

SUMMARY
Incomplete understanding of how hepatosteatosis transitions to fibrotic non-alcoholic steatohepatitis (NASH) has limited therapeutic options. Two molecules that are elevated in hepatocytes in human NASH liver are cholesterol, whose mechanistic link to NASH remains incompletely understood, and TAZ, a transcriptional regulator that promotes fibrosis but whose mechanism of increase in NASH is unknown. We now show that increased hepatocyte cholesterol upregulates TAZ and promotes fibrotic NASH. ASTER-B/C-mediated internalization of plasma membrane cholesterol activates soluble adenylyl cyclase (sAC; ADCY10), triggering a calcium-RhoA-mediated pathway that suppresses β-TrCP/proteasome-mediated TAZ degradation. In mice fed a cholesterol-rich NASH-inducing diet, hepatocyte-specific silencing of ASTER-B/C, sAC, or RhoA decreased TAZ and ameliorated fibrotic NASH. The cholesterol-TAZ pathway is present in primary human hepatocytes, and associations among liver cholesterol, TAZ, and RhoA in human NASH liver are consistent with the pathway. Thus, hepatocyte cholesterol contributes to fibrotic NASH by increasing TAZ, suggesting new targets for therapeutic intervention.
Graphical Abstract
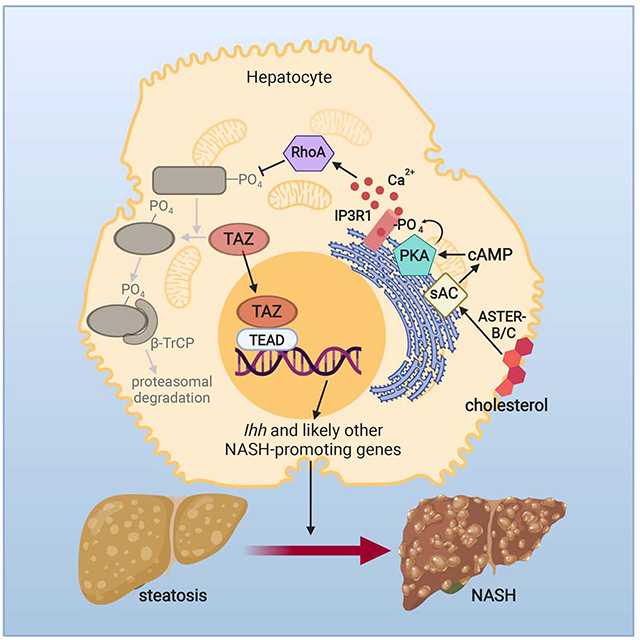
In Brief (eTOC)
Cholesterol is consistently elevated in human NASH but its mechanistic link to NASH progression remains incompletely understood. Wang et al. now define a cholesterol-TAZ connection in which increased hepatocyte cholesterol upregulates TAZ through an adenylyl cyclase-calcium-RhoA pathway and promotes fibrotic NASH.
INTRODUCTION
Nonalcoholic steatohepatitis (NASH) is emerging as the leading cause of liver disease, but incomplete understanding of pathophysiology has led to a paucity of treatment options. Fibrosis caused by activation of hepatic stellate cells (HSCs) correlates best with clinical outcome in NASH (Angulo et al., 2015a; Angulo et al., 2015b; Dulai et al., 2017; Puche et al., 2013; Vilar-Gomez et al., 2018). The transcriptional regulator TAZ (WWTR1) is markedly increased in hepatocytes in human and mouse liver during steatosis-to-NASH progression (Khajehahmadi et al., 2019; Mooring et al., 2019; Wang et al., 2016). Increased TAZ results in increased hepatocyte Indian hedgehog (Ihh) transcription and secretion, leading to HSC activation, liver fibrosis, inflammation, and cell death (Wang et al., 2016). Importantly, hepatocyte-specific silencing of TAZ in multiple mouse models at the steatosis stage blocks NASH, and TAZ silencing can also partially reverse NASH, including fibrosis (Wang et al., 2019; Wang et al., 2016) a key test of potential therapeutic potential.
Emerging from these studies is the critical question as to how TAZ is increased in hepatocytes during the progression of NASH. Previous studies have shown that TAZ and a related factor called YAP are regulated primarily by phosphorylation by so-called Hippo kinases, which act in sequence to activate the TAZ/YAP kinase LATS (Koo and Guan, 2018; Zheng and Pan, 2019). When the Hippo kinases are active, phosphorylated TAZ and/or YAP are inactive due to their sequestration in the cytoplasm by 14-3-3 or their degradation by proteasomes or autophagy (Huang et al., 2012; Lee et al., 2018). Proteasomal degradation involves recognition of certain “phosphodegrons” on TAZ/YAP by specific E3 ligases, such as β-TrCP (Liu et al., 2010; Zhao et al., 2010). Certain cues, such as a rigid extracellular matrix and RhoA activation, lead to deactivation of Hippo kinases, accumulation of non-phosphorylated TAZ/YAP in the nucleus, and induction of genes that have cognate binding sites in their promoters for the TEAD family of proteins, which are the TAZ/YAP-interacting proteins that mediate changes in gene transcription (Chang et al., 2018; Ohgushi et al., 2015; Zhao et al., 2008).
Another consistent observation in humans is the association of elevated liver cholesterol content with NASH fibrosis (Caballero et al., 2009; Ioannou, 2016; Min et al., 2012; Puri et al., 2007), but it remains unclear whether and how cholesterol might promote NASH. Hypothesized pro-NASH roles of cholesterol include perturbation of hepatocyte functions by changes in cellular membrane fluidity; triggering of inflammatory pathways in liver macrophages and hepatocytes; and activation of HSCs (Ioannou, 2016).
We now address and integrate these two critical questions in NASH by showing that cholesterol increases hepatocyte TAZ in NASH and through this pathway promotes NASH fibrosis. We demonstrate that cholesterol acts through a soluble adenylyl cyclase (sAC)–protein kinase A (PKA)–inositol triphosphate receptor (IP3R)–calcium–RhoA pathway to prevent TAZ proteasomal degradation. These findings provide new mechanistic insight into the role of hepatocyte cholesterol in NASH; elucidate how a key fibrosis inducer, TAZ, is increased in NASH; and suggest a number of new targets for therapeutic intervention against NASH fibrosis.
RESULTS
TAZ Protein Correlates with Liver Cholesterol and Fibrosis in NASH
Cholesterol content is increased in the livers of humans with NASH compared with normal liver (Caballero et al., 2009; Puri et al., 2007), and unesterified (“free”) cholesterol is correlated with disease severity (Caballero et al., 2009; Ioannou, 2016; Puri et al., 2007). In addition, high-cholesterol diets promote NASH in various mouse models (McGettigan et al., 2019; Savard et al., 2013; Van Rooyen et al., 2011; Wouters et al., 2008). To test for a possible correlation between liver cholesterol and TAZ in a mouse model of non-alcoholic fatty liver disease (NAFLD), we compared mice fed a diet rich in palmitate and fructose (NAFLD diet) that contained 0.2%, 0.5%, or 1.25% cholesterol. We have previously shown that 16 weeks of 1.25% cholesterol diet-feeding increases hepatocyte TAZ and causes multiple features of NASH, including fibrosis, in a TAZ-dependent manner (Wang et al., 2016; Zhu et al., 2018). Consistent with relatively poor absorption of dietary cholesterol in this strain of mice (Schwarz et al., 2001), only the 1.25%-cholesterol diet significantly increased liver free cholesterol content, mimicking a hallmark in human NASH liver (Puri et al., 2007) (Figure 1A), and only the 1.25% diet was able to increase TAZ (Figure 1B). Further, only the livers from the 1.25% cholesterol-fed mice had increases in fibrosis; inflammatory cells; Tgfb1 and Acta2 mRNAs, which are markers of HSC activation; and Mcp1 mRNA, which is a marker of inflammation (Figures 1C and S1A). These mice also had elevated ALT in the plasma, which indicates liver injury (Figure S1B). To test the cholesterol-TAZ association in humans, we analyzed liver biopsy specimens from humans with NASH and variable liver cholesterol values and found a strong positive correlation between free cholesterol content and TAZ protein (Figures 1D and S1C). Thus, there is an association among cholesterol content, TAZ protein, and fibrosis in the livers of mice with NAFLD, and the association between liver cholesterol and TAZ protein extends to humans with NASH.
Figure 1. TAZ Protein Correlates with Liver Cholesterol and Fibrosis, and Cholesterol Increases TAZ in Hepatocytes.
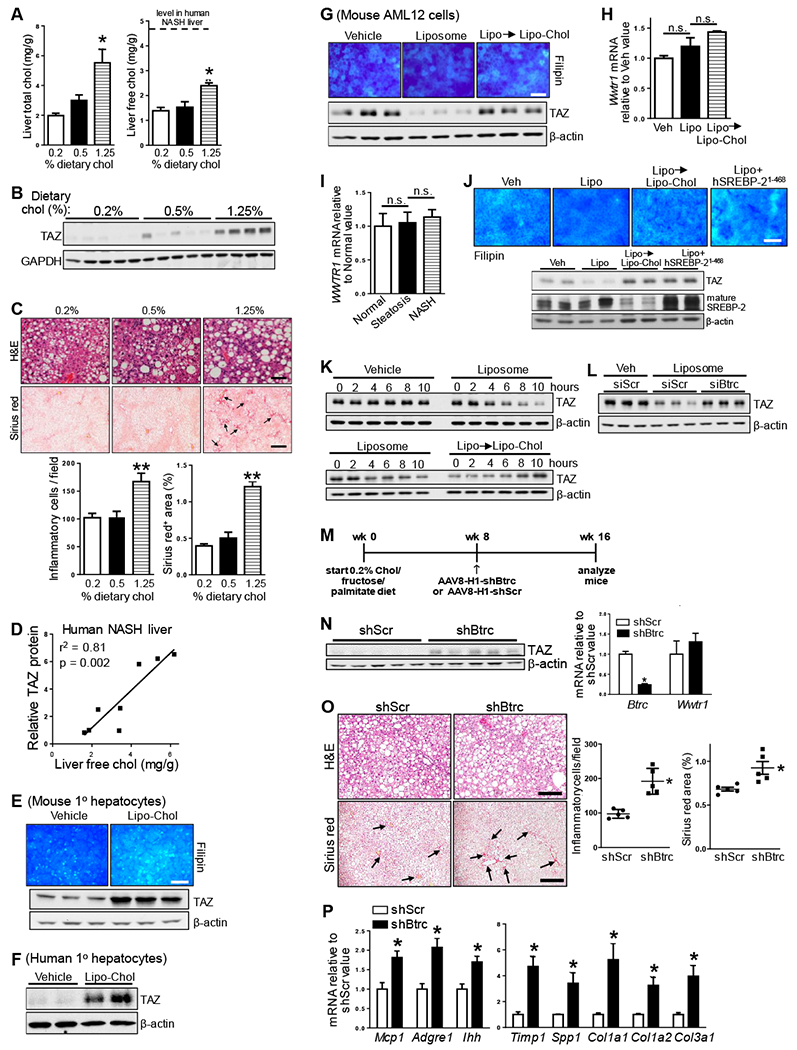
(A-C) The following parameters were measured in mice fed the NAFLD diet containing 0.2%, 0.5%, and 1.25% cholesterol (chol) for 16 weeks (n = 5 mice/group; means ± SEM; *p < 0.05, **p < 0.01, ***p < 0.001):
(A) Liver total cholesterol and free cholesterol. The dotted line indicates the free cholesterol content in liver of NASH subjects (Puri et al., 2007).
(B) Liver TAZ and GAPDH immunoblot.
(C) Liver sections stained for H&E (scale bar, 100 μm), quantified for inflammatory cells, and stained and quantified for Sirius red (arrows indicate areas of fibrosis; scale bar, 500 μm).
(D) Correlation between TAZ protein level and free cholesterol concentration in human NASH livers (n = 8 subjects).
(E) Filipin staining and TAZ immunoblot of primary mouse hepatocytes incubated for 24 h with vehicle or cholesterol-rich liposomes (Lipo-Chol). Scale bar, 100 μm.
(F) TAZ immunoblot of primary human hepatocytes incubated with 24 h with vehicle or Lipo-Chol.
(G) Filipin staining and TAZ immunoblot of AML12 cells incubated for 24 h with vehicle, or for 16 h with liposomes and then 8 h with Lipo-Chol (Lipo → Lipo-Chol). Scale bar, 100 μm.
(H) Wwtr1 mRNA of the cells in panel C (n = 4 biological replicates; means ± SEM; n.s., non-significant).
(I) WWTR1 mRNA in normal human livers (n = 7) or human livers showing features of steatosis (n =12) or NASH (n = 16; means ± SEM; n.s., non-significant).
(J) Filipin staining and TAZ and mature SREBP-2 immunoblot of AML12 cells incubated for 24 h with vehicle (Veh) or liposomes (Lipo); 16 h with liposomes followed by 8 h with Lipo-Chol); or 24 h with liposomes after transfection with human SREBP-2(1-468). Scale bar, 100 μm.
(K) TAZ immunoblot of AML12 cells treated as follows: upper blot: vehicle or liposomes for 0-10 h; lower blot: liposomes for 0-10 h, or for 16 h followed by 0-10 h with Lipo-Chol.
(L) TAZ immunoblot of AML12 cells transfected with siScr or siBtrc and then incubated for 24 h with vehicle (Veh) or liposomes.
(M-N) The following parameters were measured in mice fed the 0.2% cholesterol NAFLD diet for 16 weeks, with AAV8-H1-shBtrc or AAV8-H1-scrambled RNA (shScr) injected at the 8-week time point (n = 5 mice/group; means ± SEM; *P < 0.05):
(M) Experimental design.
(N) TAZ immunoblot; and Btrc and Wwtr1 mRNA.
(O) Liver sections stained for H&E, quantified for inflammatory cells, and stained and quantified for Sirius red (arrows indicate areas of fibrosis). Scale bar, 200 μm.
(P) Liver Mcp1, Adgre1 (F4/80), Ihh, Timp1, Spp1, Col1a1, Col1a2, and Col3a1 mRNAs.
Cholesterol Loading of Hepatocytes Increases TAZ, While Cholesterol Depletion of Hepatocytes Lowers TAZ via Proteasomal Degradation
To determine if there was a cell-autonomous, causal link between cholesterol and TAZ in hepatocytes, we incubated primary mouse and human hepatocytes with cholesterol-enriched phospholipid liposomes (Lipo-Chol) to increase their cholesterol content and found that this treatment caused a marked increase in TAZ protein (Figures 1E and 1F). We also examined AML12 cells, which are non-transformed mouse hepatocytes that have a relatively high basal content of both cholesterol and TAZ (Figure 1G, left). Incubation with phospholipid liposomes, which induces cholesterol efflux from cells, lowered filipin staining and TAZ, which could be restored by then incubating these liposome-treated cells with cholesterol-enriched liposomes (Lipo-Chol) (Figure 1G, middle and right). The mRNA encoding TAZ (Wwtr1) was unaffected by these treatments, and there were no differences in WWTR1 mRNA expression among human livers with normal histology, steatosis, and NASH (Figures 1H and 1I). Boosting endogenous cholesterol synthesis by transfecting cholesterol-depleted AML12 cells with a constitutively active form of SREBP-2 (Horton et al., 1998) also increased TAZ protein (Figure 1J). The data also show that Lipo-Chol, as expected (Brown and Goldstein, 1997), suppressed the level of the active, mature form of SREBP-2. Thus, either exogenous or endogenous cholesterol increases TAZ protein but not its mRNA in hepatocytes.
We next tested the possibility that cholesterol increased TAZ by enhancing its stability against proteasomal degradation by assaying TAZ at various times after incubation with liposomes vs. vehicle control ± the proteasome inhibitor MG132. There was a marked decrease in TAZ by 6 h of liposome incubation, and TAZ stability was restored upon cholesterol repletion (Figure 1K). The decrease in TAZ seen with cholesterol depletion was inhibited by MG132 (Figure S1D). Similar results were found when cycloheximide was used to block new protein synthesis (Figures S1E and S1F).
We next considered a role for the E3 ligase β-TrCP, which was implicated previously in TAZ proteasomal degradation in several cancer cell lines in vitro (Huang et al., 2012; Liu et al., 2010). We found that silencing β-TrCP partially blocked the decrease in TAZ in liposome-treated cells (Figures 1L, S1G, and S1H); β-TrCP level itself was not altered by cholesterol in vitro or in vivo (Figures S1I and S1J). To test the role of β-TrCP in vivo, mice fed the 0.2% diet, which does not induce NASH by itself, were injected with hepatocyte-specific AAV8-H1-shBtrc (Ghorpade et al., 2018; Wang et al., 2016; Zheng et al., 2019) (Figure 1M). This treatment did not affect body weight, percent liver:body weight, or fasting blood glucose (FBG) (Figures S1K–S1M). As hypothesized, the livers of these mice demonstrated higher TAZ protein despite unchanged Wwtr1 mRNA (Figure 1N); more fibrosis; higher expression of mRNAs encoding inflammatory cytokines and proteins associated with HSC activation, including the TAZ gene target Ihh; more inflammatory cells; and increased TUNEL+ cells; and higher plasma ALT compared with control mice (Figures 1O–1P, S1N, and S1O). Although β-TrCP can affect canonical Wnt signaling in other settings (Marikawa and Elinson, 1998), hepatocyte β-TrCP silencing did not affect liver phospho-Ser31-Ser37-Thr41-β-catenin, a measure of canonical Wnt activation (Figure S1P, with validation of the β-catenin endpoint in AML12 cells in Figure S1Q). These combined data suggest that cholesterol increases TAZ protein and TAZ-stimulated NASH by decreasing β-TrCP-mediated TAZ proteasomal degradation.
The Decrease in TAZ in Cholesterol-Depleted Hepatocytes Requires LATS1/2-Mediated TAZ Phosphorylation
LATS-mediated TAZ phosphorylation can promote its proteasomal degradation (Huang et al., 2012; Liu et al., 2010). In this context, we found that LATS2 kinase activity on recombinant TAZ was lower in the high-TAZ livers of mice fed the 1.25% vs. 0.2% or 0.5% cholesterol diets (Figure 2A). Further, phospho-TAZ was increased by cholesterol depletion of AML12 cells and decreased by cholesterol repletion of these cells and primary human hepatocytes (Figures 2B, 2C, and S2A). Importantly, the decrease in TAZ in cholesterol-depleted AML12 cells was abrogated by LATS2 silencing (Figures 2D, upper blot, and S2B). As predicted based on the coordinated regulation of TAZ and YAP (Koo and Guan, 2018; Zheng and Pan, 2019), cholesterol depletion also lowered YAP in a LATS2-dependent manner (Figure 2D, middle blot). LATS1 activity, as assessed by its phosphorylation, was also decreased in the livers of 1.25% cholesterol-fed mice, and silencing of LATS1 prevented TAZ decrease in cholesterol-depleted AML12 cells (Figures S2C–S2E). Next, we transfected AML12 cells with various hemagglutinin (HA)-tagged Ser→Ala TAZ mutants at LATS-mediated phosphodegron sites (Huang et al., 2012; Lei et al., 2008). Liposome-mediated lowering of TAZ was uniquely prevented in S117A-TAZ-transfected cells (Figures 2E and S2F–S2I), and post-cycloheximide decay of S117A-TAZ after liposome treatment was markedly slower than that of wild-type TAZ (Figure S2J). Moreover, we observed phospho-S117 in immunoprecipitated TAZ from MG132-treated, cholesterol-depleted HepG2 cells, analyzed by LC-MS/MS (Figure S2K). To investigate these findings in vivo, Wwtr1fl/fl mice fed the 0.2%-cholesterol diet for 8 weeks were injected with AAV8-TBG-Cre to deplete endogenous hepatocyte TAZ and with AAV8-TBG-HA-wild-type (WT) human TAZ or AAV8-TBG-HA-S117A human TAZ (Figure 2F). At the time of sacrifice 8 weeks later, the two groups of mice had similar body weights, percent liver:body weights, and fasting blood glucose (Figures S3A–S3C). As hypothesized, there was higher liver expression of HA-S117A versus HA-WT TAZ protein despite similar expression of human WWTR1 mRNA and similar knockdown efficiency of mouse Wwtr1 (Figures 2G and S3D). Most importantly, S117A-TAZ mice demonstrated high levels of fibrosis; inflammatory cells; and mRNAs encoding proteins associated with activated HSCs, including Ihh compared with WT-TAZ mice, concomitant with higher plasma ALT and more TUNEL+ cells in the liver (Figures 2H–J and S3E). These combined data suggest that hepatocyte cholesterol depletion promotes LATS-mediated TAZ-S117 phosphorylation and consequent TAZ proteasomal degradation and, conversely, that hepatocyte cholesterol enrichment stabilizes TAZ by preventing its phosphorylation by LATS and thereby promotes NASH.
Figure 2. Cholesterol Depletion of Hepatocytes Decreases TAZ in a LATS2-Dependent Manner.
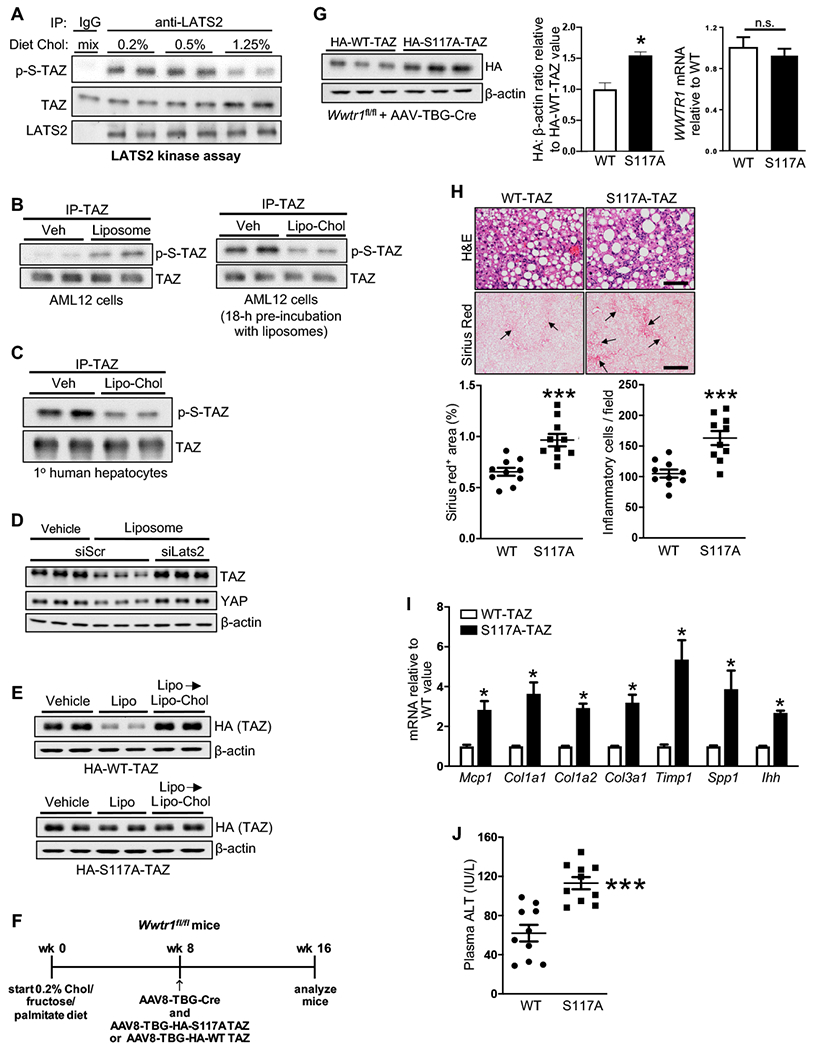
(A) LATS2 kinase assay, using rTAZ as substrate, of extracts of livers of mice fed the NAFLD diet containing 0.2%, 0.5%, or 1.25% cholesterol for 16 weeks.
(B) Immunoblots of phospho- and total TAZ in TAZ immunoprecipitates from AML12 cells incubated as follows: left blot, 4 h with MG132 plus vehicle (Veh) or liposomes; right blot, 18 h with liposomes and then 4 h with MG132 plus vehicle or Lipo-Chol.
(C) Immunoblots of phospho- and total TAZ in TAZ immunoprecipitates from primary human hepatocytes incubated for 8 h with vehicle or Lipo-Chol, with MG132 included during the last 4 h.
(D) TAZ and YAP immunoblots of siScr-treated or siLats2-treated AML12 cells that were incubated for 24 h with vehicle or liposomes.
(E) HA-TAZ immunoblot of HA-WT-human TAZ- or HA-S117A-human TAZ-transfected AML12 cells that were incubated for 24 h with vehicle, or liposomes for 16 h and then Lipo-Chol for 8 h.
(F-J) The following parameters were measured in Wwtr1fl/fl mice fed the NAFLD diet containing 0.2% cholesterol for 16 weeks, with AAV8-TBG-Cre plus either AAV8-TBG-HA-WT-hTAZ or AAV8-TBG-HA-S117A-hTAZ injected at the 8-week time point (n = 10 mice/group; means ± SEM; *p < 0.05, ***p < 0.001):
(F) Design of the experiment.
(G) Liver HA-TAZ immunoblot, with quantification, and WWTR1 mRNA.
(H) Liver sections stained for H&E, quantified for inflammatory cells, and stained and quantified for Sirius red (arrows indicate areas of fibrosis). Scale bars, 100 μm for H&E and 500 μm for Sirius red.
(I) Liver Mcp1, Col1a1, Col1a2, Col3a1, Timp1, Spp1, and Ihh mRNAs.
(J) Plasma ALT.
Cholesterol Activates RhoA Activity to Decrease LATS2 Activity and Increase TAZ
As RhoA activation can inhibit Hippo kinases (Fujimoto et al., 2015; Wada et al., 2011; Zhao et al., 2012), we hypothesized that cholesterol might stabilize TAZ by activating RhoA. Using two assays of RhoA activity, we found that cholesterol depletion of AML12 cells lowered RhoA activity, which was then restored by incubation with cholesterol-rich liposomes (Figure 3A). Cholesterol enrichment of primary human hepatocytes similarly activated RhoA (Figure 3B). Moreover, the restoration of RhoA activity by cholesterol was blocked by the C3 Rho inhibitor or by silencing RhoA with siRhoa (Figures 3C, S3F, and S3G). As expected, RhoA inhibition or silencing also lowered YAP. As a link to LATS2, we showed that siRhoa restored LATS2 kinase activity in cholesterol-repleted AML12 cells (Figure 3D).
Figure 3. Liver Cholesterol Correlates with RhoA Activity, and RhoA Silencing Decreases Liver TAZ, Inflammation, and Fibrosis in NASH Mice.
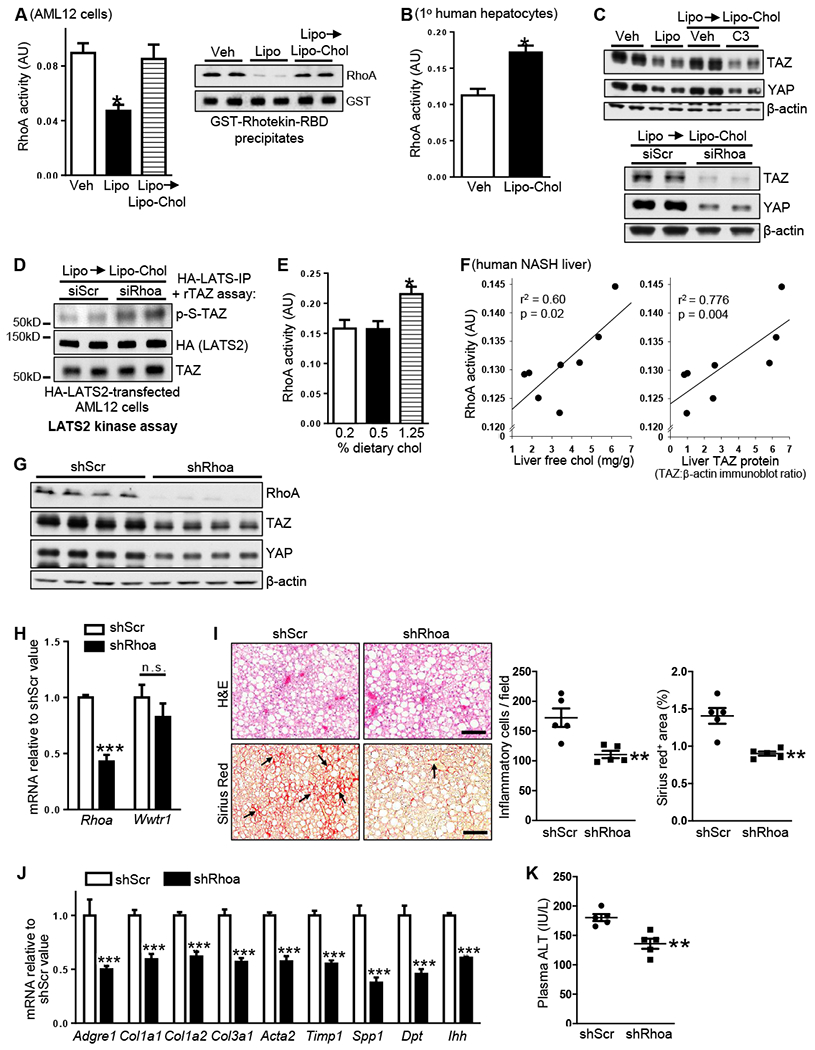
(A) RhoA activity (left graph, G-LISA® assay; right graph, GTP-RhoA precipitation using GSTRhotekin-RBD) of AML12 cells incubated for 2 h with vehicle or liposomes, or for 24 h with liposomes and then 2 h with Lipo-Chol. For the G-LISA® assay, n = 4 biological replicates; values shown are means ± SEM; *p < 0.05.
(B) RhoA activity of primary human hepatocytes incubated for 2 h with vehicle or Lipo-Chol (n = 3 biological replicates; means ± SEM; *p < 0.05).
(C) Top, TAZ and YAP immunoblots of AML12 cells incubated for 24 h with vehicle or liposomes or 16 h with liposomes and then 8 h with Lipo-Chol ± 1 μg/ml C3 transferase Rho inhibitor (C3) included during the last 4 h. Bottom, TAZ and YAP immunoblots of siScr- or siRhoa-transfected AML12 cells incubated for 16 h with liposomes and then 8 h with Lipo-Chol.
(D) LATS2 kinase assay, using rTAZ as substrate, of extracts of siScr- or siRhoa-transfected, HA-LATS-expressing AML12 cells that were incubated for 16 h with liposomes and then 2 h with Lipo-Chol.
(E) RhoA activity in liver extracts from mice fed 16 weeks with the NAFLD diet containing 0.2%, 0.5%, and 1.25% cholesterol (n = 5 mice/group; means ± SEM; *p < 0.05).
(F) Correlations of liver free cholesterol and liver TAZ with RhoA activity in liver extracts from humans subjects presented in Figure 1F (n = 8 subjects).
(G-K) The following parameters were assayed in mice fed the NASH-inducing 1.25%-cholesterol diet for 16 weeks, with AAV8-H1-shRhoa or AAV8-H1-shScr administered at the 8-week time point (n = 5 mice/group; means ± SEM; **p < 0.01, ***p < 0.001):
(G) Liver RhoA, TAZ, and YAP immunoblots.
(H) Liver Rhoa and Wwtr1 mRNAs.
(I) Liver sections stained for H&E, quantified for inflammatory cells, and stained and quantified for Sirius red (arrows indicate areas of fibrosis). Scale bar, 200 μm.
(J) Liver Adgre1 (F4/80), Col1a1, Col1a2, Col3a1, Acta2, Timp1, Spp1, Dpt, and Ihh mRNAs.
(K) Plasma ALT.
We next assayed RhoA activity in the livers from the mice fed the three diets described in Figure 1. As hypothesized, RhoA activity was higher in the livers of mice fed the NASH-inducing 1.25%-cholesterol diet than in mice fed the non-NASH-inducing 0.5%- and 0.2%- cholesterol diets (Figure 3E), and RhoA activity was strongly correlated with liver free cholesterol content and TAZ protein in the livers of humans with various degrees of NAFLD (Figure 3F and Supplemental Table 1). To test causation AAV8-H1-shRhoa, was injected into mice fed the 1.25% cholesterol diet for 8 weeks. Analysis after 8 more weeks on the diet revealed that RhoA silencing led to a decrease in liver TAZ protein, but not Wwtr1 mRNA (Figures 3G and 3H). As was observed in vitro, YAP was also lower, although the relative degree of lowering in this in vivo setting was less than that of TAZ. As predicted, the livers of shRhoa-treated mice had increased LATS1/2 activity but no change β-TrCP (Figures S3H and S3I). Despite similar body weights, percent liver:body weights, and fasting blood glucose between the two groups of mice (Figures S3J–S3L), hepatocyte RhoA silencing resulted in decreases in the liver of fibrosis, inflammatory cells, Ihh mRNA, and mRNAs encoding proteins associated with activated HSCs, and they also had lower plasma ALT and more TUNEL+ cells in the liver (Figures 3I–K and S3M). In theory, the modest decrease in YAP in these mice could have contributed to the improvement in NASH. However, in mice treated with AAV8-H1-shTaz, which markedly suppresses NASH progression (Wang et al., 2016), liver YAP was not affected (Figure S3N), suggesting that TAZ can promote NASH independently of YAP. In summary, RhoA mediates the effect of cholesterol on inactivating LATS2 and increasing TAZ in hepatocytes and thereby promotes steatosis-to-NASH progression.
Cholesterol-mediated stabilization of TAZ could be caused by cholesterol itself or a metabolite. In AML12 cells treated with an inhibitor of acyl-coenzyme A:cholesterol acyltransferase (ACAT), which converts cholesterol to cholesterol fatty acid esters, basal TAZ increased, indicating the importance of free cholesterol rather than cholesterol fatty acid esters (Figure S4A). Further, neither cholest-5-en-3β-ol (epi-cholesterol) nor enantiomeric cholesterol (ent-cholesterol) (Westover et al., 2003) was able to activate RhoA (Figure S4B). Next, we increased the expression of the mRNAs encoding two cholesterol-metabolizing enzymes—sterol 27-hydroxylase (CYP27A1) and the cholesterol 25-hydroxylase (CH25H)—by treating cholesterol-loaded AML12 cells with the LXR agonist T0901317 (Gilardi et al., 2009; Liu et al., 2018) (Figure S4C). LXR agonism lowered TAZ protein in cholesterol-loaded TAZ in these cells, and TAZ expression was restored by silencing either CYP27A1 or CH25H (Figure S4D), likely reflecting changes in cellular cholesterol content under these conditions. These data indicate that TAZ pathway is relatively specific for cholesterol.
Cholesterol Activates RhoA via a Cyclic AMP-Dependent Protein Kinase (PKA)-IP3R-Calcium Pathway
Based on findings in other cell types (Masiero et al., 1999; Zhou et al., 1991), we considered the hypothesis that cholesterol might activate RhoA by increasing cytoplasmic calcium. We transfected AML12 cells with the genetically encoded calcium indicator GCaMP6f engineered to localize to the cytoplasm or ER (Wang et al., 2017) and found that cholesterol depletion led to a decrease in cytoplasmic calcium and an increase in ER calcium (Figure 4A), consistent with cholesterol promoting ER calcium release into the cytoplasm. Moreover, the calcium ionophore ionomycin increased RhoA activity and TAZ in cholesterol-depleted AML12 cells with (Figures 4B and 4C), while the cytoplasmic calcium sequestering agent BAPTA-AM lowered RhoA activity and TAZ in cholesterol-repleted AML12 cells (Figures 4D and 4E).
Figure 4. Cholesterol Increases TAZ in Hepatocytes through an IP3R-Calcium-Protein Kinase A Pathway.
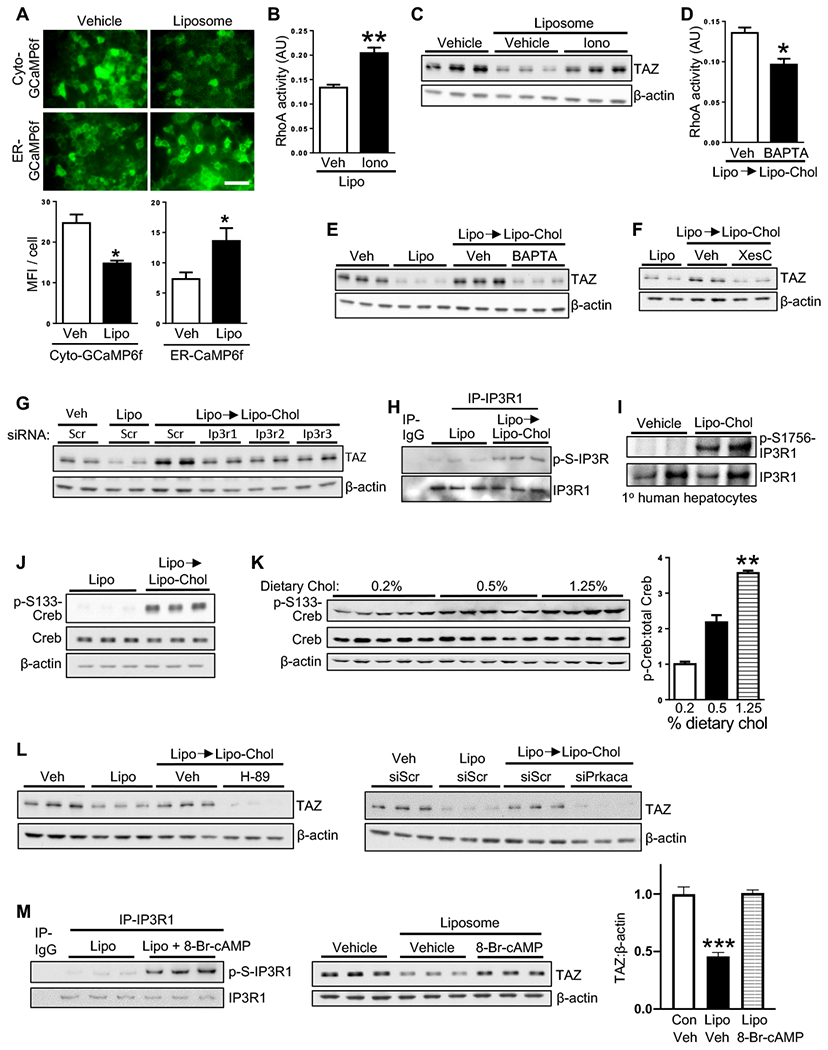
(A) Images and mean fluorescence intensity (MFI) per cell of AML12 cells transduced with cyto-GCaMP6f or ER-GCaMP6f and then incubated for 30 min with vehicle or liposomes (n = 4 biological replicates; means ± SEM; *p < 0.05). Scale bar, 100 μm.
(B) RhoA activity of AML12 cells incubated for 2 h with liposomes ± 1 μM ionomycin (Iono) included during the last hour (n = 3 biological replicates; means ± SEM; **p < 0.01).
(C) TAZ immunoblot of AML12 cells incubated with vehicle or liposomes for 24 h ± 1 μM ionomycin included during the last 4 h.
(D) RhoA activity of AML12 cells incubated for 16 h with liposomes and then 2 h with Lipo-Chol ± 5 μM BAPTA-AM (n = 3 biological replicates; means ± SEM; *p < 0.05).
(E-G) TAZ immunoblot in AML12 cells treated as follows:
(E) Vehicle or liposomes for 24 h, or liposomes for 16 h and then Lipo-Chol for 8 h ± 5 μM BAPTA-AM.
(F) Vehicle or liposomes for 24 h, or liposomes for 16 h and then Lipo-Chol for 8 h, with 2 μM xestospongin C (XesC) or vehicle included during the last 4 h.
(G) Vehicle or liposomes for 24 h, or liposomes for 16 h and then Lipo-Chol for 8 h, in AML12 cells transfected with siScr, siItpr1, siItpr2, or siItpr3.
(H) Phospho-IP3R and IP3R1 immunoblots in IP3R1 immunoprecipitates from AML12 cells incubated for 16.5 h with liposomes or 16 h with liposomes and then 30 minutes with Lipo-Chol.
(I) Phospho-S1756-IP3R1 and IP3R1 immunoblots of primary human hepatocytes incubated for 30 minutes with vehicle or Lipo-Chol.
(J) Phospho-S133-Creb and total Creb immunoblots of AML12 cells incubated for 16.5 h with liposomes or 16 h with liposomes and then 30 minutes with Lipo-Chol.
(K) Phospho-S133-Creb and total Creb immunoblots of liver extracts from mice fed 16 weeks with the NAFLD diet containing 0.2%, 0.5%, and 1.25% cholesterol, with densitometric quantification (n = 5 mice/group; means ± SEM; **p < 0.01).
(L) Left, TAZ immunoblot of AML12 cells incubated for 24 h with vehicle or liposomes or 16 h with liposomes and then 8 h with Lipo-Chol ± 10 μM H-89 PKA inhibitor for the final 4 h. Right, TAZ immunoblot of siScr- or siPrkaca-transfected AML12 cells that were incubated for 16 h with vehicle or liposomes or 16 h with liposomes and then 8 h with Lipo-Chol.
(M) Left, Phospho-S1756-IP3R1 and IP3R1 immunoblots of AML12 cells incubated for 16.5 h with liposomes or 16 h with liposomes and then 30 minutes with liposomes ± 100 μM 8-Br-cAMP. Middle, TAZ immunoblot of AML2 cells incubated for 24 h with vehicle or 24 h with liposomes ± 100 μM 8-Br-cAMP. Right, Quantification of the middle blot (n = 5 biological replicates; means ± SEM; ***p < 0.001).
Inositol triphosphate receptors (IP3R) are key mediators of ER calcium release, and we found that the IP3R inhibitor xestospongin C blocked both the increase in cytoplasmic calcium and the decrease in ER calcium in cholesterol-loaded AML12 cells (Figure S4E) and abrogated the increase in TAZ in cholesterol-loaded AML12 cells (Figure 4F). Further, increased TAZ in cholesterol-loaded AML12 cells could be suppressed by siRNA-mediated silencing any of the IP3Rs, although the suppression appeared somewhat greater with siItpr1 (Figures 4G and S4F). We also found that cholesterol enrichment of AML12 cells and human hepatocytes increased phospho-IR3R (Figures 4H and 4I), which is a marker of IP3R activation (D’Alessandro et al., 2018).
We demonstrated previously that IP3R could be serine-phosphorylated and activated by cyclic AMP-dependent protein kinase (PKA) (Wang et al., 2016; Wang et al., 2012). In this context, a marked increase in phospho-Creb, which is a marker of PKA activation, in cholesterol-repleted AML12 cells and in the livers of mice fed a high- vs. low-cholesterol diets (Figures 4J and 4K). Most importantly, inhibition of PKA by H-89 or silencing using siPrkaca prevented the increase in TAZ in cholesterol-repleted AML12 cells (Figures 4L and S4G), while PKA activation with 8-Br-cAMP increased phospho-IP3R and TAZ in cholesterol-depleted AML12 cells (Figure 4M). These combined data support a cholesterol–RhoA–TAZ pathway mediated through PKA-mediated IP3R activation, leading to activation of RhoA by cytoplasmic calcium.
Cholesterol-Mediated Induction of the PKA-TAZ Pathway in Hepatocytes Requires Adenylyl Cyclase 10
PKA is activated by cAMP, and in mammalian cells there are two families of adenylyl cyclase that synthesize cAMP: hormone-responsive transmembrane adenylyl cyclases (tmACs: ADCY1-9), which are activated by G-protein alpha subunit, group S (Gsα); and soluble adenylyl cyclase (sAC: ADCY10), which is distributed throughout the cytoplasm and inside intracellular organelles (Kamenetsky et al., 2006; Zippin et al., 2003). To determine if tmACs were involved in the TAZ pathway, we first silenced Gsα (Gnas) in cholesterol-repleted AML12 cells and found that this failed to block the increase in TAZ in cholesterol-repleted AML12 cells (Figures 5A and S5A). Next, we compared the effect of ddAdo, an inhibitor selective for tmACs relative to sAC (Bitterman et al., 2013) with LRE1, an inhibitor selective for sAC relative to tmACs (Ramos-Espiritu 2016), in primary mouse hepatocytes. LRE1, but not ddAdo, inhibited cholesterol-mediated increase in TAZ and cAMP (Figures 5B and 5C). As validation, ddAdo, but not LRE1, blocked glucagon-induced phospho-CREB, which signals via GPCR linked to Gsα and a tmAC (Figure S5B). Further, silencing sAC in hepatocytes, including human hepatocytes, lowered TAZ, p-IP3R1, and active RhoA (Figures 5D–5F, S5C, and S5D). Most importantly, treatment of Adcy10fl/fl mice fed the 1.25%-cholesterol diet with AAV8-TBG-Cre to silence hepatocyte sAC lowered liver TAZ, fibrosis, inflammatory cells, Ihh mRNA, and mRNAs encoding proteins associated with activated HSCs, TUNEL+ cells, and plasma ALT (Figures 5G–5J and S5E–S5H). Thus, sAC is the source of cAMP responsible for cholesterol-mediated activation of the PKA–IP3R–RhoA–TAZ pathway in hepatocytes.
Figure 5. Adenylyl Cyclase 10 is Necessary for Increased TAZ in Cholesterol-Repleted Hepatocytes and in NASH Mice.
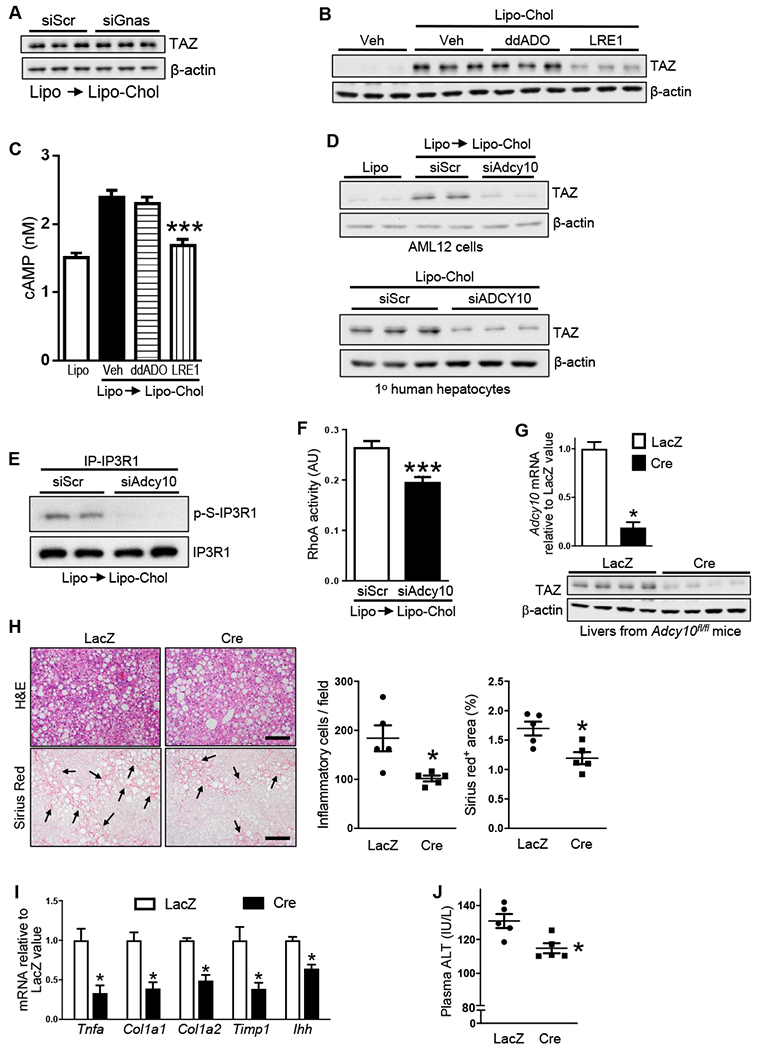
(A) TAZ immunoblot of siScr- or siGnas-transfected AML12 cells that were incubated for 16 h with liposomes and then 8 h with Lipo-Chol).
(B) TAZ immunoblot of primary mouse hepatocytes that were incubated for 24 h with vehicle or Lipo-Chol ± 10 μM 2′,5′-dideoxyadenosine (ddAdo) or 30 μM LRE1.
(C) cAMP content of AML12 cells that were incubated for 16.5 h with liposomes or 16 h with liposomes and then 30 minutes with Lipo-Chol ± 10 μM ddAdo or 80 μM LRE1 (n = 4 biological replicates; values shown are means ± SEM; ***p < 0.001 vs. Veh and ddAdo).
(D) Top, TAZ immunoblot of control AML12 cells that were incubated 24 h with liposomes, or siScr or siAdcy10-transfected AML12 that were incubated for 16 h with liposomes and then 8 h with Lipo-Chol. Bottom blot, TAZ immunoblot of siScr or siADCY10-transfected primary human hepatocytes that were incubated for 24 h with Lipo-Chol.
(E) Phospho-IP3R1 and IP3R1 immunoblots of siScr- or siAdcy10-transfected AML12 cells that were incubated for 16 h with liposomes and then 30 minutes with Lipo-Chol.
(F) RhoA activity of siScr- or siAdcy10-transfected AML12 cells that were incubated for 16 h with liposomes and then 2 h with Lipo-Chol (n = 4 biological replicates; means ± SEM; ***p < 0.001).
(G-J) The following parameters were assayed in Adcy10fl/fl mice fed the NASH-inducing 1.25%-cholesterol diet for 16 weeks, with AAV8-TBG-Cre or AAV8-TBG-LacZ administered at the 8-week time point (n = 5 mice/group; means ± SEM, *p < 0.05):
(G) Liver Adcy10 mRNA and TAZ immunoblot.
(H) Liver sections stained for H&E, quantified for inflammatory cells, and stained and quantified for Sirius red (arrows indicate areas of fibrosis). Scale bars, 200 μm.
(I) Liver Tnfa, Col1a1, Col1a2, Timp1, and Ihh mRNAs.
(J) Plasma ALT.
The Cholesterol-TAZ Pathway Requires Internalization of Plasma Membrane Cholesterol
As sAC is localized in intracellular sites, we reasoned that activation of the TAZ pathway required internalization of plasma membrane cholesterol. In support of this idea, treatment of cholesterol-loaded AML12 cells and human hepatocytes with an anthrolysin O peptide called ALOD4, which blocks plasma membrane cholesterol trafficking (Infante and Radhakrishnan, 2017), prevented the increase in RhoA activity and TAZ (Figures 6A–6C). Similar results were observed with LDL- and SREBP-2-derived cholesterol (Figures 6D–6E, S5I, and S5J), which are known to traffic through the plasma membrane (Liscum and Dahl, 1992). Note that ALOD4 increased SREBP-2 maturation, validating its ability to block cholesterol trafficking to the regulatory cholesterol pool in the ER (Infante and Radhakrishnan, 2017). As further validation, we silenced GramD1 (ASTER)-A, -B, and C, which mediate cholesterol trafficking from the plasma membrane to the cell interior (Sandhu et al., 2018). Cholesterol-induced TAZ was dependent on ASTER-B and ASTER-C (Figures 6F and S5K), and, as evidence for the role of plasma membrane cholesterol trafficking in the upstream pathway, cholesterol-induced activation of PKA (p-Creb), IP3R, ( p-IP3R1), and RhoA was blocked by ALOD4 and/or silencing Gramd1b/c (Figures 6G–I, S5L, and S5M).
Figure 6. Cholesterol Transport from the Plasma Membrane is Necessary for Increased TAZ in Cholesterol-Repleted Hepatocytes.
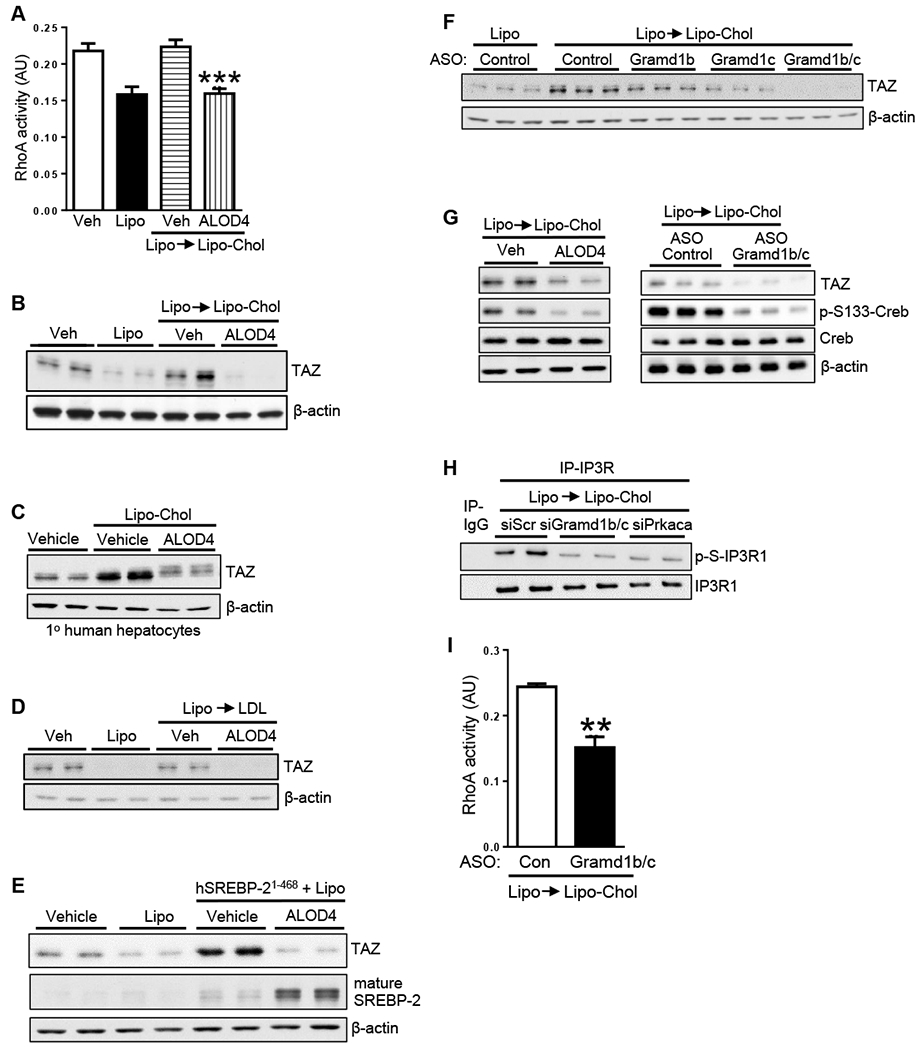
(A) RhoA activity of AML12 cells that were incubated for 24 h with vehicle or liposomes or for 2 h with liposomes and then 3 h with Lipo-Chol ± 10 μM ALOD4 (n = 6 biological replicates; means ± SEM; ***p < 0.001 vs. both Veh groups).
(B) TAZ immunoblot of AML12 cells that were incubated for 24 h with vehicle or liposomes or 24 h with liposomes and then 8 h with Lipo-Chol ± 10 μM ALOD4 included during the last 4 h.
(C) TAZ immunoblot of primary human hepatocytes that were incubated for 24 h with vehicle or Lipo-Chol ± 10 μM ALOD4 included during the last 4 h.
(D) TAZ immunoblot of AML12 cells that were incubated for 24 h with vehicle or liposomes or 24 h with liposomes and then 8 h with 100 μg/ml LDL ± 10 μM ALOD4 included during the last 4 h.
(E) TAZ and mature SREBP-2 immunoblots of control or human SREBP-2(1-468)-transfected AML12 cells that were incubated for 24 h with vehicle or liposomes ± 10 μM ALOD4 included during the last 4 h.
(F) TAZ immunoblot of AML12 cells were treated with control ASO or ASO targeting Gramd1b, Gramd1c, or both and then incubated for 24 h with liposomes or 16 h with liposomes and then 8 h with Lipo-Chol.
(G) TAZ, p-S133-Creb, total Creb immunoblots of AML12 cells were treated and incubated as in panel E for ALOD4 and panel F for ASOs targeting Gramd1b and Gramd1c.
(H) Phospho-IP3R1 and IP3R1 immunoblots of siScr-, siGramd1b- and siGramd1c-, or siPrkaca-transfected AML12 cells that were incubated for 16 h with liposomes and then 8 h with Lipo-Chol.
(I) RhoA activity of control ASO- or Gramd1b/c ASO-treated AML12 cells that were incubated for 16 h with liposomes and then 2 h with Lipo-Chol (n = 3 biological replicates; means ± SEM; **p < 0.01).
In normal liver, Gramd1c is more abundant than Gramd1b (Sandhu et al., 2018), but Gramd1b increased in the livers mice fed the 1.25%-cholesterol diet (Figure S6A). Most importantly, treatment of 8-week NASH diet-fed mice with AAV8-H1-shGramd1b/c prevented increases in liver TAZ; fibrosis, including the expression of HSC-activating TAZ gene target Ihh; inflammation; and activation of PKA (p-Creb), IP3R, (p-IP3R1), and RhoA, and LATS1/2 (p-LATS) over the ensuing 8 weeks, despite unchanged body weight, percent liver weight, or fasting blood glucose (Figures 7A–7G and S6B–S6F). Surprisingly, neither TUNEL+ cells in the liver nor plasma ALT were statistically different between the two groups of mice (Figures S6G and S6H). As expected, YAP was also lower in the livers of shGramd1b/c-treated mice, although the percent decrease of YAP was less than that of TAZ (Figure 7B). However, as noted previously, silencing hepatocyte TAZ lessens NASH without lowering YAP (Figure S3N). In summary, the combined data in this study show that internalization of plasma membrane cholesterol is required for cholesterol-induced TAZ stabilization in hepatocytes in vitro and for cholesterol-diet induced increases in liver TAZ, fibrosis, and inflammation in mice during NASH progression (Figure 7H).
Figure 7. Silencing of Hepatocyte Gramd1b/c in Mice with Hepatosteatosis Prevents TAZ Increase and Suppresses NASH Progression.
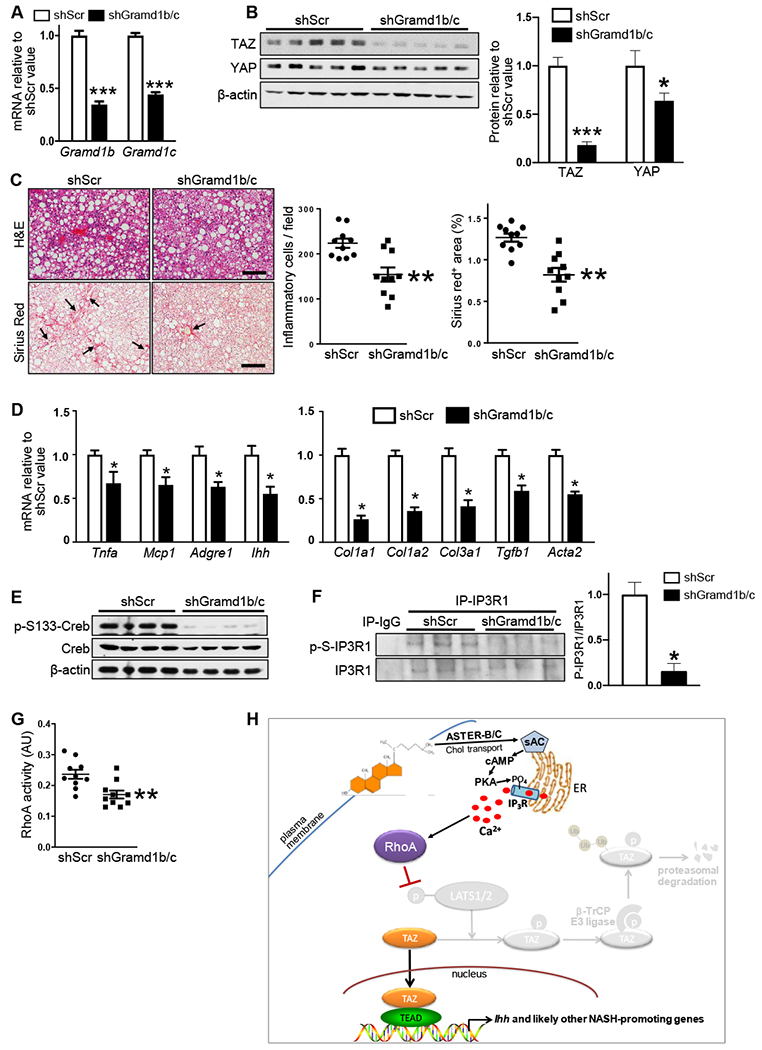
The following parameters were assayed in mice fed the NASH-inducing 1.25%-cholesterol diet for 16 weeks, with AAV8-H1-shGramd1b/c or AAV8-H1-shSrc administered at the 8-week time point (n = 10 mice/group; means ± SEM; *p < 0.05, **p < 0.01, ***p < 0.001):
(A) Liver Gramd1b and Gramd1c mRNAs.
(B) Liver TAZ and YAP immunoblots for 5 mice/group, with quantification.
(C) Liver sections stained for H&E, quantified for inflammatory cells, and stained and quantified for Sirius red (arrows indicate areas of fibrosis). Scale bars, 200 μm.
(D) Liver Tnfa, Mcp1, Adgre1 (F4/80), Ihh, Col1a1, Col1a2, Col3a1, Tgfb, and Acta2 mRNAs.
(E) Liver p-S133- and total Creb Immunoblots.
(F) Phospho-IP3R1 and total IP3R1 immunoblots of liver IP3R1 immunoprecipitates, with quantification.
(G) RhoA activity of liver extracts.
(H) Summary scheme of the hepatocyte cholesterol-TAZ pathway in NASH. In cholesterol-enriched hepatocytes, internalization of plasma membrane cholesterol by ASTER-B/C leads to sAC-mediated increase in cAMP and PKA-dependent activation of IP3R. The resulting increase in Ca2+i activates RhoA, which inhibits LATS1/2-dependent phosphorylation of TAZ. Phosphorylation of TAZ, particularly on S117, blocks β-TrCP-dependent proteasomal degradation of TAZ, resulting in an increase in TAZ-TEAD-induced genes, notably Ihh, which promotes HSC activation and liver fibrosis in NASH (Wang et al., 2016).
DISCUSSION
The cholesterol-TAZ pathway revealed herein has implications for both NASH and the fundamental process of cellular cholesterol trafficking and signaling. The findings provide plausible mechanisms linking elevated liver cholesterol to NASH and NASH to elevated TAZ, and they raise the possibility of new therapeutic targets for NASH. With regard to the cell biologic aspects of this work, the data demonstrate a new role for trafficking of plasma membrane cholesterol in the regulation of cAMP, PKA, and calcium.
The mechanisms leading to increased liver cholesterol in NASH are likely multi-factorial. A number of studies, including this one, have shown a direct link between dietary cholesterol and NASH in mice (McGettigan et al., 2019; Savard et al., 2013; Van Rooyen et al., 2011; Wouters et al., 2008) and fibrotic liver disease in humans (Ioannou et al., 2009). The C57BL/6J mouse strain has a relatively low ability to absorb dietary cholesterol (Schwarz et al., 2001) and therefore must be fed diets with high cholesterol content simply to achieve a level of liver cholesterol accumulation approaching that in human NASH (see Figure 1A). Other mechanisms proposed for liver cholesterol accumulation in NASH include dysregulated endogenous cholesterol synthesis (Min et al., 2012), including a recently described pathway that involves caspase-2 activation of SREBP-2 (Kim et al., 2018); genetic polymorphisms in lipid-related genes (Chen et al., 2017); increased cholesteryl ester hydrolysis (Min et al., 2012); and decreased cholesterol excretion into bile (Min et al., 2012). Our hepatocyte studies suggest the SREBP-2-mediated endogenous cholesterol can activate the pathway. This raises the possibility of a positive-feedback loop, as inactivation of LATS2, which we showed is downstream of cholesterol, was reported to activate SREBP-2 and cause cholesterol accumulation (Aylon et al., 2016). Future in vivo studies will be required to investigate how nondietary mechanisms of increasing liver cholesterol affect TAZ and NASH in vivo and whether the reported benefits of statins and possibly ezetimibe on NASH progression (Athyros et al., 2017) may be related to the mechanisms described here. Of note, statins may have secondary effects on TAZ by blocking the prenylation of RhoA, which is necessary for its activation (Blanco-Colio et al., 2002). Finally, it is interesting to note that plasma ALT and TUNEL+ cells in the liver were not decreased by silencing Gramd1b/c, whereas all interventions downstream of this initial step in the pathway did lower these endpoints. Thus, it is possible that cholesterol trafficking has an additional, separate role that partially protects hepatocytes from injury and death. If so, the net effect of interrupting of cholesterol trafficking may be no change in plasma ALT and TUNEL+ liver cells, i.e., lower TAZ would protect hepatocytes (Wang et al., 2016), but this effect might be negated by loss of a separate hepatocyte protective process.
There are likely multiple and complementary mechanisms linking elevated liver cholesterol to NASH, including cholesterol-mediated processes in HSCs and macrophages as well as hepatocytes. Examples of previously suggested mechanisms include: (a) activation of inflammatory and death pathways in hepatocytes and Kupffer cells (Bieghs et al., 2013; Gan et al., 2014; Ioannou, 2016) and activation of HSCs, either directly or via changes in Kupffer cells (McGettigan et al., 2019; Teratani et al., 2012). A surprising component of the pathway here relates to the role of soluble adenylyl cyclase (sAC). This finding raises a number of intriguing questions, e.g., whether there is a specific subcellular location where sAC-mediated cAMP synthesis and PKA activation occur during cholesterol repletion of hepatocytes. A previous study showed that sAC co-localizes with mitochondria in multiple cell types (Lefkimmiatis, 2014; Valsecchi et al., 2013), and mitochondria are known to associate with regions of the ER, termed mitochondria-associated membranes (MAMs), that are rich in IP3Rs (Wieckowski et al., 2009). cAMP acts locally to activate nearby PKA, which is anchored by AKAP in discrete microdomains (Rich et al., 2000; Zaccolo and Pozzan, 2002). Thus, cholesterol transferred from the plasma membrane to ER may enable sAC in ER-localized mitochondria to generate cAMP and activate nearby PKA and IP3R. In this context, analysis of NASH livers of high-cholesterol-fed diabetic mice showed filipin fluorescence in the ER and mitochondria as well as the plasma membrane of hepatocytes (Gan et al., 2014). A more fundamental question is the molecular mechanism linking cholesterol to sAC activity, which could occur through direct or indirect mechanisms (Litvin et al., 2003; Tresguerres et al., 2011). Interestingly, a recent study using a renal carcinoma cell line suggested that TAZ/YAP can actually suppress sAC activity by lowering cellular calcium (White et al., 2019). Whether this pathway is absent in cholesterol-enriched hepatocytes or counteracted by cholesterol-induced activation of IP3R remains to be determined.
A key therapeutic goal related to the epidemic of NASH is to prevent the progression of liver fibrosis before it causes liver damage. Directly targeting hepatocyte TAZ (Wang et al., 2016) or inhibiting the TAZ-inducing pathway revealed here can accomplish this goal in mice with established early fibrotic NASH. An emerging modality of therapeutic translation of these findings is hepatocyte-targeted siRNAs, e.g., siRNA conjugated to N-acetylgalactosamine (GalNAc) (Nair et al., 2014), and the first hepatocyte-targeted siRNA has been approved by the FDA for the treatment of hereditary transthyretin amyloidosis (Adams et al., 2018). In this context, we have recently shown that treatment of NASH mice with GalNAc-siTaz prevents and reverses fibrosis in NASH in mice (Wang et al., 2019). The current study now raises additional possibilities targets of TAZ, including ASTER-B/C and sAC. In view of the difficulty in predicting drug efficacy and toxicity, the availability of multiple options, especially those supported by in depth mechanistic data, in vivo molecular-genetic causation data, and human NASH data is critical.
Limitations of Study
The in vivo causation experiments were conducted in an experimental model of NASH, and thus further work is needed to show the importance of the cholesterol-TAZ pathway in human NASH. Nonetheless, human relevance is supported by the following: (1) both liver cholesterol and TAZ are associated with NASH, but not steatosis, in humans; (2) human NASH liver shows correlations among liver cholesterol, RhoA, and TAZ; and (3) key components of the cholesterol-TAZ pathway are operational in primary human hepatocytes, including the roles of cholesterol, ADCY10 (sAC), and RhoA. As predicted, the hepatocyte cholesterol-Hippo pathway revealed herein also increases YAP, and thus it is theoretically possible that hepatocyte YAP also contributes to cholesterol-induced NASH. However, as noted, shTAZ markedly suppresses NASH without affecting YAP. It is also interesting to note that interruption of cholesterol trafficking (shGramd1b/c) and RhoA in NASH mice caused less of a decrease in YAP than in TAZ. Importantly, despite their common regulation, TAZ and YAP functions can diverge, including in liver (Hagenbeek et al., 2018). In the end, the role of hepatocyte YAP in NASH must await a dedicated study in which hepatocyte YAP is silenced without altering TAZ in experimental models of NASH progression.
STAR METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Ira Tabas (iat1@columbia.edu). This study did not generate new unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal Studies
Male wild-type C57BL/6J mice (#000664, 10-11 weeks/old) were obtained from Jackson Laboratory (Bar Harbor, ME) and were allowed to adapt to housing in the Columbia University Irving Medical Center Institute of Comparative Medicine for 1 week prior to random assignment to experimental cohorts. Wwtr1fl/fl mice (TAZ gene floxed) (Xin et al., 2013) were generously provided by Dr. Eric Olson, University of Texas Southwestern and were backcrossed to C57BL/6J background and confirmed by genotyping. Adcy10fl/fl mice (Chen et al., 2013) were backcrossed to C57Bl/6 background confirmed by Mouse Universal Genotyping Array (MUGA). The mice were fed a fructose-palmitate diet containing either 1.25%, 0.5%, or 0,2% cholesterol. The 1.25%-cholesterol diet induces NASH after 16 weeks and is referred to as the “NASH diet” (Envigo, TD.160785 + sugar water) (Wang et al., 2016). The diets containing 0.5% and 0.2% cholesterol induce hepatosteatosis but not NASH and are referred to as NAFLD diets. All mice were fed with NASH/NAFLD diet and sugar water, which contains 23.1g/L of fructose and 18.9g/L of glucose. AAV8-TBG-Cre or control AAV8-TBG-LacZ (1x1011 genome copies/mouse) was administered to Wwtr1fl/fl mice by tail vein injection 1 week prior to initiation of the NASH diet or to Adcy10fl/fl mice 8 weeks after initiation of the NASH diet. AAV8-H1-shRNA, AAV8-H1-scrambled RNA, AAV8-TBG-HA-hTAZ, and AAV8-TBG-HA-hTAZ-S117A viruses were delivered by tail vein injection at 2x1011 genome copies/mouse either 1 week prior to the NASH/NAFLD diet or 8 weeks after diet initiation. Animals were housed in standard cages at 22°C in a 12-12-hour light-dark cycle in a barrier facility. All animal experiments were performed in accordance with institutional guidelines and regulations and approved by the Institutional Animal Care and Use Committee at Columbia University and, for the Adcy10fl/fl mouse experiment, at Weill-Cornell Medical Center.
Human Liver Specimens and Human Primary Hepatocytes
De-identified human liver specimens were acquired with patient consent from the Liver Tissue Cell Distribution System at the University of Minnesota. The specimens were collected on the date of liver transplantation and preserved as frozen samples. The mean age of the donors was 59 (ranging from 43 to 68 years old), and the donors included 2 males and 6 females. The diagnostic information is included in Table S1. Phenotypic and pathological characterizations were conducted by medical physicians and pathologists associated with the Liver Tissue Cell Distribution System. Human primary hepatocytes were obtained with consent from male donors at the University of Pittsburgh through the Liver Tissue Cell Distribution System. All human studies were approved by the Columbia University Institutional Review Board and were conducted in accordance with National Institutes of Health and institutional guidelines for human subject research.
Cell Culture and Cell Treatment
AML12 mouse hepatocytes were purchased from ATCC (CRL-2254) and cultured in DMEM/F12 medium (Life Technologies, #11320) with 10% (vol/vol) heat-inactivated FBS (Gibco, #16140-071) and 1X penicillin-streptomycin solution (Corning, #30-002-Cl). HepG2 cells were purchased from ATCC (HB-8065) and cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) (Corning, #10-013-CV) with 10% (vol/vol) heat-inactivated FBS and 1X penicillin-streptomycin solution. Primary mouse hepatocytes were isolated from 10-week-old wild-type C57BL/6J mice as described previously (Ozcan et al., 2012). In brief, mice were euthanized with isoflurane, the abdomen was opened, and a catheter was inserted into the vena cava. The liver was perfused with Hanks’ balanced salt solution. The portal vein was then cut and perfused with collagenase I. After the perfusion, the liver was placed in a Petri dish containing DMEM and disaggregated with forceps. Digested liver was passed through 100-μm filter and centrifuged at 50 x g for 5 min. The supernatant fraction was removed, and the hepatocytes in the pellet were resuspended and cultured in DMEM containing 10% FBS, followed by treatment as described in the figure legends. All cells were grown at 37°C and 5% CO2. For liposome treatment, 1x105 AML12 cells were plated in 24-well plate and cultured for 24-48 h until cell confluence was ~90%. Liposome solution was added to the medium at 1:10 volume ratio in a total volume of 500 μl. For the epi- and ent-cholesterol experiment, the total volume of medium was 300 μl. The cells were harvested after treatment in Laemmli Sample Buffer (Bio-Rad, #1610737) with 2-mercaptoethanol (Bio-Rad, #161-0710) for immunoblotting or in RNA lysis buffer (Qiagen, #79216) for mRNA quantification.
METHOD DETAILS
Reagents and Kits
The following plasma assay kits were used in this study: cholesterol (#439-17501), free cholesterol (#994-02501) and triglyceride (#465-09791, #461-09891) from Wako; and ALT (#A526-120) from TECO Diagnostics. Adeno-associated virus subtype 8 (AAV8)-shRNA targeting murine Rhoa was made by annealing complementary oligonucleotides (5’- CACC AgtcaagcatttctgtccaaatCTCGAGATTTGGACAGAAATGCTTGAC-3’), which were then ligated into the self-complementary (sc) AAV8-RSV-GFP-H1 vector as described previously (Wang et al., 2016). AAV8-H1-shRNA targeting murine Gramd1b was made by annealing complementary oligonucleotides (5’- CACC AgatgaaggactcgcttatcaaCTCGAGTTGATAAGCGAGTCCTTCATC-3’), which were then ligated into the scAAV-RSV-GFP-H1 vector as above. AAV8-H1-shRNA targeting murine Gramd1c was made by annealing complementary oligonucleotides (5’- CACCAgggaaagagatgagaagttctCGAAAGAACTTCTCATCTCTTTCCC-3’), which were then ligated into the scAAV-RSV-GFP-H1 vector as above. AAV8-H1-shRNA targeting murine Btrc was made by annealing complementary oligonucleotides (5’- CACCA gcgacatagtttacagagaatTCAAGAGATTCTCTGTA AACTATGTCG C-3’), which were then ligated into the scAAV-RSV-GFP-H1 vector as above. The resultant constructs were amplified by Vector Biolabs, Malvern, PA. AAV8-TBG-HA-hTAZ and AAV8-TBG-HA-S117A-hTAZ were from Vector Biolabs. AAV8 containing hepatocyte-specific TBG-Cre recombinase (AAV-TBG-Cre, 107787-AAV8) and the control vector, AAV8-TBG-LacZ (105534-AAV8), were purchased from the Addgene. Cyto-GCaMP6f and ER-GCaMP6f were subcloned into lentiviral vectors and then packaged and concentrated by ALSTEM, LLC (Wang et al., 2017). WT-HA-human TAZ plasmid was from Addgene (#32839). The human SREBP-2(1-468) plasmid (Horton et al., 1998) was a gift from Dr. Jay Horton (University of Texas-Southwestern Medical Center). The kit to carry out mutagenesis of WWTR1 was from New England Biolabs (#E0554S), and the primers are listed in Table S2. The G-LISA™ RhoA activation assay kit was from Cytoskeleton (#BK124).
Preparation of Liposomes
DMPC (1,2-dimyristoyl-sn-glycero-3-phosphocholine; Avanti Polar Lipids, 850345; molecular mass: 677.5) and cholesterol (Sigma, C8667; molecular mass: 386.6) were dissolved in chloroform. Liposomes were made by adding 40 mg of DMPC with or without 80 mg of cholesterol to a glass vessel and then removing the solvent using a stream of nitrogen gas. Ten ml PBS was added and, after mixing, the lipids were subjected to probe sonication on ice for 5 minutes using 10-second on-off intervals. The preparation was then centrifuged at 10,000 x g for 10 minutes, and the supernatant fraction was extruded through a 100-nm polycarbonate filter (Avanti, 610000-1Ea) at room temperature. Each aliquot was stored in glass vials under argon at 4°C and used within 2 weeks. For liposomes, only 40 mg of DMPC were used (without cholesterol) based on the protocol. For epi- and ent-cholesterol liposomes, the initial mixture was 1 mg DMPC and 2 mg sterol to which was added 250 μl of PBS. Epi-cholesterol (cholest-5-en-3β-ol) was purchased from Sigma-Aldrich (R207349), and ent-cholesterol was synthesized as previously described (Westover et al., 2003).
siRNA-Mediated Gene Silencing and Transfection
Scrambled siRNA control and oligo-targeting siRNAs were transfected into AML12 or primary hepatocytes using Lipofectamine RNAiMAX (Life Technologies) at 40 nM of siRNA in 24-well plates following the manufacturer’s instructions. Briefly, 2 X 105 cells at 30 - 40% confluence were incubated for 18 h with 0.5 ml of culture medium containing 1.5 μl Lipofectamine RNAiMAX and 20 pmol siRNA. The siRNA sequences are listed in Table S3. The plasmids were transfected into AML12 cells using Lipofectamine® LTX Reagent with PLUS™ Reagent (Life Technologies, #15338100). For each well in a 24-well plate, 2 μl LTX, 0.5 μl PLUS reagent, and 0.5 μg plasmid DNA are used when cells reached 30-40% confluence. After overnight incubation, the cells were switched back to normal culture medium.
Blood and Plasma Analyses
Fasting blood glucose was measured using a glucose meter (One Touch Ultra, Life- scan) in mice that were fasted for 5 h, with free access to water. Total plasma triglyceride, total cholesterol, free cholesterol, and ALT were assayed using commercially available kits, as listed in the Reagents and Kits section above.
Histopathological Analysis
Inflammatory cells in H&E-stained liver section images were quantified as the number of mononuclear cells per field (20x objective). For other parameters involving various stains, computerized image analysis (ImageJ) was used to quantify the area stained. The same threshold settings were used for all analyses. For all analyses, we quantified 6 randomly chosen fields per section per mouse. Liver fibrosis was assessed by quantifying Picrosirius (Sirius) red-stained area (Polysciences, #24901).
Filipin Staining and Immunofluorescence Microscopy
AML12 or primary cultured hepatocytes were fixed in 4% paraformaldehyde for 10 min at room temperature, rinsed using glycine/PBS, and stained with 0.25 mg/ml filipin for 2 h at room temperature. The cells were viewed in PBS by fluorescence microscopy using a UV filter set (340-380 nm excitation, 40 nm dichroic, 430-nm long pass filter). As filipin fluorescence photobleaches very rapidly, care was taken to have the same UV exposure time before image collection for all samples. TUNEL staining was conducted using a kit from Roche (#12156792910).
Measurement and Analysis of Liver Tissue Cholesterol
For liver cholesterol quantification, liver tissue (20 mg) was washed in cold PBS to remove blood, homogenized in 20x volume (400 μl) Cholesterol Assay Buffer (ab65390, Abcam), and centrifuged at 10,000 x g for 10 min. The supernatant fraction was transferred to an Eppendorf tube and mixed thoroughly. Color Reagent Solution from the Wako Total/Free Cholesterol assay kit was added at a 1:20 ratio (v/v) to these lysates, followed by incubation and quantification in a plate reader according to the manufacturer’s instructions.
Immunoblotting
Liver protein was extracted using RIPA buffer (Thermo, #89900), and the protein concentration was measured by a BCA assay (Thermo, #23227). Proteins were separated by electrophoresis on 4-20% Tris gels (Life technologies, EC60285) and transferred to nitrocellulose membranes (Bio-Rad, #1620115). The membranes were blocked for 30 min at room temperature in Tris-buffered saline and 0.1% Tween 20 (TBST) containing 5% (wt/vol) nonfat milk and then incubated with primary antibody in the same buffer at 4°C overnight, using 1:1000 dilution. The protein bands were detected with horse radish peroxidase-conjugated secondary antibodies and Supersignal West Pico enhanced chemiluminescent solution (Thermo, #34080). Cultured cells were lysed in Laemmli sample buffer (Bio-Rad, #161-0737) containing 5% 2-mercaptoethanol, heated at 100°C for 5 min, and then electrophoresed and immunoblotted as above.
Immunoprecipitation, LATS2 Kinase Assay, and GTP-RhoA Assay
AML12 cells from 100-mm culture dishes were collected into 0.4 ml 1x ice-cold cell lysis buffer (#9803S, Cell Signaling) and incubated on ice for 5 min. Then cell lysates were centrifuged at 10,000 x g for 10 min at 4°C. The supernatant fraction was pre-cleaned using 30 μl Protein G Agarose beads (Cell Signaling, #37478) and incubated overnight with anti-TAZ or anti-IP3R1 antibodies with gentle rotating at 4°C. Rabbit IgG was used as the negative control. Thirty ml of 50% protein G beads were added to the solution followed by incubation for 1 hour at 4°C. The beads were then collected by centrifugation and washed five times with 500 μl 1x cell lysis buffer. Proteins were re-suspended with 50 μl Laemmli Sample Buffer (#1610737, Bio-Rad) and then subjected to immunoblot analysis. For LATS2 kinase assay, AML12 cells transfected with HA-LATS2 plasmid (Zhao et al., 2010) were lysed in 1x ice-cold cell lysis buffer (#9803S, Cell Signaling). The extract was immunoprecipitated using anti-HA antibody, and the immunoprecipitate was incubated with recombinant TAZ (pro-1814-b, Prospec) in kinase assay buffer (#9802, Cell Signaling) containing ATP (#9804, Cell Signaling). For mouse liver, extracts were immunoprecipitated using anti-LATS2. For a control, a 1:1:1 mixture of extracts (mix) was treated with control IgG instead of anti-LATS2. The immunoprecipitates were incubated with recombinant TAZ and then immunoblotted for p-serine-TAZ using anti-phosphoserine antibody, TAZ, and LATS2. Active RhoA was assayed using either an ELISA assay (G-LISA®, #BK124, Cytoskeleton) or by precipitating GTP-bound RhoA using the RhoA Pull-down Activation Assay Biochem Kit (#BK036, Cytoskeleton). For this assay, cell lysates were incubated with beads containing GST-tagged Rho binding domain (RBD) of the human Rhotekin protein (amino acids 7-89), which binds specifically to GTP-bound Rho proteins. The beads were pelleted and immunoblotted for RhoA and GST (input).
Quantitative RT-qPCR
Total RNA was extracted from liver tissue or cultured hepatocytes using the RNeasy kit (Qiagen, 74106). The quality and concentration of the RNA was assessed by absorbance at 260 and 280 nm using a Thermo Scientific NanoDrop spectrophotometer. cDNA was synthesized from 1 μg total RNA using oligo (dT) and Superscript II (Invitrogen). qPCR was performed with a 7500 Real time PCR system (Applied Biosystems) using SYBR Green Master Mix (Life Technologies, #4367659). The primer sequences are listed in Table S4.
Cytosolic and ER Calcium Measurements
AML12 were incubated for 18 h with GCaMP6f and ER-GCaMP6-150 lentiviruses at an MOI of 5. The cells were then treated as described in the figure legends and viewed by fluorescence microscopy. Measurements of relative GFP intensity/cell, as an indicator of calcium level, were conducted on 15-20 randomly chosen cells per well, and 4-5 wells were examined for each group.
LC-MS/MS
HepG2 cells (3 x 107 cells) were treated with liposomes and MG132 for 4 h, followed by immunoprecipitation of TAZ. The immunoprecipitate was fractionated on 4-20% Tris gels (Life technologies, EC60285) and stained by GelCode™ Blue Safe Protein Stain (ThermoScientific, #1860957). The gel band containing TAZ was excised and subjected to disulfide bond reduction with 10 mM dithiothreitol (56°C for 45 min) and alkylation with 55 mM iodoacetamide (room temperature for 30 min in the dark). In-gel digestions were carried out using trypsin at 37 °C for 8 h and chymotrypsin at 25°C for 15 h. Peptides were then extracted from the gel pieces, lyophilized, and desalted using a C18 stage-tip column. Phosphopeptides were enriched with Titansphere™ TiO2 resin (particle size 10 μm, GL Sciences, Japan) and analyzed using a Thermo Fisher Scientific EASY-nLC 1200 coupled on-line to a Fusion Lumos mass spectrometer (Thermo Fisher Scientific, USA). Buffer A (0.1% formic acid in water) and buffer B (0.1% formic acid in 80 % ACN) were used as mobile phases for gradient separation. A 100-μm × 15-cm chromatography column (ReproSil-Pur C18-AQ, 3 μm, Dr. Maisch GmbH, Germany) was packed in-house for peptide separation. Peptides were separated with a gradient of 5–40% buffer B over 20 min and then 40%-100% buffer B over 5 min at a flow rate of 400 nl/min. The Fusion Lumos mass spectrometer was operated in data-dependent mode. Full MS scans were acquired in the Orbitrap mass analyzer over a range of 300-1500 m/z with resolution 60,000 at m/z 200. The top most abundant 15 precursors were selected with an isolation window of 1.4 m/z and fragmented by higher-energy collisional dissociation with normalized collision energy at 35. MS/MS scans were acquired in the Orbitrap mass analyzer with resolution 15,000 at m/z 200. The automatic gain control target value was 4e5 for full scans and 5e4 for MS/MS scans respectively, and the maximum ion injection times were 50 ms and 100 ms for full scans and MS/MS scans, respectively. The acquisition data were searched against a customized protein database (E. coli database with addition of human TAZ sequence) using MaxQuant, with phosphorylation specified as dynamic modification.
QUANTIFICATION AND STATISTICAL ANALYSIS
All results are presented as mean ± SEM. Statistical significance was determined using GraphPad Prism software. Data that passed the normality test were analyzed using Student’s t test for two groups; one-way ANOVA with Tukey’s post-hoc analysis for more than two groups; or two-way ANOVA with Sidak’s post-hoc analysis for two factors. Data that were not normally distributed were analyzed using the nonparametric Mann-Whitney U test, or, for more than two groups, by Kruskal-Wallis with post-hoc analysis by the Dunn test. The statistical details can be found in the figures and legends.
DATA AND CODE AVAILABILITY
This study did not generate any unique datasets or code.
Supplementary Material
Highlights.
Liver cholesterol and TAZ are elevated in human and mouse fibrotic NASH
Increased hepatocyte cholesterol upregulates TAZ in human and mouse hepatocytes
Cholesterol blocks TAZ proteasomal degradation via an adenylyl cyclase-RhoA pathway
Silencing the pathway in hepatocytes lowers TAZ and fibrosis in experimental NASH
Context and Significance.
Nonalcoholic steatohepatitis (NASH) is the leading cause of chronic liver disease, with liver fibrosis being the most important predictor of liver failure. Due to major gaps in understanding the mechanisms of NASH fibrosis, treatment options are markedly limited. In human NASH, there are increases in both cholesterol and a fibrotic NASH-promoting protein called TAZ, but mechanisms linking cholesterol to NASH and NASH to TAZ upregulation remain unknown. Using specimens and liver cells (hepatocytes) from human and mouse NASH livers, researchers at Columbia University show that excess cholesterol in human and mouse hepatocytes increases TAZ through a calcium-mediated signaling pathway. Importantly, silencing genes in the pathway lowers TAZ, inflammation, and fibrosis in experimental NASH, suggesting new targets for therapeutic intervention.
ACKNOWLEDGEMENTS
We thank Mengmeng Zhu and Guoan Zhang from the Proteomics and Metabolomics Core Facility at Weill Cornell Medicine for conducting the mass spectrometry analysis of TAZ S117 phosphorylation; Drs. Marion Namenwirth and Abigail Knoble (University of Minnesota) and Nicole Martik (University of Pittsburgh) for arranging and providing human liver samples and primary hepatocytes, respectively; and Dr. Jay Horton (University of Texas-Southwestern) for providing hSREBP-2(1-468) plasmid. Human liver samples and primary hepatocytes were obtained from the Liver Tissue Cell Distribution System (University of Minnesota and University of Pittsburgh), which was funded by NIH contract HHSN276201200017C. Samples for histological analysis were prepared in the Molecular Pathology Shared Resource of the Herbert Irving Comprehensive Cancer Center at Columbia University, supported by NIH/NCI grant #P30 CA013696. This work was supported by an American Liver Foundation Liver Scholar Award (to X.W.); NIH grant 1K99DK115778 (to B.C.); CAS “Light of West China” Program XAB2018AW09 (to X.Y); NIH-NHLBI Postdoctoral Fellow Institutional Training Grant T32HL007343, a Fellow Scholar Award from the American Society of Hematology, and a Career Development Award 19CDA34660043 from the American Heart Association (to Z.Z.); MyFirst Grant Associazione Italiana per la Ricerca sul Cancro (AIRC) n.16888 “EPIDEMIC-NAFLD” (to L.V.); Ricerca Finalizzata Ministero della Salute RF-2016-02364358 (to L.V.); Ricerca Corrente Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, (to L.V.); the European Union (EU) Programme Horizon 2020 (under grant agreement No. 777377) for the project LITMUS-“Liver Investigation: Testing Marker Utility in Steatohepatitis” (to L.V.); NIH grant HL20948 (to A.R. and R.E.I.); Welch Foundation grant I-1793 (to A.R.); NIH grants HD088571 and AG061290 (to J.B. & L.R.L.); the Taylor Family Institute for Innovative Psychiatric Research and NIH grant HL067773 (to D.F.C.); NIH grants DK103818 and DK119767 (to U.P.); NIH grants R35CA196878 and DE015964 (to K.L.G.); NIH grant HL136618 (to P.T.), and NIH grants DK116620, HL132412, and HL087123 (to I.T.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
Drs. Buck and Levin own equity interest in CEP Biotech, which has licensed commercialization of a panel of monoclonal antibodies directed against sAC. Drs. Tabas and Wang are co-inventors of a patent application related to the topic of this study, and Dr. Tabas is a scientific consultant for Genevant, which is developing therapies for NASH.
REFERENCES
- Adams D, Gonzalez-Duarte A, O’Riordan WD, Yang CC, Ueda M, Kristen AV, Tournev I, Schmidt HH, Coelho T, Berk JL, et al. (2018). Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med 379, 11–21. [DOI] [PubMed] [Google Scholar]
- Angulo P, Kleiner DE, Dam-Larsen S, Adams LA, Bjornsson ES, Charatcharoenwitthaya P, Mills PR, Keach JC, Lafferty HD, Stahler A, et al. (2015a). Liver fibrosis, but no other histologic features, is associated with long-term outcomes of patients with nonalcoholic fatty liver disease. Gastroenterology 149, 389–397 e310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angulo P, Machado MV, and Diehl AM (2015b). Fibrosis in nonalcoholic fatty liver disease: mechanisms and clinical implications. Semin Liver Dis 35, 132–145. [DOI] [PubMed] [Google Scholar]
- Athyros VG, Alexandrides TK, Bilianou H, Cholongitas E, Doumas M, Ganotakis ES, Goudevenos J, Elisaf MS, Germanidis G, Giouleme O, et al. (2017). The use of statins alone, or in combination with pioglitazone and other drugs, for the treatment of non-alcoholic fatty liver disease/non-alcoholic steatohepatitis and related cardiovascular risk. An Expert Panel Statement. Metabolism 71, 17–32. [DOI] [PubMed] [Google Scholar]
- Aylon Y, Gershoni A, Rotkopf R, Biton IE, Porat Z, Koh AP, Sun X, Lee Y, Fiel MI, Hoshida Y, et al. (2016). The LATS2 tumor suppressor inhibits SREBP and suppresses hepatic cholesterol accumulation. Genes Dev 30, 786–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieghs V, Hendrikx T, van Gorp PJ, Verheyen F, Guichot YD, Walenbergh SM, Jeurissen ML, Gijbels M, Rensen SS, Bast A, et al. (2013). The cholesterol derivative 27-hydroxycholesterol reduces steatohepatitis in mice. Gastroenterology 144, 167–178 e161. [DOI] [PubMed] [Google Scholar]
- Bitterman JL, Ramos-Espiritu L, Diaz A, Levin LR, and Buck J (2013). Pharmacological distinction between soluble and transmembrane adenylyl cyclases. J Pharmacol Exp Ther 347, 589–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco-Colio LM, Villa A, Ortego M, Hernandez-Presa MA, Pascual A, Plaza JJ, and Egido J (2002). 3-Hydroxy-3-methyl-glutaryl coenzyme A reductase inhibitors, atorvastatin and simvastatin, induce apoptosis of vascular smooth muscle cells by downregulation of Bcl-2 expression and Rho A prenylation. Atherosclerosis 161, 17–26. [DOI] [PubMed] [Google Scholar]
- Brown MS, and Goldstein JL (1997). The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 89, 331–340. [DOI] [PubMed] [Google Scholar]
- Caballero F, Fernandez A, De Lacy AM, Fernandez-Checa JC, Caballeria J, and Garcia-Ruiz C (2009). Enhanced free cholesterol, SREBP-2 and StAR expression in human NASH. J Hepatol 50, 789–796. [DOI] [PubMed] [Google Scholar]
- Chang L, Azzolin L, Di Biagio D, Zanconato F, Battilana G, Lucon Xiccato R, Aragona M, Giulitti S, Panciera T, Gandin A, et al. (2018). The SWI/SNF complex is a mechanoregulated inhibitor of YAP and TAZ. Nature 563, 265–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Martinez J, Milner TA, Buck J, and Levin LR (2013). Neuronal expression of soluble adenylyl cyclase in the mammalian brain. Brain Res 1518, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Du S, Lu L, Lin Z, Jin W, Hu D, Jiang X, Xin Y, and Xuan S (2017). The additive effects of the TM6SF2 E167K and PNPLA3 I148M polymorphisms on lipid metabolism. Oncotarget 8, 74209–74216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Alessandro A, El Kasmi KC, Plecita-Hlavata L, Jezek P, Li M, Zhang H, Gupte SA, and Stenmark KR (2018). Hallmarks of pulmonary hypertension: mesenchymal and inflammatory cell metabolic reprogramming. Antioxid Redox Signal 28, 230–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulai PS, Singh S, Patel J, Soni M, Prokop LJ, Younossi Z, Sebastiani G, Ekstedt M, Hagstrom H, Nasr P, et al. (2017). Increased risk of mortality by fibrosis stage in nonalcoholic fatty liver disease: Systematic review and meta-analysis. Hepatology 65, 1557–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto D, Ueda Y, Hirono Y, Goi T, and Yamaguchi A (2015). PAR1 participates in the ability of multidrug resistance and tumorigenesis by controlling Hippo-YAP pathway. Oncotarget 6, 34788–34799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan LT, Van Rooyen DM, Koina ME, McCuskey RS, Teoh NC, and Farrell GC (2014). Hepatocyte free cholesterol lipotoxicity results from JNK1-mediated mitochondrial injury and is HmGb1 and TLR4-dependent. J Hepatol 61, 1376–1384. [DOI] [PubMed] [Google Scholar]
- Ghorpade DS, Ozcan L, Zheng Z, Nicoloro SM, Shen Y, Chen E, Bluher M, Czech MP, and Tabas I (2018). Hepatocyte-secreted DPP4 in obesity promotes adipose inflammation and insulin resistance. Nature 555, 673–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilardi F, Viviani B, Galmozzi A, Boraso M, Bartesaghi S, Torri A, Caruso D, Crestani M, Marinovich M, and de Fabiani E (2009). Expression of sterol 27-hydroxylase in glial cells and its regulation by liver X receptor signaling. Neuroscience 164, 530–540. [DOI] [PubMed] [Google Scholar]
- Hagenbeek TJ, Webster JD, Kljavin NM, Chang MT, Pham T, Lee H-J, Klijn C, Cai AG, Totpal K, Ravishankar B, et al. (2018). The Hippo pathway effector TAZ induces TEAD-dependent liver inflammation and tumors. Science signaling 11, eaaj1757. [DOI] [PubMed] [Google Scholar]
- Horton JD, Shimomura I, Brown MS, Hammer RE, Goldstein JL, and Shimano H (1998). Activation of cholesterol synthesis in preference to fatty acid synthesis in liver and adipose tissue of transgenic mice overproducing sterol regulatory element-binding protein-2. J Clin Invest 101, 2331–2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Lv X, Liu C, Zha Z, Zhang H, Jiang Y, Xiong Y, Lei QY, and Guan KL (2012). The N-terminal phosphodegron targets TAZ/WWTR1 protein for SCFbeta-TrCP-dependent degradation in response to phosphatidylinositol 3-kinase inhibition. J Biol Chem 287, 26245–26253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Infante RE, and Radhakrishnan A (2017). Continuous transport of a small fraction of plasma membrane cholesterol to endoplasmic reticulum regulates total cellular cholesterol. eLife 6, e25466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioannou GN (2016). The role of cholesterol in the pathogenesis of NASH. Trends Endocrinol Metab 27, 84–95. [DOI] [PubMed] [Google Scholar]
- Ioannou GN, Morrow OB, Connole ML, and Lee SP (2009). Association between dietary nutrient composition and the incidence of cirrhosis or liver cancer in the United States population. Hepatology 50, 175–184. [DOI] [PubMed] [Google Scholar]
- Kamenetsky M, Middelhaufe S, Bank EM, Levin LR, Buck J, and Steegborn C (2006). Molecular details of cAMP generation in mammalian cells: a tale of two systems. J Mol Biol 362, 623–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khajehahmadi Z, Mohagheghi S, Nikeghbalian S, Geramizadeh B, Khodadadi I, Karimi J, Ghaffari ME, and Tavilani H (2019). Downregulation of hedgehog ligands in human simple steatosis may protect against nonalcoholic steatohepatitis: Is TAZ a crucial regulator? IUBMB Life 71, 1382–1390. [DOI] [PubMed] [Google Scholar]
- Kim JY, Garcia-Carbonell R, Yamachika S, Zhao P, Dhar D, Loomba R, Kaufman RJ, Saltiel AR, and Karin M (2018). ER stress drives lipogenesis and steatohepatitis via caspase-2 asctivation of S1P. Cell 175, 133–145 e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo JH, and Guan KL (2018). Interplay between YAP/TAZ and Metabolism. Cell Metab 28, 196–206. [DOI] [PubMed] [Google Scholar]
- Lee YA, Noon LA, Akat KM, Ybanez MD, Lee TF, Berres ML, Fujiwara N, Goossens N, Chou HI, Parvin-Nejad FP, et al. (2018). Autophagy is a gatekeeper of hepatic differentiation and carcinogenesis by controlling the degradation of Yap. Nat Commun 9, 4962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefkimmiatis K (2014). cAMP signalling meets mitochondrial compartments. Biochem Soc Trans 42, 265–269. [DOI] [PubMed] [Google Scholar]
- Lei QY, Zhang H, Zhao B, Zha ZY, Bai F, Pei XH, Zhao S, Xiong Y, and Guan KL (2008). TAZ promotes cell proliferation and epithelial-mesenchymal transition and is inhibited by the hippo pathway. Mol Cell Biol 28, 2426–2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liscum L, and Dahl NK (1992). Intracellular cholesterol transport. J Lipid Res 33, 1239–1254. [PubMed] [Google Scholar]
- Litvin TN, Kamenetsky M, Zarifyan A, Buck J, and Levin LR (2003). Kinetic properties of “soluble” adenylyl cyclase. Synergism between calcium and bicarbonate. J Biol Chem 278, 15922–15926. [DOI] [PubMed] [Google Scholar]
- Liu CY, Zha ZY, Zhou X, Zhang H, Huang W, Zhao D, Li T, Chan SW, Lim CJ, Hong W, et al. (2010). The hippo tumor pathway promotes TAZ degradation by phosphorylating a phosphodegron and recruiting the SCF{beta}-TrCP E3 ligase. J Biol Chem 285, 37159–37169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Wei Z, Ma X, Yang X, Chen Y, Sun L, Ma C, Miao QR, Hajjar DP, Han J, et al. (2018). 25-Hydroxycholesterol activates the expression of cholesterol 25-hydroxylase in an LXR-dependent mechanism. J Lipid Res 59, 439–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marikawa Y, and Elinson RP (1998). beta-TrCP is a negative regulator of Wnt/beta-catenin signaling pathway and dorsal axis formation in Xenopus embryos. Mech Dev 77, 75–80. [DOI] [PubMed] [Google Scholar]
- Masiero L, Lapidos KA, Ambudkar I, and Kohn EC (1999). Regulation of the RhoA pathway in human endothelial cell spreading on type IV collagen: role of calcium influx. J Cell Sci 112 ( Pt 19), 3205–3213. [DOI] [PubMed] [Google Scholar]
- McGettigan B, McMahan R, Orlicky D, Burchill M, Danhorn T, Francis P, Cheng LL, Golden-Mason L, Jakubzick CV, and Rosen HR (2019). Dietary lipids differentially shape nonalcoholic steatohepatitis progression and the transcriptome of kupffer cells and infiltrating macrophages. Hepatology 70, 67–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min HK, Kapoor A, Fuchs M, Mirshahi F, Zhou H, Maher J, Kellum J, Warnick R, Contos MJ, and Sanyal AJ (2012). Increased hepatic synthesis and dysregulation of cholesterol metabolism is associated with the severity of nonalcoholic fatty liver disease. Cell Metab 15, 665–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mooring M, Fowl BH, Lum SZC, Liu Y, Yao K, Softic S, Kirchner R, Bernstein A, Singhi AD, Jay DG, et al. (2019). Hepatocyte Stress Increases Expression of Yes-Associated Protein and Transcriptional Coactivator With PDZ-Binding Motif in Hepatocytes to Promote Parenchymal Inflammation and Fibrosis. Hepatology (Baltimore, Md.), 10.1002/hep.30928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair JK, Willoughby JL, Chan A, Charisse K, Alam MR, Wang Q, Hoekstra M, Kandasamy P, Kel’in AV, Milstein S, et al. (2014). Multivalent N-acetylgalactosamine-conjugated siRNA localizes in hepatocytes and elicits robust RNAi-mediated gene silencing. J Am Chem Soc 136, 16958–16961. [DOI] [PubMed] [Google Scholar]
- Ohgushi M, Minaguchi M, and Sasai Y (2015). Rho-signaling-directed YAP/TAZ activity underlies the long-term survival and expansion of human embryonic stem cells. Cell Stem Cell 17, 448–461. [DOI] [PubMed] [Google Scholar]
- Ozcan L, Wong CC, Li G, Xu T, Pajvani U, Park SK, Wronska A, Chen BX, Marks AR, Fukamizu A, et al. (2012). Calcium signaling through CaMKII regulates hepatic glucose production in fasting and obesity. Cell Metab 15, 739–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puche JE, Saiman Y, and Friedman SL (2013). Hepatic stellate cells and liver fibrosis. Compr Physiol 3, 1473–1492. [DOI] [PubMed] [Google Scholar]
- Puri P, Baillie RA, Wiest MM, Mirshahi F, Choudhury J, Cheung O, Sargeant C, Contos MJ, and Sanyal AJ (2007). A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology 46, 1081–1090. [DOI] [PubMed] [Google Scholar]
- Ramos-Espiritu L, Kleinboelting S, Navarrete FA, Alvau A, Visconti PE, Valsecchi F, Starkov A, Manfredi G, Buck H, Adura C, et al. 2016. Discovery of LRE1 as a specific and allosteric inhibitor of soluble adenylyl cyclase. Nat Chem Biol 12, 838–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich TC, Fagan KA, Nakata H, Schaack J, Cooper DM, and Karpen JW (2000). Cyclic nucleotide-gated channels colocalize with adenylyl cyclase in regions of restricted cAMP diffusion. J Gen Physiol 116, 147–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandhu J, Li S, Fairall L, Pfisterer SG, Gurnett JE, Xiao X, Weston TA, Vashi D, Ferrari A, Orozco JL, et al. (2018). Aster proteins facilitate nonvesicular plasma membrane to ER cholesterol transport in mammalian cells. Cell 175, 514–529 e520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savard C, Tartaglione EV, Kuver R, Haigh WG, Farrell GC, Subramanian S, Chait A, Yeh MM, Quinn LS, and Ioannou GN (2013). Synergistic interaction of dietary cholesterol and dietary fat in inducing experimental steatohepatitis. Hepatology 57, 81–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz M, Davis DL, Vick BR, and Russell DW (2001). Genetic analysis of intestinal cholesterol absorption in inbred mice. J Lipid Res 42, 1801–1811. [PubMed] [Google Scholar]
- Teratani T, Tomita K, Suzuki T, Oshikawa T, Yokoyama H, Shimamura K, Tominaga S, Hiroi S, Irie R, Okada Y, et al. (2012). A high-cholesterol diet exacerbates liver fibrosis in mice via accumulation of free cholesterol in hepatic stellate cells. Gastroenterology 142, 152–164. [DOI] [PubMed] [Google Scholar]
- Tresguerres M, Levin LR, and Buck J (2011). Intracellular cAMP signaling by soluble adenylyl cyclase. Kidney Int 79, 1277–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valsecchi F, Ramos-Espiritu LS, Buck J, Levin LR, and Manfredi G (2013). cAMP and mitochondria. Physiology (Bethesda) 28, 199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Rooyen DM, Larter CZ, Haigh WG, Yeh MM, Ioannou G, Kuver R, Lee SP, Teoh NC, and Farrell GC (2011). Hepatic free cholesterol accumulates in obese, diabetic mice and causes nonalcoholic steatohepatitis. Gastroenterology 141, 1393–1403, 1403 e1391-1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilar-Gomez E, Calzadilla-Bertot L, Wai-Sun Wong V, Castellanos M, Aller-de la Fuente R, Metwally M, Eslam M, Gonzalez-Fabian L, Alvarez-Quinones Sanz M, Conde-Martin AF, et al. (2018). Fibrosis severity as a determinant of cause-specific mortality in patients with advanced nonalcoholic fatty liver disease: a multi-national cohort study. Gastroenterology 155, 443–457 e417. [DOI] [PubMed] [Google Scholar]
- Wada K, Itoga K, Okano T, Yonemura S, and Sasaki H (2011). Hippo pathway regulation by cell morphology and stress fibers. Development 138, 3907–3914. [DOI] [PubMed] [Google Scholar]
- Wang X, Sommerfeld MR, Jahn-Hofmann K, Cai B, Filliol A, Remotti HE, Schwabe RF, Kannt A, and Tabas I (2019). A Therapeutic Silencing RNA Targeting Hepatocyte TAZ Prevents and Reverses Fibrosis in Nonalcoholic Steatohepatitis in Mice. Hepatol Commun 3, 1221–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zheng Z, Caviglia JM, Corey KE, Herfel TM, Cai B, Masia R, Chung RT, Lefkowitch JH, Schwabe RF, et al. (2016). Hepatocyte TAZ/WWTR1 promotes inflammation and fibrosis in nonalcoholic steatohepatitis. Cell Metab 24, 848–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Li G, Goode J, Paz JC, Ouyang K, Screaton R, Fischer WH, Chen J, Tabas I, and Montminy M (2012). Inositol-1,4,5-trisphosphate receptor regulates hepatic gluconeogenesis in fasting and diabetes. Nature 485, 128–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Subramanian M, Yurdagul A Jr., Barbosa-Lorenzi VC, Cai B, de Juan-Sanz J, Ryan TA, Nomura M, Maxfield FR, and Tabas I (2017). Mitochondrial fission promotes the continued clearance of apoptotic cells by macrophages. Cell 171, 331–345 e322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westover EJ, Covey DF, Brockman HL, Brown RE, and Pike LJ (2003). Cholesterol depletion results in site-specific increases in epidermal growth factor receptor phosphorylation due to membrane level effects. Studies with cholesterol enantiomers. J Biol Chem 278, 51125–51133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White SM, Avantaggiati ML, Nemazanyy I, Di Poto C, Yang Y, Pende M, Gibney GT, Ressom HW, Field J, Atkins MB, et al. (2019). YAP/TAZ inhibition induces metabolic and signaling rewiring resulting in targetable vulnerabilities in NF2-deficient tumor cells. Dev Cell 49, 425–443 e429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieckowski MR, Giorgi C, Lebiedzinska M, Duszynski J, and Pinton P (2009). Isolation of mitochondria-associated membranes and mitochondria from animal tissues and cells. Nat Protoc 4, 1582–1590. [DOI] [PubMed] [Google Scholar]
- Wouters K, van Gorp PJ, Bieghs V, Gijbels MJ, Duimel H, Lutjohann D, Kerksiek A, van Kruchten R, Maeda N, Staels B, et al. (2008). Dietary cholesterol, rather than liver steatosis, leads to hepatic inflammation in hyperlipidemic mouse models of nonalcoholic steatohepatitis. Hepatology 48, 474–486. [DOI] [PubMed] [Google Scholar]
- Xin M, Kim Y, Sutherland LB, Murakami M, Qi X, McAnally J, Porrello ER, Mahmoud AI, Tan W, Shelton JM, et al. (2013). Hippo pathway effector Yap promotes cardiac regeneration. Proc Natl Acad Sci U S A 110, 13839–13844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaccolo M, and Pozzan T (2002). Discrete microdomains with high concentration of cAMP in stimulated rat neonatal cardiac myocytes. Science 295, 1711–1715. [DOI] [PubMed] [Google Scholar]
- Zhao B, Li L, Tumaneng K, Wang CY, and Guan KL (2010). A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(beta-TRCP). Genes Dev 24, 72–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Li L, Wang L, Wang CY, Yu J, and Guan KL (2012). Cell detachment activates the Hippo pathway via cytoskeleton reorganization to induce anoikis. Genes Dev 26, 54–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Ye X, Yu J, Li L, Li W, Li S, Yu J, Lin JD, Wang CY, Chinnaiyan AM, et al. (2008). TEAD mediates YAP-dependent gene induction and growth control. Genes Dev 22, 1962–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, and Pan D (2019). The Hippo Signaling Pathway in Development and Disease. Dev Cell 50, 264–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Z, Nayak L, Wang W, Yurdagul A Jr., Wang X, Cai B, Lapping S, Ozcan L, Ramakrishnan R, Pestell RG, et al. (2019). An ATF6-tPA pathway in hepatocytes contributes to systemic fibrinolysis and is repressed by DACH1. Blood 133, 743–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Jimi S, Smith TL, and Kummerow FA (1991). The effect of cholesterol on the accumulation of intracellular calcium. Biochim Biophys Acta 1085, 1–6. [DOI] [PubMed] [Google Scholar]
- Zhu C, Kim K, Wang X, Bartolome A, Salomao M, Dongiovanni P, Meroni M, Graham MJ, Yates KP, Diehl AM, et al. (2018). Hepatocyte Notch activation induces liver fibrosis in nonalcoholic steatohepatitis. Science translational medicine 10, eaat0344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zippin JH, Chen Y, Nahirney P, Kamenetsky M, Wuttke MS, Fischman DA, Levin LR, and Buck J (2003). Compartmentalization of bicarbonate-sensitive adenylyl cyclase in distinct signaling microdomains. FASEB J 17, 82–84. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique datasets or code.