

Abstract
Since the introduction of the cancer stem cell (CSC) hypothesis, accumulating evidence shows that most cancers present stem-like niches. However, therapies aimed at targeting this niche have not been as successful as expected. New evidence regarding CSCs hierarchy, similarities with normal tissue stem cells and cell plasticity might be key in understanding their role in cancer biology and how to efficiently eliminate them. In this Chapter, we discuss what is known in breast and prostate CSCs from their initial discoveries to the current therapeutic efforts in the field. Future challenges towards better CSC identification and isolation strategies will be key to shed light into how CSCs could accurately be targeted in combination to traditional therapies to ultimately prolong patient survival.
1. Origin and evolution of cancer stem cells: Consensus and controversies on single vs. multi-potent progenitors and stem cell hierarchy
All of our tissues are formed thanks to the activity of stem cells. The mother of all cells is, in fact, a stem cell, that has all the requisite information necessary to generate an entire organism. The first time the term stem cell was used in the scientific literature was by Ernst Häckel (in German, ‘Stammzelle”). He employed it to refer to a common unicellular ancestor from which he imagined all multicellular organisms evolved, inspired by Darwin’s stem trees (in German, “Stammbaüme”) that represented the evolution of organisms (Häckel, 1868; Ramalho-Santos & Willenbring, 2007). Häckel later on, once again comparing evolution to embryology, proposed that the fertilized egg also be called a stem cell (Ramalho-Santos & Willenbring, 2007). Around the same time, thanks to the contributions of Ehrlich (1879), the question was raised of whether a common precursor of the various blood cell types existed. In the beginning of the 20th century, several researchers began to use the term stem cell to refer to the common precursor of the blood system (Ramalho-Santos & Willenbring, 2007).
What defines whether a single stem cell is able to regenerate a whole organism or simply regenerate one specific tissue is the stem cell hierarchy. This is what defines whether the stem cell is totipotent, pluripotent, multipotent or simply oligopotent -or even unipotent. Therefore, to be considered “stem” a cell should have these capacities: (1) Be able to self-renew; (2) Be able to regenerate certain tissues/organs/organism; (3) Generate daughter cells that have a lower regeneration capacity or hierarchy (i.e., if a stem cell can regenerate the whole mammary gland, its daughter cells can at the most regenerate certain cell layers of the gland, but are not able to regenerate the whole organ) (Kreso & Dick, 2014).
Cancer stem cells (CSCs) were first identified by John Dick in acute myeloid leukemia in the late 1990s (Bonnet & Dick, 1997; Dick, 1996). Research in CSCs dramatically increased since the beginning of the 21st century (Fig. 1A and B). In fact, publications in both breast and prostate cancer followed a similar trend, starting to spike around 2007–2008 coinciding with a renewed interest and controversies around CSCs and their potential for new therapies (Visvader & Lindeman, 2008). CSCs came back to the spotlight around that year influenced by several discoveries: (a) The discovery of a highly tumorigenic subpopulation of breast cancer cells identified as CD44+/CD24 (−/low) by the Clarke group (Al-Hajj, Wicha, Benito-Hernandez, Morrison, & Clarke, 2003; Liu et al., 2007) further confirmed as resistant to chemotherapy in the beginning of 2008 by Chang and collaborators (Li, Lewis, et al., 2008); (b) By the end of 2017, the discovery of ALDH1 as a marker of normal and malignant stem cells (Ginestier et al., 2007); (c) The identification of embryonic stem cell markers (such as Nanog, Oct4, Sox2 and c-Myc) present in poorly differentiated tumors (Ben-Porath et al., 2008); (d) A new assay to study the capacity of isolated tumor cells to re-generate the original tumor, using new more severely immunocompromised mice (NOD-SCID-IL2Rγnull or NSG) and novel implantation strategies that led to the discovery that CSCs are not as “rare” within tumors as it was previously suggested (Quintana et al., 2008); and (e) The discovery that differentiated cells could be reprogrammed into pluripotent stem cells in 2006 (Takahashi & Yamanaka, 2006) led several researchers to speculate that the tumor microenvironment could reprogram cancer cells into stem cells.
Fig. 1.
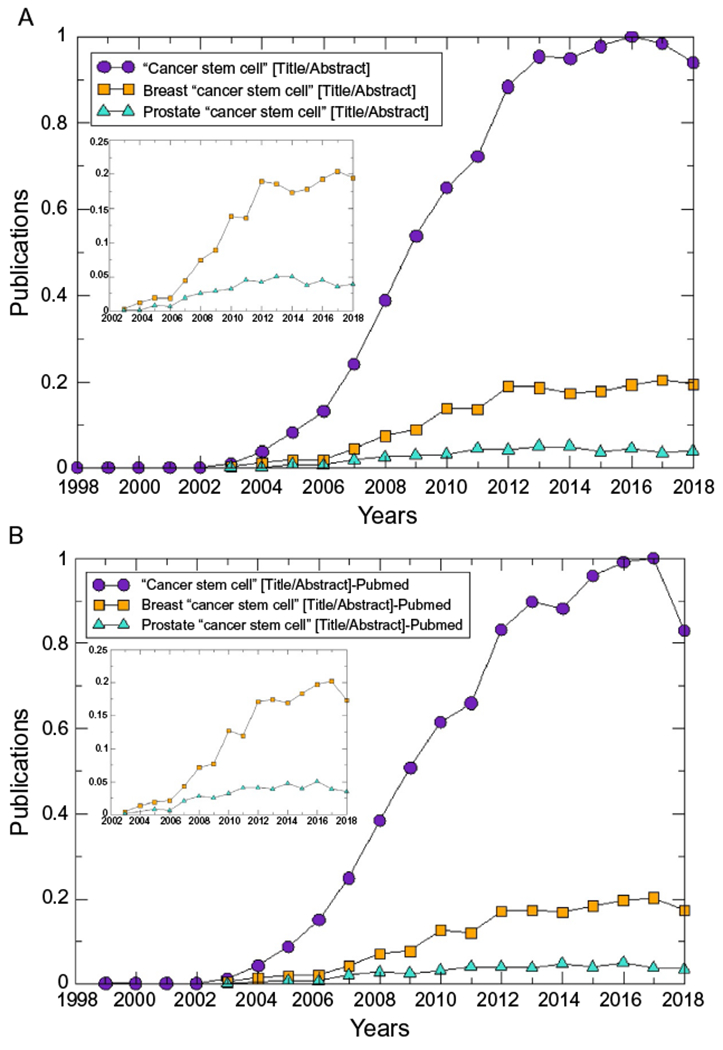
Evolution in the number of published articles in breast and prostate cancer stem cells overtime. (A) Results from the database “Dimensions” (https://www.dimensions.ai/); (B) Results from “Pubmed” (https://www.ncbi.nlm.nih.gov/pubmed/). Insets in both cases show the trends for breast and prostate CSCs with higher detail. In each case, the number of publications was corrected by the total number of publications each year and normalized to the maximum number of publications with the query “Cancer stem cell”.
As with normal stem cells, hierarchy has also been attributed to CSCs (Dick, 2009; Reya, Morrison, Clarke, & Weissman, 2001). The main distinction to normal stem cells that needs to be clarified here is that with normal stem cells, the pluripotent stem cell that gives rise to the entire tissue could phenotypically be the same as the one that maintains it. However, this is not the case for CSCs. The CSC model proposes that a small subpopulation of cells can regenerate the entire tumor heterogeneity (Shackleton, Quintana, Fearon, & Morrison, 2009). Even though this definition could be applied to both the origin of the tumor and the cells that maintain it, the first cell is almost certainty phenotypically different to the cell that maintains the tumor, since the latter have more likely accumulated a set of mutations.
Hierarchy within breast cancer has been the subject of extensive studies (Al-Hajj et al., 2003; Fridriksdottir et al., 2017; Gupta et al., 2011; Kreso & Dick, 2014; Visvader, 2009). In 2012, research from Mina Bissell and Ole Petersen’s groups using 60 primary breast tumors as well as diverse breast cancer cell lines, challenged the CSC model showing that tumor cells from different hierarchies (basal-like CD271+—p75NTR—and luminal-like MM+—milk mucin-) can initiate a tumor in vivo (Kim et al., 2012). Moreover, tumors generated by the luminal-like cells were larger and more invasive than the ones triggered by their basal-like counterpart. This indicates that while there is a differentiation hierarchy within these tumors, there does not seem to be a stem cell hierarchy since all of these cells were able to act as stem cells, capable of both self-renewal and functioning as “tumor-initiating” cells, supporting the clonal evolution model (Campbell & Polyak, 2007). However, it is true that the cells with stem markers (basal-like cancer cells) were able to recapitulate the full heterogeneity of the tumor, which might indicate that even though all of the tumor cells were able to initiate the tumor, only the more undifferentiated cells held the capacity to act as pluripotent progenitors. However, it seems clear that basal-like cells are not a requirement for breast tumor aggressiveness.
One alternative explanation would be that both differentiated and undifferentiated cancer cells can potentially convert into tumor-initiating CSCs in a reversible manner, termed cancer cell plasticity (Batlle & Clevers, 2017; Kreso & Dick, 2014). The role of the microenvironment has been shown to be key in determining tumor cell transitioning between stem and non-stem states, as will be discussed below.
The debate around a stem cell hierarchy of breast cancer stem cells remains a matter of intensive research with the potential to unravel key insights for patient care. A recent relevant study determined a novel differentiation hierarchy of breast cancer cells and found that CSCs and early progenitors correlated with metastasis and poorer outcome in a cohort of 20 breast cancer patients (Bliss et al., 2018).
In prostate, the identification and characterization of CSCs seems to be different between androgen-dependent and castrate-resistant tumors (Taylor, Toivanen, & Risbridger, 2010). In androgen-dependent prostate cancers, the normal basal stem cell marker CD133 was also identified as a marker of CSCs. A CSC population expressing the basal cell markers CD133, CD44 and/or integrin α2β1 was able to regenerate tumors that resembled the original tumor when xenografted in animals (Gu, Yuan, Wills, & Kasper, 2007; Li, Zhou, et al., 2008; Miki et al., 2007; Rybak, Bristow, & Kapoor, 2015). However, immortalized human prostate cells expressing integrin α2β1, but not CD133/AC133, which is consistent with a more differentiated proliferative progeny, also formed xenografts with malignant characteristics (Taylor, Toivanen, Frydenberg, Pedersen, & Harewood, 2012). Human prostate cancer cells derived from androgen-insensitive DU145 cells and propagated under serum-free conditions as non-adherent spheres express the surface markers CD44, integrin α2β1 and CD24, both basal and luminal cytokeratins, and display increased tumorigenicity in vivo (Rybak, He, Kapoor, Cutz, & Tang, 2011). Therefore, it is evident that CSCs from different hierarchies are capable of regenerating tumors. The recent generation of patient-derived prostate cancer organoid cultures that recapitulate the diverse mutational landscape and histology observed in their original prostate cancer tissue samples will likely further aid in the characterization of prostate CSCs, as well as improve the understanding of the molecular determinants of therapeutic and castration resistance (Gao et al., 2014; Rybak et al., 2015).
2. Are cancer stem cells related to the normal stem cells within the niche?
CSCs are different from normal stem cells as much as cancer cells are distinctive from normal cells. According to the CSC model, normal stem and progenitor cells are considered to be the most likely targets of transformation; however, no normal cells in particular are identified as such by the clonal evolution model (Campbell & Polyak, 2007). Evidence supporting the relationship between normal stem cells and cancer stem cells include the notion that normal stem cells within epithelia are “long-lived” cells compared to differentiated cells, making it more likely that these cells will acquire the multiple mutations needed to become cancer (Miller, Lavker, & Sun, 2005). Normal stem cells have enormous proliferative potential, but tend to be slow-cycling within epithelia and can divide symmetrically to self-renew or produce more differentiated daughter cells. Being long-term residents of the epithelium, stem cells are uniquely susceptible to the accumulation of oncogenic changes. These traits could also explain their proliferative capacity, and the phenotypic heterogeneity observed in tumors. These cells also share many overlapping traits, including induction of angiogenesis, resistance to apoptosis and drugs, and cell migration (Allinen et al., 2004; Bapat, 2007; Bissell & Labarge, 2005; Campbell & Polyak, 2007; Hu et al., 2005; Miller et al., 2005; Spradling, Drummond-Barbosa, & Kai, 2001; Wicha, Liu, & Dontu, 2006).
In the breast, normal stem cells give rise to the progenitors of the luminal and basal myoepithelial compartment, although controversies continue regarding the existence of a common bipotent progenitor between the luminal and myoepithelial compartments and whether stem cells that maintain both compartments are localized in each one of those (Bach et al., 2017; Visvader & Clevers, 2016; Visvader & Lindeman, 2006, 2011; Visvader & Stingl, 2014; Wang, Christin, Oktay, & Guo, 2017).
Evidence in favor of normal stem cells being the target of initial transformation include the fact that most breast cancers arise from the segment of terminal ductules where normal stem cells are believed to be located (Villadsen, 2005; Villadsen et al., 2007). In addition, a marker of normal breast cells, CD44, as mentioned above, is used to identify breast cancer stem cells (Al-Hajj et al., 2003; Shipitsin et al., 2007). Also, cancer CD44+ cells may give rise to a hierarchy of tumor cells resembling those in the normal mammary gland, since cancer CD24+ cells are similar to normal CD24+ cells and have genetic alterations not present in cancer CD44+ cells (Shipitsin et al., 2007). Interestingly, most of the surface markers used to identify CSCs in different types of tumors are derived from known normal embryonic or adult stem cell surface markers, suggesting that CSCs might originate from normal stem cells (Kim & Ryu, 2017; Tirino et al., 2013).
An alternative explanation for the similarities between CSCs in breast tumors and the normal stem cells within the tissue is that, as discussed above, plasticity of cancer cells could convert any cells within the niche into a CSC through a combination of genetic alterations and microenvironmental cues (Batlle & Clevers, 2017; Cabrera, Hollingsworth, & Hurt, 2015; Cazet et al., 2018; Garg, 2017; Meacham & Morrison, 2013). This supports the possibility that differentiated tumor cells can reversibly convert to CSCs and re-generate a tumor or conquer a metastatic niche. In this model, there is no unidirectional stem cell hierarchy within the tumor.
In prostate, there is substantial controversy related to the normal stem cells within the tissue, which has led to multiple proposed differentiation hierarchies (Taylor et al., 2010). Linear vs. bidirectional models have been proposed to explain the generation of this tissue. The linear model of differentiation was established in 1989 where stem cells within the basal compartment either self-renew to generate more basal cells or give rise to a multipotent progenitor that can then differentiate to luminal or neuroendocrine cells in a linear manner (Isaacs & Coffey, 1989). The bidirectional model proposes that a common stem cell gives rise to lineage specific progenitors that, in turn, will generate the luminal, basal and neuroendocrine compartments (Wang, Hayward, Cao, Thayer, & Cunha, 2001). There is a third model, called the independent lineage model, established upon the discovery that there are also stem cells within the luminal compartment. Therefore, each cell type (basal, luminal or neuroendocrine) will derive from their own stem progenitor cells. It is also possible that basal and/or luminal stem cells can be multipotent and also generate the opposing lineage. Overall, it is likely that cells with stem properties occur in diverse cell types and that the characteristics that define a stem cell can be turned on or off depending on their response to extrinsic or intrinsic factors.
Without a clear definition of stem cells in normal prostate (and considering there may be more than one cell type), it is difficult to determine whether the cancer cell of origin in prostate cancer is a stem cell, multipotent progenitor/transient amplifying cell, or a more differentiated progeny cell. There is evidence indicating that tumor initiating cells can include both basal (cytokeratin—CK—CK5+/CK8−) and luminal cells (CK5−/CK8+) (Taylor et al., 2010). Intermediate cells express CKs of both basal and luminal cells (CK5+/CK8+). However, a diagnostic identifier of human prostate cancer is the loss of basal cells. Therefore, prostate cancer can potentially arise from oncogenic transformation of CK5+ CK8− basal cells resulting in rapid differentiation into a luminal phenotype, or alternatively from stem or multipotent progenitor cells within the CK5+ CK8+ intermediate or CK5− CK8+ luminal populations. Accumulating evidence shows that prostate cancer initiation can occur from luminal (and potentially intermediate) cells (Ellwood-Yen et al., 2003; Iwata et al., 2010; Ma et al., 2005). Alternatively, a body of work by the Witte Laboratory provides evidence suggesting that basal cells can initiate preneoplastic and cancerous lesions (Lawson et al., 2010; Wang et al., 2006). Therefore, similar to breast cancer, there may be more than one cancer cell type capable of generating human prostate cancer, which may correlate with tumor phenotype, but this possibility requires formal experimental documentation.
3. Similarities and differences between cancer stem cells and undifferentiated aggressive cancer cells: Dormancy and beyond
During tissue and organ development, epithelial cells undergo a physiological process called epithelial-to-mesenchymal transition (EMT) through which they lose cell polarity, cell-cell contacts and cell-matrix adhesions, acquiring a more motile mesenchymal-like phenotype. Cancer cells that reside within an epithelium can acquire deregulated EMT programs that convert them into migratory cells and this can, eventually, contribute to metastasis (Larue & Bellacosa, 2005; Nistico, Bissell, & Radisky, 2012). It is believed that CSCs share molecular pathways with EMT that regulate metastatic cell dissemination and the establishment of a new tumor niche (Agliano, Calvo, & Box, 2017; Wang, Jiang, Liang, & Tang, 2015). These pathways include: activation of TGFβ, Wnt, Notch, Hedgehog and tyrosine kinase receptors (e.g., EGFR, MET among others). It is also likely that even if not occurring in the same cell, EMT mediators might contribute to modify the microenvironment and, in turn, promote CSC phenotype, further conferring resistance to different therapies and increased ability of tumor regrowth (Nistico et al., 2012).
Mitotic arrest in G0-G1—quiescence—for long periods of time is one operational definition of “dormancy”. Adult stem cells in different tissues spend certain periods of dormancy, awaiting the right signals to differentiate and repopulate the tissue (Essers & Trumpp, 2010; Fuchs, 2009). On the basis of this premise, stem cells are often identified by their propensity to retain DNA labels much longer than their rapidly proliferating off spring.
A fraction of cells within each tumor is believed to undergo certain periods of dormancy (Talukdar et al., 2019). This dormant state is reversible and tumor cells can recover the ability to proliferate once closed vessels reopen or new vasculatures reach the hypoxic areas. Chaffer and Weinberg (2011) reported that CSCs have enhanced tumor-initiating potential, but with temporary growth arrest within a tumor. A fraction of tumor cells can migrate outside the primary tumor, and the majority of disseminated tumor cells retain a state of dormancy and these are believed to be responsible for cancer metastasis and relapse (Sosa, Bragado, & Aguirre-Ghiso, 2014). In addition, some findings indicated that disseminated tumor cells could present a CSC phenotype (Gao, Zhang, Tang, & Liang, 2017; Goss & Chambers, 2010).
Dormancy may also be a mechanism for the resistance of CSCs to antiproliferative chemotherapy (Talukdar et al., 2019). Moreover, if indeed CSCs appear in a dormant state, this could explain local recurrence or distant metastasis after long lag periods (Clevers, 2011).
In breast cancer in particular, several similarities between CSCs and metastatic cancer cells have been proposed. Evidence has been provided that induction of EMT in human or mouse mammary epithelial result in the generation of cells with stem cell properties, such as self-renewal and resistance to toxins (Mani et al., 2008; Nistico et al., 2012). Also, Balic et al. showed that bone marrow metastases are enriched in CD44high/CD24low cells, markers associated with CSCs (Balic et al., 2006). Along these lines, tumorigenic CD44high/CD24/low cells have been found in metastatic pleural fluid in patients with breast cancers (Al-Hajj et al., 2003).
Also, breast tumor epithelial cells that undergo EMT express markers of CSCs (CD44high/CD24low) and display tumor-initiating potential, such as mammosphere formation, and drug resistance (Gupta et al., 2009). Furthermore, Chaffer et al. showed a ZEB-1-dependent conversion of epithelial non-CSCs to mesenchymal CSC-like state in basal breast cancer (Chaffer et al., 2013).
In order to sustain growth of the metastatic lesions, a proportion of cancer cells would need to retain CSC features to maintain the proliferative reservoir and, potentially, to recapitulate the heterogeneity of the primary tumor. Cell plasticity complicates these interactions, as cancer cells may modify their transcriptional pattern depending on the physiological conditions. In support of this viewpoint, as discussed above, it has been shown that luminal-like breast tumor cells can trigger metastasis without acquiring stem-like markers (Kim et al., 2012). This could indicate either that it is not a prerequisite condition for disseminated tumor cells to present with CSC phenotypes in order to colonize a new niche or that, due to tumor cell plasticity, a fraction of the disseminated cells can reversibly convert into CSCs and initiate the new tumor.
An additional indirect evidence of dormancy in breast CSCs relates to the gene signature of normal quiescent mammary gland stem cells present within cultured mammospheres, which correlate with CSC behavior when applied to breast cancers (Pece et al., 2010). In another study, it was found that the human breast cancer cell line MDA-MB-231 can stably survive by entering into a dormant state after several cycles of hypoxia, that selects for the CSC population (Carcereri de Prati et al., 2017). Hypoxia was also shown to induce the expression of CSC-like phenotype in dormant MCF-7 cells, promoting EMT and escape from dormancy to metastatic outgrowth (Weidenfeld et al., 2016). A role for hypoxia-inducible factors in promoting breast cancer stem cell specificities and maintenance in response to hypoxia or cytotoxic chemotherapy has also been documented (Xiang & Semenza, 2019). In prostate, CSCs are believed to be involved in metastasis and therapeutic relapse and recapitulation of the primary tumor heterogeneity in the secondary site, although their link with disseminated tumor cells is unclear (Bjerkvig, Tysnes, Aboody, Najbauer, & Terzis, 2005; Lathia, 2013; Reya et al., 2001; Schilling et al., 2012; van der Toom, Verdone, & Pienta, 2016). Several markers associated with tumor progression and therapeutic resistance can be found in disseminated tumor cells in a patient’s bone metastases. Primary tumor expression of CXCR4, EpCAM and EZH2—all associated with a CSC phenotype-correlated with increased distant metastasis and local recurrence during patient follow-up (Conley-LaComb et al., 2012; Harris & Kerr, 2017; Massoner et al., 2014; Matsika et al., 2015; Mochizuki et al., 2004) indicating that CSCs may drive metastasis.
Among the different cell types present in prostate cancers, a patient’s prostate cancer typically contains differentiated cancer cells expressing high levels of PSA (i.e., PSA+), as well as stem-like cells that express little or no PSA (i.e., PSA−/low). The tumor-regenerating ability of PSA+ cells gradually declines, suggesting that PSA−/low cells possess long-term tumor-propagating capacity, consistent with CSCs. The PSA−/low cells appear to be rare in early-stage tumors, but more abundant in high-grade and locally advanced tumors, and some tumors may even lack PSA expression (Liu et al., 2015; Qin et al., 2012; Shah et al., 2004). Low PSA expression correlates with poorer clinical outcomes, including metastasis, recurrence, and reduced patient survival.
4. How does the microenvironment determine cancer stem cell fate?
The tumor microenvironment is essential in providing a fertile ground for CSC selection and development (Aguirre-Ghiso, Bragado, & Sosa, 2013; Carcereri de Prati et al., 2017). Different signals that originate from the tumor niche regulate CSC self-renewal, survival, and ability to invade tissues and promote metastasis (Clarke et al., 2006; Jaworska, Krol, & Szliszka, 2015; Williams, Motiani, Giridhar, & Kasper, 2013; Yu et al., 2012; Zenzmaier, Untergasser, & Berger, 2008). There are several stem cell and self-renewal associated pathways that are activated in CSCs upon interaction with the surrounding tumor microenvironment that correlate with aggressive phenotypes, such as Wnt, Notch-1, PI3K, Hedgehog, CXCL12 and induction of pluripotency-associated transcription factors, such as Oct-3/4, Nanog and Sox-2 (Albini et al., 2015; Clevers, 2011; Liu et al., 2006; Mimeault & Batra, 2013; Philip, Ito, Moreno-Sanchez, & Ralph, 2013; Schwitalla et al., 2013). There are several cells and/or signals from the environment that trigger these responses. Cancer-associated fibroblasts (CAFs) or adipocytes secrete a variety of cytokines related to tumor progression, such as Platelet-Derived Growth Factor, Vascular Endothelial Growth Factor (VEGF), Hepatocyte Growth Factor (HGF), platelet-derived growth factor (PDGF) and a variety of ECM proteins that enhance tumor growth (Polanska & Orimo, 2013); also, CAFs exert a metabolic reprogramming of cancer cells by inducing a reverse Warburg phenotype. Acidity and hypoxia are often characteristics of the tumor microenvironment. Hypoxia-inducible factors, HIF1a and HIF2a, are also sensitive to cellular pH conditions (Parks, Mazure, Counillon, & Pouyssegur, 2013). Both hypoxia and changes in pH can regulate stem cell behavior by altering their metabolic status and promoting metabolic reconfiguration of cancer cells toward glycolysis, induction of the EMT phenotype (including C-X-C-chemokine receptor 4 (CXCR4), Snail and Twist gene expression) and expression of stem-prone transcription factors.
There is also compelling evidence suggesting a role of the endothelial compartment in supporting CSC phenotypes, activating Notch signaling among other pathways through the secretion of VEGF and HGF ( Jeon et al., 2014; Keith & Simon, 2007). Immune cells at the tumor site, such as NK or tumor-associated macrophages (TAMs) usually show altered phenotypes compared to those in healthy tissues. NK cells are able to modulate metastatic dissemination in different tumor types, including breast and prostate cancer (Liu et al., 2013; Wang et al., 2014). TAMs have been shown to contribute to tumor growth and metastasis by producing antiinflammatory cytokines that promote tumor cell escape from immune surveillance (Allen & Louise Jones, 2011; Solinas, Germano, Mantovani, & Allavena, 2009; Ugel, De Sanctis, Mandruzzato, & Bronte, 2015).
Additionally, cell metabolism regulates cell proliferation and differentiation in CSCs, which have different metabolic states compared to their differentiated progeny (Carey, Finley, Cross, Allis, & Thompson, 2015; Vlashi et al., 2014). Further, CSCs establish a reinforcement mechanism by secreting cytokines that help recruit and activate specific cell types that modulate their differentiation states (Plaks, Kong, & Werb, 2015). At the site of metastasis, it has been shown that the microenvironment within the bone marrow plays a crucial role in the growth of disseminated tumor “stem” cells (Pedersen, Shiozawa, Pienta, & Taichman, 2012; Rahim et al., 2014; Shiozawa et al., 2011). These cells take over the bone marrow hematopoietic stem cell niche, competing directly for the niche.
In breast cancers in particular, CAFs contribute to CSC proliferation, invasion and metastasis by producing a variety of factors including IGF, PDGF, Wnt, Notch ligand, Hedgehog ligands, and matrix metalloproteases (MMPs) (Fig. 2). Together with these factors, ECM proteins secreted primarily by activated fibroblasts are known to contribute to tumor progression (Dufraine, Funahashi, & Kitajewski, 2008; Kenny & Bissell, 2003; Pontiggia et al., 2012; Sampayo et al., 2018). In this context, increased stromal collagen content positively correlates with stemness and also enhances CSC properties of breast cancer cells in culture (Pang et al., 2016). A recent study demonstrated that stromal cues from Hedgehog-activated CAFs, including FGF and collagen-rich ECM, are able to induce and maintain a stem-like phenotype in triple negative breast cancer cells in vivo (Cazet et al., 2018).
Fig. 2.
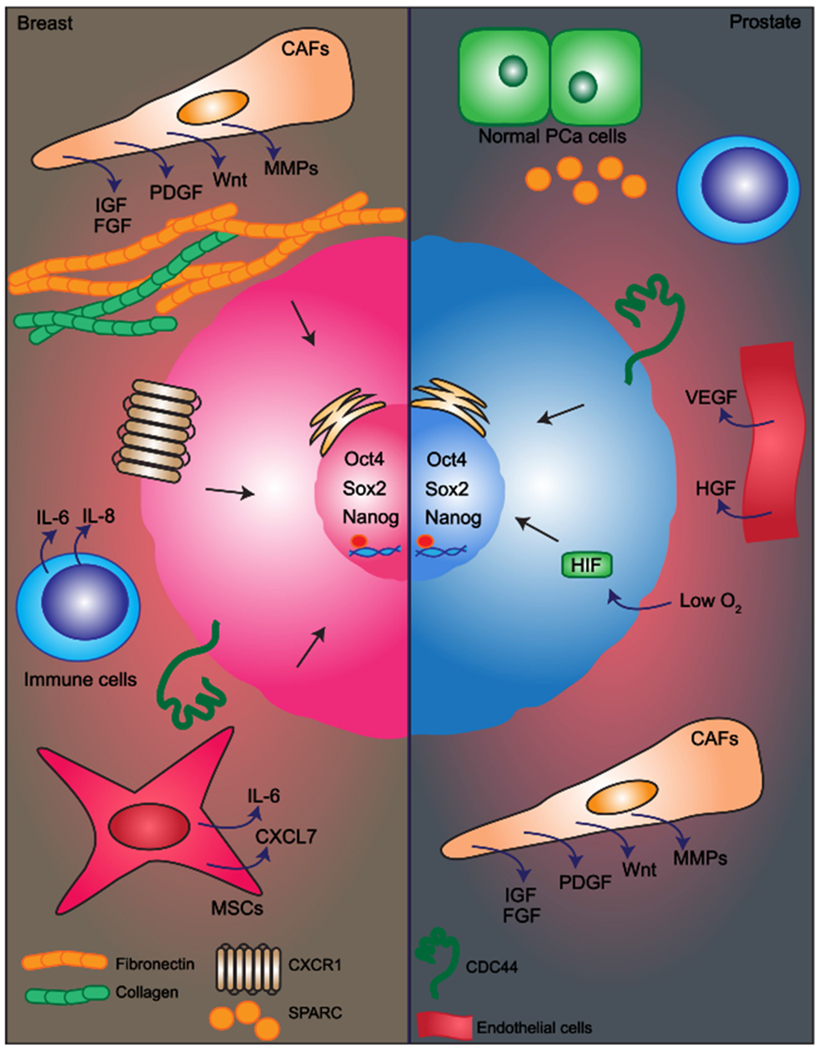
Key players in the interaction between CSCs and their microenvironment in breast and prostate cancers.
Mesenchymal stem cells (MSCs) have been shown to be recruited from the bone marrow or from the normal breast stroma (Korkaya, Liu, & Wicha, 2011; Liu et al., 2011). Within the tumor, MSCs interact with breast CSCs through cytokine loops that include IL-6 and CXCL7, to stimulate CSC self-renewal. Furthermore, MSCs can cause elevated miR-199a expression in breast cancer cells, suppressing FOXP2 expression and thereby imparting CSC properties to tumor cells (Cuiffo et al., 2014). The immune system seems to exert both inhibitory and stimulatory effects on breast tumors, and the balance between these effects dramatically influences tumor growth. Inflammatory cytokines IL-6 and IL-8 produced by immune cells promote CSC self-renewal (Sansone et al., 2007). IL-8 receptor, CXCR1, has been found to be highly expressed on breast CSCs (Ginestier et al., 2010). It has been reported that breast CSCs fail to express inhibitory NK ligands, which is consistent with metastatic spread (Wang et al., 2014).
In prostate cancer, CAFs contribute to enhance the growth potential of CSCs by increasing spheroid formation and cancer cell proliferation through different paracrine signals (Adisetiyo et al., 2014; Liao, Adisetiyo, Liang, & Roy-Burman, 2010) (Fig. 2). Moreover, co-injection of prostate cancer CSCs and CAFs into immune-compromised mice increase the number of neoplastic lesions as compared to normal fibroblasts, further supporting the significance of CAFs in regulating CSCs phenotypes (Liao et al., 2010). Cancer cell subpopulations can interact with other normal prostate cells present in the tumor environment and take advantage of these cells for tumor growth. An example of such cooperative interactions was described between two clonal subpopulations of the PC-3 prostate cancer cell line (Mateo et al., 2014). They found that the invasiveness of a CSC-enriched subpopulation is enhanced by a non-CSC subpopulation, resulting in an increase in tumorigenic and metastatic potential of CSCs. An ECM protein, SPARC (also known as osteonectin), was found to be the main factor mediating the cooperation between CSCs and non-CSCs (Mateo et al., 2014).
In addition, it has been observed that hypoxia induces, through HIF, the reprogramming of prostate cancer cells increasing the expression of stemness markers like CD44, Oct-3/4, Nanog, and drug resistance-associated molecules or anti-apoptotic proteins (Mathieu et al., 2011; Ravenna et al., 2014). Other cells within the prostate tumor microenvironment such as TAMs, endothelial cells and MSCs also support prostate cancer progression (Luo et al., 2014).
5. Biomarkers consensus: Are we there yet?
Hematopoietic stem cell markers have provided the basis for identifying and isolating CSCs in solid tumors including breast and prostate cancers (Kasper, 2008; Taylor & Risbridger, 2008). In breast, evaluation of combinatorial expression of surface markers has been proven to yield a better prognostic value for identifying breast CSCs. Al-Hajj et al. showed that less than 100 mammary CSC cells with the markers CD44+, CD24−/low, Lin− were able to generate tumors in NOD/SCID mice, whereas CD44−, CD24+ breast cancer cells, even when injected at 100-fold higher cell densities, were not able to generate tumors (Al-Hajj et al., 2003; Patrawala et al., 2005). Breast CSCs expressing CD133+ showed similar behavior, with only a few cells being able to recapitulate the original tumor (Wright et al., 2008). The CD44+, CD24−/low, Lin− subpopulation can be further subdivided based on EpCAM expression. The EpCAM+ , CD44+, CD24−/low, Lin− breast CSCs, but not EpCAM−, CD44+ , CD24−/low, Lin− cells, were capable of generating breast tumors and reconstitute the heterogeneity of the initial tumor in NOD/SCID mice (Al-Hajj et al., 2003). Both CD133+ and CD44+, CD24−/low, Lin− populations displayed markedly enhanced self-renewal capacity and shared the expression of sternness genes (OCT4, NOTCH1, ALDH1, FGFR1, SOX1) (Ginestier et al., 2007; Klonisch et al., 2008; Wright et al., 2008). However, not all are associated with aggressive metastatic growth (Lin, Zhong, Guan, Zhang, & Sun, 2012). The other markers that have been used to identify breast CSCs are CD44+ CD49fhi CD133/2hi phenotype (Meyer et al., 2010), CD49f (Cariati et al., 2008) and CD61 (Bozorgi, Khazaei, & Khazaei, 2015; Vaillant et al., 2008). Desgrosellier et al. demonstrated that CD49f and CD61 were found to be associated with tumor initiation properties through an in vivo study of breast cancer in mice (Desgrosellier et al., 2014; Lo et al., 2012; Sin & Lim, 2017). The breast CSCs markers described above are summarized in Table 1.
Table 1.
Current markers for identification of CSCs in breast and prostate cancers.
| CSCs type | Marker |
|---|---|
| Breast CSCs | EpCAM+ CD44+ CD24−/low Lin− |
| CD133+ | |
| OCT4high | |
| NOTCH1high | |
| ALDH1high | |
| FGFR1high | |
| SOX1high | |
| CD49f+ (Integrin α6) CD61+ | |
| CD44+ CD49f+ CD133/2+ | |
| Prostate CSCs | CD44+ |
| Sca-1+ | |
| CD133+ | |
| CK5/14 | |
| ALDHhigh | |
| ABCG2+ | |
| p63high | |
| Oct-3/4high | |
| β-cateninhigh | |
| CD44+ Integrin α2β1high CD133+ | |
In prostate, CD44 is a marker for normal prostatic epithelium stem cell and prostate CSCs (Tang et al., 2007; Yu et al., 2012). CD44+ prostate cancer cells are enriched in tumorigenic and metastatic progenitor cells and are more proliferative, clonogenic, tumorigenic, and metastatic than the isogenic CD44− cells (Patrawala et al., 2006). CD44+/Androgen receptor-(AR-) tumor progenitor cell population expressed stem-cell-related genes such as OCT3/4 and β-catenin and 1% of the CD44+ prostate cancer cells presented asymmetric cell division (Patrawala et al., 2006). CD44+/CD24− cells formed prostaspheres in vitro and a small number of cells was sufficient to initiate tumors in vivo (Hurt, Kawasaki, Klarmann, Thomas, & Farrar, 2008). Stem cell antigen (Sca-1)+ cells have increased proliferative capacity, with a subpopulation of Sca-1+ cells expressing Bcl-2 and integrin α6 (Xin, Lawson, & Witte, 2005). CD133+ prostate normal and cancer cells exhibit stem cell features such as prostasphere formation and they develope prostatic-like acini in immunocompromised mice (Pellacani, Oldridge, Collins, & Maitland, 2013; Richardson et al., 2004). In addition, CD133+ cells also co-express CK14 or hTERT and exhibit more developed ducts compared to CD133− cells (Mundy, 2002). The ATP-binding cassette (ABC) membrane transporter, ABCG2, expressed in a subpopulation of prostate cancer cells, enables the efflux of Hoechst 33342 dye suggesting that these cells might present multidrug resistance, which is a characteristic of CSCs (Patrawala et al., 2005). Aldehyde dehydrogenase (ALDH) is an enzyme involved in intracellular retinoic acid production. In prostate CSCs, high expression of ALDH1A1 was found to be positively correlated with Gleason score and pathologic stage and inversely correlated with overall survival, which suggests it may be a potential prostate CSC marker (Li, Cozzi, & Russell, 2010). The basal cell markers CK5/14 and p63 were found in a small fraction of cells in primary prostate carcinoma tissues and in the majority of prostate cancer metastases, suggesting that these might be markers of cells exhibiting prostate CSC features (Janssen et al., 2002; Klonisch et al., 2008).
Combining multiple markers has also improved the identification and isolation of prostate CSCs. CD44+ Integrinα2β1high CD133+ rare prostate cell populations possessed a significant capacity for self-renewal in vitro and could regenerate the mixed populations present in the tumor (Collins, Berry, Hyde, Stower, & Maitland, 2005; Moltzahn & Thalmann, 2013; Yun, Lo, & Hsieh, 2016). It was also found that ALDHhigh CD44+ cells show a higher proliferative, clonogenic, and metastatic capacity in vitro and higher tumorigenicity capacity in vivo than ALDHlow CD44− cells. However, ALDHlow CD44− cells were able to develop tumors, although they had longer latency periods (Yu et al., 2011, 2012). The variability of the different marker combinations suggests that CSC may be more than a distinct subpopulation and might be explained by the fact that CSC exhibit phenotypic plasticity which leads to dynamic phenotypes.
The prostate CSCs markers described above are summarized in Table 1.
6. Impact of the cancer stem cell hypothesis in cancer treatment and future challenges
The cancer stem-cell hypothesis, stating that tumors originate in stem or progenitor cells as a result of dysregulation of the normally tightly regulated process of self-renewal, has major implications for cancer research as well as important clinical implications for prevention and therapy (Kakarala & Wicha, 2008).
The first step in cancer prevention is the reduction of risks. In this sense, the CSC niche could be directly targeted through certain habits that can be promoted to inhibit CSC growth or that should be eliminated because they stimulate CSC proliferation. In this sense vitamin D has been shown to both promote differentiation of hematopoietic stem cells (therefore reducing the stem cell pool (Kakarala & Wicha, 2008; Nagler, Riklis, Kletter, Tatarsky, & Fabian, 1986)) and also has been recently found to directly inhibit the expression of stemness markers in glioma cells (Hu et al., 2019). On the contrary, cigarette smoke has been recently shown to promote stemness in renal cancer stem cells through activation of the Sonic Hedgehog pathway (Qian et al., 2018).
The second step in cancer prevention is early detection. Markers of cancer stem cells or their secreted factors could be used to detect early events of tumor initiation.
In terms of cancer treatment, accumulating efforts are tending to directly target the CSC compartment, either by eliminating it or promoting its differentiation, in order to prevent metastasis and prolong survival. One of the restrictions in traditional therapies is drug resistance, which is due to characteristic properties of CSCs such as high expression of drug-efflux pumps, low rate of division and high efficiency for DNA repairing (Dragu, Necula, Bleotu, Diaconu, & Chivu-Economescu, 2015). Therefore, targeting CSCs might be critical in treating cancer and preventing tumor relapse. Examples of these therapies are Notch signaling inhibitors, such as BMS-906024, which showed promising results for patients with relapsed T-cell acute lymphoblastic leukemia (Venkatesh et al., 2018), inhibition of efflux-pumps in which several generations of ABC blockers have been developed and now small-molecules are being tested to this end (Dragu et al., 2015) or targeting the interaction with the tumor microenvironment such as using CXCR4 agonists to inhibit the CXCL12-CXCR4 axis like CTCE-9908 that proved to be effective in reducing both tumor growth and metastasis in xenograft mouse models of inflammatory breast cancer (Singh et al., 2010) and in decreasing the tumor invasivity and angiogenesis in prostate cancer (Wong, Kandagatla, Korz, & Chinni, 2014).
Among the future challenges that are going to be key for CSC research in order to establish these cells as solid indicators for diagnosis and targets for therapies, the identification of better markers to unmistakably recognize this niche is going to be critical. And this identification should be done by carefully isolating tumor cell populations and testing their capacity to initiate a tumor in different types of immunocompromised mice. Research in this direction will be benefited by current state-of-the-art high-throughput sorting and screening tools.
Future research in the field should also consider increasing the specificity and efficiency of CSCs targeting, reducing toxicity of normal stem cells and also developing new strategies for delivery. These new therapies, combined with traditional therapeutic strategies, may contribute to increase their efficacy for aggressive cancers, therefore preventing tumor relapse and ultimately enhancing patient survival.
Acknowledgements
The authors would like to thank the Siebel Foundation for the Postdoctoral Fellowship awarded to R.G. Sampayo. Mina J. Bissell is supported by awards from the NCI (R01CA064786) and an award from the Breast Cancer Research Foundation.
Footnotes
Conflict of interest
The authors declare no competing financial interests.
References
- Adisetiyo H, Liang M, Liao CP, Jeong JH, Cohen MB, Roy-Burman P, et al. (2014). Dependence of castration-resistant prostate cancer (CRPC) stem cells on CRPC-associated fibroblasts. Journal of Cellular Physiology, 229, 1170–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agliano A, Calvo A, & Box C (2017). The challenge of targeting cancer stem cells to halt metastasis. Seminars in Cancer Biology, 44, 25–42. [DOI] [PubMed] [Google Scholar]
- Aguirre-Ghiso JA, Bragado P, & Sosa MS (2013). Metastasis awakening: Targeting dormant cancer. Nature Medicine, 19, 276–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albini A, Bruno A, Gallo C, Pajardi G, Noonan DM, & Dallaglio K (2015). Cancer stem cells and the tumor microenvironment: Interplay in tumor heterogeneity. Connective Tissue Research, 56, 414–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, & Clarke MF (2003). Prospective identification of tumorigenic breast cancer cells. Proceedings of the National Academy of Sciences of the United States of America, 100, 3983–3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen M, & Louise Jones J (2011). Jekyll and Hyde: The role of the microenvironment on the progression of cancer. The Journal of Pathology, 223, 162–176. [DOI] [PubMed] [Google Scholar]
- Allinen M, Beroukhim R, Cai L, Brennan C, Lahti-Domenici J, Huang H, et al. (2004). Molecular characterization of the tumor microenvironment in breast cancer. Cancer Cell, 6, 17–32. [DOI] [PubMed] [Google Scholar]
- Bach K, Pensa S, Grzelak M, Hadfield J, Adams DJ, Marioni JC, et al. (2017). Differentiation dynamics of mammary epithelial cells revealed by single-cell RNA sequencing. Nature Communications, 8, 2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balic M, Lin H, Young L, Hawes D, Giuliano A, McNamara G, et al. (2006). Most early disseminated cancer cells detected in bone marrow of breast cancer patients have a putative breast cancer stem cell phenotype. Clinical Cancer Research, 12, 5615–5621. [DOI] [PubMed] [Google Scholar]
- Bapat SA (2007). Evolution of cancer stem cells. Seminars in Cancer Biology, 17, 204–213. [DOI] [PubMed] [Google Scholar]
- Batlle E, & Clevers H (2017). Cancer stem cells revisited. Nature Medicine, 23, 1124–1134. [DOI] [PubMed] [Google Scholar]
- Ben-Porath I, Thomson MW, Carey VJ, Ge R, Bell GW, Regev A, et al. (2008). An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nature Genetics, 40, 499–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissell MJ, & Labarge MA (2005). Context, tissue plasticity, and cancer: Are tumor stem cells also regulated by the microenvironment? Cancer Cell, 7, 17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjerkvig R, Tysnes BB, Aboody KS, Najbauer J, & Terzis AJ (2005). Opinion: The origin of the cancer stem cell: Current controversies and new insights. Nat Rev Cancer.Nature Reviews Cancer, 5, 899–904. [DOI] [PubMed] [Google Scholar]
- Bliss SA, Paul S, Pobiarzyn PW, Ayer S, Sinha G, Pant S, et al. (2018). Evaluation of a developmental hierarchy for breast cancer cells to assess risk-based patient selection for targeted treatment. Scientific Reports, 8, 367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnet D, & Dick JE (1997). Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nature Medicine, 3, 730–737. [DOI] [PubMed] [Google Scholar]
- Bozorgi A, Khazaei M, &Khazaei MR (2015). Newfindings onbreastcancerstemcells:A review. Journal of Breast Cancer, 18, 303–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabrera MC, Hollingsworth RE, & Hurt EM (2015). Cancer stem cell plasticity and tumor hierarchy. World Journal of Stem Cells, 7, 27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell LL, & Polyak K (2007). Breast tumor heterogeneity: Cancer stem cells or clonal evolution? Cell Cycle, 6, 2332–2338. [DOI] [PubMed] [Google Scholar]
- Carcereri de Prati A, Butturini E, Rigo A, Oppici E, Rossin M, Boriero D, et al. (2017). Metastatic breast cancer cells enter into dormant state and express cancer stem cells phenotype under chronic hypoxia. Journal of Cellular Biochemistry, 118, 3237–3248. [DOI] [PubMed] [Google Scholar]
- Carey BW, Finley LW, Cross JR, Allis CD, & Thompson CB (2015). Intracellular alpha-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature, 518, 413–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cariati M, Naderi A, Brown JP, Smalley MJ, Pinder SE, Caldas C, et al. (2008). Alpha-6 integrin is necessary for the tumourigenicity of a stem cell-like subpopulation within the MCF7 breast cancer cell line. International Journal of Cancer, 122, 298–304. [DOI] [PubMed] [Google Scholar]
- Cazet AS, Hui MN, Elsworth BL, Wu SZ, Roden D, Chan CL, et al. (2018). Targeting stromal remodeling and cancer stem cell plasticity overcomes chemoresistance in triple negative breast cancer. Nature Communications, 9, 2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaffer CL, Marjanovic ND, Lee T, Bell G, Kleer CG, Reinhardt F, et al. (2013). Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell, 154, 61–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaffer CL, & Weinberg RA (2011). A perspective on cancer cell metastasis. Science, 331, 1559–1564. [DOI] [PubMed] [Google Scholar]
- Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CH, Jones DL, et al. (2006). Cancer stem cells—Perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Research, 66, 9339–9344. [DOI] [PubMed] [Google Scholar]
- Clevers H (2011). The cancer stem cell: Premises, promises and challenges. Nature Medicine, 17, 313–319. [DOI] [PubMed] [Google Scholar]
- Collins AT, Berry PA, Hyde C, Stower MJ, & Maitland NJ (2005). Prospective identification of tumorigenic prostate cancer stem cells. Cancer Research, 65, 10946–10951. [DOI] [PubMed] [Google Scholar]
- Conley-LaComb MK, Huang W, Wang S, Shi D, Jung YS, Najy A, et al. (2012). PTEN regulates PDGF ligand switch for beta-PDGFR signaling in prostate cancer. The American Journal of Pathology, 180, 1017–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuiffo BG, Campagne A, Bell GW, Lembo A, Orso F, Lien EC, et al. (2014). MSC-regulated microRNAs converge on the transcription factor FOXP2 and promote breast cancer metastasis. Cell Stem Cell, 15, 762–774. [DOI] [PubMed] [Google Scholar]
- Desgrosellier JS, Lesperance J, Seguin L, Gozo M, Kato S, Franovic A, et al. (2014). Integrin alphavbeta3 drives slug activation and stemness in the pregnant and neoplastic mammary gland. Developmental Cell, 30, 295–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick JE (1996). Normal and leukemic human stem cells assayed in SCID mice. Seminars in Immunology, 8, 197–206. [DOI] [PubMed] [Google Scholar]
- Dick JE (2009). Looking ahead in cancer stem cell research. Nature Biotechnology, 27, 44–46. [DOI] [PubMed] [Google Scholar]
- Dragu DL, Necula LG, Bleotu C, Diaconu CC, & Chivu-Economescu M (2015). Therapies targeting cancer stem cells: Current trends and future challenges. World Journal of Stem Cells, 7, 1185–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufraine J, Funahashi Y, & Kitajewski J (2008). Notch signaling regulates tumor angiogenesis by diverse mechanisms. Oncogene, 27, 5132–5137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich P (1879). Ueber die specifischen Granulationen des Blutes. Archivfur Anatomie und Physiologie. Physiologische Abteilung, 3, 571–579. [Google Scholar]
- Ellwood-Yen K, Graeber TG, Wongvipat J, Iruela-Arispe ML, Zhang J, Matusik R, et al. (2003). Myc-driven murine prostate cancer shares molecular features with human prostate tumors. Cancer Cell, 4, 223–238. [DOI] [PubMed] [Google Scholar]
- Essers MA, & Trumpp A (2010). Targeting leukemic stem cells by breaking their dormancy. Molecular Oncology, 4, 443–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridriksdottir AJ, Villadsen R, Morsing M, Klitgaard MC, Kim J, Petersen OW, et al. (2017). Proof of region-specific multipotent progenitors in human breast epithelia. Proceedings of the National Academy of Sciences of the United States of America, 114, E10102–E10111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs E (2009). The tortoise and the hair: Slow-cycling cells in the stem cell race. Cell, 137, 811–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao D, Vela I, Sboner A, Iaquinta PJ, Karthaus WR, Gopalan A, et al. (2014). Organoid cultures derived from patients with advanced prostate cancer. Cell, 159, 176–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao XL, Zhang M, Tang YL, & Liang XH (2017). Cancer cell dormancy: Mechanisms and implications of cancer recurrence and metastasis. OncoTargets and Therapy, 10, 5219–5228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg M (2017). Epithelial plasticity and cancer stem cells: Major mechanisms of cancer pathogenesis and therapy resistance. World Journal of Stem Cells, 9, 118–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, et al. (2007). ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell, 1, 555–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginestier C, Liu S, Diebel ME, Korkaya H, Luo M, Brown M, et al. (2010). CXCR1 blockade selectively targets human breast cancer stem cells in vitro and in xenografts. The Journal of Clinical Investigation, 120, 485–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goss PE, & Chambers AF (2010). Does tumour dormancy offer a therapeutic target? Nat Rev Cancer. Nature Reviews Cancer, 10, 871–877. [DOI] [PubMed] [Google Scholar]
- Gu G, Yuan J, Wills M, & Kasper S (2007). Prostate cancer cells with stem cell characteristics reconstitute the original human tumor in vivo. Cancer Research, 67, 4807–4815. [DOI] [PubMed] [Google Scholar]
- Gupta PB, Fillmore CM, Jiang G, Shapira SD, Tao K, Kuperwasser C, et al. (2011). Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell, 146, 633–644. [DOI] [PubMed] [Google Scholar]
- Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA, et al. (2009). Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell, 138, 645–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Häckel E (1868). Natrüliche schöpfungsgeschichte. Berlin: Georg Reimer. [Google Scholar]
- Harris KS, & Kerr BA (2017). Prostate cancer stem cell markers drive progression, therapeutic resistance, and bone metastasis. Stem Cells International, 2017, 8629234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu P, Li S, Tian N, Wu F, Hu Y, Li D, et al. (2019). Acidosis enhances the self-renewal and mitochondrial respiration of stem cell-like glioma cells through CYP24A1-mediated reduction of vitamin D. Cell Death & Disease, 10, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu M, Yao J, Cai L, Bachman KE, van denBrule F, Velculescu V, et al. (2005). Distinct epigenetic changes in the stromal cells of breast cancers. Nature Genetics, 37, 899–905. [DOI] [PubMed] [Google Scholar]
- Hurt EM, Kawasaki BT, Klarmann GJ, Thomas SB, & Farrar WL (2008). CD44+ CD24(−) prostate cells are early cancer progenitor/stem cells that provide a model for patients with poor prognosis. British Journal of Cancer, 98, 756–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacs JT, & Coffey DS (1989). Etiology and disease process of benign prostatic hyperplasia. The Prostate. Supplement, 2, 33–50. [DOI] [PubMed] [Google Scholar]
- Iwata T, Schultz D, Hicks J, Hubbard GK, Mutton LN, Lotan TL, et al. (2010). MYC overexpression induces prostatic intraepithelial neoplasia and loss of Nkx3.1 in mouse luminal epithelial cells. PLoS One, 5, e9427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen KP, El-Marjou F, Pinto D, Sastre X, Rouillard D, Fouquet C, et al. (2002). Targeted expression of oncogenic K-ras in intestinal epithelium causes spontaneous tumorigenesis in mice. Gastroenterology, 123, 492–504. [DOI] [PubMed] [Google Scholar]
- Jaworska D, Krol W, & Szliszka E (2015). Prostate cancer stem cells: Research advances. International Journal of Molecular Sciences, 16, 27433–27449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon HM, Kim SH, Jin X, Park JB, Kim SH, Joshi K, et al. (2014). Crosstalk between glioma-initiating cells and endothelial cells drives tumor progression. Cancer Research, 74, 4482–4492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakarala M, & Wicha MS (2008). Implications of the cancer stem-cell hypothesis for breast cancer prevention and therapy. Journal of Clinical Oncology, 26, 2813–2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasper S (2008). Exploring the origins of the normal prostate and prostate cancer stem cell. Stem Cell Reviews, 4, 193–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keith B, & Simon MC (2007). Hypoxia-inducible factors, stem cells, and cancer. Cell, 129, 465–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenny PA, &Bissell MJ (2003). Tumor reversion: Correction of malignant behavior by microenvironmental cues. International Journal of Cancer, 107, 688–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim WT, & Ryu CJ (2017). Cancer stem cell surface markers on normal stem cells. BMB Reports, 50, 285–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Villadsen R, Sorlie T, Fogh L, Gronlund SZ, Fridriksdottir AJ, et al. (2012). Tumor initiating but differentiated luminal-like breast cancer cells are highly invasive in the absence of basal-like activity. Proceedings of the National Academy of Sciences of the United States of America, 109, 6124–6129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klonisch T, Wiechec E, Hombach-Klonisch S, Ande SR, Wesselborg S, Schulze-Osthoff K, et al. (2008). Cancer stem cell markers in common cancers—Therapeutic implications. Trends in Molecular Medicine, 14, 450–460. [DOI] [PubMed] [Google Scholar]
- Korkaya H, Liu S, &Wicha MS (2011).Breastcancerstemcells, cytokine networks, and the tumor microenvironment. The Journal of Clinical Investigation, 121, 3804–3809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreso A, & Dick JE (2014). Evolution of the cancer stem cell model. Cell Stem Cell, 14, 275–291. [DOI] [PubMed] [Google Scholar]
- Larue L, & Bellacosa A (2005). Epithelial-mesenchymal transition in development and cancer: Role of phosphatidylinositol 3’ kinase/AKT pathways. Oncogene, 24, 7443–7454. [DOI] [PubMed] [Google Scholar]
- Lathia JD (2013). Cancerstem cells: Movingpastthe controversy. CNS Oncology, 2, 465–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson DA, Zong Y, Memarzadeh S, Xin L, Huang J, &Witte ON. (2010). Basal epithelial stem cells are efficient targets for prostate cancer initiation. Proceedings of the National Academy of Sciences of the United States of America, 107, 2610–2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Cozzi PJ, & Russell PJ (2010). Promising tumor-associated antigens for future prostate cancer therapy. Medicinal Research Reviews, 30, 67–101. [DOI] [PubMed] [Google Scholar]
- Li X, Lewis MT, Huang J, Gutierrez C, Osborne CK, Wu MF, et al. (2008). Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. Journal of the National Cancer Institute, 100, 672–679. [DOI] [PubMed] [Google Scholar]
- Li H, Zhou J, Miki J, Furusato B, Gu Y, Srivastava S, et al. (2008). Telomerase-immortalized non-malignant human prostate epithelial cells retain the properties of multipotent stem cells. Experimental Cell Research, 314, 92–102. [DOI] [PubMed] [Google Scholar]
- Liao CP, Adisetiyo H, Liang M, & Roy-Burman P (2010). Cancer-associated fibroblasts enhance the gland-forming capability of prostate cancer stem cells. Cancer Research, 70, 7294–7303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Zhong Y, Guan H, Zhang X, & Sun Q (2012). CD44+/CD24− phenotype contributes to malignant relapse following surgical resection and chemotherapy in patients with invasive ductal carcinoma. Journal of Experimental & Clinical Cancer Research, 31, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Chen X, Rycaj K, Chao HP, Deng Q, Jeter C, et al. (2015). Systematic dissection of phenotypic, functional, and tumorigenic heterogeneity of human prostate cancer cells. Oncotarget, 6, 23959–23986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Ginestier C, Ou SJ, Clouthier SG, Patel SH, Monville F, et al. (2011). Breast cancer stem cells are regulated by mesenchymal stem cells through cytokine networks. Cancer Research, 71, 614–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Lu S, Wang X, Page ST, Higano CS, Plymate SR, et al. (2013). Perturbation of NK cell peripheral homeostasis accelerates prostate carcinoma metastasis. The Journal of Clinical Investigation, 123, 4410–4422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, Wang X, Chen GY, Dalerba P, Gurney A, Hoey T, et al. (2007). The prognostic role of a gene signature from tumorigenic breast-cancer cells. The New England Journal of Medicine, 356, 217–226. [DOI] [PubMed] [Google Scholar]
- Liu ZJ, Xiao M, Balint K, Smalley KS, Brafford P, Qiu R, et al. (2006). Notch 1 signaling promotes primary melanoma progression by activating mitogen-activated protein kinase/phosphatidylinositol 3-kinase-Akt pathways and up-regulating N-cadherin expression. Cancer Research, 66, 4182–4190. [DOI] [PubMed] [Google Scholar]
- Lo PK, Kanojia D, Liu X, Singh UP, Berger FG, Wang Q, et al. (2012). CD49f and CD61 identify Her2/neu-induced mammary tumor-initiating cells that are potentially derived from luminal progenitors and maintained by the integrin-TGFbeta signaling. Oncogene, 31, 2614–2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Ok Lee S, Liang L, Huang CK, Li L, Wen S, et al. (2014). Infiltrating bone marrow mesenchymal stem cells increase prostate cancer stem cell population and metastatic ability via secreting cytokines to suppress androgen receptor signaling. Oncogene, 33, 2768–2778. [DOI] [PubMed] [Google Scholar]
- Ma X, Ziel-van der Made AC, Autar B, van der Korput HA, Vermeij M, van Duijn P, et al. (2005). Targetedbiallelic inactivation of Pten in the mouse prostate leads to prostate cancer accompanied by increased epithelial cell proliferation but not by reduced apoptosis. Cancer Research, 65, 5730–5739. [DOI] [PubMed] [Google Scholar]
- Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, et al. (2008). The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell, 133, 704–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massoner P, Thomm T, Mack B, Untergasser G, Martowicz A, Bobowski K, et al. (2014). EpCAM is overexpressed in local and metastatic prostate cancer, suppressed by chemotherapy and modulated by MET-associated miRNA-200c/205. British Journal of Cancer, 111, 955–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateo F, Meca-Cortes O, Celia-Terrassa T, Fernandez Y, Abasolo I, Sanchez-Cid L, et al. (2014). SPARC mediates metastatic cooperation between CSC and non-CSC prostate cancer cell subpopulations. Molecular Cancer, 13, 237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathieu J, Zhang Z, Zhou W, Wang AJ, Heddleston JM, Pinna CM, et al. (2011). HIF induces human embryonic stem cell markers in cancer cells. Cancer Research, 71, 4640–4652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsika A, Srinivasan B, Day C, Mader SA, Kiernan DM, Broomfield A, et al. (2015). Cancer stem cell markers in prostate cancer: An immunohistochemical study of ALDH1, SOX2 andEZH2. Pathology, 47, 622–628. [DOI] [PubMed] [Google Scholar]
- Meacham CE, & Morrison SJ (2013). Tumour heterogeneity and cancer cell plasticity. Nature, 501, 328–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer MJ, Fleming JM, Lin AF, Hussnain SA, Ginsburg E,& Vonderhaar BK (2010). CD44posCD49fhiCD133/2hi defines xenograft-initiating cells in estrogen receptor-negative breast cancer. Cancer Research, 70, 4624–4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki J, Furusato B, Li H, Gu Y, Takahashi H, Egawa S, et al. (2007). Identification of putative stem cell markers, CD133 and CXCR4, in hTERT-immortalized primary nonmalignant and malignant tumor-derived human prostate epithelial cell lines and in prostate cancer specimens. Cancer Research, 67, 3153–3161. [DOI] [PubMed] [Google Scholar]
- Miller SJ, Lavker RM, & Sun TT (2005). Interpreting epithelial cancer biology in the context of stem cells: Tumor properties and therapeutic implications. Biochimica et Biophysica Acta, 1756, 25–52. [DOI] [PubMed] [Google Scholar]
- Mimeault M, & Batra SK (2013). Hypoxia-inducing factors as master regulators of stemness properties and altered metabolism of cancer- and metastasis-initiating cells. Journal of Cellular and Molecular Medicine, 17, 30–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochizuki H, Matsubara A, Teishima J, Mutaguchi K, Yasumoto H, Dahiya R, et al. (2004). Interaction of ligand-receptor system between stromal-cell-derived factor-1 and CXC chemokine receptor 4 in human prostate cancer: A possible predictor of metastasis. Biochemical and Biophysical Research Communications, 320, 656–663. [DOI] [PubMed] [Google Scholar]
- Moltzahn F, & Thalmann GN (2013). Cancer stem cells in prostate cancer. Translational Andrology and Urology, 2, 242–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mundy GR (2002). Metastasis to bone: Causes, consequences and therapeutic opportunities. Nat Rev Cancer. Nature Reviews Cancer, 2, 584–593. [DOI] [PubMed] [Google Scholar]
- Nagler A, Riklis I, Kletter Y, Tatarsky I, & Fabian I (1986). Effect of 1,25 dihydroxyvitamin D3 and retinoic acid on normal human pluripotent (CFU-mix), erythroid (BFU-E), and myeloid (CFU-C) progenitor cell growth and differentiation patterns. Experimental Hematology, 14, 60–65. [PubMed] [Google Scholar]
- Nistico P, Bissell MJ, & Radisky DC (2012). Epithelial-mesenchymal transition: General principles and pathological relevance with special emphasis on the role of matrix metalloproteinases. Cold Spring Harbor Perspectives in Biology, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang MF, Siedlik MJ, Han S, Stallings-Mann M, Radisky DC, & Nelson CM (2016). Tissue stiffness and hypoxia modulate the integrin-linked kinase ILK to control breast cancer stem-like cells. Cancer Research, 76, 5277–5287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks SK, Mazure NM, Counillon L, & Pouyssegur J (2013). Hypoxia promotes tumor cell survival in acidic conditions by preserving ATP levels. Journal of Cellular Physiology, 228, 1854–1862. [DOI] [PubMed] [Google Scholar]
- Patrawala L, Calhoun T, Schneider-Broussard R, Li H, Bhatia B, Tang S, et al. (2006). Highly purified CD44+ prostate cancer cells from xenograft human tumors are enriched in tumorigenic and metastatic progenitor cells. Oncogene, 25, 1696–1708. [DOI] [PubMed] [Google Scholar]
- Patrawala L, Calhoun T, Schneider-Broussard R, Zhou J, Claypool K, & Tang DG (2005). Side population is enriched in tumorigenic, stem-like cancer cells, whereas ABCG2+ and ABCG2-cancer cells are similarly tumorigenic. Cancer Research, 65, 6207–6219. [DOI] [PubMed] [Google Scholar]
- Pece S, Tosoni D, Confalonieri S, Mazzarol G, Vecchi M, Ronzoni S, et al. (2010). Biological and molecular heterogeneity of breast cancers correlates with their cancer stem cell content. Cell, 140, 62–73. [DOI] [PubMed] [Google Scholar]
- Pedersen EA, Shiozawa Y, Pienta KJ, &Taichman RS. (2012). The prostate cancer bone marrow niche: More than just ‘fertile soil’. Asian Journal of Andrology, 14, 423–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellacani D, Oldridge EE, Collins AT, & Maitland NJ (2013). Prominin-1 (CD133) expression in the prostate and prostate cancer: A marker for quiescent stem cells. Adv. Exp. Med. Biol, 777, 167–184. [DOI] [PubMed] [Google Scholar]
- Philip B, Ito K, Moreno-Sanchez R, & Ralph SJ (2013). HIF expression and the role of hypoxic microenvironments within primary tumours as protective sites driving cancer stem cell renewal and metastatic progression. Carcinogenesis, 34, 1699–1707. [DOI] [PubMed] [Google Scholar]
- Plaks V, Kong N, & Werb Z (2015). The cancer stem cell niche: How essential is the niche in regulating sternness of tumor cells? Cell Stem Cell, 16, 225–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polanska UM, & Orimo A (2013). Carcinoma-associated fibroblasts: Non-neoplastic tumour-promoting mesenchymal cells. Journal of Cellular Physiology, 228, 1651–1657. [DOI] [PubMed] [Google Scholar]
- Pontiggia O, Sampayo R, Raffo D, Motter A, Xu R, Bissell MJ, et al. (2012). The tumor microenvironment modulates tamoxifen resistance in breast cancer: A role for soluble stromal factors and fibronectin through beta1 integrin. Breast Cancer Research and Treatment, 133, 459–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian W, Kong X, Zhang T, Wang D, Song J, Li Y, et al. (2018). Cigarette smoke stimulates the stemness of renal cancer stem cells via Sonic Hedgehog pathway. Oncogenesis, 7, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin J, Liu X, Laffin B, Chen X, Choy G, Jeter CR, et al. (2012). The PSA(−/lo) prostate cancer cell population harbors self-renewing long-term tumor-propagating cells that resist castration. Cell Stem Cell, 10, 556–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana E, Shackleton M, Sabel MS, Fullen DR, Johnson TM, &Morrison SJ (2008). Efficient tumour formation by single human melanoma cells. Nature, 456, 593–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahim F, Hajizamani S, Mortaz E, Ahmadzadeh A, Shahjahani M, Shahrabi S, et al. (2014). Molecular regulation of bone marrow metastasis in prostate and breast cancer. Bone Marrow Research, 2014, 405920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramalho-Santos M, & Willenbring H (2007). On the origin of the term “stem cell” Cell Stem Cell, 1, 35–38. [DOI] [PubMed] [Google Scholar]
- Ravenna L, Principessa L, Verdina A, Salvatori L, Russo MA, & Petrangeli E (2014). Distinct phenotypes of human prostate cancer cells associate with different adaptation to hypoxia and pro-inflammatory gene expression. PLoS One, 9, e96250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reya T, Morrison SJ, Clarke MF, & Weissman IL (2001). Stem cells, cancer, and cancer stem cells. Nature, 414, 105–111. [DOI] [PubMed] [Google Scholar]
- Richardson GD, Robson CN, Lang SH, Neal DE, Maitland NJ, & Collins AT (2004). CD133, a novel marker for human prostatic epithelial stem cells. Journal of Cell Science, 117, 3539–3545. [DOI] [PubMed] [Google Scholar]
- Rybak AP, Bristow RG, & Kapoor A (2015). Prostate cancer stem cells: Deciphering the origins and pathways involved in prostate tumorigenesis and aggression. Oncotarget, 6, 1900–1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybak AP, He L, Kapoor A, Cutz JC,& Tang D (2011). Characterization of sphere-propagating cells with stem-like properties from DU145 prostate cancer cells. Biochimica et Biophysica Acta, 1813, 683–694. [DOI] [PubMed] [Google Scholar]
- Sampayo RG, Toscani AM, Rubashkin MG, Thi K, Masullo LA, Violi IL, et al. (2018). Fibronectin rescues estrogen receptor alpha from lysosomal degradation in breast cancer cells. The Journal of Cell Biology, 217, 2777–2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansone P, Storci G, Tavolari S, Guarnieri T, Giovannini C, Taffurelli M, et al. (2007). IL-6 triggers malignant features in mammospheres from human ductal breast carcinoma and normal mammary gland. The Journal of Clinical Investigation, 117, 3988–4002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilling D, Todenhofer T, Hennenlotter J, Schwentner C, Fehm T, & Stenzl A (2012). Isolated, disseminated and circulating tumour cells in prostate cancer. Nature Reviews. Urology, 9, 448–463. [DOI] [PubMed] [Google Scholar]
- Schwitalla S, Fingerle AA, Cammareri P, Nebelsiek T, Goktuna SI, Ziegler PK, et al. (2013). Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell, 152, 25–38. [DOI] [PubMed] [Google Scholar]
- Shackleton M, Quintana E, Fearon ER, & Morrison SJ (2009). Heterogeneity in cancer: Cancer stem cells versus clonal evolution. Cell, 138, 822–829. [DOI] [PubMed] [Google Scholar]
- Shah RB, Mehra R, Chinnaiyan AM, Shen R, Ghosh D, Zhou M, et al. (2004). Androgen-independent prostate cancer is a heterogeneous group of diseases: Lessons from a rapid autopsy program. Cancer Research, 64, 9209–9216. [DOI] [PubMed] [Google Scholar]
- Shiozawa Y, Pedersen EA, Havens AM, Jung Y, Mishra A, Joseph J, et al. (2011). Human prostate cancer metastases target the hematopoietic stem cell niche to establish footholds in mouse bone marrow. The Journal of Clinical Investigation, 121, 1298–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shipitsin M, Campbell LL, Argani P, Weremowicz S, Bloushtain-Qimron N, Yao J, et al. (2007). Molecular definition of breast tumor heterogeneity. Cancer Cell, 11, 259–273. [DOI] [PubMed] [Google Scholar]
- Sin WC, & Lim CL (2017). Breast cancer stem cells-from origins to targeted therapy. Stem Cell Investigation, 4, 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh B, Cook KR, Martin C, Huang EH, Mosalpuria K, Krishnamurthy S, et al. (2010). Evaluation of a CXCR4 antagonist in a xenograft mouse model of inflammatory breast cancer. Clinical and Experimental Metastasis, 27, 233–240. [DOI] [PubMed] [Google Scholar]
- Solinas G, Germano G, Mantovani A, & Allavena P (2009). Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. Journal of Leukocyte Biology, 86, 1065–1073. [DOI] [PubMed] [Google Scholar]
- Sosa MS, Bragado P, & Aguirre-Ghiso JA (2014). Mechanisms of disseminated cancer cell dormancy: An awakening field. Nat Rev Cancer.Nature Reviews Cancer, 14, 611–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spradling A, Drummond-Barbosa D, & Kai T (2001). Stem cells find their niche. Nature, 414, 98–104. [DOI] [PubMed] [Google Scholar]
- Takahashi K, & Yamanaka S (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell, 126, 663–676. [DOI] [PubMed] [Google Scholar]
- Talukdar S, Bhoopathi P, Emdad L, Das S, Sarkar D, & Fisher PB (2019). Dormancy and cancer stem cells: An enigma for cancer therapeutic targeting. Advances in Cancer Research, 141, 43–84. [DOI] [PubMed] [Google Scholar]
- Tang DG, Patrawala L, Calhoun T, Bhatia B, Choy G, Schneider-Broussard R, et al. (2007). Prostate cancer stem/progenitor cells: Identification, characterization, and implications. Molecular Carcinogenesis, 46, 1–14. [DOI] [PubMed] [Google Scholar]
- Taylor RA, & Risbridger GP (2008). The path toward identifying prostatic stem cells. Differentiation, 76, 671–681. [DOI] [PubMed] [Google Scholar]
- Taylor RA, Toivanen R, Frydenberg M, Pedersen J, Harewood L, B. Australian Prostate Cancer, et al. (2012). Human epithelial basal cells are cells of origin of prostate cancer, independent of CD133 status. Stem Cells, 30, 1087–1096. [DOI] [PubMed] [Google Scholar]
- Taylor RA, Toivanen R, & Risbridger GP (2010). Stem cells in prostate cancer: Treating the root of the problem. Endocrine-Related Cancer, 17, R273–R285. [DOI] [PubMed] [Google Scholar]
- Tirino V, Desiderio V, Paino F, De Rosa A, Papaccio F, La Noce M, et al. (2013). Cancer stem cells in solid tumors: An overview and new approaches for their isolation and characterization. The FASEB Journal, 27, 13–24. [DOI] [PubMed] [Google Scholar]
- Ugel S, De Sanctis F, Mandruzzato S, & Bronte V (2015). Tumor-induced myeloid deviation: When myeloid-derived suppressor cells meet tumor-associated macrophages. The Journal of Clinical Investigation, 125, 3365–3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaillant F, Asselin-Labat ML, Shackleton M, Forrest NC, Lindeman GJ, & Visvader JE (2008). The mammary progenitor marker CD61/beta3 integrin identifies cancer stem cells in mouse models of mammary tumorigenesis. Cancer Research, 68, 7711–7717. [DOI] [PubMed] [Google Scholar]
- van der Toom EE, Verdone JE, & Pienta KJ (2016). Disseminated tumor cells and dormancy in prostate cancer metastasis. Current Opinion in Biotechnology, 40, 9–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesh V, Nataraj R, Thangaraj GS, Karthikeyan M, Gnanasekaran A, Kaginelli SB, et al. (2018). Targeting Notch signalling pathway of cancer stem cells. Stem Cell Investigation, 5, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villadsen R (2005). In search of a stem cell hierarchy in the human breast and its relevance to breast cancer evolution. APMIS, 113, 903–921. [DOI] [PubMed] [Google Scholar]
- Villadsen R, Fridriksdottir AJ, Ronnov-Jessen L, Gudjonsson T, Rank F, LaBarge MA, et al. (2007). Evidence for a stem cell hierarchy in the adult human breast. The Journal of Cell Biology, 177, 87–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visvader JE (2009). Keeping abreast of the mammary epithelial hierarchy and breast tumorigenesis. Genes and Development, 23, 2563–2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visvader JE, & Clevers H (2016). Tissue-specific designs of stem cell hierarchies. Nature Cell Biology, 18, 349–355. [DOI] [PubMed] [Google Scholar]
- Visvader JE, & Lindeman GJ (2006). Mammary stem cells and mammopoiesis. Cancer Research, 66, 9798–9801. [DOI] [PubMed] [Google Scholar]
- Visvader JE, & Lindeman GJ (2008). Cancer stem cells in solid tumours: Accumulating evidence and unresolved questions. Nat Rev Cancer.Nature Reviews Cancer, 8, 755–768. [DOI] [PubMed] [Google Scholar]
- Visvader JE,& Lindeman GJ (2011). The unmasking of novel unipotent stem cells in the mammary gland. The EMBO Journal, 30, 4858–4859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visvader JE, & Stingl J (2014). Mammary stem cells and the differentiation hierarchy: Current status and perspectives. Genes and Development, 28, 1143–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlashi E, Lagadec C, Vergnes L, Reue K, Frohnen P, Chan M, et al. (2014). Metabolic differences in breast cancer stem cells and differentiated progeny. Breast Cancer Research and Treatment, 146, 525–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Christin JR, Oktay MH, & Guo W (2017). Lineage-biased stem cells maintain estrogen-receptor-positive and -negative mouse mammary luminal lineages. Cell Reports, 18, 2825–2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Garcia AJ, Wu M, Lawson DA, Witte ON,& Wu H (2006). Pten deletion leads to the expansion of a prostatic stem/progenitor cell subpopulation and tumor initiation. Proceedings of the National Academy of Sciences of the United States of America, 103, 1480–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Hayward S, Cao M, Thayer K, & Cunha G (2001). Cell differentiation lineage in the prostate. Differentiation, 68, 270–279. [DOI] [PubMed] [Google Scholar]
- Wang SS, Jiang J, Liang XH, & Tang YL (2015). Links between cancer stem cells and epithelial-mesenchymal transition. OncoTargets and Therapy, 8, 2973–2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Wang Q, Wang Z, Jiang J, Yu SC, Ping YF, et al. (2014). Metastatic consequences of immune escape from NK cell cytotoxicity by human breast cancer stem cells. Cancer Research, 74, 5746–5757. [DOI] [PubMed] [Google Scholar]
- Weidenfeld K, Schif-Zuck S, Abu-Tayeh H, Kang K, Kessler O, Weissmann M, et al. (2016). Dormant tumor cells expressing LOXL2 acquire a stem-like phenotype mediating their transition to proliferative growth. Oncotarget, 7, 71362–71377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicha MS, Liu S, & Dontu G (2006). Cancer stem cells: An old idea—A paradigm shift. Cancer Research, 66, 1883–1890. discussion 1895–1886. [DOI] [PubMed] [Google Scholar]
- Williams K, Motiani K, Giridhar PV, & Kasper S (2013). CD44 integrates signaling in normal stem cell, cancer stem cell and (pre)metastatic niches. Experimental Biology and Medicine, 238, 324–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong D, Kandagatla P, Korz W, & Chinni SR (2014). Targeting CXCR4 with CTCE-9908 inhibits prostate tumor metastasis. BMC Urology, 14, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright MH, Calcagno AM, Salcido CD, Carlson MD, Ambudkar SV, & Varticovski L (2008). Brca1 breast tumors contain distinct CD44+/CD24− and CD133+ cells with cancer stem cell characteristics. Breast Cancer Research, 10, R10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang L, & Semenza GL (2019). Hypoxia-inducible factors promote breast cancer stem cell specification and maintenance in response to hypoxia or cytotoxic chemotherapy. Advances in Cancer Research, 141, 175–212. [DOI] [PubMed] [Google Scholar]
- Xin L, Lawson DA, & Witte ON (2005). The Sca-1 cell surface marker enriches for a prostate-regenerating cell subpopulation that can initiate prostate tumorigenesis. Proceedings of the National Academy of Sciences of the United States of America, 102, 6942–6947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu C, Shiozawa Y, Taichman RS, McCauley LK, Pienta K, & Keller E (2012). Prostate cancer and parasitism of the bone hematopoietic stem cell niche. Critical Reviews in Eukaryotic Gene Expression, 22, 131–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu C, Yao Z, Dai J, Zhang H, Escara-Wilke J, Zhang X, et al. (2011).ALDH activity indicates increased tumorigenic cells, but not cancer stem cells, in prostate cancer cell lines. In Vivo, 25, 69–76. [PubMed] [Google Scholar]
- Yun EJ, Lo UG, & Hsieh JT (2016). The evolving landscape of prostate cancer stem cell: Therapeutic implications and future challenges. Asian Journal of Urology, 3, 203–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenzmaier C, Untergasser G, & Berger P (2008). Aging of the prostate epithelial stem/progenitor cell. Experimental Gerontology, 43, 981–985. [DOI] [PubMed] [Google Scholar]