

Abstract
The SARS-CoV-2 pandemic is a healthcare crisis caused by insufficient knowledge applicable to effectively combat the virus. Therefore, different scientific discovery strategies need to be connected, to generate a rational treatment which can be made available as rapidly as possible. This relies on a solid theoretical understanding of the mechanisms of SARS-CoV-2 infection and host responses, which is coupled to the practical experience of clinicians that are treating patients. Because SARS-CoV-2 enters the cell by binding to angiotensin-converting enzyme 2 (ACE2), targeting ACE2 to prevent such binding seems an obvious strategy to combat infection. However, ACE2 performs its functions outside the cell and was found to enter the cell only by angiotensin II type 1 receptor (AT1R)-induced endocytosis, after which ACE2 is destroyed. This means that preventing uptake of ACE2 into the cell by blocking AT1R would be a more logical approach to limit entry of SARS-CoV-2 into the cell. Since ACE2 plays an important protective role in maintaining key biological processes, treatments should not disrupt the functional capacity of ACE2, to counterbalance the negative effects of the infection. Based on known mechanisms and knowledge of the characteristics of SARS-CoV we propose the hypothesis that the immune system facilitates SARS-CoV-2 replication which disrupts immune regulatory mechanisms. The proposed mechanism by which SARS-CoV-2 causes disease immediately suggests a possible treatment, since the AT1R is a key player in this whole process. AT1R antagonists appear to be the ideal candidate for the treatment of SARS-CoV-2 infection. AT1R antagonists counterbalance the negative consequences of angiotesnin II and, in addition, they might even be involved in preventing the cellular uptake of the virus without interfering with ACE2 function. AT1R antagonists are widely available, cheap, and safe. Therefore, we propose to consider using AT1R antagonists in the treatment of SARS-CoV-2.
Keywords: Coronavirus, COVID-19, Renin–angiotensin–aldosterone system, Angiotensin receptor blockers, Angiotensin-converting enzyme 2
Introduction
On 11 March 2020, the World Health Organizaton (WHO) declared the coronavirus disease COVID-19 outbreak to have evolved to a pandemic, once more emphasizing the need for preventive and curative treatments. In this COVID-19 pandemic, time pressure is extreme, and the race to find a cure and effective prevention is on. Science must be swift, and conclusions must be drawn from the limited knowledge at hand, acting on everything that has been learned from previous outbreaks that may bear similar characteristics. Human coronaviruses cause ∼30% of upper respiratory tract infections but are rarely lethal. Occasionally, however, some strains emerge that are life-threatening, having caused past epidemics such as severe acute respiratory syndrome (SARS) and Middle East respiratory syndrome (MERS), as well as the current emerging SARS-CoV-2 pandemic. Because SARS-CoV-2 enters the cell by binding to angiotensin-converting enzyme 2 (ACE2), targeting ACE2 to prevent such binding seems an obvious strategy to combat infection. However, ACE2 performs its functions outside the cell and was found to enter the cell only via angiotensin II (AngII) type 1 receptor (AT1R)-induced endocytosis, after which ACE2 is destroyed. Since ACE2 plays an important role in maintaining key biological processes, the deficiency of this enzyme may have severe consequences.
Translational perspective
We hypothesize that the immune system facilitates SARS-CoV-2 entry into the cell, while the virus is bound to angiotensin-converting enzyme 2 (ACE2). This causes extracellular ACE2 deficiency, which disrupts immune regulatory mechanisms. Since angiotensin II type 1 receptor (AT1R) appears to facilitate entry of SARS-CoV-2 into the cell and also aggravates the disrupting mechanism of angiotensin II (AngII), AT1R antagonists seem to be the ideal candidate for the treatment of SARS-CoV-2 infection. AT1R antagonists counterbalance the negative consequences of AngII and may prevent cellular uptake of the virus without interfering with ACE2 function. AT1R antagonists are widely available, cheap, and safe. Therefore, we propose to consider using AT1R antagonists in the treatment of SARS-CoV-2.
There is much discussion about the role of the renin–angiotensin system (RAS) in SARS-CoV-2 infection. Some authors claim that the use of ACE inhibitors (ACEIs) or angiotensin receptor blockers (ARBs) is detrimental to SARS-CoV-2 infection.1 This is due to the observation that among the more severe cases of SARS-CoV-2 infection, a larger number of individuals had hypertension and diabetes.2–5 A recent report in the Journal of Travel Medicine suggests the same, namely that ACEIs and ARBs might actually both increase the risk of a COVID infection, since they both increase ACE2 levels.6 This is based on the descriptive analysis of 1099 patients, with laboratory-confirmed COVID-19 infections, from Wuhan China, that found more severe disease outcomes in patients with hypertension, coronary artery disease, diabetes, and chronic renal disease. This suggested that, since all the patients with such a diagnosis have an indication for treatment with ACEIs or ARBs, the increased risk of these groups is due to these treatments. This, however, assumes that increased levels of ACE2 are associated with increased viral uptake. Considering the fact that ACE2 acts outside the cell and was found only to enter the cell via AT1R-induced endocytosis, after which ACE2 is destroyed,7 the extracellular expression of ACE2 may very well be inversely related to viral uptake. Also, Danser et al. explain that there are no data to support the notion that ACEIs or ARBs facilitate coronavirus entry by increasing ACE2 expression, either in animal experimental or in human research.8 On the contrary, animal data even support that elevated ACE2 expression, as observed in patients treated with an ARB, might confer potential protective pulmonary and cardiovascular effects.
In the New England Journal of Medicine, Vaduganathan et al. discuss the potential beneficial effects of ACEIs and ARBs in COVID-19.9 They state that SARS-CoV-2 appears to down-regulate ACE2 functions and that it has been postulated that ACE2 deficiency induces unabated AngII activity, which may be in part responsible for organ njury in COVID-19.
ACE2 and angiotensin II
AngII, as part of the RAS system, plays an important role in body fluid homeostasis and inflammatory responses. AngII has proinflammatory, proliferative, and profibrotic functions that are beneficial during infection and hypoxia but are deleterious if present in a healthy person or in the case of prolonged activation during disease.10 During pulmonary infection, AngII induces pulmonary vasoconstriction and vascular permeability, which reduces hypoxia and facilitates extravasation of cytokines to the site of inflammation, respectively. This inflammatory response, if exacerbated, causes oedema and respiratory distress.
ACE2 is mainly located at and bound to the plasma membrane of many organs, among which are the lung epithelia.11 ACE2 converts AngII to Ang(1-7) and AngI to Ang(1-9) (Figure 1). By converting AngII to Ang(1-7), it ends the AngII-induced proinflammatory response. AngII, in turn, induces cellular internalization of ACE2 by endocytosis and its degradation in lysosomes.7 ACE2 thus regulates and antagonizes AngII actions, while AngII simultaneously decreases the expression of membrane-bound ACE2. Although much is known about this homeostatic-type interaction, steering towards a healthy equilibrium, rather than an exacerbation of inflammation, a complete description of the regulatory mechanisms is still lacking.
Figure 1.
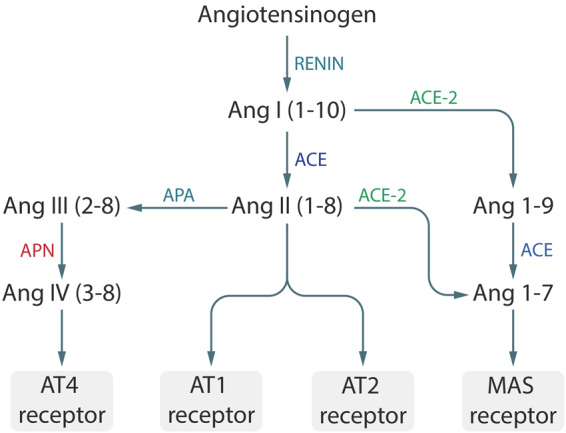
The RAS system. Coronavirus HCoV-229E binds to the APN receptor to enter the cell. SARS-CoV, NL63, and SARS-CoV-2 use the ACE2 receptor.
How does SARS-CoV-2 enter the cell?
The entry points of coronaviruses are known. and Jia et al. found that infection of human airway epithelia with SARS coronavirus correlated with ACE2 expression.12 It has also been shown that ACE2 endocytosis is dependent upon AT1R.7,13 The new SARS-CoV-2 also uses ACE2 to enter the cell14 and relies on ‘priming’ by the serine protease transmembrane protease, serine 2 (TMPRSS2).15,16
Once inside the cell, the virus escapes from the endosome, free to replicate in the cytoplasm. As one of the consequences, the normal AngII:ACE2 ratio is disrupted. Decreased ACE2 causes an unregulated inflammatory response, disproportionate to the threat posed by the initial infection.
Kuba et al.17 indeed provide the molecular explanation for how down-regulation of ACE2, through binding of SARS-CoV spike protein, contributes to the severity of lung pathologies. The fundamental problem is to understand the nature of the molecular signals that cause the ongoing induction of AngII during infection with SARS-CoV-2.
The function of the RAS in the immune response to infections
Upon infection by a previously unencountered virus, the initial immune response from the host is innate and non-specific. The adaptive, virus-specific response occurs much later. This non-specific response includes AngII activation.10,18 Active AngII inhibits the production of nitric oxide (NO), which results in a marked inflammatory reaction.19 Absence of NO induces vasoconstriction and increased vascular permeability, which prevents hypoxia and facilitates the entrance of cytokines to sites of active infection.
AngII is endogenously produced in T cells, where it plays an important role in T-cell activation and migration to the site of action.20 In SARS-CoV and MERS-CoV infections, priming of virus-specific T cells was found to be reduced, restricting the number of T cells that recognize the coronaviruses.21 Lower T-cell priming causes a delayed virus-specific immune response. In such a situation, AngII activation maintains the non-specific, innate immune reaction, prolonging the period during which the virus can enter the cell and replicate and during which ACE2 is destroyed.
AngII may engage two different receptors, AT1R and AT2R, which play opposite roles. AT1R facilitates the endocytosis of membrane-bound ACE2 at the cell surface and delivers it to the lysosomes for degradation. Upon delivery, AT1R returns to the plasma membrane to repeat this task.10 AT2R, in contrast, induces the activation of functional ACE2, which converts AngII to Ang(1-7).22
The binding of SARS-CoV-2 to ACE2 to endocytose the virus and direct the complex to lysosomes for destruction may be considered an alternative strategy to limit viral load.14 However, if a specific pH is reached, the viral envelope may fuse with the endosome before reaching the lysosome, releasing the virus into the cytoplasm for subsequent replication.23
Before ACE2 reaches the lysosomes, SARS-CoV-2 escapes by fusing with the endosomal membrane, relocating into the cytoplasm where it can replicate.14 Normally, when ACE2 reaches the lysosomes, NO is produced,24 which is thought to reduce viral replication25 and initiate a virus-specific immune response.26 This lysosomal NO down-regulates AT1R, prompting AngII to activate AT2R. AT2R then induces the production of ACE2, which in turn converts AngII into Ang(1-7), which further induces the production of NO, necessary for the adaptive immune response27 (Figure 2). In parallel, inflammation is reduced and the virus-specific immunity starts replacing non-specific inflammatory responses.27
Figure 2.

Hypothesis of the mechanism of replication of SARS-CoV-2 and treatment aims to prevent virus replication and initiating activation of the adaptive immune response. During inflammation and in the absence of NO, AngII activates the AT1R. AT1R induces cellular uptake of ACE2-bound SARS-CoV-2. Inhibition of AT1R inhibits viral uptake and facilitates AT2R-induced ACE2 expression and subsequent activation of the adaptive immune response.
Unfortunately, SARS-CoV, and most probably also SARS-CoV-2, induces the production of reactive oxygen species (ROS), in particular superoxide, which serves a function in cellular stress signalling.28 These superoxide particles react with NO to form peroxynitrite (ONOO–),29 which is unable to prevent ACE2 uptake. In the absence of NO, AngII favours the AT1R pathway,30 which in turn induces the endocytosis of membrane-bound ACE2, aggravating the inflammatory response. Therefore, it is suggested that, during active SARS-CoV-2 infection, AT1R-mediated ACE2 destruction plays a deleterious role, promoting both viral uptake and AngII-induced inflammation. Destruction of NO by superoxide radicals further induces the AngII-mediated deterioration, leading to lung injury and lung oedema.
The effect of ACE2 disruption on other organs
Since ACE2, in addition to expression in lung alveolar epithelium, is also expressed in other organs, such as the intestine, kidney, and heart,31 COVID-19 can also cause morbidity and mortality originating from disruptions of these organs. It has been shown that the SARS-CoV-2 infection was related to active myocardial injury, myocardial stress, and cardiomyopathy during the course of illness.32 Additionally, ∼25% occurrence of acute kidney injury (AKI) has been reported in several clinical settings.33,34 As regards the higher AngII levels, due to the disrupted ACE2 by the SARS-CoV-2 infection, it was recently shown that this was associated with increased coagulation and fibrinolysis, explaining the observed higher values of D-dimer and cardiovascular thrombotic events.35
Treating SARS-CoV-2 infection with AT1R antagonists
Following the above, SARS-CoV-2 infection by binding to ACE2 disrupts the normal AngII:ACE2 balance, leading to an excessive inflammatory reaction, which may ultimately lead to the ‘cytokine storm’ seen in some patients. It has indeed been reported that AT1R antagonists inhibit the activation of NF-κB and AP-1 in the lung, which mediate the release of cytokines and contribute to acute lung injury.36 Since the most important mechanism leading to pathology is the uncontrolled AngII activity, leading to increased activation of AT1R which appears to be involved in viral uptake, treatment actions should be aimed at preventing or inhibiting this. AT1R antagonists are therefore the ideal candidates.37 AT1R antagonists are expected to decrease AngII activity and reduce AT1R-induced uptake of ACE2-bound SARS-CoV-2 while preserving ACE2 functions.
Several AT1R antagonists have already been on the market as antihypertensive drugs for many years, are inexpensive, widely used, and have virtually no side effects, which make them the ideal treatment option for SARS-CoV-2 infection. We are not the only ones suggesting this hypothesis; several other researchers have come to the same conclusion.37–39
Postulating that the above hypothesis is correct, it may be expected that treatment with AT1R inhibitors (ARBs) might limit viral uptake and preserve ACE2 functions. On the other hand, the expected treatment effect will probably depend on the phase of the disease in which the treatment is initiated. In the early phase, it might be expected that treatment with an AT1R antagonist may completely prevent the excessive inflammation related to the SARS-CoV-2 infection, by restoring ACE2 function and thereby preventing viral load accumulation. On the other hand, when the disease reaches the ‘point of no return’, when viral replication has exhausted cellular metabolites and the inflammatory response has caused too much damage, AT1R antagonists may not be able to prevent further deterioration.
It has been suggested that there might also be a potential harm in blocking the RAS system in SARS-CoV-2 infection.40 It was postulated that the supposed up-regulation of ACE2 might lead to these deletorious effects. However, as already referred to, an up-regulation, if it occurs at all, is more likely to protect from severe disease, as has also been suggested by others.8,9
Other treatment options
Considering our hypothesis on the proinflammatory role of AngII and its permissive function in viral uptake in COVID-19 and the anti-inflammatory effect of ACE2, treatment should ideally prevent viral uptake and replication, while preserving ACE2 functions.
Chloroquine
The antiviral mechanism by which chloroquine exerts its action is by disruption of the glycosylation of ACE2 and by disruption of endosome recycling by increasing the pH of the cell.41 Due to disruption of ACE2 glycosylation, the binding of the virus to ACE2 will be impeded. Vincent et al. found that chloroquine was effective in preventing or diminishing viral uptake shortly after infection, by preventing SARS-CoV-2 from binding to ACE2.41 However, because glycosylation of enzymes is generally essential for their functions, we may fear that ACE2 is no longer functional after treatment with chloroquine. As soon as the host is infected, chloroquine might prevent replication. Wang et al. showed that SARS-CoV remained in the endosome, bound to its receptor, for at least 14 h, where the virus is unable to replicate.13 This raises the possibility that the virus can escape from the endosome at a later stage, starting its delayed replication after the patient is assumed to be cured.33 Therefore, it might be postulated that chloroquine appears to be unsuitable for the treatment of a SARS-CoV-2 infection, whereas it could play a modest role in its prevention. However, concerns arise regarding the possible disruption of the AngII–ACE2 equilibrium, due to the gycosylation on ACE2. Therefore, research on chloroquine as a preventive treatment option should be inititated before it can be introduced and indeed several chloroquine prevention trials in healthcare personnel have already been started.
Recombinant ACE2
Since ACE2 exerts such beneficial actions, it is tempting to assume that SARS-CoV-2 can bind to the excess of exogenous ACE2, thereby preventing it from entering the cell. Recombinant ACE2 has been developed and tested in phase I and phase II trials, and was found to be safe.42,43 Recombinant ACE2 has the capacity to convert AngII into Ang(1-7). It remains to be seen if overexpression of ACE2 disrupts the tissue-specific AngII:Ang(1-7) equilibrium, which would impair tissues in responding to hypoxia, damage, or infection by increasing AngII.
Antiviral drugs
Antiviral drugs may disrupt the viral life cycle at different stages. Some disrupt the synthesis of viral proteins and nucleic acids. Other antiviral drugs inhibit virus assembly, budding, or maturation. A separate class prevents entry into the cell.44 In the case of SARS-CoV-2, the only antiviral drugs that are being used in the treatment of the infection are the class that prevents viral replication inside the cell after the SARS-CoV-2–ACE2 complex has been endocytosed. To our knowledge, no antiviral drugs exist that decrease viral load without interfering with ACE2.
Immunomodulating therapies
Several immunomodulating therapies have been initiated to date. The best known immunomodulating therapy is steroids, but several interleukin-6 (IL-6) blocking agents such as tocilizumab are also currently being explored in clinical trials. Although the rationale seems promising, the biggest concern with these drugs are the severe side effects, of which secondary infections are the most important. In addition, these treatment options are rather expensive and, therefore, not attractive for widespread use in the early phase of the disease.
Research
Research on the hypothesis that AT1R antagonists are suitable treatment options for COVID-19 should consist of randomized controlled clinical trials, investigating the effect of an AT1R antagonist compared with usual care or placebo. Considering the proposed disease mechanism in which increasing AngII levels induce increased viral uptake, we believe that the treatment will be most effective when started within 5 days of infection. Treatment with AT1R antagonists can be easily implemented in the elderly, since they are a cheap and safe option, because of their widespread use. Once the decision is made not to admit a patient to hospital or to give treatment with mechanical ventilation, there are no other treatment options for this group of patients. Secondly, clinical data specifically concerning AT1R antagonists compared with other antihypertensive medication should be investigated with regard to their relationship with the severity of the SARS-CoV-2 infection. A recent article by Zhang et al. showed that among 1128 hypertensive individuals admitted to hospital because of SARS-CoV-2 infection, individuals using either ACEIs or an AT1R antagonist were less prone to have severe disease.45 Currently 11 randomized controlled trials with AT1R antagonists are registered. Most studies will include hospitalized patients and only two studies will include non-hospitalized patients. Most studies use modest to low dose short-acting AT1R antagonists and only three studies use higher doses of AT1R antagonists. One study is paerticularly designed for the elderly, ≥65 years of age (Table 1).
Table 1.
Registered randomized controlled trials using AT1R antagonists, up to 23 April 2020
| Trial registry number | Sponsor | Study type | Intervention | Inclusion criteria | Endpoint | Intended sample size | Recruiting |
|---|---|---|---|---|---|---|---|
| NCT04311177 | University of Minnesota | Multicentre double-blind, placebo-controlled randomized |
|
|
Admission to hospital | 580 | Yes |
| NCT04312009 | University of Minnesota | Multicentre double-blind, placebo-controlled randomized |
|
|
Difference in estimated (PEEP adjusted) P/F ratio at 7 days | 200 | Yes |
| NCT04335123 | University of Kansas Medical Center | Open label, safety |
|
|
Adverse event due to losartan | 50 | Yes |
| NCT04343001 | London School of Hygiene and Tropical Medicine | Multicentre, open label, 2 × 2 × 2 factorial, randomized |
|
|
28 day death | 10 000 | No |
| NCT04328012 | Bassett Healthcare | Multicentre, double-blind, placebo-controlled, randomized comparison |
|
|
NCOSS scores | 4000 | Yes |
| NCT04340557 | Sharp HealthCare | Multicentre open label, randomized |
|
|
ICU admission for mechanical ventilation | 200 | Yes |
| NCT04349410 | The Camelot Foundation | Clinical trial, factorial, randomized |
|
Proven SARS-CoV-2 infection | Improvement in FMTVDM measurement with nuclear imaging. | 500 | Enrolling by invitation |
| NCT04351724 | Medical University of Vienna | Multicentre, open label, randomized |
|
|
Sustained improvement (>48 h) of one point on the WHO scale | 500 | Yes |
| NCT04355936 | Laboratorio Elea S.A.C.I.F. y A. | Clinical trial, open label, randomized |
|
|
Need for supplementary oxygen | 400 | Yes |
| NCT04335786 | Radboud University | Multicentre, double-blind, placebo-controlled, randomized |
|
|
Admission to the ICU for mechanical ventilation or death | 351 | Yes |
| NCT04356495 | University Hospital, Bordeaux | Multicentre, open label, randomized |
|
|
Proportion of participants with an occurrence of Hospitalization or death | 1057 | No |
NCOSS scores, National COVID-19 Ordinal Severity Scale scores; FMTVDM, Fleming method for tissue and vascular differentiation and metabolism; t.d., twice a day.
Conclusion
Because SARS-CoV-2 enters the cell bound to ACE2, which induces ACE2 deficiency at the cell membrane, AngII is persistently activated. Increased AngII induces activation of AT1R, causing more uptake of SARS-CoV-2 and increasing ACE2 deficiency, thus maintaining and exacerbating a non-specific immune response, consisting of cytokine-induced inflammation. This non-specific immune response is an attempt to reduce the viral load, while the specific immune response is mounted. Unrestrained AngII eventually causes death by respiratory distress induced by excessive inflammation and its deleterious effects on other organs. Therefore, SARS-CoV-2-induced mortality is promoted by three mechanisms: (i) increased AngII induces endocytosis of ACE2-bound SARS-CoV-2, leading to ACE2 deficiency and viral replication; (ii) ACE2 deficiency prevents the priming of an adaptive immune response by lack of NO; and (iii) Ang II induces an increase in viral load leading to an increased innate immune response and a further increase in AngII levels. Therefore, treatments should aim at preventing the AngII ‘storm’ in an early phase of the infection, restoring the modulation of NO, and preventing the entry of SARS-CoV-2 into the cell. All these mechanisms are targeted by AT1R antagonists. They may reduce morbid inflammatory distress and provide an environment to facilitate an effective, virus-specific adaptive immune response.
Conflict of interest: none declared.
Acknowledgements
We wish to thank Otto Offringa for revising the manuscript and Julian Marquette for the illustrations.
References
- 1. Fang L, Karakiulakis G, Roth M.. Are patients with hypertension and diabetes mellitus at increased risk for COVID-19 infection? Lancet Respir Med 2020;8:e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Guan WJ, Ni ZY, Hu Y, Liang WH, Ou CQ, He JX, Liu L, Shan H, Lei CL, Hui DSC, Du B, Li LJ, Zeng G, Yuen KY, Chen RC, Tang CL, Wang T, Chen PY, Xiang J, Li SY, Wang JL, Liang ZJ, Peng YX, Wei L, Liu Y, Hu YH, Peng P, Wang JM, Liu JY, Chen Z, Li G, Zheng ZJ, Qiu SQ, Luo J, Ye CJ, Zhu SY, Zhong NS; China Medical Treatment Expert Group for Covid-19. Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med 2020;382:1708–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, Zhang L, Fan G, Xu J, Gu X, Cheng Z, Yu T, Xia J, Wei Y, Wu W, Xie X, Yin W, Li H, Liu M, Xiao Y, Gao H, Guo L, Xie J, Wang G, Jiang R, Gao Z, Jin Q, Wang J, Cao B.. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020;395:497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang D, Hu B, Hu C, Zhu F, Liu X, Zhang J, Wang B, Xiang H, Cheng Z, Xiong Y, Zhao Y, Li Y, Wang X, Peng Z.. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. JAMA 2020;doi: 10.1001/jama.2020.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhang JJ, Dong X, Cao YY, Yuan YD, Yang YB, Yan YQ, Akdis CA, Gao YD.. Clinical characteristics of 140 patients infected with SARS-CoV-2 in Wuhan, China. Allergy 2020;doi: 10.1111/all.14238.. [DOI] [PubMed] [Google Scholar]
- 6. Diaz JH. Hypothesis: angiotensin-converting enzyme inhibitors and angiotensin receptor blockers may increase the risk of severe COVID-19. J Travel Med 2020;doi: 10.1093/jtm/taaa041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Deshotels MR, Xia H, Sriramula S, Lazartigues E, Filipeanu CM.. Angiotensin II mediates angiotensin converting enzyme type 2 internalization and degradation through an angiotensin II type I receptor-dependent mechanism. Hypertension 2014;64:1368–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Danser AHJ, Epstein M, Batlle D.. Renin–angiotensin system blockers and the COVID-19 pandemic: at present there is no evidence to abandon renin–angiotensin system blockers. Hypertension 2020;doi: 10.1161/HYPERTENSIONAHA.120.15082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vaduganathan M, Vardeny O, Michel T, McMurray JJV, Pfeffer MA, Solomon SD.. Renin–angiotensin–aldosterone system inhibitors in patients with Covid-19. N Engl J Med 2020;382:1653–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Benigni A, Cassis P, Remuzzi G.. Angiotensin II revisited: new roles in inflammation, immunology and aging. EMBO Mol Med 2010;2:247–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Serfozo P, Wysocki J, Gulua G, Schulze A, Ye M, Liu P, Jin J, Bader M, Myöhänen T, García-Horsman JA, Batlle D.. Ang II (angiotensin II) conversion to angiotensin-(1-7) in the circulation is POP (prolyloligopeptidase)-dependent and ACE2 (angiotensin-converting enzyme 2)-independent. Hypertension 2020;75:173–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jia HP, Look DC, Shi L, Hickey M, Pewe L, Netland J, Farzan M, Wohlford-Lenane C, Perlman S, McCray PB Jr.. ACE2 receptor expression and severe acute respiratory syndrome coronavirus infection depend on differentiation of human airway epithelia. J Virol 2005;79:14614–14621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang H, Yang P, Liu K, Guo F, Zhang Y, Zhang G, Jiang C.. SARS coronavirus entry into host cells through a novel clathrin- and caveolae-independent endocytic pathway. Cell Res 2008;18:290–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kuba K, Imai Y, Rao S, Gao H, Guo F, Guan B, Huan Y, Yang P, Zhang Y, Deng W, Bao L, Zhang B, Liu G, Wang Z, Chappell M, Liu Y, Zheng D, Leibbrandt A, Wada T, Slutsky AS, Liu D, Qin C, Jiang C, Penninger JM.. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat Med 2005;11:875–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Matsuyama S, Aydan A, Ode H, Hata M, Sugiura W, Hoshino T.. Structural and energetic analysis on the complexes of clinically isolated subtype C HIV-1 proteases and approved inhibitors by molecular dynamics simulation. J Phys Chem B 2010;114:521–530. [DOI] [PubMed] [Google Scholar]
- 16. Hoffmann M, Kleine-Weber H, Schroeder S, Kruger N, Herrler T, Erichsen S, Hoffmann M, Kleine-Weber H, Schroeder S, Kruger N, Herrler T, Erichsen S.. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020;181:271–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kuba K, Imai Y, Penninger JM.. Multiple functions of angiotensin-converting enzyme 2 and its relevance in cardiovascular diseases. Circ J 2013;77:301–308. [DOI] [PubMed] [Google Scholar]
- 18. Nataraj C, Oliverio MI, Mannon RB, Mannon PJ, Audoly LP, Amuchastegui CS, Ruiz P, Smithies O, Coffman TM.. Angiotensin II regulates cellular immune responses through a calcineurin-dependent pathway. J Clin Invest 1999;104:1693–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhou MS, Schulman IH, Raij L.. Nitric oxide, angiotensin II, and hypertension. Semin Nephrol 2004;24:366–378. [DOI] [PubMed] [Google Scholar]
- 20. Silva-Filho JL, Souza MC, Henriques M, Morrot A, Savino W, Nunes MP, Caruso-Neves C, Pinheiro AA.. AT1 receptor-mediated angiotensin II activation and chemotaxis of T lymphocytes. Mol Immunol 2011;48:1835–1843. [DOI] [PubMed] [Google Scholar]
- 21. Channappanavar R, Zhao J, Perlman S.. T cell-mediated immune response to respiratory coronaviruses. Immunol Res 2014;59:118–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ali Q, Wu Y, Hussain T.. Chronic AT2 receptor activation increases renal ACE2 activity, attenuates AT1 receptor function and blood pressure in obese Zucker rats. Kidney Int 2013;84:931–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. White JM, Whittaker GR.. Fusion of enveloped viruses in endosomes. Traffic 2016;17:593–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li Y, Wu W, Yang J, Yuan L, Liu C, Zheng J, Yang R.. Engineering a nanolab for the determination of lysosomal nitric oxide by the rational design of a pH-activatable fluorescent probe. Chem Sci 2016;7:1920–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Akerstrom S, Gunalan V, Keng CT, Tan YJ, Mirazimi A.. Dual effect of nitric oxide on SARS-CoV replication: viral RNA production and palmitoylation of the S protein are affected. Virology 2009;395:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Niedbala W, Cai B, Liew FY.. Role of nitric oxide in the regulation of T cell functions. Ann Rheum Dis 2006;65 Suppl 3:iii37–iii40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Safari T, Nematbakhsh M.. Role of angiotensin type 2 receptor on nitric oxide production response to angiotensin II administration in ovariectomised rats treated with estradiol. Int J Prev Med 2014;5:238–240. [PMC free article] [PubMed] [Google Scholar]
- 28. Zhang L, Wei L, Jiang D, Wang J, Cong X, Fei R.. SARS-CoV nucleocapsid protein induced apoptosis of COS-1 mediated by the mitochondrial pathway. Artif Cells Blood Substit Immobil Biotechnol 2007;35:237–253. [DOI] [PubMed] [Google Scholar]
- 29. Beckman JS, Koppenol WH.. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am J Physiol 1996;271:C1424–C1437. [DOI] [PubMed] [Google Scholar]
- 30. Sharma NM, Zheng H, Li YF, Patel KP.. Nitric oxide inhibits the expression of AT1 receptors in neurons. Am J Physiol Cell Physiol 2012;302:C1162–C1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hamming I, Timens W, Bulthuis ML, Lely AT, Navis G, van Goor H.. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol 2004;203:631–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Arentz M, Yim E, Klaff L, Lokhandwala S, Riedo FX, Chong M, Lee M.. Characteristics and outcomes of 21 critically ill patients with COVID-19 in Washington state. JAMA 2020;doi: 10.1001/jama.2020.4326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yang X, Yu Y, Xu J, Shu H, Xia J, Liu H, Wu Y, Zhang L, Yu Z, Fang M, Yu T, Wang Y, Pan S, Zou X, Yuan S, Shang Y.. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: a single-centered, retrospective, observational study. Lancet Respir Med 2020;8:475–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fanelli V, Fiorentino M, Cantaluppi V, Gesualdo L, Stallone G, Ronco C, Castellano G.. Acute kidney injury in SARS-CoV-2 infected patients. Crit Care 2020;24:155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tang N, Bai H, Chen X, Gong J, Li D, Sun Z.. Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J Thromb Haemost 2020;18:1094–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wang F, Xia ZF, Chen XL, Jia YT, Wang YJ, Ma B.. Angiotensin II type-1 receptor antagonist attenuates LPS-induced acute lung injury. Cytokine 2009;48:246–253. [DOI] [PubMed] [Google Scholar]
- 37. Phadke M, Saunik S.. Rapid response: Use of angiotensin receptor blockers such as telmisartan, losartan in nCoV Wuhan Corona Virus infections—novel mode of treatment. Response to the emerging novel coronavirus outbreak. BMJ 2020;368:m406.32005675 [Google Scholar]
- 38. Sun ML, Yang JM, Sun YP, Su GH.. [Inhibitors of RAS might be a good choice for the therapy of COVID-19 pneumonia]. Zhonghua Jie He He Hu Xi Za Zhi 2020;43:219–222. [DOI] [PubMed] [Google Scholar]
- 39. Gurwitz D. Angiotensin receptor blockers as tentative SARS-CoV-2 therapeutics. Drug Dev Res 2020;doi: 10.1002/ddr.21656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Aronson JK, Ferner RE.. Drugs and the renin–angiotensin system in covid-19. BMJ 2020;369:m1313. [DOI] [PubMed] [Google Scholar]
- 41. Vincent MJ, Bergeron E, Benjannet S, Erickson BR, Rollin PE, Ksiazek TG, Seidah NG, Nichol ST.. Chloroquine is a potent inhibitor of SARS coronavirus infection and spread. Virol J 2005;2:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Khan A, Benthin C, Zeno B, Albertson TE, Boyd J, Christie JD, Hall R, Poirier G, Ronco JJ, Tidswell M, Hardes K, Powley WM, Wright TJ, Siederer SK, Fairman DA, Lipson DA, Bayliffe AI, Lazaar AL.. A pilot clinical trial of recombinant human angiotensin-converting enzyme 2 in acute respiratory distress syndrome. Crit Care 2017;21:234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Haschke M, Schuster M, Poglitsch M, Loibner H, Salzberg M, Bruggisser M, Penninger J, Krähenbühl S. Pharmacokinetics and pharmacodynamics of recombinant human angiotensin-converting enzyme 2 in healthy human subjects. Clin Pharmacokinet 2013;52:783–792. [DOI] [PubMed] [Google Scholar]
- 44. Menendez-Arias L, Gago F.. Antiviral agents: structural basis of action and rational design. Subcell Biochem 2013;68:599–630. [DOI] [PubMed] [Google Scholar]
- 45. Zhang P, Zhu L, Cai J, Lei F, Qin JJ, Xie J, Liu YM, Zhao YC, Huang X, Lin L, Xia M, Chen MM, Cheng X, Zhang X, Guo D, Peng Y, Ji YX, Chen J, She ZG, Wang Y, Xu Q, Tan R, Wang H, Lin J, Luo P, Fu S, Cai H, Ye P, Xiao B, Mao W, Liu L, Yan Y, Liu M, Chen M, Zhang XJ, Wang X, Touyz RM, Xia J, Zhang BH, Huang X, Yuan Y, Rohit L, Liu PP, Li H.. Association of inpatient use of angiotensin converting enzyme inhibitors and angiotensin II receptor blockers with mortality among patients with hypertension hospitalized with COVID-19. Circ Res 2020;doi: 10.1161/CIRCRESAHA.120.317134. [DOI] [PMC free article] [PubMed] [Google Scholar]