

Abstract
4-1BBL, a member of the tumor necrosis factor superfamily, regulates the sustained production of inflammatory cytokines in macrophages triggered by TLR signaling. In this study, we have investigated the role of 4-1BBL in macrophage metabolism and polarization, and in skin inflammation using a model of imiquimod (IMQ)-induced psoriasis in mice. Genetic ablation or blocking of 4-1BBL signaling by Ab or 4-1BB-Fc alleviated the pathology of psoriasis by regulating the expression of inflammatory cytokines associated with macrophage activation, and regulated the polarization of macrophages in vitro. We further linked this result with macrophage by finding that 4-1BBL expression during the immediate TLR response was dependent on glycolysis, mitochondrial oxidative phosphorylation and fatty acid metabolism, while the late phase 4-1BBL-mediated sustained inflammatory response was dependent on glycolysis and fatty acid synthesis. Correlating with this, administration of a fatty acid synthase inhibitor cerulenin also alleviated the pathology of psoriasis. We further found that 4-1BBL-mediated psoriasis development is independent of its receptor 4-1BB, as a deficiency of 4-1BB augmented the severity of psoriasis, linked to a reduced Treg population and increased IL-17A expression in γδ T cells. Additionally, co-blocking of 4-1BBL signaling and IL-17A activity additively ameliorated psoriasis. Taken together, 4-1BBL signaling regulates macrophage polarization and contributes to IMQ-induced psoriasis by sustaining inflammation, providing a possible avenue for psoriasis treatment in patients.
Keywords: Rodent, Monocytes/Macrophages, Cell Surface Molecules, Inflammation, Skin
INTRODUCTION
We have previously identified a novel signaling mechanism involving two distinct and sequential signaling cascades that regulate the initiation and persistence of inflammation by TLR-activated macrophages (1). 4-1BBL, a member of the TNF superfamily, allows the sustained production of inflammatory cytokines driven by TLR signaling. For example, LPS-induced production of cytokines such as TNF, IL-6, IL-12, and IL-23 occurred for 24 h in wild-type macrophages, whereas 4-1BBL-deficient macrophages produced cytokines similar to WT cells for the first few hours after LPS treatment but then ceased production. Cytokine production was also significantly reduced when 4-1BBL KO macrophages were treated with other TLR ligands, but not by other cytokines such as IL-1β, indicating the specific role of 4-1BBL in TLR responses. Expression of 4-1BBL is induced immediately upon TLR signaling and it subsequently interacts with TLRs to allow secondary late-phase signaling. Notably, genetic ablation of 4-1BBL or inhibition of the TLR-4-1BBL interaction by antagonists such as 4-1BBL Ab or 4-1BB-Fc protein significantly reduced cytokine production by macrophages and improved survival in mouse models of sepsis (1–3). These data suggest a possible role for 4-1BBL in the development of inflammatory diseases linked to macrophage activity and that inhibition of 4-1BBL might be used for the treatment of such inflammatory diseases.
Cell metabolism plays a crucial role in determining the phenotypes of macrophages, and subsequent development of inflammatory diseases (4–8). Despite the heterogeneity of macrophages associated with diverse functions, two well-established phenotypes are often referred to as classically activated pro-inflammatory M1 and alternatively activated anti-inflammatory M2 macrophages that induce TH1 and TH2 responses, respectively. In addition to distinct functions and gene expression profiles, M1 and M2 macrophages exhibit contrasting metabolic activities. M1 macrophages rely on glycolysis for ATP generation partly due to the disrupted TCA cycle, and fatty acid synthesis is activated during inflammation (9–11). In contrast, M2 macrophages have an intact TCA cycle and use oxidative phosphorylation (OXPHOS) for energy generation (12–14), indicating that different aspects of cell metabolism can regulate macrophage polarization. Although many studies have shown the role of cell metabolism in the regulation of polarization and activation of macrophages in inflammatory disease development, how cell metabolism sustains macrophage activation and if this contributes to inflammatory disease has not been fully elucidated. Moreover, how 4-1BBL activity that drives sustained macrophage responses is linked to macrophage metabolism and polarization remain unknown.
In this study, we investigated the role of 4-1BBL in the polarization and metabolism of macrophages, and in skin inflammation using an IMQ-induced psoriasis mouse model. We find that 4-1BBL induces pro-inflammatory macrophage polarization and the development of psoriasis by contributing to the expression of inflammatory cytokines. We also find that sustained inflammation mediated by 4-1BBL is regulated by glycolysis and fatty acid synthesis, indicating the distinct role of cell metabolism in the late phase of macrophage activation for sustaining inflammatory responses. Consequently, inhibition of fatty acid synthase by a pharmacological inhibitor alleviates psoriasis and reduces the expression of 4-1BBL. Additionally, co-blockade of 4-1BBL signaling and IL-17A activity additively ameliorates psoriasis, implying that combination treatment for blocking inflammatory cytokine activity could be an option for the treatment of diseases such as psoriasis.
METHODS AND MATERIALS
Reagents
2-Deoxyglucose, cerulenin, etomoxir, and rotenone were purchased from Calbiochem, and antimycin A was from Sigma. Recombinant murine IFN-γ, murine IL-4, and murine M-CSF were obtained from Peprotech, and LPS (E. coli 0111:B4) was from List Biological Laboratories. Recombinant mouse 4-1BB conjugated with human hIgG2-Fc2 (4-1BB-Fc) was prepared as previously described (3), and agonist rat anti-mouse 4-1BB (clone 3H3, rat IgG2a) antibodies were prepared in-house. Endotoxin levels were tested (< 0.01 EU/μg). Blocking anti-mouse IL-17A (clone 17F3, IgG1, κ) and anti-mouse 4-1BBL (clone TKS-1, Rat IgG2a, κ) were purchased from BioXcell. Corresponding isotype antibodies were obtained from the same source.
Animals
Sex-matched 8- to 12-week old male or female C57BL/6 mice were purchased from Jackson Laboratory and used as wildtype. 4-1BB-deficient mice ((15), C57BL/6 background) were bred at the La Jolla Institute for Immunology. Mice were housed in individually ventilated, pathogen-free cages with 12:12-h light-dark cycles, and fed ad libitum. Animal experiments were in compliance with the regulations of the Scripps Research Institute and the La Jolla Institute for Immunology Animal Care Committee in accordance with guidelines of the Association for the Assessment and Accreditation of Laboratory Animal Care.
IMQ-induced psoriasis-like skin inflammation.
To induce skin inflammation, 62.5 mg of 5% IMQ cream (Aldara; 3M Pharmaceuticals) or control cream (Vaseline) were applied to the shaved dorsal skin of mice once daily for 7 days. Mice were anesthetized in an isoflurane chamber connected to an anesthetic machine, and injected i.p. with isotype Ab, anti-4-1BBL Ab, anti-IL-17A Ab (100 μg/25g body weight), or 4-1BB-Fc (10 μg/25g body weight) daily before applying IMQ. Dorsal skin areas were scored for signs of psoriasis-like inflammation such as scaling, erythema, and thickness on a scale from 0 (none) to 4 (very severe), and a cumulative severity score was calculated (scale 0–12), as previously described (16). Sera, lymph nodes, and dorsal skin specimens were collected at the end of the experiment (day 8). Experiments were performed in a double-blinded manner.
Histology
Specimens from the dorsal skin were fixed in 10% neutral-buffered formalin, embedded in paraffin, and sectioned at 4 μm followed by H &E staining.
Cell culture
RAW 264.7 mouse macrophage cell line and immortalized WT or 4-1BBL-deficient macrophage cell lines (2) were cultured in DMEM supplemented with 10% FBS and antibiotics (Gibco). Peritoneal macrophages were obtained from peritoneum cavity of mice after the intraperitoneal injection of 4% thioglychollate. Bone marrow cells were obtained from femur and tibia of mice, and cultured in DMEM supplemented with 10% FBS, antibiotics, and 10 ng/ml M-CSF to differentiate into macrophages for 7 days. Mouse 4-1BBL-expressing adenovirus (Adv-4-1BBL) was kindly provided by Tania Watts (University of Toronto) and used to induce the overexpression of 4-1BBL in macrophages as in our previous publications (1, 2).
RAW 264.7 cells (5 × 105/ml), WT or 4-1BBL-deficient macrophages (5 × 105/ml), peritoneal macrophages (1 × 106/ml), or BMDMs (1 × 106/ml) were treated with LPS (100 ng/ml), IMQ (2 μg/ml), LPS + IFN-γ (100 g/ml), or IL-4 (10 ng/ml) for the indicated times for the preparation of RNAs, cell lysates, or culture supernatants. For the inhibition of cell metabolism, peritoneal macrophages were incubated with vehicle, 2-deoxyglucose (1 mM), rotenone (10 μM), cerulenin (20 μM), antimycin A (1 μM), etoximir (100 μM) or metformin (2 mM) for 1 h, and followed by LPS or IMQ stimulation, or Adv-4-1BBL infection (3 × 104 pfu/106 cells).
Reverse transcription and quantitative PCR
Total RNA was isolated from axillary draining lymph nodes, dorsal skins, macrophages, or cultured BMDMs using TRIzol reagent (Invitrogen). cDNA was prepared using a SuperScript IV reverse transcriptase kit (Thermo Fisher). Quantitative PCR was performed using PowerUp SYBR Green Master Mix (Thermo Fisher). Gene expression was calculated by normalizing to GAPDH mRNA levels. Primer sequences are provided in Supplemental Table 1.
Flow cytometry
For intracellular staining of cytokines, cells from axillary lymph nodes were stimulated with 20 ng/ml of PMA and 1 μg/ml of ionomycin in the presence of Monensin (Biolegend) for 6 h. Cells were incubated with anti-mouse CD16/CD32 Ab (clone 2.4G2, Tonbo Biosciences) to prevent non-specific binding, and further stained with CD45-AF700 (clone 30-F11, Biolegend), CD3-PerCP-Cy5.5 (clone 145-2C11, Biolegend), CD4-PB (clone GK1.5, Biolegend), and CD25-PE (clone PC61.5, Thermo Fisher) Abs followed by FoxP3-FITC Ab (clone FJK-16s, Thermo Fisher) using FoxP3/Transcription Factor staining kit (Thermo Fisher) to examine the intracellular FoxP3 expression, or CD45-AF700, CD3-PerCP-Cy5.5, CD4-PB, and γδ TCR-FITC (clone GL3, Biolegend) Abs followed by the intracellular staining using IL-17A-APC (clone TC11-18H10.1, Biolegend) and IFN-γ-PE (clone XMG1.2, Biolegend) Abs. Isotype antibodies were purchased from the same source.
For the analysis of macrophage polarization, WT or 4-1BBL-deficient macrophages were incubated with anti-mouse CD16/CD32 Ab and followed by cell surface staining with CD11b-FITC (clone M1/70, Thermo Fisher), F4/80-PerCPCy5.5 (clone BM8, Thermo Fisher), CD11c-AF700 (clone N418, Thermo Fisher) and CD206-APC (clone C068C2, BioLegend). After fixation and permeabilization, cells were stained intracellularly with NOS2 (iNOS)-PE eF610 (clone CXNFT, Thermo Fisher) and Egr2-PE (clone erongr2, Thermo Fisher).
Flow cytometry analysis was performed on an LSR-II Flow Cytometer and data were analyzed using FlowJo Software (version 10, FlowJo, LLC).
Extracellular flux analysis
Metabolic changes were monitored by an extracellular flux analyzer XFe24 (Seahorse Bioscience). For optimization, 3 doses of cell densities and concentrations of FCCP (carbonyl cyanide-p-trifluoromethoxyphenylhydrazone) were tested in both stress tests for the best metabolic performance of macrophages, and 125,000 cells were determined to be the best concentration along with 1 μM FCCP. Cells were resuspended in XF Base Media (DMEM, Seahorse Bioscience) supplemented with 2 mM glutamine, and seeded in a Seahorse Bioscience 24-well plate. After 3 h, cells were treated with medium, LPS + IFN-γ, IMQ, or IL-4, and incubated for 24 h at 37°C in an atmosphere of 5% CO2. During extracellular flux analysis, cells were be sequentially treated with 10 mM glucose, 2 μM oligomycin A (OA), 1 μM FCCP + 1 mM pyruvate, and 1 μM rotenone + antimycin A (R/A) to measure the extracellular acidification rate (ECAR, glycolysis indication) and the oxygen consumption rate (OCR, mitochondrial respiration indication) following a previously published protocol (17).
Short hairpin RNAs, lentivirus, and generation of Glut1 or Fasn knockdown macrophages
Lentiviral vectors expressing the short hairpin RNAs (shRNAs) that target Glut1 (clone ID TRCN0000079328) or Fasn (clone ID TRCN0000075703) genes were obtained from Sigma (MISSION® TRC shRNAs). Lentiviruses were prepared in 293T cells by co-transfection of shRNA-encoding plasmids and helper plasmids such as pRSV-REV, pMDLg and pVSV-G (Addgene).
To generate knockdown (KD) macrophage cells, RAW 264.7 cells were infected with scramble (control, cat # SHC 201), Glut1, or Fasn shRNAs lentiviruses. After infection, cells were further incubated with puromycin (5 μg/ml, Invivogen), and KD of genes was confirmed by immunoblotting using rabbit anti-GLUT1 (Abcam) or mouse anti-Fasn (Santa Cruz Biotechnology) antibodies. GAPDH levels were detected as a loading control using anti-GAPDH antibody (Santa Cruz Biotechnology).
Cytokine measurements
Concentrations of TNF, IL-23, IFN-γ and IL-17A were measured by ELISA kits (Thermo Fisher).
Intracellular succinate quantification
Succinate levels in macrophages were quantified using a colorimetric Succinate Assay Kit (Abcam) according to the manufacturer’s protocol. WT or 4-1BBL-deficient macrophages were stimulated with PBS, LPS + IFN-γ, or IMQ for 6 or 12 h, and cell lysates were prepared to determine the intracellular succinate levels.
Statistical analysis
Statistical significance was analyzed by unpaired t-test for comparison of means between two independent groups or by one-way ANOVA followed by Tukey’s post hoc multiple comparison test for differences of means between multiple groups using Prism 8 software (version 8, GraphPad, San Diego, CA). A p value <0.05 was considered statistically significant and can be found in the figure legends. For in vitro experiments, n = number of samples. For in vivo studies, n = number of animals in each group. Each experiment was repeated as indicated in the figure legends, and the representative results are shown.
RESULTS
Inhibition of 4-1BBL-mediated signaling alleviates IMQ-induced psoriasis in mice.
In our previous studies, we showed that inhibition of 4-1BBL signaling, by administration of blocking 4-1BBL Ab or antagonist 4-1BB-Fc, reduces the sustained inflammatory response of macrophages in vitro and alleviates the pathology of endotoxin-induced sepsis in mice, similar to the response of 4-1BBL-deficient macrophages or 4-1BBL-deficient mice (1–3). To extend this data, we assessed the role of 4-1BBL in skin inflammation by inhibiting 4-1BBL in a mouse model of psoriasis. Topical application of the TLR7/8 ligand IMQ induces psoriasis-like skin inflammation. In patients with psoriasis and in mice with IMQ-induced skin inflammation, inflammatory cells and mediators such as lymphocytes, macrophages, plasmacytoid DCs, IL-17A, IL-22, IL-23, and TNF all contribute to the pathology of disease (18). Histological analysis and severity scoring showed that skin inflammation was severe in IMQ-treated mice compared with control cream-treated mice, but IMQ-driven inflammation was significantly reduced by administration of 4-1BBL Ab or 4-1BB-Fc (Fig. 1 A&B). Expression of pro-inflammatory cytokines Tnf, Nos2, Il17a, and Il23 was increased in draining lymph node (LN) cells and skin of IMQ-treated control Fc-injected mice, and was reduced by 4-1BBL Ab or 4-1BB-Fc administration. mRNA expression of Il10, Arg1, and Tgfb did not change in LN cells, while expression of these molecules was reduced in the skin tissues of 4-1BBL Ab or 4-1BB-Fc-injected mice (Fig. 1 C&D). IMQ treatment also induced the expression of 4-1BBL in LN and skin tissues, and this was reduced by administration of 4-1BBL Ab or 4-1BB-Fc in LN, but not in skin. We further found that production of TNF, IL-23, and IL-17A in sera was significantly reduced by 4-1BBL Ab or 4-1BB-Fc administration, while IFN-γ production was comparable between isotype Ab-, and 4-1BBL Ab or 4-1BB-Fc-injected mice (Fig. 1E). Collectively, our results suggest that blocking of 4-1BBL signaling ameliorates the pathology of psoriasis by regulating the expression of inflammatory genes associated with skin pathology.
Figure 1.
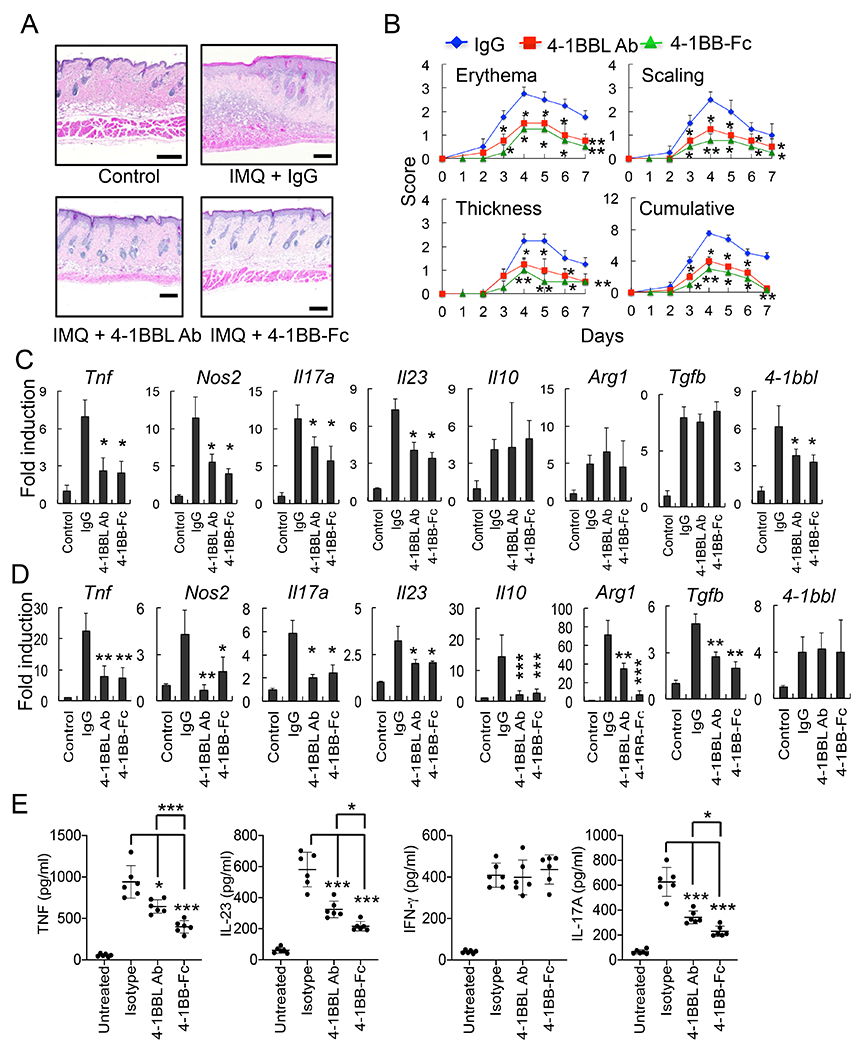
Inhibition of the 4-1BBL-mediated signaling alleviates the IMQ-induced psoriasis in mice. C57BL/6 mice (6 mice per group) were injected i.p. with isotype IgG (100 μg), 4-1BBL Ab (100 μg) or 4-1BB-Fc (10 μg) daily, and treated with IMQ cream or control cream on the shaved back skin for 7 days. A, H&E staining of the mouse back skin. B, Erythema, scaling, and thickness of the back skin was scored daily and the cumulative score is depicted. C & D, RNA samples were prepared from LNs or skin tissues on day 8, and expression of cytokine genes in LNs (C) and skin (D) was analyzed by qPCR (6 mice per group). Control cream-treated mice were used as a control. E, Sera were collected from mice after 5 days, and cytokine levels were measured by ELISA (6 mice per group). Data are shown as mean ± SD. *, p<0.05; **, p<0.01; ***p<0.005. Scale bar, 200 μm. Result shown is representative of repeated experiments.
4-1BBL regulates the polarization of macrophages.
Because we found that blocking 4-1BBL signaling reduced the expression of Tnf, Nos2, and Il23 in IMQ-treated mice, molecules that are hallmark genes of M1 macrophages, we further tested whether 4-1BBL regulates the polarization of macrophages in vitro. WT or 4-1BBL KO macrophages (2) were treated with LPS plus IFN-γ or IL-4 for 24 h. We found that a 4-1BBL-deficiency resulted in the reduced expression of Tnf, Nos2, Il23, Il6, and Cxcl10 in LPS plus IFN-γ-treated macrophages (M1). In contrast, the levels of expression of Il10, Arg1, Fizz1, Ym1, Egr2, and Mrc1 (Cd206) were increased in IL-4-treated 4-1BBL KO cells (M2) (Fig. 2A).
Figure 2.
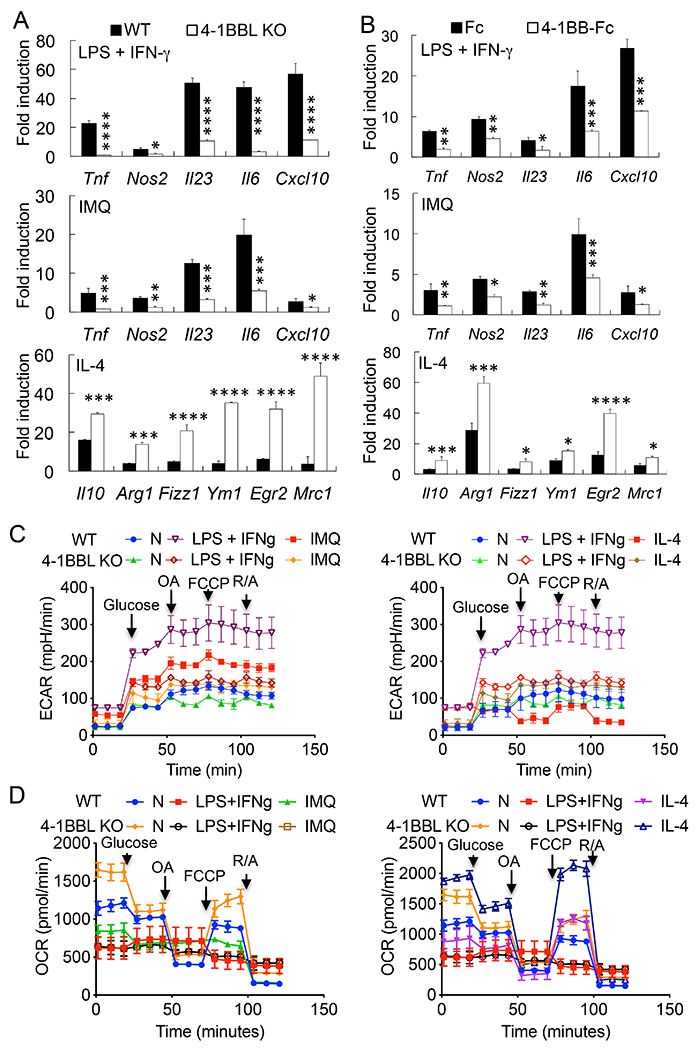
4-1BBL regulates the macrophage metabolism and polarization. A, WT or 4-1BBL-deficient macrophages were treated with LPS (100 ng/ml) + IFN-γ (100 ng/ml), IMQ (2 μg/ml), or IL-4 (10 ng/ml) for 24 h, or B, WT BMDMs were cultured in M-CSF, and treated with LPS + IFN-γ, IMQ, or IL-4 with Ig Fc or 4-1BB-Fc (5 μg/ml) for 24 h. Gene expression was analyzed by qPCR (n = 4). Fold induction was compared to each unstimulated control. C & D, Cell metabolism analysis. Glycolysis (C) and mitochondrial (D) stress tests were used to measure the contribution of 4-1BBL to macrophage metabolism. Unstimulated (N), LPS + IFN-γ-, IMQ-, or IL-4-stimulated WT or 4-1BBL-deficient (KO) macrophages were analyzed on a Seahorse XFe24 Extracellular Flux Analyzer (n=3). Data are shown as mean ± SD. *, p<0.05; **p<0.01; ***p<0.005; ****p<0.001. Statistical analysis of C & D is described in the text. Result shown is representative of 3 (A & B) or 2 (C) independent experiments.
We further tested whether blocking 4-1BBL signaling by 4-1BB-Fc regulates the polarization of macrophages. M-CSF-cultured BMDMs from WT mice were treated with LPS plus IFN-γ or IL-4. Similar to 4-1BBL KO macrophages, blocking of 4-1BBL resulted in the reduced expression of Tnf, Nos2, Il23, Il6, and Cxcl10 in LPS plus IFN-γ-treated BMDMs while IL-4-induced expression of Il10, Arg1, Fizz1, Ym1, Egr2, and Mrc1 was increased compared with control Fc-treated macrophages (Fig. 2B).
We also found that IMQ, the TLR7/8 ligand that induces psoriasis-like skin inflammation, regulates the polarization of macrophages in vitro. Similar to LPS plus IFN-γ-treated macrophages, IMQ treatment induced the expression of Tnf, Nos2, Il23, Il6, and Cxcl10 in WT macrophages. However, a deficiency of 4-1BBL or blocking of 4-1BBL resulted in the reduced the expression of these inflammatory genes (Fig. 2 A&B). Additionally, the number of F4/80+CD11b+CD11c+ cells and intracellular NOS2 levels were significantly reduced in LPS plus IFN-γ- or IMQ-treated 4-1BBL-deficient macrophages compared with WT cells, whereas those of F4/80+CD11b+CD206+ and expression of Egr2 were rather increased in IL-4-treated 4-1BBL-deficient cells, further supporting the role of 4-1BBL in the regulation of macrophage polarization (Supplemental Fig. 1A).
Since the intracellular accumulation of the TCA cycle intermediate succinate is one of the most profound metabolic changes in M1-like polarized macrophages (19), we examined the changes in succinate levels in macrophages. Treatment with LPS plus IFN-γ or IMQ increased the intracellular succinate levels in WT and 4-1BBL-deficient macrophages similarly for the first 6 h, while succinate levels were lower in 4-1BBL-deficient macrophages compared with WT cells after 12 h of stimulation (Supplemental Fig. 1B), indicating that 4-1BBL induced by TLR signaling regulates cell metabolism at the late phase of macrophage activation and polarization.
To further examine the role of 4-1BBL in macrophage polarization, we performed glycolysis stress tests using the Seahorse analyzer. Following glucose injection, a large increase in glycolysis was detected in LPS plus IFN-γ- or IMQ-treated cells, with the level of increase being higher with LPS plus IFN-γ. In contrast, 4-1BBL-deficient macrophages displayed significantly lower ECAR after glucose injection (Fig. 2C, p<0.001, unstimulated vs. LPS plus IFN-γ-stimulated; p<0.005, unstimulated vs. IMQ-stimulated; p<0.01, LPS plus IFN-γ- vs. IMQ-stimulated; p<0.005, LPS plus IFN-γ-stimulated WT vs. 4-1BBL KO cells; p<0.01, IMQ-stimulated WT vs. 4-1BBL KO cells). IL-4-polarized WT or 4-1BBL-deficient macrophages displayed lower rates of glycolysis compared with LPS plus IFN-γ- or IMQ-treated M1-like cells (Fig. 2C, p<0.001).
Next, we determined potential changes in oxidative metabolism driven by 4-1BBL. LPS plus IFN-γ- or IMQ-treated WT macrophages exhibited lower OCR compared with unstimulated or IL-4-stimulated cells and were less responsive to FCCP injection, while IL-4-stimulated macrophages displayed higher responses (Fig. 2D, p<0.01, unstimulated WT vs. LPS plus IFN-γ- or IMQ-stimulated WT or 4-1BBL KO; p<0.005, unstimulated 4-1BBL KO vs. LPS plus IFN-γ- or IMQ-stimulated WT or 4-1BBL KO; p<0.01, IL-4-stimulated WT vs. unstimulated WT; p<0.001, IL-4-stimulated 4-1BBL KO vs. unstimulated 4-1BBL KO macrophages). In this case, a deficiency of 4-1BBL did not significantly change the OCR of LPS plus IFN-γ- or IMQ-treated WT macrophages while the OCR of 4-1BBL-deficient macrophages significantly increased in response to FCCP in IL-4-stimulated cells compared with WT macrophages (Fig. 2D, p<0.001, IL-4-stimulated 4-1BBL KO vs. IL-4-stimulated WT macrophages). Collectively, our data support a role of 4-1BBL in cell metabolism associated with macrophage polarization.
Glycolysis and fatty acid synthesis regulate the 4-1BBL-mediated sustained inflammatory responses in macrophages.
Since 4-1BBL expression is initially induced in macrophages by TLR signaling (1, 2), we tested whether 4-1BBL expression driven by TLR signaling is regulated by cell metabolism. In macrophages, expression of Tnf and 4-1BBL induced by LPS was significantly reduced by inhibiting glycolysis by 2-deoxyglucose (2-DG), fatty acid synthesis by cerulenin, fatty acid oxidation by etomoxir, electron transport chain (ETC) complexes by antimycin A or rotenone, or ETC/gluconeogenesis by metformin (Fig. 3A). The optimal concentration of each inhibitor was determined in our pilot experiment (data not shown) and similar to that used in other studies. We previously showed that induction of 4-1BBL is driven by the same signaling pathway that promotes Tnf expression (1) and these inhibitors similarly reduced Tnf. Additionally, IMQ-induced expression of 4-1BBL mRNA and production of TNF were also reduced by these inhibitors in macrophages (Supplemental Fig. 2 A&B). These data indicate that cell metabolism regulates the expression of 4-1BBL during the early phase of TLR-mediated macrophage activation.
Figure 3.
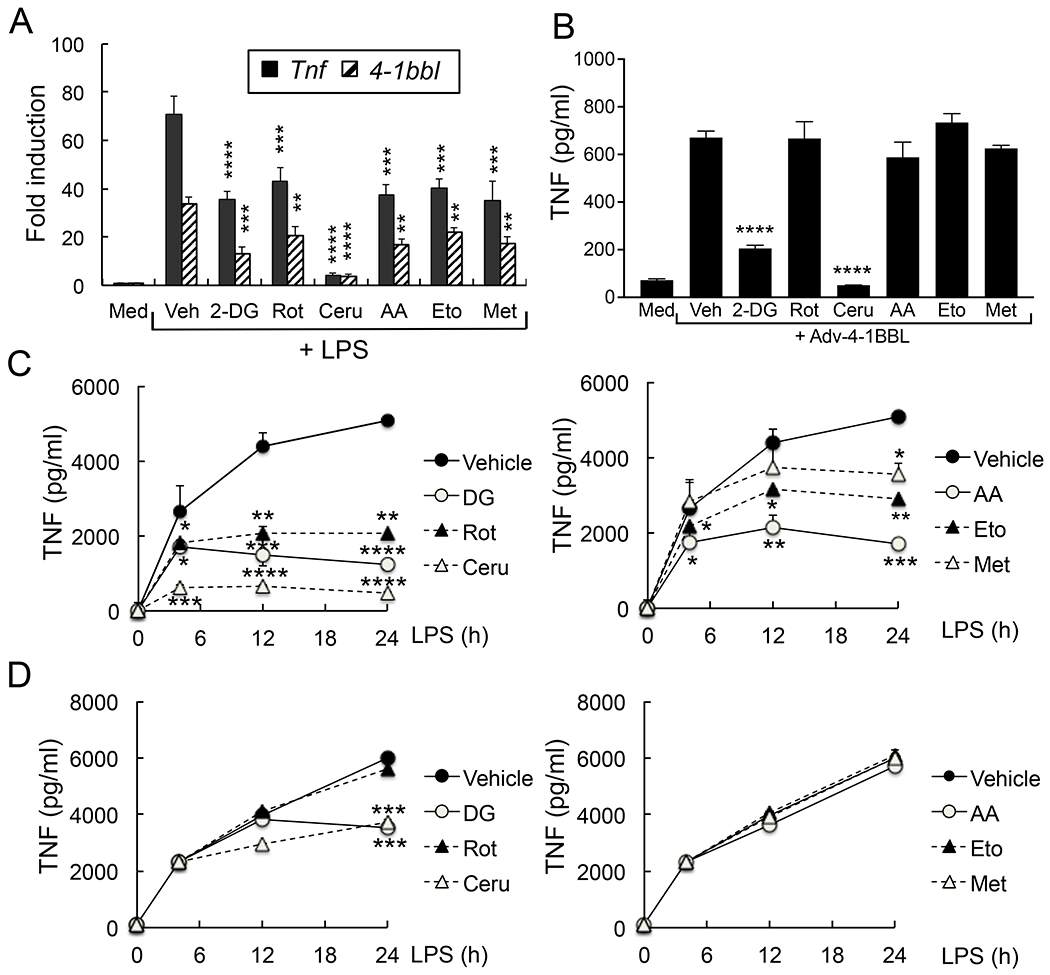
Macrophage metabolism regulates the 4-1BBL-mediated sustained inflammatory responses. A&B, Peritoneal macrophages (106 cells/ml) were incubated with vehicle, 2-deoxyglucose (2-DG, 1 mM), rotenone (Rot, 10 μM), cerulenin (Ceru, 20 μM), antimycin A (AA, 1 μM), etomoxir (Eto, 100 μM) or metformin (Met, 2 mM) for 1 h. Cells were further stimulated with LPS (100 ng/ml) and 4-1BBL expression was analyzed by qPCR after 4 h (A, n = 4), or infected with Adv-4-1BBL (3 × 104 pfu/106 cells). TNF levels in culture supernatant were measured by ELISA after 24 h (B, n = 4). C & D, Peritoneal macrophages were incubated with inhibitors 1 h before (C) or 4 h after (D) LPS stimulation. TNF levels in culture supernatants were measured at the indicated times by ELISA. Fold induction was compared to unstimulated control. Data are shown as mean ± SD. *, p<0.05; **, p<0.01; ***p<0.005; ****p<0.001. Representative result of 3 independent experiments is shown.
We further confirmed the role of glycolysis and fatty acid synthesis in the regulation of inflammatory responses by knocking down Glut1 (glucose transporter 1) or Fasn (fatty acid synthase) (Supplemental Fig. 2 D & E). LPS plus IFN-γ- or IMQ-induced expression of Tnf, Il23, Il6, and 4-1BBL was reduced in Glut1 or Fasn KD macrophages, while Nos2 expression was reduced in Glut1 KD macrophages but not in Fasn KD cells. In addition, IMQ stimulation did not induce the expression of Cxcl10, whereas LPS plus IFN-γ-induced Cxcl10 expression, which was reduced by Glut1 or Fasn KD.
When 4-1BBL is induced in macrophages it then associates directly with TLRs to allow sustained cytokine production. Overexpression of 4-1BBL by infecting cells with 4-1BBL-encoding adenovirus can also result in this late phase signaling to maintain the inflammatory response (1, 2). Thus, we tested whether cell metabolism regulates TNF production following overexpression of 4-1BBL. We found that this was significantly reduced by 2-DG or cerulenin treatment, but not by the other inhibitors (Fig. 3B), suggesting that late phase 4-1BBL-mediated response is regulated by glycolysis and fatty acid synthesis. The latter was further investigated by timed treatment with these inhibitors. While LPS- or IMQ-induced TNF production was reduced in macrophages preincubated with all of the inhibitors, addition of only 2-DG or cerulenin after 4 h of stimulation blocked TNF production (Fig. 3 C&D and Supplemental Fig. 2C).
Next, we compared the expression levels of essential genes in the cell metabolism pathways to examine whether blocking of 4-1BBL signaling affected the expression in psoriasis. Glut1 facilitates the transport of glucose across the plasma membranes of mammalian cells for the conversion of glucose to pyruvate for energy generation via citric acid cycle in mitochondria, to lactate in the presence of oxygen (Warburg Effect), or back to carbohydrate or fatty acids. Pyruvate dehydrogenase kinase 1 (Pdk1) and pyruvate dehydrogenase-alpha 1(Pdha1) regulate the further conversion of pyruvate into acetyl-CoA. ATP citrate lyase (Acly) links glycolysis and fatty acid synthesis by converting citrate from glycolysis to acetyl-CoA for fatty acid synthesis. Fasn is a critical enzyme in fatty acid synthesis that catalyzes the synthesis of palmitate from acetyl-CoA and malonyl-CoA in the presence of NADPH (5, 6, 20). Expression of Fasn was significantly reduced in LN or skin of 4-1BBL Ab- or 4-1BB-Fc-injected IMQ-treated mice, while Glut1 levels were comparable (Supplemental Fig. 3). Expression of Pdk1, Pdha1, and Acly was not induced in IMQ-treated LN or skin, and inhibition of 4-1BBL did not affect the expression (Supplemental Fig. 3). Collectively, these results suggest a role for specific macrophage metabolic pathways in sustaining inflammatory cytokine production upstream and downstream of 4-1BBL.
Inhibition of Fasn activity ameliorates psoriasis
A recent study showed the contribution of Glut1 to psoriasis development (21) as Glut1 inactivation by an inhibitor WZB117 reduced the severity of psoriasis in mice. Based on our results above, we then tested whether inhibition of Fasn activity using cerulenin can also reduce the severity of IMQ-induced psoriasis. Interestingly, IMQ-induced skin inflammation was less severe in cerulenin-administered mice compared with vehicle-injected animals, indicating the contribution of fatty acid synthesis to the pathology of psoriasis (Fig. 4 A&B). IMQ-induced expression of Tnf, Nos2, and Il17a was also significantly reduced in LN and skin by cerulenin administration, whereas expression of Il23, Il6, Il10, and Tgfb was reduced in LN, but not in the skin (Fig. 4 C&D). It should be noted that cerulenin reduced the expression of 4-1BBL in LN, but not in the skin of IMQ-treated mice, indicating the tissue-specific role of fatty acid synthesis for the expression of 4-1BBL in psoriasis. Furthermore, IMQ-induced expression of Fasn, not Glut1, was significantly reduced by cerulenin administration (Fig. 4 E&F). Cerulenin injection from day 4 onwards also reduced skin inflammation, although the effect was lower and the reduction of inflammatory cytokine expression was less than cerulenin administration from day 1 (Fig. 4 A–F). Collectively, our data suggest that fatty acid synthesis contributes to the development of psoriasis both during the initiation of disease as well as once disease is established.
Figure 4.
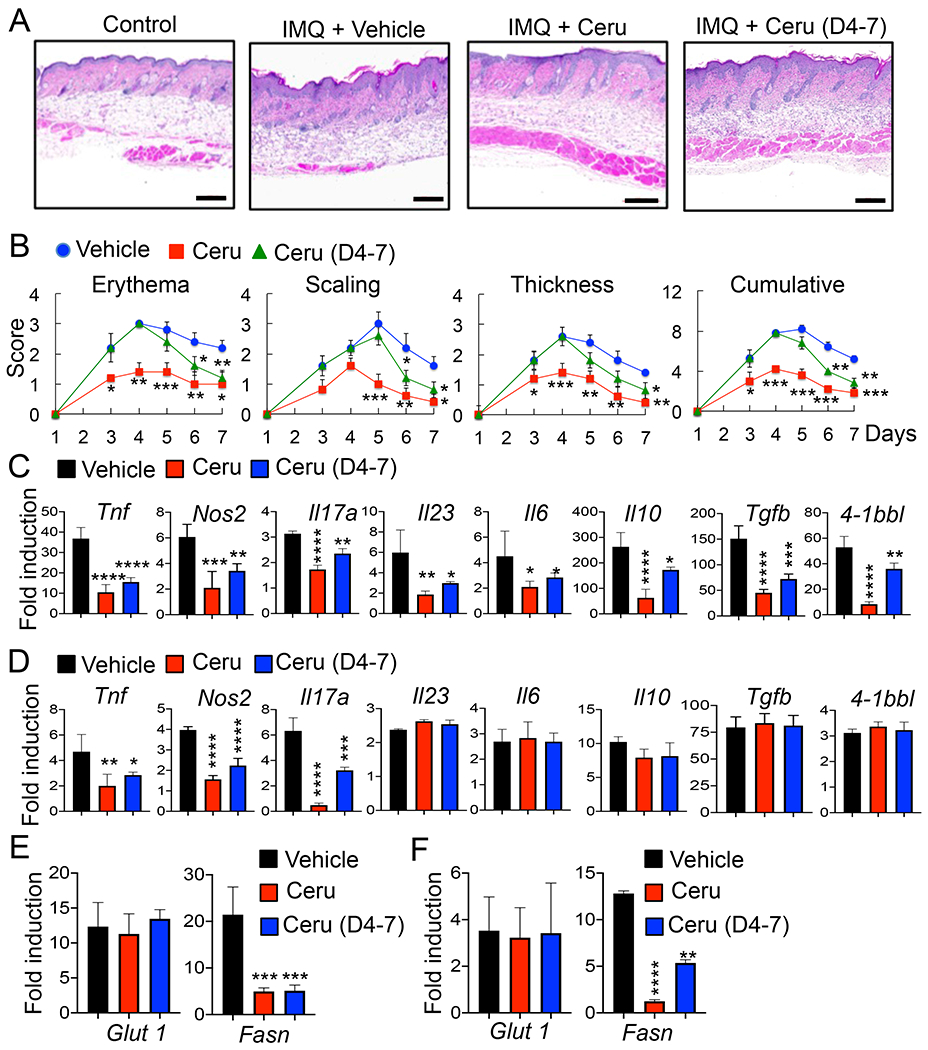
Inhibition of the fatty acid synthesis pathway alleviates the IMQ-induced psoriasis in mice. C57BL/6 mice (6 mice per group) were injected i.p. with vehicle (10% EtOH) or Ceru (30 mg/kg body weight in 10% EtOH) daily followed by control or IMQ cream application on the shaved back skin daily for 7 days. A, H&E staining of the back skin. B, Scoring of erythema, scaling, and thickness of the back skin. The cumulative score is depicted. C - F, RNA samples were prepared from LNs or skin tissues on day 8. Quantitative PCR analysis was performed to determine the expression of cytokine genes (C & D) in LNs (C) and skin (D), or Glut1 and Fasn (E & F) in LNs (E) or skin (F) (n = 4). Control cream-treated mice were used as control. Fold induction was compared to control. Data are shown as mean ± SD. *, p<0.05; **, p<0.01; ***p<0.005; ****p<0.001. Scale bar, 200 μm. Representative result of repeated experiments is shown.
4-1BB deficiency augments the severity of IMQ-induced psoriasis
Previous studies have demonstrated that 4-1BBL acts as a costimulatory molecule by binding to its receptor 4-1BB on activated T cells, but only a moderate amount of data suggests that this 4-1BB-dependent T cell activation by 4-1BBL plays a role in inflammatory disease (22, 23). In contrast, emerging evidence suggests a role for 4-1BBL in some inflammatory diseases independent of its interaction with 4-1BB (1–3). To expand on this concept, WT or 4-1BB-deficient mice were treated with IMQ, and we found that skin inflammation was actually more severe in 4-1BB KO mice compared with WT mice (Fig. 5 A&B). Expression of Nos2, Il17a, Il23, and Il6 was increased while Tnf expression was not affected in LN cells of IMQ-treated 4-1BB KO mice; however, that of Il10, Arg1, and Tgfb was reduced in LN cells of 4-1BB KO mice (Fig. 5C). In the skin tissues of 4-1BB KO mice, expression of Tnf, Nos2, Il17a, Il23, Il6, Il10, and Arg1 was significantly increased while Tgfb expression was reduced compared with WT skin (Fig. 5D).
Figure 5.
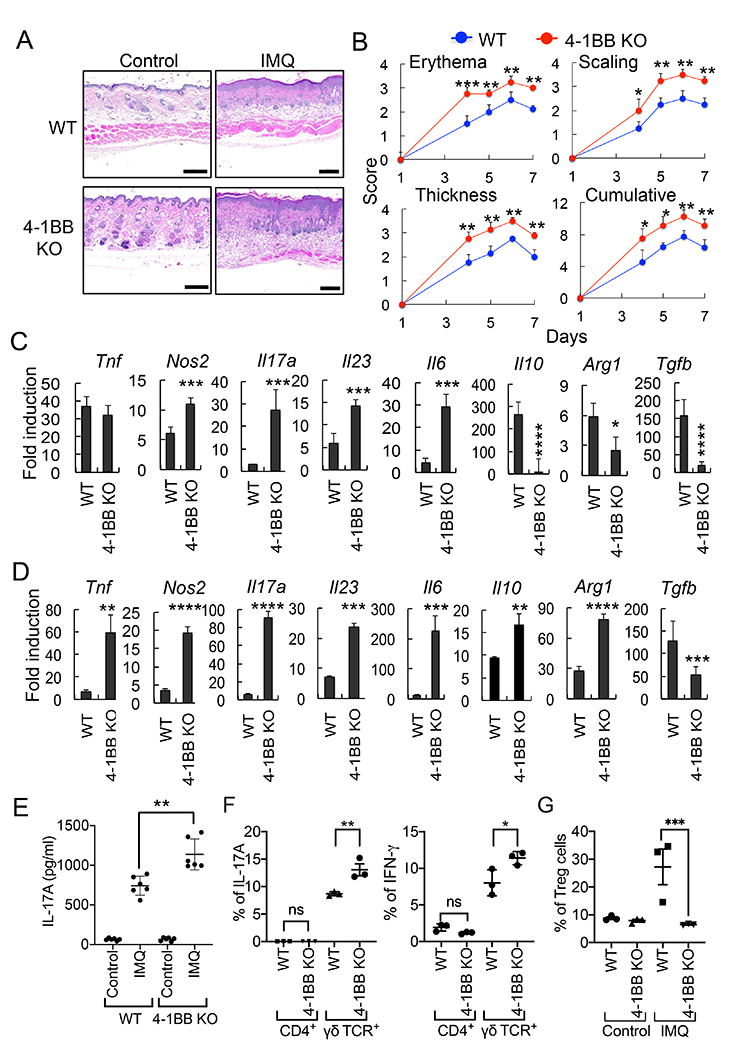
4-1BB regulates the induction of psoriasis. WT and 4-1BB-deficient mice (6 mice per group) were treated epicutaneously on the shaved back with IMQ or control cream for 7 days to induce psoriasis-like skin inflammation. A, H&E staining of the mouse dorsal skin. B, Erythema, scaling, and thickness of the back skin was scored daily from day 4 and the cumulative score is depicted. C & D, RNA samples were prepared from LN (C) or back skin (D) of mice on day 8, and the mRNA induction of inflammatory genes was analyzed by qPCR. Control cream-treated mouse samples were used as controls. E, Sera were collected from mice at day 8, and IL-17A levels were determined by ELISA (6 mice per group). F, Expression of IL-17A is regulated by 4-1BB in γδ T cells in psoriasis. Lymphocytes isolated from LN were analyzed by flow cytometry for intracellular IL-17A or IFN-γ expression. Cells were gated for CD45+CD3+CD4+ or CD45+CD3+γδ TCR+, and expression of IL-17A or IFN-γ in each population was analyzed (n = 3). G, Treg induction in LN cells was analyzed by flow cytometry. Cells were gated CD45+CD3+CD4+CD25+ cells, and the intracellular levels of FoxP3 were analyzed (n=3). Data are shown as mean score ± SD. *, p<0.05; **, p<0.01; ***p<0.005. Scale bar, 200 μm. Result shown is representative of repeated experiments.
Additionally, the serum level of IL-17A was increased in 4-1BB KO mice compared with WT mice at day 8 (Fig. 5E). Expression of IFN-γ and IL-17A in γδ T cells in LN that play a pivotal role in skin inflammation (24) were higher in IMQ-treated 4-1BB KO mice than WT mice at day 4 (Fig. 5F). Treg cells are critical for the regulation of inflammatory disease development, including in psoriasis (25, 26), and we found that the population of CD4+CD25+Foxp3+ Treg cells was significantly reduced in LN of IMQ-treated 4-1BB-deficient mice compared with WT mice (Fig. 5G). This supports the role of 4-1BB in promoting Treg that can suppress inflammatory diseases as previously reported (27, 28), and suggests that the activity of 4-1BBL in contributing to psoriasis is independent of its interaction with 4-1BB.
Administration of agonist 4-1BB Ab alleviates the severity of IMQ-induced psoriasis by increasing Treg cells.
Previous studies have shown that agonist 4-1BB Ab can improve the pathology of several inflammatory and autoimmune diseases with the prevailing view being that it expands Treg cells (28–31). For example, in EAE models, administration of agonist 4-1BB Ab prevented the development of disease by regulating the balance between TH17 and Treg cells (32, 33).
In agreement with a previous study (34), we also found that administration of agonist 4-1BB Ab significantly alleviated the severity of IMQ-induced skin inflammation (Fig. 6 A&B). Expression of Il17a in LN and skin, and production of IL-17A in sera were reduced while Tnf and Il23 expression was comparable between isotype- and 4-1BB Ab-injected mice (Fig. 6 C–E). However, expression of IL-17A in CD4+ or γδ T cells in LN was comparable between isotype- or 4-1BB Ab-administered mice at day 4 of IMQ-treatment (Fig. 6F). The number of CD4+CD25+Foxp3+ Treg cells was significantly increased in LN of 4-1BB Ab-injected mice compared with isotype Ab-administered mice (Fig. 6G), suggesting a crucial role of 4-1BB signaling in Treg activation for the regulation of IL-17A expression in γδ T cells later stage in the development of psoriasis. Taken together, our data suggest that 4-1BBL and 4-1BB play opposite roles in the development of psoriasis, as 4-1BBL promotes while 4-1BB inhibits IMQ-induced skin inflammation.
Figure 6.
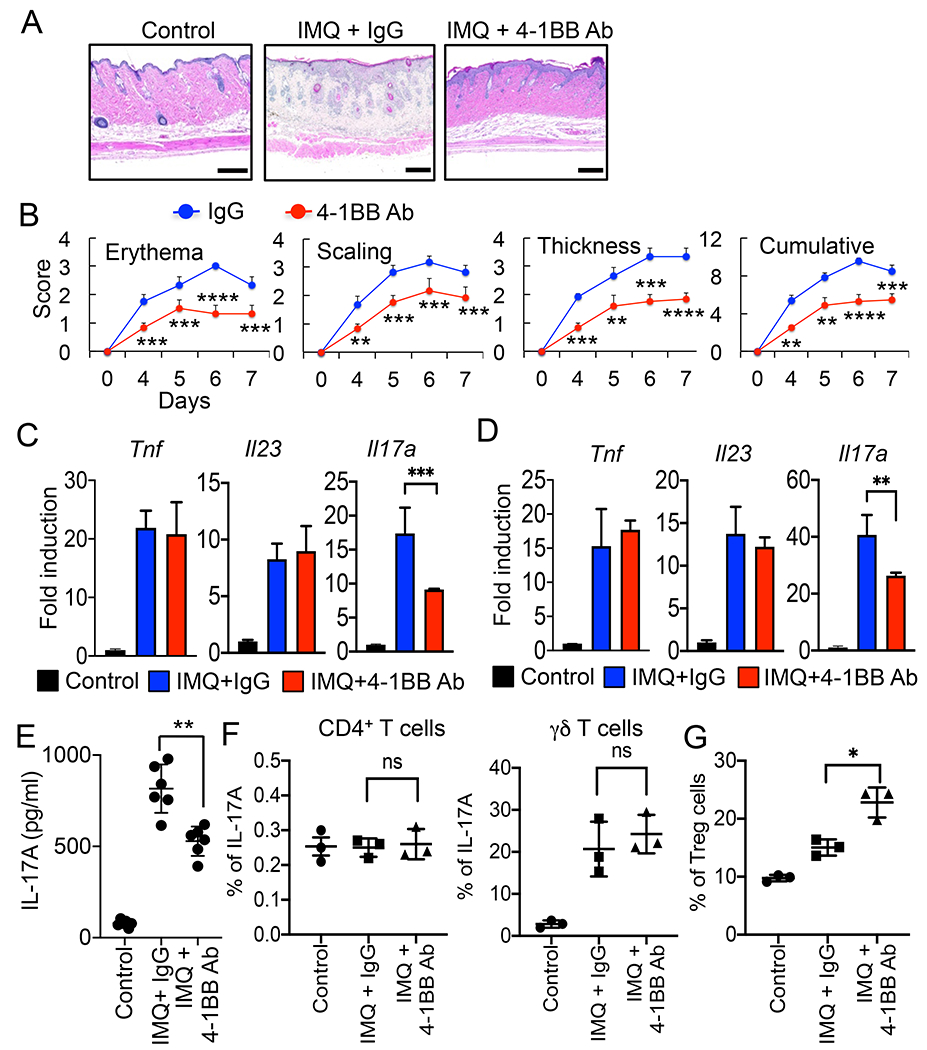
Administration of agonist 4-1BB Ab alleviates the pathology of skin inflammation. C57BL/6 mice (6 mice per group) were injected i.p. with isotype IgG (100 μg), 4-1BB Ab (100 μg) daily, and treated with IMQ cream or control cream on the shaved back skin for 7 days. A, Histology of the mouse back skin by H&E staining. B, Erythema, scaling, and thickness of the back skin was scored daily from day 4 and the cumulative score is depicted. C & D, Expression of inflammatory cytokines in LN (C) or back skin (D) of mice was analyzed by qPCR (4 mice per group). Control cream-treated mouse samples were used as controls. E, Serum IL-17A levels were determined by ELISA (n = 6). F, FACS analysis for IL-17A expression in 4-1BB Ab-treated mice. LN cells were gated for CD45+CD3+CD4+ or CD45+CD3+γδ TCR+, and intracellular level of IL-17A in each population was analyzed (n = 3). G, FACS analysis of Treg induction. LN cells were gated CD45+CD3+CD4+CD25+, and intracellular levels of FoxP3 was analyzed (n = 3). Data are shown as mean ± SD. *, p<0.05; **, p<0.01; ***p<0.005, and ns, not significant. Scale bar, 200 μm. Result shown is representative of repeated experiments.
Co-administration of 4-1BB-Fc and IL-17A Ab further ameliorates the severity of skin inflammation.
Inflammatory cytokines such as TNF, IL-23, and IL-17A play a crucial role in the pathogenesis of inflammatory diseases, and therapeutic strategies targeting cytokines have been found to be effective for the treatment of psoriasis (35–39). As we found that inhibition of 4-1BBL signaling by 4-1BB-Fc significantly alleviated the pathology of psoriasis, we further tested whether co-blockade of 4-1BBL and IL-17A could exert a greater effect on the treatment of IMQ-induced psoriasis. Histological analysis and severity scoring showed that skin inflammation was significantly reduced in 4-1BB-Fc- or IL-17A Abs-treated mice, while co-administration of 4-1BB-Fc and IL17A Ab reduced skin inflammation more effectively compared with administration of either alone (Fig. 7 A&B).
Figure 7.
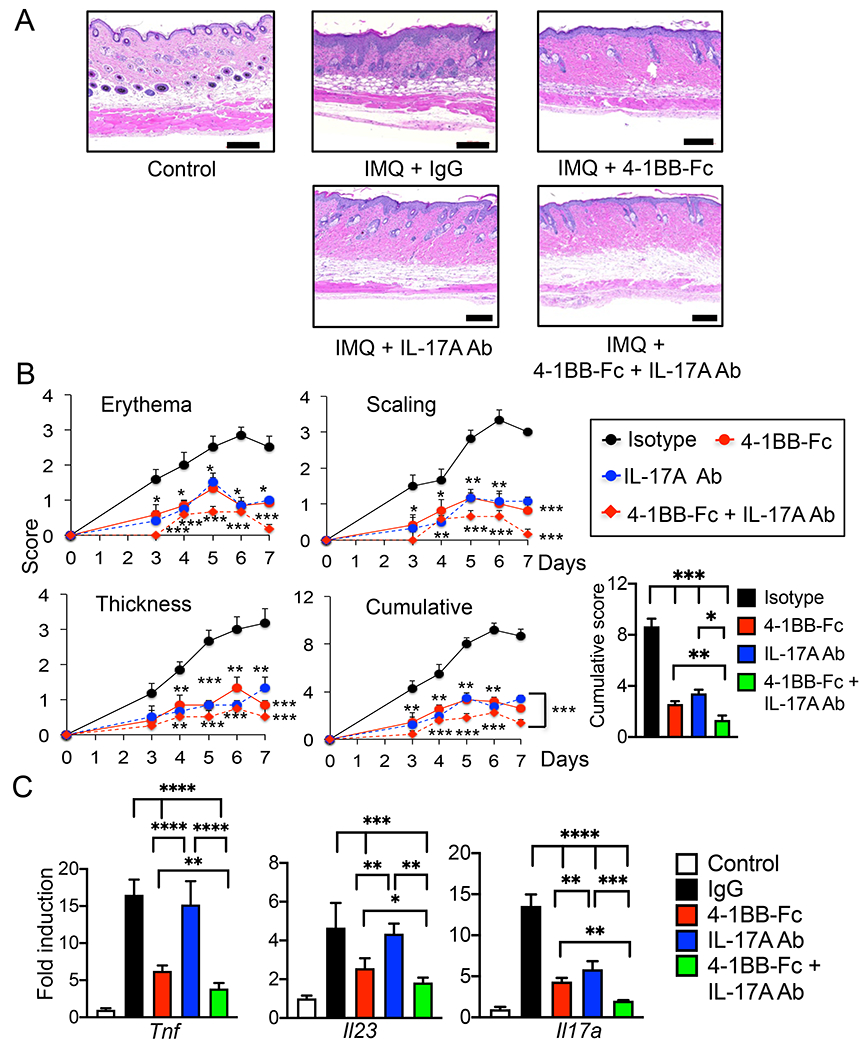
Co-administration of 4-1BB-Fc and IL-17A Ab shows greater efficacy in reduction of the psoriasis-like skin inflammation in mice. C57BL/6 mice (4 mice per group) were injected i.p. with isotype IgG (50 μg), 4-1BB-Fc (10 μg), IL-17A Ab (50 μg) or 4-1BB-Fc plus IL-17A Ab daily, and treated with IMQ cream or control cream on the shaved back skin and right ear daily for 7 days. A, H&E staining of the mouse dorsal skin. B, Erythema, scaling, and thickness of the dorsal skin was scored daily and the cumulative score is depicted, and the cumulative score at day 7 is shown. C, RNA samples were prepared from skin tissues on day 8, and expression of inflammatory cytokines was analyzed by qPCR (4 mice per group). Data are shown as mean ± SD. *, p<0.05; **, p<0.01; ***p<0.005; ****p<0.001, and ns, not significant. Scale bar, 200 μm. Result shown is representative of repeated experiments.
Administration of 4-1BB-Fc, IL-17 Ab, or the combination reduced the expression of inflammatory cytokines in skin tissues of IMQ-treated mice (Fig. 7C). 4-1BB-Fc or 4-1BB-Fc with anti-IL-17A administration was more effective than the anti-cytokine Ab in regulating the expression of Tnf, Il23, and Il17a, while IL-17A Ab administration did not affect the expression of Il23. Furthermore, expression of cytokines was reduced more by 4-1BB-Fc plus anti-IL-17A treatment than that by the treatment of either alone.
These results indicate the possibility that co-inhibition of 4-1BBL signaling and cytokine activity can be a highly effective anti-inflammation strategy for the treatment of psoriasis and possibly other inflammatory diseases.
DISCUSSION
Inflammation is a beneficial process to protect hosts from microbial infection and tissue damage, thereby maintaining homeostasis (40, 41). However, uncontrolled, excessive and sustained inflammation causes the development and progression of a range of inflammatory diseases (35, 42). Sustained inflammation is not the repetition of acute inflammation; it is distinct in a variety of pathological conditions, and its biological complexity limits the development of anti-inflammatory therapies (43). We have previously identified a novel signaling mechanism involving two distinct and sequential signaling cascades that regulate the initiation and persistence of inflammation in macrophages (1). TLR signaling drives the early response through MyD88/Trif-mediated NF-κB and MAPK signaling pathways and then 4-1BBL regulates the sustained production of inflammatory cytokines via binding to TLRs and promoting the MAPKs and protein kinase signaling pathways independently of NF-κB activation (1, 2). In line with this, here and in previous studies, we found that a deficiency or blocking 4-1BBL signaling alleviates several inflammatory conditions by dampening the sustained production of inflammatory cytokines (1–3). Thus, our results support the 4-1BBL signaling pathway as a target of anti-inflammation treatments in situations where TLR are active.
The anti-inflammatory activity of a 4-1BB-Fc fusion protein is achieved by inhibiting the oligomerization of TLRs and 4-1BBL (1–3), thereby alleviating sepsis and psoriasis in mice. Compared to a blocking anti-4-1BBL Ab, 4-1BB-Fc exhibited higher anti-inflammatory activity and binding affinity (~ 1000 fold) to 4-1BBL, while they share the same binding epitope of 4-1BBL. Thus, 4-1BB-Fc reduced the severity of skin inflammation more effectively. Most importantly, blocking of 4-1BBL significantly reduced the production of inflammatory cytokines such as TNF and IL-23 by macrophages. This is similar to the response of 4-1BBL-deficient macrophages. Thus we suggest that inhibition or blocking of 4-1BBL in innate immune cells reduces the production of pro-inflammatory cytokines that subsequently leads to dampened induction of other inflammatory cytokines, like IL-17 in psoriasis, that are key features of disease.
Cell metabolism plays a crucial role in determining the phenotypes of macrophages, and the development of inflammatory diseases (8–14). Previous studies have shown the role of cell metabolism in the polarization of macrophages in inflammatory diseases. However, how metabolic changes contribute to sustained inflammatory responses in macrophages and subsequent development of inflammatory disease are not fully elucidated. Our study demonstrates that genetic ablation of 4-1BBL or blocking of 4-1BBL signaling resulted in the reduced expression of pro-inflammatory genes associated with polarization of macrophages to the M1 phenotype. The metabolic impact of 4-1BBL in macrophages was demonstrated in that a 4-1BBL deficiency dramatically blunted glycolysis, as measured by ECAR, that was increased by TLR stimulation. OCR measurements indicated that respiration was substantially increased by the lack of 4-1BBL in the uncoupled state after FCCP injection in IL-4-stimulated 4-1BBL-deficient macrophages compared with WT cells. Thus, our data suggest that 4-1BBL contributes to the regulation of macrophage metabolism and polarization. Additionally, stimulation of macrophages with LPS or IMQ increased the intracellular levels of succinate, an indicative metabolite of M1-like polarized macrophages. 4-1BBL deficiency resulted in reduction of intracellular levels after 12 h while the accumulation of succinate was comparable between WT and 4-1BBL-deficient macrophages at early times after stimulation, further linking late 4-1BBL activity to metabolic changes and the polarization of macrophages.
Metabolic activity was also involved in TLR-mediated expression of 4-1BBL, as this was substantially reduced by a variety of inhibitors or knocking down of metabolism genes. However, the late phase of macrophage activation driven by 4-1BBL was also found to be dependent on glycolysis and fatty acid synthesis, showing the distinct role of cell metabolism in sustaining as well as initiating the inflammatory response of macrophages.
Although inhibition of glycolysis or genetic modification of Glut1 regulates the pro-inflammatory responses in macrophages, a previous study showed that treatment with the glycolysis inhibitor 2-DG reduced IL-1β, while Tnf expression was not affected in macrophages (19), which is inconsistent with our finding. Additionally, a recent study showed that IL-1β expression was rather increased while expression of TNF was not affected in LPS plus IFN-γ-stimulated Gut1-deficient macrophages (44). However, inhibition or knockdown of Glut1 reduced the expression of Tnf in LPS-treated macrophages in our system. In contrast, expression of Nos2 was significantly reduced by knockdown or genetic ablation of Glut1 in this and another study (44). Most likely this inconsistency is due to the experimental conditions of each study, such as the use of primary macrophages or cell lines, and the use of chemicals, or shRNA or genetic deletion of genes. Further studies will help us to better understand the inconsistency.
IMQ-induced expression of Fasn was downregulated by blocking of 4-1BBL signaling, implicating the fatty acid synthesis pathway in psoriasis. Fasn is expressed strongly in normal and inflamed human epidermis including psoriasis, and it may regulate epidermal differentiation (45). Additionally, inhibition of acetyl-CoA carboxylase that is an enzyme of the fatty acid synthesis pathway decreased IL-17-expressing T cell differentiation and IL-17 production by murine T cells (46), further suggesting a role of fatty acid synthesis in Th17 responses and inflammatory disease development (47). Inhibition of Fasn activity with cerulenin ameliorated psoriasis significantly by regulating the expression of inflammatory cytokines, including Tnf, Nos2, Il23, Il6, and Il17a in LN. Interestingly, cerulenin has been shown to suppress colorectal cancer development and liver metastasis (48–50), and reverse hepatic steatosis in obese mice (51). In innate immunity, cerulenin increases survival and decreases bacterial load in S. aureus-induced sepsis (52) and alleviates LPS-induced sepsis by suppressing the pro-inflammatory activation of macrophages (53, 54). However, cerulenin has side effects in cancer patients, such as anorexia and body weight loss due to a neuronal effect that reduces appetite (55). In mice, cerulenin did not show any detectable toxicity (54), but the next generation of Fasn inhibitors is currently being developed to avoid the adverse effect (56, 57).
A recent study has shown that Glut1 contributes to psoriasis development (21). Both genetic and pharmacological Glut1 inactivation by an inhibitor WZB117 reduced skin inflammation, suggesting that Glut1 is required for inflammation-associated keratinocyte proliferation. Although the expression of Glut1 was not affected by blocking 4-1BBL signaling in our study, inhibition of Glut1 might also inhibit glucose uptake in other cell types during skin inflammation, including immune cells, and thereby limit inflammation as previously reported roles for Glut1 and glucose metabolism in macrophages and T cells (44, 58, 59).
As 4-1BBL binding to its receptor 4-1BB on activated T cells has been shown to enhance T cell activity in a number of systems, this suggests that 4-1BB interaction with 4-1BBL could be involved in the development of some autoimmune or inflammatory diseases (22, 23). However, we found different phenotypes in 4-1BBL and 4-1BB knockout mice suggesting that 4-1BB and 4-1BBL can regulate the psoriasis inflammatory response independently. Specifically, genetic ablation of 4-1BB resulted in more severe skin inflammation and correspondingly stimulation of 4-1BB by agonist Ab alleviated pathology. 4-1BB deficiency increased the expression of inflammatory genes and reduced the Treg population in LN and skin of IMQ-treated mice, while the agonist Ab showed the opposite effect consistent with a previous report (34).
Inflammatory cytokines such as TNF, IL-23, and IL-17A play a crucial role in the pathogenesis of psoriasis and other diseases. However, despite the benefit of the current therapies targeting these cytokines in treating patients, a significant proportion fail to respond to the treatment and may suffer side effects (35–37, 60–65). Therefore, development of alternate therapeutic strategies is still needed. While the current therapeutics directly inhibit the activities of these individual cytokines, our strategy of targeting 4-1BBL may inhibit the production of multiple cytokines like TNF and IL-23 made by innate immune cells. Moreover, the combination of 4-1BB-Fc and IL-17A Ab was more effective in blocking psoriasis presumably by shutting down both the production of inflammatory cytokines and the activity of existing cytokines. Thus, combination therapy could be an advanced option for the treatment of psoriasis patients.
In summary our data show that 4-1BBL contributes to the pathology of psoriasis, and promotes macrophage M1 polarization, dependent on glycolysis and fatty acid synthesis. Our data suggest that fatty acid synthesis can be a potential target for the treatment of this disease, and that combination therapy with 4-1BB-Fc and anti-cytokine Abs may exhibit greater effectiveness.
Supplementary Material
KEY POINTS.
4-1BBL signaling regulates metabolism and polarization in macrophages.
Blocking 4-1BBL signaling alleviates the severity of psoriasis in mice.
Targeting 4-1BBL signaling can be an option for treating psoriasis in patients.
ACKNOWLEDGEMENT
We thank Dr. Edward E. Amento at Molecular Medicine Research Institute for helpful discussions and comments. Metabolic analysis was performed by Seahorse XFe24 extracellular flux analyzer at Cancer Metabolism Core, Sanford Burnham Prebys Medical Discovery Institute.
This work was partially supported by NIH grant AI088229 to Y. J. K., and by La Jolla Institute funds and NIH grant AR072640 to M.C.
Abbreviations used in this article,
- IMQ
imiquimod
- TCA cycle
tricarboxylic acid cycle
- OXPHOS
oxidative phosphorylation
- WT
wild-type
- KO
knockout
- BMDM
Bone marrow-derived macrophages
- M-CSF
Macrophage Colony Stimulating Factor
- 2-DG
2-deoxyglucose
- Ceru
Cerulenin
- ETO
etomoxir
- ETC
electron transport chain
- AA
antimycin A
- Rot
rotenone
- Met
metformin
Footnotes
CONFLICT OF INTEREST:
The authors have no financial conflicts of interest.
REFERENCES
- 1.Kang YJ, Kim SO, Shimada S, Otsuka M, Seit-Nebi A, Kwon BS, Watts TH, and Han J. 2007. Cell surface 4-1BBL mediates sequential signaling pathways ‘downstream’ of TLR and is required for sustained TNF production in macrophages. Nat Immunol 8: 601–609. [DOI] [PubMed] [Google Scholar]
- 2.Ma J, Bang BR, Lu J, Eun SY, Otsuka M, Croft M, Tobias P, Han J, Takeuchi O, Akira S, Karin M, Yagita H, and Kang YJ. 2013. The TNF family member 4-1BBL sustains inflammation by interacting with TLR signaling components during late-phase activation. Sci Signal 6: ra87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bang BR, Kim SJ, Yagita H, Croft M, and Kang YJ. 2015. Inhibition of 4-1BBL-regulated TLR response in macrophages ameliorates endotoxin-induced sepsis in mice. Eur J Immunol 45: 886–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.O’Neill LA 2016. A Metabolic Roadblock in Inflammatory Macrophages. Cell Rep 17: 625–626. [DOI] [PubMed] [Google Scholar]
- 5.O’Neill LA, Kishton RJ, and Rathmell J. 2016. A guide to immunometabolism for immunologists. Nat Rev Immunol 16: 553–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O’Neill LA, and Pearce EJ. 2016. Immunometabolism governs dendritic cell and macrophage function. J Exp Med 213: 15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Diskin C, and Palsson-McDermott EM. 2018. Metabolic Modulation in Macrophage Effector Function. Front Immunol 9: 270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garn H, Bahn S, Baune BT, Binder EB, Bisgaard H, Chatila TA, Chavakis T, Culmsee C, Dannlowski U, Gay S, Gern J, Haahtela T, Kircher T, Muller-Ladner U, Neurath MF, Preissner KT, Reinhardt C, Rook G, Russell S, Schmeck B, Stappenbeck T, Steinhoff U, van Os J, Weiss S, Zemlin M, and Renz H. 2016. Current concepts in chronic inflammatory diseases: Interactions between microbes, cellular metabolism, and inflammation. J Allergy Clin Immunol 138: 47–56. [DOI] [PubMed] [Google Scholar]
- 9.Posokhova EN, Khoshchenko OM, Chasovskikh MI, Pivovarova EN, and Dushkin MI. 2008. Lipid synthesis in macrophages during inflammation in vivo: effect of agonists of peroxisome proliferator activated receptors alpha and gamma and of retinoid X receptors. Biochemistry (Mosc) 73: 296–304. [DOI] [PubMed] [Google Scholar]
- 10.Feingold KR, Shigenaga JK, Kazemi MR, McDonald CM, Patzek SM, Cross AS, Moser A, and Grunfeld C. 2012. Mechanisms of triglyceride accumulation in activated macrophages. J Leukoc Biol 92: 829–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Khovidhunkit W, Kim MS, Memon RA, Shigenaga JK, Moser AH, Feingold KR, and Grunfeld C. 2004. Effects of infection and inflammation on lipid and lipoprotein metabolism: mechanisms and consequences to the host. J Lipid Res 45: 1169–1196. [DOI] [PubMed] [Google Scholar]
- 12.Lawrence T, and Natoli G. 2011. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol 11: 750–761. [DOI] [PubMed] [Google Scholar]
- 13.Mehta MM, Weinberg SE, and Chandel NS. 2017. Mitochondrial control of immunity: beyond ATP. Nat Rev Immunol 17: 608–620. [DOI] [PubMed] [Google Scholar]
- 14.Weinberg SE, Sena LA, and Chandel NS. 2015. Mitochondria in the regulation of innate and adaptive immunity. Immunity 42: 406–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kwon BS, Hurtado JC, Lee ZH, Kwack KB, Seo SK, Choi BK, Koller BH, Wolisi G, Broxmeyer HE, and Vinay DS. 2002. Immune responses in 4-1BB (CD137)-deficient mice. J Immunol 168: 5483–5490. [DOI] [PubMed] [Google Scholar]
- 16.van der Fits L, Mourits S, Voerman JS, Kant M, Boon L, Laman JD, Cornelissen F, Mus AM, Florencia E, Prens EP, and Lubberts E. 2009. Imiquimod-induced psoriasis-like skin inflammation in mice is mediated via the IL-23/IL-17 axis. J Immunol 182: 5836–5845. [DOI] [PubMed] [Google Scholar]
- 17.Van den Bossche J, Baardman J, and de Winther MP. 2015. Metabolic Characterization of Polarized M1 and M2 Bone Marrow-derived Macrophages Using Real-time Extracellular Flux Analysis. J Vis Exp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baliwag J, Barnes DH, and Johnston A. 2015. Cytokines in psoriasis. Cytokine 73: 342–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, Frezza C, Bernard NJ, Kelly B, Foley NH, Zheng L, Gardet A, Tong Z, Jany SS, Corr SC, Haneklaus M, Caffrey BE, Pierce K, Walmsley S, Beasley FC, Cummins E, Nizet V, Whyte M, Taylor CT, Lin H, Masters SL, Gottlieb E, Kelly VP, Clish C, Auron PE, Xavier RJ, and O’Neill LA. 2013. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature 496: 238–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Van den Bossche J, O’Neill LA, and Menon D. 2017. Macrophage Immunometabolism: Where Are We (Going)? Trends Immunol 38: 395–406. [DOI] [PubMed] [Google Scholar]
- 21.Zhang Z, Zi Z, Lee EE, Zhao J, Contreras DC, South AP, Abel ED, Chong BF, Vandergriff T, Hosler GA, Scherer PE, Mettlen M, Rathmell JC, DeBerardinis RJ, and Wang RC. 2018. Differential glucose requirement in skin homeostasis and injury identifies a therapeutic target for psoriasis. Nat Med 24: 617–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheung CT, Deisher TA, Luo H, Yanagawa B, Bonigut S, Samra A, Zhao H, Walker EK, and McManus BM. 2007. Neutralizing anti-4-1BBL treatment improves cardiac function in viral myocarditis. Lab Invest 87: 651–661. [DOI] [PubMed] [Google Scholar]
- 23.Seo SK, Park HY, Choi JH, Kim WY, Kim YH, Jung HW, Kwon B, Lee HW, and Kwon BS. 2003. Blocking 4-1BB/4-1BB ligand interactions prevents herpetic stromal keratitis. J Immunol 171: 576–583. [DOI] [PubMed] [Google Scholar]
- 24.Cai Y, Shen X, Ding C, Qi C, Li K, Li X, Jala VR, Zhang HG, Wang T, Zheng J, and Yan J. 2011. Pivotal role of dermal IL-17-producing gammadelta T cells in skin inflammation. Immunity 35: 596–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Buckner JH 2010. Mechanisms of impaired regulation by CD4(+)CD25(+)FOXP3(+) regulatory T cells in human autoimmune diseases. Nat Rev Immunol 10: 849–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van der Veeken J, Gonzalez AJ, Cho H, Arvey A, Hemmers S, Leslie CS, and Rudensky AY. 2016. Memory of Inflammation in Regulatory T Cells. Cell 166: 977–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Croft M 2009. The role of TNF superfamily members in T-cell function and diseases. Nat Rev Immunol 9: 271–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Croft M, Duan W, Choi H, Eun SY, Madireddi S, and Mehta A. 2012. TNF superfamily in inflammatory disease: translating basic insights. Trends Immunol 33: 144–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seo SK, Choi JH, Kim YH, Kang WJ, Park HY, Suh JH, Choi BK, Vinay DS, and Kwon BS. 2004. 4-1BB-mediated immunotherapy of rheumatoid arthritis. Nat Med 10: 1088–1094. [DOI] [PubMed] [Google Scholar]
- 30.Le NH, Kim CS, Tu TH, Choi HS, Kim BS, Kawada T, Goto T, Park T, Park JH, and Yu R. 2013. Blockade of 4-1BB and 4-1BBL interaction reduces obesity-induced skeletal muscle inflammation. Mediators Inflamm 2013: 865159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Croft M, Benedict CA, and Ware CF. 2013. Clinical targeting of the TNF and TNFR superfamilies. Nat Rev Drug Discov 12: 147–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sun Y, Lin X, Chen HM, Wu Q, Subudhi SK, Chen L, and Fu YX. 2002. Administration of agonistic anti-4-1BB monoclonal antibody leads to the amelioration of experimental autoimmune encephalomyelitis. J Immunol 168: 1457–1465. [DOI] [PubMed] [Google Scholar]
- 33.Kim YH, Choi BK, Shin SM, Kim CH, Oh HS, Park SH, Lee DG, Lee MJ, Kim KH, Vinay DS, and Kwon BS. 2011. 4-1BB triggering ameliorates experimental autoimmune encephalomyelitis by modulating the balance between Th17 and regulatory T cells. J Immunol 187: 1120–1128. [DOI] [PubMed] [Google Scholar]
- 34.Yoo JK, Choo YK, Kwak DH, Lee JM, Lim CY, Lee JH, Park MY, and Kim CH. 2016. Protective effects of agonistic anti-4-1BB antibody on the development of imiquimod-induced psoriasis-like dermatitis in mice. Immunol Lett 178: 131–139. [DOI] [PubMed] [Google Scholar]
- 35.Feldmann M 2002. Development of anti-TNF therapy for rheumatoid arthritis. Nat Rev Immunol 2: 364–371. [DOI] [PubMed] [Google Scholar]
- 36.Maini RN, Brennan FM, Williams R, Chu CQ, Cope AP, Gibbons D, Elliott M, and Feldmann M. 1993. TNF-alpha in rheumatoid arthritis and prospects of anti-TNF therapy. Clin Exp Rheumatol 11 Suppl 8: S173–175. [PubMed] [Google Scholar]
- 37.Schnitzler F, Fidder H, Ferrante M, Noman M, Arijs I, Van Assche G, Hoffman I, Van Steen K, Vermeire S, and Rutgeerts P. 2009. Long-term outcome of treatment with infliximab in 614 patients with Crohn’s disease: results from a single-centre cohort. Gut 58: 492–500. [DOI] [PubMed] [Google Scholar]
- 38.Teng MW, Bowman EP, McElwee JJ, Smyth MJ, Casanova JL, Cooper AM, and Cua DJ. 2015. IL-12 and IL-23 cytokines: from discovery to targeted therapies for immune-mediated inflammatory diseases. Nat Med 21: 719–729. [DOI] [PubMed] [Google Scholar]
- 39.Tuskey A, and Behm BW. 2014. Profile of ustekinumab and its potential in patients with moderate-to-severe Crohn’s disease. Clin Exp Gastroenterol 7: 173–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alessandri AL, Sousa LP, Lucas CD, Rossi AG, Pinho V, and Teixeira MM. 2013. Resolution of inflammation: mechanisms and opportunity for drug development. Pharmacol Ther 139: 189–212. [DOI] [PubMed] [Google Scholar]
- 41.Chen GY, and Nunez G. 2010. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol 10: 826–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vassalli P 1992. The pathophysiology of tumor necrosis factors. Annu Rev Immunol 10: 411–452. [DOI] [PubMed] [Google Scholar]
- 43.Nathan C, and Ding A. 2010. Nonresolving inflammation. Cell 140: 871–882. [DOI] [PubMed] [Google Scholar]
- 44.Freemerman AJ, Zhao L, Pingili AK, Teng B, Cozzo AJ, Fuller AM, Johnson AR, Milner JJ, Lim MF, Galanko JA, Beck MA, Bear JE, Rotty JD, Bezavada L, Smallwood HS, Puchowicz MA, Liu J, Locasale JW, Lee DP, Bennett BJ, Abel ED, Rathmell JC, and Makowski L. 2019. Myeloid Slc2a1-Deficient Murine Model Revealed Macrophage Activation and Metabolic Phenotype Are Fueled by GLUT1. J Immunol 202: 1265–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Uchiyama N, Yamamoto A, Kameda K, Yamaguchi H, and Ito M. 2000. The activity of fatty acid synthase of epidermal keratinocytes is regulated in the lower stratum spinousum and the stratum basale by local inflammation rather than by circulating hormones. J Dermatol Sci 24: 134–141. [DOI] [PubMed] [Google Scholar]
- 46.Berod L, Friedrich C, Nandan A, Freitag J, Hagemann S, Harmrolfs K, Sandouk A, Hesse C, Castro CN, Bahre H, Tschirner SK, Gorinski N, Gohmert M, Mayer CT, Huehn J, Ponimaskin E, Abraham WR, Muller R, Lochner M, and Sparwasser T. 2014. De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat Med 20: 1327–1333. [DOI] [PubMed] [Google Scholar]
- 47.Cluxton D, Petrasca A, Moran B, and Fletcher JM. 2019. Differential Regulation of Human Treg and Th17 Cells by Fatty Acid Synthesis and Glycolysis. Front Immunol 10: 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zaytseva YY, Rychahou PG, Gulhati P, Elliott VA, Mustain WC, O’Connor K, Morris AJ, Sunkara M, Weiss HL, Lee EY, and Evers BM. 2012. Inhibition of fatty acid synthase attenuates CD44-associated signaling and reduces metastasis in colorectal cancer. Cancer Res 72: 1504–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Murata S, Yanagisawa K, Fukunaga K, Oda T, Kobayashi A, Sasaki R, and Ohkohchi N. 2010. Fatty acid synthase inhibitor cerulenin suppresses liver metastasis of colon cancer in mice. Cancer Sci 101: 1861–1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Matsuura K, Canfield K, Feng W, and Kurokawa M. 2016. Metabolic Regulation of Apoptosis in Cancer. Int Rev Cell Mol Biol 327: 43–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cheng G, Palanisamy AP, Evans ZP, Sutter AG, Jin L, Singh I, May H, Schmidt MG, and Chavin KD. 2013. Cerulenin blockade of fatty acid synthase reverses hepatic steatosis in ob/ob mice. PLoS One 8: e75980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Svahn SL, Ulleryd MA, Grahnemo L, Stahlman M, Boren J, Nilsson S, Jansson JO, and Johansson ME. 2016. Dietary Omega-3 Fatty Acids Increase Survival and Decrease Bacterial Load in Mice Subjected to Staphylococcus aureus-Induced Sepsis. Infect Immun 84: 1205–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Moon JS, Lee S, Park MA, Siempos II, Haslip M, Lee PJ, Yun M, Kim CK, Howrylak J, Ryter SW, Nakahira K, and Choi AM. 2015. UCP2-induced fatty acid synthase promotes NLRP3 inflammasome activation during sepsis. J Clin Invest 125: 665–680. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 54.Lee HH, Sanada S, An SM, Ye BJ, Lee JH, Seo YK, Lee C, Lee-Kwon W, Kuper C, Neuhofer W, Choi SY, and Kwon HM. 2016. LPS-induced NFkappaB enhanceosome requires TonEBP/NFAT5 without DNA binding. Sci Rep 6: 24921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Loftus TM, Jaworsky DE, Frehywot GL, Townsend CA, Ronnett GV, Lane MD, and Kuhajda FP. 2000. Reduced food intake and body weight in mice treated with fatty acid synthase inhibitors. Science 288: 2379–2381. [DOI] [PubMed] [Google Scholar]
- 56.Turrado C, Puig T, Garcia-Carceles J, Artola M, Benhamu B, Ortega-Gutierrez S, Relat J, Oliveras G, Blancafort A, Haro D, Marrero PF, Colomer R, and Lopez-Rodriguez ML. 2012. New synthetic inhibitors of fatty acid synthase with anticancer activity. J Med Chem 55: 5013–5023. [DOI] [PubMed] [Google Scholar]
- 57.Bueno MJ, and Colomer R. 2015. A Fatty Acid Synthase Inhibitor Shows New Anticancer Mechanisms. EBioMedicine 2: 778–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Freemerman AJ, Johnson AR, Sacks GN, Milner JJ, Kirk EL, Troester MA, Macintyre AN, Goraksha-Hicks P, Rathmell JC, and Makowski L. 2014. Metabolic reprogramming of macrophages: glucose transporter 1 (GLUT1)-mediated glucose metabolism drives a proinflammatory phenotype. J Biol Chem 289: 7884–7896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, Deoliveira D, Anderson SM, Abel ED, Chen BJ, Hale LP, and Rathmell JC. 2014. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab 20: 61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Crombe V, Salleron J, Savoye G, Dupas JL, Vernier-Massouille G, Lerebours E, Cortot A, Merle V, Vasseur F, Turck D, Gower-Rousseau C, Lemann M, Colombel JF, and Duhamel A. 2011. Long-term outcome of treatment with infliximab in pediatric-onset Crohn’s disease: a population-based study. Inflamm Bowel Dis 17: 2144–2152. [DOI] [PubMed] [Google Scholar]
- 61.Cohen JD, Bournerias I, Buffard V, Paufler A, Chevalier X, Bagot M, and Claudepierre P. 2007. Psoriasis induced by tumor necrosis factor-alpha antagonist therapy: a case series. J Rheumatol 34: 380–385. [PubMed] [Google Scholar]
- 62.Collamer AN, and Battafarano DF. 2010. Psoriatic skin lesions induced by tumor necrosis factor antagonist therapy: clinical features and possible immunopathogenesis. Semin Arthritis Rheum 40: 233–240. [DOI] [PubMed] [Google Scholar]
- 63.Campa M, Mansouri B, Warren R, and Menter A. 2016. A Review of Biologic Therapies Targeting IL-23 and IL-17 for Use in Moderate-to-Severe Plaque Psoriasis. Dermatol Ther (Heidelb) 6: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fragoulis GE, Siebert S, and McInnes IB. 2016. Therapeutic Targeting of IL-17 and IL-23 Cytokines in Immune-Mediated Diseases. Annu Rev Med 67: 337–353. [DOI] [PubMed] [Google Scholar]
- 65.Wasilewska A, Winiarska M, Olszewska M, and Rudnicka L. 2016. Interleukin-17 inhibitors. A new era in treatment of psoriasis and other skin diseases. Postepy Dermatol Alergol 33: 247–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.