

Abstract
A single nucleotide polymorphism (SNP) in CHRNA5 (rs16969968, change from an aspartic acid (D) to asparagine (N) at position 398 of the human α5 nicotinic acetylcholine receptor subunit) has been associated with increased risk for nicotine dependence. Consequently, carriers of the risk variant may be at elevated risk for in utero nicotine exposure. To assess whether this gene-environment interaction might impact nicotine intake in developmental nicotine-exposed offspring, we utilized a mouse expressing this human SNP. D and N dams drank nicotine (100 μg /ml) in 0.2% saccharin water or 0.2% saccharin water alone (vehicle) as their sole source of fluid from 30 days prior to breeding until weaning of offspring. The nicotine (D Nic, N Nic) or vehicle (D Veh, N Veh) exposed offspring underwent a two-bottle choice test between postnatal ages of 30–46 days. N Nic offspring consumed the most nicotine at the highest concentration (400 μg/ml) compared to all other groups. In contrast, D Nic offspring drank the least amount of nicotine at all concentrations tested. Nicotine-stimulated dopamine (DA) release measured from striatal synaptosomes was increased in D Nic offspring, while decreased in N Nic offspring relative to their genotype-matched controls. These data suggest that the α5 variant influences the effect of developmental nicotine exposure on nicotine intake of exposed offspring. This gene-environment interaction on striatal DA release may provide motivation for increased nicotine seeking in N Nic offspring and reduced consumption in D Nic offspring.
Despite increased public awareness of the hazards of cigarette smoking during pregnancy, an estimated 400,000 fetuses in the US are exposed to nicotine each year (Law et al., 2003, Suellentrop et al., 2006, Tong et al., 2013). Declining cigarette sales have given way to increased use of alternatives including nicotine replacement and electronic nicotine delivery systems. Regardless of mode of delivery, exposure to developmental nicotine remains a relevant health concern with potential for life-changing consequences into childhood and adulthood (England et al., 2017).
Adolescence is a period of increased risk for experimentation with drugs of abuse including nicotine. Although a third of teens who start smoking in adolescence do not become daily users, initiation during the teen years is associated with greater daily consumption as well as a lower probability of quitting (Cdc, 2012). In utero nicotine exposure increases adolescent smoking initiation two-fold and also increases the likelihood for continued tobacco use compared to individuals whose mothers did not smoke during pregnancy (Difranza et al., 2004). Although genetics clearly plays a role in aspects of nicotine dependence, including smoking during pregnancy, it is not clear how genetics contributes to who does and does not become a regular smoker following nicotine exposure in utero; thus, evaluation of the effect of genotype on nicotine consumption following in utero nicotine exposure may be crucial to understanding why teens do or do not become daily nicotine users.
An intriguing gene that may be relevant for studying an interaction between genetics and in utero nicotine exposure is CHRNA5, which encodes the α5 nicotinic acetylcholine receptor (nAChR) subunit. The α5 subunit is frequently incorporated as a fifth, or “accessory” subunit in the α4β2* nAChR (*indicates other subunits are present, (Lukas et al., 1999)) in the central nervous system (Kuryatov et al., 2011). Nicotine readily crosses the placenta, interacting with nAChRs prior to onset of endogenous acetylcholine production (ACh); this premature “out-of-sequence” exposure to nicotine can disrupt normal developmental trajectories (Bailey et al., 2014, Heath et al., 2010, Heath & Picciotto, 2009). The α5 subunit is transiently incorporated into α4β2 nAChRs during embryogenesis and is expressed later in gestation in cortex and hippocampus (Azam et al., 2007, Dwyer et al., 2008, Wang et al., 1996). Neurons of both WT mice with developmental nicotine exposure and α5 knock out (KO) mice with vehicle exposure were found to have an immature morphology and reduced nicotinic currents while α5 KO mice with developmental nicotine exposure have both normal neuronal morphology and nicotinic currents (Bailey et al., 2014). , illustrating the potential for perturbation due to developmental nicotine exposure and that the α5 subunit is an important player in outcomes.
Single nucleotide polymorphisms (SNPs) in the gene cluster encoding the α5, α3, and β4 nicotinic acetylcholine receptor subunits have a strong genetic association with nicotine dependence (Bierut et al., 2008). Of particular interest is a SNP in the α5 subunit (rs16969968) which confers an increased risk for nicotine dependence, is associated with a self-reported “pleasurable buzz” during smoking, and importantly for the current study, failed quit attempts during pregnancy (Berrettini, 2008, Bierut et al., 2008, Freathy et al., 2009, Stephens et al., 2013, Weiss et al., 2008). This SNP is a missense mutation that results in the substitution of aspartate (D) with asparagine (N) at position 398 in the resulting α5 subunit protein (D398N; D= common allele; and N= risk allele). The risk variant (N398) has been shown to reduce function of α4β2α5 and α3β4α5 nAChRs in vitro (Bierut et al., 2008, George et al., 2012, Kuryatov et al., 2011, Tammimaki et al., 2012) and ex vivo (Sciaccaluga et al., 2015). In vivo, this variant leads to a partial loss of protein function and increased nicotine consumption in a self-administration paradigm (Morel et al., 2014).
The mesolimbic dopamine (DA) system has long been linked to the rewarding properties of drugs of abuse including nicotine (Pistillo et al., 2015). The cholinergic system plays a major role in the regulation of DA release from terminals (Rice & Cragg, 2004, Zhou et al., 2001). Motivation to self-administer drugs of abuse, including nicotine, despite their noxious effects is mediated by adaptations of DA transmission specifically linked to cholinergic afferents that encode reinforcing stimuli (De Kloet et al., 2015, Hilario et al., 2012, Pistillo et al., 2015). In what may serve as a “balance” to the rewarding properties of nicotine, the habenula-interpeduncular nucleus (Hb-IPN) pathway appears to play a role in aversive responses to nicotine (Antolin-Fontes et al., 2015, Lee et al., 2015, Zuo et al., 2016). A clear role for this pathway in aversion is illustrated by work by Fowler et al. (2011) that demonstrated that α5 nicotinic subunit knock out mice continue nicotine self-administration at doses that wild-type (WT) mice find aversive. The critical role for the medial habenula in this phenotype was demonstrated by selective viral re-expression of mRNA for the α5 subunit into the habenula which restored nicotine aversion in α5 KO mice and antisense-mediated knockdown of α5 mRNA in the medial habenula in WT animals which resulted in increased nicotine intake (Fowler et al., 2011).
Thus far, no studies have evaluated the interaction of a known genetic risk factor for nicotine dependence with developmental nicotine exposure. As we recently developed a mouse model that carries the rs16969968 risk variant of Chrna5, we set out to assess this interaction (Koukouli et al., 2017). In this study, we evaluated nicotine seeking during adolescence, anticipating offspring with the N variant would consume more nicotine than their D variant counterparts and that developmental nicotine exposure would further increase nicotine consumption over levels seen in vehicle exposed offspring. Increases in consumption should result in decreased nicotine aversion, as could be reflected in reduced habenular nicotinic function, and decreased nicotine reward as reflected by decreased striatal nicotinic function. Together, these data will provide us with insight to the interaction of developmental nicotine exposure with genetics.
Methods
Reagents
The radioisotopes [3H] Dopamine (7,8-3H at 20–40 Ci/mmol), carrier-free 86RbCl (initial specific activity 13.6–18.5 Ci/μg), and [125I]-epibatidine (2200 Ci/mmol) were purchased from Perkin Elmer (Waltham, MA). α-Conotoxin MII (α-CtxMII) was generously provided by J. Michael McIntosh, University of Utah, Salt Lake City, UT. NaCl, KCl, CaCl2, MgSO4, glucose, sucrose, HEPES, tetrodotoxin, (−) nicotine bitartrate, nicotine freebase, acetylcholine iodide (ACh), diisopropyl fluorophosphate (DFP), polyethelenimine, bovine serum albumin (BSA), and cytisine were purchased from Sigma Chemical Co (St. Louis, MO).
Animals
All experiments were reviewed and approved by the Animal Care and Utilization Committee at the University of Colorado Boulder and conform to the guidelines for animal care and use set by the NIH and the Guide for the Care and Use of Laboratory Animals (8th Ed.). Mice possessing the equivalent of the Chrna5 D398N SNP (rs16969968) were generated as previously described (Koukouli et al., 2017, Sciaccaluga et al., 2015). The equivalent amino acid position in the mouse Chrna5 gene is 397. Wildtype mice are described as D (possess an aspartic acid (D) residue at position 397 of CHRNA5) and mice possessing the risk variant are referred to as N (Possess an asparagine (N) residue at position 397 of CHRNA5).
Breeding females were exposed to drinking water with 0.2% saccharin or drinking water with 0.2% saccharin and nicotine (free base; 100 μg/ml) for 30 days prior to breeding. Average daily consumption of nicotine prior to mating was 6.35 ± 0.55 ml (13.72 mg/kg) for D females and 6.69 ± 0.81 ml (14.58 mg/kg) for N females. Male sires were nicotine naïve prior to mating. Mice were maintained on a standard 12h light/dark cycle with lights on at 07:00 and given food (Envigo Teklad 2914 irradiated rodent diet, Harlan, Madison, WI) and fluid (0.2% saccharin alone or nicotine in saccharin) ad libitum. Solutions were changed twice weekly; animals were maintained on their respective drinking protocol until pups were weaned. At weaning, nicotine or vehicle-exposed offspring were placed on tap water as the sole source of fluids. Both sexes of offspring were utilized. Offspring were homozygous for either the D or N variant and exposed to either saccharin vehicle or nicotine developmentally, resulting in four testing groups (D Veh, N Veh, D Nic, N Nic). The total number of animals used in this study was 230, with animals from each litter divided between drinking studies and biochemical analyses.
Ascending Two Bottle Choice Nicotine Drinking
Pups were weaned at 21 days of age and group housed by sex until 29 days of age. On day 29, mice were individually housed and presented with two glass test tubes fitted with rubber stoppers containing sipper tubes, placed equidistant from each other, both filled with tap water. Standard diet was placed across the entire hopper. On day 30, mice were given access to nicotine in one of the two bottles. For the first four days, the nicotine concentration was 100 μg/ml. Every four days the dose was increased (200, 300, and finally 400 μg/ml). Every day at least one hour after lights on (8am) within the same hour time period, water bottles were weighed and position of the nicotine bottle was reversed to adjust for side bias. Two additional empty cages with bottles were set up and rotated at the same time as test subjects to account for any loss from handling and evaporation.
Tissue preparation for functional assays and radioligand binding
A separate cohort of mice was used for all functional and binding assays to establish baseline changes resulting from the interaction of developmental nicotine and the D397N SNP. At PN day 45, whole brains were removed following cervical dislocation and decapitation and further dissected on an ice-cold platform as previously described (Marks et al., 2010). Briefly, each habenula (inclusive of lateral and medial habenula) and each pair of striata (inclusive of dorsal/ventral regions) were dissected into ice-cold, isotonic (0.32M), buffered (5mM HEPES, pH 7.5) sucrose solution and homogenized using a glass-teflon homogenizer. Samples were then centrifuged at 12,000x g for 10 minutes. This crude synaptosomal pellet was resuspended and used immediately for functional efflux and release assays. Remaining crude synaptosomes were stored frozen for subsequent membrane preparation for binding assays. On the day of membrane preparation, samples were thawed and resuspended in ice-cold hypotonic buffered salt solution (NaCl, 14 mM; KCl, 0.15 mM; CaCl2, 0.2 mM, MgSO4, 0.1 mM; HEPES 2.5 mM, pH 7.5), incubated 10 min (22°C), centrifuged at 20,000x g at 4°C for 20 min, and washed by resuspension and re-centrifugation 3x. The samples were stored as pellets under buffer frozen at −20°C until day of binding assay.
86Rb+ Efflux
Agonist-stimulated 86Rb+ efflux from crude synaptosomal preparations of habenula from D/N397 mice (45 days old) was measured as previously described (Marks et al., 1999). Briefly, crude synaptosomes were resuspended in uptake buffer (NaCl, 140 mM; KCl, 1.5 mM, CaCl2, 2 mM; MgSO4, 1 mM; HEPES, 25 mM; glucose, 20 mM; pH 7.5) containing 4 μCi 86RbCl and incubated for 30 minutes at 22°C. To inhibit acetylcholinesterase, 10 μM diisopropyl fluorophosphate was added in the final 5 minutes of incubation. Samples were then gently filtered onto 6 mm glass fiber filters (Type A/E, Gelman, Ann Arbor, MI). Following filtration samples were washed with perfusion buffer [NaCl, 135 mM; CsCl, 5 mM; KCl, 1.5 mM; CaCl2, 2 mM; MgSO4, 1 mM; HEPES, 25 mM; glucose, 20 mM; tetrodotoxin, 50 nM; atropine 1 μM; bovine serum albumin (fraction V), 0.1%; pH 7.5]. After washout, samples were exposed to varying concentrations of ACh (0.1, 0.3, 1, 3, 10, 30, 100, 300, and 1000 μM) for 5 secs to elicit nAChR mediated 86Rb+ efflux. Sample effluent was pumped through a 200 μl volume flow-through Cherenkov cell in a β -RAM Radioactivity HPLC Detector (IN/US Systems, Inc., Tampa, FL) to achieve continuous monitoring of 86Rb+ efflux from the sample.
Habenula tissue from each mouse was tested in different concentrations of ACh (group 1: 1, 10, 100, and 1000; group 2: 3, 30, 300, and 3000; or group 3: 0.1 and 0.3 μM), thus tissue from 3 animals was used to generate each 10-point concentration response curve.
DA Release
We assessed [3H]-dopamine ([3H ]-DA) release from crude synaptosomes prepared from striatal tissue, as previously described (Grady et al., 2007, Salminen et al., 2007). Briefly, synaptosomes were incubated at 37°C in uptake buffer (NaCl, 128mM; KCl, 2.4 mM; CaCl2, 3.2 mM; KH2PO4, 1.2 mM; MgSO4, 1.2 mM; HEPES, 25mM (pH 7.5); glucose, 10 mM; ascorbic acid, 1 mM; 0.01 pargyline, 0.01 mM) for 10 min prior to addition of 100 nM [3H]-DA (1 μCi per 0.20 ml synaptosomes); this suspension was incubated an additional 5 min. Thereafter, all experimental procedures were carried out at room temperature. Synaptosomal aliquots of 80 μl were placed on glass fiber filters and superfused with uptake buffer with the addition of 0.1% bovine serum albumin, 1 μM nomifensine, and 1 μM atropine at a flow rate of 0.7 ml/min for 10 minutes before fractions were collected. To inhibit α6β2*-nAChRs, some synaptosomal aliquots were exposed to α-CtxMII (50 nM) for the last 5 min of the buffer superfusion. Release was initiated by a 20 sec exposure to a range of concentrations of nicotine (0.01 to 30 μM) after 10 min of superfusion. Following release, fractions were collected (10 sec intervals, total of 4 min) into 96-well plates (Gilson FC204 fraction collector; Gilson, Inc., Middleton, WI). Radioactivity was measured using a 1450 Microbeta Trilux scintillation counter (Perkin-Elmer Life Sciences, Waltham, MA) after addition of 0.15 ml OptiPhase SuperMix scintillation cocktail (Perkin-Elmer, Waltham MA). Instrument efficiency was 40%. To differentiate contribution from α6β2*-nACh (MII-sensitive) and non-α6 containing (MII-resistant) nicotinic receptors in the striatum, MII resistant release (aliquots with MII) was subtracted from total release (without MII) to obtain a value for MII sensitive release.
[125I]-Epibatidine binding to membrane homogenates
[125I]-Epibatidine binding was used to measure nAChR expression in the synaptosomal membranes of habenula and striatum. [125I]-Epibatidine binding followed previously published methods (Marks et al., 2004, Whiteaker et al., 2000). Frozen, washed membrane pellets were resuspended in hypotonic buffer and centrifuged at 20,000x g for 20 minutes. The resulting pellets were then resuspended in ice-cold water, with volume adjusted such that less than 10% of the [125I]-epibatidine was bound to the protein. Samples were incubated for 3h at ambient temperature in 96-well polystyrene plates at a final volume of 30 μl of binding buffer containing 200 pM [125I]-epibatidine; for non-specific binding, 1 mM nicotine was added. Following incubation, samples were diluted with 200 μl ice-cold wash buffer. The diluted samples were then filtered through glass fiber filters (top-MFS type B; bottom- Gelman A/E, Gelman, Ann Arbor, MI) treated with 0.5% polyethelenimine under 0.2-atmosphere vacuum using an Inotech Cell Harvester (Inotech Systems, Rockville, MD) and washed with ice-cold buffer five times. For determination of cytisine-resistant binding sites (primarily α3β4 in the habenula), 50 nM cytisine was added to block [125I]-epibatidine binding to cytisine-sensitive sites (primarily α4β2) (Baddick & Marks, 2011, Grady et al., 2009, Marks et al., 2007). In the striatum, α-CtxMII (50 nM) was used to differentiate subpopulations (non-α6 vs α6), as described above. Sample radioactivity was measured (80% efficiency) using a Packard Cobra Auto Gamma Counter (Packard Instruments, Downers Grove, IL). Total protein concentration was determined using the Bicinchonicic Acid (BCA) protein assay (Pierce, Thermo Scientific, Watham, MA).
Statistical Analysis
All data were first analyzed using multivariate ANOVA with SPSS version 24 (IBM Analytics, Armonk, NY) to determine if there was an effect of sex. If no effect existed, data was collapsed into a mixed sex dataset and further analyzed using two-way ANOVA (repeated measures for oral intake and standard multifactorial for synaptosome assays and ligand binding) to evaluate the effects of genotype and developmental treatment, followed by Tukey’s Post Hoc analysis where appropriate. For within subjects analysis, a Greenhouse-Geisser correction was used when sphericity was violated. SigmaPlot version 12 (Systat Software, Inc., San Jose, CA) was used to derive assay parameters by nonlinear regression curve fitting.
The effect of genotype on concentration–response relations measured by 86Rb+ efflux was evaluated by curve-fitting data to a model with two Michaelis–Menten equations to calculate the components with higher sensitivity (HS) and lower sensitivity (LS) to activation by ACh as follows: 86Rb efflux data were analyzed to measure both the high sensitivity (HS) and low sensitivity (LS) receptor populations using the following general formula:
High sensitivity and low sensitivity components are denoted HS and LS, respectively. The concentration-response relationship for nicotine-stimulated [3H] DA release was adequately fit using the Michaelis-Menten equation:
Where V is the agonist-stimulated DA release at each concentration of nicotine; Vmax is the estimated maximal release and EC50 is the concentration of nicotine eliciting half-maximal response.
Results
Effect of developmental nicotine exposure and Chrna5 genotype on nicotine consumption
A major goal of these studies was to determine if developmental nicotine exposure alone or in the presence of the risk allele for nicotine dependence alters nicotine consumption during adolescence. Initial repeated measures ANOVA analysis detected no main effect of sex on the two-bottle choice test. Therefore, data were collapsed on this factor and repeated measures ANOVA was used to assess the effect of genotype and treatment on nicotine intake as measured by an ascending two-bottle nicotine choice test (Fig 1A). Analysis identified a significant main effect of genotype (F1,50 =54.80, p<0.001) and developmental treatment (F1,50 =12.31, p=0.001) as well as a significant genotype by developmental treatment interaction (F1,50= 37.21, p<0.001). By post hoc analysis, D Nic offspring drank significantly less than D Veh (p<0.001), N Veh (p<0.001), and N Nic offspring (p<0.001). Within subjects analysis also detected an effect of concentration (F2.29,105.34 = 68.66, p <0.001), a genotype x nicotine concentration interaction (F2.29,105.34 = 7.76, p <0.001) and a genotype x nicotine concentration x treatment interaction (F2.29,105.34 = 5.97, p <0.01). No other within subjects measures approached significance.
Figure 1. The effect of developmental nicotine and D397N genotype on nicotine consumption.
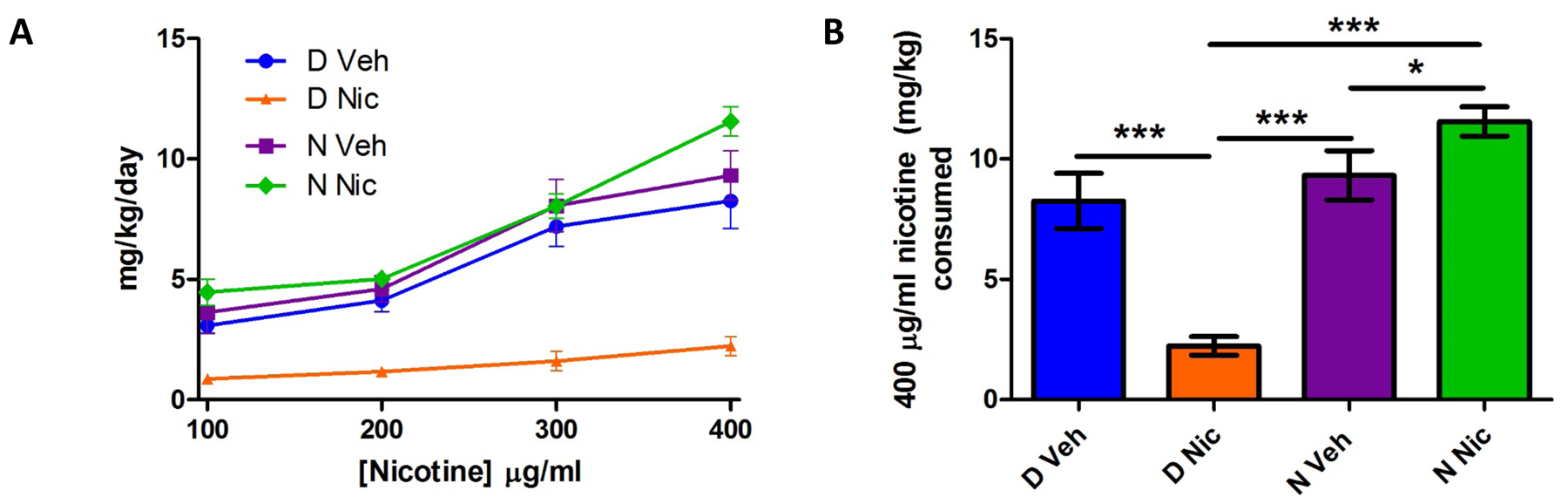
Mice were examined for nicotine consumption in a two-bottle choice test beginning at 35 days of age. (1A) D Nic mice consumed less nicotine across all concentrations tested. Analysis of nicotine consumed at the 400 μg/ml concentration only (1B) indicates N Nic mice drank more than both their N Veh counterparts and D Nic mice; D Nic mice drank the least. Data represent mean ± SEM; *= p<0.05, **=p<0.01, and ***=p<0.001; n=11–19 mice per group.
The greatest separation of nicotine consumption was observed at the highest concentration (400 μg/ml), thus we evaluated differences between groups at this concentration (Fig 1B). Results show significant main effects of genotype (F1,53= 38.98, p<0.001) and developmental treatment (F1,53= 4.08, p<0.05), as well as a significant genotype by developmental treatment interaction (F1,53= 28.76, p<0.001). In addition to reduced nicotine intake in D Nic offspring versus D Veh (p<0.001) N Veh (p<0.001), and N Nic (p<0.001) mice, post hoc analysis shows N Nic mice drank significantly more than N Veh offspring (p<0.05).
Effect of developmental nicotine exposure and Chrna5 genotype on nAChR function and expression
In order to evaluate the effects of developmental nicotine exposure and Chrna5 genotype on several measures of nicotinic function and expression, mice that had not undergone the nicotine preference studies were used. The use of this separate group of mice for all biochemical studies avoided possible effects of nicotine intake from the two-bottle choice studies on the biochemical measures.
Effect of developmental nicotine exposure and Chrna5 genotype on nAChR function and expression in the habenula
Because the α5 nAChR subunit is known to have a role in habenula for aversion to nicotine, we chose to study the impact of developmental nicotine exposure and Chrna5 genotype on function of nAChRs in this region. We assessed ACh stimulated 86Rb+ efflux from synaptosomes prepared from habenula of adolescent mice of all four groups. Initial ANOVA of the full ACh concentration-response data revealed no significant main effect of sex, therefore data for males and females were combined for further analysis. Further analysis of the combined sex data by two-way ANOVA revealed a significant main effect of group (F3,229 =5.13; p<0.01) and ACh concentration (F7,229 =135.3; p<0.001). There was no significant interaction between the two factors.
In the habenula, 86Rb+ efflux measures two populations of α4β2* nAChRs, one (HS) with high sensitivity (consisting of α42β23 and α42β22α5) to activation by ACh and another (LS) that has lower sensitivity to activation (α43β22); these components can be determined using a biphasic curve fit (Marks et al., 1999). Although β4* nAChRs make up one third of nicotinic receptors in this region, their functional contribution in this assay is negligible (Grady et al., 2009). For each treatment group, data were fit to concentration-response curves by non-linear regression. Curve fits are shown in figure 2A. Results from the curve fits were used to determine whether treatment, genotype or an interaction between these factors alters the distribution of the high and low sensitivity populations. There were no significant differences between groups in either HS or LS components (Figs 2B–C).
Figure 2. Acetylcholine-stimulated 86Rb+ efflux in the habenula.
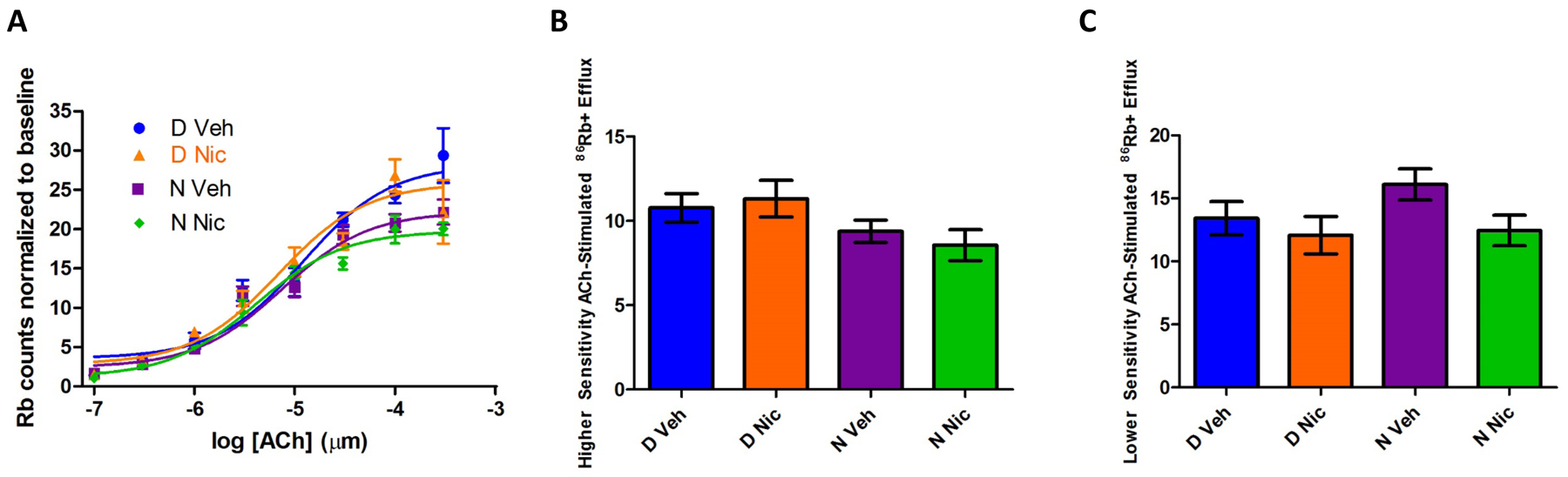
The effect of developmental nicotine on nAChR function in the habenula was determined by measuring ACh-stimulated 86Rb+ efflux from crude synaptosomal preparations of habenula from 45-day old mice. Panel 2A shows concentration-responses curves. Evaluation of maximal efflux for both the high sensitivity (HS, 2B) and the low sensitivity component (LS, 2C) found no significant differences between groups. Data represent mean ± SEM; n=4–14 mice per concentration per group.
Cytisine sensitive and resistant [125I]-epibatidine binding in habenula
Next, the potential impact of genotype, developmental nicotine exposure and the interaction of genotype and developmental nicotine exposure on nAChR expression in the habenula was assessed using [125I]-epibatidine binding in the presence and absence of cytisine to differentiate β2* (cytisine sensitive) from β4* (cytisine resistant) binding sites. This assay cannot differentiate between HS and LS forms of the α4β2 receptor. Multifactorial analysis indicated there were no sex differences thus data for male and female mice were combined for further analysis by two-way ANOVA comparing genotype and treatment group. In cytisine resistant populations (mostly α3β4* nAChRs in the habenula) (Fig 3A), no significant main effects were noted but a significant genotype and developmental treatment interaction was detected (F3,50 =6.99 p<0.05). Post hoc analysis shows binding for N Nic offspring was significantly lower than D Nic offspring (p=0.05).
Figure 3. 125I-Epibatidine binding in habenular membranes.
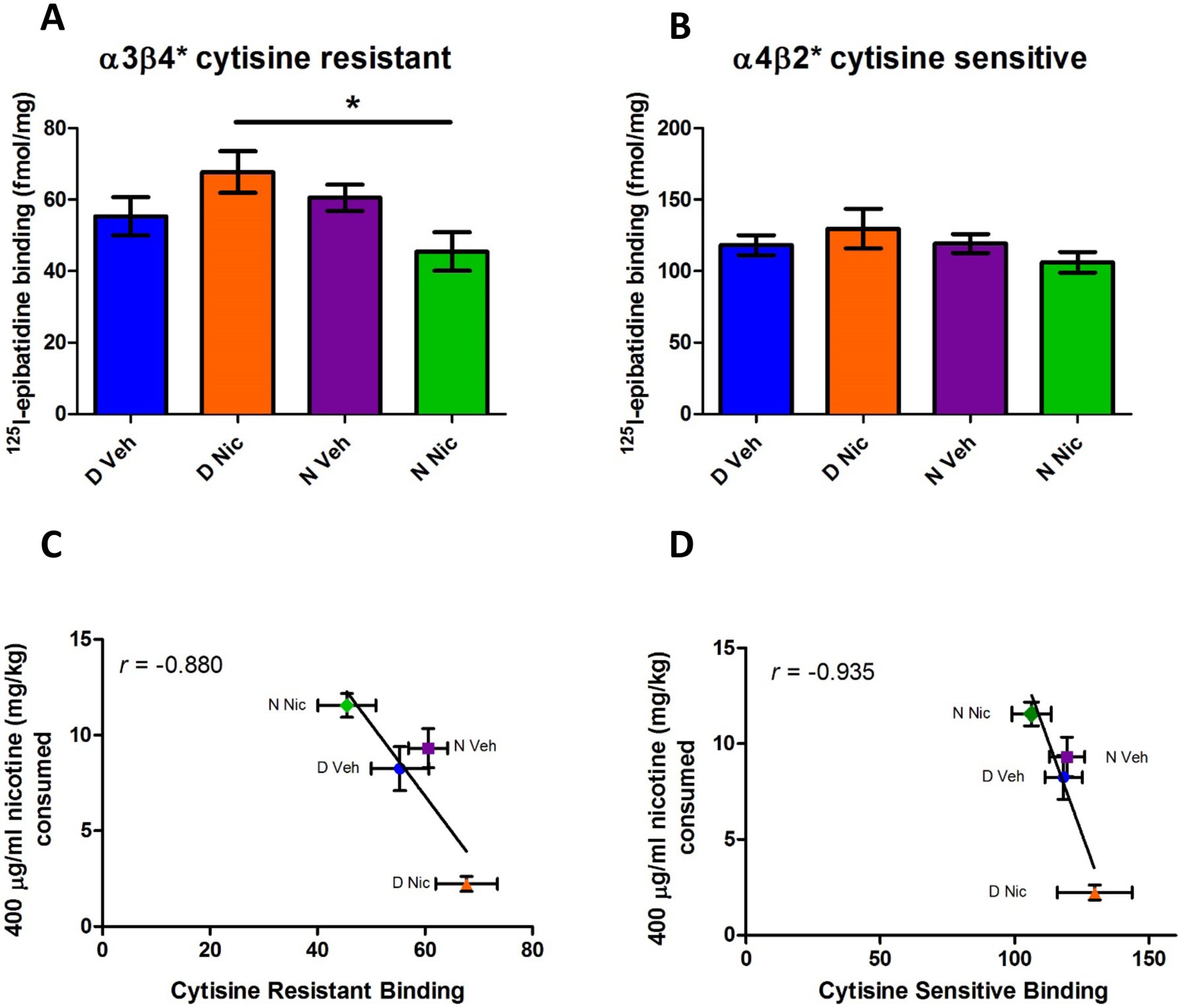
Habenula tissue was evaluated for nicotinic receptor binding sites using cytisine to separate subtypes. [125I]-epibatidine binding in cytisine resistant populations (mostly α3β4) was significantly decreased in N Nic membranes as compared to binding in D Nic membranes (3A). Populations that are sensitive to cytisine (α4β2) did not significantly differ between groups (3B). Evaluation of the correlation between nicotine consumption at the 400 μg/ml concentration and cytisine resistant (3C) or cytisine sensitive (3D) binding levels showed an inverse relationship (r=−0.88 and r=−0.94 respectively). Data represent mean ± SEM; *= p<0.05; n=8–21 mice per group.
There were no significant differences between groups in binding in cytisine-sensitive (α4β2*) populations (Fig 3B).
A simple comparison between habenular nAChR levels and nicotine consumption at the 400 μg/ml concentration showed that there is an inverse relationship between 400 μg/ml nicotine consumption and both cytisine resistant (r = −0.88; Fig 3C) and cytisine sensitive (r = −0.935) (Fig. 4D) [125I]-epibatidine binding.
Figure 4. Dopamine release in striatal synaptosomes.
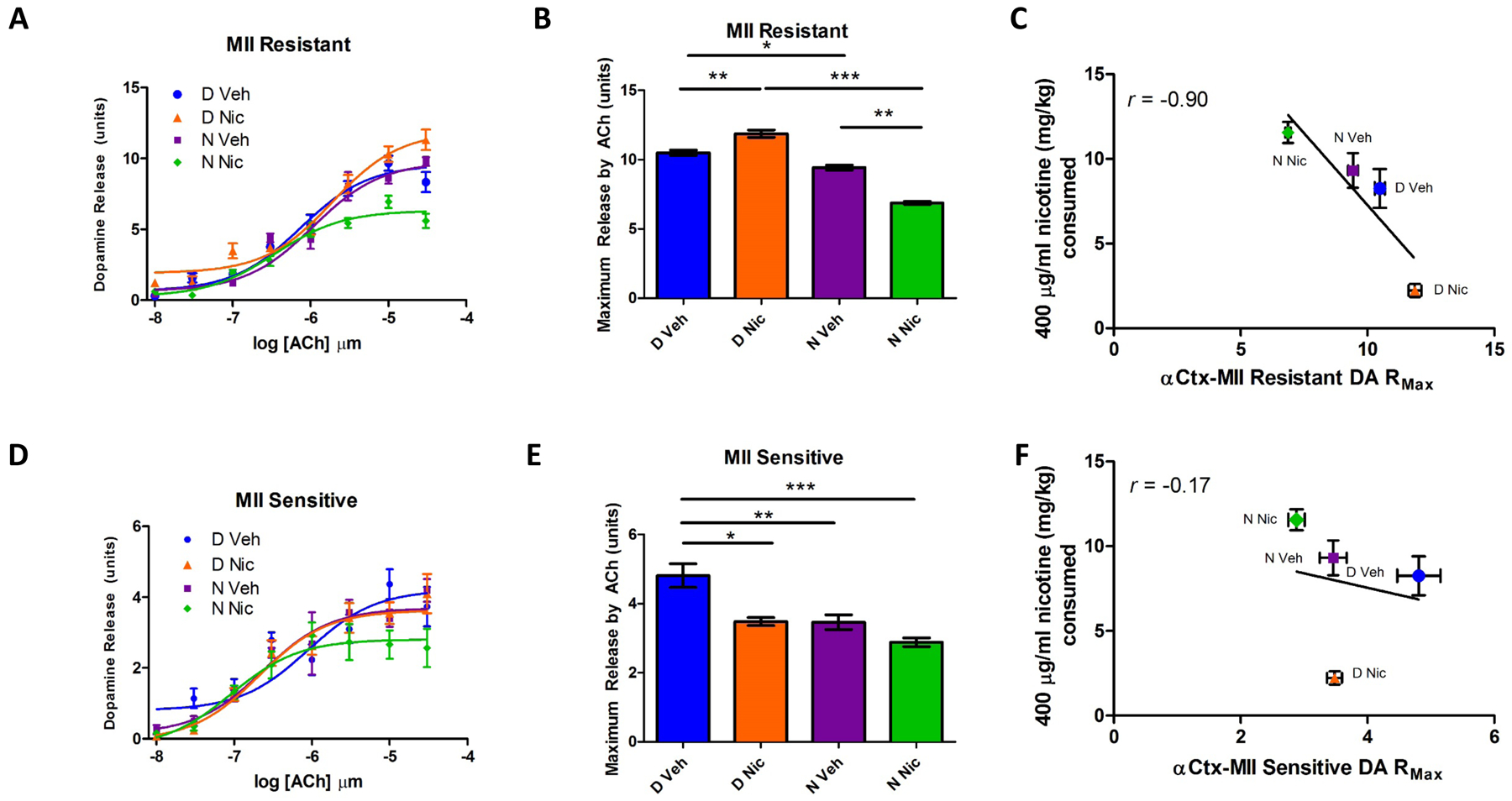
Crude striatal synaptosomal preparations were evaluated for nicotine-stimulated DA release using α-CtxMII to differentiate populations. Release data for α-CtxMII resistant (α4β2 plus α4β2α5) nAChRs (4A) showed a significant main effect of genotype (p<0.01) as well as a significant interaction between genotype and developmental treatment (p<0.01). Curves show decreased DA release in N Nic mice at higher nicotine concentrations in α-CtxMII resistant populations. The α-CtxMII resistant population (4B) showed reduced maximum release in N Veh as compared to D Veh, increased maximum release in D Nic as compared to D Veh and decreased maximum release in N Nic compared to both D Nic and N Veh groups. There was a negative correlation between nicotine consumption at the 400 μg/ml concentration and α-CtxMII resistant maximal DA release (r=−0.90) (4C). Curve fits for DA release sensitive to inhibition by α-CtxMII (α6β2*) are shown in 4D. Analysis of curve fit parameters (4E) in the α-CtxMII sensitive population found that maximal release was significantly reduced in D Nic, N Veh and N Nic groups as compared to D Veh. Relationship (4F) between α-CtxMII sensitive maximal DA release and nicotine consumption at the 400 μg/ml concentration (r = −0.17) is shown. Data represent mean ± SEM; *= p<0.05, **=p<0.01, and ***=p<0.001; n=4–20 mice per concentration per group.
Effect of developmental nicotine exposure and genotype on nAChR function and expression on striatal dopamine release
As the striatum plays a key role in the circuitry of reward and the α5 subunit is associated with α4β2-nAChRs in DA terminals, we next assessed nicotine-stimulated dopamine (DA) release to determine the impact of developmental treatment and genotype in the striatum. Using α-CtxMII we can differentiate between α6β2* populations (α-CtxMII sensitive) and HS α4β2* populations (α-CtxMII resistant; α42α5β22 and α42β23), although we cannot assay the low sensitivity component of the α4β2 response (Grady et al., 2010). Preliminary multifactorial analysis showed no significant effect of sex, therefore data for male and female offspring were combined for further analysis by two-way ANOVA.
Evaluation of the full nicotine concentration-response data for α-CtxMII resistant DA release (consisting of α4β2* subtypes, including α4β2α5) by two-way ANOVA in striatal synaptosomes revealed significant main effects of group (F3,353 =19.36; p<0.001), and of nicotine concentration (F7,353 =161.9; p<0.001); as well as a significant group by nicotine concentration interaction (F21,353 =3.30; p<0.001) (Fig 4A). Maximal MII-resistant [3H]-dopamine release derived from those curve fits was further analyzed. Significant main effects of developmental treatment and genotype (Figure 4B, F3,11= 8.96, p<0.05; F3,11=237.88, p<0.001 respectively) as well as a significant developmental treatment by genotype interaction (F3,11=100.28, p<0.001) were identified. Post hoc analysis revealed that maximal DA release for N Veh offspring was significantly lower than that for D Veh offspring (p<0.05), Maximal DA release for D Nic offspring was significantly higher than that for D Veh offspring(p<0.01), while maximal DA release for N Nic offspring was lower than that for N Veh (p<0.001). Finally, N Nic offspring maximal release was significantly lower than that for D Nic (p<0.001). Maximal DA release and the consumption of 400 μg/ml nicotine were negatively correlated (Fig 4C, r= −0.90).
Evaluation of the full nicotine concentration-response data for α-CtxMII sensitive striatal DA release (α4α6β22β3, α6β22β3 and possibly α6β2(Fig 4D) (Grady et al., 2007, Meyer et al., 2008, Salminen et al., 2007, Salminen et al., 2004) did not reveal significant differences. However, analysis of maximal release for the MII-sensitive DA release revealed significant main effects of developmental treatment (, F3,11=19.08, p<0.01) and of genotype; (F3,11=19.89, p<0.01) (Fig 4E). Post hoc analysis revealed that N Veh, D Nic, and N Nic offspring had significantly lower α-CtxMII sensitive striatal DA maximal release compared to that of D Veh offspring (p=0.01, p<0.05, and p=0.01 respectively). There was no clear relationship between consumption of 400 μg/ml nicotine and MII-sensitive DA release (Fig 4F).
α-CtxMII sensitive and resistant [125I]-epibatidine binding in striatum
Ligand binding was performed to evaluate whether developmental nicotine treatment or genotype altered nAChR expression in the striatum. To differentiate between nAChR populations, [125I]-epibatidine binding was performed in the presence and absence of α-CtxMII, a competitive antagonist that is selective for α6β2 binding sites in the striatum. Multifactorial ANOVA detected no effects of sex, genotype, or treatment on either α-CtxMII resistant [125I]-epibatidine binding (α4β2 sites) (Fig 5A) or α-CtxMII sensitive [125I]-epibatidine binding (α6β2 sites) (Fig 5B) indicating no significant differences in these nicotinic receptor population numbers in the striatum.
Figure 5. 125I-Epibatidine binding in striatal membranes.
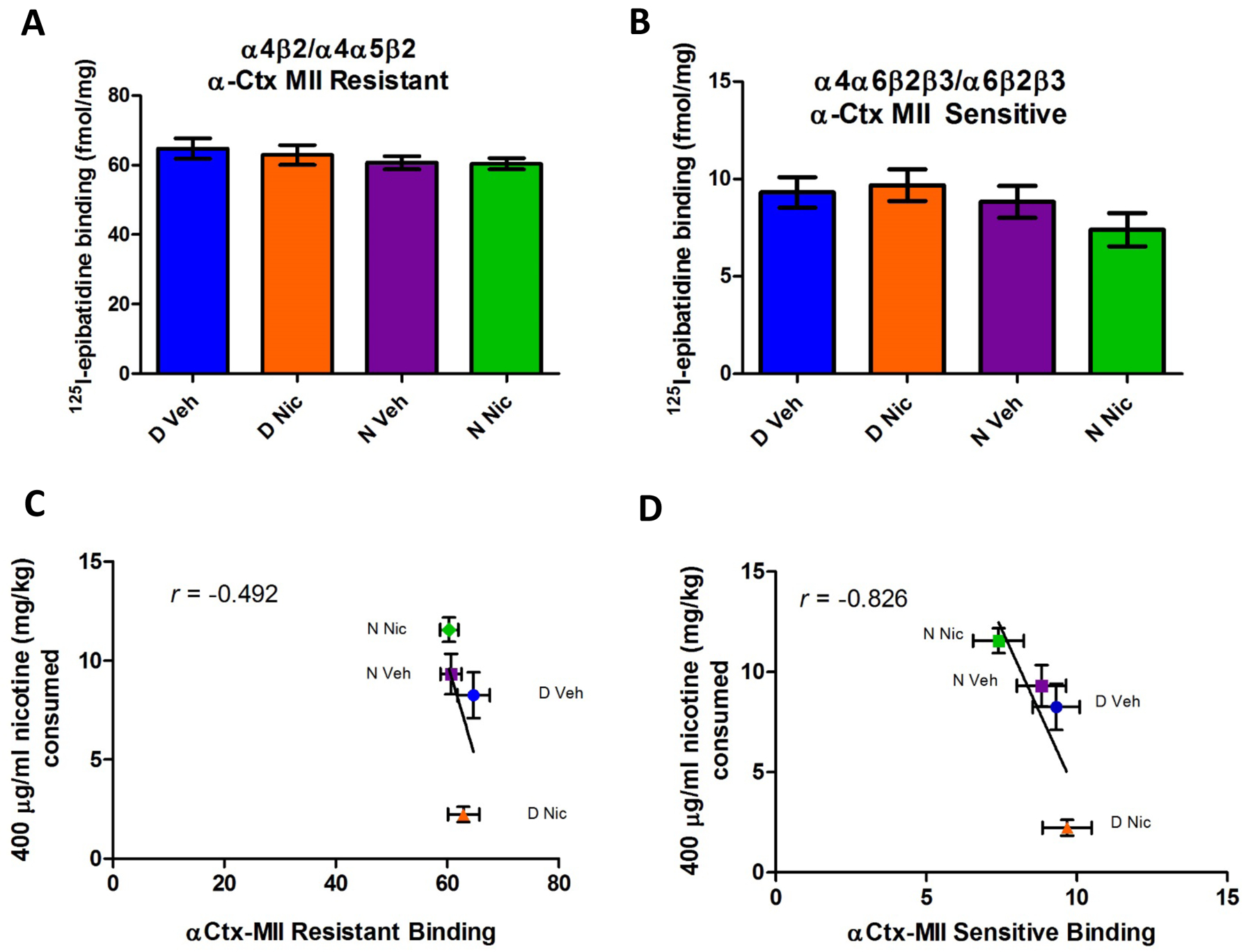
Striatal membranes were evaluated for nicotinic receptor binding using α-CtxMII to differentiate populations. Receptor binding sites in the striatum containing α4β2 without α6 are resistant to α-CtxMII (5A); no significant differences were seen between groups. Binding sites that are sensitive to α-CtxMII (α6β2) also did not significantly differ between groups (5B). Evaluation of the correlation between nicotine consumption at the 400 μg/ml concentration and either α-CtxMII resistant (4E) or α-CtxMII sensitive (4F) binding in the striatum showed an inverse relationship. Data represent mean ± SEM; n=12–24 mice per group.
Plotting [125I]-epibatidine binding versus nicotine intake at 400 μg/ml indicated a poor relationship a between nicotine consumed and α-CtxMII resistant binding in the striatum (Fig 5C) (r=−0.492); there was, however, a fairly strong inverse correlation between nicotine consumed at the 400 μg/ml concentration and α-CtxMII sensitive receptor population numbers (α6*) (Fig 5D, r= −0.826).
Discussion
Either developmental nicotine exposure or the CHRNA5 D398N variant are known to increase risk for nicotine dependence in humans (Bierut et al., 2008, Goldschmidt et al., 2012). The current studies investigated the interaction of these factors on adolescent nicotine intake as well as nicotinic receptor function and expression in adolescent mice.
Adolescent rodents are more sensitive to the rewarding effects of nicotine than adults, find nicotine less aversive (Belluzzi et al., 2004, Mojica et al., 2014) and previous (unpublished) results show offspring with the N variant will consume concentrations of nicotine than their D variant counterparts find aversive. Despite choosing higher starting concentrations than those previously reported for adolescent oral nicotine intake, both vehicle groups drank significant amounts of the nicotine solutions and had similar consumption levels (Klein et al., 2004). Consumption diverged following developmental nicotine exposure with D Nic offspring consuming significantly less oral nicotine across all nicotine concentrations and N Nic offspring consuming significantly more oral nicotine at the 400 μg/ml concentration. Thus, the current data reveal an intriguing interaction between allelic variation and developmental nicotine exposure, indicating that developmental nicotine has a differential outcome dependent upon the allele present.
Several studies have used oral exposure to nicotine during gestation, however adolescent nicotine preference was not an outcome (Bailey et al., 2014, Heath et al., 2010, Heath & Picciotto, 2009, Sparks & Pauly, 1999). To our knowledge, the only previous study to measure adolescent nicotine consumption following developmental exposure in the drinking water (50 μg/ml nicotine from gestational days 9–21) found increased nicotine consumption in male but not female C57BL/6 offspring (Klein et al., 2003). In contrast, the current study finds changes in both sexes at double the exposure concentration. Using either daily injections or osmotic minipumps to expose pups to nicotine at varying times during development results in a range from no effect on adolescent nicotine self-administration to increased intravenous self-administration in adult male rats following periadolescent exposure (Adriani et al., 2004, Matta & Elberger, 2007). To the best of our knowledge, this is the first study to report that developmental nicotine exposure can, in the case of the D398 α5 variant, lead to decreased nicotine intake among exposed offspring. It is plausible that this genotype by developmental nicotine exposure interaction differentially affects aversion to higher concentrations of nicotine.
In an attempt to establish a potential mechanism through which developmental nicotine exposure and Chrna5 genotype interact to alter nicotine intake, nicotinic receptor function and expression were measured in habenula and striatum, two brain regions that express α5 and are known to play critical roles in nicotine self-administration (Fowler et al., 2013). Developmental nicotine exposure elicited differential effects on nAChR mediated function and expression consistent with an inverse relationship between nAChR and nicotine consumption. That is, there may be a relationship between offspring expressing higher nAChR levels and reduced consumption of nicotine. Thus, it is plausible that the D398N variant plays a role in mediating differential responses to developmental nicotine exposure which may help explain why some humans exposed to nicotine in utero become smokers or consume nicotine later in life while others do not.
Some biochemical measures examined were found to exhibit an inverse relationship with nicotine consumption. In general, as receptor numbers or function increased, nicotine consumption decreased. This inverse relationship was independent of receptor population or brain region. However, the measures that may be most relevant to the effect of developmental nicotine intake are the α4β2* (MII-resistant) mediated DA release in the striatum, as measured by maximal response, and cytisine-resistant epibatidine binding in the habenula. These two measures show not only a strong inverse relationship with nicotine intake, but like nicotine intake, levels of function/expression also exhibited a genotype x environment interaction.
It has been shown that that there is an interrelationship between the function of the conotoxin MII resistant and MII sensitive components of DA release. Namely, when the function of one component is altered, the other component can compensate so that total nicotine agonist-stimulated DA release remains unaltered (Salminen et al., 2004). This compensatory response is exactly what was observed in the D variant mice after developmental nicotine exposure. The MII resistant component was increased while the MII sensitive component was reduced to produce no net change in overall maximal DA release. Interestingly, however, this was not the case for N mice- both the MII sensitive and resistant components were reduced resulting in an overall decrease in evoked DA release in N Nic offspring. This lack of compensation in N mice suggests a region of α5 containing the D398N SNP might be important for an as yet unknown intracellular communication mechanism necessary for compensatory changes between the MII sensitive and MII resistant nAChR populations. Finally, altered DA function cannot be attributed to differences in receptor numbers suggesting that the change in function is due to either changes in functional properties of the receptors or alterations in downstream signaling occurring between nAChR activation and DA release.
Two prior studies assessed DA release following developmental nicotine exposure in rats. In both cases, developmental nicotine exposure led to decreased DA release in the nucleus accumbens as measured by either in vivo microdialysis following an acute dose of nicotine (Kane et al., 2004) or ex vivo striatal synaptosomal nicotine stimulated DA release (Gold et al., 2009). In the present study, N Nic offspring had lower α4β2*-mediated maximal DA release offspring relative to N Veh offspring. However, compared to controls, α4β2*- mediated DA release in D Nic offspring was actually increased. Thus, it appears that the most common consequence of developmental nicotine exposure is to decrease nAChR-modulated DA function in striatum/nucleus accumbens. The one exception to this effect is in D398 variant offspring, which exhibit higher levels of DA release following developmental nicotine exposure. This atypical increase in α4β2*-mediated DA release may contribute, in part, to the significant decline in nicotine consumption in mice of this genotype.
Cytisine-resistant epibatidine binding in the habenula, which measures mostly α3β4 sites, also negatively correlated with nicotine intake. The finding that higher levels of α3β4* nAChRs were associated with decreased nicotine intake is consistent with a previous study which showed that overexpression of β4 in the habenula leads to decreased nicotine intake and increased aversion to nicotine (Frahm et al., 2011). Similar to the effect of developmental nicotine treatment on α4β2* function in striatum, an interaction between developmental nicotine treatment and genotype was observed resulting in D Nic offspring having higher levels of α3β4* nAChRs in the habenula than N Nic offspring. Combined, the results of the current study suggest that Chrna5 D397N genotype interacts with developmental nicotine exposure to differentially impact α4β2* nAChR function in striatum and α3β4* expression in the habenula. One shortcoming of the current methods is the inability to measure function of β4-containing function in the habenula, only function of β2 containing receptors can be discerned using 86Rb+ efflux in this region (Grady et al., 2009). Although the correlations indicate relationships between nAChR function/expression, nicotine intake and/or response to developmental nicotine exposure that are consistent with the existing literature, future studies are required to determine whether the changes in nAChR function and/or expression assessed in this study are mechanistically relevant for the genotype by developmental nicotine exposure interaction.
Previous studies have evaluated nicotinic receptor function in adolescence using 86Rb+ efflux (Gold et al., 2009), however this is the first study to evaluate habenular tissue responses in this age group. Tissue for these studies was not subdivided into medial and lateral habenula (MHb and LHb, respectively). However, it is known the densest concentration of nicotinic receptor populations are in the medial portion (Grady et al., 2009). Changes in habenular nicotinic responses were not significant when broken out into components sensitive to lower and higher concentrations of ACh (LS and HS respectively). Although current methods evaluate presynaptic responses, recently published work shows nicotine activates GABAergic inputs to the LHb, which then excite GABAergic neurons in the rostromedial tegmental nucleus (RMTg) that then in turn inhibit midbrain dopaminergic neurons (Hikosaka, 2010, Lecca et al., 2012, Zuo et al., 2016). The data are interesting in light of current studies showing changes persisting in presynaptic dopamine release. While the importance of the nicotinic system in responses to nicotine cannot be overlooked, to better understand the consequences of this gene x environmental interaction, future studies will need to investigate the interaction of other neurotransmitter systems in this region as well as afferent and efferent connections.
The α5 subunit has been shown to play a role in both morphology and function. Developmental nicotine exposure leads to an immature pattern of apical dendritic morphology in pyramidal neurons in layer VI of the medial prefrontal cortex (mPFC) in wild-type mice; this pattern is also seen in nicotine naïve α5 KO mice. Interestingly, developmental nicotine exposure reverses this deficit in α5 KO mice, leading to a mature morphology (Bailey et al., 2014). In vitro studies have shown that DA or glutamate neurons derived from induced pluripotent stem cells (iPSCs) homozygous for the CHRNA5 N398 allele exhibit increased excitatory postsynaptic potentials following nicotine exposure relative to D398 iPSCs. Also, N398 glutamatergic neurons respond to lower doses of nicotine than D398 neurons (Oni et al., 2016). Thus, future studies will investigate other brain regions as well as delve deeper into DA systems and assess GABA and glutamate responses following developmental exposure using the D397N mouse model.
Evaluation of the interaction between the Chrna5 SNP and developmental nicotine exposure is highly relevant to the human condition. Among individuals of European descent, approximately 58% of Caucasians and 28% of the population worldwide carry at least one copy of the risk allele. Studies in humans have shown that the SNP increases risk for nicotine dependence (Bierut et al., 2008) as well as decreases the ability to quit smoking during pregnancy (Freathy et al., 2009). The current data suggest that this SNP can interact with developmental nicotine exposure to produce alternative outcomes in behavioral and brain function. Therefore, it would be of interest to consider developmental nicotine exposure as a cofactor (although such information is often not available) when assessing the role of the D398N SNP in nicotine dependence. Meanwhile, studies with this mouse model will allow for further behavioral and mechanistic characterization of this gene-environment interaction that have thus far not been well characterized.
Footnotes
The authors have no conflict of interest to declare.
References
- Adriani W, Granstrem O, Macri S, Izykenova G, Dambinova S & Laviola G (2004) Behavioral and neurochemical vulnerability during adolescence in mice: studies with nicotine. Neuropsychopharmacology, 29, 869–878. [DOI] [PubMed] [Google Scholar]
- Antolin-Fontes B, Ables JL, Gorlich A & Ibanez-Tallon I (2015) The habenulo-interpeduncular pathway in nicotine aversion and withdrawal. Neuropharmacology, 96, 213–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azam L, Chen Y & Leslie FM (2007) Developmental regulation of nicotinic acetylcholine receptors within midbrain dopamine neurons. Neuroscience, 144, 1347–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baddick CG & Marks MJ (2011) An autoradiographic survey of mouse brain nicotinic acetylcholine receptors defined by null mutants. Biochem Pharmacol, 82, 828–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey CD, Tian MK, Kang L, O’Reilly R & Lambe EK (2014) Chrna5 genotype determines the long-lasting effects of developmental in vivo nicotine exposure on prefrontal attention circuitry. Neuropharmacology, 77, 145–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belluzzi JD, Lee AG, Oliff HS & Leslie FM (2004) Age-dependent effects of nicotine on locomotor activity and conditioned place preference in rats. Psychopharmacology (Berl), 174, 389–395. [DOI] [PubMed] [Google Scholar]
- Berrettini W (2008) Nicotine addiction. Am J Psychiatry, 165, 1089–1092. [DOI] [PubMed] [Google Scholar]
- Bierut LJ, Stitzel JA, Wang JC, Hinrichs AL, Grucza RA, Xuei X, Saccone NL, Saccone SF, Bertelsen S, Fox L, Horton WJ, Breslau N, Budde J, Cloninger CR, Dick DM, Foroud T, Hatsukami D, Hesselbrock V, Johnson EO, Kramer J, Kuperman S, Madden PA, Mayo K, Nurnberger J Jr., Pomerleau O, Porjesz B, Reyes O, Schuckit M, Swan G, Tischfield JA, Edenberg HJ, Rice JP & Goate AM (2008) Variants in nicotinic receptors and risk for nicotine dependence. Am J Psychiatry, 165, 1163–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CDC (2012) Preventing Tobacco Use Among Young People: A Report of the Surgeon General In Services, U.S.D.o.H.a.H. (ed), U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health, Atlanta. [Google Scholar]
- de Kloet SF, Mansvelder HD & De Vries TJ (2015) Cholinergic modulation of dopamine pathways through nicotinic acetylcholine receptors. Biochem Pharmacol, 97, 425–438. [DOI] [PubMed] [Google Scholar]
- DiFranza JR, Aligne CA & Weitzman M (2004) Prenatal and postnatal environmental tobacco smoke exposure and children’s health. Pediatrics, 113, 1007–1015. [PubMed] [Google Scholar]
- Dwyer JB, Broide RS & Leslie FM (2008) Nicotine and brain development. Birth Defects Res C Embryo Today, 84, 30–44. [DOI] [PubMed] [Google Scholar]
- England LJ, Aagaard K, Bloch M, Conway K, Cosgrove K, Grana R, Gould TJ, Hatsukami D, Jensen F, Kandel D, Lanphear B, Leslie F, Pauly JR, Neiderhiser J, Rubinstein M, Slotkin TA, Spindel E, Stroud L & Wakschlag L (2017) Developmental toxicity of nicotine: A transdisciplinary synthesis and implications for emerging tobacco products. Neurosci Biobehav Rev, 72, 176–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler CD, Lu Q, Johnson PM, Marks MJ & Kenny PJ (2011) Habenular alpha5 nicotinic receptor subunit signalling controls nicotine intake. Nature, 471, 597–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler CD, Tuesta L & Kenny PJ (2013) Role of alpha5* nicotinic acetylcholine receptors in the effects of acute and chronic nicotine treatment on brain reward function in mice. Psychopharmacology (Berl). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frahm S, Slimak MA, Ferrarese L, Santos-Torres J, Antolin-Fontes B, Auer S, Filkin S, Pons S, Fontaine JF, Tsetlin V, Maskos U & Ibanez-Tallon I (2011) Aversion to nicotine is regulated by the balanced activity of beta4 and alpha5 nicotinic receptor subunits in the medial habenula. Neuron, 70, 522–535. [DOI] [PubMed] [Google Scholar]
- Freathy RM, Ring SM, Shields B, Galobardes B, Knight B, Weedon MN, Smith GD, Frayling TM & Hattersley AT (2009) A common genetic variant in the 15q24 nicotinic acetylcholine receptor gene cluster (CHRNA5-CHRNA3-CHRNB4) is associated with a reduced ability of women to quit smoking in pregnancy. Hum Mol Genet, 18, 2922–2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George AA, Lucero LM, Damaj MI, Lukas RJ, Chen X & Whiteaker P (2012) Function of human alpha3beta4alpha5 nicotinic acetylcholine receptors is reduced by the alpha5(D398N) variant. J Biol Chem, 287, 25151–25162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold AB, Keller AB & Perry DC (2009) Prenatal exposure of rats to nicotine causes persistent alterations of nicotinic cholinergic receptors. Brain Res, 1250, 88–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldschmidt L, Cornelius MD & Day NL (2012) Prenatal cigarette smoke exposure and early initiation of multiple substance use. Nicotine Tob Res, 14, 694–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady SR, Moretti M, Zoli M, Marks MJ, Zanardi A, Pucci L, Clementi F & Gotti C (2009) Rodent habenulo-interpeduncular pathway expresses a large variety of uncommon nAChR subtypes, but only the alpha3beta4* and alpha3beta3beta4* subtypes mediate acetylcholine release. J Neurosci, 29, 2272–2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady SR, Salminen O, Laverty DC, Whiteaker P, McIntosh JM, Collins AC & Marks MJ (2007) The subtypes of nicotinic acetylcholine receptors on dopaminergic terminals of mouse striatum. Biochem Pharmacol, 74, 1235–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady SR, Salminen O, McIntosh JM, Marks MJ & Collins AC (2010) Mouse striatal dopamine nerve terminals express alpha4alpha5beta2 and two stoichiometric forms of alpha4beta2*-nicotinic acetylcholine receptors. J Mol Neurosci, 40, 91–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath CJ, King SL, Gotti C, Marks MJ & Picciotto MR (2010) Cortico-thalamic connectivity is vulnerable to nicotine exposure during early postnatal development through alpha4/beta2/alpha5 nicotinic acetylcholine receptors. Neuropsychopharmacology, 35, 2324–2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath CJ & Picciotto MR (2009) Nicotine-induced plasticity during development: modulation of the cholinergic system and long-term consequences for circuits involved in attention and sensory processing. Neuropharmacology, 56 Suppl 1, 254–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hikosaka O (2010) The habenula: from stress evasion to value-based decision-making. Nat Rev Neurosci, 11, 503–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilario MR, Turner JR & Blendy JA (2012) Reward sensitization: effects of repeated nicotine exposure and withdrawal in mice. Neuropsychopharmacology, 37, 2661–2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane VB, Fu Y, Matta SG & Sharp BM (2004) Gestational nicotine exposure attenuates nicotine-stimulated dopamine release in the nucleus accumbens shell of adolescent Lewis rats. J Pharmacol Exp Ther, 308, 521–528. [DOI] [PubMed] [Google Scholar]
- Klein LC, Stine MM, Pfaff DW & Vandenbergh DJ (2003) Laternal nicotine exposure increases nicotine preference in periadolescent male but not female C57B1/6J mice. Nicotine Tob Res, 5, 117–124. [DOI] [PubMed] [Google Scholar]
- Klein LC, Stine MM, Vandenbergh DJ, Whetzel CA & Kamens HM (2004) Sex differences in voluntary oral nicotine consumption by adolescent mice: a dose-response experiment. Pharmacol Biochem Behav, 78, 13–25. [DOI] [PubMed] [Google Scholar]
- Koukouli F, Rooy M, Tziotis D, Sailor KA, O’Neill HC, Levenga J, Witte M, Nilges M, Changeux JP, Hoeffer CA, Stitzel JA, Gutkin BS, DiGregorio DA & Maskos U (2017) Nicotine reverses hypofrontality in animal models of addiction and schizophrenia. Nat Med, 23, 347–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuryatov A, Berrettini W & Lindstrom J (2011) Acetylcholine receptor (AChR) alpha5 subunit variant associated with risk for nicotine dependence and lung cancer reduces (alpha4beta2)(2)alpha5 AChR function. Mol Pharmacol, 79, 119–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law KL, Stroud LR, LaGasse LL, Niaura R, Liu J & Lester BM (2003) Smoking during pregnancy and newborn neurobehavior. Pediatrics, 111, 1318–1323. [DOI] [PubMed] [Google Scholar]
- Lecca S, Melis M, Luchicchi A, Muntoni AL & Pistis M (2012) Inhibitory inputs from rostromedial tegmental neurons regulate spontaneous activity of midbrain dopamine cells and their responses to drugs of abuse. Neuropsychopharmacology, 37, 1164–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Kang MS, Chung JM & Noh J (2015) Repeated nicotine exposure in adolescent rats: Reduction of medial habenular activity and augmentation of nicotine preference. Physiol Behav, 138, 345–350. [DOI] [PubMed] [Google Scholar]
- Lukas RJ, Changeux JP, Le Novere N, Albuquerque EX, Balfour DJ, Berg DK, Bertrand D, Chiappinelli VA, Clarke PB, Collins AC, Dani JA, Grady SR, Kellar KJ, Lindstrom JM, Marks MJ, Quik M, Taylor PW & Wonnacott S (1999) International Union of Pharmacology. XX. Current status of the nomenclature for nicotinic acetylcholine receptors and their subunits. Pharmacol Rev, 51, 397–401. [PubMed] [Google Scholar]
- Marks MJ, Meinerz NM, Brown RW & Collins AC (2010) 86Rb+ efflux mediated by alpha4beta2*-nicotinic acetylcholine receptors with high and low-sensitivity to stimulation by acetylcholine display similar agonist-induced desensitization. Biochem Pharmacol, 80, 1238–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks MJ, Meinerz NM, Drago J & Collins AC (2007) Gene targeting demonstrates that alpha4 nicotinic acetylcholine receptor subunits contribute to expression of diverse [3H]epibatidine binding sites and components of biphasic 86Rb+ efflux with high and low sensitivity to stimulation by acetylcholine. Neuropharmacology, 53, 390–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks MJ, Rowell PP, Cao JZ, Grady SR, McCallum SE & Collins AC (2004) Subsets of acetylcholine-stimulated 86Rb+ efflux and [125I]-epibatidine binding sites in C57BL/6 mouse brain are differentially affected by chronic nicotine treatment. Neuropharmacology, 46, 1141–1157. [DOI] [PubMed] [Google Scholar]
- Marks MJ, Whiteaker P, Calcaterra J, Stitzel JA, Bullock AE, Grady SR, Picciotto MR, Changeux JP & Collins AC (1999) Two pharmacologically distinct components of nicotinic receptor-mediated rubidium efflux in mouse brain require the beta2 subunit. J Pharmacol Exp Ther, 289, 1090–1103. [PubMed] [Google Scholar]
- Matta SG & Elberger AJ (2007) Combined exposure to nicotine and ethanol throughout full gestation results in enhanced acquisition of nicotine self-administration in young adult rat offspring. Psychopharmacology (Berl), 193, 199–213. [DOI] [PubMed] [Google Scholar]
- Meyer EL, Yoshikami D & McIntosh JM (2008) The neuronal nicotinic acetylcholine receptors alpha 4* and alpha 6* differentially modulate dopamine release in mouse striatal slices. J Neurochem, 105, 1761–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mojica CY, Dao JM, Yuan M, Loughlin SE & Leslie FM (2014) Nicotine modulation of adolescent dopamine receptor signaling and hypothalamic peptide response. Neuropharmacology, 77, 285–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morel C, Fattore L, Pons S, Hay YA, Marti F, Lambolez B, De Biasi M, Lathrop M, Fratta W, Maskos U & Faure P (2014) Nicotine consumption is regulated by a human polymorphism in dopamine neurons. Mol Psychiatry, 19, 930–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oni EN, Halikere A, Li G, Toro-Ramos AJ, Swerdel MR, Verpeut JL, Moore JC, Bello NT, Bierut LJ, Goate A, Tischfield JA, Pang ZP & Hart RP (2016) Increased nicotine response in iPSC-derived human neurons carrying the CHRNA5 N398 allele. Sci Rep, 6, 34341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pistillo F, Clementi F, Zoli M & Gotti C (2015) Nicotinic, glutamatergic and dopaminergic synaptic transmission and plasticity in the mesocorticolimbic system: focus on nicotine effects. Prog Neurobiol, 124, 1–27. [DOI] [PubMed] [Google Scholar]
- Rice ME & Cragg SJ (2004) Nicotine amplifies reward-related dopamine signals in striatum. Nat Neurosci, 7, 583–584. [DOI] [PubMed] [Google Scholar]
- Salminen O, Drapeau JA, McIntosh JM, Collins AC, Marks MJ & Grady SR (2007) Pharmacology of alpha-conotoxin MII-sensitive subtypes of nicotinic acetylcholine receptors isolated by breeding of null mutant mice. Mol Pharmacol, 71, 1563–1571. [DOI] [PubMed] [Google Scholar]
- Salminen O, Murphy KL, McIntosh JM, Drago J, Marks MJ, Collins AC & Grady SR (2004) Subunit composition and pharmacology of two classes of striatal presynaptic nicotinic acetylcholine receptors mediating dopamine release in mice. Mol Pharmacol, 65, 1526–1535. [DOI] [PubMed] [Google Scholar]
- Sciaccaluga M, Moriconi C, Martinello K, Catalano M, Bermudez I, Stitzel JA, Maskos U & Fucile S (2015) Crucial role of nicotinic alpha5 subunit variants for Ca2+ fluxes in ventral midbrain neurons. FASEB J, 29, 3389–3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparks JA & Pauly JR (1999) Effects of continuous oral nicotine administration on brain nicotinic receptors and responsiveness to nicotine in C57Bl/6 mice. Psychopharmacology (Berl), 141, 145–153. [DOI] [PubMed] [Google Scholar]
- Stephens SH & Hartz SM & Hoft NR & Saccone NL & Corley RC & Hewitt JK & Hopfer CJ & Breslau N & Coon H & Chen X & Ducci F & Dueker N & Franceschini N & Frank J & Han Y & Hansel NN & Jiang C & Korhonen T & Lind PA & Liu J & Lyytikainen LP & Michel M & Shaffer JR & Short SE & Sun J & Teumer A & Thompson JR & Vogelzangs N & Vink JM & Wenzlaff A & Wheeler W & Yang BZ & Aggen SH & Balmforth AJ & Baumeister SE & Beaty TH & Benjamin DJ & Bergen AW & Broms U & Cesarini D & Chatterjee N & Chen J & Cheng YC & Cichon S & Couper D & Cucca F & Dick D & Foroud T & Furberg H & Giegling I & Gillespie NA & Gu F & Hall AS & Hallfors J & Han S & Hartmann AM & Heikkila K & Hickie IB & Hottenga JJ & Jousilahti P & Kaakinen M & Kahonen M & Koellinger PD & Kittner S & Konte B & Landi MT & Laatikainen T & Leppert M & Levy SM & Mathias RA & McNeil DW & Medland SE & Montgomery GW & Murray T & Nauck M & North KE & Pare PD & Pergadia M & Ruczinski I & Salomaa V & Viikari J & Willemsen G & Barnes KC & Boerwinkle E & Boomsma DI & Caporaso N & Edenberg HJ & Francks C & Gelernter J & Grabe HJ & Hops H & Jarvelin MR & Johannesson M & Kendler KS & Lehtimaki T & Magnusson PK & Marazita ML & Marchini J & Mitchell BD & Nothen MM, et al. (2013) Distinct loci in the CHRNA5/CHRNA3/CHRNB4 gene cluster are associated with onset of regular smoking. Genet Epidemiol, 37, 846–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suellentrop K, Morrow B, Williams L, D’Angelo D, Centers for Disease, C. & Prevention (2006) Monitoring progress toward achieving Maternal and Infant Healthy People 2010 objectives--19 states, Pregnancy Risk Assessment Monitoring System (PRAMS), 2000–2003. MMWR Surveill Summ, 55, 1–11. [PubMed] [Google Scholar]
- Tammimaki A, Herder P, Li P, Esch C, Laughlin JR, Akk G & Stitzel JA (2012) Impact of human D398N single nucleotide polymorphism on intracellular calcium response mediated by alpha3beta4alpha5 nicotinic acetylcholine receptors. Neuropharmacology, 63, 1002–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong VT, Dietz PM, Morrow B, D’Angelo DV, Farr SL, Rockhill KM, England LJ, Centers for Disease, C. & Prevention (2013) Trends in smoking before, during, and after pregnancy--Pregnancy Risk Assessment Monitoring System, United States, 40 sites, 2000–2010. MMWR Surveill Summ, 62, 1–19. [PubMed] [Google Scholar]
- Wang F, Gerzanich V, Wells GB, Anand R, Peng X, Keyser K & Lindstrom J (1996) Assembly of human neuronal nicotinic receptor alpha5 subunits with alpha3, beta2, and beta4 subunits. J Biol Chem, 271, 17656–17665. [DOI] [PubMed] [Google Scholar]
- Weiss RB, Baker TB, Cannon DS, von Niederhausern A, Dunn DM, Matsunami N, Singh NA, Baird L, Coon H, McMahon WM, Piper ME, Fiore MC, Scholand MB, Connett JE, Kanner RE, Gahring LC, Rogers SW, Hoidal JR & Leppert MF (2008) A candidate gene approach identifies the CHRNA5-A3-B4 region as a risk factor for age-dependent nicotine addiction. PLoS Genet, 4, e1000125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteaker P, Jimenez M, McIntosh JM, Collins AC & Marks MJ (2000) Identification of a novel nicotinic binding site in mouse brain using [(125)I]-epibatidine. Br J Pharmacol, 131, 729–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou FM, Liang Y & Dani JA (2001) Endogenous nicotinic cholinergic activity regulates dopamine release in the striatum. Nat Neurosci, 4, 1224–1229. [DOI] [PubMed] [Google Scholar]
- Zuo W, Xiao C, Gao M, Hopf FW, Krnjevic K, McIntosh JM, Fu R, Wu J, Bekker A & Ye JH (2016) Nicotine regulates activity of lateral habenula neurons via presynaptic and postsynaptic mechanisms. Sci Rep, 6, 32937. [DOI] [PMC free article] [PubMed] [Google Scholar]