

Sir,
Autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS) is an autosomal recessive neurodegenerative disorder characterized by a triad of early onset cerebellar ataxia, spasticity, and sensorimotor polyneuropathy (distal muscle wasting with finger and foot deformities).[1] Dysarthria, nystagmus, and retinal hypermyelination may sometimes be observed.[1] It was first described by Bouchard et al. in1978 as an uncommon cause of autosomal recessive cerebellar ataxia.[2,3] Although initially thought to be prevalent only in Canada, it has now been increasingly reported from rest of the world.[4] Exome sequencing studies identified multiple causative mutations in the sacsin molecular chaperone gene (SACS) on chromosome 13q.[5] However, new pathogenic mutations continue to be identified. We report a patient with typical clinical and imaging features of ARSACS, where targeted SACS gene testing lead to the discovery of a novel pathogenic SACS mutation.
A 28-years-old male, born of non-consanguineous marriage, presented with progressive gait ataxia and recurrent falls from 3 years of age. He also developed dysarthria and lower limb stiffness at 10 years of age, followed by thinning of feet. Family history was negative. On examination, he had bilateral pes cavus with hammer toes. Neurological examination revealed lower limb spasticity, brisk reflexes, and extensor plantar response. Sensory and fundus examination were normal. He also exhibited dysmetria, dysdiachokinesia, nystagmus, and cerebellar gait. He had no Kayser-Fleischer ring, telengiectasias, or tendon xanthomas. Systemic examination was normal. Diagnosis of an autosomal recessive ataxia syndrome was made and patient evaluated along those lines.
MRI brain revealed superior cerebellar vermian atrophy and T2 linear “striped” hypointensities in pons [Figure 1a and b]. Nerve conduction studies (NCS) demonstrated prolonged distal latencies with decreased velocities in motor nerves of all limbs while sensory NCS was not recordable. ARSACS and Refsum's disease are the ataxia syndromes associated with predominant demyelinating neuropathy. Genetic testing for Friedreich's ataxia and Spinocerebellar ataxia was negative. 2D-Echocardiography, serum albumin, lipid profile, and alpha fetoprotein were normal. Clinical exome (targeted SACS gene) sequencing revealed a heterozygous pathogenic non-sense mutation on exon 10 of SACS gene (C,4232T > G) resulting in a stop codon and premature truncation of the protein. Another mutation of uncertain significance was found on the same exon (C,8132C > T).
Figure 1.
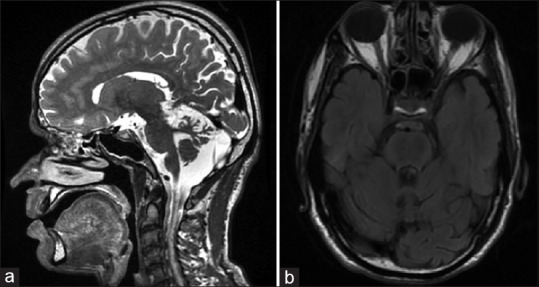
MRI brain T2 weighted image revealing superior cerebellar vermian atrophy (a) and linear “striped” hypointensities in pons (b)
ARSACS is an early onset, cerebellar ataxia first described from Quebec, Canada in 1978. Canadian patients formed a majority of these cases initially, but an increasing number have been described from other countries as well, possibly pointing toward an earlier underdiagnosis.[6] Many of these cases were initially diagnosed as spastic diplegia type of cerebral palsy.[2] Approximately 30 mutations in the SACS gene have been reported and newer ones are being continuously discovered.[7]
It usually presents in early childhood (12 months to 5.5 years of age)[3] and diagnosis is based on typical clinical manifestations (cerebellar ataxia with spasticity and peripheral neuropathy) and radiological findings (superior vermian atrophy and pontine linear T2/FLAIR hypo-intensities on T2/FLAIR). Confirmation of diagnosis is done by demonstration of pathogenic mutations in the sacsin gene located on chromosome 13q.[5]
The cardinal clinical features of ARSACS are gait instability with frequent falls, both of which were present in our patient. The disease progression is slow with patients becoming wheelchair bound around the third to fourth decade of life.[3] The initial features are a combination of early onset cerebellar signs like ataxia, dysarthria, and nystagmus, with pyramidal signs in the lower limbs like spasticity, hyperreflexia and extensor plantar response. Lower limb sensorimotor peripheral neuropathy tends to develop later in the disease course causing distal atrophy, pes cavus, and hammer toes.[8] These findings were present in our patient.
Fundoscopy may reveal hypermyelinated nerve fibres radiating from the optic disc in some patients[8] and cognition tends to be preserved. Both fundoscopy and cognition was normal in our patient.
Imaging findings described on MRI brain include early and progressive superior cerebellar vermian atrophy with the inferior vermis remaining preserved throughout.[9] However, global cerebral atrophy may occur later in the disease course. Linear T2 and FLAIR hypo-intensities have also been described.[1] Cerebral white matter remains preserved throughout. All these findings were present in our patient.
The causal mutations in the SACS gene vary between individuals. The founder mutations discovered from Quebec were a single-base deletion at position 6594 (6594delT) and a nonsense mutation 5254C > T.[10] Approximately 95% Canadian patients harbor these mutations.[5] Several novel mutations have been described with some variations in the disease phenotype. The absence of abnormal myelinated retinal fibres in our patient is probably explained by the same.
ARSACS must be suspected in early onset cerebellar ataxia combined with spasticity and peripheral neuropathy. Typical radiological features such as superior cerebellar vermian atrophy and linear pontine hypo-intensities greatly enhance the diagnosis. Ultimately, the diagnosis is confirmed by genetic studies. We describe such a case from India, and also demonstrate the occurrence of a novel pathogenic nonsense mutation in the SACS gene.
Ethical compliance statement
The study complied with all Ethical standards and did not require the consent of the Institutional Ethics Committee.
Written informed patient consent was taken.
All authors have read and complied with the journal's ethical publication guidelines.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
APPENDIX 1:
| Name | Location | Contribution | Role |
|---|---|---|---|
| Ayush Agarwal | Department of Neurology, Dr RML Institute of Medical Sciences, Lucknow | Design and conceptualized study; analyzed the data; drafted the manuscript for intellectual content | Author |
| Divyani Garg | Department of Neurology, Lady Hardinge Medical College, New Delhi | Design and conceptualized study; analyzed the data; drafted the manuscript for intellectual content | Author |
| Akash Kharat | Department of Neurology, Dr RML Institute of Medical Sciences, Lucknow | Design and conceptualized study; analyzed the data; drafted the manuscript for intellectual content | Author |
| Abdul Qavi | Department of Neurology, Dr RML Institute of Medical Sciences, Lucknow | Design and conceptualized study; analyzed the data; drafted the manuscript for intellectual content | Author |
REFERENCES
- 1.Martin MH, Bouchard JP, Sylvain M, St-Onge O, Truchon S. Autosomal recessive spastic ataxia of Charlevoix-Saguenay: A report of MR imaging in 5 patients. AJNR Am J Neuroradiol. 2007;28:1606–8. doi: 10.3174/ajnr.A0603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bouchard JP, Barbeau A, Bouchard R, Bouchard RW. Electromyography and nerve conduction studies in Friedreich's ataxia and Autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS) Can J Neurol Sci. 1979;6:185–9. doi: 10.1017/s0317167100119614. [DOI] [PubMed] [Google Scholar]
- 3.Duquette A, Brais B, Bouchard JP, Mathieu J. Clinical presentation and early evolution of spastic ataxia of Charlevoix-Saguenay. Mov Disord. 2013;28:2011–4. doi: 10.1002/mds.25604. [DOI] [PubMed] [Google Scholar]
- 4.Engert JC, Bérubé P, Mercier J, Doré C, Lepage P, Ge B, et al. ARSACS, a spastic ataxia common in northeastern Quebec, is caused by mutation in a new gene encoding an 11.5-Kb ORF. Nat Genet. 2000;24:120–5. doi: 10.1038/72769. [DOI] [PubMed] [Google Scholar]
- 5.Ogawa T, Takiyama Y, Sakoe K, Mori K, Namekawa M, Shimazaki H, et al. Identification of a SACS gene missense mutation in ARSACS. Neurology. 2004;62:107–9. doi: 10.1212/01.wnl.0000099371.14478.73. [DOI] [PubMed] [Google Scholar]
- 6.Gomez CM. ARSACS goes global. Neurology. 2004;62:10–11. doi: 10.1212/wnl.62.1.10. [DOI] [PubMed] [Google Scholar]
- 7.Takiyama Y. Autosomal recessive spastic ataxia of Charlevoix-Saguenay. Neuropathology. 2006;26:368–75. doi: 10.1111/j.1440-1789.2006.00664.x. [DOI] [PubMed] [Google Scholar]
- 8.Embirucıu EK, Martyn ML, Schlesinger D, Kok F. Autosomal recessive ataxias: 20 types and counting. Arq Neuropsiquiatr. 2009;67:1143–56. doi: 10.1590/s0004-282x2009000600036. [DOI] [PubMed] [Google Scholar]
- 9.El Euch-Fayache G, Lalani I, Amouri R, Turki I, Ouahchi K, Hung WY, et al. Phenotypic features and genetic findings in sacsin-related autosomal recessive ataxia in Tunisia. Arch Neurol. 2003;60:982–8. doi: 10.1001/archneur.60.7.982. [DOI] [PubMed] [Google Scholar]
- 10.Mercier J, Prevost C, Engert JC, Bouchard JP, Mathieu J, Richter A. Rapid detection of the sacsin mutations causing autosomal recessive spastic ataxia of Charlevoix-Saguenay. Genet Test. 2001;5:255–9. doi: 10.1089/10906570152742326. [DOI] [PubMed] [Google Scholar]