

Abstract
Objectives
Single‐cell RNA sequencing (scRNA‐Seq) is a new technique used to interrogate the transcriptome of individual cells within native tissues that have already resulted in key discoveries in auditory basic science research. Rapid advances in scRNA‐Seq make it likely that it will soon be translated into clinical medicine. The goal of this review is to inspire the use of scRNA‐Seq in otolaryngology by giving examples of how it can be applied to patient samples and how this information can be used clinically.
Methods
Studies were selected based on the scientific quality and relevance to scRNA‐Seq. In addition to mouse auditory system (inner ear including hair cells and supporting cells, spiral ganglion neurons, and inner ear organoids), recent studies using human primary cell samples are discussed. We also perform our own analysis on publicly available, published scRNA‐Seq data from oral head and neck squamous cell carcinoma (HNSCC) samples to serve as an example of a clinically relevant application of scRNA‐Seq.
Results
Studies focusing on patient tissues show that scRNA‐Seq reveals tissue heterogeneity and rare‐cell types responsible for disease pathogenesis. The heterogeneity detected by scRNA‐Seq can result in both the identification of known or novel disease biomarkers and drug targets. Our analysis of HNSCC data gives an example for how otolaryngologists can use scRNA‐Seq for clinical use.
Conclusions
Although there are limitations to the translation of scRNA‐Seq to the clinic, we show that its use in otolaryngology can give physicians insight into the tissue heterogeneity within their patient's diseased tissue giving them information on disease pathogenesis, novel disease biomarkers or druggable targets, and aid in selecting patient‐specific drug cocktails.
Keywords: head and neck squamous cell carcinoma, heterogeneity, otolaryngology, single‐cell sequencing
Uses of single‐cell RNA sequencing (scRNA‐Seq) in clinical medicine. scRNA‐Seq characterizes tissue heterogeneity including diseased and rare cell types allowing for identification of unique genes expressed by these cells that might have been obscured by traditional RNA‐Seq and that may be used as potential drug targets. Interrogation of drug target expression within each cell cluster can aid in creating a patient‐specific drug cocktail. scRNA‐Seq can also be performed before or after treatment to get a sensitive picture of how a patient responded with elimination of diseased cells, continued presence of diseased cells unaffected by treatment, or emergence of new drug‐resistant clones.
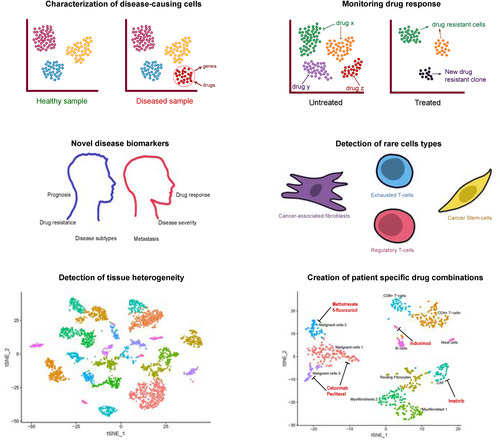
1. INTRODUCTION
Currently in otolaryngology, histology, microarray, and bulk RNA‐sequencing (RNA‐Seq) methodologies are used on patient biopsy samples to diagnose disease including microbial infections, human papilloma virus, genetic disorders such as neurofibromatosis type II, and various head and neck cancers like head and neck squamous cell carcinoma (HNSCC). These methodologies can be biased in their investigation of tissues and are limited by extremely low sensitivity, antibody or probe availability, and averaging of gene expression across the tissue of interest, ignoring tissue and cellular heterogeneity. 1 For example, the use of bulk RNA‐Seq in tumor diagnostics can obscure the importance of tumor heterogeneity and rare cell types to tumor biology and disease prognosis. 2
Single‐cell RNA‐Seq (scRNA‐Seq) is a powerful new method that allows users to obtain the transcriptome of individual cells. This technology avoids the cell averaging seen in bulk RNA‐Seq allowing for novel discoveries in tissue heterogeneity and biology at single‐cell resolution.3, 4, 5, 6, 7 The use of scRNA‐Seq in auditory basic science research has proven important in deciphering cellular heterogeneity of inner ear tissues and is just beginning to provide answers to questions regarding development of diverse cell types in this organ, how this heterogeneity relates to auditory function, and how we can use this information to understand auditory disease and mechanisms for hearing restoration. The explosive use of scRNA‐Seq in auditory research has resulted in several new discoveries in the field of otolaryngology.8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20 In endolymphatic sac, scRNA‐Seq showed that mitochondrial‐rich cells were likely responsible for the pathogenesis of enlarged vestibular aqueducts. 12 Two recent studies used scRNA‐Seq on spiral ganglion neurons (SGN) to detect four subpopulations, including three novel SGN subtypes, differing in their expression of novel marker genes and in sound intensity‐specific activation19, 20 that underlie the clinical phenomenon of “hidden hearing loss.” Another study used scRNA‐Seq of cells from the organ of Corti to prove that cochlear hair cells not only differentiate from progenitor supporting cells but also from nonsensory cell types. 8 We provide a selected list of scRNA‐Seq studies done in the mouse that are relevant to hearing for further review (Table 1). Although most scRNA‐Seq experiments related to otolaryngology are currently conducted on mice in basic science laboratories (Table 1), with the increasingly low‐cost and new high‐throughput scRNA‐Seq technologies, this exciting tool can be readily applied to human tissue to generate new discoveries in human biology and may soon be translated to clinical otolaryngology. Human vestibular and cochlear tissues, while both difficult and unethical to obtain from healthy patients, are frequently discarded as a result of standard transcranial surgical approaches to resections of acoustic neuromas, for example.21, 22 With collaborations between treating surgeons and surgeon‐scientists, sequencing of human inner ear tissue and other more easily obtained pathologic tissue, including acoustic neuromas, HNSCC, vocal cord polyps, and recurrent respiratory papillomas, could deliver valuable information on human cell types responsible for disease and possible druggable targets within these cell types. 11
TABLE 1.
Selected list of scRNA‐Seq studies from the mouse inner ear
| Study | Tissue a | Cell isolation | scRNA‐Seq technology |
|---|---|---|---|
| Burns et al 8 | Early postnatal cochlear SE, utricular SE, cochlear SC | Fluidigm C1 | Smart‐seq |
| Yamashita et al 9 | Postnatal and adult cochlear SE | Chromium (10x Genomics) | Drop‐Seq |
| Chessum et al 13 | Early postnatal IHC | Chromium (10x Genomics) | Drop‐Seq |
| McInturff et al 14 | Early postnatal and adult utricular HC | Fluidigm C1 | Smart‐seq |
| Hoa et al 15 | Adult cochlear SC | Fluidigm C1 | Smart‐seq |
| Ranum et al 16 | Early postnatal through adult cochlear IHC, OHC, SC | Micropipette | Smart‐seq2 |
| Tang et al 17 | mESC‐derived inner ear organoids | Chromium (10x Genomics) | Drop‐Seq |
| Sun et al 18 | Adult SGN | Gemcode (10x Genomics) | Drop‐Seq |
| Shrestha et al 19 | Adult SGN | Micropipette | Smart‐seq2 |
| Petitpré et al 20 | Postnatal and adult SGN | FACS | Smart‐seq |
| Sherrill et al 10 | Early postnatal SGN | Fluidigm C1 | Smart‐seq |
| Korrapati et al 11 | Adult SV | Chromium (10x Genomics) | Drop‐Seq |
| Honda et al 12 | Embryonic, early postnatal, adult ES | Fluidigm C1 | Smart‐seq |
Abbreviations: ES, endolymphatic sac; HC, hair cells; IHC, inner hair cells; mESC, mouse embryonic stem cells; OHC, outer hair cells; SC, cochlear supporting cells; scRNA‐Seq, single‐cell RNA sequencing; SE, sensory epithelium; SGN, spiral ganglion neurons, SV, stria vascularis.
All the above studies were conducted using mouse tissue.
A few studies have used scRNA‐Seq on human otolaryngologic patient samples including HNSCC,23, 24, 25, 26 sinus mucosa, 27 and melanoma. 28 These, as well as studies in other human tissues,29, 30 have shown the utility of this method in human clinical samples. These studies have generated excitement for the use of scRNA‐Seq in unraveling tissue heterogeneity with the hope of using this information to provide care that is tailored to each patient, 31 but for this goal to be attained physicians need be aware of how this technology may be applied to their clinical practice and the potential novel diagnostic information and therapeutic options it might provide in the future.32, 33 Here we give a brief overview of scRNA‐Seq methodology, suggest potential clinical applications for scRNA‐Seq in otolaryngology, and comment on potential limitations for its clinical use.
To make this review as clinically relevant as possible, we will focus on reviewing studies that have been conducted in human samples relevant to otolaryngology. While many laboratories are focusing on optimizing sequencing for human samples such as cochlear and vestibular tissues, currently it is much easier to obtain human tumor samples and the only published scRNA‐Seq studies of human tissue relevant to otolaryngology use HNSCC,23, 25, 26 melanoma, 28 or chronic rhinosinusitis 27 samples. We use these studies as examples of the use of scRNA‐Seq as a research tool in otolaryngology and its potential clinical applications. We perform our own scRNA‐Seq analysis (methods available in Data S2) of a publicly available HNSCC data set as an example of how this technique may impact clinical care in the future (Data S1).
2. scRNA‐Seq WORKFLOW
To give clinicians a general understanding of scRNA‐Seq concepts, we provide a brief overview of the scRNA‐Seq experimental workflow to inform the discussion of potential applications of this technology in otolaryngology. While a comprehensive review of this complex process is not provided here, the experimental design and analysis of scRNA‐Seq data have been extensively reviewed elsewhere and we point interested readers that would like to learn more about the details of designing scRNA‐Seq experiments to the reviews mentioned within our discussion.34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46 Most scRNA‐Seq protocols follow a similar sequence of reactions (Figure 1) including tissue dissociation to generate a single‐cell suspension, cell isolation (Figure 1A), mRNA capture and barcoding (Figure 1B), library preparation (Figure 1C), followed by next‐generation sequencing (NGS) (Figure 1D). 47 Single‐nucleus RNA‐Seq (sNuc‐Seq) protocols have also recently been developed to isolate single nuclei from difficult to dissociate or archived patient samples. 48 From a single‐cell or nuclei suspension, most methodologies require each cell or nucleus to be captured and isolated for barcoding and pooling. Cell isolation methods have been reviewed previously including a review by Qi et al on designing scRNA‐Seq experiments for head and neck cancer.34, 35 Briefly, isolation of cells into wells or microwells of a plate can involve limiting dilution, laser capture microdissection, micropipette isolation, fluorescence‐activated cell sorting (FACS), or in situ barcoding including split‐pool barcoding methods.49, 50 Isolation of cells can also be accomplished using microfluidics that either use circuits to distribute cells into nanowells (ie, Fluidigm C1 51 ) or isolate cells into nanoliter oil‐based droplets (ie, 10x Genomics Chromium 52 ). Most methods, after isolating cells into either plates (ie, STRT‐Seq, 53 CEL‐Seq2, 54 and SMART‐Seq2 55 ) or droplets (ie, inDrops, 56 Drop‐seq, 57 10x Genomics Chromium 52 ), require cell lysis to release mRNA molecules. Oligonucleotide primers with barcodes and oligo‐dT sequences are added to capture the 3′‐end mRNA poly‐A tail, give each mRNA a cell‐type‐specific barcode, and amplify mRNA. Within these primers, some methods also contain a unique molecular identifier (UMI) that labels each mRNA as a unique molecule. Recent developments in scRNA‐Seq methodologies have eliminated the need to isolate single cells or nuclei by using split‐pool barcoding.49, 50 This method allows multiple cells to be placed in individual wells containing unique barcodes and through multiple mixing, well distributions, and barcoding reactions each mRNA within each cell receives a unique cell‐specific barcode. While this technique allows for sequencing of hundreds of thousands of cells, including multiple tissue samples and fixed cells or nuclei, the large number of mixing reactions and pipetting required consumes valuable time and may perturb the native transcriptome, so precious tissue may not be amenable to this technique.
FIGURE 1.
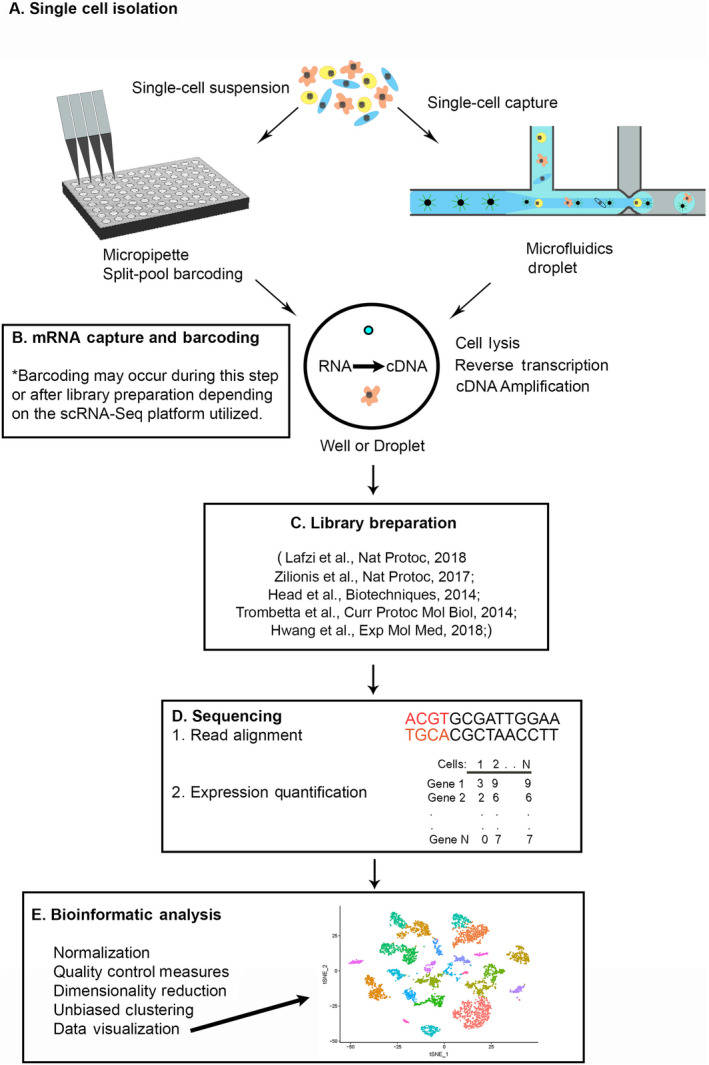
Diagram of scRNA‐Seq, single‐cell RNA sequencing (scRNA‐Seq) workflow. A, Single‐cell isolation involves generation of a single‐cell suspension. Cells are isolated by microdissection and micropipette, microfluidic circuits, droplets, or split pool barcoding. After single‐cell isolation, cells are compartmentalized into wells or droplet for library preparation depending on the scRNA‐Seq platform that is used. B, mRNA capture and barcoding involves cell lysis releasing mRNA which is barcoded and reverse transcribed into cDNA. Polymerase chain reaction or in situ transcription is used to amplify cDNA, C, library preparation involves pooling and fragmentation of cDNA and addition of adaptors used for, D, next‐generation sequencing. mRNA reads are aligned to known genes and genes are mapped back to their cell of origin. To quantify gene expression, a matrix with cells on the x‐axis and genes on the y‐axis is generated with read counts for each gene. This matrix is then used for, E, bioinformatic analysis including: quality control, filtering of unhealthy cells, normalization and scaling of mRNA read counts, principal component analysis to determine genes responsible for the most variation between cells, dimensionality reduction, unbiased clustering, followed by data visualization
After RNA is barcoded, it is reverse transcribed into cDNA and cDNA is amplified by polymerase chain reaction or in vitro transcription. In most cases, amplified cDNA from each cell is then pooled for preparation of sequencing libraries where cDNA molecules are fragmented and adaptor sequences are added for further amplification and sequencing. Methods utilized for library preparation from amplified cDNA are varied and reviewed elsewhere including a review dedicated to discussing the details of current methods for library preparation by Head et al.39, 40, 41, 42, 43 One clinically important distinction between library preparation methods is that various methodologies result in 3′‐end, 5′‐end, or full‐length sequencing data.34, 39 Most droplet‐based protocols are biased to sequencing of the 3′‐end of mRNA resulting in loss of genetic information from the 5′‐end37, 38; however, some plate‐based techniques, like SMART‐Seq2, use template switching during reverse transcription to amplify the full length strand of mRNA allowing for the detection of different mRNA isoforms, splicing variants, single‐nucleotide polymorphisms, and variant mutations that would go undetected with 3′‐end enriched sequencing. 55
Plate‐based techniques, due to lower cell numbers, allow for extremely high sensitivity and read depth (number of genes detected per cell) and this incredible depth of sequencing makes these techniques clinically useful for precious patient samples of low cell number; however, the low throughput and high cost per cell make them less clinically feasible. In contrast, due to the microscopic size of the droplets created by microfluidics, the reagent cost drops significantly and with no limit on the number of droplets created, the throughput is much higher; however, the read depth is usually lower than plate‐based methodologies because a larger number of single‐cell libraries can be pooled and loaded on the same sequencing lane. The Chromium (10x Genomics) is becoming the gold‐standard microfluidic device because of its unprecedented throughput, speed, and low cost per cell. 52 This parallels our group's experience in transitioning from circuit‐based to nanoliter droplet‐based microfluidic single‐cell RNA‐Seq systems.11, 15 The ability to sequence a greater number of cells allows for a more comprehensive picture of the transcriptomic profile as typically only 5% to 15% of the mRNA within a given single cell are captured, 57 making high‐throughput technologies an attractive platform for clinical use. Table 2 compares different single cell capture platforms. For a more in‐depth review of the intricacies of designing a single‐cell experiment and selecting the appropriate platforms readers are pointed to excellent reviews by Nguyen et al 44 and Lafzi et al. 39
TABLE 2.
Brief comparison of single‐cell RNA sequencing platforms
| Platform | inDrop | Drop‐Seq | 10x Chromium | C1 Fluidigm | SMART‐Seq | CEL‐Seq | STRT‐Seq |
|---|---|---|---|---|---|---|---|
| Type | Microfluidic | Microfluidic | Microfluidic | Microfluidic into plate | Plate‐based | Plate‐based | Plate‐based |
| Cost per cell | $0.05 | $0.06 | $0.50 | $4.70 | $1 | $3.50 | $50 |
| Cells per run | Up to 40 000 | 10 000 | 500‐80 000 | 94‐800 | 1‐1 000 | 94‐800 | 96 |
| Average read depth | 30 000‐60 000 | 30 000‐60 000 | 30 000‐60 000 | over 3 million | over 2 million | 1 70 000 | 2 40 000 |
| Read length | 3′ bias | 3′ bias | 3′ bias | Full length or 3′ bias | Full length or 3′ bias | 3′ bias | 3′ bias |
| Barcode and unique molecular identifier | Yes | Yes | Yes | No | No | Yes | Yes |
To analyze scRNA‐Seq data, computational bioinformatic analysis is necessary to convert gene expression matrices (Figure 1E) into a more interpretable graphic format. Normalization, quality control measures, dimensionality reduction, and unbiased clustering algorithms are applied to cluster cells by their potential cellular identity. Seurat (https://satijalab.org/seurat/) is a commonly used bioinformatics pipeline for scRNA‐Seq data analysis and visualization that outputs plots depicting clusters of cells grouped together based on similarities in their gene expression58, 59 (Figure 1E). These plots aid in characterizing tissue heterogeneity by identifying novel and known cell types as well as detecting expression of genes that are unique to a specific cell type or unknown to be previously expressed by a certain cell type within a given tissue. For a thorough description of computational analysis for complex scRNA‐Seq data, we encourage readers to see reviews by Hwang et al, 45 Shafer, 46 and Chen et al. 36
3. CLINICAL APPLICATIONS OF scRNA‐Seq FOR OTOLARYNGOLOGY
scRNA‐Seq techniques have already advanced the field of auditory research by identifying heterogeneity of cell types within the inner ear, including those involved in hair cell regeneration, hidden hearing loss, and the pathogenesis of enlarged vestibular aqueducts8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20 and are becoming more widespread in their utilization in human tissues.23, 25, 26, 60, 61 Fostering awareness of this technique and its applications for basic biological discovery in diseases managed by practicing otolaryngologists may assist with diagnosis, prognosis, and potentially personalized treatment recommendations in future clinical practice. To demonstrate these clinical applications, we will use three examples of diseases related to otolaryngology where scRNA‐Seq has already been applied to human samples including chronic sinusitis, oral HNSCC, and melanoma; but we encourage otolaryngologists to think about how scRNA‐Seq can be applied to their disease of interest as we walk through its potential applications.
3.1. Detecting tissue heterogeneity to understand disease pathogenesis
Diseases, like head and neck squamous cell carcinoma, are composed of a complex group of heterogenous cell types, consisting of both native and disease causing, in this case cancer causing, cells as well as cells within the microenvironment, including immune cells, extracellular matrix, fibroblasts, stem cells, and vasculature, which all communicate to play a role in tumorogenesis.62, 63, 64 Cellular heterogeneity in the setting of cancers of the head and neck and in the setting of chronic inflammation such as chronic rhinosinusitis has been implicated in disease maintenance or recurrence, patient variability in drug efficacy, and the failure of clinical drug trials.65, 66 As an example, understanding the interplay between diseased and native cell types, such as the case between tumor cells and native cell types (ie, fibroblasts, immune cells) in the tumor microenvironment (TME), will improve the knowledge base surrounding cancerous cell types and their interplay with the microenvironment. Using unbiased clustering of each individual cell followed by cell type identification of each cluster by expression of known marker genes, scRNA‐Seq data can distinguish disease‐causing cells from native tissue and reveal complex heterogeneity within diseased tissue samples.24, 25
3.1.1. Heterogeneity of malignant tumor cells
Several scRNA‐Seq studies on human tumor samples have identified tumor heterogeneity by demonstrating subpopulations within malignant tumor cells.67, 68, 69, 70 By interrogating the gene expression programs within malignant cell populations of 18 metastatic melanomas, a landmark scRNA‐Seq study by Tirosh et al 28 identified the existence of a subpopulation of malignant cells with a high level of AXL cell surface receptor tyrosine kinase program expression, a set of genes related to drug resistance and melanoma cancer stem cell (CSC) maintenance. The AXL‐high subpopulation signature could not be detected in bulk RNA‐Seq data from melanoma tumors which proves the utility of scRNA‐Seq in identifying heterogenous cells responsible for the pathogenesis of metastasis and intrinsic drug resistance. With the availability of AXL inhibitors such as BGB324 that show efficacy in decreasing human soluble AXL levels and treating human‐derived spheroid melanoma tumors,71, 72 detection of this signature by scRNA‐Seq in tumors of the head and neck may help identify which patients would respond best in human clinical trials. The influence of tumor heterogeneity on disease pathogenesis has also been demonstrated in oral HNSCC by Puram et al 23 in the first scRNA‐Seq study relevant to otolaryngology. Clustering of malignant cells from 10 HNSCC showed that malignant cells clustered based on patient, as we confirmed in our own analysis of their published data set using Seurat58, 59 (Figure 2A‐C). This suggests that clonal evolution of HNSCC malignant cells is unique to each patient and each tumor should be treated therapeutically as such. As in melanoma, malignant HNSCC cells did contain groups of cells with shared gene expression profiles or subpopulations, including one expressing high levels of extracellular matrix genes that also expressed markers related to a partial epithelial‐to‐mesenchymal transition (EMT). The authors suggest that it is possible this partial‐EMT expressing subpopulation is responsible for causing invasion and lymph node metastasis in HNSCC. 73 To confirm the presence of malignant intratumoral heterogeneity in HNSCC, we performed our own scRNA‐Seq unbiased clustering analysis of cells from one HNSCC patient, T25, which displayed three subpopulations of malignant cells within one tumor that varied in their gene expression (Figure 2D).
FIGURE 2.
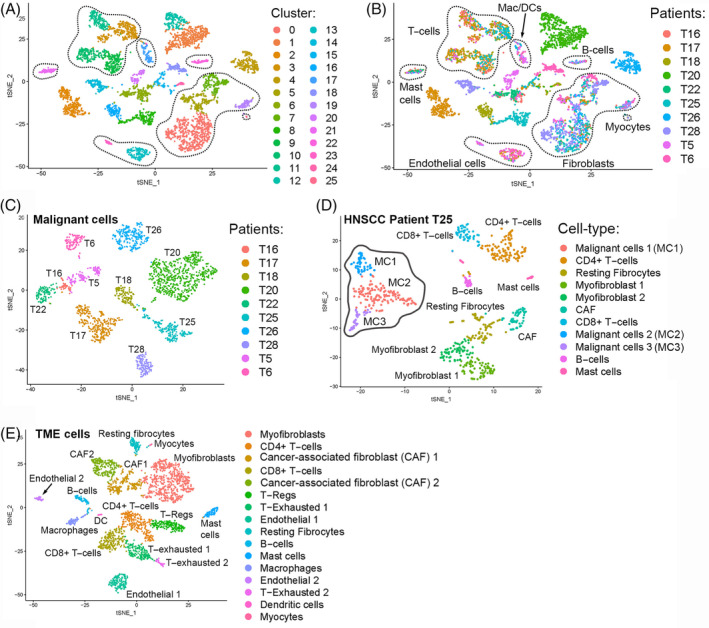
Example of our new analysis of scRNA‐Seq, single‐cell RNA sequencing (scRNA‐Seq) data from Puram et al 40 head and neck squamous cell carcinoma (HNSCC) using Seurat. A,B, Unbiased clustering of cells from 10 HNSCC patients. Dotted circles represent clusters of cells from the same cell type. Cell types were determined by cluster‐specific expression of known cell‐type markers. Seurat identified 25 independent clusters of similar cells, A. Each cell was colored by the patient of origin with malignant cell clusters consisting of cells from the same patient while tumor microenvironment (TME) cells consisted of cells from multiple HNSCC patients, B. TME cells are demarcated by the dotted outlines while the malignant cells are denoted by the absence of the dotted outlines, B. C, Unbiased clustering of only the malignant cells from the HNSCC data. Cells were colored by the patient of origin. This analysis shows that malignant cells cluster by patient. D, Unbiased clustering of all cells from HNSCC patient T25 showing that malignant cells are distributed in multiple clusters or subpopulations (malignant cells 1‐3 outlined in solid grey). Cancer associated fibroblasts (CAF) are outlined in dotted black. E, Unbiased clustering of only the TME cells from the HNSCC data. Cells are colored based on cell‐type identity demonstrating TME heterogeneity
3.1.2. Heterogeneity of cells in the microenvironment
Single‐cell profiles obtained from tumor tissue will consist of not only individual malignant tumor cells but also cells from the TME, which consists of blood vessels, immune cells, fibroblasts, signaling molecules, and extracellular matrix that surrounds malignant cells, allowing for identification of heterogeneity within these cell types. Detection and interrogation of TME subpopulations within multiple tumor types allows for insight into potential stroma‐tumor communication programs that may be playing a role in disease pathogenesis.25, 74, 75 In contrast to malignant HNSCC cells that clustered by patient, the study by Puram et al 23 found that HNSCC TME cells were not patient‐specific and clustered based on cell type as we also confirmed in our own analysis showing that each TME cluster (Figure 2E) consisted of cells from multiple patients (Figure 2B). Interestingly, HNSCC fibroblasts displayed vast heterogeneity including two subpopulations that expressed genes affiliated with cancer‐associated fibroblasts (CAF) (Figure 2E). Genes that were highly differentially expressed in CAF included mesenchymal and transforming growth factor‐beta (TGF‐β) signaling genes whose expression was shared by the malignant HNSCC partial‐EMT subpopulation, suggesting that CAF‐malignant cell signaling may be involved in promoting invasion and metastasis in HNSCC. Since most HNSCC tumors contain similar TME subpopulations, these cells could represent shared disease pathogenesis between all HNSCC patients that could be targeted by a similar drug program.
The sensitivity of scRNA‐Seq to detect TME heterogeneity also allows for the detection of rare cell types such as stem‐ or progenitor‐like cells responsible for disease maintenance70, 76, 77 or rare immune cell subpopulations involved in evasion of the immune response.74, 75, 78, 79 Ordovas‐Montanes et al 27 performed scRNA‐Seq on ethmoid sinus tissue samples from patients with chronic rhinosinusitis with (CRwNP) or without (CRsNP) nasal polyps and performed trajectory analysis on individual basal epithelial cells to computationally determine a potential pathogenic developmental cell state in CRwNP. Trajectory analysis examines gene expression and places cells along a developmental timeline based on unique gene expression. Their analysis showed that most basal cells in CRwNP are stuck in a progenitor state expressing, among other genes, increased Wnt‐signaling genes that could potentially be responsible for polyp development. Another rare cell type that can be detected by scRNA‐Seq are CSC, which have been shown to play a role in the creation of HNSCC heterogeneity, resistance to drug therapy, disease recurrence, and metastasis making the elimination of these cells critical to eradicating residual disease in HNSCC. 80 In support of this, Puram et al 23 were able to detect varying levels of epithelial gene expression within malignant HNSCC subpopulations reflecting variable levels of epithelial differentiation. Various biomarkers to identify CSC have been suggested 80 and we demonstrate how CSC may be identified within scRNA‐Seq data using these known markers in one HNSCC patient, T25 (Figure 3). Other clinically relevant rare TME subpopulations that were detected by Puram et al 23 in HNSCC and Tirosh et al 28 in melanoma were CD4+ T‐regulatory cells and exhausted T cells (Figure 2E), which likely inhibit the antitumor immune response and prevent the efficacy of immune‐modulating therapy.
FIGURE 3.
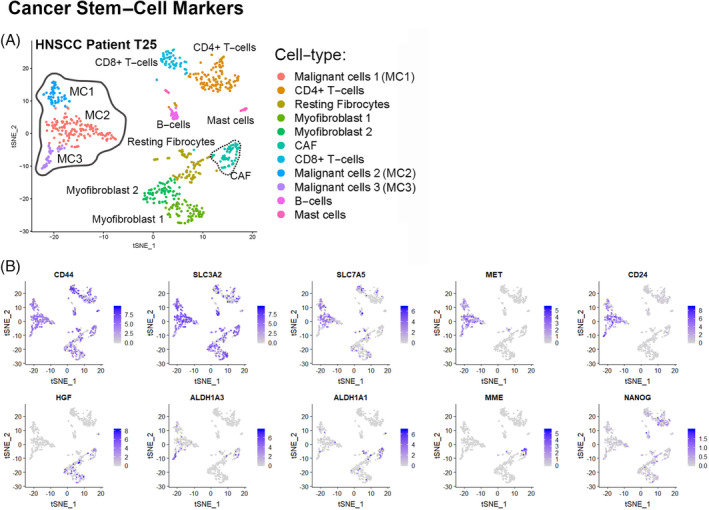
Cancer stem cell (CSC) markers present in head and neck squamous cell carcinoma (HNSCC) patient T25. The level of expression of different CSC markers are indicated on the t‐distributed stochastic neighborhood embedding (tSNE) plot of all HNSCC cells from patient T25 (Figure 2D). Cells that express CSC genes are colored in gradations of purple depending on their expression level, with blue representing the highest expression level. Malignant clusters (solid grey outline in Figure 2D) vary in their expression of CSC markers and some markers are also expressed in cancer‐associated fibroblasts (CAF) (dotted black outline in Figure 2D)
3.2. Using heterogeneity detected by scRNA‐Seq for diagnostic and prognostic evaluation
3.2.1. Identification of disease biomarkers
Obtaining single‐cell profiles from patient biopsy samples, for example HNSCC tumors or sinus mucosa, may aid in a more sensitive diagnosis by allowing physicians to identify unique cell‐type‐specific signatures or biomarkers for disease subtyping. 27 Disease subtyping is crucial in tumor diagnostic and prognostic decision making in HNSCC 81 and a few studies have shown that scRNA‐Seq‐derived signatures that account for tumor heterogeneity can more accurately classify tumor subtypes.67, 74 Conventional bulk RNA‐Seq analysis has classified HNSCC into four subtypes based on molecular gene signatures: basal, classical, atypical, or mesenchymal. 82 By scoring each HNSCC cell in their scRNA‐Seq data based on expression levels of previously defined HNSCC subtype signatures, Puram et al 23 showed that malignant HNSCC cells only mapped to basal, classical, or atypical signatures and the mesenchymal subtype was not present in any HNSCC malignant cells. However, the mesenchymal subtype scored highest in fibroblasts of the TME suggesting that this subtype truly represents HNSCC with high levels of TME gene expression highlighting the need for tumor classification systems, potentially based on compiled scRNA‐Seq data that account for tissue heterogeneity.
Investigating gene expression within heterogenous cell clusters detected by scRNA‐Seq can be used to identify cell‐type‐specific known or novel biomarkers associated with tumor metastasis, prognosis, and survival.76, 77, 83, 84 Puram et al 23 showed the novel partial‐EMT signature detected in a subset of malignant cells was present in existing bulk RNA‐Seq HNSCC tumor data and the levels of expression of this signature were associated with invasion, metastasis, and poor prognosis. Currently, most head and neck cancer patients undergo aggressive nodal dissection, a procedure associated with significant morbidity, based on unreliable markers such as tumor size or grade.85, 86 Determining the presence and extent of the partial‐EMT signature in HNSCC patients could aid in weighing the risks of nodal dissection in patients with lower risk of metastasis. Here we also show that recently published HNSCC prognostic signatures associated with decreased survival, 87 metastasis, 88 and lack of response to radiotherapy 89 can be detected in single cells from HNSCC patient T25 23 (Figure 4). Further characterization of all head and neck tumors using scRNA‐Seq could reveal other novel signatures associated with prognosis, drug response, or survival.
FIGURE 4.
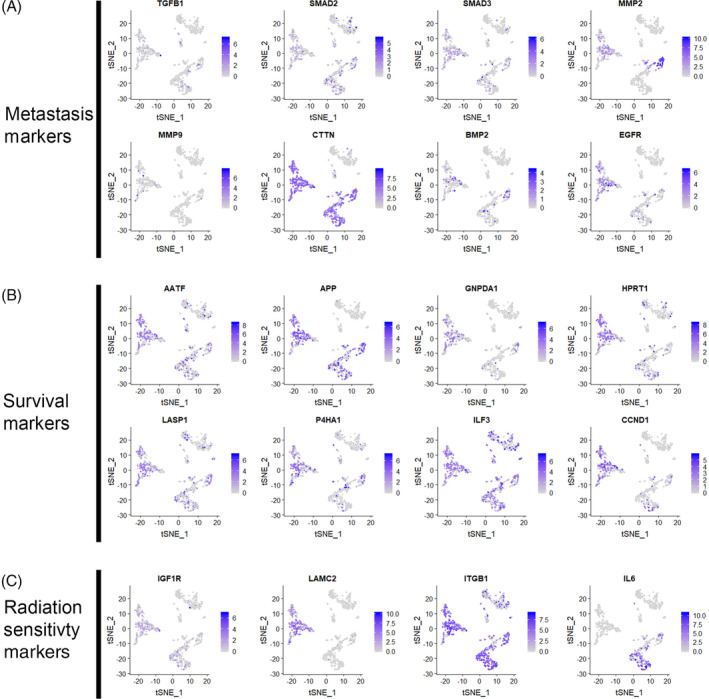
Known prognostic markers present in head and neck squamous cell carcinoma (HNSCC) patient T25. Feature plots of the original clustering from patient T25 (Figure 2D). Cells that express known metastatic, A, poor survival, B, or radiation sensitivity, C, marker genes are colored in gradations of purple depending on their expression level, with blue representing the highest expression level
scRNA‐Seq may also detect cell‐type‐specific gene expression associated with the severity of disease. For example, Ordovas‐Montanes et al 27 was also able to detect a novel cell‐type‐specific change distinguishing between severe CRwNP and less severe CRsNP. They found high interleukin (IL)‐4/IL‐13 expression within secretory epithelial cells from patients with severe disease and interferon (IFN)‐alpha/IFN‐gamma expression in less severe disease. Not only does this represent a cell‐type‐specific drug target, but also detection of the severe disease program by scRNA‐Seq in chronic rhinosinusitis (CR) patient biopsies could influence treatment decisions including surgical intervention. Ordovas‐Montanes et al 27 were also able to track changes in basal‐cell transcriptomes before and after treatment with an IL‐4Rα inhibitor. While many polyp‐specific genes were downregulated in disease‐causing basal cells, they also identified a basal‐cell polyp‐specific program that did not change with treatment revealing a drug‐resistant signature that could be interrogated for new druggable targets. This demonstrates how scRNA‐Seq can also be conducted before and after treatment to monitor drug response, emergence of resistance, and identify markers of these events.
3.3. Personalized therapy selection based on scRNA‐Seq data
3.3.1. Patient‐specific drug selection
Heterogeneity detected by scRNA‐Seq can also be used to generate novel or look for known biomarkers for drug sensitivity or resistance that will aid in selecting which patients should be treated with specific drugs.83, 90, 91 While drug targets can been analyzed in cohorts of scRNA‐Seq samples, there is a need for analysis of heterogeneity at the level of individual tumors to aid in patient‐specific drug selection, enrollment of patients in clinical trials, and analyzing ablation or alteration of specific cell types before and after drug treatment.92, 93, 94 Immunotherapy with immune checkpoint inhibitors or small‐molecule immune modulators is currently an active area of research in head and neck cancer and many clinical trials are aimed at using these drugs to subvert tumor‐induced immunosuppression.63, 95 As mentioned, both melanoma and HNSCC scRNA‐Seq studies revealed the presence of rare exhausted CD8+ T cells and developed an exhausted T‐cell signature; however, both authors found that each patient varied in their number of exhausted T cells and their expression of known exhaustion markers including receptors that inhibit the T‐cell immune response.23, 28 For example, in one melanoma tumor expression of coinhibitory CTLA4 receptor was absent from the exhausted T‐cell population; however, this patient had previously been treated with CTLA4 inhibitor, ipilimumab, and subsequently became resistant. 28 Identification of T‐regulatory and T‐exhausted subpopulations through scRNA‐Seq can lead to the creation of novel drug‐response biomarkers or potential new drug targets within these cell types. Detecting biomarkers from single‐cell TME profiles of head and neck tumor patients may aid in determining which patients will respond best to immune checkpoint inhibitors or should be considered for various immunotherapy clinical trials.
Cell clusters generated from scRNA‐Seq data can also be analyzed for expression of known drug targets to determine if or which cell types express certain drug targets and how effective the drug might be in targeting all diseased subpopulations and/or pathogenic TME cells. We show how this can theoretically be done on a patient‐specific basis by using the HNSCC data from patient T25 and displaying the cells that express the targets of current drugs used to treat HNSCC (Figure 5).86, 96 For example, epidermal growth factor receptor (EGFR) is the target of EGFR inhibitors such as cetuximab, and this gene is expressed in malignant cells from patient T25 suggesting that these cells are likely susceptible to this drug. Ideally, if a given drug does not target all subpopulations of malignant cells or a particularly pathogenic cell type of the TME such as CAF or CSC, then other drug targets could be identified within these populations and these drugs could be added to the drug cocktail until all cells are targeted.
FIGURE 5.
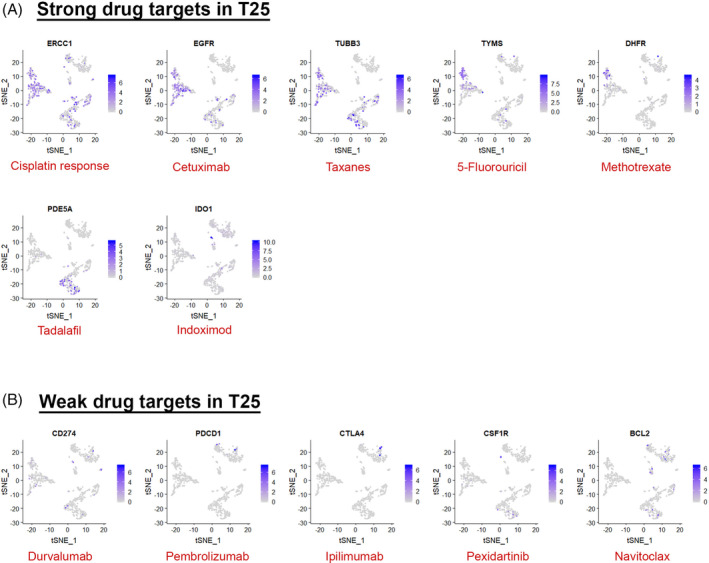
Drug targets for commonly used and new head and neck squamous cell carcinoma (HNSCC) drugs used to treat HNSCC in patient T25. Feature plots of the original clustering from patient T25 (Figure 2D). Cells that express drug target genes are colored in gradations of purple depending on their expression level, with blue representing the highest expression level. Drug target gene is written in the black in the plot title and the drug that targets it is written at the bottom of the plot in red. A, Drugs that show strong cell‐type‐specific target expression in patient T25. B, Drugs that show nonspecific or weak target expression in patient T25
One potential agnostic approach to finding new druggable targets in malignant subpopulations or TME cells is to look for the presence of genetic targets of FDA‐approved drugs or small molecules within clusters derived from scRNA‐Seq data that could be repurposed for use in head and neck cancer or other otolaryngologic disease. 97 A new database called Pharos describes 20 000 gene/protein targets and the availability of FDA‐approved drugs or small‐molecule ligands for each target. 98 To demonstrate a possible use of this resource, we used the batch search option to search for druggable targets within the top marker genes from HNSCC patient T28's one malignant and two CAF clusters (Figure 6). We found 67 genes that could be targeted with FDA‐approved drugs in the malignant population, 28 in the first CAF subpopulation, and 19 in the second CAF subpopulation (full data in Table 3). Interestingly, some drug targets overlapped between these populations suggesting shared signaling between malignant and TME cells that could be targeted by a single drug. Of note, while we only analyzed a single patient's tumor sample, this methodology could be applied across all 18 HNSCC and 5 lymph node samples in this cohort to make a more informed judgment on effective drug combinations for HNSCC and effect on patient outcomes. However, these data demonstrate the potential use of scRNA‐Seq cluster expression profiles to identify previously approved drugs and available small molecules that could have activity against specific cells for the treatment of refractory disease or generating data for new clinical drug trials.
FIGURE 6.
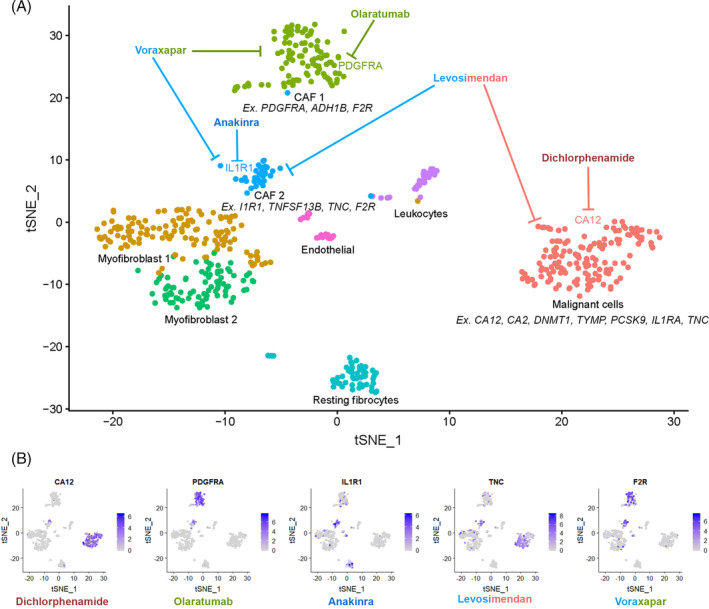
Potential drug targets and drugs that inhibit them for each malignant cell and cancer‐associated fibroblast (CAF) cluster in head and neck squamous cell carcinoma (HNSCC) patient T28. A, Unbiased clustering of HNSCC cells from patient T28. Potential cell‐type specific drug targets are shown under each cluster. Example antagonistic drugs that target each malignant and CAF cluster are shown. B, Feature plots of the original clustering of HNSCC patient T28 showing cells that cluster specific drug target genes colored in gradations of purple depending on their expression level, with blue representing the highest expression level. The drugs that target these genes are written in color under each plot and are shown in Figure 6A. Full drug target data are available in Table 3
TABLE 3.
Pharos cell‐type‐specific druggable targets from HNSCC patient T28
| Cluster | Gene Symbol | Name | Description | Development level | P value | Avg_logFC | Pct.1 | Pct.2 | P value adjusted | Cluster | Gene | Drugs |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CAF1 | PTGS1 | Prostaglandin G/H synthase 1 | Converts arachidonate to prostaglandin H2 (PGH2), a committed step in prostanoid synthesis. Involved in the constitutive production of prostanoids in particular in the stomach and platelets. In gastric epithelial cells, it is a key step in the generation of prostaglandins, such as prostaglandin E2 (PGE2), which plays an important role in cytoprotection. In platelets, it is involved in the generation of thromboxane A2 (TXA2), which promotes platelet activation and aggregation, vasoconstriction, and proliferation of vascular smooth muscle cells | Tclin | .001999 | 2.039973 | 0.278 | 0.177 | 1 | CAF 1 | PTGS1 | Piketoprofen, aminosalicylic acid, felbinac, oxaprozin, loxoprofen, dexketoprofen, oxyphenbutazone, tolmetin, suprofen, piroxicam, naproxen, ketorolac, ketoprofen, ibuprofen, flurbiprofen, fenoprofen, bromfenac, nepafenac, acetylsalicylic acid, tiaprofenic acid, fenbufen, bismuth subsalicylate, acemetacin, floctafenine, lumiracoxib, bromfenac, fenoprofen, meclofenamic acid, mefanamic acid, meloxicam, paracetamol |
| CAF1 | LEPR | Leptin receptor | Isoform E: antagonizes isoform A and isoform B‐mediated LEP binding and endocytosis | Tclin | 1.03E‐10 | 2.616011 | 0.806 | 0.717 | 2.44E‐06 | CAF 1 | LEPR | Metreleptin |
| CAF1 | FGFR1 | Fibroblast growth factor receptor 1 | Tyrosine‐protein kinase that acts as cell‐surface receptor for fibroblast growth factors and plays an essential role in the regulation of embryonic development, cell proliferation, differentiation, and migration. Required for normal mesoderm patterning and correct axial organization during embryonic development, normal skeletogenesis, and normal development of the gonadotropin‐releasing hormone (GnRH) neuronal system. Phosphorylates PLCG1, FRS2, GAB1, and SHB. Ligand binding leads to the activation of several signaling cascades. Activation of PLCG1 leads to the production of the cellular signaling molecules diacylglycerol and inositol 1,4,5‐trisphosphate. Phosphorylation of FRS2 triggers recruitment of GRB2, GAB1, PIK3R1, and SOS1, and mediates activation of RAS, MAPK1/ERK2, MAPK3/ERK1, and the MAP kinase signaling pathway, as well as of the AKT1 signaling pathway. Promotes phosphorylation of SHC1, STAT1, and PTPN11/SHP2. In the nucleus, enhances RPS6KA1 and CREB1 activity and contributes to the regulation of transcription. FGFR1 signaling is down‐regulated by IL17RD/SEF, and by FGFR1 ubiquitination, internalization, and degradation | Tclin | 1.07E‐08 | 1.567877 | 0.389 | 0.169 | .000253 | CAF 1 | FGFR1 | Levatinib, nintedanib, pazopanib, sorafenib, ponatinib, sorafenib, sunitinib |
| CAF1 | PTGS2 | Prostaglandin G/H synthase 2 | Converts arachidonate to prostaglandin H2 (PGH2), a committed step in prostanoid synthesis. Constitutively expressed in some tissues in physiological conditions, such as the endothelium, kidney, and brain, and in pathological conditions, such as in cancer. PTGS2 is responsible for production of inflammatory prostaglandins. Upregulation of PTGS2 is also associated with increased cell adhesion, phenotypic changes, resistance to apoptosis and tumor angiogenesis. In cancer cells, PTGS2 is a key step in the production of prostaglandin E2 (PGE2), which plays important roles in modulating motility, proliferation, and resistance to apoptosis | Tclin | 1.46E‐10 | 1.673831 | 0.296 | 0.085 | 3.45E‐06 | CAF 1 | PTGS2 | Piketoprofen, aminosalicylic acid, felbinac, oxaprozin, loxoprofen, dexketoprofen, oxyphenbutazone, tolmetin, suprofen, piroxicam, naproxen, ketorolac, ketoprofen, ibuprofen, flurbiprofen, fenoprofen, bromfenac, nepafenac, acetylsalicylic acid, tiaprofenic acid, fenbufen, bismuth subsalicylate, acemetacin, floctafenine, lumiracoxib, bromfenac, fenoprofen, meclofenamic acid, mefanamic acid, meloxicam, paracetamol |
| CAF1 | F2R | Proteinase‐activated receptor 1 | High affinity receptor for activated thrombin coupled to G proteins that stimulate phosphoinositide hydrolysis. May play a role in platelets activation and in vascular development | Tclin | 7.40E‐13 | 1.385996 | 0.676 | 0.402 | 1.75E‐08 | CAF 1 | F2R | Vorapaxar |
| CAF1 | UGCG | Ceramide glucosyltransferase | Catalyzes the first glycosylation step in glycosphingolipid biosynthesis, the transfer of glucose to ceramide. May also serve as a “flippase” | Tclin | .003993 | 0.447536 | 0.12 | 0.276 | 1 | CAF 1 | UGCG | Eliglustat, miglustat |
| CAF1 | PDE7B | cAMP‐specific 3′,5′‐cyclic phosphodiesterase 7B | Hydrolyzes the second messenger cAMP, which is a key regulator of many important physiological processes. May be involved in the control of cAMP‐mediated neural activity and cAMP metabolism in the brain | Tclin | 3.48E‐05 | 0.709185 | 0.38 | 0.217 | .824078 | CAF 1 | PDE7B | Flavoxate, dipyridamole |
| CAF1 | INSR | Insulin receptor | Receptor tyrosine kinase which mediates the pleiotropic actions of insulin. Binding of insulin leads to phosphorylation of several intracellular substrates, including, insulin receptor substrates (IRS1, 2, 3, 4), SHC, GAB1, CBL, and other signaling intermediates. Each of these phosphorylated proteins serve as docking proteins for other signaling proteins that contain Src‐homology‐2 domains (SH2 domain) that specifically recognize different phosphotyrosine residues, including the p85 regulatory subunit of PI3K and SHP2. Phosphorylation of IRSs proteins lead to the activation of two main signaling pathways: the PI3K‐AKT/PKB pathway, which is responsible for most of the metabolic actions of insulin, and the Ras‐MAPK pathway, which regulates expression of some genes and cooperates with the PI3K pathway to control cell growth and differentiation. Binding of the SH2 domains of PI3K to phosphotyrosines on IRS1 leads to the activation of PI3K and the generation of phosphatidylinositol‐(3,4,5)‐triphosphate (PIP3), a lipid second messenger, which activates several PIP3‐dependent serine/threonine kinases, such as PDPK1 and subsequently AKT/PKB. The net effect of this pathway is to produce a translocation of the glucose transporter SLC2A4/GLUT4 from cytoplasmic vesicles to the cell membrane to facilitate glucose transport. Moreover, upon insulin stimulation, activated AKT/PKB is responsible for: anti‐apoptotic effect of insulin by inducing phosphorylation of BAD; regulates the expression of gluconeogenic and lipogenic enzymes by controlling the activity of the winged helix or forkhead (FOX) class of transcription factors. Another pathway regulated by PI3K‐AKT/PKB activation is mTORC1 signaling pathway which regulates cell growth and metabolism and integrates signals from insulin. AKT mediates insulin‐stimulated protein synthesis by phosphorylating TSC2 thereby activating mTORC1 pathway. The Ras/RAF/MAP2K/MAPK pathway is mainly involved in mediating cell growth, survival, and cellular differentiation of insulin. Phosphorylated IRS1 recruits GRB2/SOS complex, which triggers the activation of the Ras/RAF/MAP2K/MAPK pathway. In addition to binding insulin, the insulin receptor can bind insulin‐like growth factors (IGFI and IGFII). Isoform Short has a higher affinity for IGFII binding. When present in a hybrid receptor with IGF1R, binds IGF1. PubMed: 12138094 shows that hybrid receptors composed of IGF1R and INSR isoform Long are activated with a high affinity by IGF1, with low affinity by IGF2 and not significantly activated by insulin, and that hybrid receptors composed of IGF1R and INSR isoform Short are activated by IGF1, IGF2, and insulin. In contrast, PubMed: 16831875 shows that hybrid receptors composed of IGF1R and INSR isoform Long and hybrid receptors composed of IGF1R and INSR isoform Short have similar binding characteristics, both bind IGF1 and have a low affinity for insulin | Tclin | 1.10E‐06 | 0.344001 | 0.75 | 0.579 | .026129 | CAF 1 | INSR | Insulin (glulisine, lispro, glargine, degludec, human, aspart, detemir), nintedanib, certinib, brigatinib, ceritinib |
| CAF1 | KCNK2 | Potassium channel subfamily K member 2 | Isoform 4: Does not display channel activity but reduces the channel activity of isoform 1 and isoform 2 and reduces cell surface expression of isoform 2 | Tclin | 5.54E‐25 | 3.224821 | 0.259 | 0.012 | 1.31E‐20 | CAF 1 | KCNK2 | Desflurane, isoflurane, enflurane, halothane, sevoflurane, haloperidol, flupheazine, chlorpromazine, flupentixol, amlodipine |
| CAF1 | RAMP2 | Receptor activity‐modifying protein 2 | Transports the calcitonin gene‐related peptide type 1 receptor (CALCRL) to the plasma membrane. Acts as a receptor for adrenomedullin (AM) together with CALCRL | Tclin | 9.30E‐45 | 2.001194 | 0.602 | 0.059 | 2.20E‐40 | CAF 1 | RAMP2 | Pramlintide |
| CAF1 | VKORC1 | Vitamin K epoxide reductase complex subunit 1 | Involved in vitamin K metabolism. Catalytic subunit of the vitamin K epoxide reductase (VKOR) complex which reduces inactive vitamin K 2,3‐epoxide to active vitamin K. Vitamin K is required for the gamma‐carboxylation of various proteins, including clotting factors, and is required for normal blood coagulation, but also for normal bone development | Tclin | 1.26E‐05 | 0.853782 | 0.611 | 0.47 | .298323 | CAF 1 | VKORC1 | Warfarin, phenindione, acenocoumarol, phenprocoumon, dicoumarol |
| CAF1 | AKR1B1 | Aldose reductase | Catalyzes the NADPH‐dependent reduction of a wide variety of carbonyl‐containing compounds to their corresponding alcohols with a broad range of catalytic efficiencies | Tclin | 3.20E‐10 | 1.445022 | 0.472 | 0.22 | 7.57E‐06 | CAF 1 | AKR1B1 | Tolrestat, epalrestat, gossypol, sulindac, quercetin, tolrestat |
| CAF1 | IL6ST | Interleukin‐6 receptor subunit beta | Signal‐transducing molecule. The receptor systems for IL6, LIF, OSM, CNTF, IL11, CTF1, and BSF3 can utilize IL6ST for initiating signal transmission. Binding of IL6 to IL6R induces IL6ST homodimerization and formation of a high‐affinity receptor complex, which activates Janus kinases (PubMed: 2261637). That causes phosphorylation of IL6ST tyrosine residues which in turn activates STAT3 (PubMed: 19915009, PubMed: 23294003). Mediates signals which regulate immune response, hematopoiesis, pain control, and bone metabolism (by similarity). Has a role in embryonic development (by similarity). Does not bind IL6 (PubMed: 2261637). Essential for survival of motor and sensory neurons and for differentiation of astrocytes (by similarity). Required for expression of TRPA1 in nociceptive neurons (by similarity). Required for the maintenance of PTH1R expression in the osteoblast lineage and for the stimulation of PTH‐induced osteoblast differentiation (by similarity). Required for normal trabecular bone mass and cortical bone composition (by similarity) | Tclin | .000563 | 0.420354 | 0.611 | 0.498 | 1 | CAF 1 | IL6ST | Oprelvekin, tocilizumab, sarilumab |
| CAF1 | HSD11B1 | Corticosteroid 11‐beta‐dehydrogenase isozyme 1 | Catalyzes reversibly the conversion of cortisol to the inactive metabolite cortisone. Catalyzes reversibly the conversion of 7‐ketocholesterol to 7‐beta‐hydroxycholesterol. In intact cells, the reaction runs only in one direction, from 7‐ketocholesterol to 7‐beta‐hydroxycholesterol (by similarity) | Tclin | 9.65E‐06 | 1.313712 | 0.263 | 0.066 | .228663 | CAF 2 | HSD11B1 | Carbenoxolone, enoxalone, glycyrrhizic acid |
| CAF1 | ABL1 | Tyrosine‐protein kinase ABL1 | Non‐receptor tyrosine‐protein kinase that plays a role in many key processes linked to cell growth and survival such as cytoskeleton remodeling in response to extracellular stimuli, cell motility and adhesion, receptor endocytosis, autophagy, DNA damage response, and apoptosis. Coordinates actin remodeling through tyrosine phosphorylation of proteins controlling cytoskeleton dynamics like WASF3 (involved in branch formation); ANXA1 (involved in membrane anchoring); DBN1, DBNL, CTTN, RAPH1, and ENAH (involved in signaling); or MAPT and PXN (microtubule‐binding proteins). Phosphorylation of WASF3 is critical for the stimulation of lamellipodia formation and cell migration. Involved in the regulation of cell adhesion and motility through phosphorylation of key regulators of these processes such as BCAR1, CRK, CRKL, DOK1, EFS or NEDD9. Phosphorylates multiple receptor tyrosine kinases and more particularly promotes endocytosis of EGFR, facilitates the formation of neuromuscular synapses through MUSK, inhibits PDGFRB‐mediated chemotaxis, and modulates the endocytosis of activated B‐cell receptor complexes. Other substrates which are involved in endocytosis regulation are the caveolin (CAV1) and RIN1. Moreover, ABL1 regulates the CBL family of ubiquitin ligases that drive receptor downregulation and actin remodeling. Phosphorylation of CBL leads to increased EGFR stability. Involved in late‐stage autophagy by regulating positively the trafficking and function of lysosomal components. ABL1 targets to mitochondria in response to oxidative stress and thereby mediates mitochondrial dysfunction and cell death. In response to oxidative stress, phosphorylates serine/threonine kinase PRKD2 at “Tyr‐717” (PubMed: 28428613). ABL1 is also translocated in the nucleus where it has DNA‐binding activity and is involved in DNA‐damage response and apoptosis. Many substrates are known mediators of DNA repair: DDB1, DDB2, ERCC3, ERCC6, RAD9A, RAD51, RAD52 or WRN. Activates the proapoptotic pathway when the DNA damage is too severe to be repaired. Phosphorylates TP73, a primary regulator for this type of damage‐induced apoptosis. Phosphorylates the caspase CASP9 on “Tyr‐153” and regulates its processing in the apoptotic response to DNA damage. Phosphorylates PSMA7 that leads to an inhibition of proteasomal activity and cell cycle transition blocks. ABL1 acts also as a regulator of multiple pathological signaling cascades during infection. Several known tyrosine‐phosphorylated microbial proteins have been identified as ABL1 substrates. This is the case of A36R of Vaccinia virus, Tir (translocated intimin receptor) of pathogenic Escherichia coli and possibly Citrobacter, CagA (cytotoxin‐associated gene A) of Helicobacter pylori, or AnkA (ankyrin repeat‐containing protein A) of Anaplasma phagocytophilum. Pathogens can highjack ABL1 kinase signaling to reorganize the host actin cytoskeleton for multiple purposes, like facilitating intracellular movement and host cell exit. Finally, functions as its own regulator through autocatalytic activity as well as through phosphorylation of its inhibitor, ABI1. Regulates T‐cell differentiation in a TBX21‐dependent manner. Phosphorylates TBX21 on tyrosine residues leading to an enhancement of its transcriptional activator activity (by similarity) | Tclin | 1.49E‐08 | 1.167226 | 0.491 | 0.25 | .000352 | CAF 1 | ABL1 | Radotinib, ponatinib, dasatinib, imatinib, nilotinib, bosutinib, nintedanib, crizotinib, axitinib, vandetanib |
| CAF1 | EDNRB | Endothelin receptor type B | Nonspecific receptor for endothelin 1, 2, and 3. Mediates its action by association with G proteins that activate a phosphatidylinositol‐calcium second messenger system | Tclin | 6.66E‐09 | 0.819721 | 0.463 | 0.203 | .000158 | CAF 1 | EDNRB | Macitentan, bosentan |
| CAF1 | ADH1B | Alcohol dehydrogenase 1B | Alcohol dehydrogenase 1B | Tclin | 2.41E‐77 | 7.200263 | 0.63 | 0.004 | 5.70E‐73 | CAF 1 | ADH1B | Fomepizole |
| CAF1 | PDE4D | cAMP‐specific 3′,5′‐cyclic phosphodiesterase 4D | Hydrolyzes the second messenger cAMP, which is a key regulator of many important physiological processes | Tclin | 1.16E‐13 | 1.346971 | 0.556 | 0.252 | 2.75E‐09 | CAF 1 | PDE4D | Crisaborole, apremilast, amlexanox, flavoxate, roflumilast, choline theophyllinate, bufylline, roflumilast |
| CAF1 | EGFR | Epidermal growth factor receptor | (Microbial infection) Acts as a receptor for hepatitis C virus (HCV) in hepatocytes and facilitates its cell entry. Mediates HCV entry by promoting the formation of the CD81‐CLDN1 receptor complexes that are essential for HCV entry and by enhancing membrane fusion of cells expressing HCV envelope glycoproteins | Tclin | 2.43E‐07 | 1.448342 | 0.62 | 0.411 | .005766 | CAF 1 | EGFR | Icotinib, olmutinib, necitumumab, erlotinib, gefitinib, lapatinib, cetuximab, panitumumab, vandetanib, afatinib, osimertinib, neratinib, dasatinib, sorafenib, acalabrutinib, brigatinib, gefitinib, iburtinib, |
| CAF1 | ABCA1 | ATP‐binding cassette sub‐family A member 1 | cAMP‐dependent and sulfonylurea‐sensitive anion transporter. Key gatekeeper influencing intracellular cholesterol transport | Tclin | .009897 | 0.311781 | 0.389 | 0.301 | 1 | CAF 1 | ABCA1 | Probucol |
| CAF1 | SCN7A | Sodium channel protein type 7 subunit alpha | Mediates the voltage‐dependent sodium ion permeability of excitable membranes. Assuming opened or closed conformations in response to the voltage difference across the membrane, the protein forms a sodium‐selective channel through which Na(+) ions may pass in accordance with their electrochemical gradient | Tclin | 7.43E‐45 | 3.24375 | 0.491 | 0.03 | 1.76E‐40 | CAF 1 | SCN7A | Benzocaine, ethotoin, phenazopyridine, mephenytoin, rufinamide, eslicarbazepine acetate, procainamide, tocainide |
| CAF1 | ABCC9 | ATP‐binding cassette sub‐family C member 9 | Subunit of ATP‐sensitive potassium channels (KATP). Can form cardiac and smooth muscle‐type KATP channels with KCNJ11. KCNJ11 forms the channel pore while ABCC9 is required for activation and regulation | Tclin | 9.72E‐23 | 1.087824 | 1 | 0.982 | 2.30E‐18 | CAF 1 | ABCC9 | Inoxidil, pinacidil |
| CAF1 | ERBB2 | Receptor tyrosine‐protein kinase erbB‐2 | In the nucleus is involved in transcriptional regulation. Associates with the 5′‐TCAAATTC‐3′ sequence in the PTGS2/COX‐2 promoter and activates its transcription. Implicated in transcriptional activation of CDKN1A; the function involves STAT3 and SRC. Involved in the transcription of rRNA genes by RNA Pol I and enhances protein synthesis and cell growth | Tclin | .000585 | 0.279354 | 0.102 | 0.264 | 1 | CAF 1 | ERBB2 | Trastuzumab emtansine, lapatinib, pertuzumab, afatinib, trastuzumab, neratinib, acalabrutinib, afatinib, ibrutinib, neratinib |
| CAF1 | PDGFRA | Platelet‐derived growth factor receptor alpha | Tyrosine‐protein kinase that acts as a cell‐surface receptor for PDGFA, PDGFB, and PDGFC and plays an essential role in the regulation of embryonic development, cell proliferation, survival, and chemotaxis. Depending on the context, promotes or inhibits cell proliferation and cell migration. Plays an important role in the differentiation of bone marrow‐derived mesenchymal stem cells. Required for normal skeleton development and cephalic closure during embryonic development. Required for normal development of the mucosa lining the gastrointestinal tract, and for recruitment of mesenchymal cells and normal development of intestinal villi. Plays a role in cell migration and chemotaxis in wound healing. Plays a role in platelet activation, secretion of agonists from platelet granules, and in thrombin‐induced platelet aggregation. Binding of its cognate ligands—homodimeric PDGFA, homodimeric PDGFB, heterodimers formed by PDGFA and PDGFB or homodimeric PDGFC—leads to the activation of several signaling cascades; the response depends on the nature of the bound ligand and is modulated by the formation of heterodimers between PDGFRA and PDGFRB. Phosphorylates PIK3R1, PLCG1, and PTPN11. Activation of PLCG1 leads to the production of the cellular signaling molecules diacylglycerol and inositol 1,4,5‐trisphosphate, mobilization of cytosolic Ca(2+) and the activation of protein kinase C. Phosphorylates PIK3R1, the regulatory subunit of phosphatidylinositol 3‐kinase, and thereby mediates activation of the AKT1 signaling pathway. Mediates activation of HRAS and of the MAP kinases MAPK1/ERK2 and/or MAPK3/ERK1. Promotes activation of STAT family members STAT1, STAT3, and STAT5A and/or STAT5B. Receptor signaling is downregulated by protein phosphatases that dephosphorylate the receptor and its down‐stream effectors, and by rapid internalization of the activated receptor | Tclin | 8.70E‐83 | 4.179921 | 0.861 | 0.069 | 2.06E‐78 | CAF 1 | PDGFRA | Olaratumab, lenvatinib, nintedanib, becaplermin, sunitinib, pazopanib, imatinib, dasatinib, pazopanib, tivozanib, axitinib |
| CAF1 | C1R | Complement C1r subcomponent | C1r B chain is a serine protease that combines with C1q and C1s to form C1, the first component of the classical pathway of the complement system | Tclin | 1.09E‐31 | 1.165321 | 0.991 | 0.791 | 2.57E‐27 | CAF 1 | C1R | Nafamostat |
| CAF1 | GHR | Growth hormone receptor | Isoform 2 upregulates the production of GHBP and acts as a negative inhibitor of GH signaling | Tclin | 7.95E‐14 | 1.527346 | 0.481 | 0.189 | 1.88E‐09 | CAF 1 | GHR | Pegvisomant, somatropin, somatrem |
| CAF1 | PTGFR | Prostaglandin F2‐alpha receptor | Receptor for prostaglandin F2‐alpha (PGF2‐alpha). The activity of this receptor is mediated by G proteins which activate a phosphatidylinositol‐calcium second messenger system. Initiates luteolysis in the corpus luteum (by similarity). Isoforms 2 to 7 do not bind PGF2‐alpha but are proposed to modulate signaling by participating in variant receptor complexes; heterodimers between isoform 1 and isoform 5 are proposed to be a receptor for prostamides including the synthetic analog bimatoprost | Tclin | 2.60E‐36 | 2.909864 | 0.38 | 0.018 | 6.16E‐32 | CAF 1 | PTGFR | Latanoprostene bunod, travoprost, bimatoprost, dinoprost, carboprost |
| CAF1 | ADH1A | Alcohol dehydrogenase 1A | Alcohol dehydrogenase 1A | Tclin | 3.09E‐31 | 2.023774 | 0.296 | 0.008 | 7.31E‐27 | CAF 1 | ADH1A | Fomepizole |
3.4. Limitations of scRNA‐Seq in clinical medicine
While scRNA‐Seq will help in the transition towards personalized medicine, challenges with its translation to the clinic remain including lack of large cohorts of scRNA‐Seq human patient samples, cost, user‐friendliness, and tissue preservation. The use of scRNA‐Seq on individual patient tumors to inform drug selection is currently possible as we described above and as reported in a recent case study using scRNA‐Seq of skin and blood samples from a patient with refractory drug‐induced hypersensitivity syndrome to identify patient‐specific drug targets and successfully repurpose tofacitinib to treat this difficult case. 93 While both our findings and the findings from case reports would need to be extended by performing studies on larger cohorts to make conclusions about prognostic outcomes from this type of drug selection, they demonstrate that individualized drug selection based on sequencing from a single patient sample is feasible and has been successful. More of these types of studies are needed to conclude that personalized drug selection and drug repurposing using scRNA‐Seq results in better patient outcomes. 97
The cost of scRNA‐Seq varies based on the chosen methodology and depends on the cost of equipment, reagents, and sequencing. For any platform, a higher number of cells results in higher costs. The costs of isolation and sequencing per cell have dropped significantly, but the throughput of sequencing machines has also increased, so the cost per run with more cells remains high. Most isolation platforms are also only available at basic science laboratories and would require a large upfront investment to purchase for hospital use.
In addition to cost, analysis of scRNA‐Seq data requires users to have basic bioinformatic knowledge and coding skills; however, user‐friendly bioinformatic pipelines such as the gene expression analysis resource (gEAR) (https://umgear.org/), and scRNA‐Seq workbench created to facilitate data sharing and visualization, are currently available. Further standardization of these pipelines will be necessary for clinical use. Tissue preservation is also an issue due to fragility and the small amount of time that cells remain viable. Procurement of tumor biopsies and surgical specimens may not always be predictable or fast and, currently, the use of frozen tissue samples or methanol tissue fixation 99 on scRNA‐Seq platforms is in its infancy; however, a few options to aid in tissue preservation are available. Temporary tissue stabilization buffers, PrepProtect (Miltenyi Biotec) or RNAlater (Thermo Fisher Scientific), can preserve cells for sequencing for 48 hours. While these methods may not be compatible with scRNA‐Seq if used in combination with sNuc‐Seq experiments, which have the advantage of lysing cells and only sequencing mRNA from the nucleus, 100 RNALater tissue preservation of fresh or frozen patient samples can result in useful data. 101 Cell lysis used in sNuc‐Seq allows for potentially more efficient cell type delineation including even the most interdigitated cell types and minimizes the skewing effect of degraded mRNA or cell‐stress response genes on the data.11, 60, 61, 100, 102 These advantages potentially make sNuc‐Seq an excellent option for precious patient samples including human vestibular and cochlear tissues.
4. CONCLUSIONS
Despite these limitations and the lack of widespread clinical availability, the rapid advances in scRNA‐Seq throughput and bioinformatic pipelines are making the ability for clinicians to perform scRNA‐Seq on patient tissue a reality in the next few years. We have described how this technology applied to three different otolaryngologic diseases, CR and head and neck cancers HNSCC and melanoma, can make otolaryngologists more aware of the cellular heterogeneity within their patients' diseased tissue. Further analysis of tissue heterogeneity across human patient samples can help otolaryngologist understand basic human biology and disease pathogenesis, make diagnostic and prognostic decisions, and select personalized drug therapy combinations.
We demonstrate the feasibility and importance of conducting scRNA‐Seq on human patient samples to generate new insight into healthy and diseased tissue biology and the possibility for this technique to move patient care further into the era of precision medicine where treatment can be tailored to a patient's unique transcriptome. Currently, a human single‐cell atlas project is underway with the goal of creating single‐cell profiles for every normal and pathologic cell in the human body. 103 It is not difficult to imagine a time where physicians could conduct scRNA‐Seq on each pathologic patient sample and compare it to the normal transcriptome within this atlas to determine which cells are diseased and how other patients with similar diseased‐cell transcriptomes responded to various treatments.
We hope to inform otolaryngologists of the potential of this technique to further our practice in the future and inspire ideas for how they may use it now to gain insight into both healthy and diseased tissues that may have dramatic impacts on clinical care. Because there have been so few scRNA‐Seq studies of human samples in our field, we argue for immediate use of scRNA‐Seq on more otolaryngologic patient samples to not only aid in more sensitive diagnosis and drug selection, but also to contribute single‐cell profiles to a larger database like the single‐cell atlas which will help identify new disease biomarkers associated with prognosis, subtypes, or drug resistance. This technique can be applied immediately to human auditory hair cells and spiral ganglion neurons to characterize human deafness genes and the cell types that express them. scRNA‐Seq can also be readily applied to characterize benign and malignant tumors of the head and neck, such as HNSCC and vestibular schwannomas, to identify tumor heterogeneity among cohorts of patients as these pathologies stand to benefit the most from identification of new druggable targets, selection of more effective drugs, or selection of patients for clinical trials to optimize the balance between medical and surgical treatments. As this technique becomes more readily available, other otolaryngologic problems may also be answered. These uses will not only increase our understanding of the cellular diversity involved in these diseases, but also have the potential to change the way we diagnose, monitor, and treat patients in the field of otolaryngology.
CONFLICT OF INTEREST
The authors declare no potential conflict of interest.
Supporting information
Data S1. Example R script for processing data from Puram et al's data set.
Data S2. Supplementary descriptive methods detailing bioinformatic analyses utilized to analyze data from Puram et al's data set.
ACKNOWLEDGMENTS
The authors thank Clint T. Allen, MD, Isabelle Roux, PhD, Doris Wu, PhD, and Ronna Hertzona, MD, PhD, for advice and feedback. The authors would also like to thank Puram et al and colleagues for making their oral head and neck squamous cell carcinoma single‐cell data publicly available for download, through Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/) with accession number GSE103322, so that we could use it to perform our own analysis on it to demonstrate uses for single‐cell RNA sequencing. This research was made possible through the NIH Medical Research Scholars Program, a public‐private partnership supported jointly by the NIH and contributions to the Foundation for the NIH from the Doris Duke Charitable Foundation (DDCF Grant #2014194), the American Association for Dental Research, the Colgate‐Palmolive Company, Genentech, Elsevier, and other private donors. NIDCD/NIH Intramural Research Program funds to M. H. (DC000088).
Pyle MP, Hoa M. Applications of single‐cell sequencing for the field of otolaryngology: A contemporary review. Laryngoscope Investigative Otolaryngology. 2020;5:404–431. 10.1002/lio2.388
Funding information Doris Duke Charitable Foundation, Grant/Award Number: 2014194; National Institute on Deafness and Other Communication Disorders, Grant/Award Number: DC000088; Elsevier; Genentech; Colgate‐Palmolive Company; American Association for Dental Research
REFERENCES
- 1. Potter SS. Single‐cell RNA sequencing for the study of development, physiology and disease. Nat Rev Nephrol. 2018;14(8):479‐492. 10.1038/s41581-018-0021-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. AlJanahi AA, Danielsen M, Dunbar CE. An Introduction to the Analysis of Single‐Cell RNA‐Sequencing Data. Mol Ther Methods Clin Dev. 2018;10:189‐196. 10.1016/j.omtm.2018.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Keren‐Shaul H, Spinrad A, Weiner A, et al. A unique microglia type associated with restricting development of Alzheimer's disease. Cell. 2017;169(7):1276‐1290.e17. 10.1016/j.cell.2017.05.018. [DOI] [PubMed] [Google Scholar]
- 4. Montoro DT, Haber AL, Biton M, et al. A revised airway epithelial hierarchy includes CFTR‐expressing ionocytes. Nature. 2018;560(7718):319‐324. 10.1038/s41586-018-0393-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kinchen J, Chen HH, Parikh K, et al. Structural remodeling of the human colonic mesenchyme in inflammatory bowel disease. Cell. 2018;175(2):372‐386.e17. 10.1016/j.cell.2018.08.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chu L‐F, Leng N, Zhang J, et al. Single‐cell RNA‐seq reveals novel regulators of human embryonic stem cell differentiation to definitive endoderm. Genome Biol. 2016;17(1):173 10.1186/s13059-016-1033-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fan X, Wang D, Burgmaier JE, et al. Single cell and open chromatin analysis reveals molecular origin of epidermal cells of the skin. Dev Cell. 2018;47(1):21‐36.e6. 10.1016/j.devcel.2018.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Burns JC, Kelly MC, Hoa M, Morell RJ, Kelley MW. Single‐cell RNA‐Seq resolves cellular complexity in sensory organs from the neonatal inner ear. Nat Commun. 2015;6(1):8557 10.1038/ncomms9557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yamashita T, Zheng F, Finkelstein D, et al. High‐resolution transcriptional dissection of in vivo Atoh1‐mediated hair cell conversion in mature cochleae identifies Isl1 as a co‐reprogramming factor. PLoS Genet. 2018;14(7) e1007552. 10.1371/journal.pgen.1007552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sherrill HE, Jean P, Driver EC, et al. Pou4f1 defines a subgroup of type i spiral ganglion neurons and is necessary for normal inner hair cell presynaptic Ca2+ signaling. J Neurosci. 2019;39(27):5284‐5298. 10.1523/JNEUROSCI.2728-18.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Korrapati S, Taukulis I, Olszewski R, et al. Single cell and single nucleus RNA‐Seq reveal cellular heterogeneity and homeostatic regulatory networks in adult mouse stria vascularis. Front Mol Neurosci. 2019;12:316. 10.3389/fnmol.2019.00316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Honda K, Kim SH, Kelly MC, et al. Molecular architecture underlying fluid absorption by the developing inner ear. Elife. 2017;6:1‐29. 10.7554/eLife.26851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chessum L, Matern MS, Kelly MC, et al. Helios is a key transcriptional regulator of outer hair cell maturation. Nature. 2018;563(7733):696‐724. 10.1038/s41586-018-0728-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. McInturff S, Burns JC, Kelley MW. Characterization of spatial and temporal development of Type I and Type II hair cells in the mouse utricle using new cell‐type‐specific markers. Biol Open. 2018;7(11):bio038083. 10.1242/bio.038083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hoa M, Olszewski R, Li X, et al. Characterizing adult cochlear supporting cell transcriptional diversity using single‐cell RNA‐seq: validation in the adult mouse and translational implications for the adult human cochlea. Front Mol Neurosci. 2020;13:13. 10.3389/fnmol.2020.00013.eCollection 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ranum PT, Goodwin AT, Yoshimura H, et al. Insights into the biology of hearing and deafness revealed by single‐cell RNA sequencing. Cell Rep. 2019;26(11):3160‐3171.e3. 10.1016/j.celrep.2019.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tang P‐C, Alex AL, Nie J, et al. Defective Tmprss3‐associated hair cell degeneration in inner ear organoids. Stem Cell Rep. 2019;13:1‐16. 10.1016/j.stemcr.2019.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sun S, Babola T, Pregernig G, et al. Hair cell mechanotransduction regulates spontaneous activity and spiral ganglion subtype specification in the auditory system. Cell. 2018;174(5):1247‐1263.e15. 10.1016/j.cell.2018.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shrestha BR, Chia C, Wu L, Kujawa SG, Liberman MC, Goodrich LV. Sensory neuron diversity in the inner ear is shaped by activity. Cell. 2018;174(5):1229‐1246.e17. 10.1016/J.CELL.2018.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Petitpré C, Wu H, Sharma A, et al. Neuronal heterogeneity and stereotyped connectivity in the auditory afferent system. Nat Commun. 2018;9(1):3691 10.1038/s41467-018-06033-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Carlson ML, Smadbeck JB, Link MJ, Klee EW, Vasmatzis G, Schimmenti LA. Next generation sequencing of sporadic vestibular schwannoma: necessity of biallelic NF2 inactivation and implications of accessory non‐NF2 variants. Otol Neurotol. 2018;39(9):e860‐e871. 10.1097/MAO.0000000000001932. [DOI] [PubMed] [Google Scholar]
- 22. Schrauwen I, Hasin‐Brumshtein Y, Corneveaux JJ, et al. A comprehensive catalogue of the coding and non‐coding transcripts of the human inner ear. Hear Res. 2014;333:266‐274. 10.1016/j.heares.2015.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Puram SV, Tirosh I, Parikh AS, et al. Single‐cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell. 2017;171(7):1611‐1624.e24. 10.1016/J.CELL.2017.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Neal JT, Li X, Zhu J, et al. Organoid modeling of the tumor immune microenvironment. Cell. 2018;175(7):1972‐1988.e16. 10.1016/j.cell.2018.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cillo AR, Kürten CHL, Tabib T, et al. Immune landscape of viral‐ and carcinogen‐driven head and neck cancer. Immunity. 2020;52(1):183‐199.e9. 10.1016/j.immuni.2019.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Davis‐Marcisak EF, Sherman TD, Orugunta P, et al. Differential variation analysis enables detection of tumor heterogeneity using single‐cell RNA‐sequencing data. Cancer Res. 2019;79(19):5102‐5112. 10.1158/0008-5472.CAN-18-3882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ordovas‐Montanes J, Dwyer DF, Nyquist SK, et al. Allergic inflammatory memory in human respiratory epithelial progenitor cells. Nature. 2018;560(7720):649‐654. 10.1038/s41586-018-0449-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tirosh I, Izar B, Prakadan SM, et al. Dissecting the multicellular ecosystem of metastatic melanoma by single‐cell RNA‐seq. Science. 2016;352(6282):189‐196. 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Stephenson W, Donlin LT, Butler A, et al. Single‐cell RNA‐seq of rheumatoid arthritis synovial tissue using low‐cost microfluidic instrumentation. Nat Commun. 2018;9(1):791 10.1038/s41467-017-02659-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yu J, Wu WKK, Li X, et al. Novel recurrently mutated genes and a prognostic mutation signature in colorectal cancer. Gut. 2015;64(4):636‐645. 10.1136/gutjnl-2013-306620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shalek AK, Benson M. Single‐cell analyses to tailor treatments. Sci Transl Med. 2017;9(408) eaan4730. 10.1126/scitranslmed.aan4730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sandberg R. Entering the era of single‐cell transcriptomics in biology and medicine. Nat Methods. 2014;11(1):22‐24. 10.1038/nmeth.2764. [DOI] [PubMed] [Google Scholar]
- 33. Eberwine J, Sul J‐Y, Bartfai T, Kim J. The promise of single‐cell sequencing. Nat Methods. 2014;11(1):25‐27. 10.1038/nmeth.2769. [DOI] [PubMed] [Google Scholar]
- 34. Qi Z, Barrett T, Parikh AS, Tirosh I, Puram SV. Single‐cell sequencing and its applications in head and neck cancer. Oral Oncol. 2019;99(October):104441 10.1016/j.oraloncology.2019.104441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dal Molin A, Di Camillo B. How to design a single‐cell RNA‐sequencing experiment: pitfalls, challenges and perspectives. Brief Bioinform. 2019;20(4):1384‐1394. 10.1093/bib/bby007. [DOI] [PubMed] [Google Scholar]
- 36. Chen G, Ning B, Shi T. Single‐cell RNA‐seq technologies and related computational data analysis. Front Genet. 2019;10:316. 10.3389/fgene.2019.00317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Stewart BJ, Ferdinand JR, Clatworthy MR. Using single‐cell technologies to map the human immune system ‐ implications for nephrology. Nat Rev Nephrol. 2019;16(2):112–128. 10.1038/s41581-019-0227-3. [DOI] [PubMed] [Google Scholar]
- 38. Xu G, Liu Y, Li H, Liu L, Zhang S, Zhang Z. Dissecting the human immune system with single cell RNA sequencing technology. J Leukoc Biol. 2019;107(4):613–623. 10.1002/JLB.5MR1019-179R. [DOI] [PubMed] [Google Scholar]
- 39. Lafzi A, Moutinho C, Picelli S, Heyn H. Tutorial: guidelines for the experimental design of single‐cell RNA sequencing studies. Nat Protoc. 2018;13(12):2742‐2757. 10.1038/s41596-018-0073-y. [DOI] [PubMed] [Google Scholar]
- 40. Zilionis R, Nainys J, Veres A, et al. Single‐cell barcoding and sequencing using droplet microfluidics. Nat Protoc. 2017;12(1):44‐73. 10.1038/nprot.2016.154. [DOI] [PubMed] [Google Scholar]
- 41. Head SR, Kiyomi Komori H, LaMere SA, et al. Library construction for next‐generation sequencing: overviews and challenges. Biotechniques. 2014;56(2):61‐77. 10.2144/000114133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Trombetta JJ, Gennert D, Lu D, Satija R, Shalek AK, Regev A. Preparation of single‐cell RNA‐Seq libraries for next generation sequencing. Curr Protoc Mol Biol. 2014;107(1):4.22.1‐4.22.17. 10.1002/0471142727.mb0422s107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Saliba AE, Westermann AJ, Gorski SA, Vogel J. Single‐cell RNA‐seq: advances and future challenges. Nucleic Acids Res. 2014;42(14):8845‐8860. 10.1093/nar/gku555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nguyen QH, Pervolarakis N, Nee K, Kessenbrock K. Experimental considerations for single‐cell RNA sequencing approaches. Front Cell Dev Biol. 2018;6:1‐7. 10.3389/fcell.2018.00108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hwang B, Lee JH, Bang D. Single‐cell RNA sequencing technologies and bioinformatics pipelines. Exp Mol Med. 2018;50(8):96. 10.1038/s12276-018-0071-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shafer MER. Cross‐Species Analysis of Single‐Cell Transcriptomic Data. Front Cell Dev Biol. 2019;7(September):1‐9. 10.3389/fcell.2019.00175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Haque A, Engel J, Teichmann SA, Lönnberg T. A practical guide to single‐cell RNA‐sequencing for biomedical research and clinical applications. Genome Med. 2017;9(1):75 10.1186/s13073-017-0467-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Habib N, Avraham‐Davidi I, Basu A, et al. Massively parallel single‐nucleus RNA‐seq with DroNc‐seq. Nat Methods. 2017;14(10):955–958. 10.1038/nmeth.4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rosenberg AB, Roco CM, Muscat RA, et al. Single‐cell profiling of the developing mouse brain and spinal cord with split‐pool barcoding. Science. 2018;360(6385):176‐182. 10.1126/science.aam8999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cao J, Packer JS, Ramani V, et al. Comprehensive single‐cell transcriptional profiling of a multicellular organism. Science. 2017;357(6352):661‐667. 10.1126/science.aam8940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pollen AA, Nowakowski TJ, Shuga J, et al. Low‐coverage single‐cell mRNA sequencing reveals cellular heterogeneity and activated signaling pathways in developing cerebral cortex. Nat Biotechnol. 2014;32(10):1053‐1058. 10.1038/nbt.2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zheng GXY, Terry JM, Belgrader P, et al. Massively parallel digital transcriptional profiling of single cells. Nat Commun. 2017;8:14049 10.1038/ncomms14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Islam S, Kjällquist U, Moliner A, et al. Highly multiplexed and strand‐specific single‐cell RNA 5′ end sequencing. Nat Protoc. 2012;7(5):813‐828. 10.1038/nprot.2012.022. [DOI] [PubMed] [Google Scholar]
- 54. Hashimshony T, Senderovich N, Avital G, et al. CEL‐Seq2: sensitive highly‐multiplexed single‐cell RNA‐Seq. Genome Biol. 2016;17(1):77 10.1186/s13059-016-0938-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Picelli S, Faridani OR, Björklund ÅK, Winberg G, Sagasser S, Sandberg R. Full‐length RNA‐seq from single cells using Smart‐seq2. Nat Protoc. 2014;9(1):171‐181. 10.1038/nprot.2014.006. [DOI] [PubMed] [Google Scholar]
- 56. Klein AM, Mazutis L, Akartuna I, et al. Droplet barcoding for single‐cell transcriptomics applied to embryonic stem cells. Cell. 2015;161(5):1187‐1201. 10.1016/J.CELL.2015.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Macosko EZ, Basu A, Satija R, et al. Highly parallel genome‐wide expression profiling of individual cells using nanoliter droplets. Cell. 2015;161(5):1202‐1214. 10.1016/j.cell.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single‐cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol. 2018;36(5):411‐420. 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Stuart T, Butler A, Hoffman P, et al. Comprehensive integration of single cell data. bioRxiv. 2019;177(7):1888‐1902.e21. 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lake BB, Chen S, Hoshi M, et al. A single‐nucleus RNA‐sequencing pipeline to decipher the molecular anatomy and pathophysiology of human kidneys. Nat Commun. 2019;10(1):2832. 10.1038/s41467-019-10861-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zhou Y, Song WM, Andhey PS, et al. Human and mouse single‐nucleus transcriptomics reveal TREM2‐dependent and TREM2‐independent cellular responses in Alzheimer's disease. Nat Med. 2020;26(1):131–142. 10.1038/s41591-019-0695-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Palmer C, Mulligan JK, Smith SE, Atkinson C. The role of regulatory T cells in the regulation of upper airway inflammation. Am J Rhinol Allergy. 2017;31(6):345‐351. 10.2500/ajra.2017.31.4472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Davis RJ, Van Waes C, Allen CT. Overcoming barriers to effective immunotherapy: MDSCs, TAMs, and Tregs as mediators of the immunosuppressive microenvironment in head and neck cancer. Oral Oncol. 2016;58:59‐70. 10.1016/J.ORALONCOLOGY.2016.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Schulz A, Büttner R, Hagel C, et al. The importance of nerve microenvironment for schwannoma development. Acta Neuropathol. 2016;132(2):289‐307. 10.1007/s00401-016-1583-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Prasetyanti PR, Medema JP. Intra‐tumor heterogeneity from a cancer stem cell perspective. Mol Cancer. 2017;16(1):41 10.1186/s12943-017-0600-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. McGranahan N, Swanton C. Clonal heterogeneity and tumor evolution: past, present, and the future. Cell. 2017;168(4):613‐628. 10.1016/j.cell.2017.01.018. [DOI] [PubMed] [Google Scholar]
- 67. Patel AP, Tirosh I, Trombetta JJ, et al. Single‐cell RNA‐seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344(6190):1396‐1401. 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Duan M, Hao J, Cui S, et al. Diverse modes of clonal evolution in HBV‐related hepatocellular carcinoma revealed by single‐cell genome sequencing. Cell Res. 2018;28(3):359‐373. 10.1038/cr.2018.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wang Y, Waters J, Leung ML, et al. Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature. 2014;512(7513):155‐160. 10.1038/nature13600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Tirosh I, Venteicher AS, Hebert C, et al. Single‐cell RNA‐seq supports a developmental hierarchy in human oligodendroglioma. Nature. 2016;539(7628):309‐313. 10.1038/nature20123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Flem‐Karlsen K, Nyakas M, Farstad IN, et al. Soluble AXL as a marker of disease progression and survival in melanoma. PLoS One. 2020;15(1):e0227187 10.1371/journal.pone.0227187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Flem‐Karlsen K, McFadden E, Omar N, et al. Targeting AXL and the DNA damage response pathway as a novel therapeutic strategy in melanoma. Mol Cancer Ther. 2019;19(3):895‐905. 10.1158/1535-7163.MCT-19-0290. [DOI] [PubMed] [Google Scholar]
- 73. Puram SV, Parikh AS, Tirosh I. Single cell RNA‐seq highlights a role for a partial EMT in head and neck cancer. Mol Cell Oncol. 2018;5(3):e1448244. 10.1080/23723556.2018.1448244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Li H, Courtois ET, Sengupta D, et al. Reference component analysis of single‐cell transcriptomes elucidates cellular heterogeneity in human colorectal tumors. Nat Genet. 2017;49(5):708‐718. 10.1038/ng.3818. [DOI] [PubMed] [Google Scholar]
- 75. Chung W, Eum HH, Lee H‐O, et al. Single‐cell RNA‐seq enables comprehensive tumour and immune cell profiling in primary breast cancer. Nat Commun. 2017;8:15081 10.1038/ncomms15081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Venteicher AS, Tirosh I, Hebert C, et al. Decoupling genetics, lineages, and microenvironment in IDH‐mutant gliomas by single‐cell RNA‐seq. Science. 2017;355(6332):eaai8478. 10.1126/science.aai8478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Karaayvaz M, Cristea S, Gillespie SM, et al. Unravelling subclonal heterogeneity and aggressive disease states in TNBC through single‐cell RNA‐seq. Nat Commun. 2018;9(1):3588 10.1038/s41467-018-06052-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Zheng C, Zheng L, Yoo J‐K, et al. Landscape of infiltrating T cells in liver cancer revealed by single‐cell sequencing. Cell. 2017;169(7):1342‐1356.e16. 10.1016/j.cell.2017.05.035. [DOI] [PubMed] [Google Scholar]
- 79. Savas P, Virassamy B, Ye C, et al. Single‐cell profiling of breast cancer T cells reveals a tissue‐resident memory subset associated with improved prognosis. Nat Med. 2018;24(7):986‐993. 10.1038/s41591-018-0078-7. [DOI] [PubMed] [Google Scholar]
- 80. Peitzsch C, Nathansen J, Schniewind SI, et al. Cancer stem cells in head and neck squamous cell carcinoma: identification, characterization and clinical implications. Cancers (Basel). 2019;11(5):616 10.3390/cancers11050616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Walter V, Yin X, Wilkerson MD, et al. Molecular subtypes in head and neck cancer exhibit distinct patterns of chromosomal gain and loss of canonical cancer genes. PLoS One. 2013;8(2) e56823. 10.1371/journal.pone.0056823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Network TCGA. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015;517(7536):576‐582. 10.1038/nature14129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Lee J‐K, Wang J, Sa JK, et al. Spatiotemporal genomic architecture informs precision oncology in glioblastoma. Nat Genet. 2017;49(4):594‐599. 10.1038/ng.3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Miyamoto DT, Zheng Y, Wittner BS, et al. RNA‐Seq of single prostate CTCs implicates noncanonical Wnt signaling in antiandrogen resistance. Science. 2015;349(6254):1351‐1356. 10.1126/science.aab0917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Shashanka R, Smitha BR. Head and neck melanoma. ISRN Surg. 2012;2012:948302 10.5402/2012/948302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Ausoni S, Boscolo‐Rizzo P, Singh B, et al. Targeting cellular and molecular drivers of head and neck squamous cell carcinoma: current options and emerging perspectives. Cancer Metastasis Rev. 2016;35(3):413‐426. 10.1007/s10555-016-9625-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Shen S, Bai J, Wei Y, et al. A seven‐gene prognostic signature for rapid determination of head and neck squamous cell carcinoma survival. Oncol Rep. 2017;38(6):3403‐3411. 10.3892/or.2017.6057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Chakraborty P, Karmakar T, Arora N, Mukherjee G. Immune and genomic signatures in oral (head and neck) cancer. Heliyon. 2018;4(10):e00880 10.1016/J.HELIYON.2018.E00880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. You G‐R, Cheng A‐J, Lee L‐Y, et al. Prognostic signature associated with radioresistance in head and neck cancer via transcriptomic and bioinformatic analyses. BMC Cancer. 2019;19(1):64 10.1186/s12885-018-5243-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Dago AE, Stepansky A, Carlsson A, et al. Rapid phenotypic and genomic change in response to therapeutic pressure in prostate cancer inferred by high content analysis of single circulating tumor cells. PLoS One. 2014;9(8):e101777 10.1371/journal.pone.0101777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Kim K‐T, Lee HW, Lee H‐O, et al. Application of single‐cell RNA sequencing in optimizing a combinatorial therapeutic strategy in metastatic renal cell carcinoma. Genome Biol. 2016;17(1):80 10.1186/s13059-016-0945-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Levitin HM, Yuan J, Sims PA. Single‐cell transcriptomic analysis of tumor heterogeneity. Trends Cancer. 2018;4(4):264‐268. 10.1016/j.trecan.2018.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Kim D, Kobayashi T, Voisin B, et al. Targeted therapy guided by single‐cell transcriptomic analysis in drug‐induced hypersensitivity syndrome: a case report. Nat Med. 2020;26(2):236‐243. 10.1038/s41591-019-0733-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Ocasio J, Babcock B, Malawsky D, et al. scRNA‐seq in medulloblastoma shows cellular heterogeneity and lineage expansion support resistance to SHH inhibitor therapy. Nat Commun. 2019;10(1):5829. 10.1038/s41467-019-13657-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Allen C, Clavijo P, Van Waes C, et al. Anti‐tumor immunity in head and neck cancer: understanding the evidence, how tumors escape and immunotherapeutic approaches. Cancers (Basel). 2015;7(4):2397‐2414. 10.3390/cancers7040900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Specenier P, Vermorken JB. Optimizing treatments for recurrent or metastatic head and neck squamous cell carcinoma. Expert Rev Anticancer Ther. 2018;18(9):901‐915. 10.1080/14737140.2018.1493925. [DOI] [PubMed] [Google Scholar]
- 97. Pushpakom S, Iorio F, Eyers PA, et al. Drug repurposing: progress, challenges and recommendations. Nat Rev Drug Discov. 2018;18(1):41‐58. 10.1038/nrd.2018.168. [DOI] [PubMed] [Google Scholar]
- 98. Nguyen D‐T, Mathias S, Bologa C, et al. Pharos: collating protein information to shed light on the druggable genome. Nucleic Acids Res. 2017;45(D1):D995‐D1002. 10.1093/nar/gkw1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Alles J, Karaiskos N, Praktiknjo SD, et al. Cell fixation and preservation for droplet‐based single‐cell transcriptomics. BMC Biol. 2017;15(1):44 10.1186/s12915-017-0383-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Wu H, Kirita Y, Donnelly EL, Humphreys BD. Advantages of single‐nucleus over single‐cell RNA sequencing of adult kidney: rare cell types and novel cell states revealed in fibrosis. J Am Soc Nephrol. 2019;30(1):23‐32. 10.1681/ASN.2018090912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Nguyen MQ, Le Pichon CE, Ryba N. Stereotyped transcriptomic transformation of somatosensory neurons in response to injury. Elife. 2019;8:e49679. 10.7554/eLife.49679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. O'Sullivan ED, Mylonas KJ, Hughes J, Ferenbach DA. Complementary roles for single‐nucleus and single‐cell RNA sequencing in kidney disease research. J Am Soc Nephrol. 2019;30(4):712‐713. 10.1681/ASN.2019020112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Regev A, Teichmann SA, Lander ES, et al. The human cell atlas. Elife. 2017;6:e27041. 10.7554/eLife.27041. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Example R script for processing data from Puram et al's data set.
Data S2. Supplementary descriptive methods detailing bioinformatic analyses utilized to analyze data from Puram et al's data set.