

Abstract
Cellular signaling via binding of the cytokines IL‐36α, β, and γ along with binding of the accessory protein IL‐36RAcP, to their cognate receptor IL‐36R is believed to play a major role in epithelial and immune cell‐mediated inflammation responses. Antagonizing the signaling cascade that results from these binding events via a directed monoclonal antibody provides an opportunity to suppress such immune responses. We report here the molecular structure of a complex between an extracellular portion of human IL‐36R and a Fab derived from a high affinity anti‐IL‐36R neutralizing monoclonal antibody at 2.3 Å resolution. This structure, the first of IL‐36R, reveals similarities with other structurally characterized IL‐1R family members and elucidates the molecular determinants leading to the high affinity binding of the monoclonal antibody. The structure of the complex reveals that the epitope recognized by the Fab is remote from both the putative ligand and accessory protein binding interfaces on IL‐36R, suggesting that the functional activity of the antibody is noncompetitive for these binding events.
Keywords: cytokine signaling, IL36R, monoclonal antibody, structural biology
1. INTRODUCTION
The IL‐1 cytokine family mediates important signaling events linked to both innate and adaptive immunity. 1 Understanding the mechanism by which signaling is achieved has been the focus of significant research. The first components of this mechanism to be described were the cytokines IL‐1α and IL‐1β. Subsequently, their primary cognate receptor, IL‐1R, was described. Since then, 11 members of the IL‐1 cytokine family have been identified, primarily based on sequence similarity, and classified according to their primary receptor.
A structural understanding of the mechanism by which the IL‐1 cytokines elicit physiological responses has centered on crystal structures of the IL‐1 receptor (IL‐1R) and its cognate ligands. IL‐1R is known to bind two ligands, IL‐1α and IL‐1β and also a structurally similar antagonist, IL‐1Ra. Crystal structures of the soluble, extracellular portion of IL‐1R in complex with ligand and antagonist revealed the structural adaptation of the receptor to accommodate its binding partners. A crystal structure of the complex between the extracellular portion of the type 1 IL‐1 receptor and one of its primary ligands, IL‐1β, 2 revealed that the proteins form a 1:1 complex and that all three of the receptor's immunoglobulin‐like (Ig) domains interact with the ligand. The crystal structure of the receptor with the natural antagonist IL‐1Ra 3 also contained the three Ig domains, wherein the N‐terminal two Ig domains (D1 and D2) remained in a similar relative orientation to that present in the IL‐1β bound complex, but the third Ig domain (D3) reoriented slightly upon binding IL‐1Ra.
Similar observations, that the relative orientation of the cytokine D1 and D2 domains remain fixed upon receptor binding, are provided by structures of IL‐1R family members IL‐1RII (PDB 3O4O) 4 ; and ST2 (aka IL1RL1, PDB 4KC3) 5 ; in complex with IL‐1β and IL‐1RAcP and with IL‐33, respectively.
The IL‐1R family member IL‐36R (also referred to as IL‐1Rrp2; uniprot Q9HB29) is central to innate and adaptive immune responses. 6 Three cytokine ligands: IL‐36α, IL‐36β, and IL‐36γ and one antagonist IL‐36Ra, are known to signal through IL‐36R. The relevance of the role of IL‐36 mediated signaling in inflammatory skin diseases has been highlighted by several recent publications,7, 8, 9, 10, 11, 12 providing evidence that antagonism of IL‐36R signaling provides a route to directly suppress IL‐36R driven pathways, including IL23/IL17, and TNF‐α. 13 Suppression of these signaling events, for example, by a monoclonal antibody, should result in a reduction of epithelial and immune cell‐mediated inflammation and interrupt the inflammatory response that underlies tissue pathology. Such interventions have the potential to reduce disease severity in conditions such as psoriasis.
We report here the co‐crystal structure of a Fab fragment of an IL‐36R blocking antibody in complex with the cognate portion of IL‐36R. The Fab fragment was derived from a humanized version of a murine antihuman IL‐36R monoclonal antibody. The discovery and production of the antibody, known as BI 655130, has been described previously. 14
This work represents the first reported structure of any portion of IL‐36R and shows that the structural motifs and intra‐protein domain rearrangements observed previously in IL‐1R structures are conserved for this member of the IL‐1R family. Interestingly, the Fab binds at a surface distal from the expected binding sites of the IL‐36 ligands and IL‐1RAcP (based on comparison with the IL‐1R complex structures), suggesting an allosteric mechanism of interaction and signaling inhibition.
2. RESULTS AND DISCUSSION
2.1. Molecular structure of BI 655130 Fab/IL‐36R complex
In order to understand the structural features of IL‐36R, we sought to determine the crystal structure of the ectodomain of the protein. While we were able to express and purify a protein construct containing the three Ig domains (D1, D2, and D3; residues 20–332), we were not able to crystallize the entire extra cellular domain (ECD) of the IL‐36R protein. We hypothesized that flexibility of the individual IL‐36R Ig domains with respect to each other was hindering crystallization. Two strategies were adopted to overcome this difficulty. First, based on the observation that in IL‐1R family structures (PBD 1IRA, 3 1G0Y, 15 4DEP, 16 and 4KC3 5 ) the first two Ig domains (D1 and D2) behave as a rigid unit in the various complexes, despite rearrangements in the third domain (D3), we focused on a shorter construct containing just D1 and D2 (residues 20–215). Second, it was suggested by hydrogen‐deuterium exchange mass spectrometry experiments, that the binding epitope of the anti‐IL‐36R antibody, BI 655130 was confined to the D1 and D2 domains of the receptor. 14 Therefore, the Fab fragment of BI 655130 was used as a crystallization chaperone in complex with the two‐domain IL‐36R protein construct (IL‐36R D1D2). Crystals of the two domain: Fab complex were readily obtained. Despite significant effort, crystals containing all three extracellular IgG domains of IL‐36R, either alone or in complex with BI 655130 Fab, and crystals of the 2‐domain construct alone were not obtained.
The 2.31 Å resolution crystal structure of the complex (Figure 1a) was solved by molecular replacement (MR) methods. Data collection and refinement statistics are presented in Table 1. There is one 1:1 complex of IL‐36R D1D2 and the Fab present in the asymmetric unit. The CDR loops of the variable regions from both chains of the Fab interact with the receptor fragment and the elbow angle of the Fab is 124°. The electron density is well defined throughout the structure, which allowed for unambiguous modeling of the interface between IL‐36R and the Fab (Figure S1). Glycosylation was observed on two residues of IL‐36R, Asn41, and Asn109. Neither of these residues is close to the interface with the Fab and therefore is unlikely to influence the binding.
FIGURE 1.

The structure of IL‐36R (D1D2) in complex with Fab of anti‐IL‐36R antibody, BI 655130. (a) BI 655130 (heavy chain: green and light chain: cyan) binds to the first two extracellular Ig domains (D1 and D2) of IL‐36R (orange) with the D1–D2 linker (magenta) on a surface that does not overlap with the putative binding site for the IL‐36 cytokines and antagonist protein. (b) and (c) Comparison of IL‐36R (D1D2) with IL‐1R (D1D2). (b) The overall fold and domain organization of D1 and D2 for IL‐36R (top, orange) is very similar to that of other members of the IL‐1R family (bottom, equivalent domains of IL‐1R, olive green; PDB ID 4DEP 16 ). (c) Superposition of IL‐36R D1D2 and IL‐1R D1D2, focusing on the D1–D2 interdomain interface to highlight the different conformations of the D1–D2 linker (view is rotated slightly toward the viewer along the X‐axis from the top with respect to the view in (b), colors as in (b)). The conserved disulfide bond between the linker and D2 is indicated by an arrow
TABLE 1.
X‐ray crystallographic collection and refinement statistics
| Data collection/processing | |
|---|---|
| Wavelength (Å) | 1.0 |
| Resolution range (Å) | 36.99–2.31 (2.44–2.31) |
| Space group | P 21 21 21 |
| Unit cell (a, b, c, α, β, γ) | 60.70, 70.66, 233.27, 90, 90, 90 |
| Total reflections | 302,450 (44,702) |
| Unique reflections | 45,094 (6,503) |
| Multiplicity | 6.7 (6.9) |
| Completeness (%) | 100.0 (100.0) |
| Mean I/σ (I) | 12.7 (2.5) |
| R merge | 0.09 (0.81) |
| R meas | 0.10 (0.88) |
| CC1/2 | 0.997 (0.860) |
| Wilson B‐factor | 48 |
| Refinement | |
|---|---|
| Resolution range (Å) | 36.99–2.31 (2.36–2.31) |
| Total reflections | 44,981 |
| Reflections used for R‐free | 2,280 |
| R‐work | 0.18 (0.27) |
| R‐free | 0.20 (0.29) |
| Protein residues | 636 |
| Number of non‐hydrogen atoms | 5,225 |
| Macromolecules | 4,990 |
| Ligands | 43 |
| Water | 189 |
| Average B‐factor | 55.9 |
| Macromolecules | 55.9 |
| Ligands | 81.8 |
| Solvent | 50 |
| RMS (bonds) | 0.009 |
| RMS (angles) | 1.09 |
| Ramachandran favored (%) | 96.9 |
| Ramachandran allowed (%) | 2.8 |
| Ramachandran outliers (%) | 0.3 |
| Molprobity Clashscore | 2.32 |
| Overall Molprobity score | 1.18 |
| PDB ID | 6U6U |
Note Values for the highest resolution shell are in parentheses.
The structure of the IL‐36R D1D2:Fab complex confirms that the structure and relative orientation of the first two Ig domains are conserved between IL‐36R and other IL‐1R family members (Figure 1b). The root mean squared deviation for the structural superposition 17 of the Cα‐trace of the IL‐36R D1D2 module on that of other IL‐1R family member structures, including IL‐1R1, IL‐1R2, and ST2, varies from 2.3 to 3.3 Å. The conservation of overall fold between IL‐36R and IL‐1R is not unexpected given the conservation of the Ig fold in general and the sequence similarity between the two receptors in particular (33% identity/48% similarity for 304 aligned residues spanning the entire extracellular regions; and 36% identity/52% similarity for 169 aligned residues spanning only the first two IG domains, D1 and D2). The conserved D1–D2 interdomain alignment is striking considering that the loop connecting D1 to D2 (the D1–D2 linker) in IL‐1R is four amino acids shorter than the equivalent loop in IL‐36R. The D1–D2 linker of IL‐36R is twisted relative to that of IL‐1R and appears to project further from the β‐sheets of the Ig domains, but is still contiguous with the protein surface (Figure 1c). Despite the additional residues and different conformations between the D1–D2 linkers of IL‐36R and IL‐1R, the conserved disulfide bond that pins the N‐terminus of the D1–D2 linker to the loop connecting the two β‐sheets of the D2 Ig fold is maintained in approximately the same spatial position (Figure 1c).
2.2. Interaction between BI 655130 Fab and IL‐36R D1D2
The interaction between the Fab and the receptor is primarily mediated by the CDR loops of each antibody chain. The epitope on IL‐36R is on a side of the molecule remote from the expected IL‐36 ligand/antagonist binding site. The shape complementarity statistic S c, 18 calculated for the interface between the Fab and the IL‐36R is 0.76. Considering the interface between the receptor and each chain of the Fab individually, S c for IL‐36R:V H interface is 0.79 and is 0.71 for the IL‐36R:V L interface. As is common in antibody: antigen complexes, there is a high occurrence of aromatic residues at the BI 655130 Fab:IL‐36R interface with the majority of the aromatic residues (10 of 14) being provided by the Fab portion of the complex.
The interface between BI 655130 and IL‐36R has a total buried surface area of ~1,092 Å2 (calculated using PISA 19 ). The interface with the V H chain is ~736 Å2, while the interface with the V L chain is ~356 Å2. The majority of the buried surface on IL‐36R is contributed by D2 and the D1–D2 linker; only one residue in D1, Trp87, appears to contribute significantly to the interface. On both the V H and V L chains of the Fab, all three CDR loops interact with IL‐36R. On the V L chain, only CDRL3 appears to be fully engaging the receptor, whereas only Ser30, Ser32, and Tyr33 at the C‐terminus of CDRL1 and Arg51 at the N‐terminus of CDRL2, appear to make significant contacts with the receptor. While the majority of the V L chain interactions are confined to the D1–D2 linker, the V H chain interacts extensively with both D2 and the D1–D2 linker. The IL‐36R:BI 655130 Fab interface, as well as a close‐up look at the residues involved, is depicted in Figure 2a.
FIGURE 2.
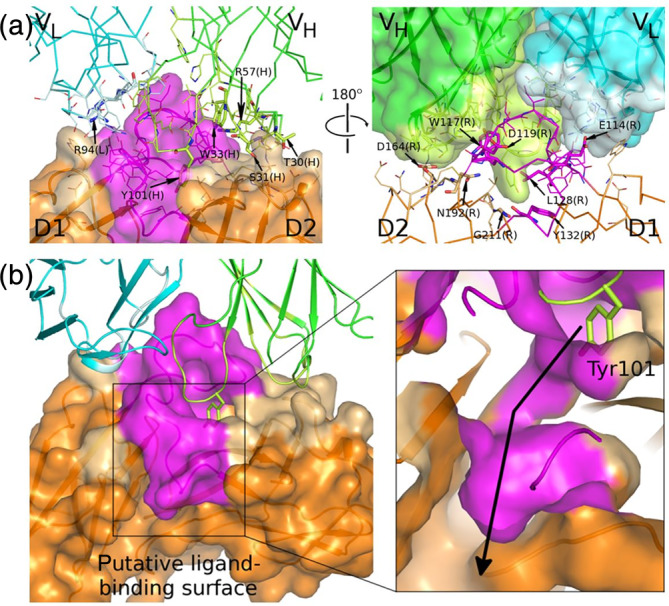
The IL‐36R:BI 655130 interface. (a) The D1–D2 linker region forms a major portion of the interface on IL‐36R and CDRH3, of BI 655130 protrudes into the interdomain space between D1 and D2 of IL‐36R (colors as described in Figure 1a). The surface that is in contact with the other proteins of the complex is indicated by a lighter shade. Interface residues are shown as lines and key residues mentioned in the text are shown as sticks and labeled (L, light chain; H, heavy chain; R, IL‐36R). (b) Tyr101 on the CDRH3 loop of BI 655130 protrudes into a deep cavity between domains D1 and D2 of IL‐36R (left). This cavity leads into a channel that continues through the IL‐36R D1–D2 interdomain space and opens up on the putative IL‐36R ligand‐binding surface (right, the view is zoomed in and sliced through the protein surface to show the channel inside of the protein). The arrow begins in the cavity on the antibody‐binding surface, travels through the channel and ends in a deep cavity on the ligand‐binding surface
Confidence that the reported crystal structure fully defines the interaction between the BI655130 antibody and full length IL‐36R is supported by the fact that no protection of the D3 domain by the antibody was observed in HDXMS studies. 14
In the linker region between the two Ig domains of the receptor, the D1–D2 linker, every amino acid in the contiguous stretch of residues between Trp117 and Leu128 interacts with the Fab. The single aromatic residue in this 11 residue stretch, Trp117, is buried deep on the surface of the V H chain of the Fab, between Tyr101 from CDRH3 and Trp33 of CDRH1. Asp119 in the linker region forms a bidentate H‐binding arrangement between its backbone carbonyl and sidechain oxygens and the sidechain Nε and Nηatoms of Arg57 in CDRH3. Trp117 also forms part of a “hydrophobic spine” of interacting residues between the Fab and the receptor. Together with Tyr132 at the C‐terminus of the D1–D2 linker, Trp117 participates in an interleaved stacking arrangement with Trp33 in CDRH1 and Tyr101 in CDRH3. Glu114 at the N‐terminus of the linker makes a salt‐bridge to Arg94 of CDRL3. Due to the extensive interaction between the D1–D2 linker of IL‐36R and the Fab, it is reasonable to assume that its conformation is at least partially influenced by Fab binding. It may adopt an alternate conformation in the absence of the Fab, particularly in the region upstream of the conserved disulfide bond.
The central role observed for the D1–D2 linker in the interaction between IL‐36R and the BI 655130 Fab is consistent with the protection of this region observed in HDXMS studies, wherein the peptide from Glu114 to Leu128 was shown to be protected upon binding of the antibody. 14
The majority of the interface between the Fab and IL‐36R is contributed by the CDR loops of the heavy chain, particularly CDRH3. Tyr101 at the tip of CDRH3 appears to be a key mediator of interactions between the Fab and IL‐36R D2. Notably, the hydroxyphenyl sidechain protrudes into a deep cavity in IL‐36R, between domains D1 and D2 (Figure 2b). Tyr101 lies on a surface formed by Gly211, Ile212, and Thr213 on the final β‐strand of D2, which makes up a large portion of the D1–D2 interdomain interface. This water‐filled cavity extends all the way through the receptor to the putative ligand‐binding surface. The cavity is much more prominent in IL‐36R than in IL‐1R and its opening is on opposite sides of the D1–D2 linker. The D1–D2 linker contributes an entire side of this cavity and, as noted above, this loop is slightly longer and appears to be displaced from the Ig domains in IL‐36R when compared to IL‐1R. The larger interdomain cavity in IL‐36R and the alternate position of its entrance could be a result of the differences in these loops. However, the loop also makes extensive contact with the Fab so there is a possibility that the nature of the cavity in IL‐36R may be influenced by Fab binding. Loop CDRH1, from Residue Tyr 27 through residue Trp33, is fully engaged with IL‐36R D2. Hydrogen bonding interactions are observed between the sidechain of Ser31 and the backbone amide nitrogen of Asn192 on the receptor, and the sidechain oxygens of Thr30 in CDRH1 and Asp164 in D2 of IL‐36R.
2.3. Structural basis for the antagonism of BI 655130
BI 655130 antagonizes the signaling events that occur downstream of IL‐36R ligand/accessory protein binding and formation of the IL‐36 signaling complex. 14 The specific mechanism by which signaling is inhibited remains unknown. While the binding sites of IL‐36 ligands and the IL‐1RAcP on IL‐36R have not been structurally determined, they can be inferred from homology to the related IL‐1R structures. Figure 3a shows a superposition, based on the conserved D1 and D2 Ig domain structures of the IL‐36R Fab complex with the trimeric complex of IL‐1R1, IL‐1β, and IL‐1RAcP (PDB 4DEP). The resulting model shows that the binding epitope of the Fab does not significantly overlap with either the putative binding sites of the IL‐36R ligands, or IL‐1RAcP. This opens the possibility that BI 655130 may bind to either the receptor alone, the cytokine complex, or even to the trimeric signaling complex and the mode of action may be noncompetitive with these binding events.
FIGURE 3.
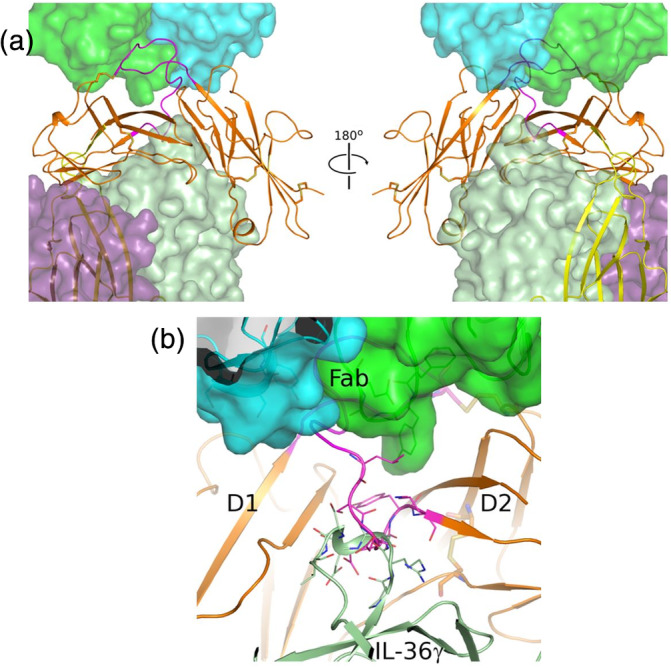
BI 655130 may act noncompetitively against IL‐36 ligand and accessory protein binding to IL‐36R. (a) Model of the quaternary complex of BI 655130 Fab:IL‐36R:IL‐36γ:IL‐36RAcP suggests that it is possible for the antagonist antibody to engage IL‐36R even when it is present as a complete signaling complex with cytokine and accessory protein. IL‐36R (D1D2) is shown as an orange cartoon and the Fab of BI 655130 is shown as a semitransparent green and cyan surface. D3 (yellow cartoon) and accessory protein (purple semitransparent surface) were placed by superposition of the IL‐1R D1D2 module from the trimeric IL‐1R:IL‐1β:IL‐1RAcP complex (PDB 4DEP 16 ) onto the D1D2 module of IL‐36R in the current structure. The bound cytokine IL‐36γ (light green semitransparent surface) was subsequently modeled by superposition of IL‐36γ (PDB 4IZE 20) onto IL‐1β in the placed IL‐1R trimeric complex. (b) Potential clash between BI 655130‐bound IL‐36R and IL‐36 ligand binding. Due to the extensive interaction between BI 655130 and the D1–D2 linker of IL‐36R, and the observed clash between the linker (magenta cartoon) and the β3–β4 loop of docked IL‐36γ (light green cartoon) in the modeled cytokine‐bound complex, an antagonist mode‐of‐action that is competitive with cytokine‐binding cannot be ruled out. This potential clash involves loops that are generally observed to be fairly mobile among IL‐receptor and cytokine structures, however, so there is uncertainty in this rigid docking pose generated by superposition and analogy with the IL‐1R signaling complex
Closer inspection of this model of IL‐36R and the modeled IL‐1β cytokine, however, reveals a possible structural clash between a portion of the IL‐36R D1–D2 linker and the loop between β‐strands 3 and 4 of the cytokine. This clash involves receptor residues 129–131 at the N‐terminal of the D2 region. These three residues do not make direct interactions with the Fab but their position could be influenced by antibody binding since, as described above, the Fab does interact extensively with the D1–D2 linker upstream of this position. Superposition of the recent structure of IL‐36γ 20 onto IL‐1β shows that the β3–β4 loop of IL‐36γ is of a similar size and occupies a similar position, suggesting that it too may clash with the D1–D2 linker of IL‐36R in the context of the antibody‐bound receptor (Figure 3b).
If the D1–D2 linker conformation is influenced by BI 655130 binding, then the possible clash between this portion of the receptor and the ligand may provide an explanation for antagonism of IL‐36 signaling since it could have an impact on cytokine‐binding. This would result in a mechanism of action that is competitive with ligand‐binding, where the formation of the IL‐36 signaling complex would be prevented. Uncertainty about such a mechanism arises from the fact that the D1–D2 linker of IL‐R family members has been shown to adopt multiple poses in crystal structures, suggesting flexibility in this region of the protein, permitting adaptation to various binding partners. Modeling the interactions of IL‐36R binding partners thus cannot rule out a noncompetitive or competitive mode of action for BI 655130 antagonism of IL‐36 signaling.
In summary, we have described the crystal structure of the Fab fragment of an anti‐IL‐36R antibody that blocks signaling by IL‐36 ligands. Modeling of the antibody in complex with the complete IL‐36 signaling complex, consisting of receptor, ligand and accessory protein, strongly suggests an antagonistic mode of action that is noncompetitive with binding of the protein partners to IL‐36R. It is not possible to rule out a competitive mode of action, however, so confirmation of how the antibody antagonizes signaling events downstream of IL‐36 signaling complex formation leading to the observed pharmacology will require additional studies.
3. MATERIALS AND METHODS
3.1. Protein expression and purification
The IL‐36R ECD D20‐S215 containing a human IgG signal sequence and a C‐terminal Hexa‐HIS tag were cloned into pcDNA2.1 expression vector.
Twenty liters of HEK293f cells were transfected with 1 mg of plasmid DNA/L using PolyPlus linear Q‐PEI at 1:1.5 (wt/vol) DNA:PEI ratio. Cultures were harvested 72 hr post transfection with a cell density of 2.7 × 106 and 89% viability. Conditioned media was clarified by centrifugation and adjusted to 0.2 M sucrose, 5% glycerol (vol/vol) and 0.01% CHAPS (wt/vol). The pH was adjusted to 7.2 using a 1 M dibasic sodium phosphate stock solution.
The media was batch bound to 60 ml of NiNTA (Qiagen) resin at 4°C overnight with stirring. Following overnight incubation, the resin was collected in 5 cm diameter Econo‐column and washed with three column volumes (CV) of Buffer A (1X PBS, [2.7 mM KCl, 1.7 mM KH2PO4, 136 mM NaCl, 10.1 mM Na2HPO4], 0.2 M sucrose, 5% glycerol [vol/vol], 0.01% CHAPS [wt/vol], 1 mM TCEP, pH 7.2 [23°C]). The washed resin was eluted in a 2.6 cm XK column over 10 CV gradient from buffer A to buffer B (Buffer A + 250 mM imidazole, pH 7.2 [23°C]) using an AKTA FPLC. Fractions (10 ml each) were collected and analyzed by SDS‐PAGE. Fractions containing IL‐36R were pooled and concentrated in a Vivacell 100 (10 K MWCO, PES membrane) centrifugal device to 70 ml and loaded onto a Superdex200 (GE Healthcare) 5 cm × 90 cm column equilibrated in SEC buffer (1X PBS, [2.7 mM KCl, 1.7 mM KH2PO4, 136 mM NaCl,10.1 mM Na2HPO4], 0.2 M sucrose, 5% glycerol [vol/vol], 0.01% CHAPS [wt/vol], 1 mM TCEP, pH 7.2 [23°C]). Fractions (13 ml) were collected over 1.1 CV and analyzed by SDS‐PAGE. Fractions containing IL‐36R were pooled in aliquots and frozen at −80°C.
IL‐36R antagonist heavy chain Fab with a C‐terminal hexa‐HIS tag and IL‐36R antagonist light chain Fab were each cloned into CMV‐promoter based (pTT5, licensed from the National Research Council of Canada) expression vectors. Two, 1 L cultures (media: F17 supplemented with 0.1% Pluronic F‐68 and 4 mM GlutaMAX) of CHO‐3E7 cells in 1 L shake flasks were transfected with 0.5 mg/L of the plasmid encoding the light chain and 0.5 mg/L of the plasmid encoding the heavy chain using Mirus TransIT‐PRO at a 1:1.5 (wt/vol) DNA:TransIT‐PRO ratio. The heterodimerization of the Fab's H & L chains are driven by the CH1‐domain/Ckappa interaction. Four hours post transfection, 5 ml/L of Anti‐Clumping Agent (ThermoFisher catalog# 0010057AE) was added to each flask. The flasks were incubated at 37°C in a humidified 5% CO2 environment with shaking at 135 rpm. The 1 L cultures were fed with CHO CD Efficient Feed B (150 ml/L) 24 hr post transfection, and transferred to 32°C.
Culture parameters were monitored using a Cedex XS for density and viability. Culture was harvested 13 days post transfection (dpt) via centrifugation for 10 min at 1,000g. The conditioned culture supernatant (CCS) was clarified by centrifugation for 2 hr at 9,100g. Density at 13 dpt was 3.7 × 106 with 51% viability.
The CCS was adjusted to 10 mM imidazole and 10 mM MgCl2. The pH of the CCS was confirmed to be pH 7.8–8.0. The target protein in the CCS was batch bound to 40 ml of Ni‐NTA resin (Novagen) at 4°C overnight with stirring. The resin was collected in a 5 cm diameter Econo‐column and washed with 15 CV of Buffer A. The protein of interest was eluted in Buffer B. Fractions (5 ml) were collected and analyzed by A280 prior to pooling fractions 6–20. Select fractions were analyzed by SDS‐PAGE. The Ni‐NTA pool was concentrated using a Vivaspin20 (10 K MWCO, PES membrane) centrifugal device to 10 ml and loaded onto a Superdex 200 (GE Healthcare) column equilibrated in destination buffer (1X PBS [2.7 mM KCl, 1.7 mM KH2PO4, 136 mM NaCl, 10.1 mM Na2HPO4], pH 7.2 [23°C]). Fractions (5 ml) were collected over 1.1 CV and analyzed by SDS‐PAGE prior to pooling fractions D7–E1. The pool was concentrated using a Vivaspin20 (10 K MWCO, PES membrane) centrifugal device to 2.7 mg/ml prior to final analysis. A total of 34 mg of protein was obtained and flash frozen in liquid nitrogen before storage at −80°C.
IL‐36R (ECD)‐6H (20–215) and IL‐36R Fab proteins were mixed together at 1:1.5 IL‐36R (ECD)‐6H (20–215): IL‐36R Fab ratio and incubated at 4°C overnight. The mixture was then concentrated to 4 ml and loaded onto a Superdex S200 10/300 (GE Healthcare) column equilibrated in SEC buffer (1X PBS, pH 7.2 [2.7 mM KCl, 1.7 mM KH2PO4, 136 mM NaCl, 10.1 mM Na2HPO4], 0.2 M sucrose, 5% glycerol, 0.01% CHAPS, and 1 mM TCEP). The SEC column was run at 1 ml/min collecting 1 ml fractions. Two peaks were obtained. The earlier eluting peak, Peak 1 contained the IL‐36R/Fab complex and the later, Peak 2, corresponded to uncomplexed Fab as judged by elution volumes relative to MW standards. Fractions were analyzed by SDS‐PAGE and those fractions containing the complex were pooled together and concentrated to 19.6 mg/ml for crystallization trials.
3.2. Crystallization, data collection, and structure determination
Purified complex of IL‐36R (20–215):BI 655130 Fab was set up in sitting drop vapor diffusion crystallization experiments consisting of 100 nl protein solution + 100 nl crystallization solution using a Mosquito liquid handling robot (TTP Labtech, Inc., Cambridge, MA). Drops were equilibrated over a 50 μl reservoir of crystallization solution. Rock Imager systems (Formulatrix, Bedford, MA) were used for incubation and imaging of the experiments. Diamond‐shaped, diffraction quality crystals grew in condition D9 (1.6 M Na3C6H5O7) of the NeXtal Classics Suite (Qiagen) within 1 week at 20°C or within 3 weeks at 4°C. Crystals were harvested from the drop with an appropriately sized CryoLoop (Hampton Research, Aliso Viejo, CA) and directly plunge frozen in liquid nitrogen.
X‐ray diffraction data were collected by Expose GmbH (Döttingen, Switzerland) at the Swiss Light Source on Paul Scherrer Institute beamline X06SA using a PILATUS 6M detector or on beamline X06DA using a PILATUS 2M detector.
Diffraction data were processed using XDS, 21 POINTLESS, 22 AIMLESS, 23 and Truncate 24 as implemented in the autoPROC pipeline. 25 The structure was solved by MR using the program Phaser24, 26 as implemented in the Phenix software package. 27 The MR search model for IL‐36R (20–215) was derived from the equivalent domains of IL‐1R (PDB 1G0Y 15 ) and the search model for BI 655130 Fab was derived from anti‐rabies glycoprotein Fab 523–11 (PDB 4M43, unpublished). The MR solution required sequentially searching for the IL‐36R domains, then the N‐terminal pair of IG domains from the heavy and light chains, and finally the pair of C‐terminal IG domains from the heavy and light chains. The initial model was greatly improved and many gaps were filled in by manual trimming, building, and refinement with Coot 28 followed by automatic building/rebuilding with autoBUILD. 29 The final model of the complex was completed by iterative rounds of building and refinement using the programs Coot and phenix.refine. 30 During refinement, each individual chain of the model was assigned to a separate translation/libration/screw group. Model validation was performed with MolProbity 31 as implemented in PHENIX. Crystallographic and refinement statistics are presented in Table 1. Superposition of structures were performed using SSM 32 within Coot or with CEAlign 17 within PyMOL. 33 Structural figures were created and rendered using PyMOL.
Coordinates and structure factors for the IL‐36R (20–215):BI 655130 Fab complex have been deposited in the PDB with accession number 6U6U.
CONFLICT OF INTEREST
The authors are, or were, employees of Boehringer Ingelheim Pharmaceuticals.
AUTHOR CONTRIBUTIONS
Eric Larson: Conceptualization; investigation; methodology; writing‐review and editing. Debra Brennan: Investigation; methodology; writing‐original draft. Eugene Hickey: Investigation; methodology; writing‐original draft. Raj Ganesan: Conceptualization; investigation; methodology; writing‐original draft. Rachel Kroe‐Barrett: Supervision; writing‐review and editing. Neil A. Farrow: Conceptualization; supervision; writing‐review and editing.
Supporting information
Figure S1. Stereo view of electron density in the interface between IL‐36R and BI 655130. The 2fo‐fc electron density (blue mesh) is contoured at 1.8σ. IL‐36R is colored orange, the heavy chain of BI 655130 is colored green, and the light chain of BI 655130 is colored cyan. The view is centered on Tyr101 of the heavy chain.
Larson ET, Brennan DL, Hickey ER, Ganesan R, Kroe‐Barrett R, Farrow NA. X‐ray crystal structure localizes the mechanism of inhibition of an IL‐36R antagonist monoclonal antibody to interaction with Ig1 and Ig2 extra cellular domains. Protein Science. 2020;29:1679–1686. 10.1002/pro.3862
REFERENCES
- 1. Garlanda C, Dinarello CA, Mantovani A. The interleukin‐1 family: Back to the future. Immunity. 2013;39:1003–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vigers GP, Anderson LJ, Caffes P, Brandhuber BJ. Crystal structure of the type‐i interleukin‐1 receptor complexed with interleukin‐1beta. Nature. 1997;386:190–194. [DOI] [PubMed] [Google Scholar]
- 3. Schreuder H, Tardif C, Trump‐Kallmeyer S, et al. A new cytokine‐receptor binding mode revealed by the crystal structure of the IL‐1 receptor with an antagonist. Nature. 1997;386:194–200. [DOI] [PubMed] [Google Scholar]
- 4. Wang D, Zhang S, Li L, Liu X, Mei K, Wang X. Structural insights into the assembly and activation of IL‐1beta with its receptors. Nat Immunol. 2010;11:905–911. [DOI] [PubMed] [Google Scholar]
- 5. Liu X, Hammel M, He Y, et al. Structural insights into the interaction of IL‐33 with its receptors. Proc Natl Acad Sci USA. 2013;110:14918–14923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gresnigt MS, van de Veerdonk FL. Biology of IL‐36 cytokines and their role in disease. Semin Immunol. 2013;25:458–465. [DOI] [PubMed] [Google Scholar]
- 7. Frey S, Derer A, Messbacher ME, et al. The novel cytokine interleukin‐36alpha is expressed in psoriatic and rheumatoid arthritis synovium. Ann Rheum Dis. 2013;72:1569–1574. [DOI] [PubMed] [Google Scholar]
- 8. Gresnigt MS, Rosler B, Jacobs CW, et al. The IL‐36 receptor pathway regulates aspergillus fumigatus‐induced th1 and th17 responses. Eur J Immunol. 2013;43:416–426. [DOI] [PubMed] [Google Scholar]
- 9. Marrakchi S, Guigue P, Renshaw BR, et al. Interleukin‐36‐receptor antagonist deficiency and generalized pustular psoriasis. N Engl J Med. 2011;365:620–628. [DOI] [PubMed] [Google Scholar]
- 10. Ramadas RA, Ewart SL, Iwakura Y, Medoff BD, LeVine AM. IL‐36alpha exerts pro‐inflammatory effects in the lungs of mice. PLoS One. 2012;7:e45784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tortola L, Rosenwald E, Abel B, et al. Psoriasiform dermatitis is driven by IL‐36‐mediated dc‐keratinocyte crosstalk. J Clin Invest. 2012;122:3965–3976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vigne S, Palmer G, Martin P, et al. IL‐36 signaling amplifies Th1 responses by enhancing proliferation and Th1 polarization of naive Cd4+ T cells. Blood. 2012;120:3478–3487. [DOI] [PubMed] [Google Scholar]
- 13. Carrier Y, Ma HL, Ramon HE, et al. Inter‐regulation of Th17 cytokines and the IL‐36 cytokines in vitro and in vivo: Implications in psoriasis pathogenesis. J Invest Dermatol. 2011;131:2428–2437. [DOI] [PubMed] [Google Scholar]
- 14. Ganesan R, Raymond EL, Mennerich D, et al. Generation and functional characterization of anti‐human and anti‐mouse IL‐36R antagonist monoclonal antibodies. MAbs. 2017;9:1143–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vigers GP, Dripps DJ, Edwards CK III, Brandhuber BJ. X‐ray crystal structure of a small antagonist peptide bound to interleukin‐1 receptor type 1. J Biol Chem. 2000;275:36927–36933. [DOI] [PubMed] [Google Scholar]
- 16. Thomas C, Bazan JF, Garcia KC. Structure of the activating IL‐1 receptor signaling complex. Nat Struct Mol Biol. 2012;19:455–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shindyalov IN, Bourne PE. Protein structure alignment by incremental combinatorial extension (CE) of the optimal path. Protein Eng. 1998;11:739–747. [DOI] [PubMed] [Google Scholar]
- 18. Lawrence MC, Colman PM. Shape complementarity at protein/protein interfaces. J Mol Biol. 1993;234:946–950. [DOI] [PubMed] [Google Scholar]
- 19. Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. [DOI] [PubMed] [Google Scholar]
- 20. Gunther S, Sundberg EJ. Molecular determinants of agonist and antagonist signaling through the IL‐36 receptor. J Immunol. 2014;193:921–930. [DOI] [PubMed] [Google Scholar]
- 21. Kabsch W. XDS. Acta Crystallogr D. 2010;66:125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Evans PR. An introduction to data reduction: Space‐group determination, scaling and intensity statistics. Acta Crystallogr D. 2011;67:282–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Evans PR, Murshudov GN. How good are my data and what is the resolution? Acta Crystallogr D. 2013;69:1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. French GS, Wilson KS. On the treatment of negative intensity observations. Acta Crystallogr A. 1978;34:517–525. [Google Scholar]
- 25. Vonrhein C, Flensburg C, Keller P, et al. Data processing and analysis with the autoproc toolbox. Acta Crystallogr D. 2011;67:293–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McCoy AJ, Grosse‐Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Cryst. 2007;40:658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Adams PD, Afonine PV, Bunkoczi G, et al. Phenix: A comprehensive python‐based system for macromolecular structure solution. Acta Crystallogr D. 2010;66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of coot. Acta Crystallogr D. 2010;66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Terwilliger TC, Grosse‐Kunstleve RW, Afonine PV, et al. Iterative model building, structure refinement and density modification with the phenix autobuild wizard. Acta Crystallogr D. 2008;64:61–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Afonine PV, Grosse‐Kunstleve RW, Echols N, et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr D. 2012;68:352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Davis IW, Murray LW, Richardson JS, Richardson DC. Molprobity: Structure validation and all‐atom contact analysis for nucleic acids and their complexes. Nucleic Acids Res. 2004;32:W615–W619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Krissinel E, Henrick K. Secondary‐structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr D. 2004;60:2256–2268. [DOI] [PubMed] [Google Scholar]
- 33.The PyMOL molecular graphics system. Version 1.6.0.0. Schrödinger, LLC; 2014. https://pymol.org/2/support.html?#citing
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Stereo view of electron density in the interface between IL‐36R and BI 655130. The 2fo‐fc electron density (blue mesh) is contoured at 1.8σ. IL‐36R is colored orange, the heavy chain of BI 655130 is colored green, and the light chain of BI 655130 is colored cyan. The view is centered on Tyr101 of the heavy chain.