

Sir,
A 32-year-old woman presented with progressively increasing difficulty in walking and gait imbalance since early childhood. She was the third born child from a non-consanguineous parentage with normal antenatal and birth history. As a child, she had a mild delay in attaining motor milestones and walking was delayed until 2 years of age. Monosyllable and bisyllable speech onset were normal, her parents recognized that her speech was unclear by around 2 years of age. Around the age of 6 years, it was noticed that she lagged behind peers while running during play. She would sway to either side while walking. In addition, she used to drop objects occasionally from her hands and had tremors in hands while reaching out for objects. Over time, her handwriting became illegible and she completed her graduation with the help of a scribe. She had no cognitive decline, sensory disturbances, or bowel/bladder dysfunction. She had two elder sisters and the eldest sister had symptoms similar to her. There was no history of affected individuals in her parents/grandparent's generation.
On examination, she was thinly built, with normal vitals. No oculocutaneous telangiectasia, Kayser-Fleischer rings or pes cavus were observed on general examination. Her mini-mental status examination score (MMSE) was 28/28 (she could not attempt writing and drawing intersecting pentagons). She had a strained, strangled speech with undue separation of syllables, suggestive of a mixed spastic-ataxic speech. She had a visual acuity of 6/12 of both eyes, and fundus examination was normal. She had slow saccades and broken pursuit movements, with gaze-evoked nystagmus. Examination of other cranial nerves was normal. She had spasticity involving both upper and lower limbs. She had MRC grade 4/5 power in all muscle groups in bilateral upper and lower limbs. Deep tendon reflexes were brisk bilaterally with bilateral extensor plantar. Cerebellar signs were notable, in the form of bilateral finger-to-nose and heel–knee incoordination, dysdiadokokinesis, past pointing, and intention tremor. She had a spastic ataxic gait. Sensory system examination revealed a loss of vibration and position sense in bilateral feet up to medial malleolus. Investigation revealed a normal hemogram, normal lipid profile, normal liver function tests, normal alpha-fetoprotein, and peripheral blood smear did not reveal any acanthocytes. An electrophysiology study showed severe sensorimotor axonal neuropathy involving the upper and lower limbs. The differentials considered for the clinical presentation included autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS), Friedrich's ataxia, spinocerebellar ataxia, Refsum's disease, abetalipoproteinemia, and ataxia with vitamin E deficiency. Echocardiography was normal. Magnetic resonance imaging (MRI) of the brain showed linear or “striped” pontine T2 hypointensities, enlarged pons, thinning of the rostral corpus callosum, superior vermian atrophy, and T2 hyperintense rim around the thalami (”bithalamic stripes”) [Figures 1 and 2]. In view of the progressive spasticity and cerebellar signs, with positive family history and with the typical imaging modalities, the probable diagnosis of an ARSACS was considered. Genomic DNA from the proband was used to perform exome sequencing and a pathogenic homozygous variant - c8793 del A in the SACS gene which was identified, confirming the diagnosis of ARSACS. Genetic testing for Friedrich's ataxia was negative. Her elder sister who had similar symptoms opted out of the evaluation including neuroimaging and genetic analysis.
Figure 1.
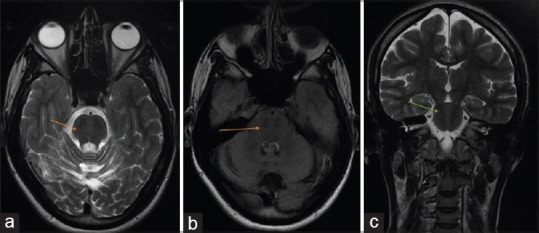
(a and b) Axial T2 and fluid-attenuated inversion recovery images show an “enlarged” pons with linear hypointense striations-”striped pons” on either side of the midline (arrows). (c) Coronal T2 magnetic resonance images showing typical, linear, “striped” pontine hypointensities (arrows)
Figure 2.
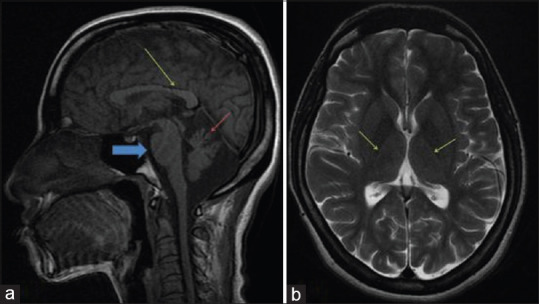
Sagittal T1 MR images showing: (a) superior vermian atrophy (red arrow), “enlarged pons” (blue arrow), and thinning of the splenium of corpus callosum (yellow arrow). (b) Axial T2-weighted image shows a rim of hyperintensity around the thalami – “bithalamic stripes” (arrows)
ARSACS was described by Bouchard and colleagues in 1978 from Quebec, Canada, as a rare cause of autosomal recessive cerebellar ataxia.[1] Subsequent reports confirmed the presence of this condition in other parts of the world including Europe and in Asia, Japan. Agarwal et al. described the first genetically proven case of ARSACS from India.[2] The first case report from Kerala was by Menon et al.[3] Agarwal et al. had identified a SACS gene duplication whereas Kuchay et al. had identified a frameshift mutation and Menon et al. a homozygous deletion in the SACS gene.[2,3,4] Whole-exome sequencing in our patient revealed a deletion - c8793 del A-in the SACS gene, which was a homozygous pathogenic variant (supplementary data).
Most ARSACS patients show a typical triad of early-onset cerebellar ataxia, lower limb spasticity, and peripheral neuropathy; however, varying phenotypes have been reported. Our patient had the classical triad described, similar to the other cases reported from India [Table 1]. Common retinal abnormalities in ARSACS include myelinated nerve fibers embedding retinal vessels and thickening of the retinal nerve fiber layer; however, this was not observed in our patient. Reports from Japan and Italy have shown that that mental retardation and retinal striations were variable features.[5]
Table 1.
Comparison of studies on ARSACS from India
| State | Sheetal et al | Agarwal et al.[2] | Kuchay et al.[4] | Menon et al.[3] |
|---|---|---|---|---|
| Kerala | Maharashtra | Jammu and Kashmir | Kerala | |
| Sex | F | M | M | M |
| Pyramidal signs | + | - | + | + |
| Cerebellar signs | + | + | + | + |
| Peripheral neuropathy | + | + | + | + |
| Intellectual disability | - | - | + | - |
| Myelinated nerve fibres on fundus | - | + | + | - |
| Retinal nerve fiber layer thickening | - | + | + | - |
| MRI brain | ||||
| 1. Striped pons | + | + | + | + |
| 2. Bithalamic stripes | + | - | + | - |
| 3. Superior vermian atrophy | + | + | + | + |
| 4.Thinning of the rostral corpus callosum | + | - | + | - |
| Genetic study (SACS gene mutation) | + | + | + | + |
Table showing comparison of studies on ARSACS from India
Characteristic imaging findings in previously described ARSACS cases include cerebellar atrophy preferentially affecting the superior vermis, linear, or “striped” pontine T2 hypointensities, thinning of the rostral corpus callosum, T2 hyperintense rim around the thalami (”bithalamic stripes”), all of which were observed in our patient[6] [Table 1]. The presence of these characteristic MRI findings in a case of cerebellar ataxia should alert the treating physician to a diagnosis of ARSACS.
An extensive PubMed search revealed only three reported cases of genetically proven ARSACS from India, of which one is from Kerala.[2,3,4] We hereby report a case of SACS gene mutation- c8793 del A- identified with whole-exome sequencing with typical clinical and neuroimaging findings of ARSACS and positive family history; the mutation being first of its kind reported from Kerala.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Bouchard JP, Barbeau A, Bouchard R, Bouchard RW. Electromyography and nerve conduction studies in Friedreich's ataxia and autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS) Can J Neurol Sci. 1979;6:185–9. doi: 10.1017/s0317167100119614. [DOI] [PubMed] [Google Scholar]
- 2.Agarwal PA, Ate-Upasani P, Ramprasad VL. Autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS)- first report of clinical and imaging features from India, and a novel SACS gene duplication. Mov Disord Clin Pract. 2017;4:775–7. doi: 10.1002/mdc3.12520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Menon MS, Shaji CV, Kabeer KA, Parvathy G. SACS gene-related autosomal recessive spastic ataxia of Charlevoix-Saguenay from South India. Arch Med Health Sci. 2016;4:122–4. [Google Scholar]
- 4.Kuchay RAH, Mir YR, Zeng X, Hassan A, Musarrat J, Parwez I, et al. ARSACS as a worldwide disease: Novel SACS mutations identified in a consanguineous family from the remote tribal Jammu and Kashmir region in India. Cerebellum. 2019;18:807–12. doi: 10.1007/s12311-019-01028-2. [DOI] [PubMed] [Google Scholar]
- 5.Ogawa T, Takiyama Y, Sakoe K, Mori K, Namekawa M, Shimazaki H, et al. Identification of a SACS gene missense mutation in ARSACS. Neurology. 2004;62:107–9. doi: 10.1212/01.wnl.0000099371.14478.73. [DOI] [PubMed] [Google Scholar]
- 6.Martin MH, Bouchard JP, Sylvain M, St-Onge O, Truchon S. Autosomal recessive spastic ataxia of Charlevoix-Saguenay: A report of MR imaging in 5 patients. Am J Neuroradiol. 2007;28:1606–8. doi: 10.3174/ajnr.A0603. [DOI] [PMC free article] [PubMed] [Google Scholar]