

Abstract
Polycyclic aromatic hydrocarbons (PAHs) are environmental toxicants primarily produced during incomplete combustion; some are carcinogens. PAHs can be safely metabolized or, paradoxically, bioactivated via specific cytochrome P450 (CYP) enzymes to more reactive metabolites, some of which can damage DNA and proteins. Among the CYP isoforms implicated in PAH metabolism, CYP1A enzymes have been reported to both sensitize and protect from PAH toxicity. To clarify the role of CYP1A in PAH toxicity, we generated transgenic Caenorhabditis elegans that express CYP1A at a basal (but not inducible) level. Because this species does not normally express any CYP1 family enzyme, this approach permitted a test of the role of basally expressed CYP1A in PAH toxicity. We exposed C. elegans at different life stages to either the PAH benzo[a]pyrene (BaP) alone, or a real-world mixture dominated by PAHs extracted from the sediment of a highly-contaminated site on the Elizabeth River (VA, USA). This site, the former Atlantic Wood Industries, was declared a Superfund site due to coal tar creosote contamination that caused very high levels (in the [mg/mL] range) of high molecular weight PAHs within the sediments. We demonstrate that CYP1A protects against BaP-induced growth delay, reproductive toxicity, and reduction of steady state ATP levels. Lack of sensitivity of a DNA repair (Nucleotide Excision Repair)-deficient strain suggested that CYP1A did not produce significant levels of DNA-reactive metabolites from BaP. The protective effects of CYP1A in Elizabeth River sediment extract (ERSE)-exposed nematodes were less pronounced than those seen in BaP-exposed nematodes; CYP1A expression protected against ERSE-induced reduction of steady-state ATP levels, but not other outcomes of exposure to sediment extracts. Overall, we find that in C. elegans, a basal level of CYP1A activity is protective against the examined PAH exposures.
Keywords: Polycyclic aromatic hydrocarbons, cytochrome P450s, carcinogenic metabolites, DNA damage, benzo[a]pyrene, Nucleotide Excision Repair
1. Introduction
PAHs are organic molecules with two or more fused benzene rings and no heteroatoms; they vary in structure and molecular weight (Moorthy, et al. 2015). PAHs are ubiquitous environmental contaminants, as they are formed through incomplete combustion of organic material (Richter and Howard 2000). The toxic, mutagenic, and carcinogenic effects of PAHs are fairly well characterized, although different PAHs have very different toxicities (Abdel-Shafy and Mansour 2016). Environmental conditions, such as biological transformation, can influence PAH toxicity because PAH metabolites are often more toxic than the parent molecules (Fallahtafti, et al. 2012).
PAHs are often studied by toxicologists in isolation as individual, pure compounds. However, PAHs invariably exist in the environment as complex mixtures (e.g., tobacco smoke and diesel exhaust). One example of a very complex mixture of high molecular weight PAHs is coal tar creosote, used in the industrial process of wood preservation (Di Giulio and Clark 2015; Hale and Aneiro 1997). The southern branch of the Elizabeth River in southeastern Virginia had large volume creosote spills (20–30,000 gallons) during the 20th century from a wood treatment facility operated by the Atlantic Wood Industries (Di Giulio and Clark 2015; Riley, et al. 2016). The PAH levels in this area remain dangerously elevated due to the long half-lives of the compounds. The risk to human and environmental health caused the site to be declared a Superfund site in the 1990s.
When humans are exposed to PAHs, a major phase I metabolic pathway for biotrasnformation involves the mono-oxygenation of the molecule to form hydroxyl groups and epoxides by CYPs 1A1, 1A2, and 1B1 (Straif, et al. 2005). Epoxides undergo epoxide hydrolysis spontaneously or via catalysis by the enzyme epoxide hydrolase, which cleaves the epoxide to create a diol. The molecule then can be conjugated by phase II enzymes including glutathione S transferases, UDP-glucuronosyltransferases, and sulphotransferases for excretion (Straif, et al. 2005). However, if instead a CYP further epoxidizes the diol, a carcinogenic diol epoxide metabolite can be created that is resistant to epoxide hydrolase. Resistance of the diol epoxide to epoxide hydrolase is dependent on the location of the epoxide on the molecule, the shape of the molecule, and the stereochemistry of the epoxide (Xue and Warshawsky 2005). The hydrolysis-resistant epoxide and other reactive metabolites can damage DNA and proteins, ultimately causing tumors in animals (Fallahtafti, et al. 2012; Moorthy, et al. 2015; Shimada, et al. 2001; Straif, et al. 2005). Thus, an important question is under what conditions CYP metabolism has protective versus damaging effects on an organism: P450s both have the capacity to facilitate elimination of PAHs, and to create carcinogenic metabolites (Fallahtafti, et al. 2012; Moorthy, et al. 2015; Shimada, et al. 2001). The answer is likely to depend not only on the specific CYP isoform, but also on the chemical characteristics of the respective PAH. The specific CYP that we tested in this study, zebrafish CYP1A, is very active in metabolizing PAHs compared to other CYP1 family enzymes, and produces a significant amount of 7,8-diol-BaP in vitro (Scornaienchi, et al. 2010b). This metabolite is an important intermediate in the pathway to the genotoxic diol epoxide, supporting the potential for toxic bioactivation of PAHs by CYP1A.
Mitochondria may be particularly sensitive to the toxicity of PAHs and/or their metabolites due at least in part to the mitochondria’s phospholipid bilayer that attracts lipophilic toxicants such as PAHs (Meyer, et al. 2018). Furthermore, mitochondria lack the nucleotide excision repair (NER) pathway, which is the pathway responsible for removing nuclear DNA adducts caused by PAH metabolites. Instead, mitochondrial genomes with such damage can be removed or replaced through fusion, fission, autophagy, and biogenesis (Bess, et al. 2012), but this process is significantly slower (Meyer, et al. 2018). Mitochondrial genomes are reported to be 2–100 times more susceptible to formation of bulky adducts than nuclear DNA (Meyer, et al. 2013).
In this study, we employed transgenic Caenorhabditis elegans nematodes expressing zebrafish CYP1A in order to test the hypothesis that CYP1A increases DNA damage caused by benzo[a]pyrene (BaP; a commonly studied “model” PAH whose metabolic activation to a DNA-reactive diol epoxide is well-characterized) and a mixture of high-molecular weight PAHs found in Elizabeth River sediment extract (ERSE; note that while this mixture does contain chemicals other than PAHs, there is previous evidence tht the very high concentrations of PAHs present in ERSE dominate the toxicity of this mixture: Fang et al., 2014). C. elegans is a particularly useful model species for this purpose because it does not naturally have family 1 P450s implicated in metabolism of high molecular weight PAHs (Harlow, et al. 2018; Leung, et al. 2010), but they can be easily genetically engineered to express the protein. After exposing wild-type (CYP1A-null) and transgenic nematodes expressing CYP1A at a basal level to BaP and ERSE, we assessed overall toxicity through larval growth and reproduction assays, and mitochondrial toxicity by measuring steady-state ATP levels. Overall, the results of this study demonstrate a protective role of basal expression of CYP1A in PAH toxicity.
2. Methods
2.1. C. elegans and culture conditions
The N2 Bristol wild-type strain and PE255 (feIs5 [sur-5p::luciferase::GFP + rol-6(su1006)]) strain were obtained from the Caenorhabditis Genetics Center (CGC). The CYP1A-expressing strain COP476 (knuIs32 [pNU193 (eft-3p::zCYP1a::tbb2utr myo2::eGFP, unc-119(+))]; unc-119(ed3) III) was generated by Knudra Transgenics (NemaMetrix, Eugene, OR). The xpa-1 mutant strain (RB864) was obtained from the CGC and out-crossed three times, as previously described (Meyer, et al. 2007). CYP1A-expressing worms from the COP476 strain were crossed into the xpa-1 and PE255 backgrounds using standard protocols (Lewis and Fleming 1995). Briefly, CYP1A-expressing males were enriched by exposing a population of 50 L4 hermaphrodites to a heat shock (5 hours at 30 degrees C). Following the heat shock, the progeny from heat shocked hermaphrodites contained an increased number of males that could be used to set up crosses. A high number of males were obtained by first setting up a cross of three CYP1A-expressing males with one L4-stage CYP1A hermaphrodite (to produce 50% male progeny). A similar approach was used to cross into the xpa-1-deficient and PE255 backgrounds: three CYP1A-expressing males were placed on a mating plate (6 cm plate seeded with a single 20 μL dot of bacteria) with one L4 hermaphrodite from the desired genetic background. Progeny were singled and homozygous strains were identified by PCR genotyping (for xpa-1 and CYP1A expression) or by visual fluorescence screening (for luciferase::GFP expression). Genotyping primers for CYP1A were: forward, 5’-ATCTTCCGTCACTCCTCCTT-3’; reverse, 5’-TGACTTGCCATTGGTTGAC-3’; annealing temperature 58 °C; product size 172 bp). Genotyping primers for xpa-1 deletion were: forward, 5’-GATAGCCGGAATAGCTGGC-3’; reverse, 5’-CTGGAGCCAATCCAACTGAT-3’; annealing temperature 59 °C; product size 2133 bp (WT) or 1220 bp (xpa-1 deletion).
Synchronized larval stage (L1) nematodes were obtained through sodium hydroxide bleach treatment and overnight starvation in complete K-medium (150 μl 1 M CaCl2, 150 μl 1 M MgSO4, 25 μl 10 mg/ml cholesterol, 50 ml sterile K medium (2.36 g KCl, 3 g NaCl, 1L ddH2O)) at 20 °C. Synchronized populations of nematodes were maintained on K-agar plates seeded with Escherichia coli OP50 at 20°C. For ATP determination, nematodes were treated with 0.6 mM fluorodeoxyuridine (FUDR) during liquid exposure to prevent reproduction, because reproduction significantly alters ATP levels. FUDR has been shown to cause minimal mitochondrial effects on C. elegans (Rooney, et al. 2014).
2.2. Assessing temperature dependence of recombinant zebrafish Cyp1a activity
The Cyp1a gene amplification, expression construction, and initial characterization of the expressed protein have been previously described (Scornaienchi, et al. 2010a; Scornaienchi, et al. 2010b). Briefly, the zebrafish Cyp1a gene was amplified from wild type zebrafish liver cDNA. The ompA(+2) signal sequence (69 nucleotides in length) was attached in frame to the CYP start site by PCR; PCR primers contained NdeI (forward) and XbaI (reverse) restriction sites. The PCR fragments with complete CYP gene and ompA(2+) sequence were digested, gel purified, and ligated into the pCW vector. The catalytic capacity of this construct, co-expressed with human reductase in bacteria, has been demonstrated for estradiol metabolism (Scornaienchi, et al. 2010a), BaP, and 11 fluorogenic substrates (Scornaienchi, et al. 2010b). Bacterially expressed Cyp1a was assessed for ethoxyresorfin-O-deethylase (EROD) activity using 50 mM Tric, 0.1 M NaCl, pH 7.8 as the buffer. Reactions were initiated with 1.3 nM NADPH and contained 2 uM 7-ethoxyresorufin and 9.3 nmol total P450. Product formation was read on a Synergy 2 fluorimeter with an excitation and emission (/bandwidth) of 540/35 and 590/20 nm, respectively. Triplicate wells were run at 20, 25 and 30°C with and without protein to assess the temperature dependence of the reactions.
2.3. Microsomes preparation and EROD activity assay
To test activity of zebrafish Cyp1a expressed in C. elegans, referred to hereafter as CYP1A (following the C. elegans protein naming convention)(Tuli 2018), microsomes were isolated from whole worms using standard differential centrifugation methods (Meyer, et al. 2002). Briefly, worms were harvested from K-agar culture plates by washing with K-medium into a centrifuge tube. The worm suspension was centrifuged for 5 min at 500 rcf (relative centrifugal force) to pellet worms, and the volume of worms was estimated and adjusted to <300 μL if necessary. An equal volume of zirconium oxide beads was then added to the tube. Homogenization Buffer (35 mM Tris, pH 7.5; 150 mM KCl; 2 mM EDTA; 0.5 mM DTT; 250 mM sucrose and Roche complete protease inhibitor cocktail) was then added to the tube (twice the volume of the worms), and the tube was placed in the Bullet Blender in an environmental room kept at 4°C. The samples were homogenized for three minutes at speed eight. Following, the samples were inspected under a microscope. If the homogenization was incomplete, the sample was homogenized for an additional two minutes at Bullet Blender speed eight and inspected again. After homogenization was complete, the supernatant was removed from the beads and transferred to new tubes, then centrifuged at 3000 rcf for five minutes. The supernatant from the 3,000 rcf centrifugation was then transferred to a new tube and centrifuged at 10,000 rcf for ten minutes. Finally, the supernatant from the 10,000 rcf centrifugation was placed in thick-wall polycarbonate ultracentrifuge tubes for 60 minutes at 100,000 rcf. The supernatant was then removed and the microsomal pellet was resuspended in 100 μL Microsomes Storage Buffer (20% glycerol; 0.5 mM DTT; 0.25 mM PMSF; 4.5 mM EDTA; 30 mM Tris, pH 7.5 and Roche Complete protease inhibitor cocktail). The protein content was assayed using a BCA assay (Thermo-Fisher Pierce) and stored at −80 degrees C until use. For EROD assays, experiments were carried out in 96-well plates as previously described (Meyer, et al. 2002). Assay wells contained 0.5 mg/mL microsomal protein, 0.1 mM NADPH, 0.12 mM NADH, 5 mM MgSO4, and 0.1M HEPES (pH 8) in a final volume of 200 μL. Reactions were initiated upon the addition of ethoxyresorufin (final concentration 1.25 mM). Blank wells contained all reagents except microsomes. Fluorescence (535 ex./595em.) was measured using a BMG LabTech Fluostar Optima Microplate Reader plate reader at 20°C for 1.5 hours; in all cases, rates were calculated only using the linear portion of the rate curve. Standards and blanks contained 50 μg bovine serum albumin (BSA). Activities were calculated as pmol of resorfun produced per min per mg protein.
2.4. Analysis of mRNA expression of CYP1A construct
mRNA was extracted by washing and centrifuging ~5,000 worms, freezing them as a pellet in liquid nitrogen, grinding in liquid nitrogen, and extracting using an RNeasy kit (Qiagen, Valencia, CA, USA, as described in detail by Leung et al., 2013. RNA was quantified with a NanoDrop 8000 spectrophotometer (Thermo Scientific/NanoDrop, Wilmington, DE, USA). Expression was measured by reverse transcription followed by quantitative real-time PCR. 250 ng of mRNA was converted to cDNA using the Qiagen Omniscript Reverse Transcription kit. Real time PCR was carried out with a 7300 Real Time PCR System; primer sequences were: forward, 5’-ATCTTCCGTCACTCCTCCTT-3’; reverse, 5’-TGACTTGCCATTGGTTGAC-3’; annealing temperature 58 °C; product size 172 bp). Three separately isolated batches per strain were tested, with triplicate technical RT-PCR reactions per sample.
2.5. BaP and ERSE exposures
Prior to each experiment, liquid BaP exposures were prepared by diluting a 10mM stock solution of BaP dissolved in DMSO according to the desired dosing concentration. The final dosing solutions were 1% DMSO:BaP mixture diluted further in complete K medium. A dosing solution containing 1% DMSO does not have toxic effects on nematodes (Leung, et al. 2013a). For toxic plate preparation for the brood size assay, a BaP:DMSO mixture was added to autoclaved liquid agar such that the total concentration of the liquid agar was 1% DMSO. The Elizabeth River sediment extract was collected, processed, and prepared as described by Fang et al., 2014. Liquid ERSE exposures were prepared by diluting ERSE with a 6ppt (parts per thousand) salt solution made from diluting commercial sea salts (60 mg/L; Instant Ocean, Foster & Smith, Rhinelander, WI, USA) to a concentration of 6 ppt, which is consistent with the salinity content of ERSE (Nishad Jayasundara and Richard Di Giulio, personal communication) and maintains a tolerable level of salinity for the nematodes. The final dosing solutions were 40% 6 ppt salt solution:ERSE mixture that was diluted further with complete K-medium. For toxic plate preparation for the brood size assay, a ERSE:salt solution mixture was added to autoclaved liquid agar such that the total concentration of the liquid agar was 20% salt solution.
2.6. ATP determination
Relative steady-state ATP levels were determined using the PE255 strain in wild-type and CYP1A-expressing backgrounds (Lagido, et al. 2008). Briefly, 50 exposed L4 larvae suspended in K-medium were loaded into each well of a 96-well plate (four wells per strain per treatment), and baseline luminescence and GFP fluorescence were measured (emissions filter: 502 nm; excitation filter: 485 nm) using a BMG LabTech Fluostar Optima Microplate Reader. Fifty microliters of luminescence buffer (140mM Na2PO4, 30mM citric acid (pH 6.5), 1% DMSO, 0.05% Triton X-100, 100μM D-luciferin) was then injected into each well and nematode bioluminescence was measured 3 minutes later. An additional 50 nematodes were plated onto 2x OP50 400 mM FUDR plates and allowed to recover for 24 hours post-exposure before ATP determination. Four technical replicates were run in individual wells and the experiment was repeated three times.
2.7. Larval Growth
Larval growth was measured by assessing size at different times post-plating of eggs, using a COPAS Biosort (Union Biometrica Inc., Somerville, MA), largely as previously described (Boyd, et al. 2012; Maurer, et al. 2015). Fifty L1 nematodes suspended in K-medium were loaded into each well of a 96-well plate (4 wells per strain per treatment). The volume of each well was brought to 150 μl with complete K-medium, UVC-killed UVRA bacteria that arrest after UVC exposure due to absence of DNA repair (Maurer, et al. 2015), and toxicant. Nematodes were allowed to develop at 20 °C for either 48 or 72 hours, and larval growth was determined using the COPAS Biosort. All time of flight data (TOF), a measure of nematode length, was normalized to percent control for each strain prior to statistical analysis to account for differences in strain growth rate. All experiments were repeated 3 times. The same larval growth method was used on nucleotide excision repair deficient nematodes to assess BaP and ERSE-induced growth delay.
2.8. Brood Size Assay
For brood size experiments, synchronized L1 populations of nematodes were obtained through sodium hydroxide bleach treatment and plating of eggs onto K-agar plates seeded with E. coli OP50 at 20 °C. Once nematodes reached L4 stage, nematodes were singled onto toxic plates of K-agar supplemented with BaP or ERSE at specific concentrations. During the first four days of reproduction, nematodes were transferred every 24 hours to fresh plates. Offspring were allowed to hatch and counted 48 hours after the worm was placed there. The brood size was determined to be the sum of all offspring from a single worm. Six technical replicates were run on a seperate set of plates and the experiment was run twice.
2.9. Statistical Analysis
ATP levels, brood size, and larval growth were analyzed using a two-way ANOVA with Sidak’s multiple comparison or unpaired t-test post-hoc analyses to compare wild-type to Cyp1a+/+ nematodes at each exposure level of PAH or ERSE. Standard error of the mean for all data was computed and reported. Full statistical results are reported in Supplemental Tables S1–S4. EROD activities were compared using Student’s t-test. Statistics were performed using GraphPad Prism 7 software (La Jolla, CA).
3. Results
3.1. Ectopic expression of zebrafish Cyp1a with ubiquitous eet-1A promoter results in a functional nematode CYP1A protein.
In order to determine the role of CYP1A in PAH toxicity, we constructed transgenic C. elegans that express zebrafish Cyp1a ectopically. C. elegans is a useful model organism for this application because although they have many CYP genes (83: Leung et al., 2010, compared to 57 in humans) they have low protein expression of P450 in general (unpublished observations, JHH), and lack homologs for CYP1A or other CYP1 family enzymes (Leung et al., 2010) as well as significant levels of apparent enzymatic CYP1A-like activity(Harlow, et al. 2018). We used zebrafish Cyp1a because their natural body temperature is closer to worms than those of mammals, and we demonstrated that purified, bacterially-expressed zebrafish Cyp1a is highly active at the typical worm cultivation temperature of 20°C based on robust EROD activity (Fig. 1A).
Figure 1: Transgenic C. elegans expressing zebrafish CYP1A were created by microinjection.
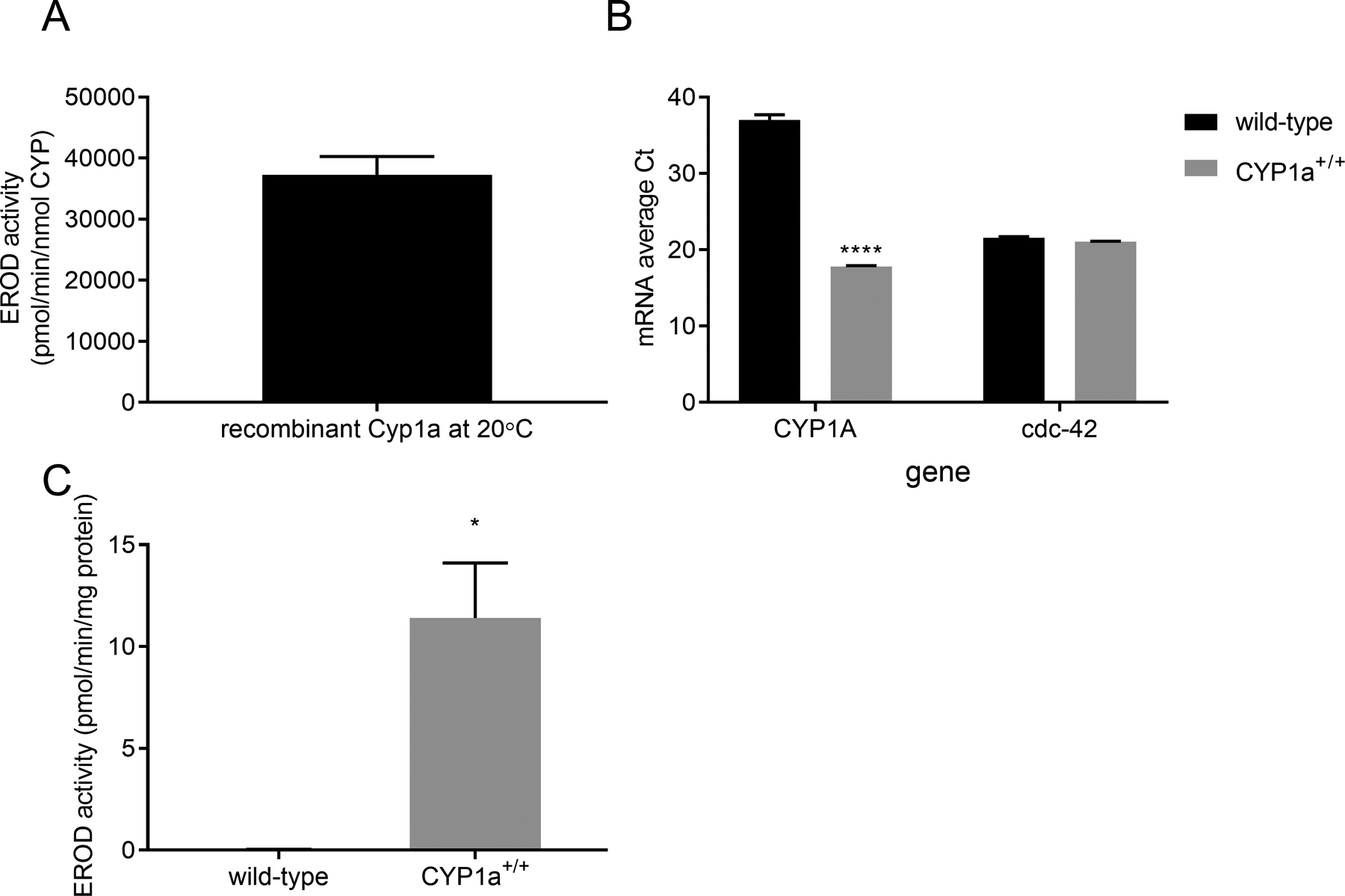
Bacterially-expressed zebrafish CYP1A shows strong EROD activity at 20° C (panel A). Transgenic worms express CYP1A mRNA as measured through mRNA average cycle threshold (Ct), while wild-type animals have a similar Ct to NTC (>36) despite similar expression of a housekeeping gene, cdc-42 (panel B). Transgenic worms also have robust CYP1A activity in isolated microsomes (panel C); microsomal EROD is undetectable in wild-type worm microsomes (panel C). Statistical significance was assessed by unpaired t-test. Asterisks represent statistical significance: *p<0.05, ****p<0.0001.
For worm expression, we inserted multiple copies of the gene under the control of a promoter (eet-2A, formerly named eft-3) that allows for ubiquitous expression in all somatic cells (Dickinson, et al. 2013). We found that the zebrafish Cyp1a transgene transcribed at high levels in transgenic animals based on mRNA content (Fig. 1B). No qPCR amplification could be detected from wild-type RNA until very high (35+) cycle number (comparable to no-template controls), and examination of product by gel electrophoresis indicated nonspecific amplification (not shown). We saw no difference in expression of cdc-42, a common housekeeping gene for worm gene expression(Hoogewijs, et al. 2008), indicating similar RNA extraction and cDNA synthesis for transgenic and wild-type worms. We also observed that the ectopically expressed Cyp1a was functional with robust EROD activity (Fig. 1C). This level of activity is comparable to the base EROD activity observed in whole-body homogenates of juvenile zebrafish (Pauka, et al. 2011). Notably, there is no detectable EROD activity in wild-type animals, allowing for a background-free negative control for CYP1A activity.
3.2. Expression of CYP1A protects against growth delay at low doses of ERSE and all doses of BaP, and against BaP-induced reproductive toxicity.
As a measure of organismal PAH toxicity, we measured larval growth of CYP1A-expressing and wild-type control worms after a 48-hour exposure to either BaP or the ERSE mixture beginning at the first larval stage (L1) (Fig. 2A). This time period encompasses development to young adulthood under control conditions, thus potentially capturing inhibition of many different developmental processes. As determined by the COPAS Biosort instrument, time of flight (TOF), a measure of nematode length, was normalized to the size of the control condition nematodes to account for any experiment- or strain-specific differences in size in controls, although strain differences between N2 and COP476 were not observed (data not shown). Both wild-type and CYP1A-expressing nematodes exhibited a dose-dependent delay in growth compared to the vehicle control. However, the CYP1A-expressing nematodes dosed with 1 and 10 μM BaP were protected against growth delay when compared to wild-type controls (Fig. 2B). CYP1A expression did not provide statistically significant protection compared to wild-type worms after exposure to the PAH mixture ERSE (Fig. 2C).
Figure 2: CYP1A protects against BaP and ERSE-induced growth delay.
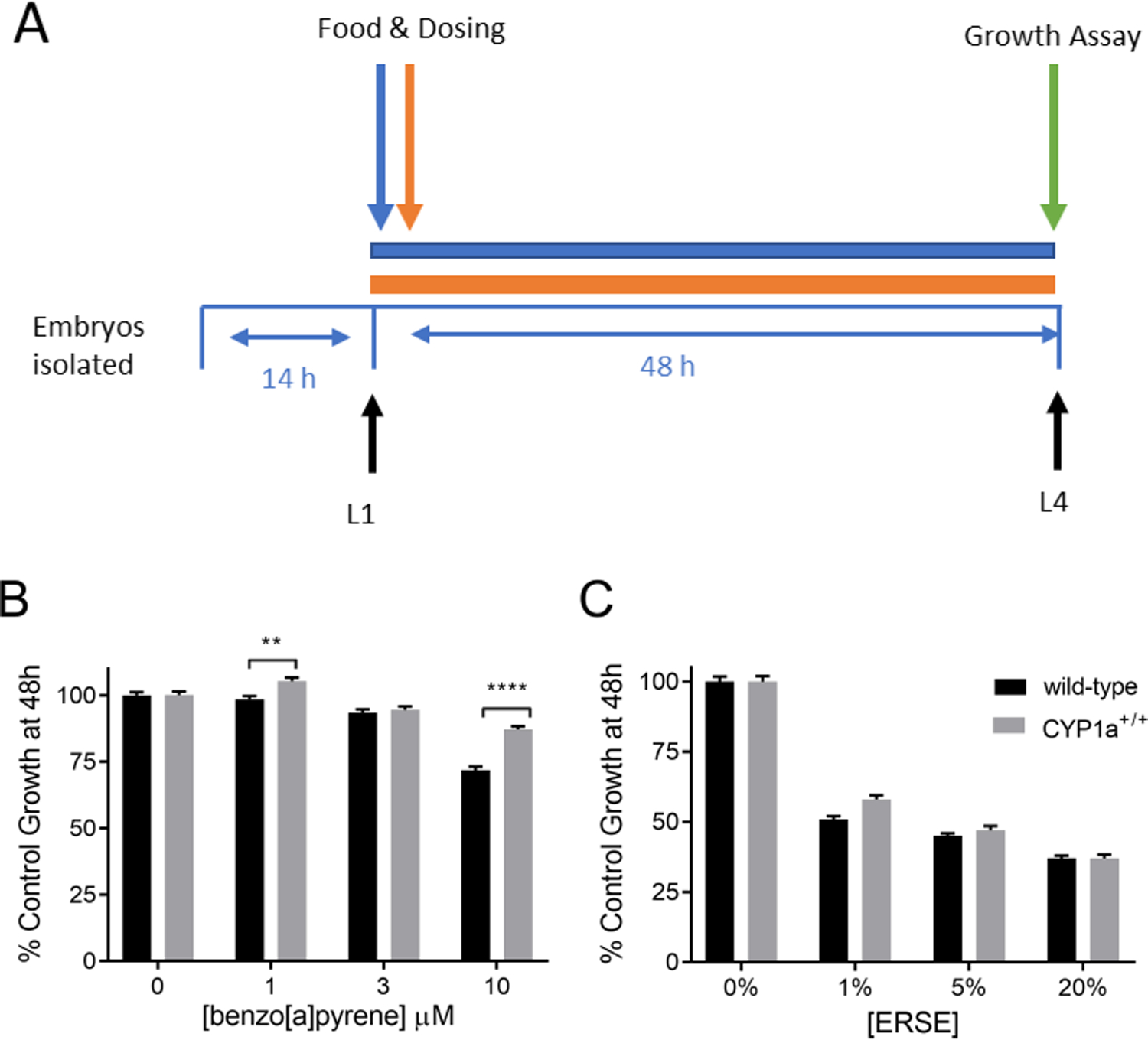
The time frame of exposure for the growth assay is shown in panel A. Fifity L1 stage C. elegans were exposed to BaP (panel B) or ERSE (panel C) in liquid for 48 hours and worm length was determined using COPAS Biosort. C. elegans were wild-type or CYP1a+/+ genotype. Four technical replicates were run in individual wells and the experiment was repeated three times. Statistical significance was assessed by a two-way ANOVA followed when appropriate by a Sidak’s multiple comparisons test comparing all groups to each other. The two-way ANOVA showed a statistically significant interaction between the effects of BaP exposure concentration and strain (p<0.0001), but not between ERSE and strain (p = 0.10). Simple main effects analysis on the BaP exposure data (panel B) showed that CYP1a+/+nematodes grew more than wild-type (p<0.0001), and the dose responses across strains was significant (p<0.0001). Main effects analysis on the ERSE exposure data (panel C) showed that the dose response across strains was significant (p<0.0001). Asterisks represent statistical significance: **p<0.01, ****p<0.0001.
In addition to the developmental exposure, we also performed an exposure beginning at the fourth larval stage (L4) to determine organismal reproductive toxicity, in the absence of developmental delay. Reproduction assays were performed by growing wild-type and CYP1A-expressing nematodes to L4 stage (32–36 hours) and transferring them to K-agar plates dosed with 0, 10, and 20 μM BaP (Fig. 3A). The nematodes were allowed to reproduce and the number of offspring was counted. While the unexposed CYP1A-expressing strain did not produce as many progeny as the wild-type control strain (~175 vs. ~250; data not shown), the CYP1A protein did provide protection against BaP-induced reduction of offspring numbers (Fig. 3B). By contrast, CYP1A-expressing worms were not protected from ERSE-induced reduction in brood size, instead trending toward an increase in sensitivity at the highest dose (Fig. 3C). Possible additional measures of reproductive toxicity, the presence of unhatched eggs or bagging in nematodes, were not observed in any of the exposed strains (data not shown).
Figure 3: CYP1A protects against BaP-induced reduction in brood size but not against ERSE-induced reduction.
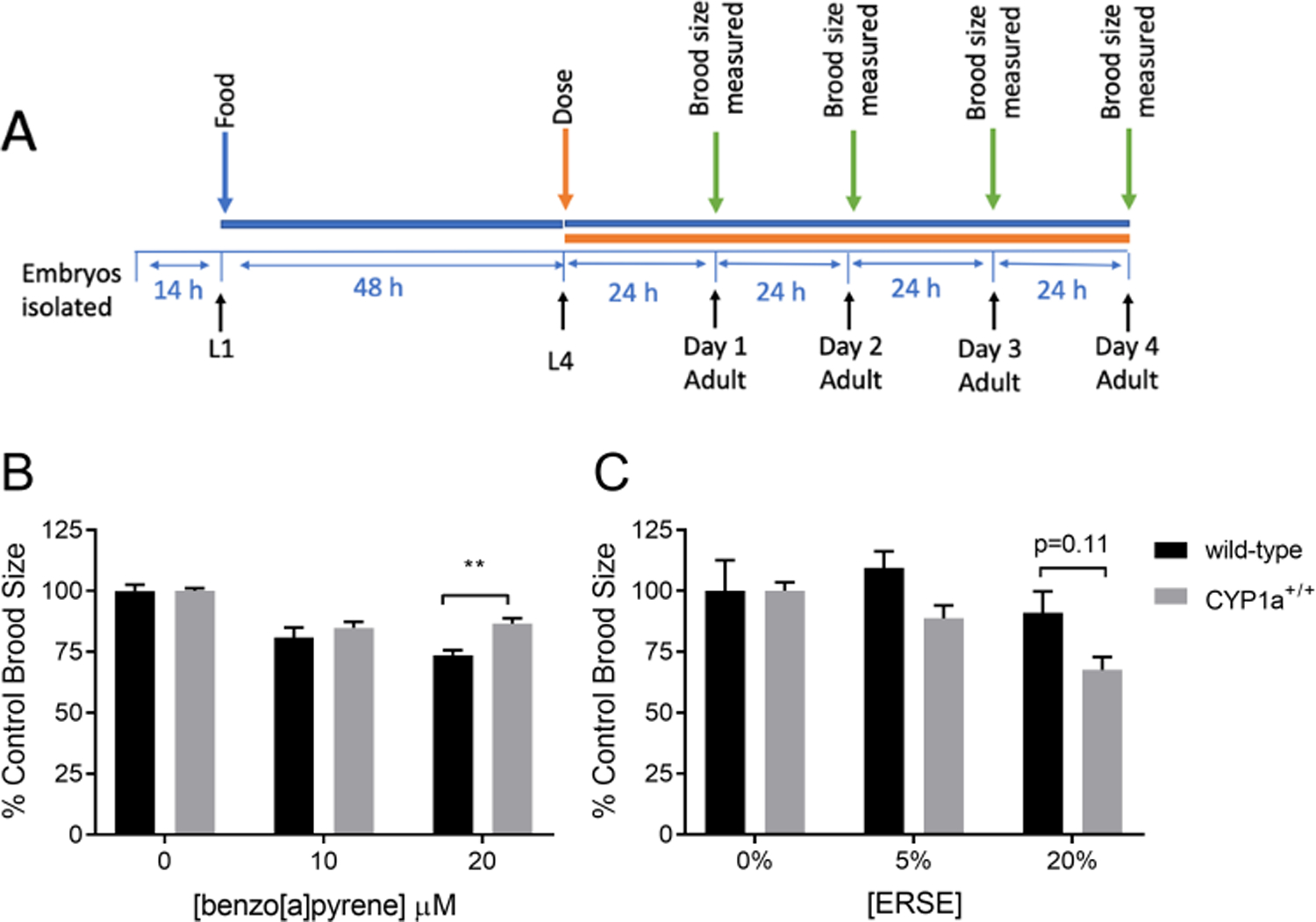
The time frame for exposure and broodsize determination is shown in panel A. L4 state C. elegans were exposed to BaP (panel B) or ERSE (panel C) during the first four days of reproduction. The offspring were counted and brood size was determined to be the sum of all offspring for an organism. Six technical replicates were run on different plates and the experiment was repeated twice. Statistical significance was assessed by a two-way ANOVA followed by a Sidak’s multiple comparisons test comparing all groups to each other. The two-way ANOVA showed a statistically significant interaction between the effects of BaP exposure and strain (p=0.03), but not between the effects of ERSE exposure and strain (p=0.26). Asterisks represent statistical significance: **p<0.01. Note that strain-related differences in brood size between Cyp1a- expressing nematodes and wildtype nematodes were observed (approximately 175 vs. 250).
3.3. Presence of CYP1A protects against decreases in steady state ATP levels caused by benzo[a]pyrene and Elizabeth River sediment extract
Depletion of steady state ATP levels can indicate that a toxicant affects energy metabolism (Lagido, et al. 2008). Due to the lipophilic nature of PAHs and mitochondrial toxicity of some PAH metabolites (Meyer, et al. 2013), we hypothesized that BaP and ERSE would cause a decrease in steady state ATP levels. Using a transgenic strain expressing firefly luciferase as a reporter of steady-state in vivo ATP levels (McLaggan, et al. 2012), we measured ATP levels in wild-type and CYP1A-expressing nematodes immediately after a 24-hour exposure to BaP or ERSE beginning at the L4 stage, or after an additional 24-hour recovery period (Fig. 4A). Supplemental Figure S1 illustrates the tissue expression of the luciferase reporter transgene, which is fused to GFP.
Figure 4: CYP1A protects against BaP and ERSE-induced ATP reduction.
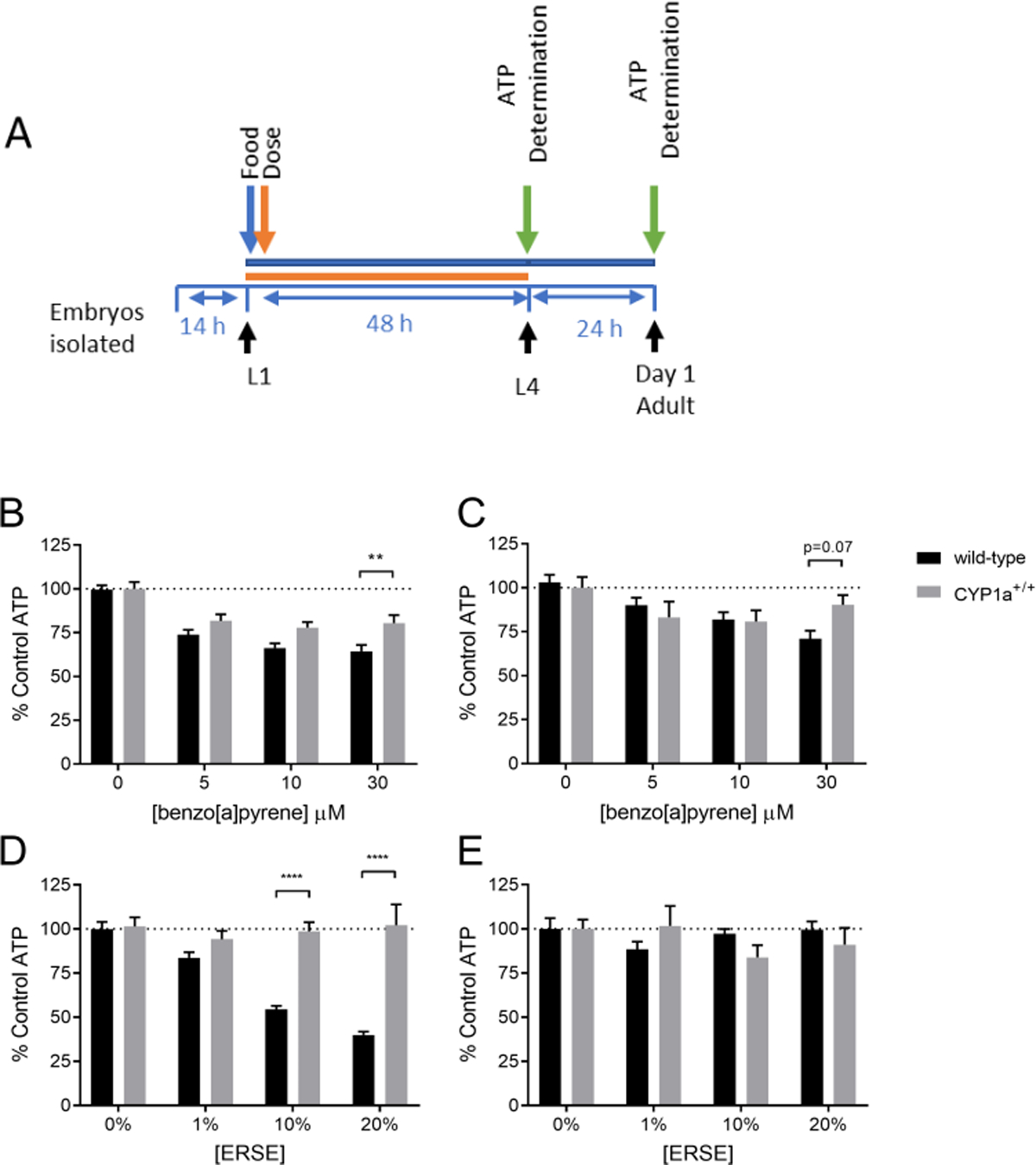
The time frame for exposure and ATP determination is shown in panel A. One hundred larval stage 4 nematodes were dosed for 24 hours in BaP (panel B) or ERSE (panel D). Steady-state ATP levels were measured using a microplate reader. Fifty of the worms were allowed a 24 hour recovery before ATP measurement (panel C & E). Four technical replicates were run in individual wells and the experiment was repeated three times. Statistical significance was assessed by a two-way ANOVA followed by an unpaired t-test comparing all groups to each other. The two-way ANOVA did not find a statistically significant interaction between the effects of BaP exposure concentration and strain (p=0.13) immediately after exposure. Dose response across strains immediately after exposure was significant (main effect of dose p<0.0001).The two-way ANOVA showed a statistically significant interaction between the effects of ERSE exposure concentration and strain (p<0.0001) immediately after exposure. Dose response immediately after ERSE exposure across strains was significant (main effect of exposure, p<0.0001). Asterisks represent statistical significance: **p<0.01, ****p<0.0001.
Exposure to BaP caused a concentration-dependent decrease in ATP levels in both wild-type and CYP1A-expressing nematodes; however, the CYP1A-expressing worms were protected compared to wild-type, particularly at the highest dose (Fig. 4B). After a 24-hour recovery period (without BaP), there was no longer a statistically significant difference between strains (Fig. 4C).
After ERSE exposure, the CYP1A-expressing worms had no change in steady-state ATP levels compared to the vehicle control, while the wild-type nematodes showed a concentration-dependent decrease in ATP levels (Fig. 4D). The recovered wild-type nematodes had similar ATP levels to the control condition nematodes (Fig. 4E).
3.4. Presence of CYP1A in nucleotide excision repair deficient nematodes protects against BaP- but not ERSE-induced growth delay
As an test of the potential for CYP1A to produce DNA-reactive metabolites from BaP or ERSE, we tested the sensitivity of nucleotide excision repair (NER) deficient nematodes with and without CYP1A expression. The metabolism of PAHs by CYP1A has the potential to produce a genotoxic PAH metabolite that can form an adduct with DNA (Moorthy, et al. 2015; Straif, et al. 2005); this form of DNA damage would be repaired in the nuclear genome by the NER pathway (Spivak 2015). Therefore, if CYP1A produces genotoxic PAH metabolites, a strain lacking NER should be sensitive to PAH exposure if CYP1A is expressed, but not without CYP1A. To indirectly test for the presence of PAH-derived DNA damage, we used a strain with a large deletion in the xpa-1 gene. XPA-1 is a critical protein in the NER pathway, and this mutant is a NER functional null (Meyer et al., 2007) that we and others have demonstrated is sensitive to DNA damage repaired by NER (Arczewska, et al. 2013; Astin, et al. 2008; Boyd, et al. 2010; Feng, et al. 2016; Leung, et al. 2010).
We crossed the NER-deficient xpa-1−/− strain with the CYP1A-expressing strain to test the effect of CYP1A expression in this NER-deficient background. Larval stage (L1) nematodes were exposed to BaP and ERSE for 48 hours (Fig. 5A). We used higher levels of BaP in this exposure than in Figure 2 in order to have a better dynamic range of growth response, enabling a clearer analysis of the role effect of CYP1A expression. CYP1A expression was protective against BaP-induced growth delay in NER-deficient worms at all exposure levels of BaP, further supporting our conclusion that CYP1A does not produce significant levels of DNA-reactive metabolites from BaP (Fig. 5B). CYP1A expression had no significant effect on ERSE sensitivity in NER-deficient animals (Fig. 5C). Furthermore, surprisingly, it appeared that the NER-deficient worms were protected against ERSE toxicity when compared to wild-type (compare Figure 5C to Figure 2C). To confirm this finding, another growth assay was performed using wildtype, CYP1A-expressing, NER-deficient, and CYP1A-expressing NER-deficient nematodes in the same assay. These strains were exposed to 0% or 20% ERSE for 48 hours prior to growth analysis. The results were consistent: CYP1A expression had no significant effect on ERSE sensitivity in any strain at the highest dose of ERSE, and xpa-1 deficiency was protective against ERSE exposure (Supplemental Figure S2).
Figure 5: CYP1A protects against BaP-induced growth delay in in nucleotide excision repair deficient nematodes, but not against ERSE-induced growth delay:
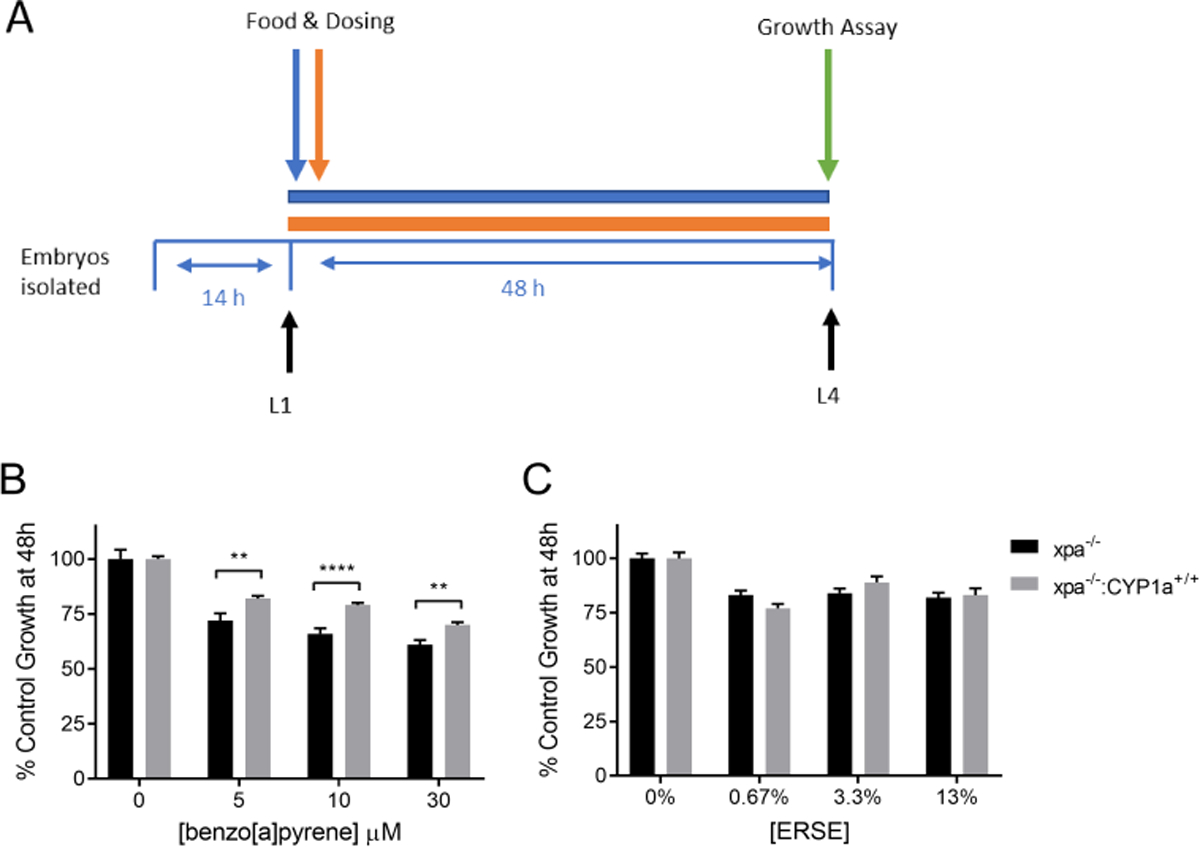
The time frame of exposure for the growth assay is shown in panel A. Fifty larval stage 1 nematodes were exposed for 48 hours in liquid to BaP (panel B) or ERSE (panel C), and worm length was determined using COPAS Biosort. C. elegans were of genotype xpa−/− or xpa−/;−CYP1a+/+. Four technical replicates were run in individual wells and the experiment was repeated three times. Statistical significance was assessed by a two-way ANOVA followed by a Sidak’s multiple comparisons test comparing all groups to each other. Two-way ANOVA showed statistically significant interactions between the effects of strain and BaP exposure concentration (p=0.008), but not between the effects of strain and ERSE esposure (p = .2016). The strain effect after ERSE esposure was significant (p<0.0001). Asterisks represent statistical significance: **p<0.01, ***p<0.0001.
4. Discussion
In this study, we utilized C. elegans, a species lacking endogenous CYP1A activity, transgenically expressing CYP1A to investigate organismal and mitochondrial PAH toxicity. We report clear protection of nematodes from exposure to both the model PAH BaP and a complex, real-world, PAH-rich (Fang et al., 2014) mixture obtained from the Elizabeth River Superfund site. Based on these protections, which were present even in the absence of NER, we conclude that CYP1A aided in safer excretion of PAHs, and did not bioactivate PAHs to genotoxic metabolites to a biologically significant extent. Furthermore, this work demonstrates a powerful new tool in mechanistic toxicity that allows for medium-throughput C. elegans assays to assess relevant metabolic pathways present in fish and mammals.
CYP1A protects against both BaP and ERSE toxicity
After exposure to BaP, we observed that growth delay, inhibition of reproduction, and decreases in ATP concentrations were all less dramatic in CYP1A-expressing nematodes than in wild-type. This finding supports the idea that CYP1A-mediated oxidation of PAHs is less harmful than permitting the buildup of BaP molecules, which may result narcotic and other forms of toxicity (Veith and Broderius 1990). Furthermore, concerns about the potential of this bioactivation to produce a carcinogenic metabolite (Straif, et al. 2005) that damages DNA were not supported, as CYP1A production protected against BaP exposure, rather than exacerbating toxicity, in NER-deficient mutants. These results suggest that although zebrafish CYP1A can generate pre-genotoxic metabolites such as the 7,8 diol of BaP substrates in vitro, these metabolites are either not be further converted to the ultimate diol epoxide carcinogen, or are produced at quantitatively lower levels than less-toxic BaP metabolites that are also formed, such as the 3,6-dione in vivo (Scornaienchi, et al. 2010b). However, as discussed in the next section, while a protective effect of CYP1A against ERSE was also observed in some cases, the degree of protection was less clear-cut than with BaP in most cases.
These findings are remarkable when compared to previous work which has found increased toxicity of PAHs through metabolism by CYP1A and/or CYP1B1 (Billiard, et al. 2008; Miller and Ramos 2001; Shimada, et al. 2001; Xue and Warshawsky 2005). The mechanism of toxicity associated with CYP1A and CYP1B1 bioactivation of BaP is well-documented, as is the incidence of nuclear and mitochondrial DNA damage that is caused by the reactive metabolites of PAHs after CYP metabolism (Moorthy, et al. 2015; Shimada, et al. 2001; Xue and Warshawsky 2005). Furthermore, earlier research on Atlantic Killifish (Fundulus heteroclitus) living in the Atlantic Wood Industries Superfund site from which the ERSE was derived reported both high levels of DNA damage in wild-caught fish, and resistance to additional DNA damage and other forms of toxicity from laboratory PAH dosing, compared to reference site fish (Chang, et al. 2007; Jung, et al. 2009; Meyer and Di Giulio 2002; Wills, et al. 2009). In addition, CYP1A inducibility is markedly reduced in the Superfund site population of killifish (Clark, et al. 2014; Meyer, et al. 2002; Meyer, et al. 2003). Overall, these findings supported the hypothesis that the presence of CYP1A increases the toxicity of PAH exposure. Our findings, in contrast, suggest CYP1A has a protective effect in PAH exposure, and support the idea that lack of CYP1A inducibility in Atlantic Killifish population in the Elizabeth River is a marker of AHR pathway inhibition rather than an evolutionary adaptation to CYP1A-induced toxicity of PAHs. A caveat to our interpretation is that although our expression system results in a relatively high level of CYP1A protein expression, it is not inducible, unlike the very highly inducible CYP1A proteins in vertebrates. Therefore, it is possible that at higher levels of CYP1A activity, more genotoxic metabolites would have been produced, or that phase II processes would have been overwhelmed. Nonetheless, our results are in accord with publications from other species, suggesting that CYP1A subfamily enzymes appear to play generally protective roles in the context of PAH exposure (Nebert, et al. 2013; Shi, et al. 2010; Shiizaki, et al. 2017; Uno, et al. 2004). A final caveat is that whle we found no evidence for a CYP1 family enzyme in worms (Leung, et al. 2010), and CYP1A-like activity measured as phenacetin metabolism is very low (Harlow, et al. 2018), others have suggested that other CYPs might metabolize PAHs in worms (Imanikia, et al. 2016) a possibility that we have not formally tested for BaP. Future studies will examine the effects of exposure to PAH on mitochondrial function and other endpoints with and without the presence of other CYP proteins that metabolize PAHs (such as 1B1) individually or in combination with CYP1A.
Finally, it is interesting to compare the toxicity values for BaP in this study to those published in previous literature. The EC50 values found in this investigation are higher than values previously reported for growth and reproduction assays, as shown in Supplemental Table S5. The reason for this discrepancy is likely differences in methods used to assess these outcomes. For example, in Sese et al, 2009 and Menzel et al, 2005 the reproductive assay method involved a liquid exposure, and Sese et al. (2009) carried out exposures in glass plates, to which BaP will sorb less than to the more typical plastic containers. The reproduction assay in this investigation was performed on agar toxic plates, so there may have been a lower available dose effecting the nematode compared to liquid exposures. In addition, we used an egg isolation/overnight starvation protocol to sunchronize L1-stage larvae; this starvation induces various defense mechanisms. Other experimental design variables such as food and nematode density and choice of carrier may also explain some differences. In terms of broader comparisons, as discussed by Sese et al. (2009), it appears that BaP is roughly similarly toxic to C. elegans compared to other invertebrates. However, BaP is toxic at much lower levels to cells in culture, presumably due to toxicokinetic and/or toxicodynamic differences.
BaP and ERSE cause toxicity by distinct mechanisms
Although the reduction of steady-state ATP levels was less dramatic in CYP1A-producing nematodes than in wild-type, there was a dose-dependent decrease in ATP levels for both strains after both exposures. Thus, ATP levels were decreased in the presence of BaP, either in its parent form or after metabolism by a non-CYP1A family enzyme (enzymatic activity of the C. elegans CYPs is not well characterized). PAHs localize to the lipophilic membrane of the mitochondria and can cause decreased membrane potential, respiration, and other forms of mitochondrial dysfunction (Meyer, et al. 2018; Raftery, et al. 2017; Xia, et al. 2004). Therefore, direct effects on mitochondria could account for the dose-dependent decrease in ATP levels caused by BaP. Notably, this is consistent with a toxicogenomic study performed in C. elegans with the PAH fluoranthene in which metabolic pathways were perturbed (Swain, et al. 2010). Since fluoranthene is not generally considered to be metabolized to a DNA-reactive form, this supports the potential for a non-DNA reactive, non-AHR mode of toxicity of PAHs, such as narcosis. Alternatively, the energetic cost of BaP detoxication and stress response or other causes may explain the decrease in ATP levels.
Other results support that BaP and ERSE act by at least somewhat different mechanisms of action. In the wild-type strain lacking CYP1A, ERSE-induced reduction of steady state ATP levels was more dramatic than the reduction after BaP exposure, despite the fact that the effect on brood size, measured at the same lifestage, was fairly similar in magnitude. Furthermore, the protective effect of CYP1A was less marked after ERSE than BaP by all measures other than ATP levels. This could be explained by the composition of PAHs in the ERSE mixture, which includes many parent PAHs that can have unexpected combinatorial effects (Billiard, et al. 2008; Liu, et al. 2013; Wassenberg and Di Giulio 2004), as well as heterocyclic compounds, quinones, and a variety of other more inherently reactive PAH-related molecules (Fang, et al. 2014). Fang et al., (2014) reported only 197 ng BaP/mL in ERSE; thus, BaP levels in the mixture were never more than 0.5nM, which is substantially lower than any of the tested concentrations of pure BaP. A lower concentration of each kind of PAH may allow each to be metabolized faster and with greater efficiency, and thus not affect ATP production. Alternatively, this combination of PAHs may have had more, or different, non-mitochondrial mechanisms of toxicity (causing similar inhibition of reproduction with less reduction in ATP levels), compared to BaP.
Finally, we found that the xpa-1-deficient nematodes were surprisingly resistant to the toxicity of ERSE, but not BaP. It is unclear why xpa-1 deficiency would confer resistance to ERSE exposure; we previously reported that this strain shows almost identical genome-wide transcriptomic patterns (by microarray) compared to wild-type under control conditions and even shortly after exposure to ultraviolet C radiation (Boyd et al., 2010). Thus, there is little evidence for either major transcriptional compensatory responses, or the presence of other mutations in the xpa-1 strain, that could explain this difference.
Improving the similarity between C. elegans and vertebrate xenobiotic metabolism
A limitation to the increased use of C. elegans as a toxicological model is existence of several differences between nematodes and vertebrates in xenobiotic metabolism. One limitation, which we begin to address here, is the absence of the CYP1 family enzymes, which are key to the metabolism of many organic toxicants. The choice to use the zebrafish Cyp1a transgene over the mammalian Cyp1A1 or Cyp1A2 transgene was based on the culture temperature of C. elegans. Zebrafish are typically cultured at 26 – 28.5°C; thus, we expected that the enzymatic activity of CYP1A would be robust at the 20°C culture temperature of C. elegans, when compared to mammalian CYP1A1 or CYP1A2 that are typically active at temperatures closer to 36°C (Avdesh, et al. 2012). Although the catalytic efficiency of the zebrafish CYP1A enzyme has not been compared directly to that of mammalian CYP1A1 and CYP1A2, it is known that the enzyme metabolizes similar substrates (Scornaienchi, et al. 2010b). For example, a diagnostic substrate for mammalian CYP1A1 metabolism is 7-ethoxyresorufin (ER) and a diagnostic substrate for mammalian CYP1A2 metabolism is 7-methoxyresorufin (MR). Zebrafish CYP1A was shown to metabolize both ER and MR at the highest rates compared to other zebrafish CYP enzymes tested, and is therefore likely homologous to both mammalian isoforms (Scornaienchi, et al. 2010b). Furthermore, the selection of zebrafish CYP1A is advantageous because CYP1A isoforms are highly variable even among mammalian species (Martignoni, et al. 2006). In general, in all mammals, CYP1A1 is not expressed basally but is inducible with xenobiotics, while CYP1A2 is expressed basally and is less inducible. However, as an example, mouse and human CYP1A2 differ 3- to 7- fold in their catalytic efficiency of ER metabolism(Aoyama, et al. 1989), while mouse and human CYP1A1 differ 10- to 100-fold in their maximal inducibility(Shi, et al. 2008). Therefore, using the single CYP1A isoform from zebrafish creates a system to study the impact of CYP1A metabolism (vs. an essentially null background) but, like mammalian models, may have additional considerations such as regulatory changes in expression that would need to be accounted for in translating to humans.
Another important difference between C. elegans and vertebrates that we have not addressed in our current transgenic system is the apparent absence in nematodes of a dioxin-like ligand-binding aryl hydrocarbon receptor (AhR)(Powell-Coffman, et al. 1998). In vertebrates, PAHs such as BaP can bind to the AhR, causing the AhR to dimerize with the aryl hydrocarbon receptor nuclear translocator (ARNT) and translocate to the nucleus where it acts as a transcription factor for xenobiotic response element -regulated genes, including the CYP1 family as well as other xenobiotic metabolism and stress response genes (Denison and Nagy 2003; Hankinson 1995; Harper, et al. 1991). While there is evidence of AhR and ARNT orthologs in C. elegans, and this heterodimer binds DNA (Powell-Coffman, et al. 1998) and regulates gene expression and physiological processes such as neuronal deveopmenta and function (Aarnio, et al. 2010; Qin and Powell-Coffman 2004; Qin, et al. 2006), the nematode AHR does not appear to bind xenobiotic ligands such as PAHs. Thus, the cycle observed in vertebrates, in which PAHs induce the enzymatic system responsible for their own metabolism, is still not fully recapitulated in our model. To some extent, this creates a caveat to the interpretation of our results: it is possible that CYP1A activity at this relatively low level of protein activity is protective, but that in a highly inducible system, the formation of reactive metabolites would be more problematic, perhaps by overwhelming phase II processes. Phase II and Phase III processes are also relatively poorly understood in C. elegans. Nonetheless, inducibility of many phase I (e.g., CYPs and flavin monooxygenases) and phase II (e.g., sulfotransferase, glutathione S-transferase, and glucuronosyl transferase) genes by PAHs and other chemicals (Jones, et al. 2013; Leung, et al. 2013b; Menzel, et al. 2001; Menzel, et al. 2005; Reichert and Menzel 2005; Swain, et al. 2010; Wu, et al. 2015) has been identified. The gene regulatory mechanisms and pathways are not well understood, however; C. elegans has a very wide repertoire of poorly-characterized nuclear hormone receptors (Taubert, et al. 2011). Furthermore, enzymatic functions and specific substrates of these enzymes remain relatively poorly characterized in C. elegans, with only a small number of reports published, focused primarily on CYPs (Chakrapani, et al. 2008; Harlow, et al. 2018; Imanikia, et al. 2016; Kulas, et al. 2008; Leung, et al. 2010; Menzel, et al. 2007; Schäfer, et al. 2009). Thus, currently, the C. elegans model can serve most appropriately as a system to test consequences of metabolism (e.g. bioactivation and cellular toxicity) rather than directly informing toxicokinetics.
C. elegans is increasingly used as a model organism in toxicological studies, and offers significant strengths for such work (Boyd, et al. 2016; Hunt 2017; Kim, et al. 2019; Leung, et al. 2008; Tejeda-Benitez and Olivero-Verbel 2016) However, there are also important limitations to the C. elegans model, including differences in xenobiotic metabolism. Transgenic manipulations that increase the toxicokinetic and toxicodynamic simliarity of C. elegans to more complex metazoans will improve extrapolability of studies of toxicity of both individual chemicals (Imanikia, et al. 2016; Luz, et al. 2017; Sese, et al. 2009; Wu, et al. 2015; Wyatt, et al. 2017) and environmental mixtures (Abbas, et al. 2018; Menzel, et al. 2009; Tejeda-Benitez and Olivero-Verbel 2016; Turner, et al. 2013)
5. Conclusions
CYP1A is an important protein for the detoxication of high molecular weight PAHs that can be metabolized to reactive forms with the potential to damage DNA and proteins. Using a species in which other potentially confounding CYP1 family enzymes are absent, we find that at least under these experimental circumstances, the protective effects of basal expression of CYP1A appear to reduce the potential danger of producing reactive metabolites, both for the well-studied model PAH BaP, and, to a lesser extent, for a real-world, complex mixture of PAHs, ERSE. We further provide evidence that the toxicity of this mixture derives from multiple mechanisms distinct from those exhibited by BaP. Finally, we provide a first step toward generating nematode strains that are more vertebrate-like in their xenobiotic metabolism, which will improve the utility of this species for toxicological studies, for which it is increasingly employed.
Supplementary Material
Acknowledgements
We thank Rakesh Bodhicharla and Kerstin Helmcke for work on early versions of the transgenic CYP1A-expressing nematodes. Funding was provided by the National Institute of Health and National Institutes of Environmental Health Sciences [R01-ES017540-01A2 and P42ES010356 to JNM, F31ES026859 to ALL, and F32ES027306 to JHH), the National Institutes of Health Office of Research Infrastructure Programs which funds the Caenorhabditis Genetics Center (P40OD010440), which provided strains, and a Natural Sciences and Engineering Research Council of Canada Discovery Grant (JYW).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Aarnio V, et al. 2010. Fatty acid composition and gene expression profiles are altered in aryl hydrocarbon receptor-1 mutant Caenorhabditis elegans. Comp Biochem Physiol C Toxicol Pharmacol 151(3):318–24. [DOI] [PubMed] [Google Scholar]
- Abbas A, et al. 2018. Ecotoxicological impacts of surface water and wastewater from conventional and advanced treatment technologies on brood size, larval length, and cytochrome P450 (35A3) expression in Caenorhabditis elegans. Environ Sci Pollut Res Int 25(14):13868–13880. [DOI] [PubMed] [Google Scholar]
- Abdel-Shafy Hussein I., and Mansour Mona S. M. 2016. A review on polycyclic aromatic hydrocarbons: Source, environmental impact, effect on human health and remediation. Egyptian Journal of Petroleum 25(1):107–123. [Google Scholar]
- Aoyama T, Gonzalez FJ, and Gelboin HV 1989. Human cDNA-expressed cytochrome P450 IA2: mutagen activation and substrate specificity. Mol Carcinog 2(4):192–8. [DOI] [PubMed] [Google Scholar]
- Arczewska Katarzyna D., et al. 2013. Active transcriptomic and proteomic reprogramming in the C. elegans nucleotide excision repair mutant xpa-1. Nucleic acids research 41(10):5368–5381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astin Jonathan W., O’Neil Nigel J., and Kuwabara Patricia E. 2008. Nucleotide excision repair and the degradation of RNA pol II by the Caenorhabditis elegans XPA and Rsp5 orthologues, RAD-3 and WWP-1. DNA Repair 7(2):267–280. [DOI] [PubMed] [Google Scholar]
- Avdesh Avdesh, et al. 2012. Regular care and maintenance of a zebrafish (Danio rerio) laboratory: an introduction. Journal of visualized experiments : JoVE (69):e4196–e4196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bess Amanda S, et al. 2012. Mitochondrial dynamics and autophagy aid in removal of persistent mitochondrial DNA damage in Caenorhabditis elegans. Nucleic acids research 40(16):7916–7931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billiard Sonya M., et al. 2008. Nonadditive effects of PAHs on Early Vertebrate Development: mechanisms and implications for risk assessment. Toxicological sciences : an official journal of the Society of Toxicology 105(1):5–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd WA, Smith MV, and Freedman JH 2012. Caenorhabditis elegans as a model in developmental toxicology. Methods Mol Biol 889:15–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd Windy A., et al. 2010. Nucleotide excision repair genes are expressed at low levels and are not detectably inducible in Caenorhabditis elegans somatic tissues, but their function is required for normal adult life after UVC exposure. Mutation research 683(1–2):57–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd Windy A., et al. 2016. Developmental Effects of the ToxCast™ Phase I and Phase II Chemicals in Caenorhabditis elegans and Corresponding Responses in Zebrafish, Rats, and Rabbits. Environmental health perspectives 124(5):586–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrapani Baby P. S., Kumar Sandeep, and Subramaniam Jamuna R. 2008. Development and evaluation of an in vivo assay in Caenorhabditis elegans for screening of compounds for their effect on cytochrome P450 expression. Journal of Biosciences 33(2):269–277. [DOI] [PubMed] [Google Scholar]
- Chang Edward Wei-Chung, et al. 2007. Quality of sleep and quality of life in caregivers of breast cancer patient. Psycho-Oncology 16(10):950–955. [DOI] [PubMed] [Google Scholar]
- Clark Bryan W., Bone AJ, and Di Giulio RT 2014. Resistance to teratogenesis by F1 and F2 embryos of PAH-adapted Fundulus heteroclitus is strongly inherited despite reduced recalcitrance of the AHR pathway. Environmental science and pollution research international 21(24):13898–13908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denison Michael S, and Nagy Scott R 2003. Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Annual review of pharmacology and toxicology 43(1):309–334. [DOI] [PubMed] [Google Scholar]
- Giulio Di, Richard T, and Clark Bryan W 2015. The Elizabeth River story: a case study in evolutionary toxicology. Journal of Toxicology and Environmental Health, Part B 18(6):259–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson Daniel J., et al. 2013. Engineering the Caenorhabditis elegans genome using Cas9-triggered homologous recombination. Nature Methods 10:1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallahtafti Shirin, et al. 2012. Toxicity of hydroxylated alkyl-phenanthrenes to the early life stages of Japanese medaka (Oryzias latipes). Aquatic toxicology 106:56–64. [DOI] [PubMed] [Google Scholar]
- Fang ML, et al. 2014. EFFECT-DIRECTED ANALYSIS OF ELIZABETH RIVER POREWATER: DEVELOPMENTAL TOXICITY IN ZEBRAFISH (DANIO RERIO). Environmental Toxicology and Chemistry 33(12):2767–2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Wei-Hong, et al. 2016. Aflatoxin B1 -Induced Developmental and DNA Damage in Caenorhabditis elegans. Toxins 9(1):9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale Robert C., and Aneiro Karen M. 1997. Determination of coal tar and creosote constituents in the aquatic environment. Journal of Chromatography A 774(1):79–95. [DOI] [PubMed] [Google Scholar]
- Hankinson Oliver 1995. The aryl hydrocarbon receptor complex. Annual review of pharmacology and toxicology 35(1):307–340. [DOI] [PubMed] [Google Scholar]
- Harlow Philippa H., et al. 2018. Comparative metabolism of xenobiotic chemicals by cytochrome P450s in the nematode Caenorhabditis elegans. Scientific Reports 8(1):13333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper Patricia A, et al. 1991. Detection and characterization of the Ah receptor for 2, 3, 7, 8-tetrachlorodibenzo-p-dioxin in the human colon adenocarcinoma cell line LS180. Archives of biochemistry and biophysics 290(1):27–36. [DOI] [PubMed] [Google Scholar]
- Hoogewijs D, et al. 2008. Selection and validation of a set of reliable reference genes for quantitative sod gene expression analysis in C. elegans. BMC Mol Biol 9:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt Piper Reid 2017. The C. elegans model in toxicity testing. Journal of applied toxicology : JAT 37(1):50–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imanikia S, et al. 2016. The application of the comet assay to assess the genotoxicity of environmental pollutants in the nematode Caenorhabditis elegans. Environ Toxicol Pharmacol 45:356–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones LM, et al. 2013. Adaptive and specialised transcriptional responses to xenobiotic stress in Caenorhabditis elegans are regulated by nuclear hormone receptors. PLoS One 8(7):e69956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung Dawoon, et al. 2009. Effects of benzo [a] pyrene on mitochondrial and nuclear DNA damage in Atlantic killifish (Fundulus heteroclitus) from a creosote-contaminated and reference site. Aquatic toxicology 95(1):44–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MinA, et al. 2019. High-throughput COPAS assay for screening of developmental and reproductive toxicity of nanoparticles using the nematode Caenorhabditis elegans. Journal of Applied Toxicology 0(0). [DOI] [PubMed] [Google Scholar]
- Kulas J, et al. 2008. Cytochrome P450-dependent metabolism of eicosapentaenoic acid in the nematode Caenorhabditis elegans. Arch Biochem Biophys 472(1):65–75. [DOI] [PubMed] [Google Scholar]
- Lagido Cristina, et al. 2008. Bridging the phenotypic gap: real-time assessment of mitochondrial function and metabolism of the nematode Caenorhabditis elegans. BMC physiology 8(1):7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung Chi K., et al. 2013a. An ultra high-throughput, whole-animal screen for small molecule modulators of a specific genetic pathway in Caenorhabditis elegans. PloS one 8(4):e62166–e62166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung Maxwell C. K., et al. 2013b. Effects of early life exposure to ultraviolet C radiation on mitochondrial DNA content, transcription, ATP production, and oxygen consumption in developing Caenorhabditis elegans. BMC Pharmacology and Toxicology 14(1):9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung Maxwell C. K., et al. 2008. Caenorhabditis elegans: an emerging model in biomedical and environmental toxicology. Toxicological sciences : an official journal of the Society of Toxicology 106(1):5–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung Maxwell CK, et al. 2010. Caenorhabditis elegans generates biologically relevant levels of genotoxic metabolites from aflatoxin B1 but not benzo [a] pyrene in vivo. Toxicological Sciences 118(2):444–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis James A., and Fleming John T. 1995. Chapter 1 Basic Culture Methods In Methods in Cell Biology. Epstein HF and Shakes DC, eds. Pp. 3–29: Academic Press. [PubMed] [Google Scholar]
- Liu Jiawang, Sridhar Jayalakshmi, and Foroozesh Maryam 2013. Cytochrome P450 family 1 inhibitors and structure-activity relationships. Molecules (Basel, Switzerland) 18(12):14470–14495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luz Anthony L., et al. 2017. Deficiencies in mitochondrial dynamics sensitize Caenorhabditis elegans to arsenite and other mitochondrial toxicants by reducing mitochondrial adaptability. Toxicology 387:81–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martignoni Marcella, Groothuis Geny M. M., and de Kanter Ruben 2006. Species differences between mouse, rat, dog, monkey and human CYP-mediated drug metabolism, inhibition and induction. Expert Opinion on Drug Metabolism & Toxicology 2(6):875–894. [DOI] [PubMed] [Google Scholar]
- Maurer Laura L., et al. 2015. Caenorhabditis elegans as a Model for Toxic Effects of Nanoparticles: Lethality, Growth, and Reproduction. Current Protocols in Toxicology 66(1):20.10.1–20.10.25. [DOI] [PubMed] [Google Scholar]
- McLaggan Debbie, et al. 2012. Impact of Sublethal Levels of Environmental Pollutants Found in Sewage Sludge on a Novel Caenorhabditis elegans Model Biosensor. PLOS ONE 7(10):e46503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menzel R, Bogaert T, and Achazi R 2001. A systematic gene expression screen of Caenorhabditis elegans cytochrome P450 genes reveals CYP35 as strongly xenobiotic inducible. Arch Biochem Biophys 395(2):158–68. [DOI] [PubMed] [Google Scholar]
- Menzel R, et al. 2005. CYP35: xenobiotically induced gene expression in the nematode Caenorhabditis elegans. Arch Biochem Biophys 438(1):93–102. [DOI] [PubMed] [Google Scholar]
- Menzel R, et al. 2009. Gene expression profiling to characterize sediment toxicity--a pilot study using Caenorhabditis elegans whole genome microarrays. BMC Genomics 10:160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menzel R, et al. 2007. Cytochrome P450s and short-chain dehydrogenases mediate the toxicogenomic response of PCB52 in the nematode Caenorhabditis elegans. J Mol Biol 370(1):1–13. [DOI] [PubMed] [Google Scholar]
- Meyer JN, et al. 2013. Mitochondria as a target of environmental toxicants. Toxicol Sci 134(1):1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer Joel, and Di Giulio Richard 2002. Patterns of heritability of decreased EROD activity and resistance to PCB 126-induced teratogenesis in laboratory-reared offspring of killifish (Fundulus heteroclitus) from a creosote-contaminated site in the Elizabeth River, VA, USA. Marine Environmental Research 54(3):621–626. [DOI] [PubMed] [Google Scholar]
- Meyer Joel N, Hartman Jessica H, and Mello Danielle F 2018. Mitochondrial toxicity. Toxicological Sciences 162(1):15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer Joel N., et al. 2007. Decline of nucleotide excision repair capacity in aging Caenorhabditis elegans. Genome biology 8(5):R70–R70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer Joel N., Nacci Diane E., and Di Giulio Richard T. 2002. Cytochrome P4501A (CYP1A) in Killifish (Fundulus heteroclitus): Heritability of Altered Expression and Relationship to Survival in Contaminated Sediments. Toxicological Sciences 68(1):69–81. [DOI] [PubMed] [Google Scholar]
- Meyer Joel N., et al. 2003. Expression and inducibility of aryl hydrocarbon receptor pathway genes in wild-caught killifish (Fundulus heteroclitus) with different contaminant-exposure histories. Environmental Toxicology and Chemistry 22(10):2337–2343. [DOI] [PubMed] [Google Scholar]
- Miller Kimberly P., and Ramos Kenneth S. 2001. IMPACT OF CELLULAR METABOLISM ON THE BIOLOGICAL EFFECTS OF BENZO[A]PYRENE AND RELATED HYDROCARBONS†. Drug Metabolism Reviews 33(1):1–35. [DOI] [PubMed] [Google Scholar]
- Moorthy Bhagavatula, Chu Chun, and Carlin Danielle J 2015. Polycyclic aromatic hydrocarbons: from metabolism to lung cancer. Toxicological Sciences 145(1):5–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nebert Daniel W., et al. 2013. Oral benzo[a]pyrene: understanding pharmacokinetics, detoxication, and consequences--Cyp1 knockout mouse lines as a paradigm. Molecular pharmacology 84(3):304–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauka Luciana M., et al. 2011. Embryotoxicity and Biotransformation Responses in Zebrafish Exposed to Water-Soluble Fraction of Crude Oil. Bulletin of Environmental Contamination and Toxicology 86(4):389–393. [DOI] [PubMed] [Google Scholar]
- Powell-Coffman Jo Anne, Bradfield Christopher A, and Wood William B 1998. Caenorhabditis elegans orthologs of the aryl hydrocarbon receptor and its heterodimerization partner the aryl hydrocarbon receptor nuclear translocator. Proceedings of the National Academy of Sciences 95(6):2844–2849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin H, and Powell-Coffman JA 2004. The Caenorhabditis elegans aryl hydrocarbon receptor, AHR-1, regulates neuronal development. Dev Biol 270(1):64–75. [DOI] [PubMed] [Google Scholar]
- Qin H, Zhai Z, and Powell-Coffman JA 2006. The Caenorhabditis elegans AHR-1 transcription complex controls expression of soluble guanylate cyclase genes in the URX neurons and regulates aggregation behavior. Dev Biol 298(2):606–15. [DOI] [PubMed] [Google Scholar]
- Raftery Tara D., Jayasundara Nishad, and Di Giulio Richard T. 2017. A bioenergetics assay for studying the effects of environmental stressors on mitochondrial function in vivo in zebrafish larvae. Comparative biochemistry and physiology. Toxicology & pharmacology : CBP 192:23–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichert K, and Menzel R 2005. Expression profiling of five different xenobiotics using a Caenorhabditis elegans whole genome microarray. Chemosphere 61(2):229–37. [DOI] [PubMed] [Google Scholar]
- Richter H, and Howard JB 2000. Formation of polycyclic aromatic hydrocarbons and their growth to soot—a review of chemical reaction pathways. Progress in Energy and Combustion Science 26(4):565–608. [Google Scholar]
- Riley Amanda K., et al. 2016. Hepatic Responses of Juvenile Fundulus heteroclitus from Pollution-adapted and Nonadapted Populations Exposed to Elizabeth River Sediment Extract. Toxicologic Pathology 44(5):738–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooney JP, et al. 2014. Effects of 5’-fluoro-2-deoxyuridine on mitochondrial biology in Caenorhabditis elegans. Experimental gerontology 56:69–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schäfer P, et al. 2009. Cytochrome P450-dependent metabolism of PCB52 in the nematode Caenorhabditis elegans. Arch Biochem Biophys 488(1):60–8. [DOI] [PubMed] [Google Scholar]
- Scornaienchi ML, et al. 2010a. Cytochrome P450-mediated 17beta-estradiol metabolism in zebrafish (Danio rerio). J Endocrinol 206(3):317–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scornaienchi ML, et al. 2010b. Functional differences in the cytochrome P450 1 family enzymes from zebrafish (Danio rerio) using heterologously expressed proteins. Arch Biochem Biophys 502(1):17–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sese BT, Grant A, and Reid BJ 2009. Toxicity of polycyclic aromatic hydrocarbons to the nematode Caenorhabditis elegans. J Toxicol Environ Health A 72(19):1168–80. [DOI] [PubMed] [Google Scholar]
- Shi Z, et al. 2008. Generation of a ‘humanized’ hCYP1A1_1A2_Cyp1a1/1a2(−/−)_Ahrd mouse line harboring the poor-affinity aryl hydrocarbon receptor. Biochem Biophys Res Commun 376(4):775–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Zhanquan, et al. 2010. Organ-Specific Roles of CYP1A1 during Detoxication of Dietary Benzo[a]pyrene. Molecular Pharmacology 78(1):46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiizaki Kazuhiro, Kawanishi Masanobu, and Yagi Takashi 2017. Modulation of benzo[a]pyrene-DNA adduct formation by CYP1 inducer and inhibitor. Genes and environment : the official journal of the Japanese Environmental Mutagen Society 39:14–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada Tsutomu, et al. 2001. Metabolic activation of polycyclic aromatic hydrocarbons and other procarcinogens by cytochromes P450 1A1 and P450 1B1 allelic variants and other human cytochromes P450 in Salmonella typhimurium NM2009. Drug metabolism and disposition 29(9):1176–1182. [PubMed] [Google Scholar]
- Spivak Graciela 2015. Nucleotide excision repair in humans. DNA repair 36:13–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straif Kurt, et al. 2005. Carcinogenicity of polycyclic aromatic hydrocarbons. The Lancet Oncology 6(12):931–932. [DOI] [PubMed] [Google Scholar]
- Swain S, et al. 2010. Linking toxicant physiological mode of action with induced gene expression changes in Caenorhabditis elegans. BMC Syst Biol 4:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taubert S, Ward JD, and Yamamoto KR 2011. Nuclear hormone receptors in nematodes: evolution and function. Mol Cell Endocrinol 334(1–2):49–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tejeda-Benitez L, and Olivero-Verbel J 2016. Caenorhabditis elegans, a Biological Model for Research in Toxicology. Rev Environ Contam Toxicol 237:1–35. [DOI] [PubMed] [Google Scholar]
- Tuli Mary Ann 2018. Wormbase Nomenclature Guide. WormBase: WormBase. [Google Scholar]
- Turner EA, et al. 2013. Assessing different mechanisms of toxicity in mountaintop removal/valley fill coal mining-affected watershed samples using Caenorhabditis elegans. PLoS One 8(9):e75329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uno Shigeyuki, et al. 2004. Oral Exposure to Benzo[a]pyrene in the Mouse: Detoxication by Inducible Cytochrome P450 Is More Important Than Metabolic Activation. Molecular Pharmacology 65(5):1225. [DOI] [PubMed] [Google Scholar]
- Veith GD, and Broderius SJ 1990. Rules for distinguishing toxicants that cause type I and type II narcosis syndromes. Environ Health Perspect 87:207–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassenberg Deena M., and Di Giulio Richard T. 2004. Synergistic embryotoxicity of polycyclic aromatic hydrocarbon aryl hydrocarbon receptor agonists with cytochrome P4501A inhibitors in Fundulus heteroclitus. Environmental health perspectives 112(17):1658–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wills Lauren P., et al. 2009. Effect of CYP1A inhibition on the biotransformation of benzo[a]pyrene in two populations of Fundulus heteroclitus with different exposure histories. Aquatic Toxicology 92(3):195–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, et al. 2015. Benzo-α-pyrene induced oxidative stress in Caenorhabditis elegans and the potential involvements of microRNA. Chemosphere 139:496–503. [DOI] [PubMed] [Google Scholar]
- Wyatt Lauren H., et al. 2017. Effects of methyl and inorganic mercury exposure on genome homeostasis and mitochondrial function in Caenorhabditis elegans. DNA Repair 52:31–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Tian, et al. 2004. Quinones and Aromatic Chemical Compounds in Particulate Matter Induce Mitochondrial Dysfunction: Implications for Ultrafine Particle Toxicity. Environmental Health Perspectives 112(14):1347–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Weiling, and Warshawsky David 2005. Metabolic activation of polycyclic and heterocyclic aromatic hydrocarbons and DNA damage: A review. Toxicology and Applied Pharmacology 206(1):73–93. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.