

Abstract
Guanidinoacetate methyltransferase (GAMT) deficiency is the second most common defect in the creatine metabolism pathway resulting in cerebral creatine deficiency syndrome (CCDS). We report three patients from two unrelated families, diagnosed with GAMT deficiency on next-generation sequencing. All the probands had happy predisposition as a predominant manifestation in addition to the reported features of global developmental delay, seizures, and microcephaly. This further expands the phenotype of CCDS. The workup for creatine deficiency disorder should be included in the diagnostic algorithm for children with nonsyndromic intellectual disability, especially in those with a happy demeanor. These cases exemplify the utility of magnetic resonance spectroscopy of the brain in the workup of nonsyndromic intellectual disability to diagnose a potentially treatable disorder. In addition, documentation of low serum creatinine may be supportive. Early diagnosis and treatment is essential for better prognosis.
Keywords: Creatine deficiency, guanidinoacetate methyltransferase, happy demeanor, intellectual disability
INTRODUCTION
Nonsyndromic intellectual disability is a diagnostic challenge. However, with an underlying etiology of a potentially treatable disorder such as guanidinoacetate methyltransferase (GAMT) deficiency, it is essential to make an early diagnosis for better outcome. GAMT is a key enzyme in the creatine metabolism pathway. GAMT deficiency results in features of cerebral creatine deficiency syndrome (CCDS). The prevalence of CCDS in patients with mild-to-severe intellectual disability is 2.7%.[1] The estimated incidence of GAMT deficiency in the general population ranges from 1:2,640,000 to 1:550,000.[2] We report three patients with nonsyndromic intellectual disability from two unrelated families with GAMT deficiency. These cases exemplify the role of careful clinical profiling, especially soft signs such as happy demeanor and easily available investigations such as serum creatinine and magnetic resonance spectroscopy (MRS) of the brain to diagnose CCDS. Next-generation sequencing (NGS) confirmed the genetic etiology in all three patients.
CASE REPORTS
Family 1: Patient A
A 3.6-year-old boy presented with a history of developmental delay and one episode of seizure at 1 year of age. He was the only child of nonconsanguineous parents, born full term by cesarean section, and the birth weight was 3.3 kg. The antenatal, perinatal, and neonatal periods were uneventful. He was exclusively breastfed for the first 6 months. The child was noted to have delay in achieving milestones at 6 months of age. He had global developmental delay when he presented at 1 year of age. Best-attained milestones at 1 year of age were sitting with support, mature palmar grasp, and babbling. He was unable to recognize parents and had no social smile. He was able to track objects and respond to sound.
The parents noticed that the child remained happy most of the times and rarely cried. The proband had one episode of fever triggered generalized seizure at one year of age. There was no history of neuroregression, irritability, altered sensorium, recurrent illness, feeding problems, or characteristic odor of urine. There was no history of looseness or tightness of the body.
On examination at 1 year, the head circumference was 2–3 standard deviation (SD) below the mean (44 cm), weight at −2 SD (8.5 kg), and length between mean and −2 SD (73 cm) as per the WHO growth charts. On examination, the child had happy demeanor with poor eye contact. He had spontaneous limb movements. He would fall within seconds when made to sit with support. He had a flat occiput with brachycephaly. There was no dysmorphism. Cranial nerves' examination was normal. There were truncal hypotonia and variable appendicular tone. The deep tendon reflexes were normal in both the upper and lower limbs. There was bilateral extensor plantar response. Rest of the systemic examination was unremarkable.
In view of global developmental delay, happy demeanor, and microcephaly, the differential diagnosis of a chromosomal microdeletion or microduplication syndromes, Angelman or Angelman-like syndromes including Mowat–Wilson syndrome, Pitt–Hopkins syndrome, Kleefstra syndrome, and adenylosuccinate lyase deficiency was considered though he lacked the characteristic syndromic features.
The investigations that were already done included a normal karyotype, electroencephalogram (EEG), auditory brain stem-evoked response, magnetic resonance imaging (MRI) of the brain, ultrasound abdomen, and echocardiography. The blood lactate, ammonia, liver function tests, creatinine phosphokinase, and urine organic acid analysis by gas chromatography-mass spectrometry (GC-MS) were normal. The chromosomal microarray analysis (CMA) and the genomic DNA testing of the child for Angelman syndrome at 15q11 region by methylation-specific multiplex ligation probe assay were normal.
On follow-up at 2 years of age, he had marked behavioral problems with frequent bursts of laughter and happy affect. There were two episodes of myoclonic seizures, and he was put on levetiracetam. There were head nodding and truncal ataxia. Further on, clinical exome sequencing test for single-gene disorders by NGS of the genomic DNA was carried out. A homozygous variant was identified in the exon 6 of GAMT gene (ENST00000252288), c.595dupG (p. Glu199GlyfsTer68) that resulted in a frameshift and premature truncation of the protein, 68 amino acids downstream to codon 199. The variant was classified as “likely pathogenic,” confirming the diagnosis of autosomal recessive CCDS type 2. This variant is novel, not reported in both the 1000 genome and ExAC databases. The in silico prediction of the variant was damaging by MutationTaster-2, PolyPhen-2, and SIFT.[3] The reference region is conserved across the species. The segregation studies of this GAMT variant by Sanger sequencing confirmed the heterozygous carrier status in parents and homozygous state in the child. The blood creatinine level done twice was low (0.15 and 0.08 mg/dl). The MRS of the brain showed an absent creatine peak at 3 ppm at TE of 135 ms [Figure 1]. The test for urine or blood guanidinoacetate was not available.
Figure 1.
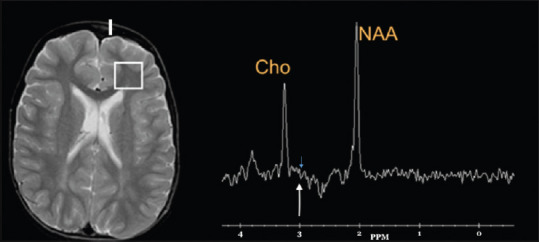
Magnetic resonance spectroscopy brain of patient A showing absent creatine peak at 3 ppm at TE of 135 ms
The child was started on treatment at 2 years of age, with oral creatine monohydrate 400 mg/kg/day and L-ornithine 400 mg/kg/day in four divided doses. On follow-up after 3 months, he was able to sit without support and stand with support but continued to have happy effect. He had complaints of polyuria. The renal function tests, glomerular filtration rate, plasma amino acids, ammonia, protein, and albumin levels were normal. The repeat MRS of the brain at 3 years of age showed a decreased creatine peak, evidence of recent supplementation of the same. At 3.6 years (1 year of treatment), he is now able to walk, run, obey commands but has not developed speech and continues to have behavioral problems. The polyuria symptom has resolved.
Family 2: Patients B and C
Patients B and C were 10-year and 8-year-old sisters born to nonconsanguineous parents [Figure 2]. The birth history was uneventful. Both had a similar phenotype; global developmental delay first was noted at 8–10 months of age when they had not achieved neck holding and social smile. At present, they could walk unsupported stooping forward with poor balance. They had no speech, poor expressive language, and cognition. They also had hyperactivity, sleep disturbances, frequent bursts of laughter, and happy predisposition. The younger sibling had myoclonic seizures twice. Both had normal vision and hearing. The weight and height of both siblings lie between −2 and −3 SD as per the WHO growth chart. The head circumferences of the girls were 50 cm and 49 cm, respectively. On examination of both siblings, there was no dysmorphism. The cranial nerve assessment, tone, and deep tendon reflexes were normal. There were no abnormal cerebellar signs except for truncal ataxia. The MRI brain, EEG, and CMA were normal. The blood tandem mass spectrometry and urine GC-MS were normal. The methylation-specific PCR study for Angelman syndrome was normal. Patient B underwent NGS-based clinical exome sequencing. A pathogenic homozygous missense variant in exon 5 of GAMT gene, c.506G>A, p.C169Y (ENST00000447102) was identified. The same homozygous variant was identified by Sanger sequencing in patient C, and their parents were confirmed to be carriers. This is a previously reported variant in a patient with GAMT deficiency and not present in 1000 genome and ExAC databases.[4] The variant is damaging by in silico prediction tools. The region is conserved across species. Reverse phenotyping with MRS brain confirmed an absent creatine peak in both siblings. The blood creatinine in patient B was 0.18 mg/dl (0.015 mmol/L) and patient C was 0.2 mg/dl (0.017 mmol/L), which were low (normal 0.55–1.3mg/dl); the urine creatinine was 2.02 mg/dl and 2.18 mg/dl in patients B and C, respectively. Both siblings were started on daily oral creatine monohydrate 400 mg/kg/day in four divided doses and L-ornithine 400 mg/kg/day in three divided doses. On follow-up after 6 months of treatment, there was a mild improvement in gait, sleep disturbance, and hyperactivity.
Figure 2.

Happy affect phenotype in patients B and C
DISCUSSION
Creatine deficiency syndromes (CDSs) comprise the arginine, glycine amidinotransferase (AGAT), and GAMT deficiencies, which are autosomal recessive disorders and the X-linked creatine transporter defect. The AGAT is involved in transfer of amidino group from arginine to glycine to yield guanidinoacetate. The guanidinoacetate is methylated by GAMT to yield creatine along with conversion of the methyl donor S-adenosylmethionine to S-adenosylhomocysteine.[5]
The creatine is utilized in the muscle and brain where it is taken up by the creatine transporter and contributes to majority of adenosine triphosphate production. Creatine is essential for proper brain function, energy storage, and neuromodulation. Creatine and creatine phosphate are nonenzymatically converted into creatinine excreted in urine, with daily turnover of 1.5% of body creatine (20–25 mg/kg/24 h).[6] The absence of creatine peak on proton MRS is a diagnostic hallmark. Urinary guanidinoacetate and the creatine-to-creatinine ratio is an important screening test for all CDSs. Increased levels of guanidinoacetate in body fluids are pathognomonic for GAMT deficiency.[7]
The GAMT gene (NM_138924.2) maps to chromosome 19p13.3 coding 236 amino acids and is found in the cytosol of the liver, pancreas, and kidney cells. At least 50 different pathogenic GAMT variants have been reported, and the most common are c.327G>A (p.K109K) and c.59G>C (p.W20S).[8]
The estimated incidence of GAMT deficiency is 1:250,000 newborns.[9] GAMT deficiency is the second most common CDS disorder, with approximately 110 patients reported worldwide.[8]
The prevalence of CDS in patients with neurological disease of unknown origin is 0.25%, and the prevalence of GAMT deficiency is 0.09%.[10] The prevalence of CDS among the children with autism is less than 7 per 1000.[11]
In 2014, Stockler-Ipsiroglu et al. studied 48 patients with GAMT deficiency. About 92% had global developmental delay, 73% patients had epilepsy, and 66% of whom had well-controlled occasional seizures, similar to patient C. The movement disorders reported in 27.5% comprising dystonia, chorea, hemiballism, ataxia, and spasticity. Behavioral problems reported in them included hyperactivity, aggressiveness, or autistic features. The patients in the current study had happy demeanor, sleep disturbances, and hyperactivity.
In the study by Stockler-Ipsiroglu et al., the treatment with creatine monohydrate resulted in improvement in 21% of individuals with developmental delay and improvement of movement disorder in 60% of patients. The seizures were eliminated in 18% and decreased in frequency of seizures in 49% patients.[8] All three patients in the current study benefitted from treatment. The variant, c.506G>A, has previously been reported in three patients, whose comparative features have been listed in Table 1.[4,8,12]
Table 1.
Clinical characteristics of patients with c.506G>A pathogenic variant in guanidinoacetate methyltransferase gene
| Clinical features | Patient B | Patient C | Caldeira et al., 2005 | Stockler et al., 2014 | Mahmutoglu et al., 2014 |
|---|---|---|---|---|---|
| Age/origin | 10 years (Indian) | 8 years (Indian) | 19 years (Portugal) | 4 years | 8 years |
| Age onset/diagnosis (years) | 1/10 | 1/8 | 2/19 | -/4 | 1/4 |
| Sex | Female | Female | Female | Male | Male |
| Intellectual disability | Severe | Severe | Severe | Mild | Mild |
| Movement disorder | Truncal ataxia | Truncal ataxia | Present | No | Extrapyramidal and pyramidal |
| Epilepsy | No | Myoclonic seizures twice | Head drop seizures on sodium valproate | No | No |
| Behavioral problems | Happy demeanor, hyperactivity | Happy demeanor, hyperactivity | Autism, disruptive aggressive | Autism, disruptive aggressive | Attention deficit hyperactivity |
| Tone (limbs, trunk) | Normal | Normal | Generalized hypertonia and rigidity | NA | Normal |
| Plasma creatinine (mmol/l) | 0.015 | 0.017 | 0.034 | NA | NA |
| Normal range | 0.048-0.114 | 0.048-0.114 | 0.035-0.122 | ||
| Plasma uric acid/creatinine ratio (control <0.65) | 2.7 | 2.25 | 0.88 | NA | NA |
| GAA level (mmol/l) (Controls 1.33–3.33) | Not done | Not done | 27 | NA | Elevated |
| Urine creatinine (mmol/l) | 0.17 (2.02 mg/dl) | 0.19 (2.18 mg/dl) | 3.15 (normal - 2.48-22.9) | NA | NA |
| GAMT enzyme assay in fibroblast | Not done | Not done | Deficient level | NA | Absent activity |
| MRI brain | Normal | Normal | Normal | Normal | Mild cortical atrophy |
| MRS brain | Absent creatine peak | Absent creatine peak | Not done | NA | Absent creatine peak |
| Treatment | |||||
| Oral creatine monohydrate (mg/kg/day) | 400 | 400 | No | 300-800 | NA |
| L-ornithine (mg/kg/day) | 400 | 400 | 200-800 | ||
| Treatment duration (months) | 6 | 6 | - | 45 | - |
| Follow-up and outcome | Stabilization | Stabilization | - | Speech delay | - |
GAMT=Guanidinoacetate methyltransferase, NA=Not availabel, GAA=Guanidinoacetic acid, MRI=Magnetic resonance imaging, MRS=Magnetic resonance spectroscopy
Stockler-Ipsiroglu et al. found that 47% (18/38) had T2 signal hyperintensities of the globus pallidus in a brain MRI/MRS done at the time of diagnosis with reversal in 89% (16/18) on treatment. The brain creatine levels were considerably higher compared to baseline levels in 79% (30/38) who had follow-up MRS upon treatment.[8] Neither the levels of creatine peak on MRS nor the guanidinoacetate acetate in blood predicts severity of the disease phenotype, as even absent levels have been found in patients with mild, moderate, and severe phenotype at diagnosis.[12]
Annual assessment of glomerular filtration rate for assessment of kidney function while on creatine supplementation therapy is essential.[13]
GAMT deficiency is a candidate disorder for newborn screening because elevated guanidinoacetate can be picked up on dried blood spot with no false-positive results using liquid GC-MS.[14] The targeted early intervention of metabolic pathophysiology provides for the opportunity to significantly impact the outcome and prognosis of an otherwise severe intellectual disability.[15]
In conclusion, there is no established genotype–phenotype correlation in GAMT gene. The understanding of the phenotypic spectrum has expanded with addition of “happy demeanor” feature from these three cases. A higher index of suspicion is required to diagnose this potentially treatable condition. Every child with nonsyndromic intellectual disability, especially with a happy disposition and a low serum creatinine, should be evaluated for CDS. MRS is virtually diagnostic of CDS. The study of larger cohorts of CDS in the Indian subcontinent is essential to lay a groundwork for establishment of newborn screening for a potentially treatable disorder with intellectual disability.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Lion-François L, Cheillan D, Pitelet G, Acquaviva-Bourdain C, Bussy G, Cotton F, et al. High frequency of creatine deficiency syndromes in patients with unexplained mental retardation. Neurology. 2006;67:1713–4. doi: 10.1212/01.wnl.0000239153.39710.81. [DOI] [PubMed] [Google Scholar]
- 2.Desroches CL, Patel J, Wang P, Minassian B, Marshall CR, Salomons GS, et al. Carrier frequency of guanidinoacetate methyltransferase deficiency in the general population by functional characterization of missense variants in the GAMT gene. Mol Genet Genomics. 2015;290:2163–71. doi: 10.1007/s00438-015-1067-x. [DOI] [PubMed] [Google Scholar]
- 3.Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat Methods. 2014;11:361–2. doi: 10.1038/nmeth.2890. [DOI] [PubMed] [Google Scholar]
- 4.Caldeira Araújo H, Smit W, Verhoeven NM, Salomons GS, Silva S, Vasconcelos R, et al. Guanidinoacetate methyltransferase deficiency identified in adults and a child with mental retardation. Am J Med Genet A. 2005;133A:122–7. doi: 10.1002/ajmg.a.30226. [DOI] [PubMed] [Google Scholar]
- 5.Sharer JD, Bodamer O, Longo N, Tortorelli S, Wamelink MM, Young S, et al. Laboratory diagnosis of creatine deficiency syndromes: A technical standard and guideline of the American College of Medical Genetics and Genomics. Genet Med. 2017;19:256–63. doi: 10.1038/gim.2016.203. [DOI] [PubMed] [Google Scholar]
- 6.Stockler-Ipsiroglu S, Mercimek-Mahmutoglu S, Salomons G. Creatine deficiency syndromes. In: Saudubray JM, van den Berghe G, Walter JH, editors. Inborn Metabolic Diseases. Diagnosis and Treatment. 5th ed. Springer-Verlag; 2012. pp. 239–47. [Google Scholar]
- 7.Longo N, Ardon O, Vanzo R, Schwartz E, Pasquali M. Disorders of creatine transport and metabolism. Am J Med Genet C Semin Med Genet. 2011;157C:72–8. doi: 10.1002/ajmg.c.30292. [DOI] [PubMed] [Google Scholar]
- 8.Stockler-Ipsiroglu S, van Karnebeek C, Longo N, Korenke GC, Mercimek-Mahmutoglu S, Marquart I, et al. Guanidinoacetate methyltransferase (GAMT) deficiency: Outcomes in 48 individuals and recommendations for diagnosis, treatment and monitoring. Mol Genet Metab. 2014;111:16–25. doi: 10.1016/j.ymgme.2013.10.018. [DOI] [PubMed] [Google Scholar]
- 9.Mercimek-Mahmutoglu S, Pop A, Kanhai W, Fernandez Ojeda M, Holwerda U, Smith D, et al. Apilot study to estimate incidence of guanidinoacetate methyltransferase deficiency in newborns by direct sequencing of the GAMT gene. Gene. 2016;575:127–31. doi: 10.1016/j.gene.2015.08.045. [DOI] [PubMed] [Google Scholar]
- 10.Cheillan D, Joncquel-Chevalier Curt M, Briand G, Salomons GS, Mention-Mulliez K, Dobbelaere D, et al. Screening for primary creatine deficiencies in French patients with unexplained neurological symptoms. Orphanet J Rare Dis. 2012;7:96. doi: 10.1186/1750-1172-7-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schulze A, Bauman M, Tsai AC, Reynolds A, Roberts W, Anagnostou E, et al. Prevalence of creatine deficiency syndromes in children with nonsyndromic autism. Pediatrics. 2016;137(1):e20152672. doi: 10.1542/peds.2015-2672. [DOI] [PubMed] [Google Scholar]
- 12.Mercimek-Mahmutoglu S, Ndika J, Kanhai W, de Villemeur TB, Cheillan D, Christensen E, et al. Thirteen new patients with guanidinoacetate methyltransferase deficiency and functional characterization of nineteen novel missense variants in the GAMT gene. Hum Mutat. 2014;35:462–9. doi: 10.1002/humu.22511. [DOI] [PubMed] [Google Scholar]
- 13.Barisic N, Bernert G, Ipsiroglu O, Stromberger C, Müller T, Gruber S, et al. Effects of oral creatine supplementation in a patient with MELAS phenotype and associated nephropathy. Neuropediatrics. 2002;33:157–61. doi: 10.1055/s-2002-33679. [DOI] [PubMed] [Google Scholar]
- 14.Pasquali M, Schwarz E, Jensen M, Yuzyuk T, DeBiase I, Randall H, et al. Feasibility of newborn screening for guanidinoacetate methyltransferase (GAMT) deficiency. J Inherit Metab Dis. 2014;37:231–6. doi: 10.1007/s10545-013-9662-7. [DOI] [PubMed] [Google Scholar]
- 15.Stockler-Ipsiroglu S, van Karnebeek CD. Cerebral creatine deficiencies: A group of treatable intellectual developmental disorders. Semin Neurol. 2014;34:350–6. doi: 10.1055/s-0034-1386772. [DOI] [PubMed] [Google Scholar]