

A combination of uridine modification and process optimization minimizes innate immune response to therapeutic mRNA.
Abstract
Messenger RNA (mRNA) represents an attractive therapeutic modality for potentially a wide range of clinical indications but requires uridine chemistry modification and/or tuning of the production process to prevent activation of cellular innate immune sensors and a concomitant reduction in protein expression. To decipher the relative contributions of these factors on immune activation, here, we compared, in multiple cell and in vivo models, mRNA that encodes human erythropoietin incorporating either canonical uridine or N1-methyl-pseudouridine (1mΨ), synthesized by either a standard process shown to have double-stranded RNA (dsRNA) impurities or a modified process that yields a highly purified mRNA preparation. Our data demonstrate that the lowest stimulation of immune endpoints was with 1mΨ made by the modified process, while mRNA containing canonical uridine was immunostimulatory regardless of process. These findings confirm that uridine modification and the reduction of dsRNA impurities are both necessary and sufficient at controlling the immune-activating profile of therapeutic mRNA.
INTRODUCTION
mRNA is a relatively new therapeutic modality with the potential for a wide range of clinical applications, including vaccines against infectious agents and cancers, cancer therapy, treatment of genetic disorders, regenerative therapeutics, and immunotherapies (1–5). Advantages of mRNA-based therapeutics compared to protein-based biologic agents include the ability to use intracellular and membrane-bound proteins as therapeutic targets, the absence of protein synthesis and purification, and the potential for rapid advancement from development to clinical manufacturing.
mRNA therapeutics comprise two key elements: the mRNA encoding the protein of interest and the delivery system. Designing an mRNA drug product requires reaching an efficacious level of protein expression at a specified dose level (6) and the development of a robust delivery system that protects the mRNA either in circulation or in the interstitium long enough to reach the desired tissue. Lipid nanoparticles (LNP) represent the most suitable system to date for mRNA delivery (7). LNP-formulated mRNA enters the cell through receptor-mediated uptake into endosomes and is subsequently released into the cytosol where it is translated into the protein of interest. Substantial advances in the development of novel lipid systems for mRNA therapeutics were recently described (8, 9).
Another important feature of an mRNA therapeutic is that it must avoid detection by the innate immune system, which can mistake therapeutic mRNA for non-self nucleic acids and thereby generate an immune response. This is of particular importance for repeat dosing of mRNA therapeutic drugs, where immune memory may limit effectiveness of the drug product (10). Identification of non-self nucleic acids is accomplished by two families of pattern recognition receptors (PRRs) that are part of the innate immune system (Fig. 1): the endosomal membrane-bound Toll-like receptors (TLR) and cytoplasmic sensors of viral nucleic acids (11). Triggering of TLR activates signal transduction pathways that selectively lead to production of proinflammatory cytokines, which collectively act to eliminate intracellular pathogens and infected cells (12, 13). In particular, TLR7 and TLR8 in humans and TLR7 in mice, which recognize single-stranded RNA (ssRNA) and activate the adaptor protein myeloid differentiation primary response 88 (MyD88) (14), and TLR3, which detects double-stranded RNA (dsRNA) and signals through Toll/interleukin-1 (IL-1) receptor domain–containing adaptor inducing interferon-β (IFNβ) (TRIF) (15), all lead to expression of a large panel of proinflammatory cytokines, including IFNα (11). The cytosolic RNA sensors retinoic acid–inducible gene I (RIG-I) and melanoma differentiation–associated protein 5 (MDA5) bind to dsRNA and induce expression of proinflammatory cytokines such as IFNβ through activation of the adaptor protein mitochondrial antiviral signaling protein (MAVS) (16, 17).
Fig. 1. Cellular innate immune pathways activated by endocytosis of foreign RNA.
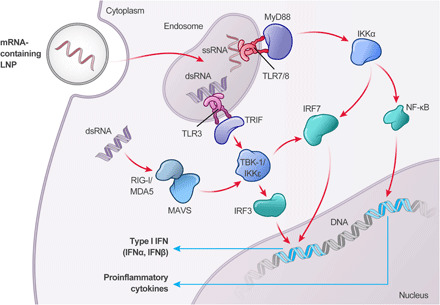
Endocytosis of mRNA in LNP is detected by the microbial-associated molecular pattern (MAMP) surveillance system, whereby detection of dsRNA, by TLR3, or ssRNA containing canonical nucleotides, by TLR7/8, activate either TRIF or MyD88, respectively. Activation of MyD88 leads to expression of proinflammatory cytokines through nuclear factor κB (NF-κB), as well as expression of type I IFN through IFN regulatory factor 7 (IRF7). Activation of TLR3 also initiates a type I IFN response, although through TRIF-mediated activation of TANK-binding kinase 1 (TBK-1) or IκB kinase epsilon (IKKε), which, in turn, activates IRF3 and IRF7. Upon escape of the exogenously delivered mRNA payload from the endosome, dsRNA impurities are detected by RIG-I or MDA5, which initiate expression of type I IFN cytokines (e.g., IFNα and IFNβ) via TBK-1/IKKε activation of IRF3.
One possible outcome of immune activation is reduced mRNA translation due to feedback between the innate immune system and translation machinery, which will decrease the potency of therapeutic mRNA (18, 19). Mechanistically, viral RNA has been shown to activate stress response genes that ultimately inhibit protein translation (19). This natural cellular defense mechanism aims to block protein expression of viral genes but has the added consequence of reducing overall cellular gene expression (19).
Therapeutic mRNA is typically synthesized using in vitro transcription (IVT) with single-subunit polymerases (e.g., T7, T3, and SP6) using a DNA template to produce multiple copies of the coded mRNA. T7 RNA polymerase can incorporate chemically modified nucleotide triphosphates (NTP), particularly uridine modifications, which have been shown to maintain translational efficiency and alter the interactions between exogenous mRNA and TLR7, TLR8, and RIG-I (20). During the enzymatic transcription process, promiscuity of T7 RNA polymerase (8, 9) can result in the generation of dsRNA impurities, which are known to trigger an innate immune response (21, 22). Purification of the IVT product by reversed-phase high-performance liquid chromatography (RP-HPLC) has been shown to reduce dsRNA impurities, decrease IFNα response, and improve protein expression from therapeutic mRNA (21). Treatment with ribonuclease III (RNase III), an RNase that recognizes and cleaves dsRNA of approximately 22 nucleotides in length, has also been shown to reduce immune-stimulatory response (23). It has also been observed that alteration of IVT components affects the purity of the mRNA, including dsRNA content, leading to greater or less innate immune activation (22, 24).
While the implications of uridine modification and dsRNA impurities in mRNA vaccines are still being understood, substantial evidence exists describing the benefits that chemical modifications and the elimination of dsRNA have on mRNA for therapeutic applications (25). To decipher the relative contributions of these factors, we comprehensively evaluated the impact of uridine chemistry and dsRNA impurities on immune activation both in vitro and in vivo. We conclude that it is necessarily the combination of both uridine modifications and the reduction of dsRNA through process optimization and/or purification that results in an mRNA drug product that is both efficacious and well tolerated for therapeutic purposes.
RESULTS
Uridine and process modifications alter dsRNA content, protein expression, and type I IFN response to mRNA
The combined impact of both uridine modification and IVT process on protein expression and innate immune activation was evaluated using human erythropoietin (hEPO) mRNA containing either unmodified uridine triphosphate (U) or N1-methylpseudo-uridine-5′-triphosphate (1mΨ) and prepared by two processes, A and B. The IVT conditions for process A contained equimolar levels of each nucleotide; process B used a custom NTP ratio observed to reduce production of dsRNA impurities. In addition to standard affinity purification using 2′-deoxy-T20 oligo resin, RP-HPLC was added to process B to ensure further removal of any residual dsRNA impurities. These four mRNA variants, which differ by uridine chemistry and mRNA purity, termed as process A (U), process A (1mΨ), process B (U), and process B (1mΨ), respectively, were used throughout further experiments.
mRNA prepared by process A resulted in higher amounts of dsRNA regardless of the substitution of U residues with 1mΨ (Fig. 2A; red and pink bars) compared to process B (Fig. 2A; light and dark blue bars). Upon transfection of BJ fibroblasts, hEPO protein production was lower with process A than process B (Fig. 2B; open bars). Protein expression was inversely correlated with the IFNβ production by these cells (Fig. 2C; open bars), as there was significant IFNβ production with process A (U) and, to a lesser extent, with process A (1mΨ) but not the process B groups (Fig. 2C; open bars). The presence of dsRNA by enzyme-linked immunosorbent assay (ELISA) and IFNβ production in BJ fibroblasts were diminished when mRNA was treated with RNase III (dsRNA-specific RNase) before transfection (Fig. 2, A and C, respectively; hatched bars), while hEPO expression was restored for groups made with U chemistry and unaffected for groups made with 1mΨ chemistry (Fig. 2B; hatched bars), confirming that the dsRNA content contributes to the IFNβ response.
Fig. 2. dsRNA levels, protein expression, and immune activation from hEPO mRNA incorporating uridine or 1mΨ, made by processes A or B, with or without RNase III treatment.
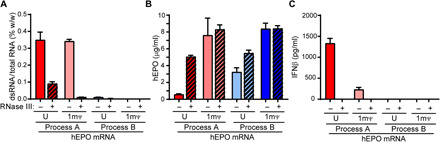
(A) dsRNA content was determined by dsRNA ELISA. (B) hEPO expression and (C) IFNβ response were measured by Ella in BJ fibroblasts at 48 hours after transfection. Data represent two independent experiments, with n = 2 to 3 technical replicates.
Type I IFN response to dsRNA impurities is RIG-I–dependent in vitro
To elucidate the mechanism by which dsRNA stimulates an innate immune response, we evaluated the ability of the mRNA preparations to activate an IFN-mediated immune response in vitro using the THP1-Dual human monocyte cell line. These cells express two stably transfected gene constructs comprising a Lucia luciferase gene reporter driven by an interferon stimulated gene 54 (ISG54) promoter and a nuclear factor κB (NF-κB)–inducible secreted alkaline phosphatase (SEAP) gene. THP1-Dual cells have functioning ssRNA- and dsRNA-sensing innate immune pathways, as demonstrated by IFN promoter stimulation with a series of agonists (Fig. 3A): RIG-I stimulation via 5′ppp-hpRNA (hairpin RNA; orange bars) and 5′ppp-dsRNA (orange hatched bars), TLR7/8 activation using R848 (green bars), TLR8 activation using TL8-506 (green hatched bars) and stimulator of IFN genes (STING) stimulation via 2′3′-cGAMP (cyclic guanosine monophosphate–adenosine monophosphate; black bars). STING is not interconnected with the TLR and RIG-I signaling pathways (26) and is unlikely to be directly affected by mRNA in these experiments; however, the agonist was a strong positive control for IFN up-regulation independent of the dsRNA signaling pathway.
Fig. 3. Immune activation in THP1-Dual, A549-Dual, and monocyte-derived macrophage cells.
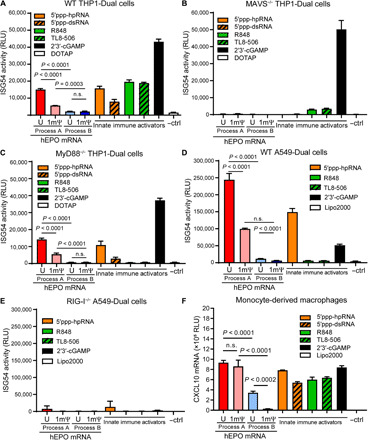
Immune activation associated with mRNA transfected in THP1-Dual, A549-Dual, and monocyte-derived macrophage (MDM) cells or TLR7/8 (R848 and TL8-506), RIG-I (5′ppp-dsRNA and 5′ppp-hpRNA), and STING (2′3′-cGAMP) agonists into (A) THP1-Dual cells, (B) MAVS−/− THP1-Dual cells, (C) MyD88−/− THP1-Dual cells, (D) A549-Dual cells, (E) RIG-I−/− A549-Dual cells, and (F) MDM was examined. ISG54 promoter activity (A to E) was measured through a Lucia luciferase reporter and reported as relative luminescence units (RLU). C-X-C motif chemokine 10 (CXCL10) mRNA induction (F) was determined by branched DNA (bDNA) analysis. Data are representative of two independent experiments, with n = 2 to 3 technical replicates. n.s., not significant; ctrl, control; Lipo2000, Lipofectamine 2000.
mRNA was introduced to wild-type (WT) THP1-Dual cells by N-[1-(2,3-Dioleoyloxy)propyl]-N,N,N-trimethylammonium methyl-sulfate (DOTAP) transfection, and ISG54 induction was measured via luciferase activity. Independent of uridine chemistry, mRNA produced by process A stimulated the IFN pathway in WT THP1-Dual cells, with process A (U) hEPO mRNA yielding a greater IFN response compared to process A (1mΨ) mRNA (Fig. 3A; red versus pink bars), whereas mRNA produced by process B did not stimulate the IFN pathway above the background control (Fig. 3A; light and dark blue bars).
MAVS, an adaptor protein that activates type I IFN through transcription factors IFN regulatory factor 3 (IRF3), IRF7, and NF-κB (27, 28), is downstream of both RIG-I and MDA5 (17). RIG-I differs from MDA5 in that it responds to shorter dsRNA or hpRNA and has the capacity to bind both blunt-end and 5′-triphosphate dsRNA, whereas MDA5 recognizes >500–base pair (bp) dsRNA with a 5′-triphosphate (29, 30). MAVS−/− THP1-Dual cells were generated via the CRISPR-Cas9 system and used to test the role of both RIG-I and MDA5 signaling in the IFN response to the mRNA preparations. Loss of MAVS, which mediates the cytosolic dsRNA sensing pathway, obliterated activation in the MAVS−/− THP1-Dual cells by both the dsRNA agonists and the induction of IFN for all mRNA preparations (Fig. 3B). Signaling through the TLR7/8 agonists was also substantially reduced in the MAVS−/− THP1-Dual cells. To determine whether the loss of IFN induction from mRNA in the MAVS−/− THP1-Dual cells was secondary to an impact on the TLR7/8 pathway, the mRNA preparations were also tested in MyD88−/− THP1-Dual cells, which should remove ssRNA signaling through TLR7/8 but not affect dsRNA sensing. While activation with the agonists R848 and TL8-506 was inhibited in these cells, there was no impact on IFN induction with mRNA (Fig. 3C). Although it is possible for the immune response downstream of dsRNA in process A mRNA to be stimulated via the dsRNA receptor TLR3 in the endosome, these data implicate the MAVS pathway, through dsRNA, in the induction of IFN in THP1-Dual cells.
To distinguish between the two MAVS-mediated dsRNA receptors RIG-I and MDA5, the mRNA preparations were transfected into WT and RIG-I−/− A549-Dual human lung epithelial carcinoma cells. The agonist panel demonstrated that WT A549-Dual cells were responsive to the RIG-I agonist 3′ppp-hpRNA and the STING agonist 2′3′-cGAMP but not to the TLR7/8 agonists R848 and TL8-506 (Fig. 3D). This is perhaps expected, as it has been previously reported that A549 cells lack TLR7 and TLR8 signaling with R848 (31). Similar to the observations in THP1-Dual cells and BJ fibroblasts, mRNA produced by process A, but not by process B, stimulated an IFN response in A549-Dual cells (Fig. 3D; red and pink versus light and dark blue bars). Removal of the RIG-I gene from these cells resulted in complete abrogation of IFN signaling in response to all mRNA variants (Fig. 3E), confirming RIG-I as the primary dsRNA sensor in vitro. Both THP1-Dual and A549-Dual cells allow for detection of NF-κB activation via SEAP. Except for process A (U), which contains the highest level of dsRNA, little to no induction of the NF-κB pathway was observed with the mRNA preparations (fig. S1, A and D). NF-κB stimulation by process A (U) was also MAVS or RIG-I mediated (fig. S1, B and E).
Last, the ability of Lipofectamine-delivered modified and unmodified mRNA produced by both processes to induce expression of C-X-C motif chemokine 10 (CXCL10; also known as IFNγ-induced protein 10) was evaluated in human primary monocyte-derived macrophages (MDMs). CXCL10 is a chemokine that plays an important role in viral and bacterial infections (32) and is a robust indicator of activation of the innate immune system. The agonist panel described above confirmed that both ssRNA (TLR7/8 agonists) and cytosolic dsRNA (RIG-I agonists) detection pathways are functional in these cells (Fig. 3F; innate immune activators). Independent of uridine chemistry, mRNA produced by process A induced high levels of CXCL10 expression (Fig. 3F; red and pink versus light and dark blue bars). In contrast, process B (U) mRNA induced moderate levels of CXCL10, while process B (1mΨ) mRNA induced very low levels of CXCL10 (Fig. 3F; light versus dark blue bars). These data demonstrate that mRNA with canonical uridine and significantly reduced dsRNA impurities may still stimulate innate immunity, possibly through ssRNA activation of the TLR7/8 pathway.
The immune response to mRNA in vivo is dependent on both uridine chemistry and dsRNA impurities
To further investigate the effects of mRNA production process and nucleotide modifications on protein expression and immune activation, LNP-encapsulated mRNA encoding hEPO was injected intravenously into C57BL/6 mice. In addition, synthetic 19-mer oligonucleotides comprising uridine [poly(U)] or 1mΨ [poly(1mΨ)] were tested to evaluate the impact of uridine modifications in the absence of IVT process. As shown in Fig. 4A, levels of hEPO protein in mouse serum at 6 hours after injection were significantly higher in mice that received mRNA prepared by process B compared with process A, regardless of uridine modification. Notably, IFNα was significantly up-regulated in mice that received mRNA made by process A but not process B, with a higher level observed in process A (U), as compared to process A (1mΨ) (Fig. 4B; red versus pink bars). These combined data confirm the findings from the in vitro experiments, showing that activation of the innate immune system with dsRNA impurities reduces protein expression from exogenous mRNA. The synthetic poly(U) or poly(1mΨ) 19-mer oligonucleotides did not elicit IFNα, while polyinosinic:polycytidylic acid [poly(I:C)], a synthetic dsRNA that primarily stimulates TLR3 when delivered gymnotically but can activate the MAVS dsRNA pathway as well (33), substantially induced serum IFNα (Fig. 4B; purple bar).
Fig. 4. Protein expression and immune activation from hEPO mRNA incorporating uridine or 1mΨ, made by processes A or B, or poly(I:C), poly(U) 19-mer, and poly(1mΨ) 19-mer in C57BL/6 mice (5 to 10 mice per group) at 6 hours after injection.
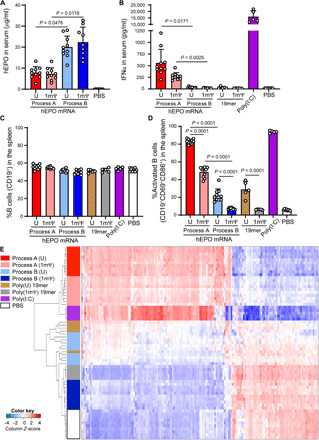
The level of (A) hEPO and (B) IFNα in serum was measured using Ella and Luminex, respectively. The percentage of (C) total (CD19+) B cells and (D) activated (CD19+CD69+CD86+) B cells in spleen was determined by flow cytometric analysis. Expression of splenic genes included in the Mouse Myeloid Innate Immunity Panel (E), represented as Z-score, was evaluated by NanoString analysis and grouped on the basis of a clustering analysis of treatment groups. PBS, phosphate-buffered saline.
To evaluate the effect of systemic administration of mRNA on a specific immune cell population, the frequency of total (Fig. 4C; CD19+ cells) and activated (Fig. 4D; CD19+CD69+CD86+ cells) B cells within the spleen was analyzed by flow cytometry. The total B cell frequency in the spleen was unaffected by any of the mRNA tested (Fig. 4C). In contrast, the frequency of activated B cells was significantly affected by mRNA process and chemistry. Process A (U), process B (U), and process A (1mΨ) all resulted in activation of B cells, and only process B (1mΨ) did not. B cell activation was significantly less with process B (U) compared to process A (U) (Fig. 4C; red versus light blue bar). A significant decrease in activated B cells was also observed between process A (1mΨ) and process B (1mΨ) (Fig. 4C; pink versus dark blue bar). Similar to CXCL10 induction in primary human MDM in vitro, process B (U) activated B cells, potentially as a result of stimulation by ssRNA through TLR7/8 activation. Consistent with this observation, the differential activation of B cells was also observed with the synthetic 19mers: poly(U) 19-mer activated B cells, while poly(1mΨ) 19-mer did not. Neither oligonucleotide should contain dsRNA. As expected, as a potent activator of multiple PRR (15, 33), poly(I:C) treatment lead to a significant increase in serum IFNα and percentage of activated B cells (Fig. 4D; purple).
Similar to the results obtained with B cell activation, an analysis of the same mouse spleen samples by NanoString with 768 splenic genes in the Mouse Myeloid Innate Immunity Panel showed differential expression of 299 genes (table S1) in five pairwise comparisons: U versus 1mΨ (in each process), process A versus process B (in each chemistry), and poly(U) versus poly(1mΨ). Notably, a nonsupervised hierarchical clustering on these 299 genes showed clear coclustering of these treatments (Fig. 4E). mRNA prepared by process A, regardless of uridine chemistry, leads to the up-regulation of 192 genes and down-regulation of 107 genes (Fig. 4E; red and pink clusters), when compared to the phosphate-buffered saline (PBS)–treated control. The presence of canonical uridine in a process that resulted in less dsRNA [process B (U)] or in the absence of IVT process [poly(U) 19mer] leads to measurable, but less, up-regulation of certain genes in this subset (Fig. 4E; light blue and brown clusters). This indicates that, despite using a highly sensitive method to detect the gene induction, the combination of process B with unmodified uridine leads to an mRNA product that does not stimulate significant gene expression in the tested genes nor, therefore, a downstream immune response greater than that generated by the poly(U) 19-mer. There was a small subset of up-regulated genes, likely downstream of TLR7 signaling since these treatments use canonical uridine. Little or no change in gene regulation was observed when animals were treated with process B (1mΨ) mRNA or poly(1mΨ) 19-mer; gene regulation with both of these treatments clustered with the PBS control (Fig. 4E; dark blue, grey, and white, respectively). This strongly reinforces the conclusion that using 1mΨ-modified uridine in process B delivers mRNA in an essentially immunologically silent manner in vivo that is almost indistinguishable from PBS at a differential gene expression level for genes involved in the myeloid innate immune response. These data show that the bulk of the myeloid immune response in C57BL/6 mice was due to process A, particularly for mRNA with 1mΨ-modified uridine.
Canonical uridine, but not modified uridine, elicits an immune response in MAVS−/− mice
To investigate the contribution in vivo of RIG-I/MDA5 signaling through MAVS in the immune activation from uridine and 1mΨ mRNA made by both processes, we intravenously administered the LNP-encapsulated mRNA preparations encoding hEPO in MAVS−/− and B6129SF2/J control mice (Fig. 5; hatched versus open bars, respectively). Process A (U), which contains the most dsRNA impurities and is the most potent immune activator, resulted in a significant reduction in hEPO protein expression in the serum of B6129SF2/J control mice when compared to all other mRNA preparations (Fig. 5A; open red versus light blue bars). However, in the MAVS−/− mice, all four mRNA preparations resulted in similar levels of hEPO expression (Fig. 5A; hatched bars).
Fig. 5. Protein expression and immune activation from hEPO mRNA incorporating uridine or 1mΨ, made by processes A or B, or poly(U) 19-mer, poly(1mΨ) 19-mer, 5′ppp-hpRNA, R848, or poly(I:C) in B6129SF2/J control and MAVS−/− mice (five mice per group) at 6 hours after injection.
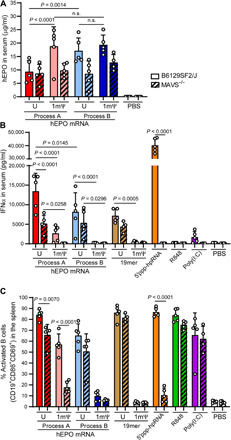
The level of (A) hEPO and (B) IFNα in serum was measured using Ella and Luminex, respectively. (C) The percentage of activated (CD19+CD69+CD86+) B cells in spleen was determined by flow cytometric analysis. Data are representative of two replicate experiments.
To ensure the complete absence of RIG-I signaling in MAVS−/− mice, we injected LNP-encapsulated 5′ppp-hpRNA, a strong specific agonist of RIG-I, and assessed the serum cytokine response (Fig. 5B; open and hatched orange bars). B6129SF2/J control mice treated with 5′ppp-hpRNA displayed a high serum IFNα response, which was undetectable in the MAVS−/− mice (Fig. 5B; open versus hatched orange bars). Poly(I:C), which mainly signals through TLR3 when delivered unformulated (but can signal though MDA5), unexpectedly led to an almost undetectable IFNα level in the B6129SF2/J control mice and no IFNα response in the MAVS−/− mice. The TLR7 agonist R848 did not induce IFNα in the MAVS−/− or B6129SF2/J control mice (Fig. 5B; open and hatched green bars). Serum IFNα was induced in response to mRNA prepared only by process A (regardless of uridine chemistry) in C57BL/6 mice (Fig. 4B) but was significantly induced in the sera of B6129SF2/J control mice in response to all treatments with canonical uridine-containing mRNA [process A (U), process B (U), and poly(U) 19-mer] in comparison to PBS-treated groups (Fig. 5B; open red, light blue, and brown bars). The mean level of serum IFNα in MAVS−/− mice injected with process A (U) was >50% lower than levels seen in B6129SF2/J control mice (Fig. 5B; open versus hatched red bars). This trend was also observed in multiple innate immune cell–produced chemokines, such as chemokine (C-C motif) ligand 4 (CCL4) and CCL5 (fig. S2, A and B). The large difference between B6129SF2/J control and MAVS−/− mice in systemic inflammatory response observed with process A (U) was not observed with process B (U) (Fig. 5B; open versus hatched light blue bars), indicating that the dsRNA impurities found in process A mainly signal though MAVS to induce IFNα. The IFNα responses in the MAVS−/− mice treated with either process A (1mΨ) or process B (1mΨ) mRNA were significantly lower than process A (U) and process B (U) (Fig. 5B; hatched red versus pink and hatched light versus dark blue, respectively). Furthermore, for both processes A and B, 1mΨ chemistry reduced the type I IFN response compared to canonical uridine in both B6129SF2/J control and MAVS−/− mice. The decrease in immune response to modified uridine mRNA-treated MAVS−/− mice indicates strongly that 1mΨ chemistry avoids TLR7 signaling that is triggered by canonical uridine and that mRNA produced using process B is essentially immunologically silent when made with 1mΨ (Fig. 5B; hatched red versus pink bars and hatched light versus dark blue bars).
To further evaluate the immune response to the mRNA variants in B6129SF2/J control and MAVS−/− mice, we compared the frequency of activated B cells in the spleen by flow cytometry. The overall B cell frequency (CD19+) was unaffected by both the targeted deletion of the MAVS gene and by the mRNA and agonist samples tested (fig. S2C; open versus hatched bars). As with serum IFNα induction, splenic B cells were activated in the B6129SF2/J control mice by mRNA prepared by process A regardless of uridine chemistry and by process B (U) but not by process B (1mΨ) (Fig. 5C; open red and pink bars). The percentage of activated B cells in the spleen was significantly lower in MAVS−/− mice injected with process A (1mΨ) mRNA (Fig. 5C; open versus hatched pink bars) compared to B6129SF2/J control mice, a trend seen in IFNα production as well, implicating the dsRNA pathway as the primary mechanism for this mRNA variant. The 19-mer poly(U), but not the 19-mer poly(1mΨ), activated splenic B cells (Fig. 5C; brown versus grey bars), which were unaffected by the absence of the MAVS gene, implicating a contribution of the ssRNA sensors such as TLR7 for the other mRNA preparations with canonical uridine produced by either process. Both processes B (1mΨ) mRNA and poly(1mΨ) 19-mer lead to a similar percentage of activated B cells at baseline as PBS (Fig. 5C; dark blue and grey versus white bars), indicating that LNP delivery of mRNA with modified uridine in the absence of dsRNA impurities is essentially immunologically silent. As expected, B cell activation in animals injected with the RIG-I agonist 5′ppp-hpRNA was completely abolished in the MAVS−/− mice (Fig. 5C; orange bars), whereas stimulation by the TLR7 activator R848 and the TLR3 activator poly(I:C) was not affected by removing the MAVS gene (Fig. 5C; green and purple bars, respectively).
To further evaluate the role of dsRNA and uridine modification on innate immunity, we analyzed differential gene expression in the spleens of B6129SF2/J control and MAVS−/− mice. Figure 6A displays the differentially expressed genes from both B6129SF2/J control and MAVS−/− mice injected with an mRNA variant, poly(U) 19-mer, poly(1mΨ) 19-mer, 5′ppp-hpRNA, or PBS. As expected, there is little difference between B6129SF2/J control and MAVS−/− mice in the PBS treatment groups, whereas differences in the gene expression pattern are marked in the 5′ppp-hpRNA–treated mice: The MAVS−/− mice showed a much lower Z-score (Fig. 6A; white and orange, respectively). This alteration was not seen with the R848 and poly(I:C) controls, which signal through TLR7 and TLR3, respectively (fig. S3A; green and purple, respectively). The greatest alteration in the gene expression pattern between the mRNA variants was observed in the process A (1mΨ)–treated mice (Fig. 6A; pink). In both 5′ppp-hpRNA and process A (1mΨ) treatment groups, the MAVS−/− mice showed a much lower Z-score across the differentially expressed genes compared to the B6129SF2/J control mice. This alteration is consistent with the MAVS-dependent activation of B cells (Fig. 5C). All groups containing canonical uridine [process A (U), process B (U), and poly(U) 19-mer] showed a marked increase in differential gene expression in both B6129SF2/J control and MAVS−/− mice (Fig. 6A; red, light blue, and grey). Cluster analysis of treatments shows that all canonical uridine–containing treatments and process A (1mΨ) cluster separately from PBS and poly(1mΨ) 19-mer in B6129SF2/J control mice but cluster closely with 5′ppp-hpRNA, indicating that the gene response in the spleen to these treatments are closely related and highly inflammatory (fig. S3B). Consistent with side-by-side comparisons, the cluster analysis of spleens from the MAVS−/− mice shows that both 5′ppp-hpRNA and process A (1mΨ), but not the canonical uridine–containing treatments, cluster with PBS and process B (1mΨ), which contains low levels of dsRNA (fig. S3C). This indicates that both modification of uridine and elimination of dsRNA play a role in the elimination or modification of the immune response.
Fig. 6. Immune activation from hEPO mRNA incorporating uridine or 1mΨ, made by processes A or B, or poly(U) 19-mer, poly(1mΨ) 19-mer, or 5′ppp-hpRNA in B6129SF2/J control and MAVS−/− mice (five mice per group) at 6 hours after injection.
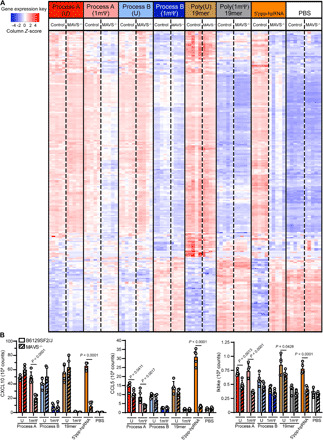
(A) Expression of splenic genes included in the Mouse Myeloid Innate Immunity Panel, represented by Z-score, was evaluated by NanoString analysis. (B) Quantitation of CXCL10, CCL5, and Ikbke levels. Data are representative of two replicate experiments.
To explore the impact of the genetic alterations on the RIG-I pathway, we compared the normalized counts from the NanoString data for genes of interest [CXCL10, CCL5, and IκB subunit epsilon (Ikbke)] between the B6129SF2/J control and MAVS−/− mice (Fig. 6B). CXCL10 and CCL5 were chosen, as they have been sensitive readouts to innate immune activation in in vitro and/or in vivo models. Ikbke was chosen for its specific regulation of dsRNA-induced NF-κB. All three genes are significantly decreased in MAVS−/− spleens in both the 5′ppp-hpRNA–treated mice and the process A (1mΨ)–treated mice compared to B6129SF2/J control mice (Fig. 6B; orange and pink bars). CCL5 and Ikbke both showed a less significant decrease in expression in MAVS−/− in process A (U)–treated mice (Fig. 6B; red bars). These combined data show that dsRNA found in process A signals via MAVS to modulate the immune system and process modifications to reduce dsRNA impurities can modify the response.
DISCUSSION
The clinical evaluation of IVT mRNA as a therapeutic has initially focused on its interface with the immune system. The intrinsic immunostimulatory profile of an IVT mRNA–based drug, coupled with coding of a target antigen, has allowed for a simplified product that coordinates immune activation and antigen presentation, mimicking a viral infection. This has led to the development of antibodies at levels indicating protection against infectious diseases and the detection of immune titers against cancer epitopes (34, 35). The ability to decouple immune system activation from protein expression should allow for the utilization of mRNA as a disruptive technology for current therapies that require protein production in the absence of an immune response. We have found that the combination of process engineering and the use of 1mΨ have a significant impact on the overall innate immune response of a therapeutic mRNA and the total amount of protein expressed.
To systematically probe how synthesis process and nucleotide chemistry affect innate immune activation, we examined four mRNA variants: process A (U), process B (U), process A (1mΨ), and process B (1mΨ). The idea that purification and reaction engineering can have a material impact on the dsRNA levels in an IVT mRNA preparation has become a recent topic of intense interest (22). Here, we showed that neither chemistry nor process alone reduced immune activation of an IVT mRNA as much as the combination of the two and that avoiding endosomal TLR–mediated microbial-associated molecular pattern (MAMP) recognition via uridine modification (1mΨ) and decreased cytosolic immune activation from 5′-triphosphorylated dsRNA impurities removed by RP-HPLC (process B) were necessary and sufficient for producing a drug product nearly indistinguishable from injected PBS with regard to innate immune activation in two mouse strains; C57BL/6 and B6129SF2/J (Figs. 4, D and E; 5C; and 6; dark blue versus white) (21, 22, 24).
TLR7 activation has been shown to occur in the presence of proximal uridines (36), while TLR8, which also recognizes ssRNA, requires a single uridine for high-affinity binding (37). To examine the hypothesis that uridine chemistry substitution (e.g., 1mΨ) achieves a reduction in immune signaling through decreased activation of TLR signaling, we transfected our mRNA variants into MyD88−/− THP1-Dual cells (Fig. 3C), which lack TLR7/8 signaling, and WT THP1-Dual cells (Fig. 3A). We expected that process A (U) and process A (1mΨ) would have similar IFN responses in MyD88−/− cells, unlike the differential response seen in the WT THP1-Dual cells (Fig. 3A). However, the response pattern to process A (U) and process A (1mΨ) in the MyD88−/− cells was equivalent to the WT cells (Fig. 3C). It is possible that formulation of mRNA with DOTAP limits engagement of the mRNA with endosomal TLR7/8 in this cell system. This is supported by a lack of stimulation seen with process B (U) in WT THP1-Dual cells. The differential response between process A (U) and process A (1mΨ) is due to either a greater amount of dsRNA in the process A (U) mRNA or the fact that dsRNA containing uridine is a far more effective activator of dsRNA PRRs compared to dsRNA that includes 1mΨ (38, 39). Unlike THP1-Dual cells, process B (U) induces the production of CXCL10 by MDM (Fig. 3F), suggesting either stimulation of the ssRNA detection pathway via TLR7/8 or increased sensitivity to any residual dsRNA in process B (U). A similar observation can be seen in vivo using LNP-encapsulated process B (U) mRNA in C57BL/6 (Fig. 4, B to D) and B6129SF2/J control mice (Fig. 5, B and C, and fig. S3B), where both strains had increased splenic IFNα, B cell activation, and up-regulation of splenic genes compared to process B (1mΨ) mRNA. This increase was likely due to MAMP-mediated TLR7 activation by uridine, as there is a significant increase in immune response in process B (U) compared to process B (1mΨ) in MAVS−/− mice, whereas there was a small, unaltered immune response between process B (1mΨ) and process A (1mΨ) (Fig. 5C; hatched bars).
To confirm the mechanistic hypothesis that process changes affect innate immune activation through cytosolic dsRNA sensors, we evaluated the mRNA variants in knockout models in vitro and in vivo. In THP1-Dual and A549-Dual cells, MAVS or RIG-I, respectively, were independently knocked out. These two cell lines showed a reduction of type I IFN promoter activity in all mRNA variants (Fig. 3, B and E), implicating dsRNA as the mediator of innate immune activation. However, an unanticipated finding was a reduction or elimination of signal in THP1-Dual cells with TLR7 or TLR8 agonists (R848 and TL8-506, respectively) and in A549 cells with a STING agonist (2′3′-cGAMP). Perhaps the failure of the controls to elicit an immune response under these conditions implies cross-talk among innate immune system sentinels (40, 41), whereby eliminating one signaling adaptor may have unintended consequences on other pathways (e.g., regulation of STING expression by RIG-I activation) (42). However, TLR7/8 agonist controls showed that TLR7 and TLR8 activate the NF-κB pathway as expected in these cells (fig. S1, B and E).
To further probe the impact of process and chemistry on innate immune activation, we used a series of WT and knockout mouse models. Clear differences in both the magnitude of innate immune response and level of protein expression can be seen, particularly comparing process A to process B (Fig. 4). We further corroborated that process and chemistry both contribute to innate immune activation using the NanoString transcript analysis of homogenized spleens (Figs. 4E and 6). Qualitative examination of the gene activation levels (Fig. 4E) shows that process A (U) and process A (1mΨ) are similar to levels seen with poly(I:C), confirming that these treatments elicit an immune response similar to this potent innate immune agonist. Gene regulation with process B (U) and process B (1mΨ) aligns with their synthetic chemistry 19-mer controls, providing confirmation of the dramatic reduction in immunostimulatory contaminants from process and purification. Furthermore, the addition of modified chemistry makes mRNA made by process B almost immunologically indistinguishable from the PBS control (Fig. 4E).
We used MAVS−/− mice to confirm the relative roles of ssRNA and dsRNA sensors in mediating innate immune activation through the four mRNA variants (Fig. 5). The comparison of poly(U) to poly(1mΨ) in B6129SF2/J control and MAVS−/− mice (Fig. 5C) revealed a strong contribution of chemistry, further supporting the role of uridine chemistry in TLR7-mediated MyD88 activation (36). As the above confirms uridine as an innate immune driver in B6129SF2/J control and MAVS−/− mice, the effects of process are most effectively elucidated via comparisons between control and MAVS−/− mice treated with either process A (1mΨ) or process B (1mΨ). The difference in immune activation between B6129SF2/J control and MAVS−/− mice is most notable with process A (1mΨ), consistent with the levels of dsRNA present in this sample (Fig. 2A).
Gene expression analysis was also performed on splenic mRNA levels in B6129SF2/J control and MAVS−/− mice injected with the mRNA variants or controls. Process A (1mΨ) shows a similar trend in gene expression as the RIG-I agonist 5′ppp-hpRNA, albeit with reduced magnitude, further suggesting the presence of dsRNA as a driver of immune activation even when uridine is replaced by 1mΨ. Comparison of gene expression trends between the two strains qualitatively shows that process B (1mΨ) but not process B (U) has a gene activation signature similar to the signature seen with PBS. There is unexpectedly little difference between B6129SF2/J control and MAVS−/− mice with process B (U) and process B (1mΨ), indicating that uridine chemistry contributes to innate immune activation of exogenous mRNA despite the process used to synthesize the mRNA. The branch migration of 5′ppp-hpRNA and process A (1mΨ) to the same cluster as PBS, observed in MAVS−/− mice, indicates the substantial difference in gene expression and downstream immune response dependent on cytosolic dsRNA sensors in vivo (fig. S3, B and C). Splenic gene expression analysis induced by short hpRNA delivered via transfection with polyethylenimine has been reported previously (29). Here, we extend these investigations with the assessment of mRNA and compare gene expression profiles in both B6129SF2/J control and MAVS−/− mice to confirm mechanism. While both reports demonstrate gene modulation through the RIG-I/MAVS pathway, differences in the gene expression profiles may be the result of the delivery vehicles used, in our case, LNP, and the gene panel analyzed (29).
Our results show that uridine chemistry and mRNA synthesis process act synergistically to reduce the immune signature of mRNA. However, we cannot say with complete certainty that inclusion of 1mΨ is solely responsible for the interaction with TLR7/8. Another possibility is that 1mΨ in the transcription reaction changes the amount or potency of dsRNA (43). Future mechanistic experiments using TLR7−/− mice could shed light on a chemistry-specific effect operating only through the TLR7/MyD88 pathway.
Strategies to engage or avoid the innate immune system will vary on the basis of the clinical application of the mRNA therapeutics (44). For vaccines, mRNA that engages the innate immune system may potentiate the response to the vaccine (44, 45); however, for nonimmunotherapy applications, activation of the innate immune response can compromise efficacy through reduction of protein expression (19, 44). Although some studies suggest that purification of unmodified mRNA is sufficient to achieve effective mRNA-based therapeutics, the measures of immune stimulation reported typically included serum activation markers and protein expression levels (46). As we have shown here, splenic B cell activation and immune-related gene expression experiments show that dsRNA removal alone is not sufficient at avoiding immune activation and that it is the combination of uridine modification and dsRNA removal that are required to generate a furtive mRNA-based drug product. This is particularly important for therapies that require repeated administrations, as immune responses have memory, which could render subsequent administrations less effective over time (47).
In summary, we found that the combination of the production process to reduce dsRNA and substitution of 1mΨ for uridine was more effective at reducing innate immune activation than either the process or nucleotide modification alone. As the field of RNA-based therapeutics advances, understanding the levers available to tune the immune signatures of therapies will be essential for developing effective and safe drugs.
MATERIALS AND METHODS
Study design
The aim of this study was to elucidate the relative contributions of uridine chemistry modification and dsRNA impurities in activation of the innate immune response. In vitro studies were carried out in duplicate or triplicate within groups, and the experiments were repeated two to three times, as indicated in the figure legends. Age-matched (8 to 10 weeks old) mice used for this study were bred or ordered from vendors and housed in the same facility at Moderna.
mRNA synthesis and formulation
mRNA was synthesized in vitro by T7 RNA polymerase–mediated transcription, using either uridine 5′-triphosphate or 100% substituted with 1mΨTP, from a linearized DNA template, which incorporates 5′ and 3′ untranslated regions and a polyadenosine tail. For process A, NTPs were included at equimolar concentrations, and the resulting mRNA was purified by 2′-deoxy-T20 oligo affinity chromatography. For process B, NTPs were included at custom molar ratios and purified by ion pair RP-HPLC. After purification, mRNA was buffer-exchanged into 2 mM sodium citrate (pH 6.5), passed through a 0.22-μm filter, and stored at −20°C until use.
LNP formulations were prepared as previously described (48). Briefly, lipids dissolved in ethanol at a molar ratio of 50:10:38.5:1.5 of ionizable:helper:structural:polyethylene glycol were mixed with acidified mRNA at a ratio of 3:1 mRNA:lipid. Formulations were dialyzed against PBS (pH 7.4) for at least 18 hours, concentrated using Amicon Ultra centrifugal filters (EMD Millipore Corp., Merck KGaA, Darmstadt, Germany), passed through a 0.22-μm filter, and stored at 4°C until use. Particle size was measured by dynamic light scattering and found to be <100 nm; encapsulation was >90%, as measured by the Quant-iT RiboGreen RNA quantitation kit (Thermo Fisher Scientific), and endotoxin was <10 endotoxin units (EU)/ml. mRNA was formulated with DOTAP (Sigma-Aldrich, St. Louis, MO) according to the manufacturer’s recommended protocol.
RNase III enzymatic treatment
mRNA was digested with RNase III (Epicentre, Madison, WI, USA) as previously described (49). Samples were incubated for 10 min at room temperature in reaction buffer [33 mM tris acetate (pH 7.5), 66 mM potassium acetate, 10 mM magnesium acetate, and 0.5 mM dithiothreitol] and RNase III (0.001 U/μg). Reactions were quenched with 50 mM EDTA.
dsRNA ELISA
dsRNA was detected by sandwich ELISA using antibodies previously reported to bind dsRNA (50). K1 mouse monoclonal antibody (SCICONS, Budapest, Hungary) was immobilized on 96-well Nunc Immuno plates (Thermo Fisher Scientific, Waltham, MA, USA) for 3 hours and then blocked with 10% nonfat dry milk in PBS overnight at 4°C. mRNA was incubated for 2 hours at room temperature, washed, and incubated with K2 mouse monoclonal antibody (SCICONS) for 1 hour at room temperature. Plates were washed and incubated for 1 hour with horseradish peroxidase–conjugated goat anti-mouse immunoglobulin G detection antibody (Thermo Fisher Scientific). After the final wash, the signal was read at 450 nm on a Synergy H1 plate reader (BioTek, Winooski, VT, USA). Determination of dsRNA concentration in total input RNA was based on a standard curve generated using a 400-bp dsRNA prepared using 1mΨ chemistry.
In vitro analysis in BJ fibroblasts
BJ fibroblasts from the American Type Culture Collection (ATCC; Manassas, VA) were cultured in EMEM (Eagle’s minimum essential medium) growth media with l-glutamine (ATCC) supplemented with 10% heat-inactivated fetal bovine serum (HI-FBS) (Life Technologies, Waltham, MA, USA). Cells were seeded in 96-well flat-bottom cell culture plates (Corning Inc., Corning, NY, USA) at 20,000 cells per well for 24 hours before transfection. Cells were transfected with mRNA (250 ng per well) using Lipofectamine 2000 (Thermo Fisher Scientific). Cell culture supernatants were harvested 48 hours after transfection for analyses.
hEPO and IFNβ analysis
hEPO and IFNβ protein levels in cell culture supernatants and mouse sera were measured with an Ella microfluidic ELISA (ProteinSimple, San Jose, CA, USA) as per the manufacturer’s recommendations.
THP1-Dual and A549-Dual cell lines
THP1-Dual, THP1-Dual MyD88−/−, A549-Dual, and A549-Dual RIG-I−/− cells were purchased from InvivoGen (San Diego, CA, USA). THP1-Dual MAVS−/− cells were generated by CRISPR-Cas9 of THP1-Dual cells using lentivirus containing single-guide RNA (GeneCopoeia, Rockville, MD, USA), targeting a specific exon on the MAVS gene and expressing mCherry and a puromycin resistance selection marker, along with Cas9 lentiviral particles expressing enhanced green fluorescent protein fluorophore and a neomycin selection marker (GeneCopoeia). Individual clones were isolated by cell sorting on a FACSAria Cell Sorting System with FACSDiva Software (BD Biosciences, San Jose, CA, USA). Knockout of the MAVS gene was confirmed by polymerase chain reaction analysis with forward and reverse primers specific to the exon flanking the MAVS gene target, using T7E1 nuclease and analyzed by agarose gel electrophoresis, and Western blotting with a MAVS antibody (Cell Signaling Technology, Danvers, MA, USA).
All THP1 cell lines were cultured in RPM1 1640 growth medium (Life Technologies) supplemented with 10% HI-FBS (Life Technologies) and selective antibiotics blasticidin (10 μg/ml; InvivoGen) and Zeocin (100 μg/ml; InvivoGen). A549 cell lines were cultured in Dulbecco’s modified Eagle’s medium (DMEM) growth media (Life Technologies) supplemented with 10% HI-FBS (Life Technologies) and selective antibiotics blasticidin (10 μg/ml; InvivoGen) and Zeocin (100 μg/ml; InvivoGen).
THP1-Dual and A549-Dual reporter assays
THP1-Dual, THP1-Dual MyD88−/−, and THP1-Dual MAVS−/− cells were seeded in 96-well V-bottom culture plates (Corning) at 75,000 cells per well in antibiotic-free RPMI 1640 growth media with 10% HI-FBS for 3 hours before transfection. A549-Dual and A549-Dual RIG-I−/− cells were seeded in 96-well flat-bottom plates (Corning) at 20,000 cells per well in antibiotic-free DMEM with 10% HI-FBS for 24 hours before transfection. Cells were transfected with mRNA (250 ng per well) or 3′ppp-hpRNA (2500 ng per well; InvivoGen), 5′ppp-dsRNA (2500 ng per well; InvivoGen), or 2′3′-cGAMP (2500 ng per well; InvivoGen) or treated with agonists R848 (2500 ng per well; InvivoGen) or TL8-506 (2500 ng per well; InvivoGen) for 24 hours. Luciferase in the supernatant was measured by the QUANTI-Luc assay (InvivoGen), and SEAP was measured by the QUANTI-Blue assay (InvivoGen) on a Synergy H1 plate reader (BioTek).
Isolation of human CD14+ monocytes
CD14+ monocytes were isolated from a 100-ml human Leukopak collected from healthy donors (STEMCELL Technologies, Cambridge, MA, USA). The peripheral blood sample was washed three times in an EasySep cell separation buffer (STEMCELL Technologies) and centrifuged at room temperature. The supernatant was aspirated, and cells were resuspended at 5 × 107 cells/ml in an EasySep buffer. An EasySep CD14+ selection kit (STEMCELL Technologies) was used to isolate monocytes. Isolation cocktail mix and magnetic particles from the kit were added to the cells and purified by magnetic separation. A series of incubation and centrifugation steps followed, resulting in an enriched cell suspension of CD14+ monocytes. The cells were counted using Vi-CELL XR (Beckman Coulter, Indianapolis, IN, USA), resuspended in CryoStor CS10 freezing media (STEMCELL Technologies), and aliquoted into 2-ml vials, followed by freezing overnight at −80°C and transferring to liquid nitrogen for long-term storage.
Culture of human MDM
Frozen aliquots of human monocytes were thawed and suspended in RPM1 1640 growth medium (Life Technologies) supplemented with 10% HI-FBS (Life Technologies) and human recombinant macrophage colony-stimulating factor (M-CSF; 20 ng/ml; Invitrogen). The monocytes were plated at 150,000 cells per well in 96-well flat-bottom culture plates (Corning) and allowed to differentiate into macrophages for 4 days. The growth medium was replaced with fresh RPMI 1640 with 10% HI-FBS without M-CSF for 24 hours before transfection. MDMs were transfected with mRNA (250 ng per well) or 3′ppp-hpRNA (2500 ng per well; InvivoGen), 5′ppp-dsRNA ((2500 ng per well; InvivoGen), or 2′3′-cGAMP ((2500 ng per well; InvivoGen) or treated with agonists R848 (2500 ng per well; InvivoGen) or TL8-506 (2500 ng per well; InvivoGen). After a 5-hour incubation, the supernatant was discarded and cells were lysed with branched DNA (bDNA) lysis buffer.
CXCL10 bDNA assay
All reagents used for the assay were purchased as a part of the QuantiGene Singleplex Assay Kit (Thermo Fisher Scientific). A working probe set was prepared with capture extender, label extender, blocking probe, and CXCL10 probe as per the manufacturer’s instructions and added to the capture plate. MDM cell lysate was added to the capture plate, and the mixture was incubated for 16 to 22 hours at 55°C for hybridization. The reaction was amplified the next day by the addition of a preamplifier, amplifier, and label probe, with intermittent incubation and wash steps. The signal was developed using a chemiluminescent substrate and read on a Synergy H1 luminometer (BioTek).
Animal studies
All animal procedures and experiments were approved by the Institutional Animal Care and Use Committee at Moderna. Female C57BL/6 mice at ~8 weeks of age were obtained from Charles River Laboratories. Mixed male and female B6129SF2/J control and MAVS−/− (B6;129-Mavstm1Zjc/J, JAX) mice at ~10 weeks of age were obtained from the Jackson laboratory. Mice were injected intravenously via the tail vein with formulated hEPO mRNA (0.5 mg/kg), poly(U) 19-mer (2 mg/kg) or poly(1mΨ) 19-mer (2 mg/kg), or control compounds at indicated doses and euthanized at 6 hours after injection. Blood was collected by cardiac puncture after euthanasia and processed for serum. Spleens were isolated to evaluate B cell activation and gene expression.
Mouse ProcartaPlex immunoassay
Cytokine levels in mouse sera were evaluated per manufacturer’s instructions using bead-based ProcartaPlex immunoassay kit (Thermo Fisher Scientific) consisting of either a custom-designed 10-plex mouse panel or a commercially available 36-plex mouse panel (Thermo Fisher Scientific). Analytes measured in the custom 10-plex included granulocyte CSF, IFNα, IFNγ, IL-12p70, IL-6, CXCL10, CCL2, CCL4, CCL7, and tumor necrosis factor–α.
B cell activation
Splenocyte single-cell suspensions were prepared by crushing half of a spleen through a 35-μm filter-capped polystyrene tube (Corning) with a 1-ml syringe plunger. Red blood cells were lysed using ammonium potassium chloride lysis buffer (Thermo Fisher Scientific) for 1 min at room temperature, and remaining cells were washed with PBS. The splenocytes were evaluated for viability by staining with LIVE/DEAD Fixable Aqua (Thermo Fisher Scientific), washed, and incubated for 30 min at 4°C with TruStain FcX (anti-mouse CD16/32) antibody (clone 93; BioLegend, San Diego, CA, USA), AF700-conjugated anti-CD19 (clone 1D3; Invitrogen), allophycocyanin (APC)–conjugated anti-CD3 (clone 145-2C11; BioLegend), phycoerythrin/cyanine-7 (PE/Cy7)–conjugated anti-CD86 (clone GL1; Thermo Fisher Scientific), or PE/CF594-conjugated anti-CD69 (clone H1.2F3; BD Biosciences) antibodies. After acquisition, cells were gated on live CD19+/CD3− cells, and percentages of CD86+/CD69+ cells were reported as activated B cells.
NanoString
Spleens were incubated in RNAlater (Thermo Fisher Scientific), followed by homogenization in a Geno/Grinder (SPEX SamplePrep, Metuchen, NJ, USA) using ZR BashingBead Lysis Tubes (Zymo Research, Irvine, CA, USA). RNA was isolated using automated Maxwell RSC Instrument with simplyRNA Tissue Kit as per the manufacturer’s recommendations (Promega Corp., Madison, WI, USA). RNA quality was assessed using TapeStation RNA ScreenTape (Agilent, Santa Clara, CA, USA) and quantitated using the Quant-iT RNA Broad Range Assay Kit (Invitrogen). RNA (200 ng) was assessed for gene expression using the nCounter Mouse Myeloid Innate Immunity v2 Panel (NanoString Technologies, Seattle, WA, USA) according to the nCounter XT CodeSet Gene Expression assay protocol. Resulting RCC files were examined with NanoString nSolver software for quality control analysis.
Statistical analysis
The sample size was determined to achieve a power of 80% to detect a twofold difference in hEPO serum levels between any chemistry or process groups, accepting a family-wise type I error rate of 0.05. Randomization and blinding were not used for analysis, as many of the assays required comparison to the negative control group. No outliers were removed, except for two samples from poly(I:C)-treated MAVS−/− mice (Fig. 5) because of a missed intravenous injection confirmed by baseline serum cytokine production. Statistical analysis for Figs. 3 and 4 was performed by nonparametric one-way analysis of variance (ANOVA) on ranks with Tukey’s multiple comparisons test. For data reported in Fig. 5, two-way ANOVA with Sidak’s multiple comparisons test was used to compare WT to MAVS−/−, and Tukey’s multiple comparisons test was used to compare treatment groups within each mouse strain. An adjusted P value of <0.05 was considered significant.
Supplementary Material
Acknowledgments
We thank A. Dricot, M. Kissai, J. Behringer, K. Winiarskyj, J. George, T. Sparrow, and C. Tunkey for technical support and G. Butora for scientific discussions. J. R. Gage (Gage Medical Writing LLC) provided medical writing support. Funding: This study was sponsored by Moderna Inc. Author contributions: J.N., E.W.S., S.M., A.E.R., G.B., N.K., S.V.S., E.J.M., W.J.I., S.H., M.G.S., and J.L.J. contributed to study design. J.N., E.W.S., S.M., A.E.R., N.K., S.V.S., and W.J.I. contributed to data acquisition. J.N., E.W.S., S.M., A.E.R., W.Z., N.K., S.V.S., and W.J.I. contributed data or analysis tools for the study. J.N., E.W.S., S.M., A.E.R., W.Z., N.K., S.V.S., and W.J.I. contributed to analyses of the data. All authors contributed to the writing of the manuscript, and all authors approved the final submitted draft. Competing interests: All authors are current or former employees and shareholders of Moderna Inc. J.N., A.E.R., E.J.M., W.J.I., and S.H. are inventors on a patent related to this work filed by Moderna Inc. (U.S. patent number 10,653,712, issued 19 May 2020). Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/26/eaaz6893/DC1
REFERENCES AND NOTES
- 1.Zhang C., Maruggi G., Shan H., Li J., Advances in mRNA vaccines for infectious diseases. Front. Immunol. 10, 594 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. RNA technologies expand tool kit for cancer immunotherapy. Cancer Discov. 9, OF5 (2019). [DOI] [PubMed] [Google Scholar]
- 3.Berraondo P., Martini P. G. V., Avila M. A., Fontanellas A., Messenger RNA therapy for rare genetic metabolic diseases. Gut 68, 1323–1330 (2019). [DOI] [PubMed] [Google Scholar]
- 4.Gan L.-M., Lagerström-Fermér M., Carlsson L. G., Arfvidsson C., Egnell A.-C., Rudvik A., Kjaer M., Collén A., Thompson J. D., Joyal J., Chialda L., Koernicke T., Fuhr R., Chien K. R., Fritsche-Danielson R., Intradermal delivery of modified mRNA encoding VEGF-A in patients with type 2 diabetes. Nat. Commun. 10, 871 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Foster J. B., Barrett D. M., Karikó K., The emerging role of in vitro-transcribed mRNA in adoptive T cell immunotherapy. Mol. Ther. 27, 747–756 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vallazza B., Petri S., Poleganov M. A., Eberle F., Kuhn A. N., Sahin U., Recombinant messenger RNA technology and its application in cancer immunotherapy, transcript replacement therapies, pluripotent stem cell induction, and beyond. Wiley Interdiscip. Rev. RNA 6, 471–499 (2015). [DOI] [PubMed] [Google Scholar]
- 7.Patel S., Ashwanikumar N., Robinson E., DuRoss A., Sun C., Murphy-Benenato K. E., Mihai C., Almarsson Ö., Sahay G., Boosting intracellular delivery of lipid nanoparticle-encapsulated mRNA. Nano Lett. 17, 5711–5718 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krupp G., Unusual promoter-independent transcription reactions with bacteriophage RNA polymerases. Nucleic Acids Res. 17, 3023–3036 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cazenave C., Uhlenbeck O. C., RNA template-directed RNA synthesis by T7 RNA polymerase. Proc. Natl. Acad. Sci. U.S.A. 91, 6972–6976 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.M. G. Stanton, K. E. Murphy-Benenato, Messenger RNA as a novel therapeutic approach, in RNA Therapeutics. Topics in Medicinal Chemistry, A. Garner, Ed. (Springer International Publishing, 2017), vol. 27, pp. 237–253. [Google Scholar]
- 11.Kawai T., Akira S., Innate immune recognition of viral infection. Nat. Immunol. 7, 131–137 (2006). [DOI] [PubMed] [Google Scholar]
- 12.G. Hartmann, Nucleic acid immunity, in Advances in Immunology, (Academic Press, 2017), vol. 133, pp 121–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roers A., Hiller B., Hornung V., Recognition of endogenous nucleic acids by the innate immune system. Immunity 44, 739–754 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Heil F., Hemmi H., Hochrein H., Ampenberger F., Kirschning C., Akira S., Lipford G., Wagner H., Bauer S., Species-specific recognition of single-stranded RNA via Toll-like receptor 7 and 8. Science 303, 1526–1529 (2004). [DOI] [PubMed] [Google Scholar]
- 15.Palchetti S., Starace D., De Cesaris P., Filippini A., Ziparo E., Riccioli A., Transfected poly(I:C) activates different dsRNA receptors, leading to apoptosis or immunoadjuvant response in androgen-independent prostate cancer cells. J. Biol. Chem. 290, 5470–5483 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gee P., Chua P. K., Gevorkyan J., Klumpp K., Najera I., Swinney D. C., Deval J., Essential role of the N-terminal domain in the regulation of RIG-I ATPase activity. J. Biol. Chem. 283, 9488–9496 (2008). [DOI] [PubMed] [Google Scholar]
- 17.Barral P. M., Sarkar D., Su Z.-z., Barber G. N., DeSalle R., Racaniello V. R., Fisher P. B., Functions of the cytoplasmic RNA sensors RIG-I and MDA-5: Key regulators of innate immunity. Pharmacol. Ther. 124, 219–234 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Anderson B. R., Muramatsu H., Nallagatla S. R., Bevilacqua P. C., Sansing L. H., Weissman D., Karikó K., Incorporation of pseudouridine into mRNA enhances translation by diminishing PKR activation. Nucleic Acids Res. 38, 5884–5892 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sonenberg N., Hinnebusch A. G., Regulation of translation initiation in eukaryotes: Mechanisms and biological targets. Cell 136, 731–745 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kormann M. S., Hasenpusch G., Aneja M. K., Nica G., Flemmer A. W., Herber-Jonat S., Huppmann M., Mays L. E., Illenyi M., Schams A., Griese M., Bittmann I., Handgretinger R., Hartl D., Rosenecker J., Rudolph C., Expression of therapeutic proteins after delivery of chemically modified mRNA in mice. Nat. Biotechnol. 29, 154–157 (2011). [DOI] [PubMed] [Google Scholar]
- 21.Karikó K., Muramatsu H., Ludwig J., Weissman D., Generating the optimal mRNA for therapy: HPLC purification eliminates immune activation and improves translation of nucleoside-modified, protein-encoding mRNA. Nucleic Acids Res. 39, e142 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mu X., Greenwald E., Ahmad S., Hur S., An origin of the immunogenicity of in vitro transcribed RNA. Nucleic Acids Res. 46, 5239–5249 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Foster J. B., Choudhari N., Perazzelli J., Storm J., Hofmann T. J., Jain P., Storm P. B., Pardi N., Weissman D., Waanders A. J., Grupp S. A., Karikó K., Resnick A. C., Barrett D. M., Purification of mRNA encoding chimeric antigen receptor is critical for generation of a robust T-cell response. Hum. Gene Ther. 30, 168–178 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hadas Y., Sultana N., Youssef E., Sharkar M. T. K., Kaur K., Chepurko E., Zangi L., Optimizing modified mRNA in vitro synthesis protocol for heart gene therapy. Mol. Ther. Methods Clin. Dev. 14, 300–305 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pardi N., Hogan M. J., Porter F. W., Weissman D., mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug Discov. 17, 261–279 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ishikawa H., Barber G. N., STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455, 674–678 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lazear H. M., Lancaster A., Wilkins C., Suthar M. S., Huang A., Vick S. C., Clepper L., Thackray L., Brassil M. M., Virgin H. W., Nikolich-Zugich J., Moses A. V., Gale M. Jr., Früh K., Diamond M. S., IRF-3, IRF-5, and IRF-7 coordinately regulate the type I IFN response in myeloid dendritic cells downstream of MAVS signaling. PLOS Pathog. 9, e1003118 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kawai T., Takahashi K., Sato S., Coban C., Kumar H., Kato H., Ishii K. J., Takeuchi O., Akira S., IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 6, 981–988 (2005). [DOI] [PubMed] [Google Scholar]
- 29.Linehan M. M., Dickey T. H., Molinari E. S., Fitzgerald M. E., Potapova O., Iwasaki A., Pyle A. M., A minimal RNA ligand for potent RIG-I activation in living mice. Sci. Adv. 4, e1701854 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hur S., Double-stranded RNA sensors and modulators in innate immunity. Ann. Rev. Immunol. 37, 349–375 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sadik C. D., Bachmann M., Pfeilschifter J., Mühl H., Activation of interferon regulatory factor-3 via Toll-like receptor 3 and immunomodulatory functions detected in A549 lung epithelial cells exposed to misplaced U1-snRNA. Nucleic Acids Res. 37, 5041–5056 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu M., Guo S., Hibbert J. M., Jain V., Singh N., Wilson N. O., Stiles J. K., CXCL10/IP-10 in infectious diseases pathogenesis and potential therapeutic implications. Cytokine Growth Factor Rev. 22, 121–130 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dauletbaev N., Cammisano M., Herscovitch K., Lands L. C., Stimulation of the RIG-I/MAVS pathway by polyinosinic:polycytidylic acid upregulates IFN-β in airway epithelial cells with minimal costimulation of IL-8. J. Immunol. 195, 2829–2841 (2015). [DOI] [PubMed] [Google Scholar]
- 34.Feldman R. A., Fuhr R., Smolenov I., Mick Ribeiro A., Panther L., Watson M., Senn J. J., Smith M., Almarsson Ö., Pujar H. S., Laska M. E., Thompson J., Zaks T., Ciaramella G., mRNA vaccines against H10N8 and H7N9 influenza viruses of pandemic potential are immunogenic and well tolerated in healthy adults in phase 1 randomized clinical trials. Vaccine 37, 3326–3334 (2019). [DOI] [PubMed] [Google Scholar]
- 35.Hewitt S. L., Bai A., Bailey D., Ichikawa K., Zielinski J., Karp R., Apte A., Arnold K., Zacharek S. J., Iliou M. S., Bhatt K., Garnaas M., Musenge F., Davis A., Khatwani N., Su S. V., MacLean G., Farlow S. J., Burke K., Frederick J. P., Durable anticancer immunity from intratumoral administration of IL-23, IL-36γ, and OX40L mRNAs. Sci. Transl. Med. 11, eaat9143 (2019). [DOI] [PubMed] [Google Scholar]
- 36.Diebold S. S., Kaisho T., Hemmi H., Akira S., Reis e Sousa C., Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science 303, 1529–1531 (2004). [DOI] [PubMed] [Google Scholar]
- 37.Tanji H., Ohto U., Shibata T., Taoka M., Yamauchi Y., Isobe T., Miyake K., Shimizu T., Toll-like receptor 8 senses degradation products of single-stranded RNA. Nat. Struct. Mol. Biol. 22, 109–115 (2015). [DOI] [PubMed] [Google Scholar]
- 38.Karikó K., Muramatsu H., Welsh F. A., Ludwig J., Kato H., Akira S., Weissman D., Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Mol. Ther. 16, 1833–1840 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Peisley A., Wu B., Yao H., Walz T., Hur S., RIG-I forms signaling-competent filaments in an ATP-dependent, ubiquitin-independent manner. Mol. Cell 51, 573–583 (2013). [DOI] [PubMed] [Google Scholar]
- 40.Tan R. S. T., Ho B., Leung B. P., Ding J. L., TLR cross-talk confers specificity to innate immunity. Int. Rev. Immunol. 33, 443–453 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Underhill D. M., Collaboration between the innate immune receptors dectin-1, TLRs, and Nods. Immunol. Rev. 219, 75–87 (2007). [DOI] [PubMed] [Google Scholar]
- 42.Liu Y., Goulet M.-L., Sze A., Hadj S. B., Belgnaoui S. M., Lababidi R. R., Zheng C., Fritz J. H., Olagnier D., Lin R., RIG-I-mediated STING upregulation restricts herpes simplex virus 1 infection. J. Virol. 90, 9406–9419 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Durbin A. F., Wang C., Marcotrigiano J., Gehrke L., RNAs containing modified nucleotides fail to trigger RIG-I conformational changes for innate immune signaling. mBio 7, e00833-16 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Devoldere J., Dewitte H., De Smedt S. C., Remaut K., Evading innate immunity in nonviral mRNA delivery: Don’t shoot the messenger. Drug Discov. Today 21, 11–25 (2016). [DOI] [PubMed] [Google Scholar]
- 45.Hoerr I., Obst R., Rammensee H. G., Jung G., In vivo application of RNA leads to induction of specific cytotoxic T lymphocytes and antibodies. Eur. J. Immunol. 30, 1–7 (2000). [DOI] [PubMed] [Google Scholar]
- 46.Thess A., Grund S., Mui B. L., Hope M. J., Baumhof P., Fotin-Mleczek M., Schlake T., Sequence-engineered mRNA without chemical nucleoside modifications enables an effective protein therapy in large animals. Mol. Ther. 23, 1456–1464 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Besin G., Milton J., Sabnis S., Howell R., Mihai C., Burke K., Benenato K. E., Stanton M. G., Smith P., Senn J. J., Hoge S., Accelerated blood clearance of lipid nanoparticles entails a biphasic humoral response of B-1 followed by B-2 lymphocytes to distinct antigenic moieties. Immunohorizons 3, 282–293 (2019). [DOI] [PubMed] [Google Scholar]
- 48.Bahl K., Senn J. J., Yuzhakov O., Bulychev A., Brito L. A., Hassett K. J., Laska M. E., Smith M., Almarsson Ö., Thompson J., Ribeiro A. M., Watson M., Zaks T., Ciaramella G., Preclinical and clinical demonstration of immunogenicity by mRNA vaccines against H10N8 and H7N9 influenza viruses. Mol. Ther. 25, 1316–1327 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lamontagne B., Larose S., Boulanger J., Elela S. A., The RNase III family: A conserved structure and expanding functions in eukaryotic dsRNA metabolism. Curr. Issues Mol. Biol. 3, 71–78 (2001). [PubMed] [Google Scholar]
- 50.Schönborn J., Oberstrass J., Breyel E., Tittgen J., Schumacher J., Lukacs N., Monoclonal antibodies to double-stranded RNA as probes of RNA structure in crude nucleic acid extracts. Nucleic Acids Res. 19, 2993–3000 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/26/eaaz6893/DC1