

Abstract
Protein degradation by aminopeptidases is involved in bacterial responses to stress. Escherichia coli produces two metal‐dependent M17 family leucine aminopeptidases (LAPs), aminopeptidase A (PepA) and aminopeptidase B (PepB). Several structures have been solved for PepA as well as other bacterial M17 peptidases. Herein, we report the first structures of a PepB M17 peptidase. The E. coli PepB protein structure was determined at a resolution of 2.05 and 2.6 Å. One structure has both Zn2+ and Mn2+, while the second structure has two Zn2+ ions bound to the active site. A 2.75 Å apo structure is also reported for PepB from Yersinia pestis. Both proteins form homohexamers, similar to the overall arrangement of PepA and other M17 peptidases. However, the divergent N‐terminal domain in PepB is much larger resulting in a tertiary structure that is more expanded. Modeling of a dipeptide substrate into the C‐terminal LAP domain reveals contacts that account for PepB to uniquely cleave after aspartate.
Keywords: aminopeptidase, Escherichia coli, hexamer, metalloprotease, PepB, X‐ray crystallography, Yersinia pestis
1. INTRODUCTION
Protein degradation is an important cellular process necessary for adaptation and survival of bacteria in response to a constantly changing environment. General proteolysis of older or damaged proteins is involved in protein quality control, while regulated proteolysis plays a signaling role in bacterial cells, including regulation of sporulation, biofilm dynamics, motility, and other processes. 1 Peptidases are classified according to the catalytic residue necessary for nucleophilic attack or by the presence of metals in the active site. 2 According to the MEROPS database, the leucine aminopeptidases (LAPs) are members of the M17 family from the MF metallopeptidase clan. 2 These peptidases remove N‐terminal amino acids from short peptide chains and are involved in a wide variety of processes, including intracellular protein turnover, bacterial stress response, and regulation of virulence. 3 Escherichia coli expresses two M17 LAPs, aminopeptidase A (PepA) and aminopeptidase B (PepB). These proteins share only 27% identity in primary amino acid sequence and differ in size, 503 and 427 amino acids, respectively. 3 Despite designation as LAPs, PepA and PepB are not limited to cleavage after leucine. Both PepA and PepB share similar substrate preference for cleavage after lysine, leucine, glycine, and methionine, although PepB is unique in its ability to cleave after an aspartate residue.3, 4 In the M17 family, the structures of PepA from E. coli 5 and Pseudomonas putida 6 have been previously solved. PepA is a homohexamer organized as a dimer of trimers with two metal ions in the active site of each monomer. The C‐terminus of the protein has a conserved LAP fold, while positively charged residues of the N‐terminal domain have a distinct function in DNA binding, consistent with PepA participation in Xer recombination.5, 7, 8 Other structures have also been solved for the Helicobacter pylori M17 aminopeptidase protein (HpM17AP) 9 and Staphylococcus aureus PepZ, 10 which cluster by sequence into groups distinct from PepA or PepB (Figure 1a). Both of these peptidases also form homohexamers.
FIGURE 1.
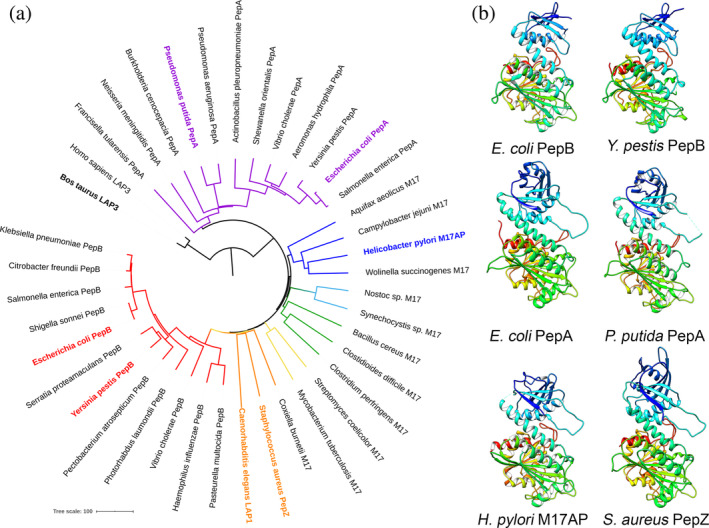
Sequence and structural comparisons of bacterial M17 LAP proteins. (a) Neighbor joining phylogenetic tree of amino acid sequences of selected M17 peptidases rooted with outlier human LAP3 M17 peptidase. Node for PepA is purple and node for PepB is red. Labels in bold represent peptidases with solved structures. (b) Rainbow ribbon diagrams of monomer subunits of bacterial M17 peptidases. E. coli PepB (PDB code 6oad, this study); Y. pestis PepB (PDB code 6cxd, this study); E. coli PepA 5 (PDB code 1gyt); P. putida PepA 6 (PDB code 3h8e); H. pylori M17AP 9 (PDB code 4zla); and S. aureus PepZ 10 (PDB code 3kzw). A multiple structure alignment of the same protein is shown in Figure S1. LAP, leucine aminopeptidase; PepA, aminopeptidase A; PepB, aminopeptidase B
No structure of an M17 peptidase from the group that clusters with PepB by sequence has been previously determined (Figure 1a). The structure of E. coli PepB is of particular interest as the gene pepB has been specifically linked to environmental stress responses. Expression of the gene pepB (also known as yfhI) is upregulated by exposure of E. coli to paraquat to induce superoxide stress. 11 In addition, PepB is linked to enhanced bacterial growth rates through adaptive laboratory evolution (ALE) experiments that identified mutations within pepB when E. coli was passaged and selected for rapid growth in glucose minimal media. 12 The abundance of PepB (also known as YPO2889) was also increased in ALE experiments conducted for Yersinia pestis. 13 The structure of PepB is particularly of interest due to the poor amino acid sequence conservation of the N‐terminal domain compared with other M17 peptidases. 4 In this article, we report two structures of bacterial PepB from E. coli and one from Y. pestis. PepB from E. coli was crystallized as a complex with metals (Zn‐ and Mn‐form), whereas PepB from Y. pestis was crystallized in an apo‐form. The structures reveal a distinct organization of the N‐terminal domain and active site, which impacts the hexameric structure and the substrate specificity of PepB compared to PepA.
2. RESULTS
2.1. Homohexamer organization of PepB
Phylogenetic analysis of the primary amino acid sequences of 37 representative M17 peptidases shows the diversity across the prokaryotes and clustering into groups (Figure 1a). No structures have been solved for the PepB‐like M17 peptidases (MEROPS M17.004). 2 For this study, crystal structures were determined for seleno‐methionine (Se‐Met)‐labeled PepB at 2.6 Å (PDB code 6ov8) and native E. coli PepB at 2.05 Å (PDB code 6oad). The two crystal structures of E. coli PepB differ from each other. The native form of E. coli PepB was crystalized in the monoclinic P21 space group with 12 chains (two hexamers) in the asymmetric unit, while the Se‐Met derivative crystalized in the orthorhombic P212121 space group with six chains (one hexamer) in the asymmetric unit. Despite these differences the two forms of PepB from E. coli overlap perfectly with the Z‐score of 8.2 and the root‐mean‐square deviation (r.m.s.d.) = 0.28 Å for all 426 compared Cα atoms using a pairwise structural alignment based on the jCE algorithm 14 (http://source.rcsb.org/). In addition, the native Y. pestis PepB structure was solved at 2.75 Å (PDB code 6cxd).
Each PepB monomer has a roughly spherical shape with a trailing end, resembling a teardrop (Figure 1b). The C‐terminal region is comprised of an LAP fold that is conserved in previously solved structures (Figure S1), which aligned closely when structures were limited to 169–190 pruned atom pairs of E. coli PepA (r.m.s.d. = 1.03 Å, 169 pairs), P. putida PepA (r.m.s.d. = 0.96 Å, 172 pairs), H. pylori HpM17A (r.m.s.d. = 0.56 Å, 190 pairs), and S. aureus PepZ (r.m.s.d. = 1.04 Å, 167 pairs). The N‐terminal domain is comprised of five β‐strands and two α‐helices. This domain has 75 fewer amino acids and is smaller when compared to the six β‐strand/four α‐helix N‐terminal domain in PepA and other M17 peptidases (Figure 1b).
Both E. coli and Y. pestis PepB assemble to form a hexamer of six identical subunits. To form the hexamers, the teardrop shaped monomers arrange into two layers of three circular disks that stack one on another with 32‐point group symmetry (Figure 2a). The layers stack in such a way that the centers of the C‐terminal sphere‐like LAP domain of each monomer in one layer aligns with the centers of the LAP domain of the monomer comprising the other layer. The N‐terminal tail‐like domain of each monomer in one layer interacts with the N‐terminal domain of the adjacent monomer in the other layer (Figure 2a). The interaction of the smaller N‐terminal domains in PepB organizes to create an overall hexagonal shape, compared to the more triangular shape of PepA (Figure 2a,b). Thus, the C‐terminal domains stabilize trimer‐to‐trimer packing, whereas N‐terminal domains form dimeric interactions, and these two types of interaction hold six monomers together. The distinct structure of PepB is not likely due to crystallization artifacts, as the two PepB structures (PDB codes 6oad and 6ov8) were highly similar and arose from distinct crystal forms.
FIGURE 2.
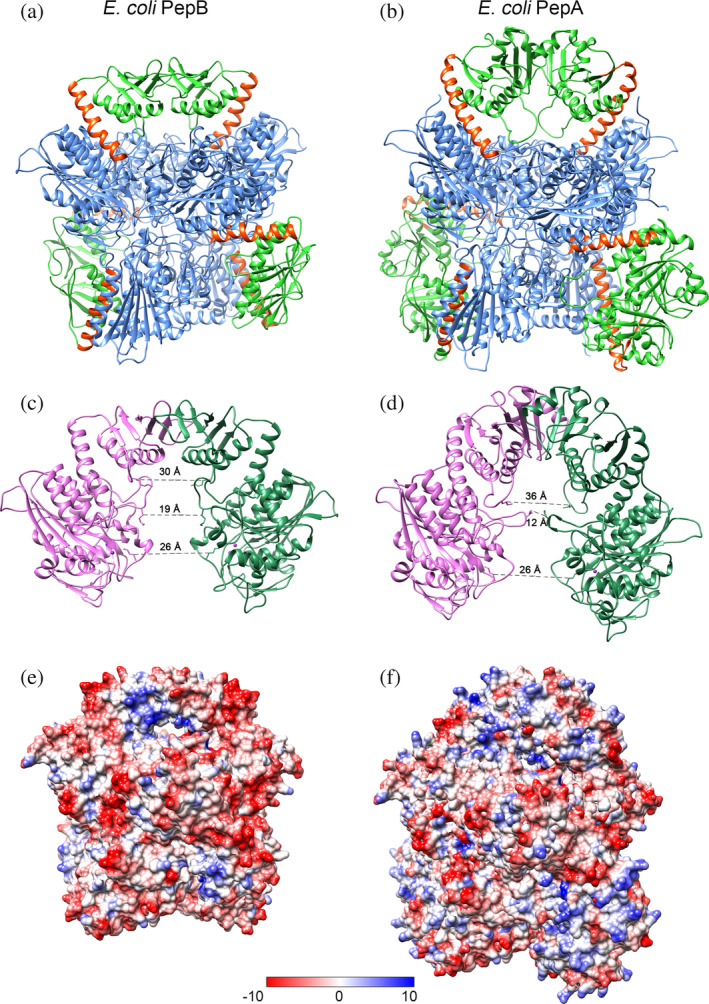
Comparison of E. coli PepB (a,c,e) and PepA (b,d,f) hexamers. (a,b) Hexamer structure of PepB (PDB code 6oad) and PepA (PDB code 1gyt): N‐terminal domain (green), connection helix (orange), and C‐terminal domain (blue). Structures were overlaid using Chain a and are shown in the same orientation. (c,d) Dimers interface of Chain a (dark green) and Chain d (pink). Distances were measured between conserved lysine residues in the loops. (e,f) Electrostatic surface projection of hexamers in the same orientation as on panel (a). LAP, leucine aminopeptidase; PepA, aminopeptidase A; PepB, aminopeptidase B
The helix α3, which connects the N‐ and C‐terminal domains, is only 22 amino acids long in E. coli PepB compared to the helix α5 in PepA, which consists of 28 amino acids. This raises the N‐terminal domain of PepA further out from the core, increasing the size of the cavity underneath the N‐terminal dimerization face. In PepB, the loop between the helix α2 and the β‐strand β5 interacts with a loop in the C‐terminal domain. This loop is six residues shorter than in PepA resulting in an expanded access to the catalytic site below. By comparison, in PepA, these loops are directed into the core creating a narrow channel for access to the active site. Despite these differences in the cavity size at the portion of the protein proximal to the N‐terminal binding interaction (middle of structures in Figure 2), the distance between the catalytic site Lys‐207 residues in the formed structure is not substantially different between PepA and PepB (Figure 2b,c). An electrostatic surface charge distribution of the hexamers shows that the surface of the PepA is more positively charged than PepB, particularly in the N‐terminal domain (Figure 2e,f), which is essential for DNA interaction during Xer recombination. These residues are not conserved in PepB.4, 8
2.2. Comparison of Mn‐ and Zn‐form of PepB from E. coli
Consistent with its classification as an M17 peptidase, the crystal structure of E. coli PepB contains two metal binding sites. In the structure of native PepB, both metal sites are occupied by Zn2+ (Zn‐form, Figure 3a,b), whereas the Se‐Met derivative contains Mn2+ in the M1 site and Zn2+ in the M2 site (Figure 3c). The LAP fold domains in the hexamer are positioned such that the metal binding and the catalytic residues are facing the interior of the hexamer and located near the core. The metal ions in the M1 sites are coordinated by Asp‐200, Asp‐277, and Glu‐279, while the M2 sites are coordinated by Lys‐195, Asp‐218, and Glu‐279. The metal ions share a water molecule, which bridges the two metal sites. Adjacent to the metal binding sites, there are canonical active site residues Lys‐207 and Arg‐281 (Figure 2a–c). The Lys‐207 residue is oriented toward the metal binding site and it is positioned to interact with the carbonyl oxygen of the scissile peptide bond for peptide bond hydrolysis. The Arg‐281 residue is involved in binding of a bicarbonate ion (BCT), which is utilized as a general acid/base and activates the conserved water in the peptide bond hydrolysis. In the Mn‐form, this place is occupied by a Cl− ion.
FIGURE 3.
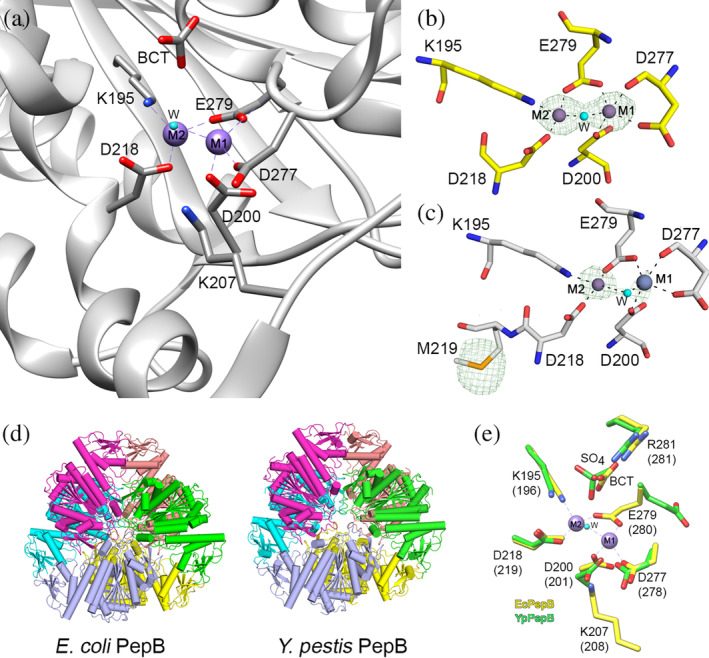
The metal binding site of PepB from E. coli and Y. pestis. (a) E. coli PepB with Zn2+ (Zn‐form) in both M1 and M2 sites. Residues that coordinate Zn2+ ions are shown. (b,c) Comparison of the Zn‐form (b) and the Mn‐form (c). Anomalous difference maps are contoured at the 3σ level and are shown as a green mesh. Zn2+ is shown in violet and Mn2+ in gray. (d) Hexameric organization of PepB proteins. (e) Superposition of the Zn‐form from E. coli (yellow) with the apo‐form from Y. pestis (green). Residue labeling is based on the sequence of PepB from E. coli with corresponding residues for Y. pestis in parenthesis. Hydrogen bond interactions are shown as black dash lines. BCT, bicarbonate ion; LAP, leucine aminopeptidase; PepA, aminopeptidase A; PepB, aminopeptidase B
2.3. Comparison of PepB apo structure from Y. pestis with Zn‐form from E. coli
For PepB from E. coli, we obtained crystals with both metal binding sites occupied, while the crystals for PepB from Y. pestis had no metal bound to either site. A pairwise structural alignment of Chain a of PepB from Y. pestis (PDB code 6cxd) with Chain a of PepB from E. coli (PDB code 6oad) reveals a high degree of structural similarity between these models (Z‐score of 8.03 and r.m.s.d. = 1.07 Å) (Figures 1b and 3d). The positions of all residues that coordinate metals overlay perfectly with the exception of Glu‐279, which in Y. pestis is oriented in the opposite direction compared to the same residue in E. coli. While the positions of Arg‐281 in these structures overlap well, the space occupied by the enzymatically important BCT is replaced by a sulfate ion in the Y. pestis PepB structure (Figure 3e).
Additional differences in the structure occur between positions of Cα atoms in the N‐terminal domain at Residues 2–4, 18–21, and 81–84 (using residue numbering from E. coli) and at the C‐terminal domain at Loops 250–259 and 365–373, which are located over 15 Å away from the metal binding sites (Figure S2). The most significant differences occur at Residues 203–212 and 307–310 where the model of PepB from Y. pestis has missing residues due to the structural disorder. Together, these data suggest that these loops become ordered only after metal binding. These “dual‐personality” regions that switch between an ordered and disordered state often play a regulating role in the protein function. 15 Notably, the disordered loop at Residues 250–259 in PepB from Y. pestis correspond exactly to a disordered loop in the apo structure of PepA from P. putida (PDB code 3h8e), suggesting that one of the roles of Zn2+ binding is structural ordering of this loop. In fact, residues Lys‐207 and Lys‐310 important for catalysis are located in these disordered regions.
2.4. Modeling binding of Asp‐Leu dipeptide to PepB from E. coli
For substrate specificity and differences between PepB and PepA, we compared the crystal structures of PepB from E. coli (PDB code 6oad) with the PepA‐bestatin complex from P. putida (PDB code 3h8g). The metal binding sites and the substrate binding residues overlay very well in these structures except Lys‐310 (Figure 4a), which is conserved in all PepB proteins but absent in PepA and other M17 peptidases. 4 In P. putida PepA, the residue in this position is Ile‐382, which together with Met‐287, Gly‐379 and Trp‐470 forms the surface of the pocket to bind the hydrophobic side chain S1 of the substrate. In the bestatin complex structure of PepA from P. putida, the pocket is occupied by the phenyl group of bestatin. The inhibitor is secured in the active site by hydrogen bond interactions of the carbonyl oxygen of phenyl with the side chain nitrogen of Lys‐279 and Mn2+ occupying the M1 site, nitrogen N of phenyl with Zn2+ located in the M2 site, and oxygen of the carboxylic group of Leu of the inhibitor with the main chain nitrogen of Gly‐379. The oxygen of the methyl carboxyl group of bestatin replaces the conserved water, which attacks the carbonyl group of the scissile peptide bond and is interacting with an oxygen of bicarbonate. Taking into consideration all these interactions, we used the position of bestatin to model the binding mode of the substrate Asp‐Leu dipeptide into the active site (Figure 4b). Then we optimized all interactions between the substrate, metals, and residues of the active site. In this model, the side chain of Asp in the S1 position is located within the hydrogen bond distance to the side chain nitrogen of the Lys‐310, which is uniquely present in PepB peptidases. This interaction is essential for the substrate specificity of PepB and makes it possible to bind acidic residues (Asp and Glu) in the P1 pocket. Positions of the main‐chain atoms of the substrate are maintained by hydrogen bond interactions between N of Asp and Zn2+ in the M2 site, O of Asp and Lys‐207, N of Leu with O of Thr‐306, and O of Leu with N of Gly‐307. In this conformation the conserved water molecule, which is activated by BCT, is located in the proper position to attack the carbonyl group of the scissile peptide bond of the Asp‐Leu dipeptide. This model provides an explanation for the unique ability of PepB to cleave after aspartate residues.
FIGURE 4.
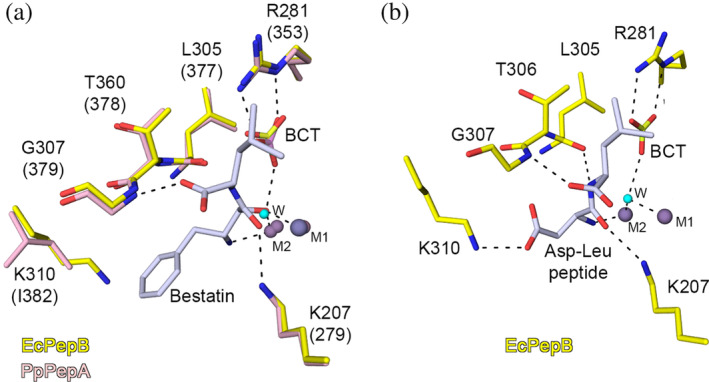
Interaction of substrate with PepB. (a) Superposition of the active sites of the Zn‐form of PepB from E. coli (PDB code 6oad, this study) and the Zn‐ and Mn‐form of the bestatin complex structure of the PepA from P. putida 6 (PDB code 3h8g). (b) Molecular modeling of the binding mode for the Asp‐Leu dipeptide in the binding pocket of the Zn‐form of E. coli PepB. Zn2+ (violet), Mn2+ (gray), and water (cyan). Carbons are colored for E. coli PepB (yellow), P. putida PepB (pink), and bestatin and Asp‐Leu dipeptide (light gray). Residue labeling is based on the sequence of PepB from E. coli and corresponding residues for P. putida are in parenthesis. Hydrogen bond interactions are shown as black dash lines. LAP, leucine aminopeptidase; PepA, aminopeptidase A; PepB, aminopeptidase B
3. CONCLUSIONS
M17 peptidases are a diverse metalloprotease family that shares the conserved LAP fold in the C‐termini. Structure analysis has thus far concentrated on members of the PepA family to elucidate the role of PepA in DNA recombination. By comparison, little is known about PepB. In this study, we determined the first structures of PepB from E. coli and from Y. pestis. Both E. coli and Y. pestis shared the conserved LAP fold, but the shorter N‐terminal domain resulted in a more compact structure, consistent with the E. coli PepB hexamer having slower mobility on size exclusion chromatography than E. coli PepA. 4 Further, the N‐terminal domain of PepB has a lower positive surface charge, which is higher in PepA from E. coli and essential for its role in Xer recombination. 8 The overall arrangement of the active sites is conserved with other M17 peptidases, including the presence of BCT in the active site. The structure of E. coli PepB was solved with Zn2+ in both the M1 and M2 sites, although Ni2+, Mn2+, Mg2+, and Co2+ are also known to occupy these sites. 16 Solving the apo structure of Y. pestis demonstrated that the binding of metals is likely important for organization of the active site, particularly the proper orientation of Glu‐279, which coordinates both metal sites. The metals are also likely necessary for organizing the loop containing Lys‐207, which is essential for catalysis, and Lys‐310, which we show is important for substrate specificity. These loops were disordered in the apo structure of PepB from Y. pestis, as well as in the previously solved apo structure of P. putida PepA (PDB code 3h8e). 6 Finally, the dipeptide substrate modeling based on the P. putida PepA‐bestatin complex showed how the variation in the substrate binding site of PepB may account for its distinct ability to cleave after aspartate.
PepB has recently been of interest as a potential point of intervention in managing antimicrobial resistance. Not all bacterial species have two aminopeptidases, although this is common in the Enterobacteriaceae and Vibrionaceae (Figure 1a). ALE experiments select for mutations that confer increased fitness during passage under stress, revealing that PepB may be essential during stress even if otherwise dispensable. The E. coli PepB Ser‐205 residue was mutated to Asn in ALE experiments. A simulated S205N mutation in PepB revealed a potential rotamer that forms hydrogen bond with residue Thr‐209 in the neighboring monomer to strengthen the interaction between monomers which may account for its role in increased fitness. With the structural information regarding the similarities and differences of PepB from PepA and other M17 peptidases, additional experiments can be conducted to determine if unique aspects of PepB could be targeted for intervention in treatment of antibiotic resistant Enterobacteriaceae.
4. MATERIALS AND METHODS
4.1. Cloning and protein purification
Both PepB proteins were expressed and purified as full‐length proteins. E. coli pepB sequence was amplified from genomic DNA of E. coli K‐12 strain MG1655 and was cloned by ligation independent cloning into vector pMCSG53. 17 The final sequenced plasmid was transformed into BL21(DE3)(Magic) and the protein was expressed in M9 media with Se‐Met or in Terrific Broth medium. Y. pestis pepB was amplified from genomic DNA of Y. pestis strain CO92 and was cloned by ligation independent cloning into vector pMCSG7. The final sequenced plasmid was transformed into BL21(DE3)(Magic) and the protein was expressed in Terrific Broth medium.
The proteins were purified by nickel affinity chromatography using the GE Healthcare ÅKTA purification system. Briefly, the cells were sonicated (pulse 5 × 10 s, 40% amplitude for 20 min) and the lysate was centrifuged at 38,800g for 40 min at 4°C. The supernatant was collected and loaded onto a nickel‐affinity column in loading buffer (10 mM Tris, 0.5 M NaCl, Tris(2‐carboxyethyl)phosphine hydrochloride [TCEP], pH 8.3), then washed twice with 10 mM Tris–HCl, 0.5 M NaCl, 25 mM imidazole, 1 mM TCEP, pH 8.3, and eluted with 10 mM Tris–HCl, 0.5 M NaCl, 500 mM imidazole, 5 mM TCEP, pH 8.3. The protein was further purified by size exclusion chromatography using the loading buffer. For PepB from E. coli, the His‐tag was cleaved using the tobacco etch virus protease with 1:20 protease:protein ratio. The cleaved protein was collected as the flow‐through from the Ni‐affinity column. For PepB from Y. pestis, the protein with the His‐tag intact was desalted after the nickel affinity column and concentrated to 19.5 mg/ml.
4.2. Crystallization
For crystallization screening of PepB from E. coli, the Se‐Met derivative (6.0 mg/ml in 10 mM Tris–HCl [pH 8.3], 0.5 M sodium chloride, 1 mM TCEP) and the native protein (8.2 mg/ml in 10 mM Tris–HCl [pH 8.3], 5 mM β‐mercaptoethanol), were incubated with 0.2 mM ZnCl2 and 0.5 mM MnCl2. Drops in 1:1 (protein:reservoir solution) ratio were equilibrated against 96 conditions/screen using commercially available Classics II, PACT, PEG's II and ComPas Suites (Qiagen). Diffraction quality native crystals of PepB grew from 0.2 M calcium acetate, 0.1 M HEPES pH 7.5, 10% (wt/vol) PEG 8000 (PEG's II Suite condition H8), and Se‐Met derivative crystals grew from 0.2 M sodium chloride, 0.1 M BIS‐TRIS pH 5.5, and 25% (wt/vol) PEG 3350 (Classics II Suite condition F10). For more details, see Table 1.
TABLE 1.
Crystallization conditions and refinement statistics
| PepB from E. coli (native) Zn‐form | PepB from E. coli (Se‐Met) Mn‐form | PepB from Y. pestis (native) apo‐form | |
|---|---|---|---|
| Crystallization conditions | |||
| Screen conditions | 0.2 M calcium acetate, 0.1 M HEPES pH 7.5, 10% (wt/vol) PEG 8000 | 0.2 M sodium chloride, 0.1 M BIS‐TRIS pH 5.5, 25% (wt/vol) PEG 3350 | 0.2 M ammonium sulfate, 0.1 M sodium cacodylate pH 6.5, 30% (wt/vol) PEG 8000 |
| Protein concentration (mg/ml) | 6.0 | 8.2 | 14.0 |
| Data collection | |||
| Space group | P21 | P212121 | H32 |
| Unit cell parameters (Å; °) | a = 150.7, b = 114.8, c = 161.2; α = 90.0, β = 92.0, γ = 90.0 | a = 114.9, b = 148.2, c = 165.0; α = β = γ = 90.0 | a = 101.6, b = 101.6, c = 240.5; α = β = 90.0, γ = 120.0 |
| Resolution range (Å) | 30.00–2.05 (2.09–2.05) | 30.00–2.60 (2.64–2.60) | 50.00–2.75 (2.80–2.75) |
| No. reflections | 338,775 (16,506) | 85,965 (4,231) | 12,763 (610) |
| R merge (%) | 9.8 (79.1) | 13.6 (79.0) | 15.2 (66.6) |
| Completeness (%) | 98.8 (96.4) | 100.0 (100.0) | 99.5 (94.8) |
| 〈I/σ(I)〉 | 11.9 (1.9) | 14.4 (2.5) | 16.5 (1.8) |
| Multiplicity | 4.1 (4.1) | 6.2 (6.3) | 10.8 (5.9) |
| Wilson B factor | 30.9 | 38.8 | 60.7 |
| Structure determination | |||
| MR initial model (PDB ID) | 3ij3 | 6oad | 3ij3 |
| Refinement | |||
| Resolution range (Å) | 29.88–2.05 (2.10–2.05) | 29.76–2.61 (2.68–2.61) | 50.00–2.75 (2.82–2.75) |
| Completeness (%) | 98.60 (94.99) | 99.6 (96.3) | 98.5 (97.04) |
| No. reflections | 321,648 (22,675) | 81,597 (5,696) | 11,976 (758) |
| R work/R free (%) | 17.2/21.5 (29.2/31.3) | 18.1/23.8 (28.9/31.3) | 17.8/24.9 (27.3/36.5) |
| Protein chains/atoms | 12/39,374 | 6/19,566 | 1/3,163 |
| Solvent atoms | 3,906 | 654 | 66 |
| Mean temperature factor (Å2) | 37.1 | 41.0 | 53.5 |
| Coordinate deviations | |||
| r.m.s.d. bonds (Å) | 0.006 | 0.003 | 0.006 |
| r.m.s.d. angles (°) | 1.473 | 1.042 | 1.032 |
| Ramachandran plot | |||
| Favored (%) | 97.0 | 97.0 | 95.2 |
| Allowed (%) | 3.0 | 3.0 | 4.6 |
| Outside allowed (%) | 0.0 | 0.0 | 0.2 |
| PDB accession code | 6oad | 6ov8 | 6cxd |
For crystallization of Y. pestis PepB, the native protein was transferred to a buffer containing 20 mM Tris–HCl pH 7.5, 150 mM sodium chloride, 10% glycerol, 0.1% sodium azide, and 0.5 mM TCEP, and concentrated to 10–14 mg/ml. Initial crystallization experiments were set up using commercially available MCSG‐1, MCSG‐2, and Top96 Screens (Anatrace) with drops in 1:1 (protein:screen solution) ratio, equilibrated against 1.5 M NaCl solution in the reservoir and later modified. Diffraction quality crystals were obtained after limited proteolysis of 14 mg/ml protein with chymotrypsin (1/40 vol/vol) in the presence of 1 mM ZnCl2; the protein sample was then mixed with 0.2 M ammonium sulfate, 0.1 M sodium cacodylate, 30% wt/vol PEG 8000 (Top96 Screen condition C5), and equilibrated against 1.5 M NaCl in the reservoir.
4.3. Data collection and processing
Data collection and data processing statistics are listed in Table 1. Prior to flash cooling in liquid nitrogen, native crystals of PepB were transferred into a 5 μl drop of 20% ethylene glycol added to the reservoir solution, whereas Se‐Met derivative crystals were frozen directly from the crystallization drop. Both data sets were collected on the LS‐CAT 21‐ID‐F beamline at the advanced photon source (APS) at the Argonne National Laboratory. A total of 350 images, which corresponded to 210° of the spindle axis rotation, were collected for each data set. All 350 images for the native data and only 250 images for the Se‐Met derivative were indexed, integrated, and scaled using HKL‐3000. 18 Y. pestis PepB crystals were harvested and flash cooled without additional cryoprotection. The data set was collected on the LS‐CAT 21‐ID‐G beamline at APS. A total of 200 images were indexed, integrated, and scaled using HKL‐3000. The diffraction images for the Y. pestis PepB data set are available on the Integrated Resource for Reproducibility in Macromolecular Crystallography server 19 at https://doi.org/10.18430/M36CXD.
4.4. Structure solution and refinement
The structure of native PepB from E. coli was solved by molecular replacement with Morda 20 from the CCP4 suite 21 using the crystal structure of the PepN cytosol aminopeptidase from Coxiella burnetii (PDB code 3ij3) as a search model. To solve the crystal structure of the Se‐Met derivative, we used the refined structure of native PepB from E. coli as a search model in Phaser 20 from the CCP4 suite. Initial solutions went through several rounds of refinement in REFMAC v.5.8.0238 and manual model corrections using Coot. 22 The water molecules were generated using ARP/wARP, 23 and metal ions and ligands were added to the model manually.
The structure of apo PepB from Y. pestis was solved within HKL‐3000 using several software packages, including POINTLESS, 24 REFMAC5, and MOLREP, 25 with the manually edited model of PepN from C. burnetii (PDB code 3ij3) as a search template for molecular replacement, where only the coordinates for the larger subunit of the protein were preserved. The optimization of side‐chain conformation was performed using Fitmunk. 26 The model was refined with HKL‐3000 using guidelines outlined elsewhere. 27 REFMAC5 was used for the reciprocal‐space refinement; Coot 22 was used for the visualization of electron density maps and manual inspection and correction of the atomic models.
At the final stages of the refinement for all models, Translation–Libration–Screw (TLS) groups were created by the TLSMD server 28 (http://skuldbmsc.washington.edu/~tlsmd/) and TLS corrections were applied during the final stages of refinement. MolProbity 29 (http://molprobity.biochem.duke.edu/) and wwPDB 30 validation servers were used for monitoring the quality of the model during refinement and for final validation of the structure. Final models and diffraction data were deposited to the Protein Data Bank (https://www.rcsb.org/) with the assigned PDB codes 6oad and 6ov8 for the native and the Se‐Met derivatives of E. coli PepB, respectively, and PDB code 6cxd for Y. pestis PepB.
The final model of the native structure of E. coli PepB consists of 12 polypeptide chains, which form two homohexamers. Chains a–c, e–h, j, and k contain 426 residues (2–427) of the PepB, whereas Chains d, i, and l contain 425 residues (3–427) of the PepB. The final model of the Se‐Met derivative consists of six polypeptide chains, which form a homohexamer. Chains d and f contain all 427 residues of PepB, Chains b and c contain 426 residues (missing first residue), and Chains a and e contain 429 residues (additional two residues of purification tag). The assignment of the metals in the active sites was confirmed using the CheckMyMetal Server. 31 Refinement statistics and the quality of the final model are summarized in Table 1.
The structure of apo PepB from Y. pestis contains one protein chain in the asymmetric unit although a hexamer was formed by symmetry. The protein consists of 432 amino acid residues and almost 97% of them were modeled. Only the Met‐1, Residues 204–213 and 308–310 were disordered and were not included in the final model.
4.5. Molecular modeling
The position of bestatin from the superposition of the crystal structure of PepA from P. putida (PDB code 3h8g) with the structure of Zn‐form of PepB from E. coli was used for manual fitting of the Asp‐Leu dipeptide into the active site using Coot. Water molecules, which match the location of the dipeptide, were removed. The optimization of the model was done using structure idealization in REFMAC. The model was restrained to retain the coordination bonds between: (a) metals and side chains of the active site; (b) bicarbonate, Arg‐281, and the conserved water molecule; (c) Asp of the substrate, Lys‐207, and Zn2+ in M2 position; and (d) Leu of the substrate and main chain atoms of Leu‐305 and Gly‐307. To remove steric clashes in the model, several cycles of structure idealization were performed using REFMAC.
4.6. Sequence and structure alignments
E. coli PepA and PepB amino acid sequences were each used as the search query against the UniProt database at the National Center for Biotechnology Information using BLASTP. Representative protein sequences from bacterial species were selected with no more than one sequence selected in the same genus. Eukaryotic M17 peptidases from Bos taurus (cow) LAP3 and Caenorhabditis elegans LAP1 were also selected as structures have been solved32, 33 and Homo sapiens (human) LAP3 was included as an outlier to root the tree. All sequences were aligned using the ClustalW algorithm 34 and the tree was generated by the Neighbor Joining Best Tree method 35 with distances based on the absolute number of differences. Analysis was done with MacVector v. 17.0.10 software package (https://macvector.com) and the tree was drawn using the Interactive Tree of Life 36 (iTOL, https://itol.embl.de). Structural diagrams were drawn from PDB files using the USCF Chimera 37 or the PyMOL Molecular Graphics System v2.0 (Schrödinger, LLC). Structural alignments were performed using POSA 38 and FATCAT 39 servers.
CONFLICT OF INTERESTS
K. J. F. S. has a significant financial interest in Situ Biosciences, LLC, a contract research organization that conducts studies unrelated to this work and holds patents on technology unrelated to this work. W. M. notes that he is a cofounder of HKL Research, Inc., and a member of the company board. Some of the structure determination software mentioned in this article has been commercialized by HKL Research, Inc.
AUTHOR CONTRIBUTIONS
Structures of E. coli PepB were solved by George Minasov and Ludmilla Shuvalova supervised by Karla J. F. Satchell. Structure of Y. pestis PepB was solved by Joanna Sławek, Ivan G. Shabalin, and Magdalena Woinska, and supervised by Wladek Minor, Adam Godzik, and Bernhard Ø. Palsson coordinated target selection and clone design. Matthew R. Lam, George Minasov, Monica Rosas Lemus, Adam Godzik, and Karla J. F. Satchell analyzed the structures and wrote the manuscript.
Supporting information
Figure S1 Multiple structure overlap of monomer subunits of bacterial M17 peptidases.
Figure S2. Displacement view of the structural changes in the PepB structure upon Zn2+ binding.
ACKNOWLEDGMENTS
The authors thank Marin Cymborowski, Sarah Grimshaw, Lukasz Jaroszewski, Olga Kiryukhina, Keewhon Kwon, Nathan Mih, Zdzislaw Warwzak, and James Windsor for target selection, cloning, crystallization, and protein purification, as well as for assistance in solving the structures. This project has been funded in whole or in part with Federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under Contract Nos. HHSN272201200026C and HHSN272201700060C and under Grant No. U01AI124316. M. R. L. was funded by an Undergraduate Research Fellowship from Northwestern University.
Minasov G, Lam MR, Rosas‐Lemus M, et al. Comparison of metal‐bound and unbound structures of aminopeptidase B proteins from Escherichia coli and Yersinia pestis . Protein Science. 2020;29:1618–1628. 10.1002/pro.3876
Funding information Undergraduate Research Fellowship from Northwestern University; National Institute of Allergy and Infectious Diseases, National Institutes of Health, Grant/Award Numbers: HHSN272201200026C, HHSN272201700060C, U01AI124316
REFERENCES
- 1. Konovalova A, Sogaard‐Andersen L, Kroos L. Regulated proteolysis in bacterial development. FEMS Microbiol Rev. 2014;38:493–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rawlings ND, Barrett AJ, Thomas PD, Huang X, Bateman A, Finn RD. The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res. 2018;46:D624–D632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Drinkwater N, Malcolm TR, McGowan S. M17 aminopeptidases diversify function by moderating their macromolecular assemblies and active site environment. Biochimie. 2019;166:38–51. [DOI] [PubMed] [Google Scholar]
- 4. Bhosale M, Pande S, Kumar A, Kairamkonda S, Nandi D. Characterization of two M17 family members in Escherichia coli, peptidase A and peptidase B. Biochem Biophys Res Commun. 2010;395:76–81. [DOI] [PubMed] [Google Scholar]
- 5. Strater N, Sherratt DJ, Colloms SD. X‐ray structure of aminopeptidase A from Escherichia coli and a model for the nucleoprotein complex in Xer site‐specific recombination. EMBO J. 1999;18:4513–4522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kale A, Pijning T, Sonke T, Dijkstra BW, Thunnissen AM. Crystal structure of the leucine aminopeptidase from Pseudomonas putida reveals the molecular basis for its enantioselectivity and broad substrate specificity. J Mol Biol. 2010;398:703–714. [DOI] [PubMed] [Google Scholar]
- 7. Alen C, Sherratt DJ, Colloms SD. Direct interaction of aminopeptidase A with recombination site DNA in Xer site‐specific recombination. EMBO J. 1997;16:5188–5197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Reijns M, Lu Y, Leach S, Colloms SD. Mutagenesis of PepA suggests a new model for the Xer/cer synaptic complex. Mol Microbiol. 2005;57:927–941. [DOI] [PubMed] [Google Scholar]
- 9. Modak JK, Rut W, Wijeyewickrema LC, Pike RN, Drag M, Roujeinikova A. Structural basis for substrate specificity of Helicobacter pylori M17 aminopeptidase. Biochimie. 2016;121:60–71. [DOI] [PubMed] [Google Scholar]
- 10. Hattne J, Dubrovska I, Halavaty A, et al. Crystal structure of cytosol aminopeptidase from Staphylococcus aureus COL. 2010. doi: 10.2210/pdb3KZW/pdb [DOI]
- 11. Pomposiello PJ, Bennik MH, Demple B. Genome‐wide transcriptional profiling of the Escherichia coli responses to superoxide stress and sodium salicylate. J Bacteriol. 2001;183:3890–3902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. LaCroix RA, Sandberg TE, O'Brien EJ, et al. Use of adaptive laboratory evolution to discover key mutations enabling rapid growth of Escherichia coli K‐12 MG1655 on glucose minimal medium. Appl Environ Microbiol. 2015;81:17–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Leiser OP, Merkley ED, Clowers BH, et al. Investigation of Yersinia pestis laboratory adaptation through a combined genomics and proteomics approach. PLoS One. 2015;10:e0142997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Prlic A, Bliven S, Rose PW, et al. Pre‐calculated protein structure alignments at the RCSB PDB website. Bioinformatics. 2010;26:2983–2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhang Y, Stec B, Godzik A. Between order and disorder in protein structures: Analysis of “dual personality” fragments in proteins. Structure. 2007;15:1141–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Suzuki H, Kamatani S, Kumagai H. Purification and characterization of aminopeptidase B from Escherichia coli K‐12. Biosci Biotechnol Biochem. 2001;65:1549–1558. [DOI] [PubMed] [Google Scholar]
- 17. Eschenfeldt WH, Makowska‐Grzyska M, Stols L, Donnelly MI, Jedrzejczak R, Joachimiak A. New LIC vectors for production of proteins from genes containing rare codons. J Struct Funct Genomics. 2013;14:135–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Minor W, Cymborowski M, Otwinowski Z, Chruszcz M. HKL‐3000: The integration of data reduction and structure solution—From diffraction images to an initial model in minutes. Acta Crystallogr. 2006;D62:859–866. [DOI] [PubMed] [Google Scholar]
- 19. Grabowski M, Langner KM, Cymborowski M, et al. A public database of macromolecular diffraction experiments. Acta Crystallogr. 2016;D72:1181–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. McCoy AJ, Grosse‐Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Winn MD, Ballard CC, Cowtan KD, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. 2011;D67:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Emsley P, Cowtan K. Coot: Model‐building tools for molecular graphics. Acta Crystallogr. 2004;D60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 23. Morris RJ, Perrakis A, Lamzin VS. ARP/wARP and automatic interpretation of protein electron density maps. Methods Enzymol. 2003;374:229–244. [DOI] [PubMed] [Google Scholar]
- 24. Evans P. Scaling and assessment of data quality. Acta Crystallogr. 2006;D62:72–82. [DOI] [PubMed] [Google Scholar]
- 25. Vagin A, Teplyakov A. MOLREP: An automated program for molecular replacement. J Appl Crystallogr. 1997;30:1022–1025. [Google Scholar]
- 26. Porebski PJ, Cymborowski M, Pasenkiewicz‐Gierula M, Minor W. Fitmunk: Improving protein structures by accurate, automatic modeling of side‐chain conformations. Acta Crystallogr. 2016;D72:266–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shabalin IG, Porebski PJ, Minor W. Refining the macromolecular model—Achieving the best agreement with the data from X‐ray diffraction experiment. Crystallogr Rev. 2018;24:236–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Painter J, Merritt EA. Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr. 2006;D62:439–450. [DOI] [PubMed] [Google Scholar]
- 29. Chen VB, Arendall WB 3rd, Headd JJ, et al. MolProbity: All‐atom structure validation for macromolecular crystallography. Acta Crystallogr. 2010;D66:12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Read RJ, Adams PD, Arendall WB 3rd, et al. A new generation of crystallographic validation tools for the protein data bank. Structure. 2011;19:1395–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zheng H, Cooper DR, Porebski PJ, Shabalin IG, Handing KB, Minor W. CheckMyMetal: A macromolecular metal‐binding validation tool. Acta Crystallogr. 2017;D73:223–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Burley SK, David PR, Taylor A, Lipscomb WN. Molecular structure of leucine aminopeptidase at 2.7‐A resolution. Proc Natl Acad Sci U S A. 1990;87:6878–6882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhan C, Patskovsky Y, Wengerter BC, et al. Structure of Caenorhabditis elegans leucine aminopeptidase‐zinc complex (LAP1). 2006. doi: 10.2210/pdb2HC9/pdb [DOI]
- 34. Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position‐specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Saitou N, Nei M. The neighbor‐joining method: A new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–425. [DOI] [PubMed] [Google Scholar]
- 36. Letunic I, Bork P. Interactive tree of life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019;47:W256–W259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pettersen EF, Goddard TD, Huang CC, et al. UCSF chimera—A visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. [DOI] [PubMed] [Google Scholar]
- 38. Li Z, Natarajan P, Ye Y, Hrabe T, Godzik A. POSA: A user‐driven, interactive multiple protein structure alignment server. Nucleic Acids Res. 2014;42:W240–W245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ye Y, Godzik A. FATCAT: A web server for flexible structure comparison and structure similarity searching. Nucleic Acids Res. 2004;32:W582–W585. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Multiple structure overlap of monomer subunits of bacterial M17 peptidases.
Figure S2. Displacement view of the structural changes in the PepB structure upon Zn2+ binding.