

Abstract
While cell transplantation presents a potential strategy to treat the functional deficits of neurodegenerative diseases or central nervous system injuries, the poor survival rate of grafted cells in vivo is a major barrier to effective therapeutic treatment. In this study, we investigated the role of a peptide-based nanofibrous scaffold composed of the self-assembling peptide RADA16-I to support the reprogramming and maturation of human neurons in vitro and to transplant these neurons in vivo. The induced human neurons were generated via the single transcriptional factor transduction of induced pluripotent stem cells (iPSCs), which are a promising cell source for regenerative therapies. These neurons encapsulated within RADA16-I scaffolds displayed robust neurite outgrowth and demonstrated high levels of functional activity in vitro compared to that of 2-D controls, as determined by live cell calcium imaging. When evaluated in vivo as a transplantation vehicle for adherent, functional networks of neurons, monodisperse RADA16-I microspheres significantly increased survival (over 100-fold greater) compared to the conventional transplantation of unsupported neurons in suspension. The scaffold-encapsulated neurons integrated well in vivo within the injection site, extending neurites several hundred microns long into the host brain tissue. Overall, these results suggest that this biomaterial platform can be used to successfully improve the outcome of cell transplantation and neuro-regenerative therapies.
Keywords: nanofiber, self-assembling peptide, neuronal reprogramming, brain transplantation, human induced pluripotent stem cell, neural tissue engineering
Graphical Abstract
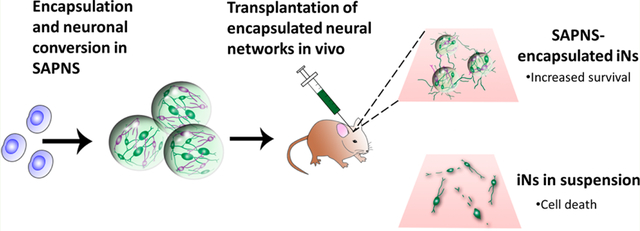
INTRODUCTION
Traumatic injury to the central nervous system (CNS) or neurodegenerative diseases can lead to extensive neuronal death and subsequent loss of function.1,2 Cell transplantation therapies hold promise for regeneration and functional recovery; however, poor in vivo survival rates of traditionally transplanted cells which are injected in suspension limit the efficacy of such therapies.3–7 This highlights the need for a strategy to improve the survival of transplanted neurons in vivo, which can be achieved via a neural tissue engineering approach of using a biomaterial scaffold as a transplantable cell support system.
In order to prepare for conventional transplantation procedures, cells must be first enzymatically detached from a 2-D culture surface and dissociated into a single cell suspension, thereby disrupting cell–cell and cell–material interactions, and impairing any previously formed neuronal connections. Anchorage-dependent cells have been shown to undergo apoptosis due to detachment from the extracellular matrix (ECM) and neighboring cells, known as anoikis.8 In comparison, culturing cells within a transplantable 3-D biomaterial scaffold confers the advantage of a matrix milieu that can maintain these cell–cell and cell–biomaterial interactions, leaving adherent neuronal networks intact, during the transition to a potentially hostile in vivo environment. Additionally, an appropriate 3-D scaffold is able to mimic the in vivo microenvironment more closely than traditional 2-D culture substrates.
A promising biomaterials platform for the support of 3-D neuronal cultures involves the design of self-assembling peptides, which are fabricated from natural amino acids and can undergo spontaneous assembly into nanofiber scaffolds when exposed to physiological media or salt solutions.9,10 One example of these self-assembling peptide nanofiber scaffolds (SAPNS) is RADA16-I, formed from alternating sequences of the amino acids arginine (R), alanine (A), and aspartic acid (D). When exposed to millimolar amounts of monovalent cations, the peptides undergo self-assembly to form stable β-sheets via ionic interactions between the positively charged arginine and negatively charged aspartic acid, and hydrophobic interactions between the alanine. These β-sheets have two distinct sides, with the exterior of the sheet formed by hydrophilic arginine and aspartic acid, and the interior of the sheet formed by hydrophobic alanine.9,11,12 After self-assembly, RADA16-I resembles a hydrogel with greater than 99% water content, with fibers approximately 10 nm in diameter and pores between 5 and 200 nm, surrounding cells in a manner similar to the native nanofibrous ECM.9,10 This synthetic peptide has been shown to support the in vitro attachment and growth of a wide variety of mammalian cells, particularly neural stem/progenitor cells.13–17 In vivo, RADA16-I has been shown to promote host axonal regeneration and blood vessel ingrowth, and also reduced the lesion size and glial reaction within the acutely injured brain.18,19 Self-assembled peptide scaffolds have not been previously used to transplant human neurons into the CNS.
With the advent of stem cell reprogramming technologies, human neurons can now be derived from human induced pluripotent stem cells (iPSCs), which have emerged as a promising cell source for transplantation, in vitro disease modeling, and drug screening.20–22 Mature, functional neuronal cells can be directly generated from iPSCs via transfection with one or more key transcription factors.23,24 Additionally, iPSCs are highly expandable and give us the ability to generate large numbers of neurons, whereas an extremely limited amount of induced neurons can be generated by directly reprogramming fibroblasts. This approach is advantageous due to rapid neuronal conversion (neurons are generated in approximately 2 weeks) and the ability to generate patient-specific neurons and specific subtypes of neurons. In comparison, traditional methods of differentiation can take months to generate neurons.
In this study, we investigated the role of self-assembled peptide nanofibrous scaffolds (SAPNS) based on the RADA16-I peptide as a 3-D in vitro culture environment for the reprogramming and functional maturation of neurons derived from iPSCs. Neurons were generated via the lentiviral-mediated transfection of human iPSCs, inducing these cells to express the single neuronal transcription factor NeuroD1 and undergo subsequent neuronal conversion. Further, we fabricated monodisperse RADA16-I microspheres using a custom-made microfluidic T-junction device and evaluated these microspheres as a vehicle for the minimally invasive in vivo delivery of iPSC-derived neurons into a mouse brain. This is the first report of using SAPNS for the culture and in vivo transplantation of human neurons.
MATERIALS AND METHODS
Lentiviral Infection of iPSCs.
Human iPSCs (HFF1-iPSC) were obtained from the Rutgers University Cell and DNA Repository (RUCDR). iPSCs were infected with lentiviruses as previously described.24,25 Briefly, iPSCs were treated with Accutase (Life Technologies) and plated as single cells in mTeSR-1 medium (Stem Cell Technologies, Vancouver, Canada) supplemented with 5 μM Y-27632 (R&D Systems, Minneapolis, MN). The following day, the medium was replaced with mTeSR-1 containing 5 μM Y-27632 and 2 μM Polybrene (Sigma-Aldrich, St. Louis, MO), and the concentrated virus was added (rtTA-FUW, Tet-O-FUW NeuroD1, and, for some infections, Tet-O-FUW EGFP). The lentiviral constructs and packaging plasmids were generated by Marius Wernig’s laboratory at Stanford University. The following day, the medium was replaced with mTeSR-1. For cell expansion and maintenance, infected iPSCs (termed iPSC-RN) were cultured under feeder-free conditions on tissue culture polystyrene dishes coated with Matrigel (BD Biosciences, Franklin Lakes, NJ) in mTeSR-1 medium at 37 °C and 5% CO2.
Cell Culture and Neuronal Induction.
To initiate neuronal induction in cells cultured in 2-D, iPSC-RNs were treated with Accutase and plated as single cells in mTeSR-1 supplemented with 5 μM Y-27632 on a Matrigel-coated dish. The following day, the medium was replaced with NBM medium: Neurobasal medium (Life Technologies) supplemented with 1× B27 supplement without Vitamin A (Life Technologies), 2 mM l-glutamine, 0.5% penicillin/streptomycin, 10 ng/mL each of BDNF, GDNF, and NT-3 (Peprotech), and 2 μg/mL doxycycline, to induce NeuroD1 and EGFP gene expression. The medium was replaced every 2–3 days for the duration of the experiments.
Encapsulation of Cells within Bulk RADA16-I Hydrogels.
RADA16-I, a self-assembling peptide with the amino acid sequence [COCH3]-RADARADARADARADA-[CONH2] (PuraMatrix), was kindly provided by 3-D Matrix, Inc. (Waltham, MA) at a concentration of 1% (w/v). RADA16-I (diluted to 0.5% in 10% sucrose) was sonicated in a bath sonicator for 30 min and vortexed prior to gelation. To encapsulate cells within RADA16-I, iPSC-RNs were treated with Accutase and washed once in 10% sucrose prior to resuspending in fresh 10% sucrose at a concentration of 10 × 106 or 20 × 106 cells/mL. The cell suspension and RADA16-I peptide were mixed by gentle pipetting in a 1:1 ratio for a final concentration of 5 × 106 or 10 × 106 cells in 0.25% RADA16-I, prior to transferring the mixture onto a UV-sterilized coverslip in a 24 well TCPS plate (100 μL of mixture per well). DMEM/F12 medium (0.5 mL) was gently added to the well to induce gelation, and the gels were placed into an incubator. After 10 min, approximately ~2/3 of the DMEM/F12 was removed and replaced with fresh DMEM/F12. After an additional 30 min, all of the DMEM/F12 was replaced with 0.5 mL of mTeSR-1 containing 5 μM Y-27632. The following day, the medium was replaced with NBM medium and was changed every 2–3 days for the duration of the experiments.
Microfluidic T-Junction Setup and RADA16-I Microsphere Fabrication.
Cells were encapsulated within RADA16-I microspheres using a custom-made microfluidic T-junction. The microfluidic T-junction was fabricated using commercially available fluorinated ethylene propylene (FEP) tubing (inlet tubing, 0.0625 in OD, 0.02 in ID; outlet tubing, 0.0625 in OD, 0.01 in ID) and polyether ether ketone (PEEK) micro-T-junction connectors (0.006 in through hole) from Upchurch Scientific (IDEX Health and Science). Female-to-female luer lock connectors and fingertight fittings were used to connect the polymer tubing to the syringes. Figure 4 shows the setup of the microfluidic T-junction device. The horizontal 10 mL syringe, containing hexadecane with 2% Span 80 (Sigma), was connected to a syringe pump (NE-1000, New Era Pump Systems). The vertical 1 mL syringe, containing the peptide mixture for gelation (0.5% RADA16-I mixed with 10% sucrose or cell suspension in 10% sucrose, 1:1), was connected to a second syringe pump (KD Scientific). For microsphere fabrication, the RADA16-I syringe pump flow rate (Qd) was set to 0.01 mL/min, and the hexadecane + Span 80 syringe pump flow rate (Qc) was set to 0.05 mL/min, resulting in a flow rate ratio of Qc/Qd = 5. RADA16-I microspheres exiting the outlet tube were collected in a 1.5 mL microcentrifuge tube containing DMEM/F12 medium with 0.25% Tween 20, centrifuged at 200g for 30 s to collect the microspheres, and washed once with DMEM/F12 medium. After centrifuging again at 200g for 1 min, the DMEM/F12 supernatant was removed, and the microspheres were transferred to mTeSR-1 medium containing 5 μM Y-27632 in a 24-well TCPS plate. RADA16-I microspheres were maintained in culture in a manner identical to that of the bulk RADA16-I hydrogels.
Figure 4.

Monodisperse RADA16-I microspheres were fabricated using a custom-made microfluidic T junction. (A) Schematic of the microsphere fabrication process. (B) Phase contrast images of blank and cell-containing microspheres. Scale bar: 100 μm. (C) Frequency distribution of blank microsphere diameter fabricated at a flow rate ratio of 5. (D) Graph illustrating the inverse relationship of microsphere diameter and flow rate ratio. Error bars: standard deviation.
Cell Viability.
Viability of cells encapsulated within the RADA16 peptide hydrogel was measured by the Live/Dead assay. Hydrogels containing encapsulated cells were incubated in a dye solution containing 4 μM Calcein AM (Life Technologies) and 1 μg/mL propidium iodide (Life Technologies) for 30 min at 37 °C. The stained cells were analyzed by fluorescence microscopy (Leica TCS-SP2 confocal microscope, Leica Microsystems, Wetzlar, Germany) and quantified using ImageJ software (NIH, Bethesda, MD). Quantitative viability data are presented as the mean ± SD.
Calcium Imaging.
iN cells cultured on glass coverslips or encapsulated within RADA16-I hydrogels were labeled with 3 μM Fluo-4 AM for 30 min at room temperature in an extracellular bath solution (147 μM NaCl, 4 μM KCl, 2 mM CaCl2, 2 mM MgCl2, 10 mM HEPES, and 10 μM glucose, pH 7.4) containing 0.02% Pluronic F-127 (Sigma-Aldrich). The cells were incubated in bath solution for an additional 30 min at room temperature, prior to transferring them to a custom-made electrical stimulation chamber. To assess calcium channel activity, an external electrical stimulus was applied using a function generator (Global Specialties) to deliver a 5 s stimulus at 7.5 V/cm with 5 ms square pulses at 40 Hz. Time-lapse imaging was performed on a Leica TCS-SP2 confocal microscope at a resolution of 512 × 512 pixels and a time resolution of 1 Hz. Image analysis was performed using custom ImageJ macros and MATLAB programs for region of interest (ROI) selection and fluorescence peak detection. Cells were considered to be responsive to the external electrical stimuli if they displayed an increase in fluorescence at least 3 standard deviations above baseline fluorescence in response to every delivered stimulus. Quantitative data are presented as the mean ± SD of the percentage of cells responding to electrical stimuli (5 frames and blank cells analyzed per replicate).
In vivo Transplantation.
All animal experiments were carried out according to the Rutgers University Policy on Animal Welfare and were approved by the Institutional Animal Care and Use Committee (IACUC) at Rutgers University Robert Wood Johnson Medical School.
NOD-SCID IL2Rγc null mice (20–35g; Jackson Laboratory) were anesthetized with isoflurane (induction at 4% and maintained at 0.5–1% inhalation). At day 8 after initiating neuronal induction, GFP+ iN cells on a TCPS plate were treated with Accutase and resuspended in ice cold MEM (Life Technologies). RADA16-encapsulated GFP+ iN cells were collected and resuspended in ice cold MEM. The dissociated GFP+ iN cells (100 × 103 cells; 5 μL volume) or RADA16-encapsulated GFP+ iN cells (5 μL) were injected stereotactically into the striatum using a 100 μL gastight Hamilton syringe and 26G Hamilton needle. Bilateral injections were made at the following coordinates (in mm): AP, 1.3 (from bregma); ML, ±1.5; DV, −3.0 (from dura). Mice were sacrificed and perfused with 4% paraformaldehyde for fixation, and the brains were dissected and processed for immunohistochemistry 3 weeks after transplantation. Quantification of human iN cells within the graft was performed by visualization of GFP. Neurite outgrowth was quantified by measurement of GFP+ neurites.
Immunohistochemistry.
iN cells cultured in vitro were fixed in 4% paraformaldehyde for 30 min at room temperature and washed three times in PBS. The fixed samples (cells cultured in vitro or free floating slices from in vivo experiments) were blocked/permeabilized in a solution containing 0.1% Triton X-100 (Sigma-Aldrich), 10% normal goat serum (source), and 1% bovine serum albumin (Sigma-Aldrich) in PBS for 1 h at room temperature. Samples were incubated with primary antibodies (diluted in blocking buffer) overnight at 4 °C and washed three times in PBS. Primary antibodies used include: anti-βIII-tubulin (Tuj1, 1:1000 mouse IgG2a, Covance), anti-MAP-2 (1:500 mouse IgG1, BD Biosciences), anti-synaptophysin (1:250 rabbit IgG, Millipore), anti-GFP (1:1000 chicken IgG, Abcam), anti-GFAP (1:500 rabbit IgG, Dako), anti-ED1 (1:100 mouse IgG1, Abcam), and antihuman specific nuclei (1:100 mouse IgG1, Millipore). Appropriate Alexa Fluor secondary antibodies (1:500 dilution in blocking buffer; Life Technologies) were applied for 1 h at room temperature, and samples were then counterstained with 1 μg/mL DAPI (Sigma-Aldrich) for 5 min. Samples were washed at least three times with PBS after incubation. Brain slices were mounted on glass slides and coverslips with Prolong Gold Anti-Fade reagent (Life Technologies) prior to imaging.
To show the morphology of the injection site and surrounding areas, fixed and blocked tissue sections were stained with a fluorescent Nissl stain solution (NeuroTrace 530/615, Life Technologies) for 20 min at room temperature. Sections were washed at least three times with PBS prior to imaging.
Statistical Analysis.
Statistical analysis was performed using GraphPad Prism 5 software (GraphPad). Data are presented as the mean ± standard deviation. Quantitative analysis was performed using one-way ANOVA and Tukey’s posthoc test, with p < 0.05 considered statistically significant.
RESULTS
Human iPSC-RNs were encapsulated within SAPNS hydrogels 24 h prior to initiating neuronal induction. Approximately 2 h after encapsulation, cell viability was quantified to be 80.7 ± 1.9%, significantly greater (p < 0.0001) than the viability of 57.5 ± 0.8% for induced neurons (iNs) encapsulated after initiating neuronal induction (at day 4 postinduction). The viability of the iPSC-RNs increased to 85.2 ± 2.0% at 24 h postencapsulation since these cells were still in a proliferative state, while the viability of the postmitotic iNs did not recover and continued to decrease at 24 h after encapsulation (48.4 ± 5.8% viability). In all conditions, cells can be seen evenly distributed throughout the SAPNS hydrogel (Figure 1A).
Figure 1.

SAPNS hydrogels support in situ neuronal reprogramming and growth. (A) Live/dead assay of cells encapsulated within RADA16-I SAPNS hydrogels. Green, live cells (calcein AM); red, dead cells (propidium iodide). Scale bar: 200 μm. (B) Quantification of cell viability within RADA16-I SAPNS hydrogels. Error bars: standard deviation. *** indicates p < 0.0001. (C) Immunocytochemistry displaying neurite outgrowth of iN cells encapsulated prior to induction. Scale bar: 50 μm. (D) Immunocytochemistry displaying neurite outgrowth of iN cell encapsulated postinduction. Scale bar: 50 μm.
For cells encapsulated prior to neuronal induction, robust neurite outgrowth was observed within SAPNS hydrogels 8 days after induction, and these encapsulated iN cells expressed the neural markers βIII-tubulin (Tuj1) and MAP-2 as seen after immunofluorescence staining (Figure 1C). SAPNS hydrogel-encapsulated iNs also expressed the mature neuronal marker synaptophysin, a synaptic vesicle protein, suggesting the formation of synapses and neuronal networks within the nanofibrous hydrogel (Figure 3A). Cells encapsulated after neuronal induction displayed very little neurite outgrowth, in addition to their poor viability (Figure 1D), and were not used for further experiments.
Figure 3.

RADA16-I SAPNS hydrogel supports the functional maturation of encapsulated iN cells. (A) Immunocytochemistry of maturing encapsulated iN cells expressing the synaptic marker synaptophysin. Scale bar: 50 μm. (B) Still images from time lapse videos during live cell calcium imaging. Scale bar: 50 μm. (C) Quantification of the change in intracellular fluorescence in response to external electrical stimuli. Error bars: standard deviation.
The RADA16-I hydrogel does not contain bioactive domains but was still able to support the proliferation of iPSCs encapsulated prior to neuronal induction and subsequent neurite outgrowth. These cells were shown to secrete the ECM proteins collagen I and laminin within the hydrogel, while cells encapsulated after neuronal induction displayed a relative lack of ECM secretion along with limited viability and neurite outgrowth (Figure 2).
Figure 2.

iPSCs encapsulated within SAPNS hydrogels secrete ECM proteins collagen I and laminin, shown in red. Green, BIII-tubulin (Tuj1); red, collagen I or laminin; blue, DAPI. Scale bar: 50 μm.
A population-wide analysis of the functional activity of SAPNS hydrogel-encapsulated iNs versus conventional control cultures of iNs was performed by loading cells with the calcium indicator dye Fluo-4 AM and quantifying the change in intracellular fluorescence in response to exogenous electrical stimuli (Figure 3B). Optical recording of neuronal circuit dynamics is noninvasive, which permits simultaneous recording from many neurons at once, and can be applied to track the functional activity of neuronal populations in vitro and in vivo.26 At day 12 after neuronal induction, electrical stimuli caused a significant increase in fluorescence in both SAPNS hydrogel-encapsulated iNs and control iNs cultured on glass coverslips, as seen in still images taken from time lapse videos (Figure 3B). 89.0 ± 3.4% of SAPNS hydrogel-encapsulated iNs responded to the electrical stimuli compared with 90.9 ± 6.7% of iNs cultured conventionally in 2D (p > 0.05, Figure 3C), indicating that the 3-D SAPNS hydrogel is able to support the functional maturation of iNs in a manner comparable to a conventional in vitro culture system.
Given the limited survival and neurite outgrowth of iN cells encapsulated in SAPNS hydrogels, we chose an alternative approach for encapsulation followed by neuronal induction. The protocol involved encapsulation of iPSCs within SAPNS microspheres prior to initiating neuronal induction and culturing the microsphere-encapsulated cells in vitro for 8 days prior to transplantation in vivo. A custom-made microfluidic T-junction device was used to produce monodisperse RADA16-I based SAPNS microspheres via the merging flow of RADA16-I peptide and an immiscible organic phase of hexadecane containing Span 80 as a water-in-oil droplet stabilizer (Figure 4A).27
Microspheres produced at a flow rate ratio (flow rate of continuous organic phase (Qc)/flow rate of discontinuous peptide phase (Qd)) of 5 had an average diameter of 106.9 ± 5.1 μm, with a coefficient of variation of 4.79%, indicating that the microspheres were monodisperse in size (Figure 4C).28 The diameter of the microspheres was not significantly different when comparing blank microspheres or those containing cells at a concentration of 5 × 106 cells/mL (110.8 ± 5.3 μm) or 10 × 106 cells/mL (108.5 ± 5.4 μm). The cell-containing microspheres also maintained monodispersity, with C.V. of 4.82% and 4.94%, respectively. Microsphere diameter was inversely proportional to the flow rate ratio, as shown in Figure 4D. As the flow rate ratio increased, the diameter of the microspheres decreased, ranging in size from 122.4 ± 6.2 μm at a flow rate ratio of 2 to 86.0 ± 5.9 μm at a flow rate ratio of 15 for blank microspheres (Figure 4D).
Cells encapsulated within the SAPNS microspheres had 72.7 ± 2.8% viability at 2 h postencapsulation, which is not significantly different from viability within the bulk RADA16-I hydrogel also gelled using microsphere collection buffer (DMEM/F12 containing 0.25% Tween 20) as a control (74.9 ± 4.0%) (Figure 5A,B). This represents a slight decrease in viability (p < 0.05) compared to the cells within bulk RADA1-I gelled in normal DMEM/F12 medium, suggesting that the only factor of the microsphere fabrication process contributing to cell death was the presence of the water-soluble surfactant Tween 20 in the collection buffer. It should be noted that the viability levels were still significantly greater than those obtained by encapsulation of preprogrammed neurons. Patterns of neurite outgrowth and expression of βIII-tubulin (Tuj1) and MAP-2 were also observed within the microspheres, similar to those in the bulk hydrogel (Figure 5C).
Figure 5.

Cell viability and neurite outgrowth in RADA16-I microspheres. (A) Live/dead assay of cells encapsulated within RADA16-I SAPNS bulk hydrogels and microspheres. Green, live cells (calcein AM); red, dead cells (propidium iodide). Scale bar: 200 μm. (B) Quantification of cell viability within RADA16-I SAPNS hydrogels and microspheres. Error bars: standard deviation. (C) Immunocytochemistry of neurite outgrowth within a single microsphere. Scale bar: 50 μm.
To evaluate whether the SAPNS microspheres could be used as a neuronal transplantation vehicle, iPSCs were encapsulated within RADA16-I microspheres and injected into the striatum of NOD-SCID IL2Rγc mice at day 8 after neuronal induction. Three weeks after transplantation, immunohistochemical analysis revealed that encapsulation of iNs within SAPNS microspheres significantly increased survival in vivo (Figure 6). Cells were counted within three representative 60 μm brain slices from each mouse, quantifying colocalization of GFP and the human-specific nuclei marker HuNu. The average total cell count in this volume was 1502.7 ± 390.5 for SAPNS-encapsulated iNs (initial number injected: 2 × 103 cells) and 219.7 ± 82.6 for iNs transplanted in suspension (initial number injected: 100 × 103 cells). Thus, the fractional retention of injected cells is 2 orders of magnitude greater for the SAPNS-encapsulated iN system than that of suspended iNs. The viability of cells within this volume was significantly higher for RADA16-I encapsulated iNs compared to iNs in suspension (p = 0.0047), indicating a much higher survival rate of encapsulated cells based on the initial numbers injected. GFAP-positive astrocytes were visible surrounding both iNs transplanted in suspension and RADA16-I-encapsulated iNs (Figure 6B).
Figure 6.

RADA16-I SAPNS microspheres support the in vivo transplantation of GFP+ iNs. (A) GFP+ iNs within the mouse brain. Scale bar: 500 μm. (B) Inset from A, showing GFP, human-specific nuclei (huNu), and GFAP staining. Scale bar: 100 μm. (C) Number of viable cells within 0.306 mm3 brain tissue. Error bars: standard deviation. ** indicates p < 0.01.
The injection site of RADA16-I-encapsulated iNs is visible via GFP, Nissl, and DAPI counterstaining shown in Figure 7. The fluorescent Nissl stain used here is selective for the Nissl bodies associated with ribosomes and the rough endoplasmic reticulum within neurons, and is seen throughout the host brain neurons and also within the GFP+ transplanted iNs. Additionally, no cavities or cysts were observed at the injection site, indicating excellent integration of the RADA16-I microspheres and incorporated iNs within the host brain tissue.
Figure 7.

RADA16-I-encapsulated iNs integrate well within the host brain tissue at the injection site. (A) GFP+ iNs showing neurite extension; (B) fluorescent Nissl staining showing the presence of neurons; (C) Nissl and DAPI double staining showing integration into the host tissue without cavities. The needle track is indicated by white dotted lines. Scale bar: 100 μm.
DISCUSSION
Maintaining cell survival after transplantation into the injured or neurodegenerative brain presents a critical challenge since this is the first step toward integration into the host tissue and realization of subsequent functional recovery. The objective of this study was to determine whether a 3-D nanofibrous hydrogel could serve as an integrated neuronal reprogramming and transplantation platform in order to improve the survival of human neurons in vivo. Here, we evaluated the ability of hydrogels self-assembled from the β-sheet forming peptide RADA16-I to support the induction and functional maturation of iPSC-derived neurons in vitro. We also report on the high levels of efficacy of RADA16-I microspheres as vehicles for the in vivo transplantation of adherent networks of induced human neurons.
The material properties and nanofiber formation of RADA16-I have been previously characterized,29 along with its ability to support mammalian cell growth in vitro.10,15 However, this is the first report of using RADA16-I hydrogels to support the growth of iPSC-derived human neurons. The iN cells used here were produced by the direct neuronal conversion of iPSCs through the doxycycline-inducible ectopic expression of a single neuronal transcription factor, NeuroD1.25 Neuronal reprogramming is conventionally achieved on a 2-D substrate and has not been previously attempted within a 3-D hydrogel culture environment. Neuronal reprogramming of iPSCs in 2-D is performed on tissue culture polystyrene dishes coated with Matrigel, a basement membrane matrix extracted from the Englebreth-Holm-Swarm mouse sarcoma that contains several ECM proteins and growth factors. Although Matrigel has been effectively used as a cell carrier for in vivo transplantation, it cannot be used clinically due to its murine sarcoma origins.30 In contrast, the synthetic SAPNS RADA16-I contains no biologically active domains to promote cell attachment or neurite outgrowth. Despite the lack of any bioactive domains in the substrate, we found that iPSCs encapsulated in RADA16-I prior to initiating neuronal induction led to the development of a population of iN cells with robust neurite outgrowth. The high surface area-to-volume ratio provided by nanofiber scaffolds has been shown to promote cell adhesion, proliferation, and differentiation.31 In addition to providing a large surface area for cell growth, the 3-D nanofibrous substrate can provide spatial cues to extending neurites. The SAPNS supported the initial proliferation and migration/clustering of iPSCs while still in a proliferative state and the extension of neurites after direct neuronal induction. These iNs displayed neuronal phenotypes and population-wide functional activity comparable to that of NeuroD1-reprogrammed iNs on 2-D surfaces abundant in ECM matrix proteins. In addition to the biomaterial substrate providing physical support for the neurons, the ECM proteins secreted by iPSCs encapsulated prior to neuronal induction promote neurite growth.
Many studies have demonstrated that ECM proteins are important for neuron survival and neurite outgrowth, providing anchorage points and guidance cues for extending neurites.32,33 In particular, laminin has been widely investigated as a substrate for neural growth, influencing adhesion, neurite growth, and movement of growth cones.34,35 Although type I collagen is not considered a primary adhesive substrate for neural cells, it has also been investigated for use in neural tissue engineering scaffolds.36,37 In combination, cogels of collagen I and laminin were found to be optimal for promoting dorsal root ganglia neurite extension.38 Here, encapsulated iPSCs produced more laminin and collagen I within the SAPNS hydrogels than iN cells encapsulated after neuronal induction, supporting a higher degree of cell viability and neurite outgrowth. Although these common ECM components of systemic organs are beneficial for neural growth, they are mostly absent from the adult brain ECM,39,40 and therefore not abundantly secreted by adult neural cells. However, some of these ECM components are present in the embryonic brain, playing major roles in nervous tissue development and patterning.40 iPSCs, which have similar properties as embryonic stem cells, are capable of producing a broad spectrum of ECM molecules41 in higher quantities than neurons, as seen within our SAPNS hydrogels.
Low viability and poor neurite outgrowth was observed when iNs were encapsulated within the hydrogels, leading us to pre-encapsulate the cells in RADA16-I microspheres prior to neuronal induction in order to maintain viability and allow the neurites to grow prior to transplantation. These monodisperse RADA16-I microspheres can be fabricated over a wide range of diameters, enabling minimally invasive injection through narrow needles. This process also allowed us to transplant a robust adherent neuronal network into the brain, rather than attempting to gel a poorly surviving iN/RADA16-I mixture in situ. Further, preculturing the cell-seeded microspheres prior to injection also allows us to avoid a potential inflammatory response in the CNS, which has been caused by transplantation of the acidic untreated RADA16-I, leading to gaps and cysts around the injection site.42 It would not be feasible to induce neuronal conversion of the hiPSC-RNs in vivo primarily because the conversion process is induced by the addition of the drug doxycycline. Also, if an attempt were made to trigger neuronal induction in vivo, any cells that failed to convert into neurons would remain proliferative and could contribute to the formation of a teratoma.
When transplanted into the mouse brain, the RADA16-I-encapsulated neurons displayed robust survival and neurite outgrowth in vivo. In contrast, transplantation of neurons in suspension led to poor viability in vivo, with survival rates comparable to those seen by Zhang et al., who performed brain injections of neurons generated in a similar manner (direct neuronal induction of iPSCs via transfection with a single transcription factor).20 In comparing these outcomes, the significantly increased survival of RADA16-I encapsulated neurons can likely be attributed to the prevention of dissociation-induced cell death. RADA6-I microspheres support the transplantation of adherent neuronal networks, avoiding the need for enzymatic dissociation and the ensuing loss of cell–cell and cell-material interactions.8,43 We have previously used microfibrous electrospun scaffolds to transplant iPSC-derived neurons into the mouse brain, in order to achieve the purpose of transplanting adherent neuronal networks in vivo.25 The RADA16-I SAPNS microspheres significantly outperformed the electrospun scaffolds as neuronal transplantation vehicles (with in vivo neuron survival rates over 2 orders of magnitude greater).25 In addition to surrounding encapsulated cells in a manner similar to natural ECM, the RADA16-I SAPNS has a greater than 99% water content and elastic modulus comparable to that of brain tissue,44,45 which makes this biomaterial more applicable for CNS transplantation than comparatively stiff polymer fibers. These characteristics provide an appropriate microenvironment for encapsulated cells, effectively mimicking the natural in vivo microenvironment, and acting as a suitable milieu for transition between in vitro culture and the in vivo CNS injection site. The biocompatibility of this substrate is illustrated by the integration with host brain tissue and lack of obvious gaps or cysts near the injection site.
The SAPNS system can also allow for the incorporation of specific neuroadhesive and neuroprotective cues. In addition to anoikis, oxidative stress is a major contributor to the death of grafted and host neurons, particularly in neurodegenerative diseases such as Parkinson’s Disease.46 Future transplantation strategies to further enhance cell survival in the neurodegenerative brain may include modification of the RADA16-I peptide with various functional motifs to provide neuroprotection against oxidative stress and inflammation. RADA16-I-based SAPNS can also be used for the reprogramming and transplantation of multiple neuronal subtypes, applying this construct to various diseases or injury sites within the CNS space. The only design differences necessary would be to functionalize the SAPNS with applicable neuroprotective or neurotrophic peptides and to encapsulate a specific subtype of neuron, pertinent to the injury site or neurodegenerative disease in question. Overall, this study indicates that the SAPNS RADA16-I is effective as a reprogramming and transplantation vehicle for neurons and shows promise for in vivo regenerative therapies.
ACKNOWLEDGMENTS
Research reported here was supported by NIH P41 EB001046, NIH R21 NS095082, NIH T32EB005583, and NSF-IGERT grant 0801620. We gratefully acknowledge the RUCDR for the gift of the iPSC line used in this study and 3-D Matrix for the gift of the RADA16-I self-assembling peptide (PuraMatrix).
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Clark RSB; Kochanek PM; Watkins SC; Chen MZ; Dixon CE; Seidberg NA; et al. Caspase-3 mediated neuronal death after traumatic brain injury in rats. J. Neurochem 2000, 74, 740–53. [DOI] [PubMed] [Google Scholar]
- (2).Gorman AM Neuronal cell death in neurodegenerative diseases: recurring themes around protein handling. J. Cell Mol. Med 2008, 12, 2263–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Cooke MJ; Vulic K; Shoichet MS Design of biomaterials to enhance stem cell survival when transplanted into the damaged central nervous system. Soft Matter 2010, 6, 4988–98. [Google Scholar]
- (4).Bakshi A; Keck CA; Koshkin VS; LeBold DG; Siman R; Snyder EY; et al. Caspase-mediated cell death predominates following engraftment of neural progenitor cells into traumatically injured rat brain. Brain Res. 2005, 1065, 8–19. [DOI] [PubMed] [Google Scholar]
- (5).Einstein O; Ben Menachem O; Mizrachi-Kol R; Reinhartz E; Grigoriadis N; Ben-Hur T Survival of neural precursor cells in growth factor poor environment: Implications for transplantation in chronic disease. Glia 2006, 53, 449–455. [DOI] [PubMed] [Google Scholar]
- (6).Cordeiro KK; Cordeiro JG; Furlanetti LL; Salazar JAG; Tenorio SB; Winkler C; et al. Subthalamic nucleus lesion improves cell survival and functional recovery following dopaminergic cell transplantation in parkinsonian rats. Eur. J. Neurosci 2014, 39, 1474–84. [DOI] [PubMed] [Google Scholar]
- (7).Nakaji-Hirabayashi T; Kato K; Iwata H In vivo Study on the Survival of Neural Stem Cells Transplanted into the Rat Brain with a Collagen Hydrogel That Incorporates Laminin-Derived Polypeptides. Bioconjugate Chem. 2013, 24, 1798–804. [DOI] [PubMed] [Google Scholar]
- (8).Marchionini DM; Collier TJ; Camargo M; McGuire S; Pitzer M; Sortwell CE Interference with anoikis-induced cell death of dopamine neurons: Implications for augmenting embryonic graft survival in a rat model of Parkinson’s disease. J. Comp. Neurol 2003, 464, 172–9. [DOI] [PubMed] [Google Scholar]
- (9).Zhang S; Holmes T; Lockshin C; Rich A Spontaneous assembly of a self-complementary oligopeptide to form a stable macroscopic membrane. Proc. Natl. Acad. Sci. U. S. A 1993, 90, 3334–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Zhang S; Holmes TC; DiPersio CM; Hynes RO; Su X; Rich A Self-complementary oligopeptide matrices support mammalian cell attachment. Biomaterials 1995, 16, 1385–93. [DOI] [PubMed] [Google Scholar]
- (11).Yanlian Y; Ulung K; Xiumei W; Horii A; Yokoi H; Shuguang Z Designer self-assembling peptide nanomaterials. Nano Today 2009, 4, 193–210. [Google Scholar]
- (12).Cormier AR; Pang X; Zimmerman MI; Zhou HX; Paravastu AK Molecular structure of RADA16-I designer self-assembling peptide nanofibers. ACS Nano 2013, 7, 7562–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Holmes TC; de Lacalle S; Su X; Liu G; Rich A; Zhang S Extensive neurite outgrowth and active synapse formation on self-assembling peptide scaffolds. Proc. Natl. Acad. Sci. U. S. A 2000, 97, 6728–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Cheng TY; Chen MH; Chang WH; Huang MY; Wang TW Neural stem cells encapsulated in a functionalized self-assembling peptide hydrogel for brain tissue engineering. Biomaterials 2013, 34, 2005–16. [DOI] [PubMed] [Google Scholar]
- (15).Yla-Outinen L; Joki T; Varjola M; Skottman H; Narkilahti S Three-dimensional growth matrix for human embryonic stem cell-derived neuronal cells. J. Tissue Eng. Regener. Med 2014, 8, 186–94. [DOI] [PubMed] [Google Scholar]
- (16).Thonhoff JR; Lou DI; Jordan PM; Zhao X; Wu P Compatibility of human fetal neural stem cells with hydrogel biomaterials in vitro. Brain Res. 2008, 1187, 42–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Zhang ZX; Zheng QX; Wu YC; Hao DJ Compatibility of Neural Stem Cells with Functionalized Self-assembling Peptide Scaffold In vitro. Biotechnol. Bioprocess Eng 2010, 15, 545–51. [Google Scholar]
- (18).Guo J; Leung KK; Su H; Yuan Q; Wang L; Chu TH; et al. Self-assembling peptide nanofiber scaffold promotes the reconstruction of acutely injured brain. Nanomedicine 2009, 5, 345–51. [DOI] [PubMed] [Google Scholar]
- (19).Ellis-Behnke RG; Liang YX; You SW; Tay DK; Zhang S; So KF; et al. Nano neuro knitting: peptide nanofiber scaffold for brain repair and axon regeneration with functional return of vision. Proc. Natl. Acad. Sci. U. S. A 2006, 103, 5054–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Zhang Y; Pak C; Han Y; Ahlenius H; Zhang Z; Chanda S; et al. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron 2013, 78, 785–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Marchetto MCN; Carromeu C; Acab A; Yu D; Yeo GW; Mu YL; et al. A Model for Neural Development and Treatment of Rett Syndrome Using Human Induced Pluripotent Stem Cells. Cell 2010, 143, 527–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Inoue H; Yamanaka S The Use of Induced Pluripotent Stem Cells in Drug Development. Clin. Pharmacol. Ther 2011, 89, 655–61. [DOI] [PubMed] [Google Scholar]
- (23).Vierbuchen T; Ostermeier A; Pang ZP; Kokubu Y; Sudhof TC; Wernig M Direct conversion of fibroblasts to functional neurons by defined factors. Nature 2010, 463, 1035–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Pang ZP; Yang N; Vierbuchen T; Ostermeier A; Fuentes DR; Yang TQ; et al. Induction of human neuronal cells by defined transcription factors. Nature 2011, 476, 220–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Carlson AL; Bennett NK; Francis NL; Halikere A; Clarke S; Moore JC; et al. Generation and transplantation of reprogrammed human neurons in the brain using 3D microtopographic scaffolds. Nat. Commun 2016, 7, 10862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Smetters D; Majewska A; Yuste R Detecting action potentials in neuronal populations with calcium imaging. Methods 1999, 18, 215–21. [DOI] [PubMed] [Google Scholar]
- (27).Tsuda Y; Morimoto Y; Takeuchi S Monodisperse cell-encapsulating peptide microgel beads for 3D cell culture. Langmuir 2010, 26, 2645–9. [DOI] [PubMed] [Google Scholar]
- (28).Takeuchi S; Garstecki P; Weibel DB; Whitesides GM An axisymmetric flow-focusing microfluidic device. Adv. Mater 2005, 17, 1067–1072. [Google Scholar]
- (29).Yokoi H; Kinoshita T; Zhang S Dynamic reassembly of peptide RADA16 nanofiber scaffold. Proc. Natl. Acad. Sci. U. S. A 2005, 102, 8414–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Uemura M; Refaat MM; Shinoyama M; Hayashi H; Hashimoto N; Takahashi J Matrigel Supports Survival and Neuronal Differentiation of Grafted Embryonic Stem Cell-Derived Neural Precursor Cells. J. Neurosci. Res 2010, 88, 542–51. [DOI] [PubMed] [Google Scholar]
- (31).Vasita R; Katti DS Nanofibers and their applications in tissue engineering. International journal of nanomedicine 2006, 1, 15–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Yu L; Leipzig ND; Shoichet MS Promoting neuron adhesion and growth. Mater. Today 2008, 11, 36–43. [Google Scholar]
- (33).Srinivasan P; Zervantonakis IK; Kothapalli CR Synergistic effects of 3D ECM and chemogradients on neurite outgrowth and guidance: a simple modeling and microfluidic framework. PLoS One 2014, 9, e99640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Ford-Holevinski TS; Hopkins JM; McCoy JP; Agranoff BW Laminin supports neurite outgrowth from explants of axotomized adult rat retinal neurons. Dev. Brain Res 1986, 28, 121–6. [DOI] [PubMed] [Google Scholar]
- (35).Liu W; Xing S; Yuan B; Zheng W; Jiang X Change of laminin density stimulates axon branching via growth cone myosin II-mediated adhesion. Integrative biology: quantitative biosciences from nano to macro 2013, 5, 1244–52. [DOI] [PubMed] [Google Scholar]
- (36).Phillips JB; Bunting SC; Hall SM; Brown RA Neural tissue engineering: a self-organizing collagen guidance conduit. Tissue Eng. 2005, 11, 1611–7. [DOI] [PubMed] [Google Scholar]
- (37).Swindle-Reilly KE; Papke JB; Kutosky HP; Throm A; Hammer JA; Harkins AB; et al. The impact of laminin on 3D neurite extension in collagen gels. Journal of neural engineering 2012, 9, 046007. [DOI] [PubMed] [Google Scholar]
- (38).Deister C; Aljabari S; Schmidt CE Effects of collagen 1, fibronectin, laminin and hyaluronic acid concentration in multi-component gels on neurite extension. J. Biomater. Sci., Polym. Ed 2007, 18, 983–97. [DOI] [PubMed] [Google Scholar]
- (39).Bonneh-Barkay D; Wiley CA Brain extracellular matrix in neurodegeneration. Brain Pathol. 2009, 19, 573–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Ruoslahti E Brain extracellular matrix. Glycobiology 1996, 6, 489–92. [DOI] [PubMed] [Google Scholar]
- (41).Baraniak PR; McDevitt TC Stem cell paracrine actions and tissue regeneration. Regener. Med 2010, 5, 121–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Guo J; Su H; Zeng Y; Liang YX; Wong WM; Ellis-Behnke RG; et al. Reknitting the injured spinal cord by self-assembling peptide nanofiber scaffold. Nanomedicine 2007, 3, 311–21. [DOI] [PubMed] [Google Scholar]
- (43).Raff MC Social controls on cell survival and cell death. Nature 1992, 356, 397–400. [DOI] [PubMed] [Google Scholar]
- (44).Genove E; Shen C; Zhang S; Semino CE The effect of functionalized self-assembling peptide scaffolds on human aortic endothelial cell function. Biomaterials 2005, 26, 3341–51. [DOI] [PubMed] [Google Scholar]
- (45).Green MA; Bilston LE; Sinkus R In vivo brain viscoelastic properties measured by magnetic resonance elastography. NMR Biomed. 2008, 21, 755–64. [DOI] [PubMed] [Google Scholar]
- (46).Hwang O Role of oxidative stress in Parkinson’s disease. Experimental neurobiology 2013, 22, 11–7. [DOI] [PMC free article] [PubMed] [Google Scholar]