs3
- suppressor of cytokine signaling 3
- SPR
- surface plasmon resonance
- TRIM
- tripartite motif-containing protein
- USP18
- ubiquitin specific peptidase 18
- VSV
- vesicular stomatitis virus
- WT
- wild-type.
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Underhill D. M., Ozinsky A., Smith K. D., Aderem A. 1999. Toll-like receptor-2 mediates mycobacteria-induced proinflammatory signaling in macrophages. Proc. Natl. Acad. Sci. USA 96: 14459–14463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gilleron M., Nigou J., Nicolle D., Quesniaux V., Puzo G. 2006. The acylation state of mycobacterial lipomannans modulates innate immunity response through toll-like receptor 2. Chem. Biol. 13: 39–47. [DOI] [PubMed] [Google Scholar]
- 3.Watson R. O., Bell S. L., MacDuff D. A., Kimmey J. M., Diner E. J., Olivas J., Vance R. E., Stallings C. L., Virgin H. W., Cox J. S. 2015. The cytosolic sensor cGAS detects Mycobacterium tuberculosis DNA to induce type I interferons and activate autophagy. Cell Host Microbe 17: 811–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wassermann R., Gulen M. F., Sala C., Perin S. G., Lou Y., Rybniker J., Schmid-Burgk J. L., Schmidt T., Hornung V., Cole S. T., Ablasser A. 2015. Mycobacterium tuberculosis differentially activates cGAS- and inflammasome-dependent intracellular immune responses through ESX-1. Cell Host Microbe 17: 799–810. [DOI] [PubMed] [Google Scholar]
- 5.Collins A. C., Cai H., Li T., Franco L. H., Li X.-D., Nair V. R., Scharn C. R., Stamm C. E., Levine B., Chen Z. J., Shiloh M. U. 2015. Cyclic GMP-AMP synthase is an innate immune DNA sensor for Mycobacterium tuberculosis. Cell Host Microbe 17: 820–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Watson R. O., Manzanillo P. S., Cox J. S. 2012. Extracellular M. tuberculosis DNA targets bacteria for autophagy by activating the host DNA-sensing pathway. Cell 150: 803–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Manzanillo P. S., Shiloh M. U., Portnoy D. A., Cox J. S. 2012. Mycobacterium tuberculosis activates the DNA-dependent cytosolic surveillance pathway within macrophages. Cell Host Microbe 11: 469–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Gent M., Sparrer K. M. J., Gack M. U. 2018. TRIM proteins and their roles in antiviral host defenses. Annu. Rev. Virol. 5: 385–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rajsbaum R., García-Sastre A., Versteeg G. A. 2014. TRIMmunity: the roles of the TRIM E3-ubiquitin ligase family in innate antiviral immunity. J. Mol. Biol. 426: 1265–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Versteeg G. A., Rajsbaum R., Sánchez-Aparicio M. T., Maestre A. M., Valdiviezo J., Shi M., Inn K.-S., Fernandez-Sesma A., Jung J., García-Sastre A. 2013. The E3-ligase TRIM family of proteins regulates signaling pathways triggered by innate immune pattern-recognition receptors. Immunity 38: 384–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koliopoulos M. G., Esposito D., Christodoulou E., Taylor I. A., Rittinger K. 2016. Functional role of TRIM E3 ligase oligomerization and regulation of catalytic activity. EMBO J. 35: 1204–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sanchez J. G., Okreglicka K., Chandrasekaran V., Welker J. M., Sundquist W. I., Pornillos O. 2014. The tripartite motif coiled-coil is an elongated antiparallel hairpin dimer. Proc. Natl. Acad. Sci. USA 111: 2494–2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stremlau M., Owens C. M., Perron M. J., Kiessling M., Autissier P., Sodroski J. 2004. The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in old world monkeys. Nature 427: 848–853. [DOI] [PubMed] [Google Scholar]
- 14.Sanchez J. G., Chiang J. J., Sparrer K. M. J., Alam S. L., Chi M., Roganowicz M. D., Sankaran B., Gack M. U., Pornillos O. 2016. Mechanism of TRIM25 catalytic activation in the antiviral RIG-I pathway. Cell Rep. 16: 1315–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao C., Jia M., Song H., Yu Z., Wang W., Li Q., Zhang L., Zhao W., Cao X. 2017. The E3 ubiquitin ligase TRIM40 attenuates antiviral immune responses by targeting MDA5 and RIG-I. Cell Rep. 21: 1613–1623. [DOI] [PubMed] [Google Scholar]
- 16.Sparrer K. M. J., Gableske S., Zurenski M. A., Parker Z. M., Full F., Baumgart G. J., Kato J., Pacheco-Rodriguez G., Liang C., Pornillos O., et al. 2017. TRIM23 mediates virus-induced autophagy via activation of TBK1. Nat. Microbiol. 2: 1543–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Porritt R. A., Hertzog P. J. 2015. Dynamic control of type I IFN signalling by an integrated network of negative regulators. Trends Immunol. 36: 150–160. [DOI] [PubMed] [Google Scholar]
- 18.Patrick K. L., Bell S. L., Watson R. O. 2016. For better or worse: cytosolic DNA sensing during intracellular bacterial infection induces potent innate immune responses. J. Mol. Biol. 428: 3372–3386. [DOI] [PubMed] [Google Scholar]
- 19.Zhang Y., Yeruva L., Marinov A., Prantner D., Wyrick P. B., Lupashin V., Nagarajan U. M. 2014. The DNA sensor, cyclic GMP-AMP synthase, is essential for induction of IFN-β during Chlamydia trachomatis infection. J. Immunol. 193: 2394–2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Woodward J. J., Iavarone A. T., Portnoy D. A. 2010. c-di-AMP secreted by intracellular Listeria monocytogenes activates a host type I interferon response. Science 328: 1703–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fiskin E., Bhogaraju S., Herhaus L., Kalayil S., Hahn M., Dikic I. 2017. Structural basis for the recognition and degradation of host TRIM proteins by Salmonella effector SopA. Nat. Commun. 8: 14004–14014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kamanova J., Sun H., Lara-Tejero M., Galán J. E. 2016. The Salmonella effector protein SopA modulates innate immune responses by targeting TRIM E3 ligase family members. PLoS Pathog. 12: e1005552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ye W., Hu M.-M., Lei C.-Q., Zhou Q., Lin H., Sun M.-S., Shu H.-B. 2017. TRIM8 negatively regulates TLR3/4-mediated innate immune response by blocking TRIF-TBK1 interaction. J. Immunol. 199: 1856–1864. [DOI] [PubMed] [Google Scholar]
- 24.Nagre N., Cong X., Ji H.-L., Schreiber J. M., Fu H., Pepper I., Warren S., Sill J. M., Hubmayr R. D., Zhao X. 2018. Inhaled TRIM72 protein protects ventilation injury to the lung through injury-guided cell repair. Am. J. Respir. Cell Mol. Biol. 59: 635–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu X., Wang J., Wang S., Wu F., Chen Z., Li C., Cheng G., Qin F. X.-F. 2019. Inhibition of influenza A virus replication by TRIM14 via its multifaceted protein-protein interaction with NP. Front. Microbiol. 10: 344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tan G., Xu F., Song H., Yuan Y., Xiao Q., Ma F., Qin F. X.-F., Cheng G. 2018. Identification of TRIM14 as a type I IFN-stimulated gene controlling hepatitis B virus replication by targeting HBx. Front. Immunol. 9: 1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang S., Chen Y., Li C., Wu Y., Guo L., Peng C., Huang Y., Cheng G., Qin F. X.-F. 2016. TRIM14 inhibits hepatitis C virus infection by SPRY domain-dependent targeted degradation of the viral NS5A protein. Sci. Rep. 6: 32336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou Z., Jia X., Xue Q., Dou Z., Ma Y., Zhao Z., Jiang Z., He B., Jin Q., Wang J. 2014. TRIM14 is a mitochondrial adaptor that facilitates retinoic acid-inducible gene-I-like receptor-mediated innate immune response. Proc. Natl. Acad. Sci. USA 111: E245–E254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen M., Meng Q., Qin Y., Liang P., Tan P., He L., Zhou Y., Chen Y., Huang J., Wang R.-F., Cui J. 2016. TRIM14 inhibits cGAS degradation mediated by selective autophagy receptor p62 to promote innate immune responses. Mol. Cell 64: 105–119. [DOI] [PubMed] [Google Scholar]
- 30.West K. O., Scott H. M., Torres-Odio S., West A. P., Patrick K. L., Watson R. O. 2019. The splicing factor hnRNP M is a critical regulator of innate immune gene expression in macrophages. Cell Rep. 29: 1594–1609.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stanley S. A., Raghavan S., Hwang W. W., Cox J. S. 2003. Acute infection and macrophage subversion by Mycobacterium tuberculosis require a specialized secretion system. Proc. Natl. Acad. Sci. USA 100: 13001–13006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stanley S. A., Johndrow J. E., Manzanillo P., Cox J. S. 2007. The type I IFN response to infection with Mycobacterium tuberculosis requires ESX-1-mediated secretion and contributes to pathogenesis. J. Immunol. 178: 3143–3152. [DOI] [PubMed] [Google Scholar]
- 33.Andreu N., Phelan J., de Sessions P. F., Cliff J. M., Clark T. G., Hibberd M. L. 2017. Primary macrophages and J774 cells respond differently to infection with Mycobacterium tuberculosis. Sci. Rep. 7: 42225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Quesniaux V., Fremond C., Jacobs M., Parida S., Nicolle D., Yeremeev V., Bihl F., Erard F., Botha T., Drennan M., et al. 2004. Toll-like receptor pathways in the immune responses to mycobacteria. Microbes Infect. 6: 946–959. [DOI] [PubMed] [Google Scholar]
- 35.Schoggins J. W., MacDuff D. A., Imanaka N., Gainey M. D., Shrestha B., Eitson J. L., Mar K. B., Richardson R. B., Ratushny A. V., Litvak V., et al. 2014. Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. [Published erratum appears in 2015 Nature 525: 144.] Nature 505: 691–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stetson D. B., Medzhitov R. 2006. Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity 24: 93–103. [DOI] [PubMed] [Google Scholar]
- 37.Li X., Shu C., Yi G., Chaton C. T., Shelton C. L., Diao J., Zuo X., Kao C. C., Herr A. B., Li P. 2013. Cyclic GMP-AMP synthase is activated by double-stranded DNA-induced oligomerization. Immunity 39: 1019–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shu C., Sankaran B., Chaton C. T., Herr A. B., Mishra A., Peng J., Li P. 2013. Structural insights into the functions of TBK1 in innate antimicrobial immunity. Structure 21: 1137–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Larabi A., Devos J. M., Ng S.-L., Nanao M. H., Round A., Maniatis T., Panne D. 2013. Crystal structure and mechanism of activation of TANK-binding kinase 1. Cell Rep. 3: 734–746. [DOI] [PubMed] [Google Scholar]
- 40.Helgason E., Phung Q. T., Dueber E. C. 2013. Recent insights into the complexity of Tank-binding kinase 1 signaling networks: the emerging role of cellular localization in the activation and substrate specificity of TBK1. FEBS Lett. 587: 1230–1237. [DOI] [PubMed] [Google Scholar]
- 41.Weidberg H., Elazar Z. 2011. TBK1 mediates crosstalk between the innate immune response and autophagy. Sci. Signal. 4: pe39. [DOI] [PubMed] [Google Scholar]
- 42.Wang W.-B., Levy D. E., Lee C.-K. 2011. STAT3 negatively regulates type I IFN-mediated antiviral response. J. Immunol. 187: 2578–2585. [DOI] [PubMed] [Google Scholar]
- 43.Tsai M.-H., Pai L.-M., Lee C.-K. 2019. Fine-tuning of type I interferon response by STAT3. Front. Immunol. 10: 1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hemmi H., Takeuchi O., Sato S., Yamamoto M., Kaisho T., Sanjo H., Kawai T., Hoshino K., Takeda K., Akira S. 2004. The roles of two IkappaB kinase-related kinases in lipopolysaccharide and double stranded RNA signaling and viral infection. J. Exp. Med. 199: 1641–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hsia H.-C., Hutti J. E., Baldwin A. S. 2017. Cytosolic DNA promotes signal transducer and activator of transcription 3 (STAT3) phosphorylation by TANK-binding kinase 1 (TBK1) to restrain STAT3 activity. J. Biol. Chem. 292: 5405–5417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Perry A. K., Chow E. K., Goodnough J. B., Yeh W.-C., Cheng G. 2004. Differential requirement for TANK-binding kinase-1 in type I interferon responses to toll-like receptor activation and viral infection. J. Exp. Med. 199: 1651–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fitzgerald K. A., McWhirter S. M., Faia K. L., Rowe D. C., Latz E., Golenbock D. T., Coyle A. J., Liao S.-M., Maniatis T. 2003. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 4: 491–496. [DOI] [PubMed] [Google Scholar]
- 48.Tanaka Y., Chen Z. J. 2012. STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci. Signal. 5: ra20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ivashkiv L. B., Donlin L. T. 2014. Regulation of type I interferon responses. Nat. Rev. Immunol. 14: 36–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Honke N., Shaabani N., Zhang D.-E., Hardt C., Lang K. S. 2016. Multiple functions of USP18. Cell Death Dis. 7: e2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hutchins A. P., Diez D., Takahashi Y., Ahmad S., Jauch R., Tremblay M. L., Miranda-Saavedra D. 2013. Distinct transcriptional regulatory modules underlie STAT3’s cell type-independent and cell type-specific functions. Nucleic Acids Res. 41: 2155–2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ramadoss P., Unger-Smith N. E., Lam F. S., Hollenberg A. N. 2009. STAT3 targets the regulatory regions of gluconeogenic genes in vivo. Mol. Endocrinol. 23: 827–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Penn B. H., Netter Z., Johnson J. R., Von Dollen J., Jang G. M., Johnson T., Ohol Y. M., Maher C., Bell S. L., Geiger K., et al. 2018. An Mtb-human protein-protein interaction map identifies a switch between host antiviral and antibacterial responses. Mol. Cell 71: 637–648.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Roberts A. W., Popov L. M., Mitchell G., Ching K. L., Licht D. J., Golovkine G., Barton G. M., Cox J. S. 2019. Cas9+ conditionally-immortalized macrophages as a tool for bacterial pathogenesis and beyond. eLife 8: e45957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhuang S. 2013. Regulation of STAT signaling by acetylation. Cell. Signal. 25: 1924–1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang R., Cherukuri P., Luo J. 2005. Activation of Stat3 sequence-specific DNA binding and transcription by p300/CREB-binding protein-mediated acetylation. J. Biol. Chem. 280: 11528–11534. [DOI] [PubMed] [Google Scholar]
- 57.Wiejak J., Dunlop J., Gao S., Borland G., Yarwood S. J. 2012. Extracellular signal-regulated kinase mitogen-activated protein kinase-dependent SOCS-3 gene induction requires c-Jun, signal transducer and activator of transcription 3, and specificity protein 3 transcription factors. Mol. Pharmacol. 81: 657–668. [DOI] [PubMed] [Google Scholar]
- 58.Yang J., Huang J., Dasgupta M., Sears N., Miyagi M., Wang B., Chance M. R., Chen X., Du Y., Wang Y., et al. 2010. Reversible methylation of promoter-bound STAT3 by histone-modifying enzymes. Proc. Natl. Acad. Sci. USA 107: 21499–21504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Isobe A., Takeda T., Sakata M., Yamamoto T., Minekawa R., Hayashi M., Auernhammer C. J., Tasaka K., Murata Y. 2006. STAT3-mediated constitutive expression of SOCS3 in an undifferentiated rat trophoblast-like cell line. Placenta 27: 912–918. [DOI] [PubMed] [Google Scholar]
- 60.Grivennikov S. I., Karin M. 2010. Dangerous liaisons: STAT3 and NF-kappaB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev. 21: 11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kesanakurti D., Chetty C., Rajasekhar Maddirela D., Gujrati M., Rao J. S. 2013. Essential role of cooperative NF-κB and Stat3 recruitment to ICAM-1 intronic consensus elements in the regulation of radiation-induced invasion and migration in glioma. Oncogene 32: 5144–5155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yu Z., Kone B. C. 2004. The STAT3 DNA-binding domain mediates interaction with NF-kappaB p65 and inducible nitric oxide synthase transrepression in mesangial cells. J. Am. Soc. Nephrol. 15: 585–591. [DOI] [PubMed] [Google Scholar]
- 63.Huang L., Hu B., Ni J., Wu J., Jiang W., Chen C., Yang L., Zeng Y., Wan R., Hu G., Wang X. 2016. Transcriptional repression of SOCS3 mediated by IL-6/STAT3 signaling via DNMT1 promotes pancreatic cancer growth and metastasis. J. Exp. Clin. Cancer Res. 35: 27. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 64.Lu Y., Fukuyama S., Yoshida R., Kobayashi T., Saeki K., Shiraishi H., Yoshimura A., Takaesu G. 2006. Loss of SOCS3 gene expression converts STAT3 function from anti-apoptotic to pro-apoptotic. J. Biol. Chem. 281: 36683–36690. [DOI] [PubMed] [Google Scholar]
- 65.Siveen K. S., Sikka S., Surana R., Dai X., Zhang J., Kumar A. P., Tan B. K. H., Sethi G., Bishayee A. 2014. Targeting the STAT3 signaling pathway in cancer: role of synthetic and natural inhibitors. Biochim. Biophys. Acta 1845: 136–154. [DOI] [PubMed] [Google Scholar]
- 66.Resemann H. K., Watson C. J., Lloyd-Lewis B. 2014. The Stat3 paradox: a killer and an oncogene. Mol. Cell. Endocrinol. 382: 603–611. [DOI] [PubMed] [Google Scholar]
- 67.Chen W., Choi Y. J., Jung J. U. 2017. Unexpected alliance of WHIP-TRIM14-PPP6C to combat viruses. Mol. Cell 68: 259–261. [DOI] [PubMed] [Google Scholar]
- 68.Jia X., Zhou H., Wu C., Wu Q., Ma S., Wei C., Cao Y., Song J., Zhong H., Zhou Z., Wang J. 2017. The ubiquitin ligase RNF125 targets innate immune adaptor protein TRIM14 for ubiquitination and degradation. J. Immunol. 198: 4652–4658. [DOI] [PubMed] [Google Scholar]
- 69.Queval C. J., Song O.-R., Deboosère N., Delorme V., Debrie A.-S., Iantomasi R., Veyron-Churlet R., Jouny S., Redhage K., Deloison G., et al. 2016. STAT3 represses nitric oxide synthesis in human macrophages upon Mycobacterium tuberculosis infection. Sci. Rep. 6: 29297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Perelman S. S., Abrams M. E., Eitson J. L., Chen D., Jimenez A., Mettlen M., Schoggins J. W., Alto N. M. 2016. Cell-based screen identifies human interferon-stimulated regulators of Listeria monocytogenes infection. PLoS Pathog. 12: e1006102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.