

Abstract
The inflammatory responses in chronic airway diseases leading to emphysema are not fully defined. We hypothesized that lung eosinophilia contributes to airspace enlargement in a mouse model, and emphysema in patients with COPD.
A transgenic mouse model of chronic type 2 pulmonary inflammation (I5/hE2) was used to examine eosinophil-dependent mechanisms leading to airspace enlargement.
Human sputum samples were collected for translational studies examining eosinophilia and MMP-12 levels in patients with chronic airways disease.
Airspace enlargement was identified in I5/hE2 mice and was dependent on eosinophils. Examination of I5/hE2 bronchoalveolar lavage identified elevated MMP-12, a mediator of emphysema. We showed, in vitro, that eosinophil-derived IL-13 promoted alveolar macrophage MMP-12 production. Airspace enlargement in I5/hE2 mice was dependent on MMP-12 and eosinophil-derived IL-4/13. Consistent with this, MMP-12 was elevated in patients with sputum eosinophilia and CT-evidence of emphysema, and also negatively correlated with FEV1.
A mouse model of chronic type 2 pulmonary inflammation exhibited airspace enlargement dependent on MMP-12 and eosinophil-derived IL-4/13. In chronic airways disease patients, lung eosinophilia was associated with elevated MMP-12 levels, which was a predictor of emphysema. These findings suggest an underappreciated mechanism by which eosinophils contribute to the pathologies associated with asthma and COPD.
Introduction:
Chronic obstructive pulmonary disease (COPD) is a chronic respiratory condition associated with cigarette smoking and characterized by irreversible airflow obstruction. It is often accompanied by the presence of airway inflammation and emphysema. COPD is among the leading causes of death worldwide and is increasing in prevalence (1). There is no cure for COPD but a better understanding of disease subtypes has led to improved therapeutic options (2-4). The lung inflammatory cell infiltrate in COPD is key to disease pathogenesis. Alveolar macrophages and infiltrating neutrophils mediate destructive activities on the architecture and function of the lung in part through the release of proteases and inflammatory cytokines (5). Despite the canonical classification of COPD as a neutrophilic disease, blood and airway eosinophilia is found in roughly a third of COPD patients (6-8). Clinical studies suggest an association between eosinophilia and increased risk of exacerbation (9, 10). Sputum eosinophil levels, in particular, correlate with increased exacerbations, worsening fixed airflow obstruction, and poorer quality of life scores in COPD patients (11). In this large cohort, significantly higher emphysema indices (as measured by quantitative CT) were noted in patients stratified to the sputum eosinophil-high group.
Eosinophils produce a wide array of mediators including toxic granule proteins (such as eosinophil peroxidase (EPX)), potent lipid derivatives (such as leukotrienes), and numerous cytokines and chemokines (12). In mouse models of chronic lung inflammation eosinophil mediators have been shown to contribute to pathologies observed in patients including bronchoconstriction, fibrosis, and mucus production (13, 14). Eosinophil-derived IL-13 is of particular interest as it has been shown to contribute to all of these pathologies (15, 16). IL-13 mediates these activities through numerous cell types including alveolar macrophages which play a key role in the lung remodeling associated with severe asthma and COPD.
Alveolar macrophage production of proteases including matrix metalloproteases (MMPs) contributes to alveolar destruction in COPD (17). MMPs are extracellular matrix degrading enzymes. MMP-12, in particular, has been shown to be a critical mediator of alveolar destruction in a mouse model of COPD (18) and evidence points to its importance in COPD patients (17). IL-13 signaling can polarize macrophages to an M2 phenotype, a feature found in chronic asthma and COPD (19), and induce MMP-12 production. Notably, IL-13 stimulated mouse alveolar macrophages produce MMP-12 in vitro (20) and overproduction of IL-13 in the lungs of transgenic mice was sufficient to induce MMP-12 production and destruction of alveolar spaces (21).
We hypothesized that eosinophils are an important source of IL-13 in chronic lung disease thereby contributing to remodeling of the airways resulting in emphysema. To explore this, we utilized a mouse model of chronicTh2 pulmonary inflammation and also analyzed sputum samples from patients with asthma or COPD. Identification of the pathways by which emphysema develops/progresses may present new therapeutic targets and strategies to prevent or stabilize this chronic respiratory disease feature.
Methods:
Mice
Studies involving mice were performed in accordance with National Institutes of Health and Mayo Clinic Institutional Animal Care and Use Committee guidelines. Strains of mice employed include C57BL/6J wild type (WT) (Jackson Laboratory, Bar Harbor, ME), eoCRE (22), NJ.1638 (23), I5/hE2 (14), PHIL (13), MMP-12−/− (Jackson Laboratory, B6.129X-Mmp12tm1Sds/J), floxed IL-4/13 (Jackson Laboratory, C.129P2(Cg)-Il4/Il13tm1.1Lky/J) and IL-13−/− (gift from Andrew McKenzie (24)). Mice were analyzed between 2-3 months of age. Mice were maintained in the Mayo Clinic Arizona Small Animal Facility (a specific pathogen-free facility).
Cell isolation and culture
Eosinophils were isolated as described previously (25) from NJ.1638 or IL-13−/−/NJ.1638 (23, 24) mice. IL-33 (Peprotech) was added to activate the eosinophils to 50ng/mL (IL-33 was omitted from the resting eosinophil experimental group). Cells were incubated at 37°C, 5%CO2 and 24 hours later were washed three times to remove added cytokines. Macrophages were isolated from C57BL/6J or MMP-12−/− mice as described by Lasbury et al (26). Cells were plated at 150,000 cells per well in 24 well culture plates (Corning, Corning, NY) and cultured for 48 hours with the addition of either eosinophils (750,000 cells) or media alone in 500μl total volume. To assess contact dependency the above was performed in transwell plates (0.4μm pore) (Corning).
Histology
Lung samples were processed for histological analysis as previously described (27). Hematoxylin and eosin (H&E) stained sections were assessed for alveolar space characteristics as previously described (28).
ELISA and multiplex
MMP-12 was determined by mouse MMP-12 PicoKine ELISA (Boster Bio, Pleasanton, CA). EPX was measured as described previously (29). Other mouse and human cytokines were assessed by bead-based multiplexing technology (Eve Technologies, Calgary, Canada).
Bronchoalveolar lavage, lung homogenate, cytospins, cell counts, and differentials
Bronchoalveolar lavage (BAL), lung collection, and homogenate preparation were performed as described previously (14, 30). Cytospins, cell counts, and differentials were performed as described previously (31, 32).
Assessment of clinical sputa
We used sputum supernatants (non-cellular fraction) from 43 patients with a diagnosis of either asthma or COPD which were previously stored based on observational protocols approved by the local Hospital Research Ethics Board in St. Joseph’s Hospital, Hamilton, Ontario. These sputum samples were induced and processed, as described previously (33). Samples from asthmatics were randomly selected from patients who had clinically-indicated sputum testing between January and November 2017. Asthma diagnosis was based on bronchodilator reversibility and/or PC20 value <8 mg/mL upon a methacholine challenge test. The sputa of patients with COPD were previously collected as part of a local observational study during a period of clinical stability (from November 2016 to November 2017). Inclusion criteria included fixed airflow obstruction (FEV1/FVC<0.7) and/or evidence of emphysema on imaging with a ≥10 pack-year history of smoking. Those with a previous diagnosis of asthma or a significant improvement in post-bronchodilator spirometry were excluded. COPD was assessed based on Global Initiative for Chronic Obstructive Lung Disease (GOLD) criteria (34). Computed tomography (CT) scans of the chest were acquired during routine clinical work-up and as such had a variety of protocols applied. The most common CT scan available was of normal resolution without contrast. Emphysema grouping was confirmed by a three-step process: 1) Extrapolation from the clinical radiologist’s summary report; 2) Applying a quantitative CT metric – low attenuation area for Hounsfield units less than 950 %LAA −950HU (using the open source Chest Imaging Platform provided by 3D Slicer); and 3) Confirmation of the presence of emphysema by a blinded radiologist. Eosinophilic patients were defined as those with presence of sputum eosinophils >3%, and/or evidence of blood eosinophils >300/μL, as well as those with historical evidence of eosinophils which were now suppressed due to high-dose inhaled or oral corticosteroid treatment.
Statistical analyses
Data were analyzed with GraphPad Prism 7 (GraphPad Software, Inc., La Jolla, CA, USA). Statistical comparisons of mouse model and cell culture data were performed with student’s t-test. Results are represented as mean ± S.E.M. Statistical comparisons between human subject groups were performed by Analysis of Variance (ANOVA) / Kruskal-Wallis non-parametric tests, and associations were determined by Spearman’s rank/ Pearson correlation test based on the distribution of the respective data sets (D'Agostino & Pearson omnibus normality tests). IBM® SPSS® Statistical software (version 23.0) was used for multivariate regression analysis. P values ≤ 0.05 were considered to be significant.
Results:
I5/hE2 mice exhibit eosinophil-dependent airspace enlargement
To explore the role of eosinophils in the chronically inflamed lung we employed the I5/hE2 mouse model of chronic Th2 pulmonary inflammation. The I5/hE2 mouse has been shown to develop marked airway remodeling dependent on lung eosinophilia (14, 27). Examination of I5/hE2 vs. wild type H&E stained lung sections revealed airspace enlargement in the lung parenchyma of the I5/hE2 mice (Figure 1A). We crossed the I5/hE2 mouse with the eosinophil-deficient PHIL mouse (13) to examine eosinophil-dependent changes in lung structure. The cross of I5/hE2 with PHIL revealed that these lesions were entirely eosinophil dependent. Quantification of the mean intercept length between alveolar septa showed a greater than two-fold increase (~30um vs. 70um) in size in I5/hE2 mice as compared to I5/hE2/PHIL mice (Figure 1B).
Figure 1. Eosinophil-dependent alveolar destruction and modulation of MMP-12.

(A) H&E stained lung sections demonstrate increased alveolar spaces in I5/hE2 lungs relative to wild type and, their eosinophil deficient counterparts, I5/hE2/PHIL. Lung parenchyma 200x. (B) Quantification of average alveolar space revealed a two-fold increase in mean intercept length in I5/hE2 lungs (panel B, n=5 mice). (C) BAL fluid was collected and assessed by ELISA for MMP-12, a lung remodeling mediator with dramatically increased expression in I5/hE2 lungs (panel C, n=6 mice). Scale bar = 50μm. *:p<0.05. ****:p<0.0001. ns: not significant.
Our previous investigations into eosinophil dependent changes in gene expression in the I5/hE2 lung focused attention on MMP-12 as a candidate for mediating the airspace enlargement observed in I5/hE2. MMP-12 has been shown to cause the emphysema-associated breakdown of alveolar septa in a COPD model (18) and we found MMP-12 was the most upregulated eosinophil-dependent gene in the I5/hE2 lung (27). Consistent with this observation, MMP-12 protein levels in the BAL fluid were elevated in I5/hE2 mice and nearly undetectable in I5/hE2/PHIL mice (Figure 1C). Together these results suggest that eosinophils promote MMP-12 production and airspace enlargement.
MMP-12 is a mediator of alveolar destruction in I5/hE2 mice
To explore the significance of MMP-12 to the airspace enlargement observed in I5/hE2 we compared I5/hE2 mice with their MMP-12 deficient counterpart I5/hE2/MMP-12−/−. Histological examination of the lungs revealed reduced lesions in the absence of MMP-12 (Figure 2A). Quantification of the mean intercept length between alveolar septa showed a significant reduction in I5/hE2/MMP-12−/− mice (Figure 2B). However, I5/hE2/MMP-12−/− mice had an increased mean intercept length relative to wild type mice suggesting other eosinophil-dependent mediators are contributing to these lesions. For example, other matrix metalloproteinases (MMP-2 and 9) have been linked with emphysema (35, 36) and we found these were also elevated in I5/hE2 lungs (Supplementary Figure 1). BAL cellularity (I5/hE2 (46.85 ± 6.45 x105) and I5/hE2/MMP-12−/− (35.23 ± 4.25 x105)) and eosinophil numbers (I5/hE2 (43.79 ± 5.53 x105) and I5/hE2/MMP-12−/− (34.6 ± 4.85 x105)) were not affected by the loss of MMP-12 in this model.
Figure 2. MMP-12 mediates alveolar destruction in I5/hE2 model.

(A) H&E stained lung sections demonstrate enlarged airspaces in I5/hE2 mice relative to wild type and MMP-12−/− mice. (B) H&E stained lung sections assessed for mean intercept length (Lm) demonstrate increased alveolar spaces in I5/hE2 mice relative to their MMP-12 deficient counterparts I5/hE2/MMP-12−/−. Lung parenchyma 200x. (C). n≥3 mice. Scale bar = 50μm. **: p<0.01, ***:p<0.001, ****:p<0.0001.
Eosinophil-derived IL-13 promotes alveolar macrophage MMP-12 production in vitro
To investigate the relationship between eosinophils and MMP-12 production we performed in vitro experiments with eosinophils and known airway MMP-12 producing cells, alveolar macrophages (37, 38). In vitro experiments revealed that MMP-12 was not detectable in eosinophil culture supernatants whereas MMP-12 could be detected in alveolar macrophage culture supernatants. Eosinophil and macrophage co-culture resulted in significantly increased levels of MMP-12 compared to macrophage culture alone (Figure 3A). Co-culture of wild type eosinophils with MMP-12−/− alveolar macrophages showed that the MMP-12 in these culture experiments is derived from macrophages (Figure 3A).
Figure 3. Eosinophils directly modulate alveolar macrophage production of MMP-12 through IL-13 dependent mechanisms.
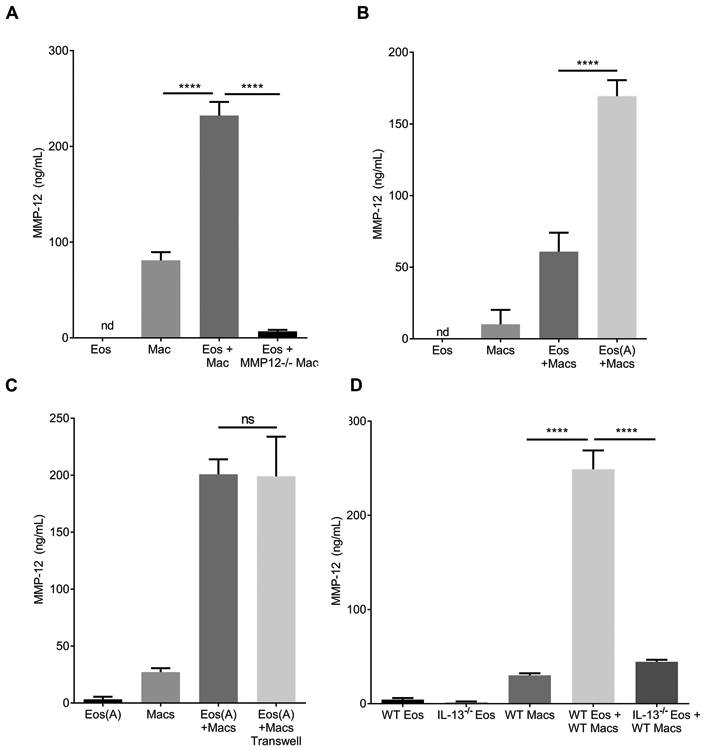
ELISA measurement of MMP-12 in cell culture supernatant from in vitro 48h cultures of eosinophils (Eos) and/or alveolar macrophages (Macs). (A) Eosinophils cultured alone, macrophages cultured alone, and co-culture (Eos+Mac). Wild type eosinophils were activated (IL-33 for 24 hours then washed) and cultured with WT macrophages or MMP-12−/− macrophages showing the MMP-12 is from macrophages. (B) Assessment of co-culture supernatants from resting eosinophils (Eos) vs. activated eosinophils (Eos(A)) with macrophages (Macs) demonstrated that pre-activation of eosinophils (IL-33 for 24 hours then washed) enhances macrophage MMP-12 production. (C) Transwell co-culture of activated eosinophils with macrophages shows that contact is not required for the enhanced macrophage MMP-12 production. (D) ELISA measurements from co-culture supernatants of IL-13−/− vs. Wild Type (WT) eosinophils with WT macrophages demonstrates that eosinophil-derived IL-13 is promoting macrophage MMP-12 production. ***:p<0.001, ****:p<0.0001, ns: not significant.
To explore the mechanism of eosinophil mediated macrophage MMP-12 production we first assessed the role of eosinophil activation with IL-33, a cytokine associated with chronic lung disease and elevated in the I5/hE2 mouse lung (27, 39). Eosinophil activation by IL-33 has been demonstrated to increase release of soluble mediators that may influence the production of MMP-12 from macrophages (40, 41). Activation of the eosinophils with IL-33 prior to co-culture with alveolar macrophages enhanced macrophage MMP-12 production relative to co-culture with non-activated eosinophils (Figure 3B). We next examined contact dependency by transwell co-culture of eosinophils with alveolar macrophages. We showed that the increased MMP-12 from macrophages is not contact-dependent (Figure 3C) suggesting that macrophages produce MMP-12 in response to an eosinophil-secreted mediator.
Previous studies have demonstrated IL-13 promotes MMP-12 production by macrophages (20, 36) and IL-33 is known to induce IL-13 from eosinophils (15, 42). We assessed the role of eosinophil-derived IL-13 by co-cultures of wild type alveolar macrophages (WT macs) with eosinophils sufficient or deficient for IL-13 (WT vs. IL-13−/− eos). We found that eosinophil-derived IL-13 promoted alveolar macrophage MMP-12 production (Figure 3D).
I5/hE2 mice exhibit airspace enlargement dependent on eosinophil-derived IL-13 and MMP-12
To examine if the lung eosinophilia in our mouse model of chronic Th2 pulmonary inflammation induces alveolar destruction via IL-13-mediated MMP-12 production we generated conditional knockout mice (43) by crossing the eosinophil-specific Cre recombinase mouse, eoCRE, with the available floxed IL-4/13 mouse (44) resulting in eosinophil-specific loss of IL-13 expression (as well as IL-4) (Supplementary Figure 2). We then crossed these mice with I5/hE2 to create I5/hE2/eoCRE/4/13fl/fl which was compared with I5/hE2/eoCRE as a control. Histological examination of the lungs again showed airspace enlargement in I5/hE2/eoCRE which was dependent upon eosinophil-derived IL-4/13 (Figure 4A). Notably, alveolar spaces in I5/hE2/eoCRE/4/13fl/fl were equivalent to those of wild type mice suggesting that the enlarged alveolar spaces in I5/hE2 mice were entirely dependent on eosinophil-derived IL-4/13 (Figure 4B). BAL cellularity (I5/hE2/eoCRE 39.1 ± 8.74 x105) and I5/hE2/eoCRE/4/13fl/fl (22.98 ± 4.31 x105)) and eosinophil cellularity (I5/hE2/eoCRE 26.1 ± 4.04 x105) and I5/hE2/eoCRE/4/13fl/fl (41.53 ± 8.28 x105)) were not affected by the loss of eosinophil-derived IL-4/13 in this model. Significantly, BAL MMP-12 levels were entirely dependent on eosinophil-derived IL-4/13 (Figure 4C). Combined, our data suggest eosinophil-derived IL-13 is a key mediator of alveolar macrophage MMP-12 production and alveolar destruction in this chronic pulmonary inflammation model.
Figure 4. Eosinophil-derived IL-4/13 induces MMP-12 production and alveolar destruction in I5/hE2 mice.

(A) A genetic cross of I5/hE2 with the eosinophil-specific Cre mouse, eoCRE, x the available floxed IL-4/13 mouse (I5/hE2/eoCRE/4/13fl/fl) revealed that enlarged alveolar spaces in I5/hE2 mice are dependent on eosinophil-derived IL-4/13. (B) H&E stained lung sections demonstrate enlarged airspaces in I5/hE2 mice relative to eosinophil-derived IL-4/13 deficient mice. (C) ELISA measurement of MMP-12 in BAL showed MMP-12 in I5/hE2 mice is dependent on eosinophil-derived IL-4/13. Lung parenchyma 200x. n≥3 mice. Scale bar = 50μm. *:p<0.05, **: p<0.01, ***:p<0.001, ****:p<0.0001.
Elevated MMP-12 is associated with eosinophilia, emphysema, and decreased lung function in human subjects
Next, we investigated the relationship between MMP-12, eosinophilia, emphysema, and lung function in humans. Out of the 44 patient sputa randomly selected, 5 were rejected (3 rejected for co-morbidities/other inflammatory abnormalities, 1 for not meeting criteria for asthma or COPD, and 1 for age < 18 years). Of the remaining subjects 16 had asthma, 23 had COPD, and their clinical characteristics are represented in Tables 1 and 2. Sputum EPX levels were higher in the eosinophilic group (p=0.002, Figure 5A) and correlated significantly with sputum IL-13 levels (r=0.536, P=0.0004, Spearman rank). Irrespective of diagnosis, the MMP-12 levels detected in the sputa were higher in the eosinophilic group (Figure 5B, P=0.01) and modestly correlated with sputum IL-13 levels (r=0.35, P=0.03, Figure 5C). The significant correlation of MMP-12 with airway IL-13 levels and not EPX (r=0.05, P=0.7) suggested the possibility that MMP-12 was not directly associated with eosinophilia per se, but instead associated with eosinophil-derived, and potentially other sources of, IL-13 inflammation.
Table 1:
Demographic table of recruited patients: Asthma and COPD
| Asthma | COPD | |
|---|---|---|
| N | 16 | 23 |
| Age (mean, SD) | 62 (10) | 64 (11) |
| Sex (F, n, % of n) | 9 (56) | 10 (43) |
| FEV1 %Pred (mean, SD) | 65 (17)* | 49 (17)* |
| FEV1/FVC (mean, SD) | 0.62 (0.13)* | 0.49 (0.13)* |
| DLCO %Pred (mean, SD) | 99 (33)*† | 61 (18) *†† |
| Current Smoking (n, % of n) | 1 (6)* | 12 (52)* |
| Previous Smoking (n, % of n) | 0 (0) | 7 (30) |
| Smoking History in pack-years (mean, SD) | N/A | 46 (24) |
| Emphysema (n, % of n) | 1 (6)* | 15 (65)* |
| Centrilobular (n) | 1 | 7 |
| Paraseptal (n) | 0 | 1 |
| Both (n) | 0 | 7 |
| Eosinophilia (n, % of n) | 11 (69) | 11 (48) |
| Sputum eosinophil% (mean, SD) | 7.0 (9.1) | 5.9 (15) |
| Sputum EPX (ng/mL-g; mean, SD) | 375 (587)* | 91.4 (144)* |
| ICS dose (median, max-min) | 750 (2000, 0) | 750 (3500, 0) |
| ICS use (n, % of n) | 15 (94) | 20 (87) |
| OCS use (n, % of n) | 5 (31) | 2 (9) |
| LABA use (n, % of n) | 15 (94) | 18 (83) |
| LAMA use (n, % of n) | 0 (0)* | 12 (52)* |
denotes statistically significant difference between groups
based on n=3
based on n=9
SD – standard deviation; FEV1 – forced expiratory volume in 1 second; FVC – forced vital capacity; EPX – eosinophil peroxidase; ICS – inhaled corticosteroid dose, daily mcg equivalent for fluticasone; OCS – on maintenance oral corticosteroid; LABA – long acting beta-agonists; LAMA : long acting muscarinic antagonists. All COPD patients are current smokers or ex-smokers with >10 pack year history.
Table 2:
Demographic table of recruited patients: Eosinophilic and Non-eosinophilic
| Eosinophilic | Non-Eosinophilic | |
|---|---|---|
| n | 22 | 17 |
| Asthma (n, % of n) | 11 (50) | 5 (29) |
| Age (mean, SD) | 63 (7.5) | 64 (14) |
| Sex (F, n, % of n) | 10 (46) | 9 (53) |
| FEV1 %Pred (mean, SD) | 52 (18) | 60 (19) |
| FEV1/FVC (mean, SD) | 0.53 (0.15) | 0.56 (0.14) |
| DLCO %Pred (mean, SD) | 71 (32)† | N/A |
| Current Smoking (n, % of n) | 5 (23) | 9 (53) |
| Previous Smoking (n, % of n) | 5 (23) | 2 (12) |
| Smoking History in pack-years (mean, SD) | 25 (32) | 42 (24) |
| Emphysema (n, % of n) | 8 (36) | 8 (47) |
| Centrilobular (n) | 5 | 3 |
| Paraseptal (n) | 1 | 0 |
| Both (n) | 2 | 5 |
| Sputum eosinophil% (mean, SD) | 11 (15)* | 0.25 (0.40)* |
| Sputum EPX (ng/mL-g; mean, SD) | 293 (506)* | 81 (118)* |
| ICS dose (median, max-min) | 750 (2000, 0) | 500 (3500, 0) |
| ICS use (n, % of n) | 19 (86) | 16 (94) |
| OCS use (n, % of n) | 5 (23) | 2 (12) |
| LABA use (n, % of n) | 20 (91) | 14 (82) |
| LAMA use (n, % of n) | 7 (32) | 5 (29) |
denotes statistically significant difference between groups
based on n=7
Figure 5. MMP-12 expression in eosinophilic emphysematous airways.
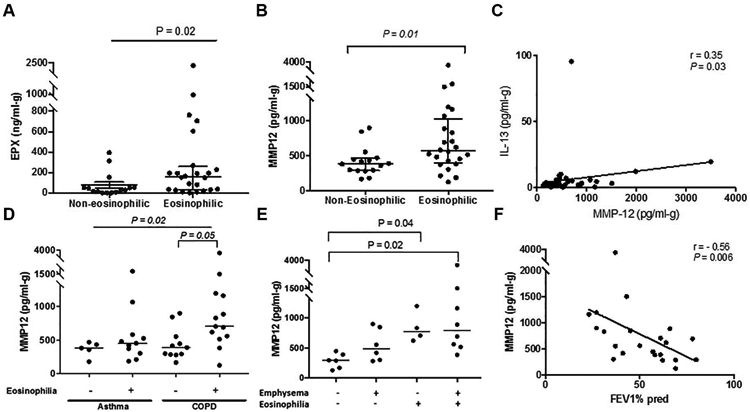
(A) EPX (B) MMP-12 expression in airways of n=40 patients with chronic airways disease (diagnosis of either asthma or COPD) classified based on eosinophilia. Mann Whitney Test . (C) Correlation of sputum MMP-12 with IL-13 (n=40). Spearman rank. (D) MMP-12 expression in airways of both asthma (n=16) and COPD (n=24), classified based on eosinophilia and (E) COPD samples only, classified with respect to eosinophilia and emphysema. Kruskal Wallis with Dunn’s multiple correction. (F) Correlation of sputum MMP-12 with FEV1 % predicted in n=24 COPD patients. Spearman rank. Each symbol represents individual patient data, and median is indicated for each subset. Figure 5D, grey open circle indicates asthmatic patient with evidence of airspace enlargement. P≤0.05 considered as significant.
In the sub-group analysis, differential expression of MMP-12 in the asthma subset with respect to eosinophilia was unremarkable (P>0.99, Figure 5D); however, increased detection of MMP-12 was evident in the eosinophilic COPD airways (P=0.05, Figure 5D). In fact, MMP-12 levels in COPD patients were higher in those with concomitant eosinophilia and emphysema compared to those who did not exhibit either (P= 0.02, Figure 5E). Comparing %LAA −950HU by eosinophilic status revealed a higher value in those with eosinophilia (Median: 1.4% versus 0.08%, P=0.04 by Mann Whitney test). Emphysema grouping was confirmed by the three steps described in the Methods section. The accuracy of grouping (by clinical radiologist report) was supported by a significantly higher %LAA −950HU in the emphysema group (Median: 1.97% versus 0.18%, P=0.0073 by Mann Whitney test), and confirmation of emphysema by a blinded radiologist for each subject. Of interest, the patient in the eosinophilic asthma group who had the highest level of MMP-12 (marked as open grey circle in Figure 5D) had CT-evidence of air space enlargement. If this data-point is removed, MMP-12 levels are significantly higher in the eosinophilic COPD patient sputa compared to eosinophilic asthmatics (P=0.01, Mann Whitney), which was otherwise comparable (P=0.43, Figure 5D). In a multivariate regression model, MMP-12 was computed to be a predictor (beta coefficient = 0.361, R squared value = 0.13, Standard error of estimate: 0.47, P=0.02) for the presence of emphysema (CT evidence) and not eosinophilia, EPX, or IL-13 (Supplementary Table 1). Finally, MMP-12 levels were negatively associated with FEV1% predicted in patients with COPD (r= −0.56, P= 0.006, Figure 5F), and this association remained significant with the inclusion of asthmatic participants (r= −0.45, P=0.005).
Discussion:
Though type 2 inflammation is more frequently associated with asthma than COPD, it can be present or absent in either condition. In this study, we used a mouse model of chronic type 2 pulmonary inflammation (I5/hE2) to explore the link between eosinophils and alveolar destruction, without any connotation regarding the physiologic features required for a diagnosis of asthma or COPD or involvement of smoking-induced pathways. These mice develop eosinophil-dependent lung pathologies that are absent when crossed with eosinophil-deficient mice (I5/hE2/PHIL) but may be restored with eosinophil-sufficient bone marrow engraftment (14). We found that I5/hE2 mice develop eosinophil-dependent airspace enlargement. Significantly, MMP-12, a protease known to be a critical mediator of alveolar destruction, was elevated in I5/hE2 lungs and this was entirely dependent upon the presence of eosinophils. We demonstrated ex vivo that eosinophil-derived IL-13 stimulated alveolar macrophages to produce MMP-12. To confirm these pathways in vivo we crossed I5/hE2 with various genetically engineered mice and showed that eosinophils promote airspace enlargement dependent on IL-13 (and possibly IL-4 as we utilized the available floxed IL-4/13 mouse) and MMP-12. Consistent with these observations, it has been shown previously that MMP-12 is induced by IL-13 in the lung (20, 36) and eosinophils are a known source of IL-13 (15, 42). Further, overexpression of IL-13 in the mouse lung resulted in increased MMP-12 levels and airspace enlargement (36). Together, these data reveal a clear path by which eosinophils may contribute to the destruction of airspaces in chronic respiratory diseases.
Eosinophil-derived IL-13 has been demonstrated to promote M2 macrophage differentiation as well as accumulation in a mouse model of allergic pulmonary inflammation (15). M2 alveolar macrophages have been identified as MMP-12 producing cells (reviewed in (38)). Notably, we found that other proteases associated with alveolar destruction, MMP-2 and MMP-9, are also elevated in the lungs of I5/hE2 mice dependent on eosinophil-derived IL-4/13. Eosinophils have been shown to express MMP-9 in asthmatic airways (45) and it is likely that eosinophils are indirectly linked, through interactions with other cell types such as macrophages, to the production of numerous remodeling agents including MMP-2 and 9. Indeed, we showed that removal of MMP-12 only partially reduced the alveolar destruction in I5/hE2 lungs and observed that in older MMP-12 deficient I5/hE2 mice the extent of alveolar destruction approached that of I5/hE2 (data not shown) suggesting a role for other mediators. Key contributors likely include other matrix metalloproteases and cathepsins previously observed to contribute to these changes in a transgenic mouse with overexpression of IL-13 in the lung (21). Future studies are needed to determine other key eosinophil-associated mediator(s) of alveolar destruction in the I5/hE2 lung. Together, these interactions highlight the role of eosinophils as regulators of local immune responses and remodeling events (46).
The I5/hE2 model enabled us to isolate the contribution of eosinophils to airspace enlargement. It is likely that other cell types can participate in the pathways explored in this work. In particular, ILC2s and Th2 T cells produce IL-13 in the lung (reviewed in (47, 48); moreover, the airway epithelium and smooth muscle have been identified as producers of MMP-12 (49, 50). Nevertheless, our finding that eosinophils can mediate these activities is intriguing. While IL-13 is a secreted factor, it is possible that eosinophils are a particularly potent source for these interactions. Clearly, the interactions between eosinophils and alveolar macrophages deserve further investigation as it may be possible that a spacial, temporal, and/ or multi-factor association (e.g. IL-13 in combination with other signals) is important for these activities.
A pathogenic role of eosinophils in COPD has long been considered. Despite their potential importance in the pathophysiology of COPD, efforts to target eosinophils in clinical trials of COPD have met with limited success. A very modest clinical benefit has been noted in patients with blood eosinophilia post treatment with subcutaneous Mepolizumab (an anti-IL-5 antibody that results in eosinophil depletion) (METREX) (51). In the parallel study evaluating two doses of Mepolizumab as an add-on treatment for frequently exacerbating COPD patients characterized by eosinophil level (METREO), the annual rate of exacerbations compared to the placebo arm was deemed insignificant. These findings corroborated those reported in a smaller study where the drug reduced both sputum and blood eosinophils, and yet failed to show any significant improvement in the rate of exacerbations, lung function, or radiologic evidence of disease severity (52). Similarly, targeting eosinophils in COPD using Benralizumab (an anti-IL5Rα antibody that results in eosinophil depletion) showed no clinical improvement in the rate of exacerbations compared to placebo. However, a modest improvement in FEV1 was reported in the drug arm, indicating eosinophils, or by extension, eosinophilic factors might be affecting lung function (53).
The limited improvement in clinical trials examining anti-eosinophil agents in COPD may be due to irreversible airspace enlargement. Our experiments suggest a role of eosinophils in the initiation/development of airspace enlargement, albeit indirectly through stimulation of MMP-12 (and possibly other protease) production by alveolar macrophages. Indeed, in COPD patients with evidence of eosinophilia there was a detectable increase in MMP-12, which was computed to be a predictor for the presence of CT-evidence of emphysema. These findings suggest strategies targeting eosinophils in eosinophilic COPD may be beneficial but that long-term studies with lung function as the primary outcome may be needed to appreciate this benefit. Alternative strategies to IL-5 blockade that target eosinophils and other cells (e.g. ILC2 cells) could potentially reduce exacerbations and should be considered for future studies. One such strategy suggested by previous observations and the findings from this study is a combination of IL-33 and IL-13 blockade.
The concept of eosinophil-driven emphysema has been studied previously within COPD cohorts, which identified associations between blood eosinophils and markers of connective tissue destruction (54), and between sputum eosinophils and emphysema indices on quantitative CT (11). Furthermore, a network analysis incorporating measures of the blood, bone marrow, as well as clinical variables, demonstrated that the capacity for immune-mediated repair was impaired in the presence of elevated blood eosinophils and/or emphysema (55). The current study provides mechanistic, mouse-model evidence and corroborating data from a “real life” COPD cohort which lends support to the previous findings.
In summary, our findings reveal a role for eosinophil-derived IL-13 in alveolar destruction through induction of MMP-12. Asthma and COPD are diagnosed based on physiologic criteria, with the former demonstrating reversible airflow obstruction or airway hyper-responsiveness, and the latter demonstrating fixed airflow obstruction. We translated our findings to patients with eosinophilic COPD, suggesting a contribution of eosinophil-mediated induction of MMP-12 in the development of emphysema. Sputa was tested in patients with asthma or COPD, given that eosinophilic inflammation can be a shared component between these diseases, but emphysema is not classically associated with asthma. It is interesting to speculate that our findings may extend to other chronic inflammatory respiratory diseases. Notably, asthma disease progression and severity is associated with eosinophilia and may develop into/overlap with COPD. Indeed, the asthmatic patient with highest MMP-12 detection (levels comparable to the COPD subset) had persistent eosinophilia as well as CT-evidence of emphysema. A recent report from Gelb et. al. builds on evidence of asthma subjects with type 2 phenotype developing mild emphysema and loss of lung elasticity (56). In addition, a case report of a non-smoking eosinophilic bronchitis patient developing emphysema highlights this possibility (57). Together, these findings suggest that a focus on therapeutic management of lung eosinophilia, IL-13, and proteases including MMP-12 may be effective strategies for slowing the progression of emphysema in certain patient populations.
Supplementary Material
Acknowledgments:
We thank all members of the Lee laboratory group for their support and we recognize the excellent administrative support provided by Stefanie Brendle, Linda Mardel, and Shirley “Charlie” Kern. We also recognize Melanie Kjarsgaard, Nicola LaVigne, Katherine Radford, and Dr. Colm Boylan for their expert technical assistance. This work is dedicated to the memory of our dear friend and colleague James “Jamie” Lee whose larger than life character was matched only by his scientific genius. We miss him dearly.
Support:
This work was supported by Mayo Foundation for Medical Education and Research, NIH NHLBI R01 HL058723, HL065228, and NIH NCRR K26 RR0109709. ADD was supported by the Mayo Graduate School, the Mayo Clinic Sidney Luckman Family Predoctoral Fellowship, the Donald R. Levin Family Foundation, and NIH NHLBI F31HL124959. PN is supported by the Frederick E. Hargreave Teva Innovation Chair in Airway Diseases.
Footnotes
This article has an online data supplement
References:
- 1.May SM, Li JT. Burden of chronic obstructive pulmonary disease: healthcare costs and beyond. Allergy Asthma Proc 2015; 36: 4–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Turner AM, Tamasi L, Schleich F, Hoxha M, Horvath I, Louis R, Barnes N. Clinically relevant subgroups in COPD and asthma. Eur Respir Rev 2015; 24: 283–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bel EH, Ten Brinke A. New Anti-Eosinophil Drugs for Asthma and COPD: Targeting the Trait! Chest 2017; 152: 1276–1282. [DOI] [PubMed] [Google Scholar]
- 4.Segal LN, Martinez FJ. Chronic obstructive pulmonary disease subpopulations and phenotyping. J Allergy Clin Immunol 2018; 141: 1961–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barnes PJ. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol 2016; 138: 16–27. [DOI] [PubMed] [Google Scholar]
- 6.Saha S, Brightling CE. Eosinophilic airway inflammation in COPD. Int J Chron Obstruct Pulmon Dis 2006; 1: 39–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bafadhel M, Pavord ID, Russell REK. Eosinophils in COPD: just another biomarker? Lancet Respir Med 2017; 5: 747–759. [DOI] [PubMed] [Google Scholar]
- 8.Postma DS, Rabe KF. The Asthma-COPD Overlap Syndrome. N Engl J Med 2015; 373: 1241–1249. [DOI] [PubMed] [Google Scholar]
- 9.Vedel-Krogh S, Nielsen SF, Lange P, Vestbo J, Nordestgaard BG. Blood eosinophils and exacerbations in chronic obstructive pulmonary disease. The Copenhagen General Population Study. Am J Respir Crit Care Med 2016; 193: 956–974. [DOI] [PubMed] [Google Scholar]
- 10.Siddiqui SH, Guasconi A, Vestbo J, Jones P, Agusti A, Paggiaro P, Wedzicha JA, Singh D. Blood eosinophils: a biomarker od response to extrafine beclomethasone/formoterol in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2015; 192: 523–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hastie AT, Martinez FJ, Curtis JL, Doerschuk CM, Hansel NN, Christenson S, Putcha N, Ortega VE, Li X, Barr RG, Carretta EE, Couper DJ, Cooper CB, Hoffman EA, Kanner RE, Kleerup E, O'Neal WK, Paine R 3rd, Peters SP, Alexis NE, Woodruff PG, Han MK, Meyers DA, Bleecker ER, investigators S Association of sputum and blood eosinophil concentrations with clinical measures of COPD severity: an analysis of the SPIROMICS cohort. Lancet Respir Med 2017; 5: 956–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jacobsen EA, Helmers RA, Lee JJ, Lee NA. The expanding role(s) of eosinophils in health and disease. Blood 2012; 120: 3882–3890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee JJ, Dimina D, Macias MP, Ochkur SI, McGarry MP, O'Neill KR, Protheroe C, Pero R, Nguyen T, Cormier SA, Lenkiewicz E, Colbert D, Rinaldi L, Ackerman SJ, Irvin CG, Lee NA. Defining a link with asthma in mice congenitally deficient in eosinophils. Science 2004; 305: 1773–1776. [DOI] [PubMed] [Google Scholar]
- 14.Ochkur SI, Jacobsen EA, Protheroe CA, Biechele TL, Pero RS, McGarry MP, Wang H, O'Neill KR, Colbert DC, Colby TV, Shen H, Blackburn MR, Irvin CC, Lee JJ, Lee NA. Coexpression of IL-5 and eotaxin-2 in mice creates an eosinophil-dependent model of respiratory inflammation with characteristics of severe asthma. J Immunol 2007; 178: 7879–7889. [DOI] [PubMed] [Google Scholar]
- 15.Jacobsen EA, Doyle AD, Colbert DC, Zellner KR, Protheroe CA, LeSuer WE, Lee NA, Lee JJ. Differential activation of airway eosinophils induces IL-13-mediated allergic Th2 pulmonary responses in mice. Allergy 2015; 70: 1148–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jacobsen EA, Ochkur SI, Doyle AD, LeSuer WE, Li W, Protheroe CA, Colbert D, Zellner KR, Shen HH, Irvin CG, Lee JJ, Lee NA. Lung Pathologies in a Chronic Inflammation Mouse Model Are Independent of Eosinophil Degranulation. Am J Respir Crit Care Med 2017; 195: 1321–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gharib SA, Manicone AM, Parks WC. Matrix metalloproteinases in emphysema. Matrix Biol 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hautamaki RD, Kobayashi DK, Senior RM, Shapiro SD. Requirement for macrophage elastase for cigarette smoke-induced emphysema in mice. Science 1997; 277: 2002–2004. [DOI] [PubMed] [Google Scholar]
- 19.Shaykhiev R, Krause A, Salit J, Strulovici-Barel Y, Harvey BG, O'Connor TP, Crystal RG. Smoking-dependent reprogramming of alveolar macrophage polarization: implication for pathogenesis of chronic obstructive pulmonary disease. J Immunol 2009; 183: 2867–2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pouladi MA, Robbins CS, Swirski FK, Cundall M, McKenzie AN, Jordana M, Shapiro SD, Stampfli MR. Interleukin-13-dependent expression of matrix metalloproteinase-12 is required for the development of airway eosinophilia in mice. Am J Respir Cell Mol Biol 2004; 30: 84–90. [DOI] [PubMed] [Google Scholar]
- 21.Zheng T, Zhu Z, Wang Z, Homer RJ, Ma B, Riese RJ Jr., Chapman HA Jr., Shapiro SD, Elias JA. Inducible targeting of IL-13 to the adult lung causes matrix metalloproteinase- and cathepsin-dependent emphysema. J Clin Invest 2000; 106: 1081–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Doyle AD, Jacobsen EA, Ochkur SI, Willetts L, Shim K, Neely J, Kloeber J, Lesuer WE, Pero RS, Lacy P, Moqbel R, Lee NA, Lee JJ. Homologous recombination into the eosinophil peroxidase locus generates a strain of mice expressing Cre recombinase exclusively in eosinophils. J Leukoc Biol 2013; 94: 17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee NA, McGarry MP, Larson KA, Horton MA, Kristensen AB, Lee JJ. Expression of IL-5 in thymocytes/T cells leads to the development of a massive eosinophilia, extramedullary eosinophilopoiesis, and unique histopathologies. J Immunol 1997; 158: 1332–1344. [PubMed] [Google Scholar]
- 24.McKenzie GJ, Emson CL, Bell SE, Anderson S, Fallon P, Zurawski G, Murray R, Grencis R, McKenzie AN. Impaired development of Th2 cells in IL-13-deficient mice. Immunity 1998; 9: 423–432. [DOI] [PubMed] [Google Scholar]
- 25.Jacobsen EA, Zellner KR, Colbert D, Lee NA, Lee JJ. Eosinophils regulate dendritic cells and Th2 pulmonary immune responses following allergen provocation. J Immunol 2011; 187: 6059–6068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lasbury ME, Durant PJ, Lee CH. Numbers of alveolar macrophages are increased during Pneumocystis pneumonia in mice. J Eukaryot Microbiol 2003; 50 Suppl: 637–638. [DOI] [PubMed] [Google Scholar]
- 27.Ochkur SI, Protheroe CA, Li W, Colbert DC, Zellner KR, Shen HH, Luster AD, Irvin CG, Lee JJ, Lee NA. Cys-leukotrienes promote fibrosis in a mouse model of eosinophil-mediated respiratory inflammation. Am J Respir Cell Mol Biol 2013; 49: 1074–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Knudsen L, Weibel ER, Gundersen HJ, Weinstein FV, Ochs M. Assessment of air space size characteristics by intercept (chord) measurement: an accurate and efficient stereological approach. J Appl Physiol (1985) 2010; 108: 412–421. [DOI] [PubMed] [Google Scholar]
- 29.Ochkur SI, Kim JD, Protheroe CA, Colbert D, Condjella RM, Bersoux S, Helmers RA, Moqbel R, Lacy P, Kelly EA, Jarjour NN, Kern R, Peters A, Schleimer RP, Furuta GT, Nair P, Lee JJ, Lee NA. A sensitive high throughput ELISA for human eosinophil peroxidase: a specific assay to quantify eosinophil degranulation from patient-derived sources. J Immunol Methods 2012; 384: 10–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jacobsen EA, Ochkur SI, Pero RS, Taranova AG, Protheroe CA, Colbert DC, Lee NA, Lee JJ. Allergic pulmonary inflammation in mice is dependent on eosinophil-induced recruitment of effector T cells. J Exp Med 2008; 205: 699–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Doyle AD, Jacobsen EA, Ochkur SI, McGarry MP, Shim KG, Nguyen DT, Protheroe C, Colbert D, Kloeber J, Neely J, Shim KP, Dyer KD, Rosenberg HF, Lee JJ, Lee NA. Expression of the secondary granule proteins major basic protein 1 (MBP-1) and eosinophil peroxidase (EPX) is required for eosinophilopoiesis in mice. Blood 2013; 122: 781–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McGarry MP, Protheroe CA, Lee JJ. Mouse hematology : a laboratory manual. Cold Spring Harbor, N.Y.: Cold Spring Harbor Laboratory Press; 2010. [Google Scholar]
- 33.Pizzichini E, Pizzichini MM, Efthimiadis A, Hargreave FE, Dolovich J. Measurement of inflammatory indices in induced sputum: effects of selection of sputum to minimize salivary contamination. Eur Respir J 1996; 9: 1174–1180. [DOI] [PubMed] [Google Scholar]
- 34.Global Initiative for Chronic Obstructive Lung Disease (GOLD) 2011. 2011. February 20, 2018]. Available from: http://www.goldcopd.org/. [Google Scholar]
- 35.Baraldo S, Bazzan E, Zanin ME, Turato G, Garbisa S, Maestrelli P, Papi A, Miniati M, Fabbri LM, Zuin R, Saetta M. Matrix metalloproteinase-2 protein in lung periphery is related to COPD progression. Chest 2007; 132: 1733–1740. [DOI] [PubMed] [Google Scholar]
- 36.Lanone S, Zheng T, Zhu Z, Liu W, Lee CG, Ma B, Chen Q, Homer RJ, Wang J, Rabach LA, Rabach ME, Shipley JM, Shapiro SD, Senior RM, Elias JA. Overlapping and enzyme-specific contributions of matrix metalloproteinases-9 and -12 in IL-13-induced inflammation and remodeling. J Clin Invest 2002; 110: 463–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shapiro SD, Kobayashi DK, Ley TJ. Cloning and characterization of a unique elastolytic metalloproteinase produced by human alveolar macrophages. J Biol Chem 1993; 268: 23824–23829. [PubMed] [Google Scholar]
- 38.Boorsma CE, Draijer C, Melgert BN. Macrophage heterogeneity in respiratory diseases. Mediators Inflamm 2013; 2013: 769214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao J, Zhao Y. Interleukin-33 and its Receptor in Pulmonary Inflammatory Diseases. Crit Rev Immunol 2015; 35: 451–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stolarski B, Kurowska-Stolarska M, Kewin P, Xu D, Liew FY. IL-33 exacerbates eosinophil-mediated airway inflammation. J Immunol 2010; 185: 3472–3480. [DOI] [PubMed] [Google Scholar]
- 41.Willebrand R, Voehringer D. IL-33-Induced Cytokine Secretion and Survival of Mouse Eosinophils Is Promoted by Autocrine GM-CSF. PLoS One 2016; 11: e0163751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schmid-Grendelmeier P, Altznauer F, Fischer B, Bizer C, Straumann A, Menz G, Blaser K, Wuthrich B, Simon HU. Eosinophils express functional IL-13 in eosinophilic inflammatory diseases. J Immunol 2002; 169: 1021–1027. [DOI] [PubMed] [Google Scholar]
- 43.Doyle A, McGarry MP, Lee NA, Lee JJ. The construction of transgenic and gene knockout/knockin mouse models of human disease. Transgenic Res 2012; 21: 327–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Voehringer D, Wu D, Liang HE, Locksley RM. Efficient generation of long-distance conditional alleles using recombineering and a dual selection strategy in replicate plates. BMC Biotechnol 2009; 9: 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ohno I, Ohtani H, Nitta Y, Suzuki J, Hoshi H, Honma M, Isoyama S, Tanno Y, Tamura G, Yamauchi K, Nagura H, Shirato K. Eosinophils as a source of matrix metalloproteinase-9 in asthmatic airway inflammation. Am J Respir Cell Mol Biol 1997; 16: 212–219. [DOI] [PubMed] [Google Scholar]
- 46.Lee JJ, Jacobsen EA, McGarry MP, Schleimer RP, Lee NA. Eosinophils in health and disease: the LIAR hypothesis. Clin Exp Allergy 2010; 40: 563–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Drake LY, Kita H. Group 2 innate lymphoid cells in the lung. Adv Immunol 2014; 124: 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wills-Karp M Immunologic basis of antigen-induced airway hyperresponsiveness. Annu Rev Immunol 1999; 17: 255–281. [DOI] [PubMed] [Google Scholar]
- 49.Lagente V, Le Quement C, Boichot E. Macrophage metalloelastase (MMP-12) as a target for inflammatory respiratory diseases. Expert Opin Ther Targets 2009; 13: 287–295. [DOI] [PubMed] [Google Scholar]
- 50.Lavigne MC, Eppihimer MJ. Cigarette smoke condensate induces MMP-12 gene expression in airway-like epithelia. Biochem Biophys Res Commun 2005; 330: 194–203. [DOI] [PubMed] [Google Scholar]
- 51.Pavord ID, Chanez P, Criner GJ, Kerstjens HAM, Korn S, Lugogo N, Martinot JB, Sagara H, Albers FC, Bradford ES, Harris SS, Mayer B, Rubin DB, Yancey SW, Sciurba FC. Mepolizumab for Eosinophilic Chronic Obstructive Pulmonary Disease. N Engl J Med 2017; 377: 1613–1629. [DOI] [PubMed] [Google Scholar]
- 52.Dasgupta A, Kjarsgaard M, Capaldi D, Radford K, Aleman F, Boylan C, Altman LC, Wight TN, Parraga G, O'Byrne PM, Nair P. A pilot randomised clinical trial of mepolizumab in COPD with eosinophilic bronchitis. Eur Respir J 2017; 49. [DOI] [PubMed] [Google Scholar]
- 53.Brightling CE, Bleecker ER, Panettieri RA Jr., Bafadhel M, She D, Ward CK, Xu X, Birrell C, van der Merwe R. Benralizumab for chronic obstructive pulmonary disease and sputum eosinophilia: a randomised, double-blind, placebo-controlled, phase 2a study. Lancet Respir Med 2014; 2: 891–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bihlet AR, Karsdal MA, Sand JM, Leeming DJ, Roberts M, White W, Bowler R. Biomarkers of extracellular matrix turnover are associated with emphysema and eosinophilic-bronchitis in COPD. Respir Res 2017; 18: 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Toledo-Pons N, Noell G, Jahn A, Iglesias A, Duran MA, Iglesias J, Rios A, Scrimini S, Faner R, Gigirey O, Agusti A, Cosio BG. Bone marrow characterization in COPD: a multi-level network analysis. Respir Res 2018; 19: 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gelb AF, Yamamoto A, Verbeken EK, Schein MJ, Moridzadeh R, Tran D, Fraser C, Barbers R, Elatre W, Koss MN, Glassy EF, Nadel JA. Further Studies of Unsuspected Emphysema in Nonsmoking Patients With Asthma With Persistent Expiratory Airflow Obstruction. Chest 2018; 153: 618–629. [DOI] [PubMed] [Google Scholar]
- 57.Brightling CE, Woltmann G, Wardlaw AJ, Pavord ID. Development of irreversible airflow obstruction in a patient with eosinophilic bronchitis without asthma. Eur Respir J 1999; 14: 1228–1230. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.