

Abstract
Valvular heart disease occurs as either a congenital or acquired condition and advances in medical care have resulted in valve disease becoming increasingly prevalent. Unfortunately, treatments remain inadequate due to our limited understanding of the genetic and molecular etiology of diseases affecting the heart valves. Therefore, surgical repair or replacement remains the most effective option, which comes with additional complications and no guarantee of life-long success. Over the past decade, there have been significant advances in our understanding of cardiac valve development and not surprisingly mutations in these developmental genes have been identified in humans with congenital valve malformations. Concurrently, there has been a greater realization that acquired valve disease is not simply a degenerative process. Molecular investigation of acquired valve disease has identified that numerous signaling pathways critical for normal valve development are re-expressed in diseased valves. This review will discuss recent advances in our understanding of the development of the heart valves along with the implications of these findings on the genetics of congenital and acquired valvular heart disease.
Keywords: heart valve development, valvular heart disease, bicuspid aortic valve, mitral valve prolapse, aortic valve calcification
INTRODUCTION
Valvular heart disease (VHD), which includes both congenital and acquired forms, is an important and growing public health problem. Based on epidemiologic studies in the United States, it has an overall prevalence of 2.5%, and the incidence increases with age.1 With the projected increases in the elderly population worldwide, acquired forms of VHD are likely to rise in prevalence in all industrialized countries.2 Similarly, advances in the care of infants born with congenital heart defects (CHD) will add to the growing VHD population as congenital valve abnormalities are identified in over 50% of CHD.3 Despite the prevalence, pharmacologic therapies for VHD are limited, and progressive valve dysfunction often requires surgical repair or replacement as the primary treatment.4,5 In the United States, the annual direct cost for VHD is estimated at $1 billion and the need to develop novel therapeutic strategies is becoming increasingly imperative.6
Essential to developing new non-invasive therapies to treat VHD is the elucidation of the primary etiologic contributors for disease development and progression. Congenital valve malformations are primarily the result of perturbation of genes that regulate normal heart valve development. For acquired valve disease, exposure to non-genetic risk factors such as hypercholesterolemia, hypertension, and tobacco use along with rheumatic heart disease are proposed to be primary disease contributors.6 While the mechanisms of congenital and acquired valve disease are not fully understood, the pathogenesis is thought to stem from the interplay between genetic and environmental influences. However, there is increasing evidence to suggest that genes critical for normal valve development may play roles in the development of acquired VHD.
Development and maintenance of the cardiac valves is a complex process and requires formation of intricate valve structures that must open and close over 100,000 times a day to maintain unidirectional blood flow through the heart. During embryogenesis, endocardial cushions (EC) are the primordia of mature valve leaflets and composed of a highly organized extracellular matrix (ECM) consisting of three distinct layers made of collagens, proteoglycans and elastin that together, provide all the necessary biomechanical properties to withstand constant changes in the hemodynamic environment.7 Turnover of the valve ECM is tightly regulated by valve interstitial cells (VICs) that are largely quiescent and fibroblast-like in the absence of disease. Overlying valve endothelial cells (VECs) form an uninterrupted protective endothelium over the surface of the leaflets and molecularly communicate with underlying VICs to regulate their behavior in response to changes in the hemodynamic environment (Figure 1).8 In contrast to healthy valves, disruption of normal valve development or acquired valve disease is characterized by alterations in the organization and composition of ECM, VIC activation and endothelial disruption.7 Together, these pathological changes alter valve mechanics and hinder opening or closing of the valve leaflets, leading to dysfunction and eventual heart failure.
Figure 1. Atrioventricular and semilunar valve structure.
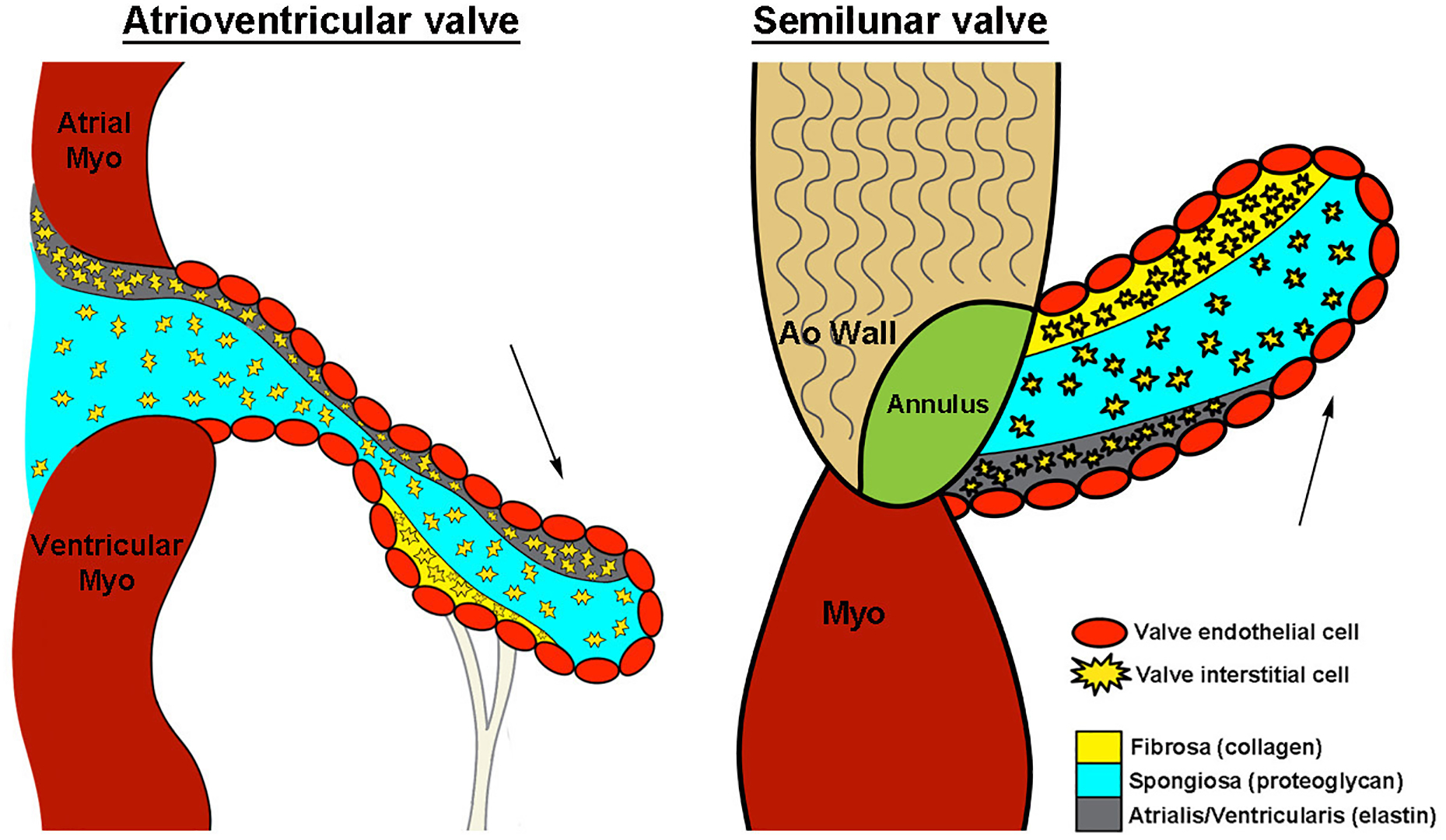
Schematic demonstrating cellular architecture of atrioventricular and semilunar valves. Arrows, blood flow direction. Myo, myocardium; Ao, aortic. Reproduced with permission from Tao G, et al.8
In this review, we will describe the anatomy of heart valves and discuss the key cellular and molecular processes required for establishing healthy valve structures in the embryo. We review the genetic contributors to the most common congenital valve malformations and have focused on how alterations in valve development genes are being discovered to be important for the onset and progression of the most prevalent acquired VHD.
VALVE STRUCTURE
The mature heart valves, consisting of the atrioventricular (AV) (mitral and tricuspid) and semilunar (SL) (aortic and pulmonic), are primarily composed of an outer layer of VECs that surround three stratified layers of specialized ECM, interspersed with differentiated VICs.8 Each ECM layer is organized according to blood flow and works together to withstand the continual hemodynamic changes of the cardiac cycle.9 The fibrosa layer, which is predominantly composed of collagen, provides tensile strength to the valve leaflet during opening, while transmitting forces to promote leaflet coaptation when closed (Figure 1).10,11 Adjacent to the fibrosa is the spongiosa layer, with a lower collagen abundance and higher proteoglycan prevalence. This composition provides a more compressible matrix, allowing the valve to ‘flex’ and absorb high force. The layer adjacent to blood flow is termed the atrialis or ventricularis in AV or SL valves, respectively. It largely consists of elastin fibers that facilitate extension of the leaflets during closure.12,13 This is further supported by the fibrous annulus structure between the leaflets/cusps and myocardium and external tendinous-chordae in the AV position. The VIC and VEC populations also play essential roles in maintaining connective tissue homeostasis in valve leaflets/cusps. Although originally thought of as a homogenous population of fibroblast-like cells, VICs are now considered highly heterogeneous with quiescent, activated and progenitor-like phenotypes reported in development and disease.12 In addition to VICs, VECs are also highly responsive to changes in the hemodynamic environment and molecularly communicate with underlying VICs to regulate their behavior. As normal valve function is dependent on the complex arrangement of connective tissue and overall valve morphology, it is not surprising that alterations in localization and/or contribution of matrix components leads to functional VHD.
VALVE DEVELOPMENT & REMODELING
Endocardial cushion formation
The primitive beating heart tube consists of an outer layer of myocardial cells and an inner layer of endocardial cells, separated by cardiac jelly.14 Following rightward looping, a subset of endocardial cells localized within the AV canal and outflow tract (OFT) regions secrete a hyaluronan-rich ECM to form ‘swellings’ known as ECs. In response to signals emanating mostly from the adjacent myocardium, endocardial cells overlying the cushions loose cell-cell contact, transform into mesenchymal cells, and migrate into the underlying cushion jelly and proliferate.15 This process of endothelial-to-mesenchymal transformation (EMT) is essential for generating the pool of precursor cells that will give rise to the mature valves. Complex networks of signaling pathways tightly regulate each step of EMT. Mice with EC defects caused by aberrations in EMT usually die around E10.5, highlighting the requirement of EMT for valvulogenesis. Furthermore, gene-targeting studies have been insightful in identifying the critical regulators of EMT and include secreted growth factors, transcription factors and signaling molecules (discussed below and Table 1).
Table 1.
Mouse models of endocardial cushion defects associated with gene targeting of Tgf-β, Wnt, Notch and VEGF signaling.
| Family | Gene | Phenotype in mice | Reference (#) |
|---|---|---|---|
| Tgfβ | |||
| Tgfβ2 | Tgfβ2−/−: Hypoplastic endocardial cushions | Reviewed in Kruithof et al., 2012 (ref. 20) | |
| TgfβRI | TgfβRI−/−: Hypoplastic endocardial cushions due to impaired EMT | Reviewed in Kruithof et al., 2012 (ref. 20) | |
| Snai1 | Endocardial Tie2cre;Snai1+/−: EMT initiated, but reduced mesenchyme cell proliferation | Tao et al., 2012 (ref. 8) | |
| Snai2 | Snai2I−/−: Hypoplastic endocardial cushions due to impaired EMT | Niessen et al., 2008 (ref. 24) | |
| Bmp2 | Myocardial deletion; Reduced cardiac jelly and impaired EMT initiation | Reviewed in Kruithof et al., 2012 (ref. 20) | |
| Bmp4 | Myocardial or anterior heart field deletion; Reduced mesenchyme cell proliferation | Reviewed in Kruithof et al., 2012 (ref. 20) | |
| BMPRI | Endocardial deletion; Hypoplastic endocardial cushions due to impaired EMT | Reviewed in Kruithof et al., 2012 (ref. 20) | |
| BMPRIA | Endocardial deletion; Hypoplastic endocardial cushions due to impaired EMT | Reviewed in Kruithof et al., 2012 (ref. 20) | |
| Wnt | |||
| β-catenin | Endocardial deletion; Hypoplastic endocardial cushions due to impaired EMT | Leibner et al., 2004 (ref. 27) | |
| Notch | |||
| RBPJk | RBPJk−/−: Hypoplastic endocardial cushions due to impaired EMT | Reviewed de la Pompa and Epstein, 2012 (ref. 30) | |
| Notch1 | Notch1−/−: Hypoplastic endocardial cushions due to impaired EMT | Reviewed de la Pompa and Epstein, 2012 (ref. 30) | |
| MAML | Isl1-cre; dom neg MAML: Hypoplasia of OFT endocardial cushions | Reviewed de la Pompa and Epstein, 2012 (ref. 30) | |
| Vegf | |||
| VEGF | Myocardial transgenic; EMT inhibited (50% penetrance) | Dor et al., 2001 (ref. 36) | |
Due to limitations, relevant review articles are cited.
Contribution of non-endothelial cell lineages to valve structures
Formation of ECs requires contribution of several cell lineages from multiple sources. Early cell lineage studies using Tie2cre;ROSA26R mice that allowed for marking of endocardial cells and their progenitors suggested that the majority of mesenchymal cells within the AV canal cushions are derived from endocardial cells following EMT.16,17 Recent studies have shown that migrating epicardial cells also contribute to the parietal leaflets of the AV valves.18 In the OFT valves, there appears to be little epicardial contribution but lineage tracing studies using Wnt1-Cre, suggest that neural crest-derived cells contribute to OFT valves and aortopulmonary septum.19 The valve mesenchymal cell population is derived from multiple sources, and collectively, each may play essential roles throughout valvulogenesis.
Regulators of EMT
The number of signaling pathways known to contribute to EC formation is expansive. This review will focus on the most common, including signaling molecules Tgfβ, Bmp, Wnt, Notch and VEGF as they have been linked to human HVD (Table 1).
Transforming Growth Factor- β
TGFβs and their downstream signaling mediators are derived from the myocardium and regulate several EMT-related processes.20,21,22 During avian EC formation, Tgfβ2 and Tgfβ3, via TgfβRII are required for VECs to initiate EMT and promote migration of newly transformed cells. Mice with deletion of Tgfβ, latent Tgfβ binding protein, or endocardial-deletion of TgfβRI (Alk5) develop valve abnormalities including hypoplastic ECs.20 Tgfβ-mediated activation of the downstream effectors, phosphorylated (p)Smad2/3, is sufficient to induce expression of the transcription factors Snai1(Snail) and Snai2(Slug). Studies have shown that Snai1 and Snai2 are essential for EMT in vivo, and mice with null alleles develop hypoplastic cushions, similar to Tgfβ mutants.23,24,25,26 This collection of studies indicate that Tgfβ receptors, ligands and downstream effectors are important for several biological processes required for EC formation including EMT initiation, breakdown of cell-cell contacts, mesenchymal cell motility and proliferation.
As members of the TGFβ super-family, BMP signaling through canonical SMADs (pSmad1/5/8) also plays a critical role in cushion formation. Numerous studies in primary cells and animal models show that BMPs similar to Tgfβs, are a major source of myocardial-derived signals for EMT initiation.20 Among BMP family members, Bmp2 and 4 are most predominantly expressed in the AV and OFT myocardium adjacent to developing cushions, respectively.20 As with Tgfβ, mutations that target decreased BMP signaling in mice commonly result in hypoplastic ECs as a result of impaired EMT.20 The significance of EC defects observed in BMP mutants is likely the result of compromised pSmad1/5/8 activity, which in healthy hearts is required to sustain BMP signaling from the myocardium to VECs during EMT. Proper BMP signaling is also required for sufficient functioning of Tgfβ, as well as the expression of transcription factors Twist1 and Msx2, which are well-known positive regulators of EMT.26 These studies highlight the complex transcriptional networks activated from growth factor signaling during EMT and EC formation.
Wnt
Tgfβ signaling pathways cross-talk with several signaling pathways including β-catenin, a mediator of canonical Wnt signaling that is an important regulator of EC formation. Endothelial deletion of β-catenin inhibits Tgfβ-mediated induction of EMT in mice, suggesting a requirement for canonical Wnt signaling in EMT.27 In zebrafish, overexpression of the secreted Wnt inhibitor, Dickkopf1, blocks cushion formation while truncation mutants develop hyperplastic cushions due to increased cell proliferation.28 The notion that Wnt signaling regulates VIC proliferation is continued in the avian system where the Wnt receptor Frzb and Wnt9a promote mesenchyme cell number in the AV cushions.29 These studies identify a role for β-catenin during early stages of EMT, while downstream effectors of the Wnt signaling pathway appear to be important for mesenchyme cell proliferation and establishing the pool of valve precursor cells.
Notch
Notch signaling is an activator of EMT, predominantly through Notch1 that is expressed in VECs. As ligands (Delta and Jagged) and receptors (Notch) in the Notch signaling pathway are membrane-bound proteins, Notch sets up an efficient intercellular local signaling system that binds to the DNA via co-repressor RBPJ or recruits the coactivator, MAML1, to regulate transcriptional activity of multiple downstream targets.30 Deletion of Notch1 or RBPJ in mice leads to EC defects that lack significant numbers of transformed mesenchymal cells.31 In contrast to reduced Notch signaling, overexpression of constitutively active Notch1 in the endocardium delays EC formation and ectopic EMT is observed in the ventricular chambers.32 Target genes affected by reduced Notch signaling in developing cushions include Snai2 and Hey family of transcription factors.33 In chick and mice, Hey1 and Hey2 are highly expressed in the VECs and are important for restricting expression of Bmp2 and Tbx2 in the AV canal region.34,35 These studies highlight the essential role(s) that Notch-related genes play in EMT and EC formation.
VEGF
Vascular endothelial growth factor A (VEGF) is a potent cytokine highly expressed by the myocardium and VECs before cushion formation. However, expression of the ligand and its receptors (VEGFR) become restricted to the endocardium once cushion formation is initiated.36 Collectively, studies in multiple animal models have demonstrated a tightly controlled role for VEGF in EC formation as too much VEGF inhibits EMT, while too little attenuates VEC proliferation, thereby decreasing VEC availability for transformation.37 Recently, it was shown that VEGF signaling through VEGFR2 in endocardial cells is required for transformation into mesenchymal cells in the OFT, but not AV cushion.38 This is one of the few studies to identify discrepancies between EMT regulation between the OFT and AV valves. NFATc1 (nuclear factor of activated T-cells cytoplasmic 1) is a downstream target of VEGF in both developing and mature VECs and promotes cell proliferation.37 While Nfatc1−/− mice undergo successful EMT, a new role has recently emerged for an Nfatc1 enhancer region in fate decisions of VEC during transformation stages.39
Valve remodeling, maturation and maintenance
Once EC formation is complete, overlying VECs stop undergoing EMT, re-gain cell-cell contacts and form an uninterrupted endothelium.15 Around this time, the EC fuse and elongate into mitral and tricuspid valve primordia in AV region, and primitive aortic and pulmonic structures in OFT (Figure 2).8 The mechanisms of elongation are not clear but cell proliferation is localized to the distal tips of developing valves, and VEGF has been suggested to play a role.7,17,38 As the primordia develop, mesenchyme cells within the developing valves proliferate less and lose expression of mesenchymal markers but maintain smooth muscle α-actin (SMA) expression, suggesting a VIC-like phenotype. These embryonic VICs actively remodel the ECM by expressing matrix degradation enzymes to break down the hyaluronan, and secrete new ECM that that will later form the three stratified layers.6,40 Compared to EMT, much less is known about valve remodeling, but some of the key regulators of EMT have differential roles at this stage. BMP2 has been shown to be important for promoting expression of proteoglycans including versican and hyaluronan in the remodeling valves.41 In addition, Sox9 and Scleraxis regulate expression of other ECM components associated with the fibrosa and spongiosa layers, respectively. In mice, Sox9 is required during remodeling stages for expression of known cartilaginous downstream target genes Col2a1 and Cartilage Link Protein (CLP) within the leaflets.42 Scleraxis is both sufficient and required for proteoglycan expression and is positively regulated by canonical Tgfβ2 with cross talk from MAPK signaling.43
Figure 2. Heart valve development and progression to calcific disease.
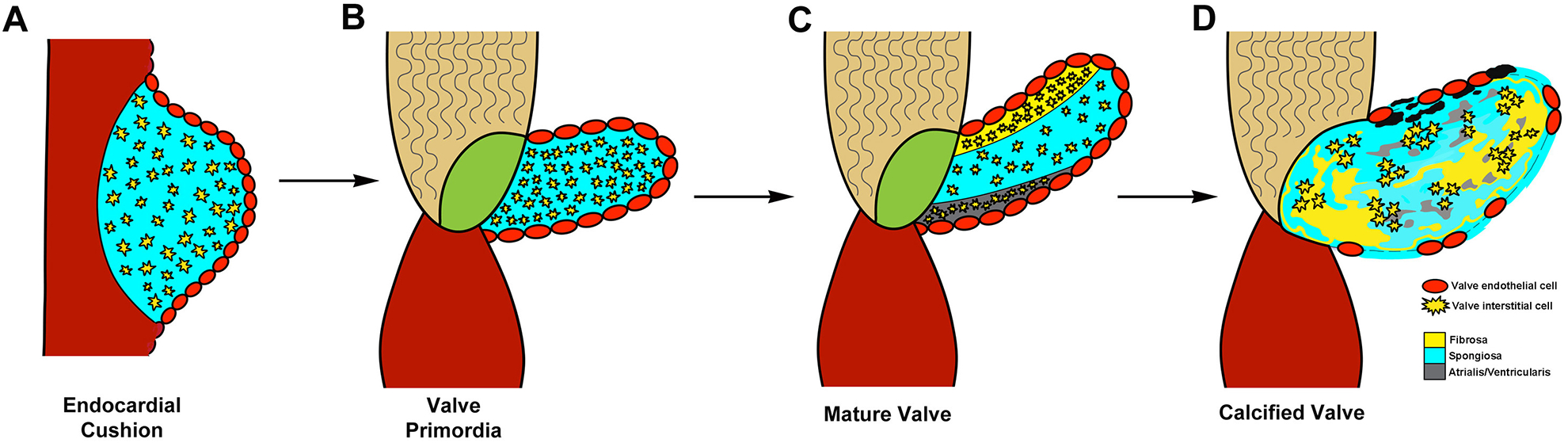
Valve development begins with endocardial cushion formation (A). Cushions grow to form valve primordial (B), which remodel into mature valve leaflet (C). Calcification of the valve leaflets occurs with disease (D) Reproduced with permission from Tao G, et al.8
It is not until postnatal stages that the elastin fibers are laid down in the atrialis/ventricularis layers and the valve becomes tri-stratified.7 At this time, VICs downregulate smooth muscle α-actin and become quiescent and fibroblast-like and remain this way throughout life in the absence of injury/disease.12 Little is known about the pathways that maintain tissue homeostasis and integrity in the postnatal valve, but disturbances in VEC or VIC function are proposed to have detrimental effects on valve ECM organization and therefore biomechanical properties.
GENETICS OF VALVE DISEASE
The sequencing of the human genome and advances in genetic technologies has contributed to the discovery of VHD-causing genes. Congenital anomalies of each four heart valves have been described, and these malformations disrupt normal valvular function. Not surprisingly, the same genes and molecular pathways implicated in heart valve development have been found to be mutated in humans with congenital VHD and are being found to play a role in acquired VHD. Here, we will focus on genetic basis of two of most common congenital valve anomalies, bicuspid aortic valve (BAV) and mitral valve prolapse (MVP) and in addition discuss genetic contributors to the most common form of acquired HVD, calcific aortic valve disease (CAVD). The genetic contributors to less common types of VHD are reviewed elsewhere.44
Bicuspid aortic valve
With an estimated prevalence in the population of 1–2%, bicuspid aortic valve (BAV) is one of the most common valve malformations.45 While the normal aortic valve has three cusps, BAV is the result of fusion of the two of leaflets during development. BAV is associated with significant long-term morbidity due to aortic valve dysfunction primarily through calcification (discussed below) and is thought to have a strong genetic etiology.46,47 BAV can be categorized based on the type of cusp fusion, which is proposed to have different embryologic origins. Fusion of the right-left cusps is due to abnormal septation of the OFT and right-noncoronary fusion due to abnormal OFT cushion development and these subtypes are associated with differing clinical outcomes.48
The first gene found to be mutated in human BAV was NOTCH1, a member of the Notch signaling pathway (discussed above). In families with autosomal dominant BAV, heterozygous loss-of-function NOTCH1 mutations were found to segregate with disease. Subsequently, NOTCH1 mutations have been found in other individuals and families with aortic valve disease.49,50 Further support of this link was identification that mice haploinsufficient for Notch1 in the setting of endothelial nitric oxide synthase (Nos3) deletion display a near 100% penetrance of BAV as compared to the lower incidence (~25–30%) of BAV in Nos3−/− mice.51 Recently, deletion of Gata5, a zinc finger transcription factor, in the valve endothelium was found to result in a partially penetrant right-noncoronary BAV phenotype in mice with associated dysregulation of the Notch signaling pathway in the developing OFT.52 Examination of humans with BAV have identified rare non-synonymous variants in GATA5 but individuals harboring mutations were found with both right-left and right-noncoronary BAV.53,54 While these finding support a genetic association between NOTCH1 and GATA5 and BAV, the underlying mechanisms remain unclear hindering the ability to link genetic etiologies to long-term morbidities.
Mitral Valve Prolapse
Mitral valve prolapse (MVP), which affects about 2–3% of the population, occurs when there is systolic displacement of a thickened mitral valve leaflet into the left atrium, and MVP is associated with valve regurgitation, congestive heart failure, arrhythmias, and infective endocarditis.55 MVP is often identified in adulthood and is characterized by fibromyxomatous degeneration of the leaflets which results abnormally thickened and lengthened leaflets that bulge into the atrium, leading to mitral regurgitation.55 Diseased myxomatous valves have an expanded spongiosa layer which is the result of excess proteoglycan deposition. This pathological remodeling is also associated with diminished collagen fibers, elastin fragmentation, myofibroblast activation, and overexpression of proteolytic enzymes such as MMP-1, MMP-2, and MMP-13.43,56
The best-studied etiology of MVP is mitral valve disease associated with Marfan syndrome (MFS), a connective tissue disorder affecting multiple tissues. Mutations in Fibrillin-1 (FBN1), a key component of ECM microfibrils cause MFS.57 As has been reported with ascending aortic aneurysms associated with MFS, the molecular basis of MVP has been demonstrated to be increased Tgf-β signaling, which is a critical regulator of valve development (discussed above).58 A mouse model of MVP that carries a human disease-causing Fbn1 mutation, displays thickened valves soon after birth associated with increased Tgf-β expression which can be rescued by pharmacologic inhibition of Tgf-β signaling during embryogenesis. Further supporting a role for Tgf-β signaling in the pathogenesis of MVP is the identification of mutations in TGF-β receptors 1 and 2 (TGFBR1 and TGFBR2) are a cause of Loeys-Dietz (LDS), which has a similar MVP phenotype.59
Additional human genetic evidence linking abnormal Tgf-β signaling with MVP was identified by investigating families with an X-linked form of valvular dysplasia, which affects the mitral and aortic valves. Mutations in Filamin A (FLNA), which encodes a widely expressed protein that interacts with ECM bound cell-surface integrins to regulate the actin cytoskeleton.60,61 Mice null for Flna display a spectrum of valve abnormalities and defects in cardiac and OFT septation and loss of endothelial Flna in mice leads to myxomatous mitral valve disease.62 Similar to Fbn1, Flna was shown to regulate the Tgf-β signaling pathway through its interactions with the Smad proteins.60,61 These studies suggest a critical role for Tgf-β signaling in the development of syndromic forms of MVP and also suggest a role for Tgf-β in the remodeling and maintenance of the mitral valve. In addition, further studies are needed to determine if similar mechanisms are responsible for non-syndromic MVP.
Calcific valve disease
Among acquired valve diseases, calcification of the aortic valve and the mitral annulus have been described. Calcific aortic valve disease (CAVD) and mitral annular calcification (MAC) are characterized by calcium deposition on the aortic valve cusps and mitral annulus, respectively and share environmental risk factors similar to atherosclerosis. Human genetic studies, by evidence of familial clustering and genome-wide association, have demonstrated a genetic component to the development of CAVD and MAC.63,64 Recently, a large genome-wide association study including individuals with both CAVD and MAC led to the identification of a single nucleotide polymorphism (SNP) in the lipoprotein(a) (LPA) locus that was linked to only CAVD in multiple ethnicities highlighting the differences for these acquired calcific valve diseases.65 As the SNP correlated with Lp(a) levels, it is a potentially exciting finding that may lead to novel therapies for CAVD.
CAVD is associated with endothelial dysfunction, lipid accumulation and inflammatory cell infiltration.6 Environmental factors are though to increase the risk for development of CAVD by a process initiated by endothelial cell dysfunction that results in activation of VICs leading to the expression of bone development genes including Runx2, Osteopontin and Osteocalcin, as found in calcified human aortic valves.6 The first suggestion of this concept came from human genetic studies on BAV families discussed above, which suggested a role for NOTCH1 in CAVD as some family members harboring a NOTCH1 mutation did not have a BAV but developed CAVD.66 Constitutively active Notch1 along with Hey1 and Hey2 inhibited Runx2 activation in cell culture assays. Further in vivo evidence of this inhibitory role has been demonstrated as mice heterozygous for Notch1, Rbpj, Jagged1 and Hey2 develop aortic valve calcification.67,68,69 These studies demonstrated that valve development genes may potentially play a role in this adult-onset disease and the mechanisms by which loss of Notch signaling leads to calcification are being investigated.70
Sox9, which encodes an SRY-related transcription factor well-known for its requirement in cartilage development, was also implicated in CAVD.71 Sox9 is required early for EC development.42 While later, targeted reduced function in mice leads to early onset CAVD by 3 months of age and this is associated with increased expression of osteogenic genes seen in human calcified valves, partly due to lost repression of the target gene, Osteopontin (Spp1).71,72 In addition to increased calcification, heart valves from Col2a1-cre;Sox9fl/+ mice show reduced expression of cartilage-associated ECM proteins that are highly expressed in healthy valves. This imbalance between osteo- and cartilage-like phenotypes likely occurs as Sox9 is known to transcriptionally activate chondrogenic genes and repress osteogenic markers, and therefore this regulatory hierarchy is lost in Sox9 mutant mice.42,71
Similar to Notch1 and Sox9, additional valve developmental pathways have been linked to the development and progression of CAVD. Wnt/β-catenin signaling has been implicated in CAVD as Lrp5, a coreceptor in this pathway, is overexpressed in diseased valves.73 Similarly, NFATc1 has been shown to be upregulated in CAVD.74 The developmental transcription factors, Twist1, Sox9 and Msx2, which are important in EMT during valvulogenesis have also been shown to be expressed in calcified aortic valves.75 In combination, these findings suggest that CAVD is correlated with the expression of valve development genes. The significance of this in disease initiation and progression remains unclear and is an active area of investigation.
CONCLUSIONS AND FUTURE PERSPECTIVES
In summary, advances in our understanding of the molecular pathways that regulate valve development are beginning to have broad implications. As expected, identification of genes important for valve morphogenesis has assisted in the discovery of genes responsible for congenital valve malformations, and will accelerate gene discovery in the future.76 Ultimately, this increased understanding will impact the long-term morbidities associated with common forms of VHD including BAV and MVP. Specifically, the importance of Tgf-β signaling in MVP offers a therapeutic target to halt the progression of disease as has been demonstrated for ascending aortic aneurysms in MFS.77 While the human genetic studies on CAVD and identification of embryonic pathways active in pathogenesis are beginning to offer insights into the development of therapies. To date, it remains unclear why the mitral and aortic valves are susceptible to differential pathogenic programs, but this highlights the complexity of HVD that involves the interplay of genetic, hemodynamic and environmental factors.
Funding support:
Supported by grants from NIH/NHLBI R01HL091878 (JL), NIH/NHLBI R01HL109758 (VG) and Saving tiny Hearts Society (VG)
REFERENCES
- 1.Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB, et al. Executive Summary: Heart disease and stroke statistics--2013 update: a report from the American Heart Association. Circulation 2013;127:143–52. [DOI] [PubMed] [Google Scholar]
- 2.d’Arcy JL, Prendergast BD, Chambers JB, Ray SG, Bridgewater B. Valvular heart disease: the next cardiac epidemic. Heart 2011;97:91–93. [DOI] [PubMed] [Google Scholar]
- 3.Le Gloan L, Mercier LA, Dore A, Marcotte F, Ibrahim R, Mongeon FP, et al. Recent advances in adult congenital heart disease. Circ J 2011;75:2287–2295. [DOI] [PubMed] [Google Scholar]
- 4.Itagaki S, Adams DH, Anyanwu AC. Triggers for surgical referral in degenerative mitral valve regurgitation. Circ J 2013:77:28–34. [DOI] [PubMed] [Google Scholar]
- 5.Maeda K, Kuratani T, Mizote I, Shimamura K, Takeda Y, Torikai K, Nakatani S, Nanto S, Sawa Y. Early experiences of transcatheter aortic valve replacement in Japan. Circ J 77:359–362. [DOI] [PubMed] [Google Scholar]
- 6.Rajamannan NM, Evans FJ, Aikawa E, Grande-Allen KJ, Demer LL, Heistad DD, et al. Calcific aortic valve disease: not simply a degenerative process: A review and agenda for research from the National Heart and Lung and Blood Institute Aortic Stenosis Working Group. Executive summary: Calcific aortic valve disease-2011 update. Circulation 2011;124:1783–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hinton RB Jr., Lincoln J, Deutsch GH, Osinska H, Manning PB, Benson DW, et al. Extracellular matrix remodeling and organization in developing and diseased aortic valves. Circ Res 2006;98:1431–1438. [DOI] [PubMed] [Google Scholar]
- 8.Tao G, Kotick JD, Lincoln J. Heart valve development, maintenance, and disease: the role of endothelial cells. Curr Top Dev Biol 2012;100:203–232. [DOI] [PubMed] [Google Scholar]
- 9.Lincoln J, Florer JB, Deutsch GH, Wenstrup RJ, Yutzey KE. ColVa1 and ColXIa1 are required for myocardial morphogenesis and heart valve development. Dev Dyn 2006;235:3295–3305. [DOI] [PubMed] [Google Scholar]
- 10.Grande-Allen KJ, Liao J. The heterogeneous biomechanics and mechanobiology of the mitral valve: implications for tissue engineering. Curr Cardiol Rep 2011;13:113–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sacks MS, David Merryman W, Schmidt DE. On the biomechanics of heart valve function. J Biomech 2009;42:1804–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schoen FJ. Evolving concepts of cardiac valve dynamics. Circulation 2008;118:1864–80. [DOI] [PubMed] [Google Scholar]
- 13.Hinton RB, Yutzey KE. Heart valve structure and function in development and disease. Annu Rev Physiol 2011;73:29–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kodo K, Yamagishi H. A decade of advances in the molecular embryology and genetics underlying congenital heart defects. Circ J 2011;75:2296–2304. [DOI] [PubMed] [Google Scholar]
- 15.Person AD, Klewer SE, Runyan RB. Cell biology of cardiac cushion development. Int Rev Cyt 2005;243:287–335. [DOI] [PubMed] [Google Scholar]
- 16.deLange FJ, Moorman AFM, Anderson RH, Manner J, Soufan AT, deGier-deVries C, et al. Lineage and morphogenetic analysis of the cardiac valves. Circ Res 2004;95:645–654. [DOI] [PubMed] [Google Scholar]
- 17.Lincoln J, Alfieri CM, Yutzey KE. Development of heart valve leaflets and supporting apparatus in chicken and mouse embryos. Dev Dyn 2004;230:239–250. [DOI] [PubMed] [Google Scholar]
- 18.Wessels A, van den Hoff MJ, Adamo RF, Phelps AL, Lockhart MM, Sauls K, et al. Epicardially derived fibroblasts preferentially contribute to the parietal leaflets of the atrioventricular valves in the murine heart. Dev Biol 2012;366:111–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiang X, Rowitch DH, Soriano P, McMahon AP, Sucov HM. Fate of the mammalian cardiac neural crest. Development 2000;127:1607–1616. [DOI] [PubMed] [Google Scholar]
- 20.Kruithof BP, Duim SN, Moerkamp AT, Goumans MJ. TGFβ and BMP signaling in cardiac cushion formation: lessons from mice and chicken. Differentation 2012. 84:89–102. [DOI] [PubMed] [Google Scholar]
- 21.Yamagishi T, Ando K, Nakamura H. Roles of TGFβ and BMP during valvulo-septal endocardial cushion formation. Anat Sci Int 2009. 84:77–87. [DOI] [PubMed] [Google Scholar]
- 22.Azhar M, Schultz JJ, Grupp I, Dorn GW, Meneton P, Molin DG, et al. Transforming growth factor beta in cardiovascular development and function. Cytokine Growth Factor Rev 2003. 14:391–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cano A, Perez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, et al. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol 2000;2:76–83. [DOI] [PubMed] [Google Scholar]
- 24.Niessen K, Fu Y, Chang L, Hoodless PA, McFadden D, Karsan A. Slug is a direct Notch target required for initiation of cardiac cushion cellularization. J Cell Biol 2008;182:315–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tao G, Levay AK, Gridley T, Lincoln J. Mmp15 is a direct target of Snai1 during endothelial to mesenchymal transformation and endocardial cushion development. Dev Biol 2011;359:209–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Combs MD, Yutzey KE. Heart valve development: regulatory networks in development and disease. Circ Res 2009;105:408–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liebner S, Cattelino A, Gallini R, Rudini N, Iurlaro M, Piccolo S, et al. b-catenin is required for endothelial-mesenchymal transformation during heart cushion development in the mouse. J Cell Biol 2004;166:359–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hurlstone AF, Haramis AP, Wienholds E, Begthel H, Korving J, Van Eeden F, et al. The Wnt/beta-catenin pathway regulates cardiac valve formation. Nature 2003;425:633–637. [DOI] [PubMed] [Google Scholar]
- 29.Person AD, Garriock RJ, Krieg PA, Runyan RB, Klewer SE. Frzb modulates Wnt-9a-mediated b-catenin signaling during avian atrioventricular cardiac cushion development. Dev Biol 2005;278:35–48. [DOI] [PubMed] [Google Scholar]
- 30.de la Pompa JL, Epstein JA. Coordinating tissue interactions: Notch signaling in cardiac development and disease. Dev Cell 2012;22:244–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Luna-Zurita L, Prados B, Grego-Bessa J, Luxan G, del Monte G, Benguria A, et al. Integration of a Notch-dependent mesenchymal gene program and Bmp2-driven cell invasiveness regulates murine cardiac valve formation. J Clin Invest 2010;120:3493–3507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Venkatesh DA, Park KS, Harrington A, Miceli-Libby L, Yoon JK, Liaw L. Cardiovascular and hematopoietic defects associated with Notch1 activation in embryonic Tie2-expressing populations. Circ Res 2008;103:423–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Niessen K, Karsan A. Notch signaling in cardiac development. Circ Res 2008;102:1169–1181. [DOI] [PubMed] [Google Scholar]
- 34.Kokubo H, Tomita-Miyagawa S, Hamada Y, Saga Y. Hesr1 and Hesr2 regulate atrioventricular boundary formation in the developing heart through the repression of Tbx2. Development 2007;134:747–755. [DOI] [PubMed] [Google Scholar]
- 35.Rutenberg JB, Fischer A, Jia H, Gessler M, Zhong TP, Mercola M. Developmental patterning of the cardiac atrioventricular canal by Notch and Hairy-related transcription factors. Development 2006;133:4381–4390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dor Y, Camenisch TD, Itin A, Fishman GI, McDonald JA, Carmeliet P, et al. A novel role for VEGF in endocardial cushion formation and its potential contribution to congenital heart defects. Development 2001;128:1531–1538. [DOI] [PubMed] [Google Scholar]
- 37.Combs MD, Yutzey KE. VEGF and RANKL regulation of NFATc1 in heart valve development. Circ Res 2009;105:565–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stankunas K, Ma GK, Kuhnert FJ, Kuo CJ, Chang CP. VEGF signaling has distinct spatiotemporal roles during heart valve development. Dev Biol 2010;347:325–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu B, Wang Y, Lui W, Langworthy M, Tompkins KL, Hatzopoulos AK, et al. Nfatc1 coordinates valve endocardial cell lineage development required for heart valve formation. Circ Res 2011;109:183–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rabkin E, Aikawa M, Stone JR, Fukumoto Y, Libby P, Schoen FJ. Activated interstitial myofibroblasts express catabolic enzymes and mediate matrix remodeling in myxomatous heart valves. Circulation 2001;104:2525–2532. [DOI] [PubMed] [Google Scholar]
- 41.Inai K, Burnside JL, Hoffman S, Toole BP, Sugi Y. BMP-2 induces versican and hyaluronan that contribute to post-EMT AV cushion cell migration. PLoS One 2013;8:e77593, doi: 10.1371/journal.pone.0077593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lincoln J, Kist R, Scherer G, Yutzey KE. Sox9 is required for precursor cell expansion and extracellular matrix organization during mouse heart valve development. Dev Biol 2007;305:120–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Barnette DN, Hulin A, Ahmed AS, Colige AC, Azhar M, Lincoln J. Tgfbeta-Smad and MAPK signaling mediate scleraxis and proteoglycan expression in heart valves. J Mol Cell Cardiol 2013;65:137–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lahaye S, Lincoln J, Garg V. Genetics of valvular heart disease. Curr Cardiol Rep 2014;16:487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McBride KL, Garg V. Heredity of bicuspid aortic valve: is family screening indicated? Heart 2011;97:1193–1195. [DOI] [PubMed] [Google Scholar]
- 46.Garg V Molecular genetics of aortic valve disease. Curr Opin Cardiol 2006;21:180–184. [DOI] [PubMed] [Google Scholar]
- 47.Fernandez B, Duran AC, Fernandez-Gallego T, Fernandez MC, Such M, Arque JM, et al. Bicuspid aortic valves with different spatial orientations of the leaflets are distinct etiological entities. J Am Coll Cardiol 2009;54:2312–2318. [DOI] [PubMed] [Google Scholar]
- 48.Verma S, Siu SC. Aortic dilatation in patients with bicuspid aortic valve. N Engl J Med 2014;370:1920–1929. [DOI] [PubMed] [Google Scholar]
- 49.McBride KL, Riley MF, Zender GA, Fitzgerald-Butt SM, Towbin JA, et al. NOTCH1 mutations in individuals with left ventricular outflow tract malformations reduce ligand-induced signaling. Hum Mol Genet 2008;17:2886–2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Foffa I, Ait Ali L, Panesi P, Mariani M, Festa P, Botto N, et al. Sequencing of NOTCH1, GATA5, TGFBR1 and TGFBR2 genes in familial cases of bicuspid aortic valve. BMC Med Genet 2013;14:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bosse K, Hans CP, Zhao N, Koenig SN, Huang N, Guggilam A, et al. Endothelial nitric oxide signaling regulates Notch1 in aortic valve disease. J Mol Cell Cardiol 2013;60:27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Laforest B, Andelfinger G, Nemer M. Loss of Gata5 in mice leads to bicuspid aortic valve. J Clin Invest 2011;121:2876–2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Padang R, Bagnall RD, Richmond DR, Bannon PG, Semsarian C. Rare non-synonymous variations in the transcriptional activation domains of GATA5 in bicuspid aortic valve disease. J Mol Cell Cardiol 2012;53:277–281. [DOI] [PubMed] [Google Scholar]
- 54.Bonachea EM, Chang SW, Zender G, LaHaye S, Fitzgerald-Butt S, McBride KL, Garg V GATA5 Sequence Variants Identified in Individuals with Bicuspid Aortic Valve. Pediatr Res 2014. doi: 10.1038/pr.2014.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Freed LA, Levy D, Levine RA, Larson MG, Evans JC, Fuller DL, et al. Prevalence and clinical outcome of mitral-valve prolapse. N Engl J Med 1999;341:1–7. [DOI] [PubMed] [Google Scholar]
- 56.Gupta V, Barzilla JE, Mendez JS, Stephens EH, Lee EL, Collard CD, et al. Abundance and localization of proteoglycans and hyaluronan within normal and myxomatous mitral valves. Cardiovasc Path 2009;18:191–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dietz HC, Cutting GR, Pyeritz RE, Maslen CL, Sakai LY, Corson GM, et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature 1991;352:337–339. [DOI] [PubMed] [Google Scholar]
- 58.Ng CM, Cheng A, Myers LA, Martinez-Murillo F, Jie C, Bedja D, et al. TGF-beta-dependent pathogenesis of mitral valve prolapse in a mouse model of Marfan syndrome. J Clin Invest 2004;114:1586–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Judge DP, Markwald RR, Hagege AA, Levine RA. Translational research on the mitral valve: from developmental mechanisms to new therapies. J Cardiovasc Transl Res 2011;4:699–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kyndt F, Gueffet JP, Probst V, Jaafar P, Legendre A, Le Bouffant F, et al. Mutations in the gene encoding filamin A as a cause for familial cardiac valvular dystrophy. Circulation 2007;115:40–49. [DOI] [PubMed] [Google Scholar]
- 61.Lardeux A, Kyndt F, Lecointe S, Marec HL, Merot J, Schott JJ, et al. Filamin-a-related myxomatous mitral valve dystrophy: genetic, echocardiographic and functional aspects. J Cardiovasc Transl Res 2011;4:748–756. [DOI] [PubMed] [Google Scholar]
- 62.Sauls K, de Vlaming A, Harris BS, Williams K, Wessels A, Levine RA, et al. Developmental basis for filamin-A-associated myxomatous mitral valve disease. Cardiovasc Res 2012;96:109–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Probst V, Le Scouarnec S, Legendre A, Jousseaume V, Jaafar P, Nguyen JM,et al. Familial aggregation of calcific aortic valve stenosis in the western part of France. Circulation. 2006;113:856–860. [DOI] [PubMed] [Google Scholar]
- 64.Bella JN, Tang W, Kraja A, Rao DC, Hunt SC, Miller MB, et al. Genome-wide linkage mapping for valve calcification susceptibility loci in hypertensive sibships: the Hypertension Genetic Epidemiology Network Study. Hypertension. 2007;49:453–460. [DOI] [PubMed] [Google Scholar]
- 65.Thanassoulis G, Campbell CY, Owens DS, Smith JG, Smith AV, Peloso GM, et al. Genetic associations with valvular calcification and aortic stenosis. N Engl J Med. 2013;368:503–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Garg V, Muth AN, Ransom JF, Schluterman MK, Barnes R, King IN, et al. Mutations in NOTCH1 cause aortic valve disease. Nature 2005;437:270–274. [DOI] [PubMed] [Google Scholar]
- 67.Hofmann JJ, Briot A, Enciso J, Zovein AC, Ren S, Zhang ZW, et al. Endothelial deletion of murine Jag1 leads to valve calcification and congenital heart defects associated with Alagille syndrome. Development 2012;139:4449–4460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kokubo H, Miyagawa-Tomita S, Nakashima Y, Kume T, Yoshizumi M, Nakanishi T, et al. Hesr2 knockout mice develop aortic valve disease with advancing age. Arterioscl Thromb Vasc Biol 2013;33:e84–92, doi: 10.1161/ATVBAHA.112.300573. [DOI] [PubMed] [Google Scholar]
- 69.Nus M, MacGrogan D, Martinez-Poveda B, Benito Y, Casanova JC, Fernandez-Aviles F, et al. Diet-induced aortic valve disease in mice haploinsufficient for the Notch pathway effector RBPJK/CSL. Arterioscl Thromb Vasc Biol 2011;31:1580–1588. [DOI] [PubMed] [Google Scholar]
- 70.Acharya A, Hans CP, Koenig SN, Nichols HA, Galindo CL, Garner HR, et al. Inhibitory role of Notch1 in calcific aortic valve disease. PLoS One 2011;6:e27743, doi: 10.1371/journal.pone.0027743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Peacock JD, Levay AK, Gillaspie DB, Tao G, Lincoln J. Reduced sox9 function promotes heart valve calcification phenotypes in vivo. Circ Res 2010;106:712–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Peacock JD, Huk DJ, Ediriweera HN, Lincoln J. Sox9 transcriptionally represses Spp1 to prevent matrix mineralization in maturing heart valves and chondrocytes. PLoS One 2011;6:e26769, doi: 10.1371/journal.pone.0026769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wirrig EE, Hinton RB, Yutzey KE. Differential expression of cartilage and bone-related proteins in pediatric and adult diseased aortic valves. J Mol Cell Cardiol 2011;50:561–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Alexopoulos A, Bravou V, Peroukides S, Kaklamanis L, Varakis J, Alexopoulos D, et al. Bone regulatory factors NFATc1 and Osterix in human calcific aortic valves. Int J Cardiol 2010;139:142–149. [DOI] [PubMed] [Google Scholar]
- 75.Caira FC, Stock SR, Gleason TG, McGee EC, Huang J, Bonow RO, et al. Human degenerative valve disease is associated with up-regulation of low-density lipoprotein receptor-related protein 5 receptor-mediated bone formation. J Am Coll Cardiol 2006;47:1707–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Morita H Human genomics in cardiovascular medicine: implications and perspectives. Circ J 2013;77:876–885. [DOI] [PubMed] [Google Scholar]
- 77.Habashi JP, Judge DP, Holm TM, Cohn RD, Loeys B, Cooper TK, et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science 2006;312:117–121. [DOI] [PMC free article] [PubMed] [Google Scholar]