

Abstract
Mitochondria are dynamic organelles that can change shape and size depending on the needs of the cell through the processes of mitochondrial fission and fusion. In this work, we investigated the role of mitochondrial dynamics in organismal stress response. By using C. elegans as a genetic model, we could visualize mitochondrial morphology in a live organism with well‐established stress assays and well‐characterized stress response pathways. We found that disrupting mitochondrial fission (DRP1/drp‐1) or fusion (OPA1/eat‐3, MFN/fzo‐1) genes caused alterations in mitochondrial morphology that impacted both mitochondrial function and physiologic rates. While both mitochondrial fission and mitochondrial fusion mutants showed increased sensitivity to osmotic stress and anoxia, surprisingly we found that the mitochondrial fusion mutants eat‐3 and fzo‐1 are more resistant to both heat stress and oxidative stress. In exploring the mechanism of increased stress resistance, we found that disruption of mitochondrial fusion genes resulted in the upregulation of multiple stress response pathways. Overall, this work demonstrates that disrupting mitochondrial dynamics can have opposite effects on resistance to different types of stress. Our results suggest that disruption of mitochondrial fusion activates multiple stress response pathways that enhance resistance to specific stresses.
Keywords: C. elegans, genetics, mitochondria, mitochondrial dynamics, stress resistance
Abbreviations
- ANOVA
analysis of variance
- ATP
adenosine triphosphate
- cDNA
complementary deoxyribose nucleic acid
- cytoUPR
cytosolic unfolded protein response
- ER‐UPR
endoplasmic reticulum unfolded protein response
- EV
empty vector
- FUdR
5‐Fluoro‐2'‐deoxyuridine
- GFP
green fluorescent protein
- GTP
guanosine triphosphate
- IPTG
isopropyl β‐D‐1‐thiogalactopyranoside
- mitoUPR
mitochondrial unfolded protein response
- mRNA
messenger RNA
- NGM
nematode growth media
- PCR
polymerase chain reaction
- qPCR
quantitative PCR
- RNA
ribonucleic acid
- RNAi
RNA interference
- ROS
reactive oxygen species
- RT‐PCR
reverse transcriptase PCR
1. INTRODUCTION
Mitochondria are double‐membrane organelles responsible for many processes within the cell. The mitochondria are highly networked and can change shape and size in response to stress and energy demands of the cell. 1 , 2 Although most frequently known as the powerhouse of the cell due to their large ATP‐producing capacity, the mitochondria also have a role in responding to stress. 3 Mitochondrial fusion is the process of one or more mitochondria joining together; while mitochondrial fission is the process of one or more mitochondria breaking off of the network. Both processes are highly conserved and governed by a group of well‐conserved dynamin‐family GTPases. 4 These processes can occur in very short time frames, and one event is often followed by the other. 5
Mitochondrial fission is primarily governed by the GTPase DRP1, along with mitochondrial receptor proteins which recruit cytosolic DRP1 to the mitochondrial membrane. These mitochondrial receptor proteins include mitochondrial fission factors (MFF‐1/MFF‐2) and mitochondrial fission proteins (FIS‐1 and FIS‐2). Mitochondrial fission is important for growing and dividing cells to provide new mitochondria and is also important for removing damaged mitochondria from the cell, as it is a precursor to mitophagy. 6 , 7
Mitochondrial fusion is governed by the inner‐membrane fusion protein optic‐atrophy 1 (OPA1 in mammals, EAT‐3 in C. elegans) and the outer‐membrane fusion proteins mitofusin‐1 and mitofusion‐2 (MFN1 and MFN2 in mammals, FZO‐1 in C. elegans). Mitochondrial fusion allows for complementation to compensate for damage to individual mitochondria, functions as an antiapoptotic mechanism, and helps optimize mitochondrial bioenergetic capacity. 8 , 9 It should be noted that while EAT‐3 and FZO‐1 are both involved in the process of mitochondrial fusion, they perform different parts of this process. EAT‐3 is involved in the fusion of the inner‐mitochondrial membrane, while FZO‐1 is involved in the fusion of the outer‐mitochondrial membrane. In addition to its role in inner‐mitochondrial membrane fusion, EAT‐3 is also required for maintaining the structure of the mitochondrial cristae, which in turn allows for increased respiratory efficiency and prevents cytochrome c release.
The importance of mitochondrial dynamics to organismal function is indicated by the fact that primary mutations in mitochondrial fission and fusion genes cause a variety of neurological diseases. 10 Autosomal dominant optic atrophy is caused by a mutation in the OPA1 gene, 11 while mutations in MFN2 cause the most common form of Charcot‐Marie‐Tooth Disease. 12 Similarly, a mutation in DRP1 has been shown to cause encephalopathy and lactic acidosis, and is often lethal. 13
Alterations in mitochondrial dynamics have also been found in many neurodegenerative diseases and cancers. 10 , 14 For example, there is evidence of increased mitochondrial fragmentation in Alzheimer's, Parkinson's, and Huntington's disease. 15 , 16 However, it is still largely unknown whether disruptions in mitochondrial dynamics are causal to cellular dysfunction or secondary to other deficits. 17
The nature of how mitochondrial dynamics affect the cellular stress response is still unclear. It is also unclear how mitochondrial dynamics affect physiological rates in a whole organism and the extent to which changes in mitochondrial function and physiological rates contribute to stress resistance. While there have been elegant time‐course studies in live cells, it is still difficult to image mitochondrial dynamics in vivo in a whole organism. In this work, we use C. elegans to explore the relationship between mitochondrial dynamics and stress. Due to its transparent body, mitochondrial morphology can be visualized in a living organism by fluorescently labeling mitochondrially targeted proteins. In addition, C. elegans have well‐defined stress assays and well‐characterized stress response pathways.
We find that disruption of genes involved in mitochondrial fission and fusion alters mitochondrial morphology leading to changes in both mitochondrial function and physiologic rates. In addition, we find that mitochondrial fission and fusion mutants have increased resistance to specific stresses resulting from upregulation of stress response pathways. Together, this work highlights the importance of mitochondrial dynamics in organismal stress response.
2. MATERIALS AND METHODS
2.1. Strains
The following strains were utilized in these experiments:
WT/N2
eat‐3(tm1107)
drp‐1(tm1108)
fzo‐1(tm1133)
bcIs78(pMyo‐3::mitoGFP(matrixGFP) + pRF4)
fzo‐1(tm1133); bcIs78(pMyo‐3::mitoGFP(matrixGFP) + pRF4)
eat‐3(tm1107); bcIs78(pMyo‐3::mitoGFP(matrixGFP) + pRF4)
drp‐1(tm1108);bcIs78(pMyo‐3::mitoGFP(matrixGFP) + pRF4)
fis‐1(tm1867)
fis‐2(gk414)
fis‐1(tm1867);fis‐2(gk414)
mff‐1(tm2955)
mff‐2 (tm3041)
mff‐1(tm2955);mff‐2(tm3041)
zcIs13[Phsp‐6::GFP]
dvIs70[Phsp‐16.2::GFP,rol‐6(su1006)]
dvIs19[Pgst‐4::GFP::NLS]
iaIs7[Pnhr‐57::GFP]
muEx336[Pmtl‐1::RFP + rol‐6(su1006)]
daf‐16(mu86)
eat‐3(tm1107); daf‐16(mu86)
drp‐1(tm1108); daf‐16(mu86)
fzo‐1(tm1133); daf‐16(mu86)
Strains were grown on OP50 bacteria at 20°C. Strains were outcrossed a minimum of 5 generations to reduce nonspecific background mutations. All of the strains were genotyped to confirm the presence of the mutation in the gene of interest. tm1108 is a 424 bp deletion in the drp‐1 gene along with an 18 bp insertion. The deletion affects exons 2 and 3 of 8 exons of the drp‐1 gene and is predicted to affect all drp‐1 transcripts. tm1133 is a 419 bp deletion in the fzo‐1 gene along with a 14 bp insertion. The deletion affects exons 2‐4 of 8 exons of the fzo‐1 gene. tm1107 is a 417 bp deletion in the eat‐3 gene, which affects exons 5 and 6 of 9 exons.
2.2. Confocal imaging and quantification
Mitochondria were imaged and quantified as previously described. 18 Briefly, to image the mitochondria, a z‐stack of 17 images spaced 0.125 µm apart was collected. For the representative images shown in the figures, a maximum image projection was created in Nikon Elements to compress z‐stacks into a single image. All imaging conditions were kept the same for all images. For imaging worms under stress, worms were taken directly from stress plates and put on slides to avoid mitochondrial recovery. For quantification, a single representative slice for each z‐stack was used to avoid the complication of mitochondria being present in two planes. This slice was made binary using the Nikon Elements thresholding tool. First, a background subtraction of a constant 50 was applied. A threshold for the GFP signal was determined by applying the average threshold for mask creation of control images and was applied to all images. Mitochondrial circularity, number, and area were measured using the measure objects tool in Nikon Elements after the threshold was applied.
2.3. Oxygen consumption
Basal oxygen consumption rate was measured using a Seahorse XFe96 analyzer (Seahorse bioscience Inc, North Billerica, MA, USA). Synchronized worms were aged to the time point of interest and then cleaned in M9 buffer (22 mM KH2PO3, 34 mM NA2HPO3, 86 mM NaCl, and 1 mM MgSO4). Cleaned nematodes were pipetted in calibrant (at 30‐90 worms per well) into a Seahorse 96‐well plate. Oxygen consumption was measured six times and rates of respiration were normalized to the number of worms in each individual well. The plate readings were begun within 20 minutes of introduction of the worms into the well. Reading from each well was normalized relative to the number of animals per well. Well probes were hydrated in a 175 µL Seahorse calibrant overnight before this assay was begun. We found it is important to turn off the heating incubator to allow the Seahorse machine to reach room temperature before placing nematodes inside the machine.
2.4. ATP levels
Approximately 200 worms were age synchronized by a limited lay. Worms were collected in de‐ionized water, washed, and freeze‐thawed three times. The resulting pellet was sonicated in a Bioruptor (Diagenode) with 30 cycles of 30 seconds on, 30 seconds off. The pellet was boiled for 15 minutes to release ATP, and then spun at 4°C at 11,000 g for 10 minutes. The supernatant was collected and measured using a Molecular Probes ATP determination Kit (Life Technologies). Luminescence was normalized to protein content, which was measured with a Pierce BCA protein determination kit (Thermo Scientific). A minimum of three biological replicates per strain were performed.
2.5. Embryonic lethality
Approximately 20‐30 gravid adult worms were transferred to freshly seeded NGM plates to ensure a thin bacterial lawn. Worms were allowed to lay eggs for a limited period of time (~4‐6 hours) and then all adults were removed. The number of unhatched eggs and live progeny were quantified at 24 and 48 hours after the removal of adults.
2.6. Brood size
Brood size was determined by placing individual prefertile young adult animals onto NGM plates. Worms were transferred daily to new plates until progeny production ceased. The resulting progeny was allowed to develop to adulthood before quantification. Three replicates of 5 animals each were completed.
2.7. Postembryonic development and developmental arrest
Postembryonic development (PED) was assessed by moving ~ 100‐200 eggs to agar plates. After 3 h, newly hatched L1 worms were transferred to a new plate. The hours from hatching to the L4 to young adult transition was measured as the PED time. Worms that never reached the young adult stage were scored as undergoing developmental arrest. For developmental arrest, worms were checked for an additional 2 weeks after the average worm for the particular strain being examined developed to adulthood. For quantification of PED time, only animals that eventually reached adulthood were included. Three replicates of 20 animals each were completed.
2.8. Rate of movement
The rate of movement was assessed by measuring thrashing rate in liquid. Thrashing rates were determined manually by transferring 20 worms onto an unseeded agar plate. One milliliter of M9 buffer was added and the number of body bends per 30 seconds was counted for 5‐10 worms per strain for each of 3 biological replicates.
2.9. Defecation cycle
Defecation cycle length was determined by measuring the time between consecutive pBoc contractions in day 1 young adult worms with 5‐10 worms per replicate for each of three biological replicates. To minimize the effects of ambient laboratory temperature, defecation was measured on water filled chambers that had been incubated at 20°C and the lids of the plates containing the worms were not removed.
2.10. Stress assays
All stress assays were performed on prefertile, young adults, and at least 3 replicates for each assay were performed. Stress assays, especially the heat stress assay, can be very sensitive to the exact conditions during the assay (eg, the amount of time out of the incubator to check the worms, how long the door of the incubator was open, the exact temperature of the incubator). 19 As it is not uncommon to observe large variation in survival even within an experiment, all of the controls for each stress assay were always run at the same time as the experimental animals.
2.10.1. Oxidative stress
Resistance to acute oxidative stress was performed by transferring 20‐30 prefertile young adult worms to freshly prepared plates containing 300 µM juglone and survival was monitored hourly. Resistance to chronic oxidative stress was performed by transferring 20 prefertile young adult worms to freshly prepared plates containing 4 mM paraquat, and survival was monitored daily. For imaging, worms were exposed to 2 mM paraquat for 24 hours. Worms were then removed from paraquat plates and immediately transferred to agar pads.
2.10.2. Heat stress
Heat stress resistance was measured by transferring 20‐30 worms to a clean, seeded NGM plate in a 37°C incubator. Survival was monitored hourly. For imaging, worms were exposed to 37°C for 2 hours and immediately transferred to agar pads.
2.10.3. Osmotic stress
Osmotic stress was performed by transferring 20‐30 worms to freshly made plates containing 500mM NaCl. Survival was scored after 48 hours. For imaging, worms were exposed to 400 mM NaCl for 24 hours.
2.10.4. Anoxia
We assessed nematode resistance to low oxygen environments by utilizing the Becton‐Dickinson Bio‐Bag Environmental Chamber. Anaerobic atmosphere is achieved within the sealed Bio‐Bag using a self‐contained generator and resazurin indicator made up of an ampule of HCl solution and two gas‐generating tablets. The ampule was crushed, and the acid reacted with the tablets resulting in a gas mixture that absorbs oxygen in the presence of palladium. For imaging assays and qPCR, 20 animals were moved to freshly seeded NGM plates and placed in the anoxic chamber for 24 hours. Worms were transferred immediately after removal from the Bio‐Bag to agar pads for imaging. For survival assays, worms were removed from the Bio‐Bag after 48 hours and allowed to recover in normoxic conditions on seeded NGM plates for 24 hours. Worms were then scored for survival.
2.11. Measurement and quantification of reporter activity
Reporter activity in adult worms was assessed through measurement of whole worm fluorescence as previously described. 20 For each biological replicate, 8 animals were paralyzed with 2 mM levamisole, mounted on an NGM plate. Fluorescent images were captured using an AVT Stingray F145B camera and VimbaViewer 1.1.2 software and integrated density was quantified using ImageJ. The threshold for imaging was set independently for each reporter strain and thus the magnitude of increase cannot be directly compared between the different genes.
2.12. RNAi
All RNAi clones in these experiments were sequence verified. We have previously shown that the atfs‐1 RNAi clone knocks down the expression of atfs‐1 and its target gene hsp‐6. 21 We have also shown that the daf‐16 RNAi clone phenocopies a daf‐16 mutation. 22 To knockdown expression of genes, the RNAi clones were grown approximately 12 hours in LB with 50 μg/mL carbenicillin. Cultures were concentrated (5x) and seeded onto NGM plates containing 5 mM IPTG and 50 μg/mL carbenicillin. Plates were incubated to induce RNAi for 2 d at room temperature. RNAi was performed at 20°C. For daf‐16, atfs‐1, and hif‐1, treatment with RNAi was begun at the L4 stage of the parental generation, worms were transferred to a new plate the following day as gravid adults, and then removed after 24 hours. The resulting F1 progeny from these worms were used for analysis. In the case of hsf‐1 and skn‐1, this paradigm resulted in high embryonic lethality and poor growth among those worms that hatched. For these RNAi clones, RNAi treatment was begun at the egg stage of the experimental generation.
2.13. Quantitative real‐time RT‐PCR
mRNA was collected from prefertile young adult worms using Trizol as previously described. 20 We collected three biological replicates each for WT, drp‐1, fzo‐1, and eat‐3 worms. The mRNA was converted into cDNA using a High‐Capacity cDNA Reverse Transcription kit (Life Technologies/Invitrogen) according to the manufacturer's directions. qPCR was performed using a FastStart Universal SYBR Green kit (Roche) in an AP Biosystems RT‐PCR machine. 23 Primer sequences for the DAF‐16 target genes were published previously. 22 Primer sequences for other genes tested include: hsp‐6 (L‐CGCTGGAGATAAGATCATCG, R‐TTCACGAAGTCTCTGCATGG), gst‐4 (L‐CTGAAGCCAACGACTCCATT, R‐GCGTAAGCTTCTTCCTCTGC), hsp‐16.2 (L‐CCATCTGAGTCTTCTGAGATTGTT, R‐CTTTCTTTGGCGCTTCAATC), and nhr‐57 (L‐GACTCTGTGTGGAGTGATGGAGAG, R‐ GTGGCTCTTGGTGTCAATTTCGGG).
2.14. Statistical analysis
For all experiments, we completed a minimum of three biological replicates (independent population of worms tested on a different day). Statistical significance of differences between groups was determined by one‐way, two‐way, or repeated measures ANOVA using Graphpad Prism. For survival analysis, a log‐rank test was used. Error bars indicate standard error of the mean. Survival on paraquat plates was graphed using a Kaplan‐Meier plot and significance assessed using the log‐rank tested.
3. RESULTS
3.1. Disruption of mitochondrial fission and fusion genes causes alterations in mitochondrial morphology and function
In order to explore the relationship between mitochondrial dynamics and stress resistance, we obtained mutants with deletions in genes responsible for mitochondrial fission (drp‐1) or mitochondrial fusion (eat‐3, fzo‐1). Before examining stress resistance in these strains, we first sought to confirm the effect of these mutations on mitochondrial morphology and function.
To determine the effects of mutations in fission and fusion genes on mitochondrial morphology, we crossed worms with loss‐of‐function mutations in drp‐1(tm1108), eat‐3(tm1107), and fzo‐1(tm1133) to a strain that expresses GFP connected to the mitochondrial targeting sequence of the outer‐mitochondrial membrane protein TOM20 under the body wall muscle myo‐3 promoter. 24 We chose to examine mitochondrial morphology in body wall muscle as these are large cells in which we can visualize mitochondrial morphology in a living worm.
On day 1 of adulthood, we found that both mitochondrial fusion mutants exhibit altered mitochondrial morphology (Figure 1A). While wild‐type worms have tubular, interconnected networks of mitochondria, eat‐3 and fzo‐1 worms have round, fragmented mitochondria, as has been reported previously. 25 , 26 , 27 , 28 In these worms, the average area of individual mitochondria is significantly decreased compared to wild‐type worms (Figure 1B) and the total number of mitochondria is increased (Figure 1C), consistent with mitochondrial fragmentation. In addition, mitochondrial shape in eat‐3 and fzo‐1 worms is significantly more circular than in wild‐type worms (Figure 1D). While some groups have reported that disruption of drp‐1 results in hyperfused mitochondrial networks, 29 , 30 , 31 , 32 other groups have found that the mitochondria in drp‐1 mutants are similar to wild‐type. 27 , 28 In agreement with the latter observation, we did not observe any significant difference in mitochondrial morphology in drp‐1 mutants compared to wild‐type worms on day 1 of adulthood.
FIGURE 1.
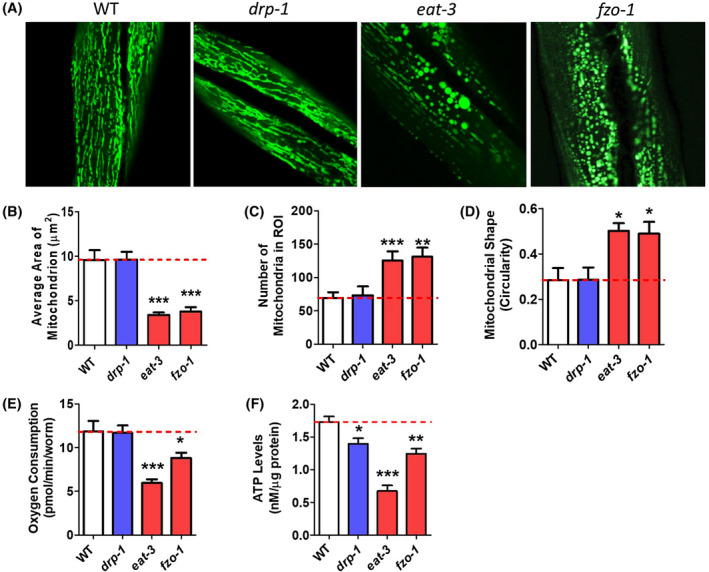
Disruption of mitochondrial fission and fusion genes causes alterations in mitochondrial morphology and function at day 1 of adulthood. A, Mitochondrial fusion mutants eat‐3 and fzo‐1 exhibit increased mitochondrial fragmentation compared to wild‐type worms. As a result, these worms have decreased mitochondrial area (B), increased number of mitochondria (C), and increased mitochondrial circularity (D) compared to wild‐type worms. eat‐3 and fzo‐1 mutants also have decreased oxygen consumption at day 1 of adulthood compared to drp‐1 or wild‐type worms (E). ATP production is decreased in mitochondrial fission and fusion mutants at day 1 of adulthood compared to wild‐type worms (F). Error bars represent SEM. Significance indicates difference from wild‐type worms. *P < .05, **P < .01, ***P < .001
In addition to morphological defects, we investigated basal mitochondrial function in these strains by assessing oxygen consumption and ATP levels in worms at day 1 of adulthood. We found that eat‐3 and fzo‐1 mutants exhibit decreased oxygen consumption per worm (Figure 1E) and decreased ATP levels (Figure 1F) compared to wild‐type worms. While drp‐1 mutants exhibited normal oxygen consumption, ATP levels show a small reduction compared to wild‐type worms.
To examine the effect of aging on mitochondrial morphology and function in the mitochondrial dynamics mutants, we aged worms to day 7 of adulthood. We observed age‐dependent changes in mitochondrial morphology in all strains (Figure 2A). Compared to mitochondria in worms on day 1 of adulthood, mitochondria in day 7 adult worms have decreased area (Figure 2B) and increased circularity (Figure 2D), as others have previously noted. 33 We also observed age‐dependent changes that were genotype specific. Mitochondrial area in aged drp‐1 worms exhibited a trend toward increase compared to wild‐type worms (Figure 2B). Compared to wild‐type worms, aged eat‐3 mutants have an increase in both number of mitochondria (Figure 2C) and mitochondrial circularity (Figure 2D), both of which are consistent with increased fragmentation.
FIGURE 2.
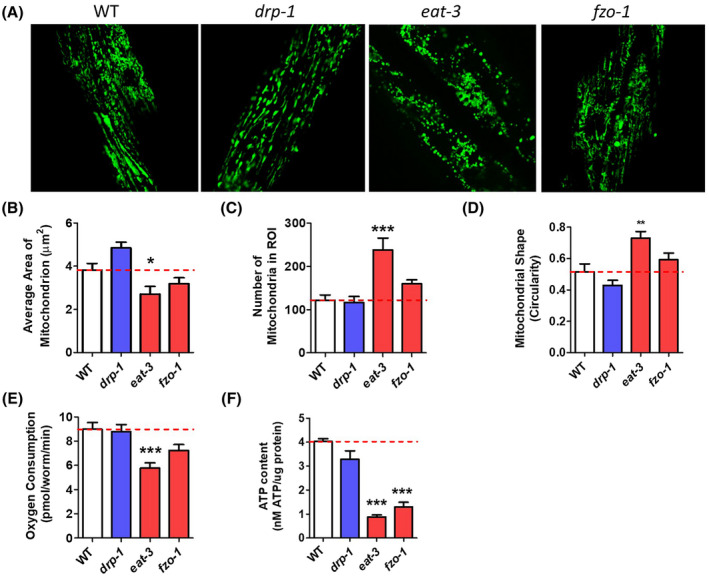
Disruption of mitochondrial fission and fusion genes causes alterations in mitochondrial morphology and function at day 7 of adulthood. A, Mitochondrial fragmentation increases with age in all strains. At day 7, eat‐3 mutants still have decreased mitochondrial size (B), increased mitochondrial number (C), increased mitochondrial circularity (D), decreased oxygen consumption (E), and decreased levels of ATP (F). fzo‐1 mutants show a trend toward similar changes but only the deficit in ATP levels is significant. Quantification of mitochondrial morphology in drp‐1 mutants shows no significant differences from wild‐type worms, although there is a trend toward increased mitochondrial area. Error bars represent SEM. Significance indicates difference from wild‐type worms. *P < .05, **P < .01, ***P < .001
To investigate mitochondrial function on day 7 of adulthood, we measured oxygen consumption and ATP levels. We found that although oxygen consumption decreased in all strains with age, the overall pattern of decreased oxygen consumption in the mitochondrial fusion mutants, eat‐3 and fzo‐1, was observed at both time points (Figure 2E). Similarly, eat‐3 and fzo‐1 worms also have decreased ATP levels compared to wild‐type worms at the aged time point (Figure 2F). Combined, these results show that disrupting genes involved with mitochondrial fission and fusion cause predictable changes in mitochondrial morphology and that these changes affect mitochondrial function.
3.2. Disruption of mitochondrial fission and fusion genes causes a slowing of physiologic rates
Having shown that mitochondrial morphology and function are disrupted in mitochondrial fission and fusion mutants, we investigated whether these deficits lead to an alteration of physiologic rates. Previous studies have demonstrated that mutations affecting the mitochondria often cause a slowing of physiologic rates. 20 , 21 , 34 , 35 , 36 While wild‐type worms exhibit essentially no embryonic lethality, we found that almost 50% of drp‐1 eggs and over 10% of eat‐3 and fzo‐1 eggs fail to hatch indicating an increased rate of embryonic lethality (Figure 3A). Of those eggs that hatch, 10%‐15% of eat‐3 mutants arrest during development—these worms never develop to adulthood (Figure 3B). Examination of self‐brood size revealed that drp‐1, eat‐3 and fzo‐1 worms all have markedly decreased fertility, producing less than 100 progeny per worm, whereas wild‐type worms produce approximately 300 progeny per worm (Figure 3C). The postembryonic development time was also impacted in all three strains: drp‐1 worms developed slower than wild‐type worms, fzo‐1 worms developed slower than drp‐1 worms, and eat‐3 worms developed markedly slower than even fzo‐1 worms (Figure 3D) (note that these development times do not include the worms that arrested, which were quantified in Figure 3B). Once these mutants reach adulthood, we observed a mild deficit in the rate of movement, as measured by thrashing in liquid, in drp‐1 and eat‐3 worms (Figure 3E). This is consistent with what others have reported. 26 , 28 Finally, we found that defecation cycle length is prolonged in all of the fission and fusion mutants (Figure 3F). As with development time, the deficit was greatest in eat‐3 mutants and mildest in drp‐1 worms. Overall, characterization of the physiologic rates of these worms demonstrates that the deficits observed in mitochondrial function are large enough to impact worm physiology.
FIGURE 3.
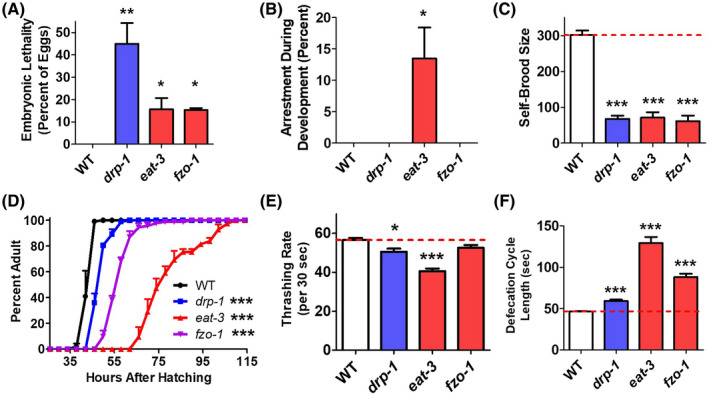
Disruption of mitochondrial fission and fusion genes causes a slowing of physiologic rates. A, Mitochondrial fission and fusion mutants show an increase in embryonic lethality compared to wild‐type worms. B, After hatching, a significantly increased proportion of eat‐3 mutants fail to develop to adulthood. C, The total number of progeny produced from drp‐1, eat‐3, and fzo‐1 worms is markedly decreased compared to wild‐type worms. D, The mitochondrial fission and fusion mutant strains also exhibit slow postembryonic development. E, The rate of movement (thrashing rate) is decreased in drp‐1 and eat‐3 mutants compared to wild‐type worms. F, Defecation cycle is significantly elongated in mitochondrial fission and fusion mutants. Overall, both the mitochondrial fission and fusion mutant strains exhibit slow physiologic rates. Error bars represent SEM. Significance indicates difference from wild‐type worms. *P < .05, **P < .01, ***P < .001
3.3. Exposure to stress causes mitochondrial fragmentation and increased expression of mitochondrial fission genes
Having shown that mutations in drp‐1, eat‐3, and fzo‐1 disrupt mitochondrial dynamics in a predictable manner, we could then utilize these mutants to examine the role of mitochondrial fission and fusion in responding to various types of stress. Before quantifying stress resistance in these mutant strains, we examined how mitochondrial morphology is affected by different types of stress.
To characterize how mitochondrial morphology changes in response to stress, we treated worms with a mitochondrial matrix targeted GFP (Pmyo‐3::mitoGFP) with different stresses that are known to influence the function and survival of worms. 37 We utilized four different stresses: oxidative stress through exposure to 2 mM paraquat for 24 hours, heat stress through exposure to 37°C heat for 2 hours, osmotic stress through exposure to 400 mM NaCl for 24 hours, and complete anoxia for 24 hours. These specific stress paradigms have been previously utilized by our laboratory as well as several others. 38 , 39 , 40 , 41 , 42 , 43 , 44 In this experiment, we chose to expose worms to a shorter duration or smaller dose of stress than we would use in a test of stress resistance to ensure that all worms would survive, and so that we could observe the effect of stress, rather than dying, on mitochondrial morphology. Importantly, we have previously used fluorescent reporter strains to determine which stress response pathways are activated by each of these stress response paradigms and found that these stresses activated the predicted stress response pathways (ie, oxidative stress activated a Psod‐3::GFP reporter strain). 45 However, it should be noted that some stresses activated multiple stress activated reporter strains, and some stress‐responsive reporter strains were activated by multiple stresses.
Because mitochondrial morphology can change rapidly in response to stress, we transferred worms directly from stress plates to the slide for imaging of live worms. We found that all four types of stress resulted in clear changes in mitochondrial morphology. While there are differences in the way that mitochondrial morphology was altered in response to the different types of stress, all four stresses increased mitochondrial fragmentation (Figure 4A). Quantification of mitochondrial morphology revealed an increase in the number of mitochondria (Figure 4B), a decrease in the average cross‐sectional area of mitochondria (Figure 4C), and an increase in mitochondrial circularity (Figure 4D), all of which are consistent with increased mitochondrial fragmentation.
FIGURE 4.
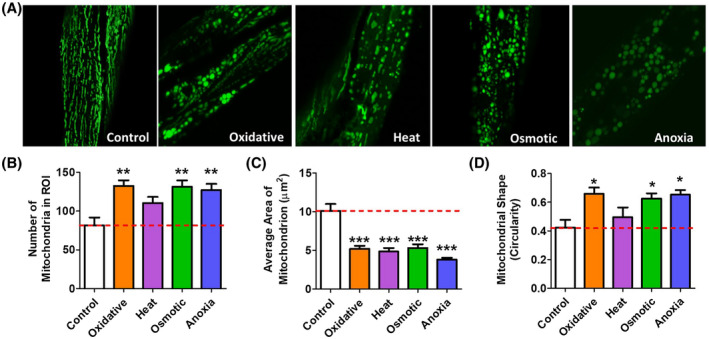
Exposure to multiple types of stress causes mitochondrial fragmentation. Worms were exposed to four different stresses and the morphology of the mitochondria was immediately quantified. Oxidative stress was induced by exposing worms to 2 mM paraquat for 24 hours. Heat stress was induced at 37°C for 2 hours, osmotic stress was induced at 400 mM NaCl for 24 hours. Anoxia was induced for 24 hours. A. All four types of stress caused mitochondrial fragmentation. Quantification of mitochondrial morphology revealed that exposure to exogenous stress can cause an increase in mitochondrial number (B), a decrease in mitochondrial size (C), and an increase in mitochondrial circularity (D). Error bars represent SEM. Significance indicates difference from wild‐type worms. *P < .05, **P < .01, ***P < .001
3.4. Disruption of mitochondrial fission and fusion alters resistance to stress
Exogenous stress can cause damage to the mitochondria. Mitochondrial fusion can mitigate the effects of that damage by forming mitochondrial networks that facilitate complementation, while mitochondrial fission can help to remove damaged mitochondria by facilitating mitophagy. Accordingly, we sought to determine the effect of disrupting mitochondrial fission or fusion on resistance to different types of stress. Based on the importance of mitochondrial fission and fusion for mitophagy and complementation, respectively, we hypothesized that mutations in these genes would decrease stress resistance in worms. To test this hypothesis, we treated drp‐1, eat‐3, and fzo‐1 worms with different types of stress and measured survival.
First, we examined resistance to oxidative stress by exposing worms to 300 µM juglone. Surprisingly, we found that all three mutants showed increased resistance to oxidative stress compared to wild‐type worms (Figure 5A). Resistance to oxidative stress was mildly increased in drp‐1 mutants but much increased in fzo‐1 and eat‐3 mutants. To ensure that this result was not specific to how oxidative stress was administered, we treated worms with 4 mM paraquat. Again, under these conditions we found that eat‐3 and fzo‐1 worms had markedly increased resistance to oxidative stress, while oxidative stress resistance was mildly increased in drp‐1 worms (Figure 5B).
FIGURE 5.
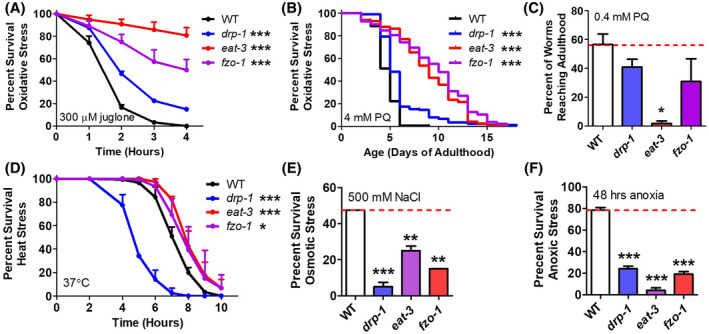
Disruption of mitochondrial fission and fusion genes alters resistance to stress. A. Mitochondrial fission and fusion mutants exhibit increased resistance to oxidative stress compared to wild‐type worms in acute (A, 300 µM juglone) and chronic (B, 4 mM paraquat) assays during adulthood. C. In contrast, eat‐3 worms fail to develop to adulthood under conditions of oxidative stress (0.4 mM paraquat) that are well tolerated by wild‐type worms. D. While mitochondrial fusion mutants show increased resistance to heat stress, mitochondrial fission mutants are markedly more sensitive than wild‐type worms. Both mitochondrial fission and fusion mutants are more sensitive to osmotic stress (E, 500 mM NaCl) and anoxia (F, 0% oxygen, 48 hours) than wild‐type worms. Error bars represent SEM. Significance indicates difference from wild‐type worms. *P < .05, **P < .01, ***P < .001
As a previous study reported increased sensitivity to oxidative stress in eat‐3 mutants, 25 we measured sensitivity to oxidative stress during development using a similar paradigm to what was previously published. Consistent with the previous report, we found that although eat‐3 adults are more resistant to oxidative stress during adulthood, eat‐3 worms are sensitive to oxidative stress during development, as far fewer eat‐3 worms develop to adulthood under conditions of oxidative stress (0.4 mM paraquat) than wild‐type worms (Figure 5C). It should be noted, however, that the development time of eat‐3 worms is much longer than wild‐type worms, and thus to develop to adulthood they are exposed to oxidative stress for a longer duration than wild‐type worms.
To determine whether the increased stress resistance in the fission and fusion mutants is specific to oxidative stress, we examined survival after exposure to other types of stress. Heat stress resistance was assessed by exposing worms to 37°C heat stress. Under these conditions, we found that eat‐3 and fzo‐1 worms show increased resistance to heat stress, while drp‐1 show markedly decreased resistance (Figure 5D). In contrast to other types of stress, but consistent with our initial hypothesis, both fission and fusion mutants were sensitive to 500 mM NaCl osmotic stress and 48 hours of anoxia (Figure 5E, F). Taken together, these results suggest that the requirement of mitochondrial fission and fusion for the proper stress response is different for different stresses.
To determine the extent to which the changes in stress resistance in drp‐1 mutants resulted from a disruption of mitochondrial fission, we examined resistance to stress in mutants with deletions in other genes involved in mitochondrial fission: fis‐1, fis‐2, mff‐2, and mff‐2. As in drp‐1 mutants, we found that disrupting the genes encoding either the mitochondrial fission proteins (fis genes) or the mitochondrial fission factors (mff genes) resulted in increased resistance to oxidative stress (Figure 6A,B). Similar to drp‐1 worms, fis and mff mutants also exhibited increased sensitivity to heat stress (Figure 6C), osmotic stress (Figure 6D), and anoxia (Figure 6E). Combined these results indicate that disruption of mitochondrial fission or fusion causes altered stress resistance.
FIGURE 6.
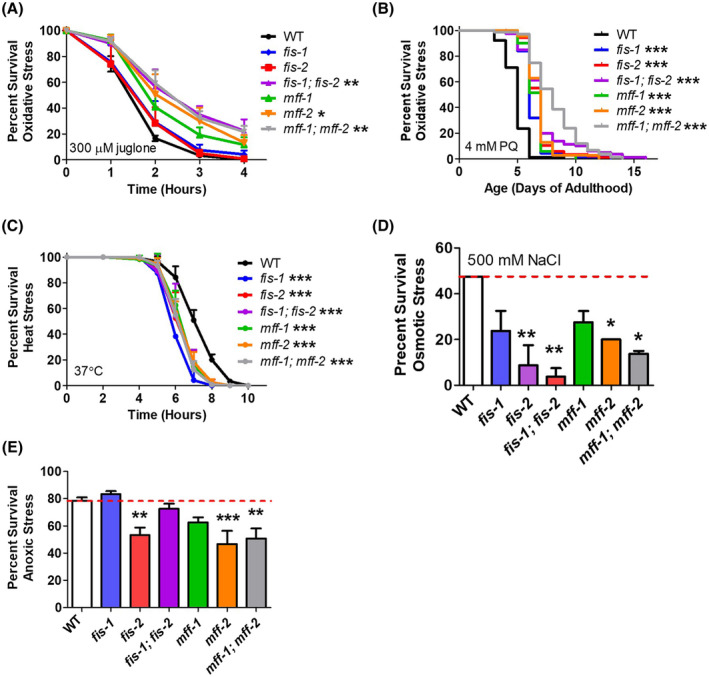
Mutants of fission accessory genes have altered resistance to stress. As with the mitochondrial fission gene drp‐1, eletion of mitochondrial fission accessory genes results in increased resistance to oxidative (A,B) stress, but increase sensitivity to heat stress (C), osmotic stress (D), and anoxia (E). Error bars represent SEM. Significance indicates difference from wild‐type worms. *P < .05, **P < .01, ***P < .001
3.5. Disruption of mitochondrial fission and fusion genes activates stress response pathways
In order to explore the mechanism underlying the increased resistance to stress in the mitochondrial fission and fusion mutants, we hypothesized that the disruption of mitochondrial function triggered an upregulation of stress response pathways as we have observed in long‐lived mitochondrial mutants. 20 , 21 , 34 To examine the activation of stress response pathways, we crossed drp‐1, eat‐3, and fzo‐1 to fluorescent reporter strains. We examined the mitochondrial unfolded protein response (mitoUPR; Phsp‐6::GFP), 46 the SKN‐1‐mediated oxidative stress response (Pgst‐4::GFP), 47 the cytosolic unfolded protein response (cytoUPR; also known as the heat‐shock response) using the Phsp16.2::GFP reporter, 48 the hypoxia response (Pnhr‐57::GFP), 49 and the DAF‐16‐mediated stress response (Pmtl‐1::RFP). 50
We observed increased activation of the Phsp‐6::GFP reporter in both eat‐3 and fzo‐1 worms indicating an activation of the mitoUPR (Figure 7A). Similarly, we found that drp‐1, eat‐3, and fzo‐1 worms exhibit increased fluorescence from the Pgst‐4::GFP reporter suggesting activation of the SKN‐1‐mediated oxidative stress response (Figure 7B) (although it is also possible that the gst‐4 promoter is activated by epidermal growth factor signaling 51 ). While we did not observe any fluorescence from the Phsp‐16.2::GFP reporter in any of the mutants under basal conditions, we did observe increased activation of this reporter in response to heat in eat‐3 and fzo‐1 worms compared to wild‐type worms (Figure 7C). We also observed increased activation of the Pnhr‐57::GFP reporter in eat‐3 and fzo‐1 worms, which is consistent with activation of the hypoxia stress response pathway (Figure 7D). Finally, Pmtl‐1::RFP reporter activity was increased in eat‐3 and fzo‐1 mutants indicating activation of the DAF‐16‐mediated stress response (Figure 7E).
FIGURE 7.
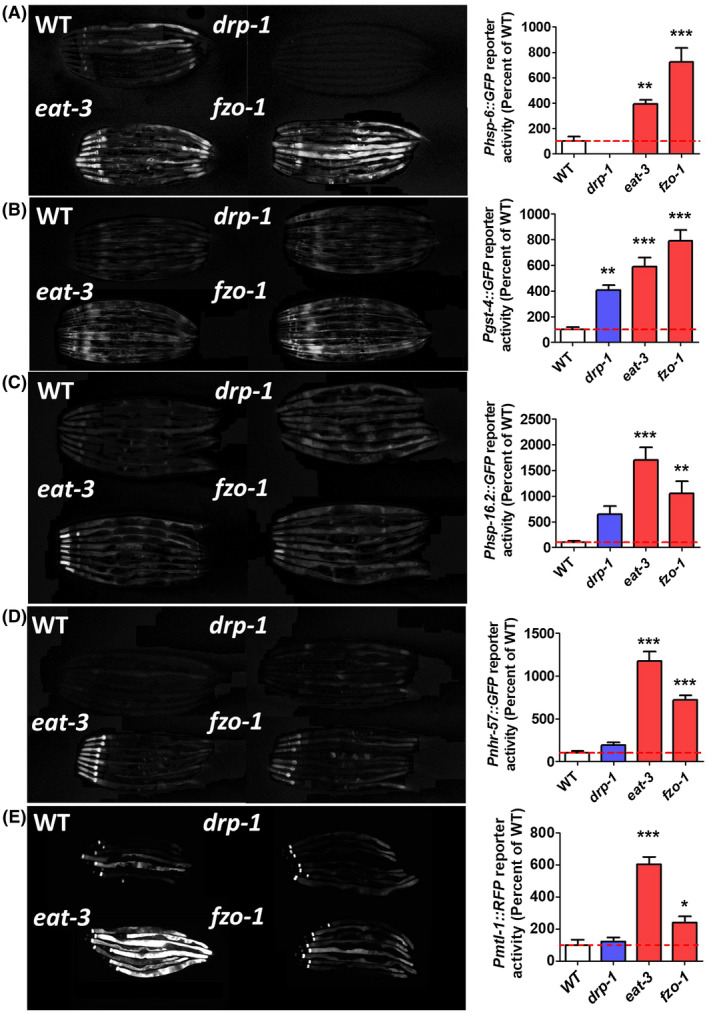
Disruption of mitochondrial fission and fusion genes activates stress response pathways. To examine the activation of stress response pathways, mitochondrial fission and fusion mutants were crossed to fluorescent reporter strains for the mitochondrial unfolded protein response (A, Phsp‐6::GFP), the SKN‐1‐mediated oxidative stress response (B, Pgst‐4::GFP), the cytosolic unfolded protein response (C, Phsp‐16.2::GFP), the HIF‐1‐mediated hypoxia response (D, Pnhr‐57::GFP), and the DAF‐16‐mediated stress response (E, Pmtl‐1::RFP). For all of these stress response pathways, both mitochondrial fusion mutants, eat‐3 and fzo‐1, showed increased activation compared to wild‐type worms. Note that the Phsp‐16.2::GFP reporter was induced by a mild 35°C heat stress. Error bars represent SEM. Significance indicates difference from wild‐type worms. **P < .01, ***P < .001
In order to confirm the results that we obtained using the fluorescent reporter strains, we measured the expression levels of the same genes in prefertile young adult worms using quantitative real‐time RT‐PCR. As with the fluorescent reporter strains, we found that eat‐3 worms have significantly increase levels of hsp‐6, gst‐4, nhr‐57, and mtl‐1 (Figure S1). Similarly, fzo‐1 worms showed a significant increase in the expression of gst‐4 and mtl‐1 (Figure S1). Combined with the results from the fluorescent reporter strains, these results indicate that multiple stress response pathways are activated in the mitochondrial fusion mutants.
3.6. Specific stress response pathways are required for enhanced stress resistance in mitochondrial fission and fusion mutants
To determine the extent to which each stress response pathway contributes to the increased resistance to oxidative and heat stress observed in the mitochondrial fission and fusion mutant strains, we used RNAi to knockdown the transcription factors known to mediate each stress response pathway and measured sensitivity to stress. ATFS‐1 (activating transcription factor associated with stress) is the transcription factor that is responsible for the activation of the mitoUPR. 52 Similarly, SKN‐1, HSF‐1, HIF‐1, and DAF‐16 are stress‐responsive transcription factors that mediate the SKN‐1‐mediated oxidative stress response, the cytoUPR, the hypoxia response, and the DAF‐16‐mediated stress response, respectively. 53 , 54 , 55 , 56
First, we examined the effect of knocking down these transcription factors on resistance to oxidative stress by exposing worms to 300 µM juglone. We found that inhibiting the mitoUPR (atfs‐1 RNAi) decreased oxidative stress resistance in drp‐1, eat‐3, and fzo‐1 worms (Figure 8) (a trend towards decrease was also observed in wild‐type worms). Inhibition of the SKN‐1‐mediated oxidative stress response (skn‐1 RNAi) paradoxically resulted in increased resistance to oxidative stress in wild‐type, drp‐1, and fzo‐1 worms, while significantly decreasing oxidative stress resistance in eat‐3 worms (Figure 8). RNAi against hsf‐1 to inhibit the cytoUPR caused decreased resistance to oxidative stress in WT, drp‐1, and eat‐3 worms (Figure 8). Blocking the hypoxia response through hif‐1 RNAi decreased resistance to oxidative stress in fzo‐1 worms (Figure 8). Finally, RNAi against daf‐16 significantly decreased resistance to oxidative stress in fzo‐1 worms (Figure 8), although a trend toward decreased stress resistance was also observed in eat‐3 worms. Since RNAi can be variable and knockdown can be incomplete, we also examined the effect of a daf‐16 null mutation (mu86). In this case we found that the disruption of daf‐16 significantly decreased resistance to oxidative stress in both eat‐3 and fzo‐1 worms (Figure S2).Combined, these results indicate that inhibition of specific stress response pathways results in decreased resistance to oxidative stress, and suggests that the upregulation of these pathways may contribute to the enhanced resistance to oxidative stress observed in the mitochondrial fission and fusion mutants.
FIGURE 8.
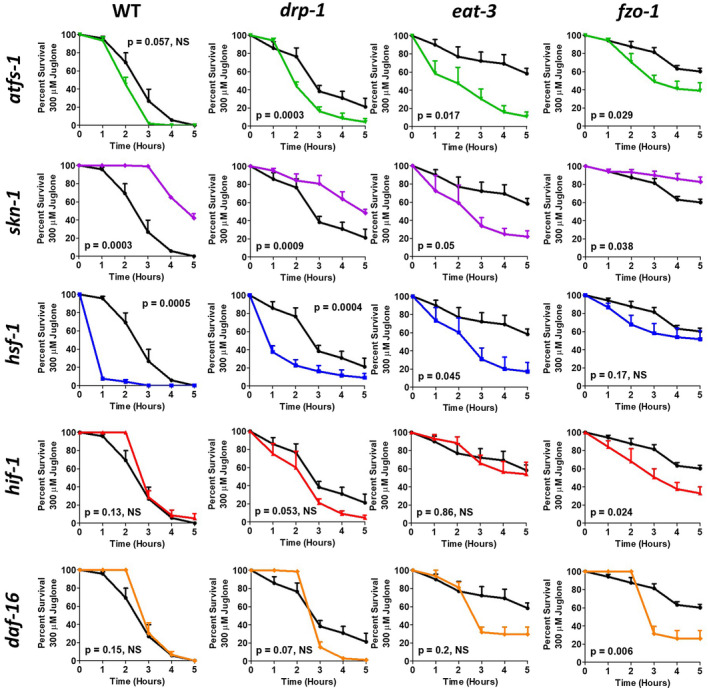
Increased resistance to oxidative stress in mitochondrial fission and fusion mutants requires stress‐responsive transcription factors. To assess the contribution of different stress response pathways to the enhanced oxidative stress resistance in drp‐1, eat‐3, and fzo‐1 worms, worms were treated with RNAi targeting the transcription factor response for mediating each pathway. atfs‐1 RNAi decreased oxidative stress resistance in drp‐1, eat‐3, and fzo‐1 worms. RNAi against skn‐1 increased oxidative stress resistance in wild‐type, drp‐1, and fzo‐1 worms but decreased it in eat‐3 worms. hsf‐1 RNAi decreased resistance to oxidative stress in all strains, except fzo‐1, which only exhibited a trend toward decreased resistance. RNAi against hif‐1 significantly decreased resistance to oxidative stress in fzo‐1 worms. daf‐16 RNAi only significantly decreased oxidative stress resistance in fzo‐1 worms. Combined this indicates that there are multiple stress response pathways contributing to the enhanced oxidative stress resistance in the mitochondrial fission and fusion mutants. Error bars indicate SEM. Black line indicates empty vector (EV), green line indicates atfs‐1 RNAi, purple line indicates skn‐1 RNAi, blue line indicates hsf‐1 RNAi, red line indicates hif‐1 RNAi, and orange line indicates daf‐16 RNAi
Second, we studied the impact of knocking down the stress‐responsive transcription factors on resistance to heat stress at 37°C. Inhibition of the mitoUPR decreased heat stress resistance in fzo‐1 worms (Figure 9). RNAi against skn‐1 did not decrease resistance to heat stress in any strain (Figure 9). As might be predicted based on the function of HSF‐1, disrupting the cytoUPR through hsf‐1 RNAi decreased resistance to heat stress in wild‐type, eat‐3, and fzo‐1 worms (Figure 9). As with skn‐1 RNAi, RNAi against hif‐1 did not reduce heat stress resistance in any strain (Figure 9). Similarly, daf‐16 RNAi did not affect heat stress resistance in any strain; however, daf‐16 deletion did result in a small but significant decrease in heat stress resistance in eat‐3 worms (Figure S2). Combined these results indicate that the mitoUPR, the cytoUPR, and to a lesser extent the DAF‐16‐mediated stress response help protect against heat stress, and suggest that the upregulation of these pathways may contribute to the enhanced heat stress resistance in the mitochondrial fusion mutants.
FIGURE 9.
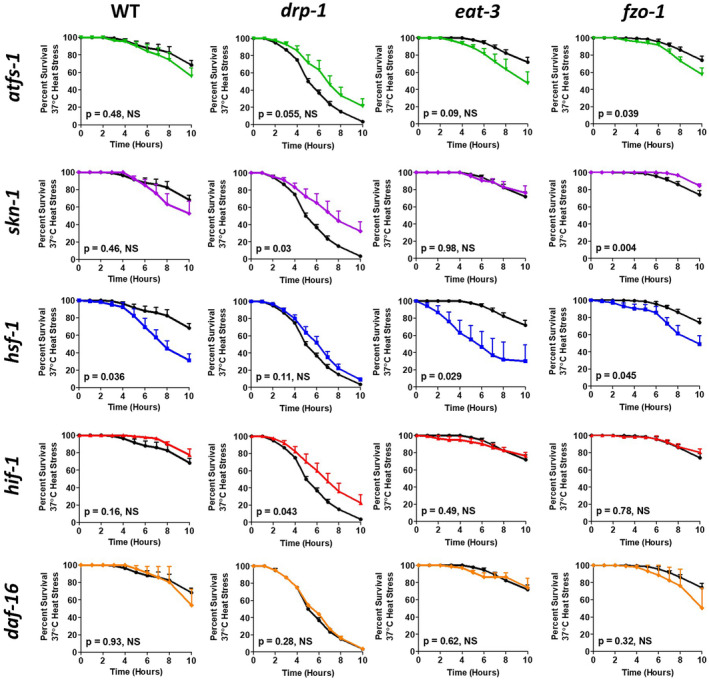
Increased resistance to heat stress in mitochondrial fusion mutants requires stress‐responsive transcription factors. To assess the contribution of different stress response pathways to the enhanced heat stress resistance in eat‐3 and fzo‐1 worms, worms were treated with RNAi targeting the transcription factor response for mediating each pathway. atfs‐1 RNAi decreased heat stress resistance in fzo‐1 worms. skn‐1 RNAi increased resistance to heat stress in drp‐1 mutants. RNAi against hsf‐1 decreased heat stress resistance in wild‐type, eat‐3, and fzo‐1 worms but did not affect drp‐1 worms. Neither hif‐1 RNAi nor daf‐16 RNAi decreased resistance to heat stress in any strain. Combined this indicates that there are multiple stress response pathways contributing to the enhanced heat stress resistance in the mitochondrial fusion mutants. Error bars indicate SEM. Black line indicates empty vector (EV), green line indicates atfs‐1 RNAi, purple line indicates skn‐1 RNAi, blue line indicates hsf‐1 RNAi, red line indicates hif‐1 RNAi, and orange line indicates daf‐16 RNAi
4. DISCUSSION
4.1. Exogenous stress causes mitochondrial fragmentation
In this work, we have characterized in a live organism how mitochondrial morphology changes in response to different stresses. We show that in response to multiple types of stresses, mitochondrial fragmentation occurs. Consistent with our findings, it has been shown that hypoxia also causes mitochondrial fragmentation. 57 Mitochondrial fragmentation may help the organism recover from stress by allowing for the removal of damaged mitochondria through mitophagy. Mitochondrial fragmentation may also be beneficial by facilitating transport of mitochondria in the cell possibly to compartments of the cell that require additional energy to recover from the stress.
In contrast to our findings, it has previously been reported that in response to oxidative stress, mitochondria can undergo stress‐induced hyperfusion in a mouse myoblast cell line. 58 Mitochondrial hyperfusion may help in responding to stress by allowing for complementation to compensate for damage that occurs in individual mitochondria. While our work did not address this, it is possible that both mitochondrial fragmentation and hyperfusion can occur in response to stress depending on the dose, duration, and nature of the stress. One possibility is that when the exogenous stress damages the mitochondria past the point at which complementation will be beneficial, the mitochondria undergoes fission and mitophagy to remove it from the cell. It is also possible that both mitochondrial fission and fusion occur after stress but at different time points or in different cell types.
4.2. Disruption of mitochondrial fission or fusion affects mitochondrial morphology and function leading to slow physiologic rates
While mitochondria have long been known to have an essential role in energy production, the effect of mitochondrial fission and fusion on mitochondrial function is not well understood. To address this, we examined mitochondrial morphology and function throughout age. Consistent with what was observed previously, 25 , 26 , 27 , 28 we found that disrupting genes involved in mitochondrial fusion resulted in an increase in mitochondrial fragmentation. The increase in mitochondrial fragmentation in the mitochondrial fusion mutants resulted in a decrease in oxygen consumption and decreased ATP production, suggesting that isolated mitochondria are less efficient than networked mitochondria or that continuous mixing of mitochondrial contents through sequential mitochondrial fusion and fission is important in optimizing mitochondrial function.
However, it should be noted that there have been varying results in the literature with respect to oxygen consumption and ATP levels in the mitochondrial fission and fusion mutants. Previous work reported that basal oxygen consumption is either normal or increased in drp‐1 worms and normal in fzo‐1 mutants, and that ATP levels are normal in drp‐1 and fzo‐1 mutants. 26 , 27 , 59 The reason for the differences could result from the fact that many of the experimental parameters were different between our study and previous studies. For example, different stages of worms were used (L4 vs young adult), different normalization was used (per protein vs per worm), and different methods of quantifying ATP were used (lucigenin‐based assay vs HPLC). Combined, this suggests that these outcome measures are sensitive to experimental conditions and future work will need to establish the precise conditions under which these mutants show normal or decreased oxygen consumption and ATP levels.
Disruption of genes involved in mitochondrial dynamics also had marked effects on physiologic rates. During development new mitochondria must be produced as cell number is increased and this requires mitochondrial fission. Consistent with this, we observed a high rate of embryonic lethality and low brood size in drp‐1 mitochondrial fission mutants. We also observed increased embryonic lethality, and decreased brood size in both mitochondrial fusion mutants, suggesting that mitochondrial fusion is also important during development. The mitochondrial dynamics mutants also showed slow development, slow movement (thrashing rate), and a slow rate of defecation. These phenotypes have also been observed in other mitochondrial mutants such as clk‐1, isp‐1, and nuo‐6, 20 , 21 , 34 , 35 , 36 and may be at least partially explained by the observed reduction in ATP levels.
4.3. Disrupting mitochondrial dynamics causes changes in resistance to stress
While our work and the work of others has shown that mitochondrial morphology changes in response to stress, 7 , 57 , 60 the precise role of mitochondrial fission and fusion in responding to different types of stress is not completely understood. Surprisingly, we found that disrupting mitochondrial dynamics has a complicated impact on resistance to stress. It can increase resistance to some stresses, including oxidative stress and heat stress, while decreasing resistance to other stresses, including osmotic stress and anoxia. The differential effect on different types of stress could potentially be explained by the observation that increasing resistance to one type of stress can result in decreased resistance to other types, 61 perhaps due to redistribution of limited cellular resources. It was also surprising to note that disrupting mitochondrial fission and mitochondrial fusion only affected stress resistance in opposite directions for heat stress but similarly affected resistance to oxidative, osmotic, and anoxic stress.
4.4. Mutations in mitochondrial fusion genes increase resistance to stresses through activation of stress response pathways
Having observed an increase in resistance to stress following disruption of mitochondrial fusion genes, we hypothesized that disrupting mitochondrial fusion might result in an activation of stress response pathways. There are a number of genetic pathways that are activated in response to stress that alter gene expression to help the organism survive. While some types of stress, such as oxidative stress, activate many stress response pathways, others activate few. 45 We examined five different stress response pathways including the mitoUPR, the SKN‐1‐mediated oxidative stress response, the cytoUPR (also known as the heat‐shock response), the HIF‐1‐mediated hypoxia response, and the DAF‐16‐mediated stress response. We found that all of these stress response pathways are activated by the disruption of mitochondrial fusion genes. We found that the SKN‐1‐mediated oxidative stress response is also activated in drp‐1 mutants, which may explain the increased resistance to oxidative stress in these worms.
Our work and others have shown that mutations leading to mild impairment of mitochondrial function can result in increased lifespan 35 , 62 , 63 , 64 and increased resistance to stress. 20 , 21 , 34 , 65 At least in some of these mutants, our results indicated that the activation of stress response pathways, namely, the mitoUPR, is required for both the increase in lifespan and resistance to stress. 21
Our results suggest that the mitochondrial and cytosolic unfolded protein responses have the greatest impact on stress resistance as RNAi against atfs‐1 or hsf‐1 decreased resistance to both oxidative and heat stress in most strains. This is interesting as our previous work suggested that the mitoUPR is activated in response to oxidative stress but not heat stress and the cytoUPR is activated in response to heat stress but not oxidative stress. 45
Finally, we found that inhibiting the SKN‐1 oxidative stress response can increase resistance to both oxidative stress and heat stress. This was a surprising observation since SKN‐1 protects against oxidative stress. 66 This could potentially be explained if skn‐1 RNAi leads to a mild increase in reactive oxygen species, which we and others have shown to be sufficient to increase lifespan and resistance to stress. 21 , 35 , 67 Combined our results show that knocking down stress response pathways can decrease resistance to heat and oxidative stress and suggest that the mitoUPR, the cytoplasmic UPR, and the DAF‐16‐mediated stress response all contribute to the increased stress resistance in the mitochondrial fusion mutants.
5. CONCLUSIONS
In this work we explore the relationship among mitochondrial dynamics, morphology, and function, and how disrupting these processes affect physiologic rates and resistance to stress. Although disruption of mitochondrial fission has little impact on mitochondrial morphology and function, this process is important for development and surviving multiple types of stress. Disrupting mitochondrial fusion induces mitochondrial fragmentation and decreases energy production leading to slow physiologic rates. Surprisingly, inhibiting mitochondrial fusion can increase resistance to specific stresses through the activation of stress response pathways. Overall, our work demonstrates the importance of mitochondrial dynamics for mitochondrial function as well as physiologic rates and stress resistance of the whole organism.
CONFLICT OF INTEREST
The authors have declared that no competing interests exist.
AUTHOR CONTRIBUTIONS
Conceptualization: E. Machiela, J.M. Van Raamsdonk. Methodology: E. Machiela, M.M. Senchuk, J.M. Van Raamsdonk. Investigation: E. Machiela, T. Liontis, D.J. Dues, P.D. Rudich, A. Traa, L. Wyman, J.F. Cooper, L. Lew, S. Nadarajan, M.M. Senchuk, J.M. Van Raamsdonk. Writing – original draft: E. Machiela. Writing – review and editing: E. Machiela, T. Liontis, M.M. Senchuk, J.M. Van Raamsdonk. Visualization: E. Machiela, T. Liontis, D.J. Dues, P.D. Rudich, A. Traa, L. Wyman, J.F. Cooper, L. Lew, M.M. Senchuk, J.M. Van Raamsdonk. Supervision: E. Machiela, J.F. Cooper, M.M. Senchuk, J.M. Van Raamsdonk.
Supporting information
Fig S1‐S2
ACKNOWLEDGMENTS
This work was supported by the National Institute of General Medical Sciences (NIGMS; https://www.nigms.nih.gov/; JVR) by grant number R01 GM121756, the Canadian Institutes of Health Research (CIHR; http://www.cihr‐irsc.gc.ca/; JVR), and the Natural Sciences and Engineering Research Council of Canada (NSERC; https://www.nserc‐crsng.gc.ca/index_eng.asp; JVR). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. Some strains were provided by the CGC, which is funded by NIH Office of Research Infrastructure Programs (P30 OD010440). We would also like to acknowledge the C. elegans knockout consortium and the National Bioresource Project of Japan for providing strains used in this research.
Machiela E, Liontis T, Dues DJ, et al. Disruption of mitochondrial dynamics increases stress resistance through activation of multiple stress response pathways. The FASEB Journal. 2020;34:8475–8492. 10.1096/fj.201903235R
Emily Machiela, Thomas Liontis, Dylan J. Dues contributed equally to this work.
REFERENCES
- 1. Liesa M, Palacin M, Zorzano A. Mitochondrial dynamics in mammalian health and disease. Physiol Rev. 2009;89:799‐845. [DOI] [PubMed] [Google Scholar]
- 2. Westermann B. Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol. 2010;11:872‐884. [DOI] [PubMed] [Google Scholar]
- 3. Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science. 2012;337:1062‐1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. van der Bliek AM, Shen Q, Kawajiri S. Mechanisms of mitochondrial fission and fusion. Cold Spring Harb Perspect Biol. 2013;5:a011072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wang S, Xiao W, Shan S, et al. Multi‐patterned dynamics of mitochondrial fission and fusion in a living cell. PLoS ONE. 2012;7:e19879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Frank S, Gaume B, Bergmann‐Leitner ES, et al. The role of dynamin‐related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell. 2001;1:515‐525. [DOI] [PubMed] [Google Scholar]
- 7. Toyama EQ, Herzig S, Courchet J, et al. Metabolism. AMP‐activated protein kinase mediates mitochondrial fission in response to energy stress. Science. 2016;351:275‐281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Karbowski M, Youle RJ. Dynamics of mitochondrial morphology in healthy cells and during apoptosis. Cell Death Differ. 2003;10:870‐880. [DOI] [PubMed] [Google Scholar]
- 9. Okamoto K, Shaw JM. Mitochondrial morphology and dynamics in yeast and multicellular eukaryotes. Annu Rev Genet. 2005;39:503‐536. [DOI] [PubMed] [Google Scholar]
- 10. Burte F, Carelli V, Chinnery PF, Yu‐Wai‐Man P. Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat Rev Neurol. 2015;11:11‐24. [DOI] [PubMed] [Google Scholar]
- 11. Delettre C, Lenaers G, Griffoin JM, et al. Nuclear gene OPA1, encoding a mitochondrial dynamin‐related protein, is mutated in dominant optic atrophy. Nat Genet. 2000;26:207‐210. [DOI] [PubMed] [Google Scholar]
- 12. Zuchner S, Mersiyanova IV, Muglia M, et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot‐Marie‐Tooth neuropathy type 2A. Nat Genet. 2004;36:449‐451. [DOI] [PubMed] [Google Scholar]
- 13. Archer SL. Mitochondrial dynamics–mitochondrial fission and fusion in human diseases. N Engl J Med. 2013;369:2236‐2251. [DOI] [PubMed] [Google Scholar]
- 14. Maycotte P, Marin‐Hernandez A, Goyri‐Aguirre M, Anaya‐Ruiz M, Reyes‐Leyva J, Cortes‐Hernandez P. Mitochondrial dynamics and cancer. Tumour Biol. 2017;39:1010428317698391. [DOI] [PubMed] [Google Scholar]
- 15. Lu B. Mitochondrial dynamics and neurodegeneration. Curr Neurol Neurosci Rep. 2009;9:212‐219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zorzano A, Claret M. Implications of mitochondrial dynamics on neurodegeneration and on hypothalamic dysfunction. Front Aging Neurosci. 2015;7:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. van Horssen J, van Schaik P, Witte M. Inflammation and mitochondrial dysfunction: A vicious circle in neurodegenerative disorders? Neurosci Lett. 2017. [DOI] [PubMed] [Google Scholar]
- 18. Cooper JF, Machiela E, Dues DJ, Spielbauer KK, Senchuk MM, Van Raamsdonk JM. Activation of the mitochondrial unfolded protein response promotes longevity and dopamine neuron survival in Parkinson's disease models. Sci Rep. 2017;7:16441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zevian SC, Yanowitz JL. Methodological considerations for heat shock of the nematode Caenorhabditis elegans . Methods. 2014;68:450‐457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schaar CE, Dues DJ, Spielbauer KK, et al. Mitochondrial and cytoplasmic ROS have opposing effects on lifespan. PLoS Genet. 2015;11:e1004972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wu Z, Senchuk MM, Dues DJ, et al. Mitochondrial unfolded protein response transcription factor ATFS‐1 promotes longevity in a long‐lived mitochondrial mutant through activation of stress response pathways. BMC Biol. 2018;16:147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Senchuk MM, Dues DJ, Schaar CE, et al. Activation of DAF‐16/FOXO by reactive oxygen species contributes to longevity in long‐lived mitochondrial mutants in Caenorhabditis elegans . PLoS Genet. 2018;14:e1007268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Machiela E, Dues DJ, Senchuk MM, Van Raamsdonk JM. Oxidative stress is increased in C. elegans models of Huntington's disease but does not contribute to polyglutamine toxicity phenotypes. Neurobiol Dis. 2016;96:1‐11. [DOI] [PubMed] [Google Scholar]
- 24. Rolland SG, Motori E, Memar N, et al. Impaired complex IV activity in response to loss of LRPPRC function can be compensated by mitochondrial hyperfusion. Proc Natl Acad Sci USA. 2013;110:E2967‐2976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kanazawa T, Zappaterra MD, Hasegawa A, et al. The C elegans Opa1 homologue EAT‐3 is essential for resistance to free radicals. PLoS Genet. 2008;4:e1000022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Johnson D, Nehrke K. Mitochondrial fragmentation leads to intracellular acidification in Caenorhabditis elegans and mammalian cells. Mol Biol Cell. 2010;21:2191‐2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Luz AL, Rooney JP, Kubik LL, Gonzalez CP, Song DH, Meyer JN. Mitochondrial morphology and fundamental parameters of the mitochondrial respiratory chain are altered in Caenorhabditis elegans strains deficient in mitochondrial dynamics and homeostasis processes. PLoS ONE. 2015;10:e0130940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Byrne JJ, Soh MS, Chandhok G, et al. Disruption of mitochondrial dynamics affects behaviour and lifespan in Caenorhabditis elegans . Cell Mol Life Sci. 2019;76(10):1967–1985. 10.1007/s00018-019-03024-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tan FJ, Husain M, Manlandro CM, Koppenol M, Fire AZ, Hill RB. CED‐9 and mitochondrial homeostasis in C. elegans muscle. J Cell Sci. 2008;121:3373‐3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rolland SG, Lu Y, David CN, Conradt B. The BCL‐2‐like protein CED‐9 of C. elegans promotes FZO‐1/Mfn1,2‐ and EAT‐3/Opa1‐dependent mitochondrial fusion. J Cell Biol. 2009;186:525‐540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Labrousse AM, Zappaterra MD, Rube DA, van der Bliek AM. C. elegans dynamin‐related protein DRP‐1 controls severing of the mitochondrial outer membrane. Mol Cell. 1999;4:815‐826. [DOI] [PubMed] [Google Scholar]
- 32. Breckenridge DG, Kang BH, Xue D. Bcl‐2 proteins EGL‐1 and CED‐9 do not regulate mitochondrial fission or fusion in Caenorhabditis elegans . Curr Biol. 2009;19:768‐773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Regmi SG, Rolland SG, Conradt B. Age‐dependent changes in mitochondrial morphology and volume are not predictors of lifespan. Aging (Albany NY). 2014;6:118‐130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dues DJ, Schaar CE, Johnson BK, et al. Uncoupling of oxidative stress resistance and lifespan in long‐lived isp‐1 mitochondrial mutants in Caenorhabditis elegans . Free Radic Biol Med. 2017;108:362‐373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Van Raamsdonk JM, Hekimi S. Deletion of the mitochondrial superoxide dismutase sod‐2 extends lifespan in Caenorhabditis elegans . PLoS Genet. 2009;5:e1000361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Van Raamsdonk JM, Meng Y, Camp D, et al. Decreased energy metabolism extends life span in Caenorhabditis elegans without reducing oxidative damage. Genetics. 2010;185:559‐571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Park HH, Jung Y, Lee SV. Survival assays using Caenorhabditis elegans . Mol Cells. 2017;40:90‐99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. de Castro E, Hegi de Castro S, Johnson TE. Isolation of long‐lived mutants in Caenorhabditis elegans using selection for resistance to juglone. Free Radic Biol Med. 2004;37:139‐145. [DOI] [PubMed] [Google Scholar]
- 39. Rea SL, Wu D, Cypser JR, Vaupel JW, Johnson TE. A stress‐sensitive reporter predicts longevity in isogenic populations of Caenorhabditis elegans . Nat Genet. 2005;37:894‐898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lamitina ST, Morrison R, Moeckel GW, Strange K. Adaptation of the nematode Caenorhabditis elegans to extreme osmotic stress. Am J Physiol Cell Physiol. 2004;286:C785‐791. [DOI] [PubMed] [Google Scholar]
- 41. Mendenhall AR, LaRue B, Padilla PA. Glyceraldehyde‐3‐phosphate dehydrogenase mediates anoxia response and survival in Caenorhabditis elegans . Genetics. 2006;174:1173‐1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dues DJ, Andrews EK, Senchuk MM, Van Raamsdonk JM. Resistance to stress can be experimentally dissociated from longevity. J Gerontol A Biol Sci Med Sci. 2019;74:1206‐1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Senchuk MM, Dues DJ, Van Raamsdonk JM. Measuring oxidative stress in Caenorhabditis elegans: Paraquat and Juglone sensitivity assays. Bio‐Protoc. 2017;7(1). 10.21769/BioProtoc.2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cooper JF, Spielbauer KK, Senchuk MM, Nadarajan S, Colaiacovo MP, Van Raamsdonk JM. Alpha‐synuclein expression from a single copy transgene increases sensitivity to stress and accelerates neuronal loss in genetic models of Parkinson's disease. Exp Neurol. 2018;310:58‐69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dues DJ, Andrews EK, Schaar CE, Bergsma AL, Senchuk MM, Van Raamsdonk JM. Aging causes decreased resistance to multiple stresses and a failure to activate specific stress response pathways. Aging (Albany NY). 2016;8:777‐795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yoneda T, Benedetti C, Urano F, Clark SG, Harding HP, Ron D. Compartment‐specific perturbation of protein handling activates genes encoding mitochondrial chaperones. J Cell Sci. 2004;117:4055‐4066. [DOI] [PubMed] [Google Scholar]
- 47. Link CD, Johnson CJ. Reporter transgenes for study of oxidant stress in Caenorhabditis elegans . Methods Enzymol. 2002;353:497‐505. [DOI] [PubMed] [Google Scholar]
- 48. Link CD, Cypser JR, Johnson CJ, Johnson TE. Direct observation of stress response in Caenorhabditis elegans using a reporter transgene. Cell Stress Chaperones. 1999;4:235‐242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhang Y, Shao Z, Zhai Z, Shen C, Powell‐Coffman JA. The HIF‐1 hypoxia‐inducible factor modulates lifespan in C. elegans . PLoS ONE. 2009;4:e6348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhang P, Judy M, Lee SJ, Kenyon C. Direct and indirect gene regulation by a life‐extending FOXO protein in C. elegans: Roles for GATA factors and lipid gene regulators. Cell Metab. 2013;17:85‐100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Detienne G, Van de Walle P, De Haes W, Schoofs L, Temmerman L. SKN‐1‐independent transcriptional activation of glutathione S‐transferase 4 (GST‐4) by EGF signaling. Worm. 2016;5:e1230585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Nargund AM, Pellegrino MW, Fiorese CJ, Baker BM, Haynes CM. Mitochondrial import efficiency of ATFS‐1 regulates mitochondrial UPR activation. Science. 2012;337:587‐590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hajdu‐Cronin YM, Chen WJ, Sternberg PW. The L‐type cyclin CYL‐1 and the heat‐shock‐factor HSF‐1 are required for heat‐shock‐induced protein expression in Caenorhabditis elegans . Genetics. 2004;168:1937‐1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hwang AB, Lee SJ. Regulation of life span by mitochondrial respiration: the HIF‐1 and ROS connection. Aging (Albany NY). 2011;3:304‐310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Inoue H, Hisamoto N, An JH, et al. The C. elegans p38 MAPK pathway regulates nuclear localization of the transcription factor SKN‐1 in oxidative stress response. Genes Dev. 2005;19:2278‐2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Henderson ST, Johnson TE. daf‐16 integrates developmental and environmental inputs to mediate aging in the nematode Caenorhabditis elegans . Curr Biol. 2001;11:1975‐1980. [DOI] [PubMed] [Google Scholar]
- 57. Kaufman DM, Crowder CM. Mitochondrial proteostatic collapse leads to hypoxic injury. Curr Biol. 2015;25:2171‐2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Redpath CJ, Bou Khalil M, Drozdzal G, Radisic M, McBride HM. Mitochondrial hyperfusion during oxidative stress is coupled to a dysregulation in calcium handling within a C2C12 cell model. PLoS ONE. 2013;8:e69165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Luz AL, Godebo TR, Smith LL, Leuthner TC, Maurer LL, Meyer JN. Deficiencies in mitochondrial dynamics sensitize Caenorhabditis elegans to arsenite and other mitochondrial toxicants by reducing mitochondrial adaptability. Toxicology. 2017;387:81‐94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Tondera D, Grandemange S, Jourdain A, et al. SLP‐2 is required for stress‐induced mitochondrial hyperfusion. EMBO J. 2009;28:1589‐1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Frazier HN 3rd, Roth MB. Adaptive sugar provisioning controls survival of C. elegans embryos in adverse environments. Curr Biol. 2009;19:859‐863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lakowski B, Hekimi S. Determination of life‐span in Caenorhabditis elegans by four clock genes. Science. 1996;272:1010‐1013. [DOI] [PubMed] [Google Scholar]
- 63. Feng J, Bussiere F, Hekimi S. Mitochondrial electron transport is a key determinant of life span in Caenorhabditis elegans . Dev Cell. 2001;1:633‐644. [DOI] [PubMed] [Google Scholar]
- 64. Yang W, Hekimi S. Two modes of mitochondrial dysfunction lead independently to lifespan extension in Caenorhabditis elegans . Aging Cell. 2010;9:433‐447. [DOI] [PubMed] [Google Scholar]
- 65. Labbadia J, Brielmann RM, Neto MF, Lin YF, Haynes CM, Morimoto RI. Mitochondrial stress restores the heat shock response and prevents proteostasis collapse during aging. Cell Rep. 2017;21:1481‐1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Tullet JMA, Green JW, Au C, et al. The SKN‐1/Nrf2 transcription factor can protect against oxidative stress and increase lifespan in C. elegans by distinct mechanisms. Aging Cell. 2017;16:1191‐1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Yang W, Hekimi S. A mitochondrial superoxide signal triggers increased longevity in Caenorhabditis elegans . PLoS Biol. 2010;8:e1000556. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S2