

Abstract
Memory disruption in mild cognitive impairment (MCI) and Alzheimer's disease (AD) is poorly understood, particularly at early stages preceding neurodegeneration. In mouse models of AD, there are disruptions to sharp wave ripples (SWRs), hippocampal population events with a critical role in memory consolidation. However, the microcircuitry underlying these disruptions is under-explored. We tested whether a selective reduction in parvalbumin-expressing (PV) inhibitory interneuron activity underlies hyperactivity and SWR disruption. We employed the 5xFAD model of familial AD crossed with mouse lines labeling excitatory pyramidal cells (PCs) and inhibitory PV cells. We observed a 33% increase in frequency, 58% increase in amplitude, and 8% decrease in duration of SWRs in ex vivo slices from male and female three-month 5xFAD mice versus littermate controls. 5xFAD mice of the same age were impaired in a hippocampal-dependent memory task. Concurrent with SWR recordings, we performed calcium imaging, cell-attached, and whole-cell recordings of PC and PV cells within the CA1 region. PCs in 5xFAD mice participated in enlarged ensembles, with superficial PCs (sPCs) having a higher probability of spiking during SWRs. Both deep PCs (dPCs) and sPCs displayed an increased synaptic E/I ratio, suggesting a disinhibitory mechanism. In contrast, we observed a 46% spike rate reduction during SWRs in PV basket cells (PVBCs), while PV bistratified and axo-axonic cells were unimpaired. Excitatory synaptic drive to PVBCs was selectively reduced by 50%, resulting in decreased E/I ratio. Considering prior studies of intrinsic PV cell dysfunction in AD, these findings suggest alterations to the PC-PVBC microcircuit also contribute to impairment.
SIGNIFICANCE STATEMENT We demonstrate that a specific subtype of inhibitory neuron, parvalbumin-expressing (PV) basket cells, have selectively reduced activity in a model of Alzheimer's disease (AD) during activity critical for the consolidation of memory. These results identify a potential cellular target for therapeutic intervention to restore aberrant network activity in early amyloid pathology. While PV cells have previously been identified as a potential therapeutic target, this study for the first time recognizes that other PV neuronal subtypes, including bistratified and axo-axonic cells, are spared. These experiments are the first to record synaptic and spiking activity during sharp wave ripple (SWR) events in early amyloid pathology and reveal that a selective decrease in excitatory synaptic drive to PV basket cells (PVBCs) likely underlies reduced function.
Keywords: Alzheimer's, hippocampus, hyperactivity, interneuron, parvalbumin, sharp wave ripple
Introduction
Alzheimer's disease (AD) is the leading cause of dementia, and a growing public health crisis as worldwide life expectancy increases (Mattson, 2004). AD is characterized by learning and memory deficits, the pathologic accumulation of amyloid β (Aβ) plaques and neurofibrillary tangles, and synaptic and neuronal degeneration (Serrano-Pozo et al., 2011). The cause of memory disruption in the disease is poorly understood, particularly at early ages before widespread neurodegeneration. The hippocampus, a region particularly important for the encoding and consolidation of spatial memory, is one of the first regions impaired in AD (Braak and Braak, 1991). Hyperactivity within the hippocampus is observed in mouse models of AD (Palop et al., 2007; Busche et al., 2008, 2012; Palop and Mucke, 2010), as well as in clinical populations, where seizures are an increasingly recognized co-morbidity of AD (Hauser et al., 1986; Amatniek et al., 2006; Palop and Mucke, 2009). While it is well appreciated that Aβ impairs the synaptic function of excitatory pyramidal cells (PCs) in later disease progression (Kamenetz et al., 2003; Shankar et al., 2008; Pozueta et al., 2013), there is growing evidence of early deficits to inhibitory GABAergic cells (Li et al., 2016), potentially explaining this shift to hyperactivity through disinhibition. In particular, several functional impairments are observed in inhibitory parvalbumin-expressing (PV) fast-spiking interneurons (Verret et al., 2012; Mahar et al., 2016; Yang et al., 2018; Hijazi et al., 2019). However, there are at least three distinct PV cell subtypes within the CA1 region of hippocampus with varying anatomic connections and function (Varga et al., 2014), and the separate impact of AD pathology on these subtypes is unknown.
PV cells play a critical role in hippocampal sharp wave ripples (SWRs; Ellender et al., 2010; Schlingloff et al., 2014; Ognjanovski et al., 2017), spontaneous neuronal population events characterized by a low frequency SW (1–30 Hz) and a high-frequency ripple (120–250 Hz; Buzsáki, 1986, 2015; Colgin, 2016). SWRs principally originate in the CA3 region and propagate to CA1 along the Schaffer collaterals, occurring in all mammalian species studied to date (Buzsáki et al., 2013). Even following decortication in brain slices, SWRs spontaneously arise in hippocampus (Buzsáki et al., 1983). SWRs have been extensively studied in large part due to their proposed role in memory consolidation (Wilson and McNaughton, 1994; Kudrimoti et al., 1999; O'Neill et al., 2008; Karlsson and Frank, 2009). Sequences of place cells activated during spatial learning replay in temporally compressed neuronal ensembles within SWRs during rest (Nádasdy et al., 1999; Lee and Wilson, 2002). Online interruption of SWRs through both electrical (Girardeau et al., 2009; Ego-Stengel and Wilson, 2010) and optogenetic stimulation (Ven et al., 2016; Roux et al., 2017) leads to learning and memory deficits, demonstrating their critical role in memory consolidation. Notably, in several mouse models of AD, SWRs are disrupted (Gillespie et al., 2016; Iaccarino et al., 2016; Nicole et al., 2016; Xiao et al., 2017; Hollnagel et al., 2019; Jones et al., 2019; Jura et al., 2019). However, the microcircuitry underlying these disruptions has yet to be explored in detail. In this study, we employed the 5xFAD mouse model of familial AD crossed with mouse lines that selectively fluoresce in excitatory PCs and inhibitory PV cells. We performed patch clamp recordings of deep PCs (dPCs) and superficial PCs (sPCs) and three distinct PV cell subtypes to record the spiking activity and synaptic input during SWR events in ex vivo slices. Our findings support the hypothesis that a preferential reduction in synaptic input and activity of PV basket cells (PVBCs) underlies downstream network alterations and suggest that long-term alterations to PC-PVBC microcircuitry contribute to dysfunction in early amyloid pathology.
Materials and Methods
Experimental animals
To record the activity of excitatory PCs and inhibitory PV cells in amyloid pathology, we employed a combined breeding strategy of transgenic and targeted knock-in mice. Transgenic 5xFAD mice (RRID:MMRRC_034840-JAX; Oakley et al., 2006) were back-crossed for over five generations to the C57BL/6J (RRID:IMSR_JAX:000664) background, which was common to all other strains used. To target the calcium activity of PCs under confocal microscopy, transgenic homozygous Thy1-GCaMP6f-GP5.5 (RRID:IMSR_JAX:024276; Dana et al., 2014) were crossed with hemizygous 5xFAD mice to yield litters with both 5xFAD/+;Thy1-GCaMP6f/+ and Thy1-GCaMP6f/+ littermate controls. PV cells were identified by crossing double homozygous knock-in PVCre/PVCre;tdTom/tdTom (RRID:IMSR_JAX:008069, RRID:IMSR_JAX:007914; Hippenmeyer et al., 2005; Madisen et al., 2010) with hemizygous 5xFAD mice to yield litters with both 5xFAD/+;PVCre/+;tdTom/+ and PVCre/+;tdTom/+ littermate controls. In a subset of experiments, the reporter lines were crossed, yielding quadruple transgenic cohorts of 5xFAD/+;Thy1-GCaMP6f/+;PVCre/+;tdTom/+ and Thy1-GCaMP6f/+;PVCre/+;tdTom/+ littermate controls. The initial intention was to use a consistent cohort of quadruple transgenic mice for all experiments, yet the breeding strategy proved inhibitive for the number of experiments, thus, patch clamp data were pooled across reporter genotype (Table 1). 5xFAD genotype was assessed at post-natal day 7 by tail biopsy via automated genotyping services (Transnetyx). For all experiments, experimenters were blind to 5xFAD genotype until after data collection and analysis were fully complete. Mice were weaned at P21 and group housed in cages with three to five separated by sex. As a model of early amyloid pathology before neuronal or synaptic loss (Oakley et al., 2006), two experimental cohorts were chosen (each including both males and females) at one and three months of age. Mice were kept on a standard 12/12 h light/dark cycle, food and water were provided ad libitum, with all experimental procedures performed in accordance with the guidelines of the Georgetown University Animal Care and Use Committee.
Table 1.
Mouse cohorts used in study
| Control 1 month | 5xFAD 1 month | Control 3 months | 5xFAD 3 months | Control 1 month | 5xFAD 1 month | Control 3 months | 5xFAD 3 months | |
|---|---|---|---|---|---|---|---|---|
| nmice | 10 (6f) | 10 (4f) | 27 (17f) | 29 (12f) | 10 (2f) | 10 (6f) | 15 (7f) | 12 (8f) |
| Age (postnatal days) | 40.2 ± 4.4 | 40.4 ± 4.6 | 95.8 ± 6.0 | 94.5 ± 5.8 | 42.0 ± 5.2 | 43.8 ± 4.2 | 91.8 ± 5.2 | 92.5 ± 5.4 |
| nslice (range 2–8) | 5.6 ± 0.8 | 4.0 ± 1.8 | 3.9 ± 1.3 | 3.2 ± 1.4 | ||||
| % Slices with SWRs | 93.8 ± 8.1% | 70.8 ± 22.0% | 72.4 ± 19.8% | 66.3 ± 26.1% | ||||
| Thy1-GCaMP6f(Figs. 3–5) | PVCre-tdTom(Figs. 6–8) | Thy1-GCaMP6f; PVCre-tdTom(Figs. 4–8) | ||||||
| Control | 5xFAD | Control | 5xFAD | Control | 5xFAD | |||
| nmice | 10 (5f) | 11 (5f) | 13 (8f) | 11 (5f) | 4 (4f) | 7 (2f) | ||
| Age (postnatal days) | 95.0 ± 7.4 | 93.4 ± 3.2 | 96.8 ± 5.7 | 97.4 ± 6.0 | 94.8 ± 3.2 | 91.6 ± 2.1 | ||
| nslice (range 2–8) | 3.9 ± 1.1 | 2.8 ± 1.3 | 3.8 ± 1.5 | 4.9 ± 2.1 | 4.5 ± 1.3 | 3.6 ± 1.8 | ||
| % Slices with SWRs | 75.5 ± 20.2% | 67.9 ± 26.2% | 67.4 ± 20.5% | 68.2 ± 25.3% | 80.8 ± 16.4% | 60.7 ± 30.5% | ||
| ncell-attached | 39 (PC) | 39 (PC) | 13 (PV) | 18 (PV) | 11 (PV) | 5 (PC); 14 (PV) | ||
| nwhole-cell | 21 (PC) | 15 (PC) | 11 (PV) | 16 (PV) | 10 (PV) | 1 (PC); 13 (PV) | ||
Top, Electrophysiology/imaging cohorts were distinct from behavioral cohorts. For both experiments, mice were studied at an age of one and three months. Bottom, The three-month electrophysiology/imaging cohort consisted of three subcohorts with different reporter genes.
Amyloid staining
Mice were anesthetized with isoflurane, transcardially perfused in iced (0°C) PBS and fixed for 48 h in paraformaldehyde (PFA) at 4°C. Brains were then given four 15-min PBS washes, and sliced horizontally at 100-μm thickness with a Vibratome Series 1000. In free floating slices, antigen retrieval was performed for 20 min in a steamer with citrate buffer containing 10 mm Na3C6H5O7, 0.05% Tween 20, and pH adjusted to 6.0 with HCl. Slices were cooled to room temperature in PBS, then permeabilized for 30 min with 0.5% Triton X-100 in PBS, given two more PBS rinses, blocked for 2 h with 10% normal goat serum (NGS) and 5% bovine serum albumin (BSA) in PBS, and incubated overnight at 4°C with the primary monoclonal mouse antibody MOAB-2 against β amyloid (1:500; Abcam catalog #ab126649; Youmans et al., 2012), 0.1% Tween 20, 1% NGS, 1% BSA in PBS. The following day, slices were given four 15-min PBS washes and incubated in secondary containing Alexa Fluor 647-conjugated goat anti-mouse IgG (1:500; Jackson ImmunoResearch catalog #115-605-003, RRID:AB_2338902), Thioflavin-S (1:2000; Sigma-Aldrich catalog #T1892), and 1% NGS in PBS for 2 h at room temperature. Slices were given three more 15-min PBS washes, rinsed in ddH20, and mounted on slides with Vectashield antifade mounting medium with DAPI (Vector Laboratories catalog #H-1200, RRID:AB_2336790).
Behavioral testing
Hippocampal-dependent learning and memory deficits were assessed by the Barnes maze (Barnes, 1979), with some modifications. The Barnes maze was performed on a white plastic apparatus (San Diego Instruments) 0.914 m in diameter, with overhead bright illumination (286 lx) serving as the aversive stimulus. The target hole was randomly selected, with four distal visual clues present for visuospatial learning. The training phase consisted of four 180-s trials per day for four consecutive days. The probe trial, in which the target hole was inaccessible, was conducted on the fifth consecutive day, consisting of one 90 s trial. Mice were tracked using the ANY-maze tracking system, which was used for distance and speed measurements. The primary measures of latency and number of entries to target hole were hand-scored for increased reliability. Anxiety-like behavior was tested on the elevated plus maze (Pellow et al., 1985). The mice could explore the maze for 5 min, in which time the number of entries and fraction of time in the open arms were assessed by the ANY-maze tracking system. All tests were conducted in the light cycle, at consistent times of the day for each mouse, in an enclosed behavior room with 50-dB ambient sound and 23-lx ambient illumination. Males and females were run on the same day, but in separate groups. Cohorts were not balanced by both sex and genotype, but as much as possible testing order was prepared so that control and 5xFAD mice were alternated.
Acute slice preparation
Brain slices were prepared using NMDG and HEPES-buffered artificial CSF (aCSF) following a protective recovery protocol (Ting et al., 2014, 2018). Briefly, mice were anesthetized with isoflurane, transcardially perfused, dissected, and sliced in iced (0°C) NMDG-aCSF containing the following: 92 mm NMDG, 2.5 mm KCl, 1.25 mm NaH2PO4, 30 mm NaHCO3, 20 mm HEPES, 25 mm glucose, 10 mm sucrose, 5 mm ascorbic acid, 2 mm thiourea, 3 mm sodium pyruvate, 5 mm N-acetyl-L-cysteine, 10 mm MgSO4, and 0.5 mm CaCl2, pH to 7.3–7.4 with HCl (300–310 Osm). All common reagents were obtained from Thermo Fisher Scientific. The brains were sliced horizontally at 500-μm thickness with a Vibratome Series 3000 to preserve hippocampal microcircuitry and spontaneous SWRs. Three to four slices were typically obtained per brain, which were bisected so that six to eight hemislices in total were studied per animal. Slices spanned the dorsal-ventral axis, although were primarily medial, as only horizontal slices with intact DG, CA3, and CA1 were retained, ranging from bregma 2–4 mm. The slices were transferred together to heated (33°C) NMDG-aCSF, in which Na+ was gradually introduced along an increasing concentration gradient every 5 min before transferring to room temperature HEPES-aCSF containing the following: 92 mm NaCl, 2.5 mm KCl, 1.25 mm NaH2PO4, 30 mm NaHCO3, 20 mm HEPES, 25 mm glucose, 5 mm ascorbic acid, 2 mm thiourea, 3 mm sodium pyruvate, 5 mm N-acetyl-L-cysteine, 2 mm MgSO4, and 2 mm CaCl2, pH 7.3–7.4 with NaOH (300–310 Osm). Slices recovered for 4 h in a custom-built 150 ml incubation chamber with circulating oxygenated HEPES-aCSF.
Slice electrophysiology
Slices were transferred to a Siskiyou PC-H perfusion chamber with a custom-built suspended Lycra thread grid to allow perfusion below and above slice, modeled after (Hájos et al., 2009). Submerged slices were anchored with Warner Instruments slice anchors so that they were sandwiched between two grids, and perfused at a rate of 5 ml/min with heated (30°C) oxygenated aCSF containing the following: 124 mm NaCl, 3.5 mm KCl, 1.2 mm NaH2PO4, 26 mm NaHCO3, 10 mm glucose, 1 mm MgCl2, and 2 mm CaCl2, pH 7.3–7.4 (300–310 Osm). Recordings were conducted with a Multiclamp 700B amplifier (Molecular Devices), digitized at 20 kHz and low-pass Bessel-filtered at 2 kHz with a personal computer running Clampex 11 and a DigiData 1440 (Molecular Devices). Two concurrent channels were captured: the local field potential (LFP) was recorded with 0.5–1 MΩ borosilicate pipettes filled with aCSF, paired with 3- to 5-MΩ borosilicate pipettes for cellular recordings. Cell-attached recordings were performed with aCSF + 5 μm Alexa Fluor 488 nm (Invitrogen catalog #A-10440) or 594 nm (Invitrogen catalog #A-10442) for pipette localization under confocal microscopy. The selected concentration of Alexa Fluor dye was below reported alterations to synaptic transmission (Maroteaux and Liu, 2016). Cell-attached recordings were followed with whole-cell recordings of the same cell with a new pipette filled with a cesium internal, containing the following: 120 mm CsMeSO3, 5 mm NaCl, 10 mm TEA•Cl, 10 mm HEPES, 1.1 mm EGTA, 4 mm QX314, 4 mm ATP•Na, and 0.3 mm GTP•Na, pH to 7.2 with CsOH (285 Osm). For pipette localization and post hoc morphologic reconstruction, 5 μm Alexa Fluor (either 488 or 594 nm) and 0.5% wt/vl biocytin were added to the internal solution on the day of experiment.
The LFP electrode was placed in CA1 on the border of stratum pyramidale (str. pyr.) and oriens, a location where both high amplitude SWs and ripples are simultaneously detectable. Consistent placement of the electrode was attempted in all slices at a depth of ∼20 µm. Recordings began 10 min after LFP electrode placement to allow slice to recover. If visually detectable SWRs were not observed, the slice was logged as non-SWR producing (Table 1) and discarded. A fluorescent cell was targeted for a loose (20- to 40-MΩ seal resistance) cell-attached recording of 3–5 min in duration. For Thy1-GCaMP6f slices, Ca2+ ensemble activity was recorded concurrently with a laser scanning confocal microscope system (Thor Imaging Systems Division) equipped with 488/561/642-nm lasers and green/red/far-red filters and dichroics mounted on an upright Eclipse FN1 microscope (Nikon Instruments). One thousand 512 × 512-pixel frames were captured at a sample rate of 7.5 Hz. A 40× water immersion objective was used, covering an imaging field of 350 × 350 μm, as a balance between maximizing the imaging field while providing sufficient magnification for patch clamp electrophysiology.
Following the cell-attached recording, the same cell was targeted with a new cesium internal electrode. Upon reaching 1-GΩ seal resistance, the membrane was broken by voltage pulse and quick negative pressure. Access resistance was monitored periodically and recordings with a change >20% were discarded. Putative EPSCs were measured in voltage-clamp at a holding voltage of −70 mV, and putative IPSCs in the same cell at 0 mV. Glutamatergic and GABAergic events were not pharmacologically isolated, as the primary goal was to correlate synaptic activity with spontaneous SWRs, which would be affected by drug administration. The reversal potentials of putative EPSCs and IPSCs matched closely with the expected glutamate and GABA reversal potentials of 0 and −70 mV. This was determined by holding the cell from +20 to −100 mV in 10-mV voltage steps and monitoring the current polarity inversion. Each voltage-clamp recording ranged from 1 to 3 min. Following PV cell voltage-clamp recordings, the cell was then switched to current-clamp, with current injection to offset the leak current and maintain a membrane potential of −70 mV. Thirty-five hyperpolarizing/depolarizing steps of 5-pA increments were delivered to fill the cell with biocytin. A total duration of at least 15 min of whole-cell configuration was maintained, after which an outside-out patch was formed by slowly withdrawing the pipette. Recordings were attempted at −70 and 0 mV for all PCs, while only a subset of PV cells were clamped at 0 mV. The PV cell protocol was initially designed to minimize cell disruption and ensure proper biocytin-filling; however, the recording protocol was revised during the experiment to also record inhibitory input.
Post hoc staining and microscopy
Slices with biocytin-filled PV cells were returned to the HEPES-aCSF incubation chamber for the remainder of the day, then fixed overnight in 4% PFA, 4% glucose in PBS at 4°C. Slices then received four 15-min PBS washes, 2 h of permeabilization with 0.5% Triton X-100 in PBS, two PBS rinses, 3 h in fluorescein-avidin (1:500; Vector Laboratories catalog #A-2001, RRID:AB_2336455) in PBS, and four additional 15-min PBS washes. Free floating slices were imaged with a laser scanning confocal microscope system (Thor Imaging Systems Division). Under 20× magnification, z-stacks were obtained covering the span of visible cellular processes (40–80 µm in 1-µm steps), for both green (biocytin) and red (PVCre-tdTom) channels. In a subset of 14 slices [nslice = 5 control (CT), 9 5xFAD from nmice = 4 CT, 5 5xFAD], an additional round of immuno-staining was performed for Ankyrin G, which labels the axon initial segment (AIS) of PCs. A separate subset of Thy1-GCaMP6f slices (nslice = 2 CT, 2 5xFAD each from a separate animal) were stained for calbindin (CB), with differential expression between sPCs and dPCs. Antigen retrieval was performed on the 500-μm slices for 20 min in a steamer with citrate buffer. Slices were cooled to room temperature for 20 min in PBS, then blocked overnight with 10% NGS at 4°C. The following day the slices were incubated with a primary monoclonal mouse antibody against Ankyrin G (1:100; Thermo Fisher Scientific catalog # 33-8800, RRID:AB_2533145) or CB D-28k (1:1000; Swant catalog #300, RRID:AB_10000347), 0.1% Tween 20, 1% NGS in PBS. After 48 h of primary incubation at 4°C, slices were given four 15-min PBS washes and incubated in secondary containing Alexa Fluor 647-conjugated goat anti-mouse IgG (1:500; Jackson ImmunoResearch catalog #115-605-003, RRID:AB_2338902) and 1% NGS in PBS for 3 h at room temperature. Slices were given four more 15-min PBS washes, rinsed in ddH20, and mounted on slides between silicone isolators with Vectashield mounting medium.
Pre-processing of electrophysiology data and event detection
Pre-processing of files was conducted in Clampfit 11 (pClamp, Molecular Devices). Files for calcium imaging experiments were trimmed around the confocal laser trigger signal for alignment of SWR and calcium transients. Spikes were detected in cell-attached recordings with a 3- or 1.75-ms template search for PCs or PV cells, respectively. A threshold of 6–8 was used (Clements and Bekkers, 1997) and false negatives minimized (confirmed by manual inspection). False positives were removed by plotting peak versus anti-peak amplitude to segregate noise from true spikes. Bursts were detected with the built-in burst analysis, defined as three or more successive spikes, each within 60 or 40 ms (intraburst interval) for PCs or PV cells, respectively. Spike start, peak, and end times, burst start and end times, number of events in burst, and intraburst interval were exported from Clampfit for coincidence detection in MATLAB.
Whole-cell recordings of PSC signals were low-pass filtered below 1000 Hz with a zero-phase Gaussian finite impulse response (FIR) filter. PSCs were detected with template searches. Multiple template categories (3–4) of varying duration (ranging from 3 to 20 ms) were used to improve detection for overlapping PSCs often seen around SWRs. Shorter duration templates used increasingly higher thresholds, between 5 and 8, to minimize false negatives. Parameters were kept constant for each cell type. PSC results including start, peak, and end times, amplitude, rise tau and decay tau were exported from Clampfit and processed in Microsoft Excel to remove duplicate events with identical peak times. Limits were set on rise and decay tau to remove false positives due to noise. Accepted EPSCs fell within a range of 0.05- to 5-ms rise tau and 1- to 50-ms decay tau, while IPSCs fell within a range of 0.1- to 10-ms rise tau and 3- to 100-ms decay tau.
Local field analysis and SWR detection
All analysis for coincidence detection between the LFP and cellular events was conducted with custom-built MATLAB functions. Raw traces were imported using the abfload protocol (https://github.com/fcollman/abfload). All applied filters were FIR Gaussian filter with constant and corrected phase delays. A wide bandpass (1–1000 Hz) was first applied to remove both low-frequency DC drift and high-frequency instrument noise. The detection of SWR events was based on prior approaches (Siapas and Wilson, 1998; Csicsvari et al., 1999; Eschenko et al., 2008), with refinements to minimize false positives and permit additional analyses. The LFP was filtered in both the SW (1–30 Hz) and ripple (120–220 Hz) ranges, and the root mean square (RMS) was computed every 5 ms in a 10-ms sliding window. The threshold for peak detection was set to four SD above the baseline (lower 0.95 quantile) RMS mean. Event start and end times were set at 2 SD crossings. SWR events were defined as the intersection of concurrent SW and ripple events. The duration of SWR events was determined from the union of concurrent SW and ripple events. The peak of the SWR event was defined as the peak of the SW-RMS signal, and the amplitude as the difference between the peak and baseline values of the SW signal. The power of the SW and ripple were determined by the bandpower MATLAB function of the relevant filtered signal, which computes an approximation of the integral of the power spectral density between the start and end times of the SWR event. Additional filters were applied in the slow γ (20–50 Hz) and fast/pathologic ripple (250–500 Hz) ranges, and the power computed on a SWR-event basis, as the power of these SWR-nested oscillations has been implicated in memory performance (Carr et al., 2012) and epileptogenesis (Foffani et al., 2007), respectively.
To visualize the spectral components of the LFP as a function of time, spectral analysis was performed for the duration of the recording as well as per SWR event via a short-time Fourier transform (STFT) between 1 and 500 Hz. To better observe deviations from baseline power, the z-score for each 1-Hz frequency band was calculated. A fast Fourier transform (FFT) was also computed for a 200-ms window centered around each SWR peak and averaged across all events as an additional visualization of spectral power. The determination of the phase of slow γ and ripple oscillations during SWRs was based on the analysis of Varga et al. (2012). Within a 200-ms window centered around the SWR peak, the extrema of the filtered signal of interest were identified, and a piece-wise linear function fit with values from 0° to 180° between a maximum and minimum, 180–360° between a minimum and maximum, and then resetting to 0°. The number of cycles within the duration of the SWR event was recorded, from which the peak frequency was calculated as ncycle/SWRduration.
Pre-processing of Ca2+ imaging data
Raw time series were converted to the change in fluorescence normalized to baseline (ΔF/F) with custom-built ImageJ (FIJI) macros, in which batches of raw TIF images were imported with the Bio-Formats plugin (Open Microscopy Environment) and saved as TIF stacks. The TIF stacks were corrected for photograph-bleaching via two iterations of the built-in Correct-Bleach plugin. Photograph-bleaching was modeled as a sum of two exponentials, a fast 5-s decay and a slower decay over the duration of the recording (133 s). To assist in region of interest (ROI) placement, a semi-automated algorithm was applied to highlight regions of highest fluorescence change. For each TIF stack, the squared coefficient of variation (SCV) image was calculated, defined as the variance divided by the average squared for each pixel, or equivalently:
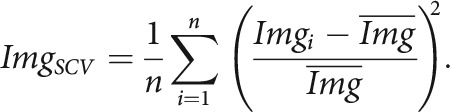 |
Circular ROIs were manually drawn over all identified cells in the SCV image and confirmed with the time series to encompass active cells. Notably, in this way only cells with variable fluorescence were identified, and static highly fluorescent Ca2+-loaded cells were excluded. The ΔF/F for each cell was calculated by first subtracting the background fluorescence Fb, defined as the lowest intensity pixel across the entire time series. F0 for each ROI was defined as the average of the ten images with the lowest intensity. The ΔF/F was thus calculated as:
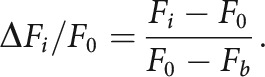 |
These values were exported from ImageJ, along with a separately calculated timing file for each image, computed from a threshold search in Clampfit of the confocal laser trigger channel.
Calcium transient detection and SWR coincidence analysis
Automatic detection of calcium transients was performed by first correcting for slow changes in fluorescence, either from remaining photograph-bleaching or gradual drift of the imaging plane. A smoothed moving average, calculated with robust locally-weighted regression (Cleveland, 1979) with a window 25% of the file duration (33 s), was subtracted from each ΔF/F trace. The baseline-corrected ΔF/F traces were interpolated from the raw sampling rate of 7.5 Hz to 2 kHz, and the LFP downsampled from 20 to 2 kHz for coincidence detection. Automatic threshold detection for each cell was set at 4 SD above baseline, with the start and end times for each event set at 2 SD. The baseline of each cell was determined by an iterative algorithm of Gaussian fitting to the histogram of all data points, which for most cells was a skewed one-tailed distribution with a large baseline peak at zero and a long positive tail representing transient events. For uncommon cells without a skewed distribution (kurtosis < 0), a single Gaussian fit was applied to the entire histogram, providing an estimate of the baseline mean and SD. However, for most cells, two iterations were employed, with the first a double-Gaussian fit to the entire histogram. The higher amplitude Gaussian SD was then used to constrain the upper limit on a second iteration single-Gaussian refinement fit of only the baseline. Special logic was necessary in rare situations. For excessively skewed distributions (high ΔF/F, kurtosis > 5) the double-Gaussian fit was constrained to the lower 0.95 quantile of data. For extremely active cells, because of the slow decay kinetics of GCaMP, the signal peak rivaled or exceeded the baseline peak in amplitude. For these cells, the more negative rather than the higher amplitude peak was used as an estimate of the baseline. In the situation where two peaks were not clearly differentiated (peak separation < 0.1 ΔF/F), the Gaussian means and SDs were averaged together to arrive at an estimate for constraining the second iteration.
Once the baseline mean, SD, and thresholds were determined for each cell, events were detected on a cell-by-cell basis, and characteristics calculated including start, peak, and end times, inter-event interval, duration, amplitude, and frequency. The interpolated calcium traces were trimmed and aligned with the down-sampled LFP trace. Each calcium transient was classified as SWR-coincident if there was any overlap between the start and end of the calcium transient and the start and end of the SWR, otherwise it was classified as spontaneous. Cellular participation during SWRs was assessed by constructing a simplified event matrix with SWRs in one dimension, and cells in the other, with a zero or one if the cell reached threshold during the SWR. For fields with five or more active cells, the ensemble diversity was assessed by calculating the pairwise Jaccard Similarity index between SWR events, ranging from a value of zero if two SWR events had no cells in common, to a value of one if all cellular participants were identical, modeled after the analysis of Miyawaki et al. (2014). The cell-cell pairwise index was also calculated, ranging from zero if two cells never participated in the same SWR events, to one if they participated in precisely the same SWRs. The cumulative distribution functions were determined for each recording by considering all off-diagonal values in one half of the symmetric similarity matrices.
Spike and SWR coincidence analysis
Each spike/burst was classified as SWR-coincident if there was any overlap between the start and end of the spike/burst and the start and end of the SWR, otherwise it was classified as spontaneous. To examine spike rate in more detail around SWR events, the peri-SWR spike probability was calculated by sorting all spikes that occurred within a 200-ms window centered around each SWR peak into 2-ms bins and normalizing to all SWR events. Spike-phase coupling was determined by identifying the previously calculated γ and ripple phases at the spike peak time. Spike phase times were only considered for further analysis if trough-peak amplitude exceeded four SD of the γ or ripple signal.
Postsynaptic current and SWR coincidence analysis
Each EPSC/IPSC was classified as a SWR-coincident EPSC/IPSC (swrEPSC/swrIPSC) if there was any overlap between the start and end of the event and a 100-ms window centered around each SWR peak,otherwise it was classified as a spontaneous EPSC/IPSC (sEPSC/sIPSC). The more conservative window to classify spontaneous events (compared with the calcium and spike analysis) better captured the buildup of EPSCs/IPSCs preceding SWRs (Schlingloff et al., 2014). As events overlap and complicate detection during SWRs, for quantification the total excitatory/inhibitory post-synaptic charge (EPSQ/IPSQ) was examined. The EPSQ/IPSQ was calculated by integrating the current in a 100 ms window centered around the SWR peak (swrEPSQ/swrIPSQ). To examine the distribution of sEPSCs/sIPSCs, the cumulative distribution function of all events was computed for each cell and averaged across all cells in a group. For a view of the temporal progression of synaptic input during SWRs, the EPSQ/IPSQ was also calculated by integrating a sliding 100-ms window every 2 ms in a 200-ms window centered around the SWR peak.
Experimental design and statistical analyses
Most experiments in this study have been presented in a case-control experimental design, in which data from 5xFAD mice are compared with littermate controls. With behavioral learning on the Barnes maze, we have employed a longitudinal repeated measures (RM) design. In analyzing the impact of factors on LFP activity in a large sample of ex vivo slices, we have taken a factorial design. All data were tested for normality and lognormality via Shapiro–Wilk tests. If all groups were normally distributed, they were analyzed with parametric tests (unpaired t test, n-way ANOVA, n-way RM ANOVA) and have been displayed on bar plots with error bars representing the mean ± SEM. If any group was not normally distributed, the data have been presented on box-whisker or violin plots, with lines indicating median and quartiles, and full error bars representing range. If all groups were lognormally distributed, the data were log-transformed (LT), the results of which were analyzed with parametric tests, with the log-means compared. For clarity, the original non-transformed data have been displayed in plots. If any group was neither normal nor lognormal, non-parametric statistical tests of rank were employed. Circular data (phase angles) were analyzed with a similar approach. Data were tested for non-uniformity via a Raleigh's test to determine whether they could be sampled from a von Mises (circular normal) distribution. If the Raleigh's test reached significance for all groups, means were compared with the Watson–Williams test, otherwise a circular-median test was performed.
The particular statistical tests used are listed in Results. Any values cited in the text are mean ± SEM. All post hoc multiple comparisons used the Šidák correction (ŠC). Within each plot all individual data points are presented. No data were excluded based on their values, but only for experimental reasons (e.g., no SWRs present, excessive slice movement, unstable patch clamp recording). The n is indicated in the text and figure legends and differs between experiment, either nmice, nslice, or ncell. Raw p values are displayed in plots; if less than a significance level of 0.05, they are bold. Data originating from male or female mice are presented as closed or open circles, respectively. Sex differences were examined for some endpoints, but in general, the experiments were insufficiently powered to determine sex differences. A power analysis of the principal experiments was performed based on preliminary data, guiding the number of animals/cells chosen.
GraphPad Prism 8 was used for all one- and two-sample statistical tests. Microsoft Excel and MATLAB 2019 were used for some simple calculations of mean, SD, SEM, ratio, and error propagation; n-way ANOVAs were performed in the Statistics and Machine Learning Toolbox of MATLAB 2019. Non-parametric factorial data were aligned and ranked with ARTool (Wobbrock et al., 2011), before running ANOVAs. Circular statistics were run in the Circular Statistics Toolbox for MATLAB (Berens, 2009).
Code accessibility
All code is open-source and available in public repositories, including versions under active development (GitHub) as well as archival copies used for this manuscript (Zenodo). MATLAB functions are available at https://github.com/acaccavano/SWR-Analysis (archival copy: doi:10.5281/zenodo.3625236). ImageJ (FIJI) macros are available at https://github.com/acaccavano/deltaFoF (archival copy: doi:10.5281/zenodo.3625130).
Results
Three-month 5xFAD mice deposit amyloid and display minor impairment in spatial memory
The 5xFAD model of familial AD is an aggressive although useful model of amyloid pathology, as it exhibits heavy amyloid accumulation in hippocampus and associated cortices, and is accompanied with memory impairment (Oakley et al., 2006). 5xFAD mice are unimpaired in performance on the T-maze at two months but become impaired by four to five months when amyloid burden is greater (Oakley et al., 2006). Interestingly, at three months, there is no evidence for neuronal or synaptic degeneration, while several synaptic markers begin to decline at four months and are significantly reduced from controls by nine months. We observed intracellular amyloid accumulation in the subiculum and CA1 region of hippocampus in one-month 5xFAD mice without the presence of extracellular plaques (nmice = 2 CT, 2 5xFAD), while in three-month 5xFAD mice, we observed multiple extracellular plaques in subiculum, with sparse plaques in CA1 (nmice = 2 CT, 2 5xFAD; Fig. 1A). Prior work indicates that amyloid burden continues to increase throughout the lifespan of 5xFAD mice (Oakley et al., 2006; Youmans et al., 2012).
Figure 1.

Three-month 5xFAD mice deposit amyloid and display minor impairment in spatial memory. A, Representative IHC in the CA1 – subiculum region in one month (top) and three months (bottom) for control (CT, left) and 5xFAD (right) mice. Red, MOAB-2, labeling both intraneuronal and extracellular Aβ, including unaggregated, oligomeric, and fibrillar Aβ42 and unaggregated Aβ40. Cyan, Thioflavin-S, labeling fibrils in extracellular plaques. Staining was repeated in two mice of each genotype and age. Scale bar = 100 μm. Inset scale bars = 50 μm. B, Cohorts of one-month (nmice = 10 CT, 10 5xFAD; top) and three-month (nmice = 15 CT, 12 5xFAD; bottom) mice, were trained on the Barnes maze, consisting of 4 d of training, each with four 180 s trials. The latency to find the target escape hole was averaged over the four trials. C, Latency to find target on the final 90-s probe trial on the fifth day for one-month (top) and three-months (bottom) cohorts. D, Representative heat map for three-month animals on the final probe trial. Warmer colors represent longer duration. Control mice spent more time at the target and surrounding region than 5xFAD mice. For all plots, individual data points represent an animal. Closed circles represent males, open circles females. Box-whisker plots represent non-normal data as Median and IQRs; p values indicated above brackets.
In separate one-month (nmice = 10 CT, 10 5xFAD) and three-month (nmice = 15 CT, 12 5xFAD; Table 1) cohorts, we examined the performance of 5xFAD mice and littermate controls on the Barnes maze. Both age cohorts learned the task, as seen by decreased latency to find escape hole over progressive days of training, and a significant effect of training day [one month: F(3,54) = 23.9, p = 5.7 × 10−10; three months: F(3,75) = 31.6, p = 2.7 × 10−13; two-way RM ANOVA, align-rank transformed (ART); Fig. 1B]. There were no differences observed in training between genotype for either the one-month cohort (F(1,18) = 0.096, p = 0.760) or the three-month cohort (F(1,25) = 1.086, p = 0.307), nor were there interactions of genotype × training day (one month: F(3,54) = 0.496, p = 0.687; three months: F(3,75) = 0.983, p = 0.405). On the probe day, one-month 5xFAD mice had similar latencies to controls (U = 46, p = 0.781; Mann–Whitney; Fig. 1C), while three-month 5xFAD mice had a longer latency to find the escape hole from 18.4 ± 7.6 to 37.4 ± 9.8 s (U = 48, p = 0.040; Mann–Whitney; Fig. 1C,D). The number of entries to the area of the escape hole, another commonly reported endpoint, was not significantly different for either cohort (one month: t(18) = 0.735, p = 0.472; three months: t(25) = 0.691, p = 0.496; unpaired t tests). 5xFAD mice displayed no obvious motor impairments, as the total distance traveled did not differ between genotype at either age [one month: 3.98 ± 0.91 m (CT), 3.77 ± 0.49 m (5xFAD), t(18) = 0.203, p = 0.842; three months: 3.98 ± 0.49 m (CT), 3.40 ± 0.49 m (5xFAD), t(25) = 0.816, p = 0.422; unpaired t tests), neither did the mean speed [one month: 4.4 ± 1.0 cm/s (CT), 4.2 ± 0.5 cm/s (5xFAD), t(18) = 0.186, p = 0.856; three months: 4.4 ± 0.6 cm/s (CT), 3.8 ± 0.5 cm/s (5xFAD), t(25) = 0.819, p = 0.420; unpaired t tests].
To test whether this observed memory impairment could be attributed to altered anxiety-like behavior, the cohorts were also tested on the Elevated Plus Maze. Neither age cohort showed a significant difference in fraction of time spent in open arms [one month: 15.4 ± 2.2% (CT), 15.1 ± 0.9% (5xFAD), t(18) = 0.129, p = 0.900; three months: 10.7 ± 1.0 (CT), 9.3 ± 1.5 (5xFAD), t(25) = 0.848, p = 0.404; unpaired t tests), nor in number of open arm entries (one month: 7.9 ± 0.6 (CT), 7.4 ± 0.6 (5xFAD), t(18) = 0.570, p = 0.576; three months: 9.2 ± 0.7 (CT), 7.5 ± 0.6 (5xFAD), t(25) = 1.863, p = 0.074; unpaired t tests]. Therefore, we concluded that three-month 5xFAD mice on our genetic background had a mild spatial memory impairment. As memory impairment has been widely reported in 5xFAD mice at later ages (Oakley et al., 2006; Ohno, 2009; Tohda et al., 2012), no further memory tasks were performed.
SWRs are increased in three-month 5xFAD mice
As spatial memory relies heavily on the activity of the hippocampus, and SWRs are critical for the consolidation of new memories (Buzsáki, 1986; Wilson and McNaughton, 1994), we next recorded spontaneous SWRs in hippocampal slices from control and 5xFAD mice (Fig. 2A,B). SWRs were recorded in the CA1 region in multiple slices for each animal and averaged in both a one-month cohort (nmice = 10 CT, 10 5xFAD) and a three-month cohort (nmice = 27 CT, 29 5xFAD; Table 1). While the electrode placement was kept as consistent as possible across recordings (see Materials and Methods), slight deviations in placement can result in large variance in the LFP. We attempted to control for this by recording from many slices (one month: nslice = 51 CT, 45 5xFAD; three months: nslice = 101 CT, 88 5xFAD) and then averaging across slices for each animal. In the one-month cohort, the SWR event frequency did not differ (t(18) = 0.946, p = 0.357; unpaired t test; Fig. 2C), nor were there changes for any other LFP endpoints (data not shown). However, in the three-month cohort, SWR event frequency was increased in 5xFAD mice versus controls from 0.94 ± 0.07 to 1.25 ± 0.08 Hz (t(54) = 2.89, p = 0.006; unpaired t test; Fig. 2C). We next verified whether there was an altered percentage of slices exhibiting SWRs in 5xFAD mice (Table 1), as this could underlie observed differences in event frequency. While a two-way ANOVA revealed a significant effect of 5xFAD genotype (F(1,72) = 6.51, p = 0.013) and age (F(1,72) = 5.21, p = 0.0254) on percentage of slices with SWRs, the only significant difference observed when corrected for multiple comparisons was between control slices at one and three months (p = 0.039, ŠC), while three-month slices did not differ between genotype (p = 0.759, ŠC). Therefore, the observed differences in three-month mice are not likely due to altered viability of the slices.
Figure 2.

SWRs are increased in three-month 5xFAD mice. A, SWRs were recorded in acute horizontal slices with the LFP electrode located in CA1. B, Example traces of three-month control (left) and 5xFAD (right) slices. First trace, LFP filtered between 1 and 1000 Hz, raster below indicates detected SWR events as overlap of SW and ripple events. Second trace, SW, filtered between 1 and 30 Hz, raster below indicates detected SW events. Third trace, Low γ, filtered between 20 and 50 Hz. Fourth trace, Ripple, filtered between 120 and 220 Hz, raster below indicates detected ripple events. Bottom, Z-scored time-frequency spectrogram from 1 to 250 Hz. C, Summary plot of SWR event frequency for 5xFAD and littermate controls for both one-month (nmice = 10 CT, 10 5xFAD; left) and three-month cohorts (nmice = 27 CT, 29 5xFAD; right). D, SWR event amplitude and duration. E, Peak ripple frequency and the number of ripple cycles within SWR duration. F, Example SWR events for traces displayed in B, with average z-scored time-frequency spectrogram of all events below. G, Average FFT of 200-ms window around all SWR events from characteristic subset of nslice = 7 CT, 7 5xFAD from nmice = 4 CT, 5 5xFAD. Shaded region represents SEM. H–J, SWR-locked oscillation power in the low γ (20–50 Hz; H), ripple (120–220 Hz; I), and fast ripple (250–500 Hz; J) ranges. Note that in H, one value was identified as an outlier by the ROUT method but was retained in analysis, as the data point appeared valid, and even with removal did not alter the observed increase (U = 193, p = 0.0015; Mann–Whitney). For all plots, individual data points represent the average of all slices recorded from an animal (nslice/animal in Table 1). Closed circles represent males, open circles females. Bar plots indicate normal data with mean ± SEM. Box-whisker plots represent non-normal data with Median and IQRs; p values indicated above brackets.
In addition to an increased event frequency, SWR amplitude was increased by 58 ± 17% in three-month 5xFAD mice (t(48.9) = 3.59, p = 0.0008; Welch's t test) and surprisingly were 3.7 ± 1.2 ms shorter in duration (t(54) = 3.18, p = 0.0025; unpaired t test; Fig. 2D). This decrease in duration was likely attributable to both increased peak ripple frequency (t(54) = 3.422 p = 0.0012; unpaired t test) and decreased number of complete ripple cycles (t(54) = 3.48, p = 0.016; unpaired t test; Fig. 2E), and is of interest as longer duration SWRs have been demonstrated to improve memory (Fernández-Ruiz et al., 2019). Spectral features of SWRs also differed between genotype (Fig. 2F–J). Low-γ (20–50 Hz) nested within SWRs, speculated to play a role in coordinating CA3 and CA1 memory replay (Carr et al., 2012), was increased in 5xFAD mice (U = 193, p = 0.0009, Mann–Whitney; Fig. 2H). While the ripple peak frequency was shifted (Fig. 2E,G), the total power within the ripple frequency range from 120 to 220 Hz was unchanged (U = 358, p = 0.591; Mann–Whitney; Fig. 2I). The power of fast or pathologic ripples (250–500 Hz) did not differ between 5xFAD and control slices (U = 379, p = 0.845; Mann–Whitney; Fig. 2J), suggesting the increased activity in 5xFAD slices was distinct from epileptiform activity (Foffani et al., 2007).
Spontaneous SWRs recordings were repeated in three separate three-month subcohorts with different reporter mouse lines for subsequent patch-clamp and Ca2+ imaging experiments (Table 1). SWR frequency increased in 5xFAD mice in both larger cohorts: PVCre/+;tdTom/+ mice (135 ± 18%, nmice = 13 CT, 11 5xFAD) and Thy1-GCaMP6f mice (156 ± 21%, nmice = 10 CT, 11 5xFAD). In the third smaller Thy1-GCaMP6f;PVCre/+;tdTom/+ cohort, there was no significant increase observed (93 ± 18%, nmice = 4 CT, 7 5xFAD). A two-way ANOVA for 5xFAD genotype and reporter line revealed only a significant effect of 5xFAD genotype (F(1,50) = 5.41, p = 0.024) and not reporter line (F(2,50) = 1.50, p = 0.233) nor interaction term (F(2,50) = 2.06, p = 0.138). The data for the three subcohorts were therefore pooled into the results presented in Figure 2, and in subsequent patch clamp experiments, with the lack of phenotype in the quadruple transgenic cohort likely attributable to smaller sampling. To test whether sex, brain hemisphere, and dorsal-ventral slice position had an effect, an additional four-way ANOVA of all slices was performed. A small effect of 5xFAD genotype was found (nslice = 101 CT, 88 5xFAD; F(1,178) = 10.4, p = 0.0015), and a small effect of dorsal-ventral position (F(1,178) = 10.46, p = 0.0015), with ventral slices (bregma 3–4 mm) 126 ± 12% the frequency of more dorsal slices (bregma 2–3 mm) for both control and 5xFAD slices. Correcting for multiple comparisons, within each genotype the difference between dorsal and ventral slices did not reach significance [D versus V: p = 0.078 (CT), p = 0.235 (5xFAD), ŠC]. No significant effects were observed for sex (F(1,178) = 0.728, p = 0.395), brain hemisphere (F(1,178) = 0.873, p = 0.351), nor any interaction terms.
Altered ensembles of PCs are recruited in 5xFAD slices
The replay of PC ensembles during SWRs is critical for the consolidation of spatial memory (Wilson and McNaughton, 1994; Skaggs and McNaughton, 1996; Lee and Wilson, 2002; O'Neill et al., 2008). Given the observed alterations to SWRs in three-month 5xFAD mice, we next sought to determine whether ensembles of PCs were altered via calcium imaging (Fig. 3A). PCs differentiate into sPCs (closer to str. radiatum) and dPCs (closer to str. oriens) cells, with different function, connectivity, and molecular profiles (Lee et al., 2014; Valero et al., 2015). We distinguished sPCs and dPCs via post hoc staining of imaged slices for CB, which is more highly expressed in sPCs. While dorsal hippocampus exhibits a clear delineation of CB+ sPCs and CB– dPCs (Lee et al., 2014), we observed a CB bilayer in our slices (Fig. 3B), as previously reported in more ventral hippocampus (Baimbridge and Miller, 1982; Slomianka et al., 2011). In nslice = 4 from nmice = 2 CT, 2 5xFAD, we counted the total number of GCaMP (GC)+ CB– (539), GC– CB+ (292) and GC+ CB+ (146) cells, and for each calculated the distance from the border of str. radiatum and pyramidale (Fig. 3C). At 30 µm, there was a switch from majority CB+ to GC+ cells, with a non-trivial fraction of double-labeled GC+ CB+ cells: 17.9% from 0 to 30 µm and 15.7% from 30 to 90 µm. At depths >90 µm, there was lower co-expression of 7.9%. All subsequent experiments were performed on GC+ cells with a cutoff of 30 µm between putative sPCs and dPCs.
Figure 3.

Altered ensembles of PCs are recruited in 5xFAD slices. A, Confocal imaging of PC ensembles were recorded concurrently with SWRs. Scale bars = 50 μm. B, Post hoc IHC for CB of imaged Thy1-GCaMP6f (GC) slices. Dashed gray lines approximately distinguish layers of hippocampus, str. rad. = stratum radiatum, str. pyr. = stratum pyramidale, str. or. = stratum oriens. Scale bar = 50 μm. C, Histogram of the distance from the center of the cell body to the str. pyr./rad. border for 539 GC+ CB–, 292 GC– CB+, and 146 GC+ CB+ cells pooled from nslice = 4 from nmice = 2 CT, 2 5xFAD. D, LFP and ΔF/F for identified cells in slices from three-month control (left) and 5xFAD (right) mice. Top trace, LFP trace with identified SWR events in raster below. Middle traces, Individual ΔF/F for each cell, sPCs in magenta, and dPCs in green. Bottom raster, Identified Ca2+ events above threshold. Dark colored event indicate concurrence with SWR, light gray indicates spontaneous event. E, The total number of active sPCs and dPCs for CT (nslice = 25) and 5xFAD (nslice = 23) slices. F, The number of sPCs/dPCs active during SWRs. Frequency (G), amplitude (H), and duration (I) of Ca2+ transient events averaged across sPCs and dPCs for each slice. J, top, Event matrix displaying for one example recording, the SWR events on the x-axis, and cells on the y-axis (magenta = sPC, green = dPC). Bottom left, Pairwise Jaccard similarity between SWR events (columns in event matrix). Similarity matrix is symmetric and the diagonal = 1 by definition - these have not been plotted for clarity. Bottom right, Pairwise Jaccard similarity between cells (rows in event matrix). Dashed lines indicate borders between sPC-sPC, sPC-dPC, and dPC-dPC comparisons. Quantification of cell similarity was performed by grouping all sPC-PC comparisons (sPC-sPC and sPC-dPC), and all dPC-PC comparisons (dPC-dPC and sPC-dPC). Cumulative distribution functions were calculated from all pairwise comparisons for (K) SWR-SWR similarity, (L) sPC-PC similarity, and (M) dPC-PC similar. K–M, All showed significant genotype differences via two-way ANOVAs; asterisks indicate regions surviving multiple comparisons; *p < 0.05, **p < 0.01, ***p < 0.001. Box-whisker plots represent non-normal data with median and IQRs. Individual data points represent a slice. Closed circles represent slices from males, open circles females; p values of pairwise comparisons indicated above brackets.
The ensemble activity of PCs was recorded under confocal microscopy in slices from 5xFAD/+;Thy1-GCaMP6f mice and Thy1-GCaMP6f littermate controls (Fig. 3D). Active cells were detected with a semi-automated algorithm and both spontaneous and SWR-coincident Ca2+ transient events were detected. Within 5xFAD slices, the total number of active PCs detected per imaging field was increased from 17.0 ± 2.6 to 24.0 ± 1.6 PCs in nslice = 25 CT, 23 5xFAD (t(31.2) = 3.091, p = 0.0042; Welch's t test, LT), with a greater number of PCs active during SWRs (CT: 3.6 ± 0.6, 5xFAD: 4.9 ± 0.8 cells, t(40.1) = 2.063, p = 0.046; Welch's t test, LT). When delineated by PC subtype, there was a significant effect of genotype on the number of active cells (F(1,92) = 11.8, p = 0.00,088; two-way ANOVA, ART; Fig. 3E), as well as an effect of cell type (F(1,92) = 20.6, p = 1.7 × 10−5). The number of sPCs was increased in 5xFAD slices from 5.7 ± 0.8 to 8.7 ± 1.0 cells (p = 0.067, ŠC) and dPCs from 11.3 ± 2.0 to 15.6 ± 1.2 cells (p = 0.039, ŠC), with significantly more dPCs than sPCs for both genotypes (CT: p = 0.014; 5xFAD: p = 0.00,041, ŠC). During SWRs, more dPCs participated than sPCs (F(1,87) = 12.19, p = 0.00,076; two-way ANOVA, ART; Fig. 3F) in both CT (p = 0.0063, ŠC) and 5xFAD slices (p = 0.00,095, ŠC). However, no effect of genotype was observed (F(1,87) = 0.747, p = 0.390), in contrast to the increase observed when considering all PCs together.
We next asked whether characteristics of the Ca2+ events differed between genotype, as an indirect measure PC firing activity. Averaging across cells from each slice, no differences were found in the frequency (F(1,87) = 0.094, p = 0.759; two-way ANOVA, LT; Fig. 3G), amplitude (F(1,87) = 0.093, p = 0.761; two-way ANOVA, LT; Fig. 3H), nor duration of Ca2+ transient events (F(1,87) = 0.289, p = 0.593; two-way ANOVA, LT; Fig. 3I). This suggests that on an individual cell level, PCs exhibit similar activity in 5xFAD mice as compared with controls, with differences only becoming apparent on the ensemble level. Finally, we sought to determine whether the cellular composition of PC ensembles during SWR events was altered. Ensemble diversity was assessed by calculating the pairwise Jaccard similarity of cellular participation between all SWR events (Fig. 3J). Additionally, the Jaccard similarity of SWR participation between cells was computed between sPCs/dPCs and all other PCs (Fig. 3J, bottom right). The cumulative distribution functions of all pairwise comparisons were calculated for each slice and averaged across genotype, revealing a reduced degree of similarity in PC ensembles during SWRs in 5xFAD, as compared with control slices (F(1,4100) = 85.6, p < 10−15; two-way ANOVA; Fig. 3K), although with no bins surviving multiple comparisons. This suggests there may be an increased repertoire of ensembles in 5xFAD slices. The similarity between sPCs and all other PCs was reduced (F(1,3600) = 25.2, p = 5.5 × 10−7; two-way ANOVA; Fig. 3L), with similarities between 0.1 and 0.15 surviving multiple comparisons, as was the similarity between dPCs and all other PCs (F(1,3800) = 15.88, p = 6.9 × 10−5; two-way ANOVA; Fig. 3M), suggesting aberrant cell participation may be disrupting ensembles.
PC spiking is relatively unchanged in 5xFAD mice
To test more directly whether PC activity was altered, GCaMP6f+ sPCs (nsPC = 13 CT, 9 5xFAD) and dPCs (ndPC = 26 CT, 35 5xFAD) were targeted for loose cell-attached recordings (Fig. 4A). Most cells (nPC = 39 CT, 39 5xFAD) were from 5xFAD;Thy1-GCaMP6f and Thy1-GCaMP6f littermate controls. A small number (nsPC = 1, ndPC = 4) were from 5xFAD;Thy1-GCaMP6f;PVCre/+;tdTom/+ mice, which labeled both excitatory PCs in green and inhibitory PV cells in red. Spikes and bursts (three or more spikes each within 60 ms) were delineated as spontaneous or SWR coincident (Fig. 4B). Consistent with prior studies (Mizuseki and Buzsáki, 2013), the distribution of spike rates was lognormal for both sPCs and dPCs. Both sPCs (Fig. 4C) and dPCs (Fig. 4D) increased their spike rate during SWRs, with a significant effect of spontaneous/SWR time period (sPCs: F(1,20) = 11.0, p = 0.0034; dPCs: F(1,59) = 18.5, p = 6.4 × 10−5; two-way RM ANOVAs, ART, zero values preclude LT), with only sPCs/dPCs from 5xFAD mice showing a significant spike rate increase when correcting for multiple comparisons [sPCs: p = 0.272 (CT), p = 0.023 (5xFAD); dPCs: p = 0.085 (CT), p = 0.00,013 (5xFAD); Wilcoxon post hoc, ŠC]. sPCs displayed a significant genotype difference (F(1,20) = 4.64, p = 0.044), and interaction of genotype × period (F(1,20) = 5.59, p = 0.028), although neither the spontaneous nor SWR spike rate survived multiple comparisons (Spontaneous p = 0.934, SWR: p = 0.301; Mann–Whitney post hoc, ŠC). In contrast, dPCs displayed no genotype difference (F(1,59) = 0.914, p = 0.343), nor interaction of genotype × period (F(1,59) = 1.803, p = 0.184).
Figure 4.

PC spiking is relatively unchanged in 5xFAD mice. A, Diagram of a loose cell-attached recording of spikes from a Thy1-GCaMP6f+ PC paired with LFP recording of SWRs. B, Example traces from three-month control (left) and 5xFAD (right) slice. Top trace, LFP with SWR events in raster below. Gray shaded events indicate spontaneous SWR. Green shading indicates SWR coincident with at least one spike. Dark green shading indicates SWR coincident with burst, defined as three or more spikes each within 60 ms. Bottom trace, Cell-attached recording from PC. Detected spikes and bursts indicated in raster below. Dark shading indicates they are coincident with SWR event, light gray shading indicates spontaneous events. Spike rates were calculated separately during spontaneous and SWR periods for (C) sPCs, nsPC = 13 CT, 9 5xFAD, and (D) dPCs, ndPC = 26 CT, 35 5xFAD. The peri-SWR spike probability was calculated for (E) sPCs and (F) dPCs, by binning spikes into 100 2-ms bins and normalizing by total number of SWRs. Dark lines indicate average of all cells within a genotype, shaded regions indicate SEM, 0 ms indicates time of SWR peak. G, The rate of bursts for sPCs and dPCs, defined as at least three spikes each within 60 ms. H, Intraburst interval for sPCs and dPCs, defined as the average time between successive spikes in a burst. I, The fraction of spikes that occurred during SWRs. J, The fraction of SWRs that had one or more spikes. K, Example of SWR (top trace), with filtered slow γ (20–50 Hz) and ripple (120–220 Hz) signals (middle traces). The phase of oscillations was set at 0° at peaks and 180° at troughs. Bottom trace, Cell-attached recording, with spike times marked by vertical dashed lines. L–O, Polar phase plots of phase-locking of spikes to SWR-nested slow γ (L–M) and ripple (N–O) for sPCs (L, N) and dPCs (M, O). The angles of individual data points represent the average phase of all spikes for a cell. Length from origin (0–1) indicates the degree of phase-locking. A length of 1 signifies perfect phase-locking (every spike at same phase); a length of 0 indicates random or no phase-locking. Lines with arrowheads represent cell average, solid colored = CT, dashed gray = 5xFAD. Asterisks indicate result of Raleigh's test for non-uniformity, *p < 0.05, **p < 0.01, ***p < 0.001. p Values indicate results of circular mean comparison. For all plots, individual data points represent a cell. Closed circles represent cells from males, open circles females. Box-whisker plots represent non-normal data as median and IQRs; p values of pairwise comparisons indicated above brackets.
To examine spike rate in more detail around SWR events, the peri-SWR spike probability was averaged across all cells within each genotype, showing an increased spiking probability in 5xFAD sPCs (F(1,2000) = 37.73, p = 9.8 × 10−10; two-way ANOVA; Fig. 4E), with significant differences when correcting for multiple comparisons from −4 to +8 ms around the SWR peak (p < 0.01, ŠC). In contrast, the dPC spike probability did not differ between genotype (F(1,5900) = 2.38, p = 0.123; two-way ANOVA; Fig. 4F). There was no difference in the rate of spike bursts (three or more spikes each within 60 ms) between genotype (F(1,79) = 1.00, p = 0.319; two-way ANOVA, ART; Fig. 4G) or PC type (F(1,79) = 2.37, p = 0.127). There was a tendency for a significant effect of genotype on the intraburst interval (F(1,77) = 3.26, p = 0.075; two-way ANOVA, ART; Fig. 4H), with dPCs spiking somewhat faster from 14.4 ± 1.6 to 11.1 ± 0.8 ms (p = 0.045, ŠC) and with no change for sPCs (p = 0.997, ŠC). The percentage of cells with bursts did not differ between genotype (97.6% CT, 96.1% 5xFAD; χ2(3) = 0.158, p = 0.691; χ2 test). In particular, 100% of sPCs had at least one burst in both CT and 5xFAD mice, an important consideration given prior work indicating dPCs burst more than sPCs (Mizuseki et al., 2011). As we only targeted cells with visible calcium transients for localization under confocal microscopy, it is possible the population of sPCs and dPCs studied are a more highly active subset of all PCs. We also examined whether the fractional participation of PCs in SWRs differed, quantified as the fraction of spikes that occurred during SWRs (Fig. 4I), and the fraction of SWRs that had one or more spikes (Fig. 4J). There was a tendency for an effect of genotype for both endpoints (F(1,79) = 3.90, p = 0.052; F(1,79) = 3.11, p = 0.082; two-way ANOVA, ART), although with neither cell type surviving multiple comparisons (Fig. 4I,J).
Finally, we examined the phase-locking of spikes in both the slow γ and ripple ranges (Fig. 4K). Phase-locking of PCs to the trough of ripples has been widely reported in vivo (Ylinen et al., 1995; Csicsvari et al., 1999; Le Van Quyen et al., 2008). Additionally, phase-locking of spikes to SWR-nested slow γ has been reported to be reduced in 5xFAD mice (Iaccarino et al., 2016). We observed that sPCs showed broad phase preference in the γ range, with averages following the trough at 223° for controls (Z = 0.365, p = 0.706; Raleigh's test), and following the peak at 63° in 5xFAD mice (Z = 1.88, p = 0.154; Raleigh's test), with no significant genotype difference (p = 2.86, p = 0.091, circular-median test; Fig. 4L). In contrast, control dPCs were significantly phase-locked at 143° (110−177° 95%; Z = 5.79, p = 0.0023; Raleigh's test), while 5xFAD dPCs showed a broader phase preference with a mean of 177° (110−245° 95%; Z = 2.25, p = 0.105; Raleigh's test), and no significant genotype difference (p = 0.147, p = 0.701, circular-median test; Fig. 4M). In the ripple range, cells displayed broad phase preference, with neither sPCs (Z = 0.355, p = 0.712; Z = 1.58, p = 0.212; Raleigh's test; Fig. 4N) nor dPCs (Z = 0.981, p = 0.340; 5xFAD: Z = 1.09, p = 0.339; Raleigh's test; Fig. 4O) significantly phase-locked, nor different between genotypes (sPC: p = 0.254, p = 0.614; dPC: p = 0.528, p = 0.467; circular-median test). However, in control dPCs, the mean phase at the trough of 191° more closely matches prior in vivo findings than for 5xFAD mice, with a mean phase of 224° following the trough (Fig. 4O). These results indicate that spike-phase coupling may be impaired in 5xFAD mice, although the lack of significant ripple phase-locking for either genotype suggests there are limitations to this analysis in our slice preparation, as this finding differs from the robust ripple phase-locking observed in vivo (Ylinen et al., 1995; Csicsvari et al., 1999; Le Van Quyen et al., 2008). One difference between our study and prior in vivo recordings is that the spikes and LFP were recorded from different electrodes, while in vivo LFP and spikes are typically recorded from the same set of channels. As ripples are highly localized events, the distance between electrodes may confound spatial phase locking. Taken together with the Ca2+ imaging data, these results indicate relatively minor alterations to the spiking activity of PCs in 5xFAD slices. However, the large variability in spiking rate may mask small differences in activity.
PCs receive increased synaptic E/I ratio
Following the cell-attached recording of PC spiking activity, the electrode was replaced with one containing a cesium-based internal solution and the same cell targeted for a whole-cell voltage-clamp recording to detect EPSCs and IPSCs at −70 mV (nsPC = 7 CT, 5 5xFAD; ndPC = 12 CT, 9 5xFAD; Fig. 5A,B) and 0 mV (nsPC = 7 CT, 5 5xFAD; ndPC = 14 CT, 11 5xFAD; Fig. 5C,D), respectively. Events were sorted as spontaneous (sEPSCs/sIPSCs) or SWR-coincident (swrEPSCs/swrIPSCs). sEPSC frequency and amplitude were unchanged across genotype [frequency: F(1,29) = 0.296, p = 0.591 (Fig. 5E); amplitude: F(1,29) = 2.26, p = 0.143 (Fig. 5F); two-way ANOVA, LT], as were the kinetics of sEPSCs [rise tau: F(1,29) = 0.540, p = 0.468 (Fig. 5G); decay tau: F(1,29) = 0.268, p = 0.608 (Fig. 5H); two-way ANOVA]. The excitatory charge during spontaneous periods (sEPSQ) was also unchanged across genotype (F(1,29) = 0.835, p = 0.369; two-way ANOVA, LT; Fig. 5I). However, there was a significant effect of genotype on the excitatory charge during SWRs (swrEPSQ; F(1,29) = 6.56, p = 0.016; two-way ANOVA, LT; Fig. 5J), with dPCs seeing a 114 ± 41% increase (p = 0.043, ŠC), while sPCs were not significantly different (p = 0.368, ŠC). Considering we observed that SWRs in 5xFAD mice were larger in amplitude with more total PCs active, this was not an altogether surprising result. This increase indicates that the enlarged SWRs were accompanied with increased excitatory synaptic activity, likely originating from the CA3 region. The lack of any increase in spontaneous excitatory activity is also consistent with our cell-attached results showing that at least locally in CA1, both sPCs and dPCs had no changes in firing rate during spontaneous periods (Fig. 4C,D). Together, these results indicate that spontaneous excitatory synaptic input is unchanged, but increased during SWRs in 5xFAD mice, particularly for dPCs, consistent with expectations from our LFP experiments.
Figure 5.

PCs receive increased synaptic E/I ratio. A, Example traces of LFP + whole-cell recording voltage-clamped at −70 mV in Thy1-GCaMP6f littermate controls and 5xFAD/+;Thy1-GCaMP6f mice. Excitatory postsynaptic potentials (EPSCs) were sorted as sEPSCs or swrEPSCs. B, Example swrEPSCs. C, Example traces of LFP + whole-cell recording voltage-clamped at 0 mV to detect sIPSCs and swrIPSCs. D, Example swrIPSCs. E–H, Summary plots of sEPSC frequency (E), amplitude (F), rise tau (G), and decay tau (H) for sPCs and dPCs in control and 5xFAD mice (nsPC = 7 CT, 5 5xFAD; ndPC = 12 CT, 9 5xFAD). I, sEPSQ = normalized spontaneous excitatory charge (integrated current per unit time). J, swrEPSQ = total excitatory charge during SWRs, integrated over a 100-ms window centered around the SWR peak. K–N, Summary plots of sIPSC frequency (K), amplitude (L), rise tau (M), and decay tau (N; nsPC = 7 CT, 5 5xFAD; ndPC = 14 CT, 11 5xFAD). O, P, Cumulative distribution functions, averaged over all cells for sIPSC amplitude (O) and decay tau (P), both for sPCs (top) and dPCs (bottom). Dark lines represent cell average, shaded region represents SEM. Q, sIPSQ = normalized spontaneous inhibitory charge (integrated current per unit time). R, swrIPSQ = total inhibitory charge during SWRs. S, Spontaneous synaptic E/I ratio of normalized charge for each cell (sEPSQ/sIPSQ). T, Synaptic E/I ratio for 100-ms window centered around SWR peak (swrEPSQ/swrIPSQ). U, V, During SWRs, the charge for each cell (EPSQ and IPSQ) was calculated in 2-ms bins by integrating the current in a sliding 100-ms window centered around that bin, for both sPCs (U) and dPCs (V). Summary data in J and R thus represent these curves at the y-intercept. Asterisks indicate regions that survived Šidák's multiple comparisons; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. W, X, The ratios of curves in U, V yield the synaptic E/I ratio during SWRs. Summary data in T represent these curves at the y-intercept. For all plots, individual data points represent a cell. Closed circles represent cells from males, open circles females. Box-whisker plots represent non-normal data with median and IQRs. Bar plots represent normal data with mean ± SEM; p values of pairwise comparisons indicated above brackets.
In contrast, for sIPSCs, there was an effect of genotype on the frequency (F(1,33) = 9.38, p = 0.0043; two-way ANOVA, LT; Fig. 5K), as well as an effect of PC type (F(1,33) = 8.19, p = 0.0073), with sPCs seeing a preferential 52 ± 10% decrease (p = 0.049, ŠC), while dPCs were unchanged (p = 0.466, ŠC). There was a tendency for an effect of genotype on the amplitude of sIPSCs (F(1,33) = 3.40, p = 0.074; two-way ANOVA, LT; Fig. 5L), and no effect for the rise tau (F(1,33) = 1.73, p = 0.197; two-way ANOVA; Fig. 5M). However the decay tau of sIPSCs saw an effect of genotype (F(1,33) = 9.36, p = 0.0044; two-way ANOVA; Fig. 5N), cell type (F(1,33) = 10.7, p = 0.0025), and interaction of genotype × cell type (F(1,33) = 7.61, p = 0.0094), with sPCs from 5xFAD mice displaying a 5-ms longer decay than control sPCs (p = 0.030, ŠC) and dPCs (p = 0.00072, ŠC). This specific alteration prompted us to examine in more detail the distribution of sIPSCs. For each cell, the cumulative distribution function of all events was calculated and averaged across cells. This analysis revealed for sPCs a significantly lower sIPSC amplitude (F(1,1000) = 39.8, p = 4.2 × 10−15; two-way ANOVA; Fig. 5O, upper) and greater decay tau (F(1,1000) = 469, p < 10−15; two-way ANOVA; Fig. 5P, upper), with events of amplitude 10–65 pA and decay tau 9–22 ms surviving multiple comparisons. This suggests that for sPCs, fast and high amplitude inhibitory input, typically attributed to fast-spiking cells, is preferentially reduced. In contrast, dPCs saw no significant range of bins survive multiple comparisons (Fig. 5O,P, lower). This analysis was performed for all sEPSC and sIPSC end-points, and no other end-points saw significant genotype differences surviving multiple comparisons.
The normalized inhibitory charge during spontaneous periods (sIPSQ) was also preferentially reduced for 5xFAD sPCs (genotype effect: F(1,33) = 10.3, p = 0.0029; two-way ANOVA, LT; sPC: p = 0.044, dPC: p = 0.331, ŠC; Fig. 5Q). During SWRs, the total inhibitory charge (swrIPSQ) was differentially altered between cell types, with a significant interaction of genotype and cell type (F(1,33) = 7.14, p = 0.0116; two-way ANOVA, LT; Fig. 5R), a non-significant reduction for sPCs (p = 0.294, ŠC) and a significant increase for dPCs (p = 0.022, ŠC). These results suggest a selective impairment in inhibition in sPCs that is not seen in dPCs. Despite the observed increase in SWR activity in 5xFAD mice, the inhibition did not scale proportionally with the increased excitation, thus shifting the synaptic E/I balance. We assessed this for both spontaneous and SWR-driven currents. During spontaneous periods, there was a significant effect of genotype on the E/I ratio, defined as the ratio of sEPSQ/sIPSQ (F(1,27) = 14.7, p = 0.00,069; two-way ANOVA; Fig. 5S), as well as an effect of cell type (F(1,27) = 7.39, p = 0.0113). Both sPCs and dPCs saw an increase (sPC: p = 0.055, dPC: p = 0.049, ŠC). During SWRs, the synaptic E/I ratio (swrEPSQ/swrIPSQ) was affected by both genotype (F(1,27) = 7.59, p = 0.0104; two-way ANOVA; Fig. 5R) and a genotype × cell type interaction (F(1,27) = 5.12, p = 0.0319), but only in sPCs was there an increase (sPC: p = 0.048, dPC: p = 0.877). To examine the temporal progression of synaptic input during SWRs, we calculated the EPSQ and IPSQ in a sliding 100-ms window across the SWR peak (Fig. 5U,V), from which a time course of the synaptic E/I ratio could be determined (Fig. 5W,X). While for both the EPSQ and IPSQ there was a significant effect of genotype for sPCs (EPSQ: F(1,1000) = 184, p < 10−15; IPSQ: F(1,1000) = 129, p < 10−15; two-way ANOVA; Fig. 5U) and dPCs (EPSQ: F(1,1900) = 295, p < 10−15; IPSQ: F(1,2300) = 708, p < 10−15; two-way ANOVA; Fig. 5V), only for the IPSQ in dPCs did individual bins from −18 to +62 ms (relative to the SWR peak) survive multiple comparisons. The ratio of the EPSQ/IPSQ time course revealed a significant effect of genotype for both sPCs (F(1,1000) = 159, p < 10−15; two-way ANOVA; Fig. 5W) and dPCs (F(1,1800) = 24.3, p = 9.1 × 10−7; two-way ANOVA; Fig. 5X). For both sPCs and dPCs, there was an early peak in the synaptic E/I ratio for 5xFAD mice that was not present in controls (Fig. 5W,X), which can be attributed to a build-up in excitation with a delayed increase in inhibition. Only for dPCs did individual bins from −70 to −60 ms survive multiple comparisons, in part likely due to the greater sampling of dPCs.
PVBCs have selectively reduced spiking
While there are numerous inhibitory cell subtypes in the CA1 region that could underlie a shift in E/I synaptic input to PCs (Pelkey et al., 2017), we focused on PV interneurons, as they are the most highly active during SWR events (Somogyi et al., 2014). We performed cell-attached recordings in 5xFAD/+;PVCre/+;tdTom/+ and PVCre/+;tdTom/+ littermate controls (ncell = 13 CT, 18 5xFAD; Fig. 6A). Cells were also recorded from 5xFAD;Thy1-GCaMP6f;PVCre/+;tdTom/+ and Thy1-GCaMP6f;PVCre/+;tdTom/+ littermate controls (ncell = 11 CT, 14 5xFAD), which were pooled together. One complication with the PV cell population is that there are at least three distinct subtypes within CA1 str. pyr., which vary in function and axonal target: basket cells (PVBCs), which target perisomatic regions of PCs, bistratified cells (PVBSCs), which target both apical and basal dendrites of PCs, and axo-axonic cells (PVAACs), which selectively target the AIS (Fig. 6B). To distinguish these, we morphologically reconstructed the cells post hoc after filling with biocytin in whole-cell configuration (Fig. 6C) and sorted them by axonal target. Of the total reconstructed cells (ncell = 24 CT, 32 5xFAD), PVBSCs (Fig. 6C2) were easily distinguished from both PVBCs (Fig. 6C1) and PVAACs (Fig. 6C3), with their axonal arbor avoiding str. pyr. (nPVBSC = 5 CT, 10 5xFAD). While PVBCs and PVAACs have overlapping axonal targets, some PVBCs were easily distinguished with axonal terminals directly targeting PC somas as visualized through the PVCre-tdTom and/or Thy1-GCaMP6f fluorescence (nPVBC = 9 CT, 9 5xFAD). Likewise, some PVAACs exhibited the characteristic “chandelier” phenotype and lack of somatic targeting (nPVAAC = 4 CT, 4 5xFAD). However, there were some cells with ambiguous PVBC/PVAAC morphology based solely on axonal targets (ncell = 6 CT, 9 5xFAD). To sort these cells, we examined the spike rate. Both PVBCs and PVBSCs are known to strongly increase their spike rate during SWRs (Lapray et al., 2012; Katona et al., 2014), while the PVAAC spike rate does not increase (Viney et al., 2013). Thus, for cells with ambiguous PVBC/PVAAC morphology, those with a spike rate increase during SWRs (53 ± 11 Hz) were sorted as putative PVBCs, while those with no increase (0.6 ± 0.6 Hz) were sorted as putative PVAACs. In a subset of 14 slices (5 CT, 9 5xFAD from nmice = 4 CT, 5 5xFAD), we additionally stained for Ankyrin G, which labels the AIS, and confirmed colocalization with two putative PVAACs (Fig. 6C4). Based on this sorting methodology, we identified a total of nPVBC = 13 CT, 16 5xFAD, nPVBSC = 5 CT, 10 5xFAD, and nPVAAC = 6 CT, 6 5xFAD. The proportion of cells did not significantly differ from prior published findings of 60% PVBC, 25% PVBSC, 15% PVAAC in CA1 str. pyr. (χ2(2) = 0.754, p = 0.686; Baude et al., 2007), and did not significantly differ between genotype (χ2(2) = 1.09, p = 0.580; Fig. 6D).
Figure 6.

PV cells in CA1 delineate into basket (PVBCs), bistratified (PVBSCs), and axo-axonic (PVAACs) cells. A, Diagram of LFP + PV cell recording in three-month 5xFAD/+; PVCre/+;tdTom/+ and PVCre/+;tdTom/+ littermate controls. B, Diagram of PV cell subtypes and their axonal targets in CA1 str. pyr. Examples of (C1) PVBCs, (C2) PVBSCs, and (C3) PVAACs. Green fluorescence indicates avidin-fluorescein bound to the biocyin in the filled PV cell. Red fluorescence indicates PVCre-dTom expression. Dashed gray lines approximately distinguish layers of hippocampus, str. l.m. = stratum lacunosum-moleculare, str. rad. = stratum radiatum, str. pyr. = stratum pyramidale, str. or. = stratum oriens. Notice that str. pyr. is dimly red due to the expression of other non-filled PVBCs targeting perisomatic regions of PCs. Scale bars = 100 μm. Bottom traces, Examples of LFP and cell-attached recording of spikes, which was used to aid in distinguishing PVBCs and PVAACs. PVAACs are unique in that they reduce their firing during SWRs. C4, Ankyrin G immunostaining showing co-localization of synaptic boutons with AIS of PCs for identified PVAAC. Scale bar = 10 μm. D, Proportions of sorted cells in each genotype.
Based on these delineations, we performed PV cell-attached + LFP recordings in control and 5xFAD mice for the identified populations of PVBCs (Fig. 7A1), PVBSCs (Fig. 7A2), and PVAACs (Fig. 7A3). We observed an effect of genotype on PVBC spike rate (F(1,27) = 10.4, p = 0.0033; two-way RM ANOVA, ART; Fig. 7B1), with a selective and robust reduction during SWR periods, from 62.9 ± 10.6 to 34.0 ± 6.2 Hz (p = 0.044; Mann–Whitney post hoc, ŠC), whereas the spontaneous spike rate did not significantly differ (p = 0.209; Mann–Whitney post hoc, ŠC). In contrast, there was no effect of genotype on the spike rate of PVBSCs (F(1,13) = 1.35, p = 0.267; two-way RM ANOVA, ART; Fig. 7B2) or PVAACs (F(1,10) = 3.83, p = 0.079; two-way RM ANOVA, ART; Fig. 7B3). As expected from the cell-sorting methodology, spike rates increased during SWRS for PVBCs, with an effect of period (F(1,27) = 179, p = 1.9 × 10−13; Fig. 7B1), and increases of 5.6 ± 1.2-fold in control (p = 0.00,049; Wilcoxon post hoc, ŠC) and 5.9 ± 2.4-fold in 5xFAD mice (p = 0.00012; Wilcoxon post hoc, ŠC). Additionally, there was an interaction of genotype × period (F(1,27) = 16.8, p = 0.00034), indicating that PVBC modulation of spiking during SWRs differed between genotype. Similarly, there was an effect of period for PVBSCs (F(1,13) = 30.6, p = 9.7 × 10−5; Fig. 7B2), with a non-significant 4.4 ± 1.2-fold increase in control (p = 0.121; Wilcoxon post hoc, ŠC) and a 6.8 ± 5.0-fold increase in 5xFAD mice (p = 0.019; Wilcoxon post hoc, ŠC), and no interaction of genotype × period (F(1,13) = 2.56, p = 0.133). While PVAACs did see an effect of period (F(1,10) = 16.7, p = 0.0022; Fig. 7B3), neither in control nor 5xFAD mice was the increase significant (CT: 1.0 ± 0.4-fold, p = 0.527, 5xFAD: 2.4 ± 0.8-fold, p = 0.062; Wilcoxon post hoc, ŠC), nor was there an interaction of genotype × period (F(1,10) = 3.21, p = 0.104). However, the low number of PVAACs recorded from may mask small alterations in this cell population.
Figure 7.

PVBCs have selectively reduced spiking. Spiking data for PVBCs (panel subheading 1), PVBSCs (panel subheading 2), and PVAACs (panel subheading 3). A, Example LFP and cell-attached traces for CT (left) and 5xFAD (right), for a PVBC (A1), PVBSC (A2), and PVAAC (A3). Spike and SWR events are color-coded as in Figure 4B. B, Summary spike rate data for PVBCs (nPVBC = 13 CT, 16 5xFAD, B1), PVBSCs (nPVBSC = 5 CT, 10 5xFAD, B2), and PVAACs (nPVAAC = 6 CT, 6 5xFAD, B3) during spontaneous and SWR periods. C, Peri-SWR spike probability. Asterisks indicate regions surviving Šidák's multiple comparisons correction. D, Burst rate, defined as 3 or more spikes each within 40 ms. E, The fraction of SWRs with one or more spike. F, The fraction of SWRs with a burst. G-H, Polar phase plots of phase-locking of spikes to SWR-nested slow γ (G) and ripple (H). The angles of individual data points represent the average phase of all spikes for a cell. Length from origin (0–1) indicates the degree of phase-locking. Lines with arrowheads represent cell average, solid colored = CT, dashed gray = 5xFAD. Asterisks indicate result of Raleigh's test for non-uniformity, p values indicate results of Watson–Williams or circular mean test; *p < 0.05, **p < 0.01, ***p < 0.001. For all plots, individual data points represent a cell. Closed circles represent cells from males, open circles females. Box-whisker plots represent non-normal data with Median and IQRs; p values indicated above brackets.
The peri-SWR spike probability revealed that in 5xFAD mice, PVBCs spikes fell in a significantly narrower window (Fig. 7C1), with a reduced full-width half-maximum value from 44 ± 19 to 18 ± 5 ms, and a significant effect of genotype when analyzed via two-way ANOVA (F(1,2700) = 267, p < 10−15), with bins from −20 to −16 and 0 to +20 ms relative to the SWR peak surviving multiple comparisons. Intriguingly, this narrower window of spiking in 5xFAD mice was accompanied by shorter duration SWRs (Fig. 2D), suggesting the activity of these cells is critical for normal ripple progression. In contrast, PVBSCs appeared to increase their firing after the SWR peak in 5xFAD mice (Fig. 7C2), perhaps playing a compensatory role, with a significant effect of genotype (F(1,1300) = 33.9, p = 7.3 × 10−9; two-way ANOVA). PVAACs appeared to have decreased probability of spiking in 5xFAD mice, particularly before the SWR peak (Fig. 7C3), with a significant effect of genotype (F(1,1000) = 30.15, p = 5.1 × 10−8; two-way ANOVA). However, unlike PVBCs, neither PVBSCs nor PVAACs showed significant genotype differences when corrected for multiple comparisons (Fig. 7C).
Considering the rate of bursts, defined as three or more spikes each within 40 ms, there was a tendency for a reduction for PVBCs (U = 60, p = 0.056; Mann–Whitney; Fig. 7D1) and no change for PVBSCs (U = 22.5, p = 0.793; Mann–Whitney; Fig. 7D2) or PVAACs (U = 24, p > 0.999; Mann–Whitney; Fig. 7D3). No differences were observed in the proportion of cells that exhibited bursts for PVBCs (100% CT, 93.8% 5xFAD; χ2(1) = 0.842, p = 0.359; χ2 test), PVBSCs (80.0% CT, 90.0% 5xFAD; χ2(1) = 0.288, p = 0.591), or PVAACs (50.0% CT, 83.3% 5xFAD; χ2(1) = 1.500, p = 0.221). The fraction of SWRs that coincided with a PV spike was no different between genotypes for PVBCs (U = 72, p = 0.170; Mann–Whitney; Fig. 7E1), PVBSCs (U = 21, p = 0.655; Mann–Whitney; Fig. 7E2), or PVAACs (U = 6, p = 0.065; Mann–Whitney; Fig. 7E3). However, in PVBCs, the fraction of SWRs that coincided with a PV burst was significantly reduced from 65.4 ± 8.5% to 32.9 ± 8.5% (U = 53, p = 0.025; Mann–Whitney; Fig. 7F1), while there was no genotype difference for PVBSCs (U = 16.5, p = 0.323; Mann–Whitney; Fig. 7F2) or PVAACs (U = 10.5, p = 0.182; Mann–Whitney; Fig. 7F3).
We also examined the spike phase-locking of these three PV cell types, as these have been carefully studied in vivo for θ (8–12 Hz, during mobility) and ripple oscillations (Varga et al., 2014). The precise temporal ordering of PV cell subtypes during network oscillations has been proposed to play a critical role in the spatiotemporal control of PCs. During ripples, PVBCs have been observed to fire just after the trough of the ripple, followed by PVBSCs and then PVAACs. Less studied is the phase-locking of PV cells during SWR-nested slow γ oscillations, which we examined as we observed alterations in this endpoint for the PC population (Fig. 4M). We found significant γ phase-locking of PVBCs for 5xFAD mice at 273° (247−298° 95%; Z = 7.94, p = 0.00,012; Raleigh's test), with broader phase preference in control mice (Z = 2.48, p = 0.082; Raleigh's test), and with no difference in median phase angle (p = 0.144, p = 0.705; circular-median test; Fig. 7G1). PVBSCs were similarly γ phase-locked for 5xFAD mice at 292° (249−336° 95%; Z = 3.54, p = 0.024; Raleigh's test), with broader phase preference in control mice (Z = 1.57, p = 0.216; Raleigh's test), and with no difference in median phase angle (p = 0.311, p = 0.577; circular-median test; Fig. 7G2). In contrast, in PVAACs we observed phase-locking in control mice at 246° (224−368° 95%; Z = 4.43, p = 0.005; Raleigh's test), with broader phase preference in 5xFAD mice (Z = 0.913, p = 0.421; Raleigh's test), and with a tendency for a difference in median phase angle (p = 3.60, p = 0.058; circular-median test; Fig. 7G3). While the precise significance of γ phase-locking has yet to be demonstrated, these genotype differences point to a temporal disordering of PV cell inhibition.
Within the ripple range, in PVBCs, there was a significant phase-locking in control mice at 228° (198−259° 95%; Z = 6.01, p = 0.0014; Raleigh's test), and in 5xFAD mice at 208° (186−231° 95%; Z = 9.35, p = 1.7 × 10−5; Raleigh's test), with no significant genotype difference (F(1,27) = 1.34, p = 0.258; Watson–Williams; Fig. 7H1). These values are in line with prior in vivo studies, where 0° in our study was defined as the peak of the ripple cycle and 180° as the trough. PVBSCs exhibited more varied phase-locking in the ripple range, although the average angles are in line with in vivo studies (CT: 241°, Z = 1.63, p = 0.202; 5xFAD: 225°, Z = 1.33, p = 0.270; Raleigh's test; Fig. 7H2). PVAACs exhibited ripple phase preference for control mice at 257° (219−296° 95%; Z = 3.75, p = 0.0015; Raleigh's test), in line with in vivo studies. However, in 5xFAD mice, PVAACs spiked earlier, at 204° (164−245° 95%; Z = 3.54, p = 0.0020; Raleigh's test), with a significant genotype difference in mean phase angle (F(1,9) = 5.68, p = 0.044; Watson–Williams; Fig. 7H3). Although we observed no significant change in PVAAC spike rate, this disruption in temporal ordering may still have downstream network consequences.
PVBCs have selective decrease in excitatory synaptic drive and decreased E/I ratio
Following cell-attached recording of spiking activity, the electrode was replaced with a cesium internal and the same PV cell was targeted for a whole-cell voltage-clamp recording. EPSCs were recorded at −70 mV for PVBCs (nPVBC = 12 CT, 16 5xFAD; Fig. 8A1), PVBSCs (nPVBSC = 4 CT, 6 5xFAD; Fig. 8A2), and PVAACs (nPVAAC = 5 CT, 6 5xFAD; Fig. 8A3). None of the three PV cell subtypes had any change in sEPSC frequency (PVBC: t(26) = 1.30, p = 0.206; PVBSC: t(8) = 0.990, p = 0.351; PVAAC: t(9) = 0.129, p = 0.900; unpaired t tests), or amplitude (PVBC: t(26) = 0.545, p = 0.591; PVBSC: t(8) = 0.058, p = 0.956; PVAAC: t(9) = 2.11, p = 0.064; unpaired t tests; Fig. 8B1–B3). The kinetics of sEPSCs in 5xFAD PVBCs were altered however, with a similar rise tau (t(26) = 0.307, p = 0.761; unpaired t test), but a reduction in the decay tau (t(26) = 2.41, p = 0.024; unpaired t test; Fig. 8C1). There were no changes to sEPSC kinetics in PVBSCs (rise: t(8) = 0.671, p = 0.521; decay: t(8) = 1.02, p = 0.340; Fig. 8C2) or PVAACs (rise: t(9) = 0.857, p = 0.414; decay: t(9) = 1.79, p = 0.107; Fig. 8C3). To examine the decreased sEPSC decay tau in PVBCs, the cumulative distribution function of all sEPSCs was calculated and averaged across cells, revealing an effect of genotype (F(1,2500) = 2165, p < 10−15; two-way ANOVA; Fig. 8D), with events of decay tau 1–7 ms surviving multiple comparisons. This decrease in decay tau was somewhat unexpected, considering that in PV cells, neuronal pentraxins are associated with an acquisition of GluA4 subunits, which speeds up the kinetics of AMPA receptor-mediated EPSCs (Pelkey et al., 2015). Both pentraxins and GluA4 are selectively reduced in human AD (Xiao et al., 2017), suggesting that in AD, a longer decay of AMPA-mediated EPSCs may underlie PV cell dysfunction. Our results indicate that in three-month 5xFAD mice, this does not appear to be a prominent mechanism.
Figure 8.

PVBCs have selective decrease in excitatory synaptic input and decreased E/I ratio. Whole-cell data for PVBC EPSCs (panel subheading 1), PVBSC EPSCs (panel subheading 2), PVAAC EPSCs (panel subheading 3), and PVBC IPSCs (panel subheading 4). A, Example recordings of LFP + whole-cell PV cell recording in three-month 5xFAD/+; PVCre/+;tdTom/+ and PVCre/+;tdTom/+ littermate controls, recording synaptic input during spontaneous and SWR period. EPSCs were recorded at −70 mV in PVBCs (A1), PVBSCs (A2), and PVAACs (A3), and IPSCs recorded at 0 mV in a subset of PVBCs (A4). B, Summary data for spontaneous PSC frequency and amplitude for PVBC sEPSCs (nPVBC = 12 CT, 16 5xFAD, B1), PVBSC sEPSCs (nPVBSC = 4 CT, 6 5xFAD, B2), PVAAC sEPSCs (nPVAAC = 5 CT, 6 5xFAD, B3), and PVBC sIPSCs (nPVBC = 8 CT, 8 5xFAD, B4). C, Spontaneous PSC kinetics: rise and decay tau. D, Cumulative distribution function of PVBC decay tau. Dark lines represent cell average, shaded region represents SEM. E, swrEPSQ and swrIPSQ = total excitatory and inhibitory charge during SWRs, integrated over a 100-ms window centered around the SWR peak. F, left, PVBC spontaneous synaptic E/I ratio of normalized charge (sEPSQ/sIPSQ). F, right, PVBC synaptic E/I ratio for 100-ms window centered around SWR peak (swrEPSQ/swrIPSQ). G, The charge for each PVBC (EPSQ and IPSQ), calculated in 2-ms bins by integrating the current in a sliding 100-ms window centered around that bin. Summary data in E1, E4 thus represent these curves at the y-intercept. Asterisks indicate regions that survived Šidák's multiple comparisons; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. H, The ratios of curves in G yield the synaptic E/I ratio during SWRs. Summary data in F represents these curves at the y-intercept. I, Linear regression of spike rate during SWRs versus the E/I ratio. For all plots, individual data points represent a cell. Closed circles represent cells from males, open circles females. Bar plots represent normal data with Mean ± SEM; p values indicated above brackets.
During SWRs, PVBCs from 5xFAD mice saw a 50.1 ± 10.7% reduction in the total excitatory synaptic charge in a 100-ms window centered around the SWR peak (swrEPSQ) (t(26) = 3.20, p = 0.0036, unpaired t test; Fig. 8E1), whereas there was no change for PVBSCs (t(8) = 0.997, p = 0.348; unpaired t test; Fig. 8E2) or PVAACs (t(9) = 1.63, p = 0.139; unpaired t test; Fig. 8D3). In a subset of cells (nPVBC = 8 CT, 8 5xFAD), we also recorded IPSCs at 0 mV (Fig. 8A4; PVBCs, IPSCs were not recorded in every cell, thus there were insufficient numbers of PVBSCs and PVAACs for statistical analysis). Spontaneous inhibitory input to PVBCs was unchanged, including sIPSC frequency (t(14) = 0.448, p = 0.661; unpaired t test), amplitude (t(14) = 0.339, p = 0.740; unpaired t test; Fig. 8B4), and kinetics (rise: t(14) = 0.263, p = 0.796; decay: t(14) = 0.672, p = 0.513; Fig. 8C4). The total inhibitory charge during SWRs (swrIPSQ), was also no different between genotype (t(14) = 0.0083, p = 0.994; unpaired t test; Fig. 8E4). This selective decrease in excitation to PVBCs during SWRs, in contrast to the observation in PCs (Fig. 5J), resulted in a significant decrease in the synaptic E/I ratio during SWRs (t(14) = 2.70, p = 0.017, unpaired t test), although not during spontaneous periods (t(14) = 0.929, p = 0.368, unpaired t test; Fig. 8F). As with PCs, we examined the time course of synaptic charge during SWRs in PVBCs, and found a significant effect of genotype on the excitatory charge (F(1,2500) = 664, p < 10−15; two-way ANOVA), with bins from −32 to +42 ms relative to the SWR peak surviving multiple comparisons (Fig. 8G). There was no effect of genotype on inhibitory charge (F(1,1400) = 2.12, p = 0.145, two-way ANOVA; Fig. 8G). This resulted in a significant effect of genotype on the E/I ratio (F(1,1300) = 339, p < 10−15, two-way ANOVA), with an early peak that survived multiple comparisons in control above 5xFAD cells from −66 to −58 ms relative to the SWR peak (Fig. 8H), precisely the opposite effect observed in PCs (Fig. 5W,X)
Since we recorded the cell-attached spiking activity from the same PVBCs, we were next interested whether reduced SWR spike rate (Fig. 7B1) was correlated with altered synaptic E/I ratio during SWRs. We observed a moderate positive correlation for control PVBCs (nPVBC = 8, R2 = 0.625, F(1,6) = 9.98, p = 0.020, linear regression), but no correlation between spike rate and E/I ratio for 5xFAD PVBCs (nPVBC = 8, R2 = 0.0092, F(1,6) = 0.056, p = 0.822; Fig. 8I), suggesting that not only is excitatory synaptic input during SWRs reduced, potentially through altered PC-PVBC connectivity, but there are also deficits in PVBC input-output function, consistent with prior studies of intrinsic PV cell dysfunction (Verret et al., 2012).
Discussion
Here, we identified a selective reduction in PVBC activity in a model of AD, while PVBSCs, PVAACs, and excitatory PCs were relatively spared. By investigating the synaptic input and spike output of these cell types, we present a careful analysis of hippocampal microcircuitry alterations in early amyloid pathology (Fig. 9) during activity critical for memory consolidation (i.e., SWRs). PVBCs displayed a reduced synaptic E/I ratio during SWRs (Fig. 8F), driven by a reduction in excitatory synaptic input (Fig. 8E1). PVBCs spiked less during SWRs (Fig. 7B1), thus providing reduced inhibitory control to excitatory PCs. In contrast, PCs displayed an increased synaptic E/I ratio (Fig. 5S,T), with differences between sPCs and dPCs. sPCs saw a greater reduction in inhibitory input than dPCs (Fig. 5Q,R) and also displayed an increased probability of spiking during SWRs (Fig. 4E,F). As the strong inhibition PVBCs provide is critical for the selection of PC ensembles (Klausberger and Somogyi, 2008; Ellender et al., 2010), this selective reduction may explain the enlarged PC ensembles (Fig. 3E), aberrant cellular participation (Fig. 3L,M), and more frequent and larger amplitude SWRs in 5xFAD mice (Fig. 2C,D). Intriguingly, the increase in SWRs appears detrimental, despite their role in memory consolidation, as we also observed spatial memory deficits (Fig. 1C). Considering the surprising role SWRs play in downregulating synapses and reducing memory-irrelevant activity (Norimoto et al., 2018), these aberrant SWRs may be interfering with memory-relevant reactivations. Several mechanisms likely contribute to the increase in SWRs. The reduction of sIPSCs that sPCs receive between SWRs (Fig. 5K) may permit the buildup of excitation necessary for SWR initiation. The decreased duration of SWRs (Fig. 2D) may more quickly reset the system for subsequent events. A more complete description would also require investigation of CA3 and CA2 microcircuitry.
Figure 9.

Schematic of alterations to the CA1 microcircuit in 5xFAD mice. In PVBCs, the synaptic E/I ratio during SWRs was reduced, with a reduction in spike rate during SWRs. In sPCs, there was a reduction in sIPSCs, an increased E/I ratio during SWRs and an increased probability of spiking during SWRs. In dPCs, there was an increase in synaptic E/I during spontaneous periods (between SWRs), no change in the E/I ratio during SWRs (with concomitant increases in excitation and inhibition), and enlarged ensembles.
Hyperactivity and inhibitory disruption in AD
Our findings are consistent with growing evidence linking hyperactivity and Aβ aggregation (Zott et al., 2018). Hippocampal hyperactivity is seen in mouse models of early amyloid pathology as increased seizure risk (Palop et al., 2007) and enlarged ensemble activity (Busche et al., 2012). Our results extend these findings to the study of SWRs. In addition, several studies have demonstrated that a preferential disruption to inhibitory cells underlies hyperactivity (Verret et al., 2012; Hazra et al., 2013; Mahar et al., 2016; Hijazi et al., 2019). In the clinical population, seizures are more prevalent and associated with earlier onset of cognitive decline in amnestic mild cognitive impairment (MCI) patients (Vossel et al., 2013). Additionally, task-related engagement of the hippocampus, as tested via event-related fMRI, indicates hippocampal hyperactivation in MCI patients relative to aged controls, while more progressed AD patients experience hypoactivation (Dickerson et al., 2005; Pariente et al., 2005). These studies suggest our observed phenotype may better model MCI rather than fully progressed AD.
SWR alterations in aging and disease
In normal aging, SWR event and ripple frequency decrease (Wiegand et al., 2016; Cowen et al., 2020), contrasting with the phenotype we observed in younger mice. While it is accepted that AD is distinct from accelerated aging (Toepper, 2017), similar decreases in SWRs have been observed in aged AD models. In an apoE-ε4 knock-in model of sporadic AD, SWR event frequency and γ power are reduced in 12- to 18-month mice (Gillespie et al., 2016; Jones et al., 2019). Similarly, in 9- to 12-month TgF344-AD rats, SWR event frequency, power, and γ power are reduced (Stoiljkovic et al., 2019). In younger adults, the findings are somewhat mixed. As seen in a cohort of six three-month 5xFAD mice, SWR event frequency and γ power during non-θ periods are reduced (Iaccarino et al., 2016), the opposite phenotype we observe in slice. In two- to four-month rTg4510 mice, SWR event frequency is unchanged while amplitude and ripple power are reduced (Ciupek et al., 2015). Another study has suggested a failure of PC-PV circuits in amyloid pathology is due to regulation of the GluA4 AMPA receptor, resulting in more frequent SWRs with reduced ripple frequency in three- to four-month APPswe/PS1ΔE9 mice (Xiao et al., 2017). Propagation of SWRs also appears disrupted; three-month APP-PS1 mice show impaired propagation from CA3 to CA1 correlated with increased immunoreactivity for PV (Hollnagel et al., 2019).
One notable difference between our study and others is the acute slice versus in vivo preparation. Slice electrophysiology permits a careful study of the synaptic inputs to different neuronal subtypes during SWRs, an infeasible task in vivo. It is unknown whether awake versus asleep SWRs are differentially affected in AD, and it is unclear which, if either, are better modeled in slice. The choice of AD model also surely has implications. The 5xFAD model has several advantages over other mouse models in replicating human disease: a high ratio of Aβ42 over Aβ40, memory impairment, and neurodegeneration in later disease progression. However, it also has limitations. As with most AD models, it only models the familial variant of the disease, while the sporadic variant accounts for most human cases. Moreover, the presence of five APP/PS1 gene mutations would certainly never be observed in a patient. Another disadvantage is APP overexpression, common to all first-generation transgenic models. Some have noted electrophysiological alterations are more related to APP overexpression than Aβ aggregation (Born et al., 2014). We attempted to address this by examining a one-month cohort (preceding plaque aggregation), in which APP overexpression would still presumably have an effect, a cohort in which we observed no alterations (Figs. 1C, 2C). Fewer studies exist of second-generation APP knock-in models which address the APP overexpression problem, yet have less pronounced disease phenotypes (Sasaguri et al., 2017). In one relevant study, sIPSC amplitude from putative PV cells is decreased in parietal cortex PCs from 18- to 20-month AppNL-F mice, although synaptic E/I balance is unchanged (Chen et al., 2018). It will be critical to study hippocampal network alterations, both at the level of the microcircuit and behaving animal, in novel AD mouse models with greater validity.
Potential mechanisms for PVBC deficit
Several mechanisms may underlie PVBC dysfunction in amyloid pathology. Intrinsic factors including downregulation of the voltage-gated Nav1.1 channel can explain decreased PV cell excitability and cortical network hyperactivity (Verret et al., 2012). It remains to be shown whether different PV subtypes are differentially affected, but according to the Allen Brain Institute Cell Types Database, all identified murine PV clusters express the Scn1a gene encoding Nav1.1 (Lein et al., 2007). Another potential mechanism is loss of perineuronal nets (PNNs), part of the extracellular matrix surrounding soma and proximal dendrites that preferentially ensheathe PV cells (Kwok et al., 2011; Sorg et al., 2016). The function of PNN is incompletely understood, but both enhances PV cell excitability and activity (Slaker et al., 2015; Balmer, 2016). In a prior study, we observed degradation of PNN increases SWR event frequency by ∼50% (Sun et al., 2018), a similar magnitude effect as the current study. Additionally, the selective reduction of excitatory input to PV cells (Fig. 8E1) is consistent with the decrease in miniature EPSCs in PV cells in a brevican knock-out, a major component of PNN (Favuzzi et al., 2017). Moreover, PNN staining is reduced in three-month Tg2576 mice (Cattaud et al., 2018). PNNs are more selectively located around PVBCs (>90%) than PVBSCs (25–50%) or PVAACs (<10%; Yamada and Jinno, 2015), potentially explaining the specificity we observe. However, our experiments would also be consistent with upstream deficits in excitatory input to PV cells. In four-month Tg2576 mice, there is a preferential degeneration of direct entorhinal input to CA1 PV cells; optogenetic restoration of this input rescues synaptic and spatial learning deficits (Yang et al., 2018). The overall cause of disruption to PV cells is likely from several contributing factors, and further studies are required to identify the most salient impairment, and thus the most promising avenue for intervention.
Implications of PVBC-specific deficit
Here, we identified a preferential impairment in PVBC function, concurrent with altered network activity. The selectivity of synaptic alterations in PVBCs suggest they may be a promising target for intervention to restore hippocampal network activity in early amyloid pathology. Given the rapid evolution of tools to manipulate activity in a cell-type-specific manner, this finding is of particular importance considering optogenetic (Iaccarino et al., 2016) and chemogenetic (Hijazi et al., 2019) approaches to ameliorate memory decline in AD. A limitation of these strategies is thatthe PVCre driver will also target PVBSCs and PVAACs, potentially leading to unintended off-target consequences in temporal sequencing. Novel combinatorial approaches using both Creand Flp (He et al., 2016) provide a promising avenue to selectively manipulate distinct neuronal subtypes. For example, the Nkx2.1CreER;LSLFlp mouse would provide an efficient means to record PVAAC activity, in which we saw small alterations that did not reach significance. In conclusion, this study investigated the synaptic input and spiking output of distinct PC and PV cell-types within CA1 microcircuitry over the course of SWR events, providing insight into synaptic deficits in early amyloid pathology,and informing future attempts to manipulate the hippocampal microcircuit.
Footnotes
A.C. was supported by the National Institutes of Health (NIH)/National Institute on Aging (NIA) Grant F31 AG062030, the NIH/National Center for Advancing Translational Sciences Grant TL1 TR001431, and the ARCS Foundation. P.L.B. was supported by the NIH/National Institute of Neurological Disorders and Stroke (NINDS) Grant T32 NS041218. D.T.S.P. and S.V. were partially supported by the NIH/NIA Grant RF1 AG056603. K.C. and S.V. were partially supported by the NIH/NINDS Grant R01 NS108810.
The authors declare no competing financial interests.
References
- Amatniek JC, Hauser WA, DelCastillo-Castaneda C, Jacobs DM, Marder K, Bell K, Albert M, Brandt J, Stern Y (2006) Incidence and predictors of seizures in patients with Alzheimer's disease. Epilepsia 47:867–872. 10.1111/j.1528-1167.2006.00554.x [DOI] [PubMed] [Google Scholar]
- Baimbridge KG, Miller JJ (1982) Immunohistochemical localization of calcium-binding protein in the cerebellum, hippocampal formation and olfactory bulb of the rat. Brain Res 245:223–229. 10.1016/0006-8993(82)90804-6 [DOI] [PubMed] [Google Scholar]
- Balmer TS. (2016) Perineuronal nets enhance the excitability of fast-spiking neurons. eNeuro 3:ENEURO.0112-16.2016 10.1523/ENEURO.0112-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes CA. (1979) Memory deficits associated with senescence: a neurophysiological and behavioral study in the rat. J Comp Physiol Psychol 93:74–104. 10.1037/h0077579 [DOI] [PubMed] [Google Scholar]
- Baude A, Bleasdale C, Dalezios Y, Somogyi P, Klausberger T (2007) Immunoreactivity for the GABAA receptor alpha1 subunit, somatostatin and Connexin36 distinguishes axoaxonic, basket, and bistratified interneurons of the rat hippocampus. Cereb Cortex 17:2094–2107. 10.1093/cercor/bhl117 [DOI] [PubMed] [Google Scholar]
- Berens P. (2009) CircStat: a MATLAB toolbox for circular statistics. J Stat Soft 31:1–21. 10.18637/jss.v031.i10 [DOI] [Google Scholar]
- Born HA, Kim J-Y, Savjani RR, Das P, Dabaghian YA, Guo Q, Yoo JW, Schuler DR, Cirrito JR, Zheng H, Golde TE, Noebels JL, Jankowsky JL (2014) Genetic suppression of transgenic APP rescues Hypersynchronous network activity in a mouse model of Alzeimer's disease. J Neurosci 34:3826–3840. 10.1523/JNEUROSCI.5171-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82:239–259. 10.1007/BF00308809 [DOI] [PubMed] [Google Scholar]
- Busche MA, Eichhoff G, Adelsberger H, Abramowski D, Wiederhold K-H, Haass C, Staufenbiel M, Konnerth A, Garaschuk O (2008) Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer's disease. Science 321:1686–1689. 10.1126/science.1162844 [DOI] [PubMed] [Google Scholar]
- Busche MA, Chen X, Henning HA, Reichwald J, Staufenbiel M, Sakmann B, Konnerth A (2012) Critical role of soluble amyloid-β for early hippocampal hyperactivity in a mouse model of Alzheimer's disease. Proc Natl Acad Sci USA 109:8740–8745. 10.1073/pnas.1206171109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsáki G. (1986) Hippocampal sharp waves: their origin and significance. Brain Res 398:242–252. 10.1016/0006-8993(86)91483-6 [DOI] [PubMed] [Google Scholar]
- Buzsáki G. (2015) Hippocampal sharp wave-ripple: a cognitive biomarker for episodic memory and planning. Hippocampus 25:1073–1188. 10.1002/hipo.22488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsáki G, Leung LW, Vanderwolf CH (1983) Cellular bases of hippocampal EEG in the behaving rat. Brain Res 287:139–171. 10.1016/0165-0173(83)90037-1 [DOI] [PubMed] [Google Scholar]
- Buzsáki G, Logothetis N, Singer W (2013) Scaling brain size, keeping timing: evolutionary preservation of brain rhythms. Neuron 80:751–764. 10.1016/j.neuron.2013.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr MF, Karlsson MP, Frank LM (2012) Transient slow gamma synchrony underlies hippocampal memory replay. Neuron 75:700–713. 10.1016/j.neuron.2012.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattaud V, Bezzina C, Rey CC, Lejards C, Dahan L, Verret L (2018) Early disruption of parvalbumin expression and perineuronal nets in the hippocampus of the Tg2576 mouse model of Alzheimer's disease can be rescued by enriched environment. Neurobiol Aging 72:147–158. 10.1016/j.neurobiolaging.2018.08.024 [DOI] [PubMed] [Google Scholar]
- Chen L, Saito T, Saido TC, Mody I (2018) Novel quantitative analyses of spontaneous synaptic events in cortical pyramidal cells reveal subtle parvalbumin-expressing interneuron dysfunction in a knock-in mouse model of Alzheimer's disease. eNeuro 5: ENEURO.0059-18.2018 10.1523/ENEURO.0059-18.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciupek SM, Cheng J, Ali YO, Lu H-C, Ji D (2015) Progressive functional impairments of hippocampal neurons in a tauopathy mouse model. J Neurosci 35:8118–8131. 10.1523/JNEUROSCI.3130-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements JD, Bekkers JM (1997) Detection of spontaneous synaptic events with an optimally scaled template. Biophys J 73:220–229. 10.1016/S0006-3495(97)78062-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleveland WS. (1979) Robust locally weighted regression and smoothing scatterplots. J Am Stat Assoc 74:829–836. 10.1080/01621459.1979.10481038 [DOI] [Google Scholar]
- Colgin LL. (2016) Rhythms of the hippocampal network. Nat Rev Neurosci 17:239–249. 10.1038/nrn.2016.21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowen SL, Gray DT, Wiegand JP, Schimanski LA, Barnes CA (2020) Age-associated changes in waking hippocampal sharp-wave ripples. Hippocampus 30:28–38. 10.1002/hipo.23005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csicsvari J, Hirase H, Czurkó A, Mamiya A, Buzsáki G (1999) Oscillatory coupling of hippocampal pyramidal cells and interneurons in the behaving rat. J Neurosci 19:274–287. 10.1523/JNEUROSCI.19-01-00274.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dana H, Chen TW, Hu A, Shields BC, Guo C, Looger LL, Kim DS, Svoboda K (2014) Thy1-GCaMP6 transgenic mice for neuronal population imaging in vivo. PLoS One 9:e108697. 10.1371/journal.pone.0108697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickerson BC, Salat DH, Greve DN, Chua EF, Rand-Giovannetti E, Rentz DM, Bertram L, Mullin K, Tanzi RE, Blacker D, Albert MS, Sperling RA (2005) Increased hippocampal activation in mild cognitive impairment compared to normal aging and AD. Neurology 65:404–411. 10.1212/01.wnl.0000171450.97464.49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ego-Stengel V, Wilson MA (2010) Disruption of ripple-associated hippocampal activity during rest impairs spatial learning in the rat. Hippocampus 20:1–10. 10.1002/hipo.20707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellender TJ, Nissen W, Colgin LL, Mann EO, Paulsen O (2010) Priming of hippocampal population bursts by individual perisomatic-targeting interneurons. J Neurosci 30:5979–5991. 10.1523/JNEUROSCI.3962-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eschenko O, Ramadan W, Mölle M, Born J, Sara SJ (2008) Sustained increase in hippocampal sharp-wave ripple activity during slow-wave sleep after learning. Learn Mem 15:222–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favuzzi E, Marques-Smith A, Deogracias R, Winterflood CM, Sánchez-Aguilera A, Mantoan L, Maeso P, Fernandes C, Ewers H, Rico B (2017) Activity-dependent gating of parvalbumin interneuron function by the perineuronal net protein brevican. Neuron 95:639–655.e10. 10.1016/j.neuron.2017.06.028 [DOI] [PubMed] [Google Scholar]
- Fernández-Ruiz A, Oliva A, Oliveira E. d, Rocha-Almeida F, Tingley D, Buzsáki G (2019) Long-duration hippocampal sharp wave ripples improve memory. Science 364:1082–1086. 10.1126/science.aax0758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foffani G, Uzcategui YG, Gal B, Menendez de la Prida L (2007) Reduced spike-timing reliability correlates with the emergence of fast ripples in the rat epileptic hippocampus. Neuron 55:930–941. 10.1016/j.neuron.2007.07.040 [DOI] [PubMed] [Google Scholar]
- Gillespie AK, Jones EA, Lin Y-H, Karlsson MP, Kay K, Yoon SY, Tong LM, Nova P, Carr JS, Frank LM, Huang Y (2016) Apolipoprotein EIV causes age-dependent disruption of slow gamma oscillations during hippocampal sharp-wave ripples. Neuron 90:740–751. 10.1016/j.neuron.2016.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girardeau G, Benchenane K, Wiener SI, Buzsáki G, Zugaro MB (2009) Selective suppression of hippocampal ripples impairs spatial memory. Nat Neurosci 12:1222–1223. 10.1038/nn.2384 [DOI] [PubMed] [Google Scholar]
- Hájos N, Ellender TJ, Zemankovics R, Mann EO, Exley R, Cragg SJ, Freund TF, Paulsen O (2009) Maintaining network activity in submerged hippocampal slices: importance of oxygen supply. Eur J Neurosci 29:319–327. 10.1111/j.1460-9568.2008.06577.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser WA, Morris ML, Heston LL, Anderson VE (1986) Seizures and myoclonus in patients with Alzheimer's disease. Neurology 36:1226–1230. 10.1212/wnl.36.9.1226 [DOI] [PubMed] [Google Scholar]
- Hazra A, Gu F, Aulakh A, Berridge C, Eriksen JL, Ziburkus J (2013) Inhibitory neuron and hippocampal circuit dysfunction in an aged mouse model of Alzheimer's disease. PLoS One 8:e64318. 10.1371/journal.pone.0064318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He M, Tucciarone J, Lee S, Nigro MJ, Kim Y, Levine JM, Kelly SM, Krugikov I, Wu P, Chen Y, Gong L, Hou Y, Osten P, Rudy B, Huang ZJ (2016) Strategies and tools for combinatorial targeting of GABAergic neurons in mouse cerebral cortex. Neuron 91:1228–1243. 10.1016/j.neuron.2016.08.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hijazi S, Heistek TS, Scheltens P, Neumann U, Shimshek DR, Mansvelder HD, Smit AB, Kesteren RE van (2019) Early restoration of parvalbumin interneuron activity prevents memory loss and network hyperexcitability in a mouse model of Alzheimer's disease. Mol Psychiatry. Advance online publication. Retrieved Aug 20, 2019. doi: 10.1038/s41380-019-0483-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hippenmeyer S, Vrieseling E, Sigrist M, Portmann T, Laengle C, Ladle DR, Arber S (2005) A developmental switch in the response of DRG neurons to ETS transcription factor signaling. PLoS Biol 3:e159. 10.1371/journal.pbio.0030159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollnagel J-O, Elzoheiry S, Gorgas K, Kins S, Beretta CA, Kirsch J, Kuhse J, Kann O, Kiss E (2019) Early alterations in hippocampal perisomatic GABAergic synapses and network oscillations in a mouse model of Alzheimer's disease amyloidosis. PLoS One 14:e0209228. 10.1371/journal.pone.0209228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iaccarino HF, Singer AC, Martorell AJ, Rudenko A, Gao F, Gillingham TZ, Mathys H, Seo J, Kritskiy O, Abdurrob F, Adaikkan C, Canter RG, Rueda R, Brown EN, Boyden ES, Tsai LH (2016) Gamma frequency entrainment attenuates amyloid load and modifies microglia. Nature 540:230–235. 10.1038/nature20587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones EA, Gillespie AK, Yoon SY, Frank LM, Huang Y (2019) Early hippocampal sharp-wave ripple deficits predict later learning and memory impairments in an Alzheimer's disease mouse model. Cell Rep 29:2123–2133.e4. 10.1016/j.celrep.2019.10.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jura B, Macrez N, Meyrand P, Bem T (2019) Deficit in hippocampal ripples does not preclude spatial memory formation in APP/PS1 mice. Sci Rep 9:20129 10.1038/s41598-019-56582-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R (2003) APP processing and synaptic function. Neuron 37:925–937. 10.1016/s0896-6273(03)00124-7 [DOI] [PubMed] [Google Scholar]
- Karlsson MP, Frank LM (2009) Awake replay of remote experiences in the hippocampus. Nat Neurosci 12:913–918. 10.1038/nn.2344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katona L, Lapray D, Viney TJ, Oulhaj A, Borhegyi Z, Micklem BR, Klausberger T, Somogyi P (2014) Sleep and movement differentiates actions of two types of somatostatin-expressing GABAergic interneuron in rat hippocampus. Neuron 82:872–886. 10.1016/j.neuron.2014.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klausberger T, Somogyi P (2008) Neuronal diversity and temporal dynamics: the unity of hippocampal circuit operations. Science 321:53–57. 10.1126/science.1149381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudrimoti HS, Barnes CA, McNaughton BL (1999) Reactivation of hippocampal cell assemblies: effects of behavioral state, experience, and EEG dynamics. J Neurosci 19:4090–4101. 10.1523/JNEUROSCI.19-10-04090.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwok JCF, Dick G, Wang D, Fawcett JW (2011) Extracellular matrix and perineuronal nets in CNS repair. Dev Neurobiol 71:1073–1089. 10.1002/dneu.20974 [DOI] [PubMed] [Google Scholar]
- Lapray D, Lasztoczi B, Lagler M, Viney TJ, Katona L, Valenti O, Hartwich K, Borhegyi Z, Somogyi P, Klausberger T (2012) Behavior-dependent specialization of identified hippocampal interneurons. Nat Neurosci 15:1265–1271. 10.1038/nn.3176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AK, Wilson MA (2002) Memory of sequential experience in the hippocampus during slow wave sleep. Neuron 36:1183–1194. 10.1016/s0896-6273(02)01096-6 [DOI] [PubMed] [Google Scholar]
- Lee SH, Marchionni I, Bezaire M, Varga C, Danielson N, Lovett-Barron M, Losonczy A, Soltesz I (2014) Parvalbumin-positive basket cells differentiate among hippocampal pyramidal cells. Neuron 82:1129–1144. 10.1016/j.neuron.2014.03.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, Chen L, Chen L, Chen TM, Chin MC, Chong J, Crook BE, Czaplinska A, Dang CN, Datta S, Dee NR, et al. (2007) Genome-wide atlas of gene expression in the adult mouse brain. Nature 445:168–176. 10.1038/nature05453 [DOI] [PubMed] [Google Scholar]
- Le Van Quyen M, Bragin A, Staba R, Crépon B, Wilson CL, Engel J (2008) Cell type-specific firing during ripple oscillations in the hippocampal formation of humans. J Neurosci 28:6104–6110. 10.1523/JNEUROSCI.0437-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Sun H, Chen Z, Xu H, Bu G, Zheng H (2016) Implications of GABAergic neurotransmission in Alzheimer's disease. Front Aging Neurosci 8:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, Lein ES, Zeng H (2010) A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci 13:133–140. 10.1038/nn.2467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahar I, Albuquerque MS, Mondragon-Rodriguez S, Cavanagh C, Davoli MA, Chabot J-G, Williams S, Mechawar N, Quirion R, Krantic S (2016) Phenotypic alterations in hippocampal NPY- and PV-expressing interneurons in a presymptomatic transgenic mouse model of Alzheimer's disease. Front Aging Neurosci 8:327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroteaux M, Liu SJ (2016) Alteration of AMPA receptor-mediated synaptic transmission by Alexa Fluor 488 and 594 in cerebellar stellate cells. eNeuro 3: ENEURO.0109-15.2016 10.1523/ENEURO.0109-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP. (2004) Pathways towards and away from Alzheimer's disease. Nature 430:631–639. 10.1038/nature02621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyawaki T, Norimoto H, Ishikawa T, Watanabe Y, Matsuki N, Ikegaya Y (2014) Dopamine receptor activation reorganizes neuronal ensembles during hippocampal sharp waves in vitro. PLoS One 9:e104438. 10.1371/journal.pone.0104438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuseki K, Buzsáki G (2013) Preconfigured, skewed distribution of firing rates in the hippocampus and entorhinal cortex. Cell Rep 4:1010–1021. 10.1016/j.celrep.2013.07.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuseki K, Diba K, Pastalkova E, Buzsáki G (2011) Hippocampal CA1 pyramidal cells form functionally distinct sublayers. Nat Neurosci 14:1174–1181. 10.1038/nn.2894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nádasdy Z, Hirase H, Czurkó A, Csicsvari J, Buzsáki G (1999) Replay and time compression of recurring spike sequences in the hippocampus. J Neurosci 19:9497–9507. 10.1523/JNEUROSCI.19-21-09497.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicole O, Hadzibegovic S, Gajda J, Bontempi B, Bem T, Meyrand P (2016) Soluble amyloid beta oligomers block the learning-induced increase in hippocampal sharp wave-ripple rate and impair spatial memory formation. Sci Rep 6:22728. 10.1038/srep22728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norimoto H, Makino K, Gao M, Shikano Y, Okamoto K, Ishikawa T, Sasaki T, Hioki H, Fujisawa S, Ikegaya Y (2018) Hippocampal ripples down-regulate synapses. Science 359:1524–1527. 10.1126/science.aao0702 [DOI] [PubMed] [Google Scholar]
- Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Eldik LV, Berry R, Vassar R (2006) Intraneuronal β-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J Neurosci 26:10129–10140. 10.1523/JNEUROSCI.1202-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ognjanovski N, Schaeffer S, Wu J, Mofakham S, Maruyama D, Zochowski M, Aton SJ (2017) Parvalbumin-expressing interneurons coordinate hippocampal network dynamics required for memory consolidation. Nat Commun 8:15039. 10.1038/ncomms15039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno M. (2009) Failures to reconsolidate memory in a mouse model of Alzheimer's disease. Neurobiol Learn Mem 92:455–459. 10.1016/j.nlm.2009.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neill J, Senior TJ, Allen K, Huxter JR, Csicsvari J (2008) Reactivation of experience-dependent cell assembly patterns in the hippocampus. Nat Neurosci 11:209–215. 10.1038/nn2037 [DOI] [PubMed] [Google Scholar]
- Palop JJ, Mucke L (2009) Epilepsy and cognitive impairments in Alzheimer disease. Arch Neurol 66:435–440. 10.1001/archneurol.2009.15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Mucke L (2010) Amyloid-beta-induced neuronal dysfunction in Alzheimer's disease: from synapses toward neural networks. Nat Neurosci 13:812–818. 10.1038/nn.2583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N, Yoo J, Ho KO, Yu GQ, Kreitzer A, Finkbeiner S, Noebels JL, Mucke L (2007) Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer's disease. Neuron 55:697–711. 10.1016/j.neuron.2007.07.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pariente J, Cole S, Henson R, Clare L, Kennedy A, Rossor M, Cipoloti L, Puel M, Demonet JF, Chollet F, Frackowiak RSJ (2005) Alzheimer's patients engage an alternative network during a memory task. Ann Neurol 58:870–879. 10.1002/ana.20653 [DOI] [PubMed] [Google Scholar]
- Pelkey KA, Barksdale E, Craig MT, Yuan X, Sukumaran M, Vargish GA, Mitchell RM, Wyeth MS, Petralia RS, Chittajallu R, Karlsson RM, Cameron HA, Murata Y, Colonnese MT, Worley PF, McBain CJ (2015) Pentraxins coordinate excitatory synapse maturation and circuit integration of parvalbumin interneurons. Neuron 85:1257–1272. 10.1016/j.neuron.2015.02.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelkey KA, Chittajallu R, Craig MT, Tricoire L, Wester JC, McBain CJ (2017) Hippocampal GABAergic inhibitory interneurons. Physiol Rev 97:1619–1747. 10.1152/physrev.00007.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellow S, Chopin P, File SE, Briley M (1985) Validation of open:closed arm entries in an elevated plus-maze as a measure of anxiety in the rat. J Neurosci Methods 14:149–167. 10.1016/0165-0270(85)90031-7 [DOI] [PubMed] [Google Scholar]
- Pozueta J, Lefort R, Shelanski ML (2013) Synaptic changes in Alzheimer's disease and its models. Neuroscience 251:51–65. 10.1016/j.neuroscience.2012.05.050 [DOI] [PubMed] [Google Scholar]
- Roux L, Hu B, Eichler R, Stark E, Buzsáki G (2017) Sharp wave ripples during learning stabilize the hippocampal spatial map. Nat Neurosci 20:845–853. 10.1038/nn.4543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaguri H, Nilsson P, Hashimoto S, Nagata K, Saito T, De Strooper B, Hardy J, Vassar R, Winblad B, Saido TC (2017) APP mouse models for Alzheimer's disease preclinical studies. EMBO J 36:2473–2487. 10.15252/embj.201797397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlingloff D, Káli S, Freund TF, Hájos N, Gulyás AI (2014) Mechanisms of sharp wave initiation and ripple generation. J Neurosci 34:11385–11398. 10.1523/JNEUROSCI.0867-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT (2011) Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med 1:a006189. 10.1101/cshperspect.a006189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ (2008) Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med 14:837–842. 10.1038/nm1782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siapas AG, Wilson MA (1998) Coordinated interactions between hippocampal ripples and cortical spindles during slow-wave sleep. Neuron 21:1123–1128. [DOI] [PubMed] [Google Scholar]
- Skaggs WE, McNaughton BL (1996) Replay of neuronal firing sequences in rat hippocampus during sleep following spatial experience. Science 271:1870–1873. 10.1126/science.271.5257.1870 [DOI] [PubMed] [Google Scholar]
- Slaker M, Churchill L, Todd RP, Blacktop JM, Zuloaga DG, Raber J, Darling RA, Brown TE, Sorg BA (2015) Removal of perineuronal nets in the medial prefrontal cortex impairs the acquisition and reconsolidation of a cocaine-induced conditioned place preference memory. J Neurosci 35:4190–4202. 10.1523/JNEUROSCI.3592-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slomianka L, Amrein I, Knuesel I, Sørensen JC, Wolfer DP (2011) Hippocampal pyramidal cells: the reemergence of cortical lamination. Brain Struct Funct 216:301–317. 10.1007/s00429-011-0322-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somogyi P, Katona L, Klausberger T, Lasztóczi B, Viney TJ (2014) Temporal redistribution of inhibition over neuronal subcellular domains underlies state-dependent rhythmic change of excitability in the hippocampus. Philos Trans R Soc Lond B Biol Sci 369:20120518. 10.1098/rstb.2012.0518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorg BA, Berretta S, Blacktop JM, Fawcett JW, Kitagawa H, Kwok JCF, Miquel M (2016) Casting a wide net: role of perineuronal nets in neural plasticity. J Neurosci 36:11459–11468. 10.1523/JNEUROSCI.2351-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoiljkovic M, Kelley C, Stutz B, Horvath TL, Hajós M (2019) Altered cortical and hippocampal excitability in TgF344-AD rats modeling Alzheimer's disease pathology. Cereb Cortex 29:2716–2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun ZY, Bozzelli PL, Caccavano A, Allen M, Balmuth J, Vicini S, Wu JY, Conant K (2018) Disruption of perineuronal nets increases the frequency of sharp wave ripple events. Hippocampus 28:42–52. 10.1002/hipo.22804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting JT, Daigle TL, Chen Q, Feng G (2014) Acute brain slice methods for adult and aging animals: application of targeted patch clamp analysis and optogenetics. Methods Mol Biol 1183:221–242. 10.1007/978-1-4939-1096-0_14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting JT, Lee BR, Chong P, Soler-Llavina G, Cobbs C, Koch C, Zeng H, Lein E (2018) Preparation of acute brain slices using an optimized N-methyl-D-glucamine protective recovery method. J Vis Exp. Advance online publication. Retrieved Feb 26, 2018. doi: 10.3791/53825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toepper M. (2017) Dissociating normal aging from Alzheimer's disease: a view from cognitive neuroscience. J Alzheimers Dis 57:331–352. 10.3233/JAD-161099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tohda C, Urano T, Umezaki M, Nemere I, Kuboyama T (2012) Diosgenin is an exogenous activator of 1,25D3-MARRS/Pdia3/ERp57 and improves Alzheimer's disease pathologies in 5XFAD mice. Sci Rep 2:535. 10.1038/srep00535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valero M, Cid E, Averkin RG, Aguilar J, Sanchez-Aguilera A, Viney TJ, Gomez-Dominguez D, Bellistri E, de la Prida LM (2015) Determinants of different deep and superficial CA1 pyramidal cell dynamics during sharp-wave ripples. Nat Neurosci 18:1281–1290. 10.1038/nn.4074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga C, Golshani P, Soltesz I (2012) Frequency-invariant temporal ordering of interneuronal discharges during hippocampal oscillations in awake mice. Proc Natl Acad Sci USA 109:E2726–E2734. 10.1073/pnas.1210929109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga C, Oijala M, Lish J, Szabo GG, Bezaire M, Marchionni I, Golshani P, Soltesz I (2014) Functional fission of parvalbumin interneuron classes during fast network events. Elife 3:e04006 10.7554/eLife.04006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ven GM, van de Trouche S, McNamara CG, Allen K, Dupret D (2016) Hippocampal offline reactivation consolidates recently formed cell assembly patterns during sharp wave-ripples. Neuron 92:968–974. 10.1016/j.neuron.2016.10.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verret L, Mann EO, Hang GB, Barth AMI, Cobos I, Ho K, Devidze N, Masliah E, Kreitzer AC, Mody I, Mucke L, Palop JJ (2012) Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell 149:708–721. 10.1016/j.cell.2012.02.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viney TJ, Lasztoczi B, Katona L, Crump MG, Tukker JJ, Klausberger T, Somogyi P (2013) Network state-dependent inhibition of identified hippocampal CA3 axo-axonic cells in vivo. Nat Neurosci 16:1802–1811. 10.1038/nn.3550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vossel KA, Beagle AJ, Rabinovici GD, Shu H, Lee SE, Naasan G, Hegde M, Cornes SB, Henry ML, Nelson AB, Seeley WW, Geschwind MD, Gorno-Tempini ML, Shih T, Kirsch HE, Garcia PA, Miller BL, Mucke L (2013) Seizures and epileptiform activity in the early stages of Alzheimer disease. JAMA Neurol 70:1158–1166. 10.1001/jamaneurol.2013.136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiegand JP, Gray DT, Schimanski LA, Lipa P, Barnes CA, Cowen SL (2016) Age is associated with reduced sharp-wave ripple frequency and altered patterns of neuronal variability. J Neurosci 36:5650–5660. 10.1523/JNEUROSCI.3069-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MA, McNaughton BL (1994) Reactivation of hippocampal ensemble memories during sleep. Science 265:676–679. 10.1126/science.8036517 [DOI] [PubMed] [Google Scholar]
- Wobbrock JO, Findlater L, Gergle D, Higgins JJ (2011) The aligned rank transform for nonparametric factorial analyses using only ANOVA procedures. Proceedings of the SIGCHI Conference on Human Factors in Computing Systems, pp 143–146. Accessed December 12, 2019. Available at http://doi.acm.org/10.1145/1978942.1978963 10.1145/1978942.1978963 [DOI]
- Xiao MF, Xu D, Craig MT, Pelkey KA, Chien CC, Shi Y, Zhang J, Resnick S, Pletnikova O, Salmon D, Brewer J, Edland S, Wegiel J, Tycko B, Savonenko A, Reeves RH, Troncoso JC, McBain CJ, Galasko D, Worley PF (2017) NPTX2 and cognitive dysfunction in Alzheimer's disease. Elife 6:e23798 10.7554/eLife.23798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada J, Jinno S (2015) Subclass-specific formation of perineuronal nets around parvalbumin-expressing GABAergic neurons in Ammon's horn of the mouse hippocampus. J Comp Neurol 523:790–804. 10.1002/cne.23712 [DOI] [PubMed] [Google Scholar]
- Yang X, Yao C, Tian T, Li X, Yan H, Wu J, Li H, Pei L, Liu D, Tian Q, Zhu LQ, Lu Y (2018) A novel mechanism of memory loss in Alzheimer's disease mice via the degeneration of entorhinal-CA1 synapses. Mol Psychiatry 23:199–210. 10.1038/mp.2016.151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ylinen A, Bragin A, Nádasdy Z, Jandó G, Szabó I, Sik A, Buzsáki G (1995) Sharp wave-associated high-frequency oscillation (200 Hz) in the intact hippocampus: network and intracellular mechanisms. J Neurosci 15:30–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youmans KL, Tai LM, Kanekiyo T, Stine WB, Michon SC, Nwabuisi-Heath E, Manelli AM, Fu Y, Riordan S, Eimer WA, Binder L, Bu G, Yu C, Hartley DM, LaDu MJ (2012) Intraneuronal Aβ detection in 5xFAD mice by a new Aβ-specific antibody. Mol Neurodegener 7:8. 10.1186/1750-1326-7-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zott B, Busche MA, Sperling RA, Konnerth A (2018) What happens with the circuit in Alzheimer's disease in mice and humans? Annu Rev Neurosci 41:277–297. 10.1146/annurev-neuro-080317-061725 [DOI] [PMC free article] [PubMed] [Google Scholar]