

Abstract
RNA-chromatin interactions represent an important aspect of transcriptional regulation of genes and transposable elements. However, analyses of chromatin-associated RNAs (caRNA) are often limited to one caRNA at a time. Here, we describe the iMARGI (in situ Mapping of RNA-Genome jnteractome) technique used to discover caRNAs and reveal their respective genomic interaction loci. iMARGI starts with in situ crosslinking and genome fragmentation, followed by converting each proximal RNA-DNA pair into an RNA-linker-DNA chimeric sequence. These chimeric sequences are subsequently converted into a sequencing library suitable for paired-end sequencing. A standardized bioinformatic software package called iMARGI-Docker is provided to decode the paired-end sequencing data into caRNA-DNA interactions (https://sysbio.ucsd.edu/imargi_pipeline ). Compared to its predecessor MARGI, in iMARGI the number of input cells is 3-5 million, which is reduced by 100-fold, experimental time is reduced, and clear checkpoints have been established. It takes a few hours a day and a total of 8 days to complete the construction of an iMARGI sequencing library and one day to carry out data processing with iMARGI-Docker.
Introduction
Chromatin-associated RNAs (caRNAs) are proposed to be a layer of the epigenome1. Interactions of caRNA with chromatin are essential for diverse molecular and cellular functions, including X chromosome silencing2, anchoring nucleolus-chromosome interactions3, homology-directed repair of telomeres4, and RNA-mediated epigenetic inheritance5. On specific genomic loci, caRNA-DNA interactions also contribute to de novo DNA methylation6, promotion7, 8 and suppression of transcription9, as well as demarcation of active and silent chromatin domains10.
The genomic interaction loci of a specific caRNA can be determined by a number of technologies, including Capture Hybridization Analysis of RNA Targets (CHART)11, Chromatin Isolation by RNA Purification (ChIRP)12 and RNA Antisense Purification (RAP)13. However, these one-RNA-versus-the-genome technologies are not applicable to the discovery of new caRNAs. This challenge was addressed by a recent cohort of assays that can reveal caRNAs-genomic interactions in an all-RNA-versus-the-genome fashion. These genome-wide RNA-chromatin interaction assays include Mapping RNA-Genome Interactions (MARGI)14 and its improved version called in situ MARGI (iMARGI)15, Chromatin-Associated RNA Sequencing (ChAR-seq)16, and Mapping Global RNA Interactions With DNA by Deep Sequencing (GRID-seq)17. Both GRID-seq and ChAR-seq revealed a range of chromatin-bound RNAs including nascent transcripts, chromosome-specific dosage compensation non-coding RNAs (ncRNAs), and trans-associated RNAs16, 17 GRID-seq also revealed extensive interactions between mRNAs and enhancers 17
MARGI and iMARGI revealed thousands of caRNAs including both coding and noncoding RNAs14, 15, 18 These caRNAs are not only associated with the genomic sequences from which they are transcribed (to form proximal interactions), but can also attach to distal genomic sequences (to form distal interactions) on the same chromosomes or to other chromosomes (to form inter-chromosomal interactions). Surprisingly, transcription start sites (TSS) have been identified to be preferred genomic loci targeted by non-coding caRNAs through distal and inter-chromosomal interactions14. A subsequent effort revealed that many non-coding caRNAs are tethered to chromatin by RNA polymerase II (Pol-II)-associated U1 snRNP, which may offer a mechanistic explanation to the accumulation of caRNA on TSS19. The amount of accumulated caRNAs at each TSS correlated with the expression level of the gene whose transcription starts at this TSS14, suggesting a regulatory role of the TSS-accumulated caRNAs. Consistent with this notion, suppression of TSS-accumulated caRNA leads to lower expression of the gene transcribed from the TSS7. Another unexpected consequence of the accumulation of distal and inter-chromosomal caRNAs at TSS is that the TSS-associated caRNAs become susceptible to be trans-spliced with their nearby nascent transcripts and, thus, create fusion RNAs15. These data highlight the power of iMARGI to generate novel hypotheses related to functions of caRNA. Comparisons of MARGI and iMARGI data in terms of genome-wide read pair distribution and numbers of discovered caRNAs were given in Reference15.
Development of the protocol
The central idea of the iMARGI technology is to convert each RNA-DNA interaction preserved in the crosslinked chromosomes into a unique DNA sequence and leverage paired-end sequencing as the high-throughput readout for mapping RNA-DNA interactions (Fig. 1a). RNA and DNA are first fragmented and then a specifically designed linker sequence is introduced to ligate with RNA on one end and with DNA on the other end, thus forming RNA-linker-DNA chimeric sequences. The RNA-linker-DNA chimeric sequence is subsequently converted to double-stranded DNA suitable for paired-end sequencing. Each end of a read pair is separately mapped to the genome, and pairing information of the two read ends is used to infer the original RNA-DNA interaction.
Figure 1.
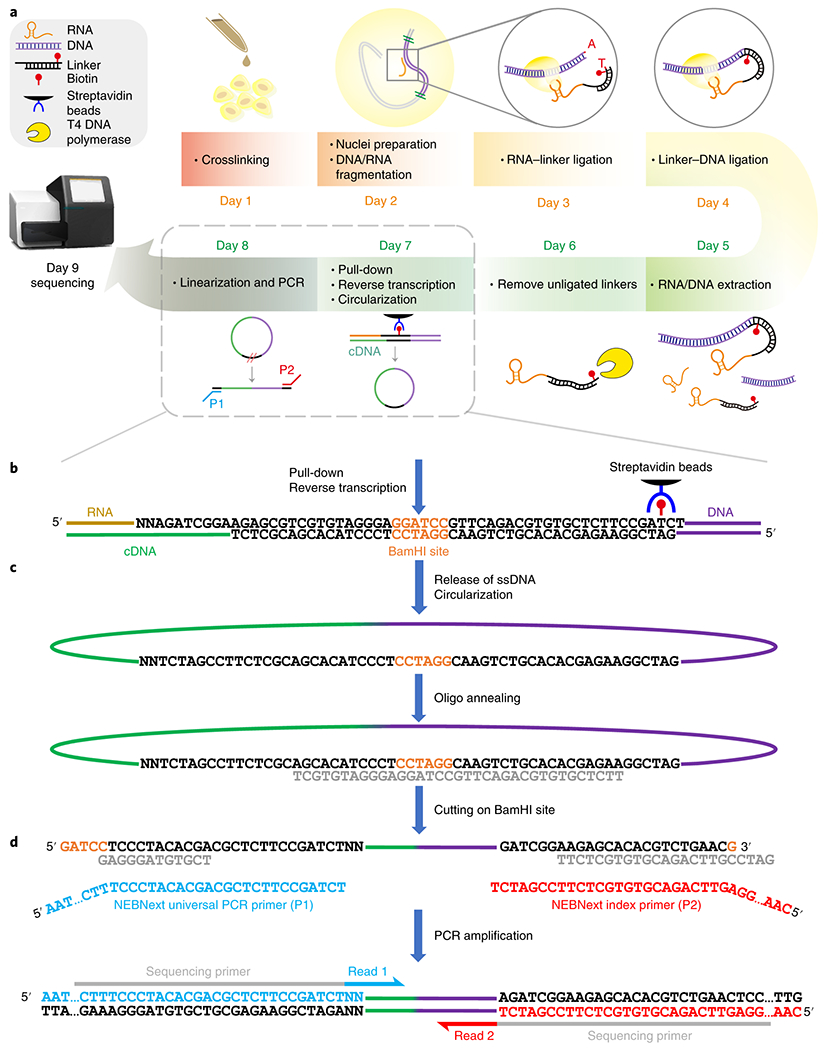
iMARGI protocol and linker design, (a) A schematic overview of the iMARGI protocol. Experimental steps are carried out in nuclei (day 1 to day 4) (Step 1 to 54) and subsequently in solution (day 5 to day 8) (Step 55 to 113). [AU: Editor has changed text. Please check if OK. I think this can be removed as the contents of the box are self-explanatory.] (b-d) An expanded view of the experimental steps in day 7 and day 8. The linker sequence is composed of two strands of DNA with different lengths (black characters). The double-stranded region of the linker sequence contains a BamHI restriction site (orange). The top strand of the linker sequence is biotin-labeled (red pin). The double stranded linker is ligated to genomic DNA (purple lines). cDNA (green line) is synthesized in the 3’ direction of the bottom strand, (c) The bottom strand is released as single-stranded DNA (ssDNA), circularized, and hybridized with an annealing oligo (grey characters). A cut on the BamHI site linearizes the circular DNA, and puts the two halves of the bottom strand of the linker at the two ends of the ssDNA. (d) The half linker at the 5’ end is identical to a fraction of the NEBNext universal PCR primer (blue characters), which contains Illumina’s sequencing primer (grey bar) for Read 1 (blue arrow). The other half linker at the 3’ end is complementary to a fraction of the NEBNext index primer (red characters), which contains Illumina’s sequencing primer (grey bar) for Read 2 (red arrow).
Critical to this experimental design is keeping track of which side of the linker corresponds to RNA and which side corresponds to DNA. To record this orientation information, the RNA has to be specifically ligated to one designated side of the linker, and the fragmented DNA has to be specifically ligated to the other side. These orientation-specific ligations are ensured by the design of linker sequence (Fig. 1b).
In order to extract the orientation information, the linker sequence itself has to be sequenced. However, because the linker resides in the middle of the desired ligation products in the form of RNA-linker-DNA, typical next-generation sequencing is not always able to sequence through the linker region, resulting in uninformative reads which do not have the necessary orientation information. To address this challenge, iMARGI incorporates a strategy whereby the reverse transcribed strand (cDNA) of the ligation product (cDNA-linker-DNA) is circularized and then re-linearized by cutting at the BamHI site within the linker sequence (Fig. 1c–d). As a result, the linker is cut into two halves, which are positioned at the two ends of the chimeric sequence, forming a left.half.Linker-cDNA-DNA-right.half.Linker (Fig. 1d). The two halves of the linker (left.half.Linker and right.half.Linker) are designed to be parts of the two NEBNext PCR primers for Illumina. Thus, the re-linearized sequences can be directly amplified with the same PCR primers (blue and red sequences, Fig. 1d) during the preparation of a typical sequencing library.
Other important considerations of the iMARGI protocol include performing the ligation steps in intact crosslinked nuclei, maximizing ligation efficiencies, minimizing side products, and removal of uninformative sequences including un-ligated or partially ligated products. Compared to ligation in vitro, ligation in intact crosslinked nuclei avoids non-specific interactions between RNAs and streptavidin beads. These are achieved by preparation and permeabilization of crosslinked nuclei, a combination of RNA and DNA end-modifications, and specifically designed and modified ends of the linker sequence.
To remove incomplete ligation products in the form of RNA-linker, Exol exonuclease and T4 DNA polymerase with exonuclease activity are applied to cleave the biotinylated nucleotide in the linker sequence. The cleaved linker sequences cannot be pulled down with streptavidin and are thus discarded. Finally, to minimize the sequence bias of RNA-linker ligation, the 5’ end of the top strand of the linker sequence is composed of two random nucleotides (“NN” in Fig. 1b). Different RNA sequences tend to preferentially ligate to the linkers in the randomized pool with which they have the potential to form particular structures20–22.
To minimize variations in data processing, we developed a complete data processing pipeline with full documentation (https://sysbio.ucsd.edu/imargi_pipeline) (Fig. 2). This pipeline called iMARGI-Docker helps to improve analysis reproducibility by standardizing the data processing steps. Based on the Docker technology, iMARGI-Docker can be executed on all mainstream Linux distributions without system-specific configuration, thus simplifying the computer technology requirements of the data analysis.
Figure 2.
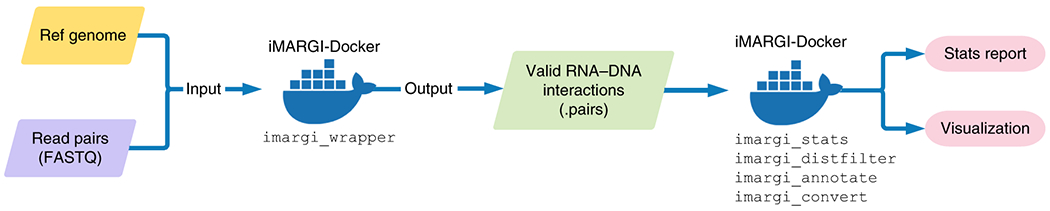
Computational workflow of iMARGI data analysis.
Applications of the protocol
iMARGI does not require genome-engineering or introduction of any exogenous molecules to cells before the cells are fixed. Thus, iMARGI is applicable to both cell lines and primary cells. Several millions of cells are sufficient for carrying out iMARGI analysis. Although we recommend starting with 5 million cells, our tests with 2-3 million cells generally produced high-quality data as well. We anticipate the minimum required cell number to vary among cell types. Please refer to Step 113 for determining the minimum amount of input cells.
Comparison to other methods
The three genome-wide RNA-chromatin interaction assays include iMARGI (and its predecessor MARGI14), GRID-seq17, and ChAR-seq16 iMARGI and its predecessor MARGI share the linker design and most of the experimental steps. However, in iMARGI the ligation steps are carried out in the nuclei instead of in solution. As a result, iMARGI requires fewer cells for the experiment while generating a larger number of informative sequence read pairs (see “Comparison of iMARGI and MARGI section”15). We will devote the rest of this section to comparison of iMARGI, GRID-seq, and CHAR-seq.
iMARGI has been tested on mammalian cells. MARGI and GRID-seq have been tested on fruit-fly and mammalian cells, whereas ChAR-seq has only been tested with fruit-fly cells16 (Table 1). iMARGI and GRID-seq require several millions of cells, whereas ChAR-seq require more than 100 million cells (Table 1). Considering these differences, hereafter we will emphasize the comparison between iMARGI and GRID-seq.
Table 1.
| iMARGI | GRID-seq | ChAR-seq | Pros and Cons | |
|---|---|---|---|---|
| Applications | ||||
| Tested species | Human | Human, mouse, fruit fly | Fruit fly | iMARGI and GRID-seq are tested in human cells. |
| Input cell number | 2-5 million | ~ 2 million | 100 million | |
| Data format | ||||
| Sequencing type | Paired-end | Single-end | Single-end | |
| Usable sequence length for mapping RNA | Up to 100 bp | 18–23 bp | 65 bp on average | Short sequence length can result in ambiguity in mapping. |
| Usable sequence length for mapping DNA | Up to 100 bp | 18–23 bp | 65 bp on average | |
| Experimental design | ||||
| Ligation | In nucleus | In nucleus | In nucleus | |
| Reverse transcription | In solution, with RNase inhibitor | In nucleus | In nucleus | Reverse transcriptase is less likely to be blocked by RNA binding proteins or RNA secondary structure in solution (iMARGI). |
| Second strand synthesis | n/a | Yes | Yes | iMARGI’s ssDNA product can be directly PCR amplified to produce sequencing library. |
| Removal of incomplete ligation products | Exonuclease treatment combined with biotin selection | Gel-based size selection | No relevant experimental steps | iMARGI’s enzyme-based method is easier to carry out than gel-based selection, which may cause loss of material. |
| Ligation of adaptors | n/a | Y-shaped adaptors required | NEBNext hairpin adaptors required. | iMARGI does not require adaptor ligation steps. |
A main advantage of iMARGI is the maximized usable sequence length of the reads. Because the linker does not appear in the final read sequences, the entire sequence of each read pair can be used for sequence mapping, thus minimizing ambiguity in sequence alignment. In contrast, in GRID-seq the linker has to be read through to determine which portion of the read sequence corresponds to RNA and which corresponds to DNA. In order to ensure that the linker sequence is included within the read sequence, GRID-seq deploys an experimental strategy that trims the RNA-side and the DNA-side of the ligation product into 18-23 bp17. Thus, only 18-23 bp of the sequence can be used for mapping. Sequences in this size range cannot always be uniquely mapped to the human genome. Even without allowing for any mismatch, more than 25% of the 20-bp sequences have ambiguous mapping to the human genome (See Figure 1 in 23).
The second advantage of iMARGI is the minimized potential bias. iMARGI has a unique design where the linker contains two random bases on the 5’ end of the top strand (Fig. 1b–c), which minimizes the sequence bias of the RNA ligase15. Linker sequences used in GRID-seq and ChAR-seq do not contain these random bases (Table 1). Furthermore, in iMARGI the first strand cDNA synthesis is carried out in solution, whereas in GRID-seq and ChAR-seq this process is carried out in the nuclei. It is possible that performing these enzymatic reactions in situ may result in partial inhibition by RNA structure or RNA binding proteins (Table 1).
In addition to the two above-noted advantages, the iMARGI experimental procedure is simplified in comparison to GRID-seq or ChAR-seq. While iMARGI’s linker (composed of two DNA strands) can be easily prepared by annealing, GRID-seq’s RNA-DNA chimeric linker requires special synthesis (Table 2). Furthermore, in iMARGI an enzyme-based removal of un-ligated products is deployed, whereas GRID-seq relies on gel-based size selection, which can be time-consuming and can cause loss of materials. Another notable advantage is that iMARGI does not require 2nd strand DNA synthesis, which is required by GRID-seq and ChAR-seq. iMARGI converts the first synthesized DNA strand into a suitable form for PCR-based library construction, thus simplifying the experimental process. Finally, iMARGI does not require ligation of sequencing adaptors, which is required by GRID-seq and ChAR-seq. After splitting the linker, the two halves of iMARGI’s linker sequences are compatible with library construction PCR primers. Using standard library preparation primers, iMARGI’s sequence products can be directly amplified into a sequencing library.
Table 2.
Comparison of the linker sequences in iMARGI15, GRID-seq17, and ChAR-seq16 ssDNA: single strand DNA. dsDNA: double strand DNA.
| iMARGI | GRID-seq | ChAR-seq | Comment | |
|---|---|---|---|---|
| Biotinylation | Biotinylated | Biotinylated | Biotinylated | Allows for selection of ligation products. |
| Strands | A longer ssDNA (top strand) annealed with a shorter ssDNA (top strand) | ssDNA annealed with an RNA-DNA chimeric sequence | dsDNA with a 5’-overhang and a 3’-three carbon spacer | GRID-seq’s linker sequence requires special synthesis |
| 5’ adenylation | 5’ adenylated | 5’ adenylated | 5’ adenylated | For ligation to 3’-end of RNA |
| 5’ random bases | Contains 2 random bases on the 5’ of linker top strand | None | None | 5’ random bases (NN) in the linker top strand can minimize biases of RNA ligase. |
| Restriction sites | BamHI | 2 × Mmel | Dpn II | Dpn II allows for carrying out Hi-C experiments in parallel. |
| Complementarity to adaptors | Yes | No | No | Without sequencing the linker sequence, iMARGI is able to resolve the orientation of the RNA-linker-DNA. There is no need to ligate library construction adaptors in iMARGI. |
iMARGI/MARGI, GRID-seq and ChAR-seq used different methods to assess false positive rates. MARGI and GRID-seq mixed fruit-fly and mammalian cells as input and used the ligated sequences of the two species as false positives14,17. ChAR-seq used spike-in RNAs to estimate false positives16.
For statistical analyses, GRID-seq provided a data normalization method to correct for background noises and identify specific RNA-DNA interactions17. In comparison, iMARGI/MARGI and ChAR-seq did not provide a one-size-fits-all statistical method for identifying all RNA-DNA interactions. Instead, the authors opted for the statistical methods that best fit each biological question, including calling chromatin enriched RNAs16; assessing enrichments of RNA-DNA interactions at TAD boundaries16, TSS14, and the chromosomal regions near nuclear speckles18; and comparing global RNA-DNA interactions with genome-wide distributions of histone modifications14, 16 transcription factor binding intensities18, and fusion RNAs15.
Limitations of the protocol
The major limitation of the iMARGI technology is the requirement of millions of input cells. Another limitation is the relatively long experimental process, which requires 3 to 6 hours per day and a total of 8 days to complete the library construction. Furthermore, due to scarcity of independent experiments, it is difficult to globally evaluate the sensitivity and specificity of interactions detected by iMARGI. In addition, iMARGI does not distinguish ‘active’ interactions such as the RNAs bound to a chromatin binding protein complex24 and ‘passive’ proximity such as nascent RNA-chromatin interactions introduced by DNA looping15 [AU: Editor has changed text. Please check if it is OK.] Finally, we have not provided a unified statistical method for identifying all the chromatin-associated RNAs. We suspect that a unified statistical method cannot simultaneously optimize the sensitivity and the specificity, in light of the diverse modes of RNA-DNA interactions. It falls onto the users to choose the suitable statistical methods based on their biological questions.
Experimental design
iMARGI starts with crosslinking cells and collecting nuclei, followed by fragmenting RNA and DNA in nuclei (Fig. 1a). A specifically designed linker sequence is introduced to the permeabilised nuclei to first ligate it with the fragmented RNA and subsequently with spatially proximal DNA. After these ligation steps, nuclei are lysed and crosslinks are reversed. Nucleic acids are purified and subsequently treated with exonucleases to remove any linker sequences that are not successfully ligated with both RNA and DNA. The desired ligation products in the form of RNA-linker-DNA are pulled down with streptavidin beads. The RNA part of the pulled down sequence is reverse transcribed into cDNA, resulting in a complementary strand of (5’)DNA-linker-cDNA(3’) (Fig. 1b). Single-stranded DNA-linker-cDNA is released from streptavidin beads, circularized and re-linearized, producing single-stranded DNA in the form of left.half.Linker-cDNA-DNA-right.ha If .Linker (Fig. 1c–d). The two halves of the linker (left.half.Linker and right.half.Linker) are templates for PCR amplification into the final sequencing library (Fig. 1d).
Collecting nuclei
Approximately 5 million cells are fixed in 1% (wt/vol) formaldehyde. We have successfully performed iMARGI with human embryonic kidney (HEK293T), human foreskin fibroblast (HFF), and HUVEC cells. To isolate cell nuclei, cells are incubated with a lysis buffer containing mild non-ionic detergent, which selectively disrupts the plasma membrane while keeping nuclear membrane intact25–27. Nuclei are subsequently obtained by centrifugation. White nuclear pellet can be observed by eye at the bottom of the tube after centrifugation. As a checkpoint for nuclear integrity, the nuclei are stained with DAPI and imaged. Confined DAPI signals in the nuclei are an indication of nuclear integrity (Fig. 3a–c).
Figure 3.
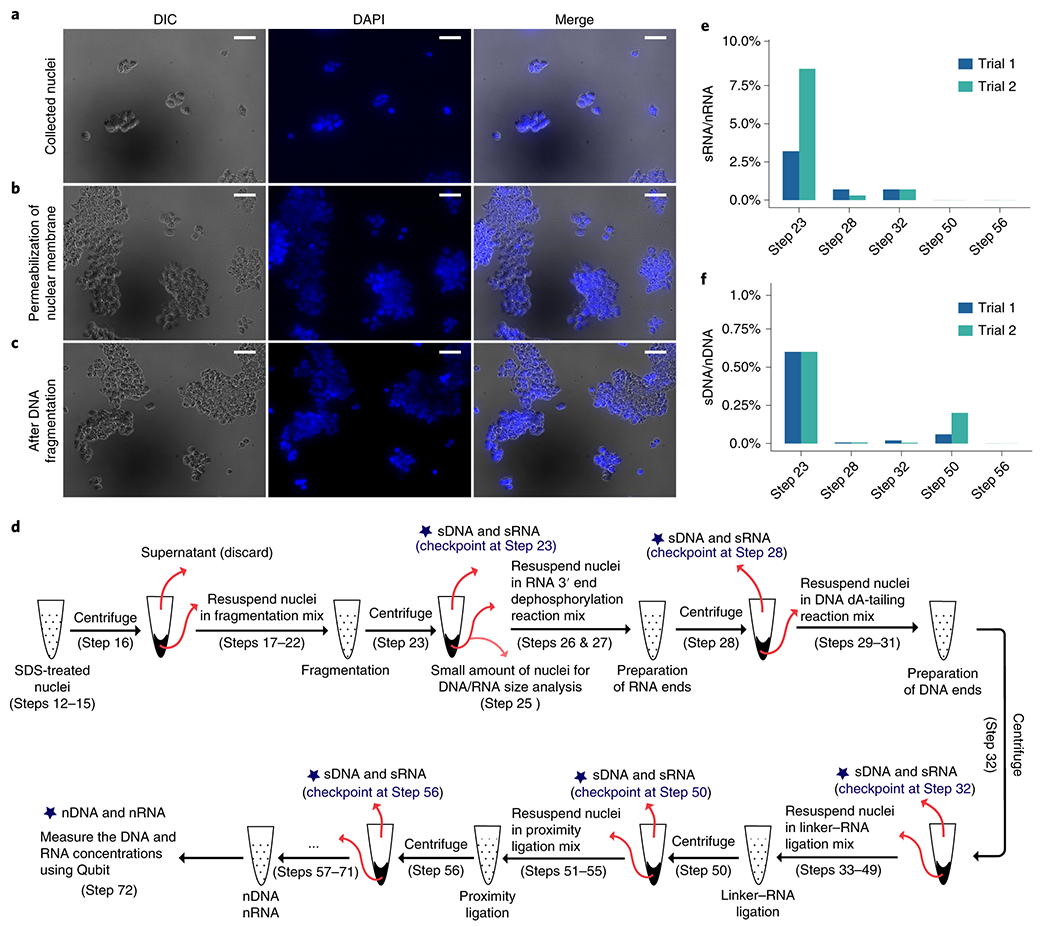
Checkpoints for nuclear integrity, (a-c) Image-based checkpoints (Box 1). DAPI staining (blue) and DIC images (grey) were taken after collection of nuclei (Step 9) (a), permeabilization of nuclear membrane (Step 17) (b), and DNA fragmentation (Step 23) (c). Confined DAPI staining in DIC defined nuclei is an indication of non-leaking nuclei. Scale bar: 50 μm. (d-f) Quantitative assessment of nuclear integrity (Box 2). (d) A schematic view of the experimental steps where supernatant RNA (sRNA) and supernatant DNA (sDNA) are quantified. Nuclear RNA (nRNA) and nuclear DNA (nDNA) are quantified at Step 72. (e) Ratios (y axis) between the amount of sRNA at each step (x axis) to the amount of nRNA measured at Step 72. (f) Ratios (y axis) between the amount of sDNA at each step (x axis) to the amount of nDNA measured at Step 72. Trials 1 and 2 are two separate experiments that start with approximately 5 million and 3 million HEK293T cells, respectively. The HEK293T cell line used in panels in this figure has been authenticated and tested to ensure its identity and that it is free from mycoplasma contamination.
Fragmentation of DNA and RNA in nuclei
The harvested nuclei are permeabilized by SDS treatment and subsequently incubated with RNase I and the restriction enzyme Alul for RNA and DNA fragmentation, respectively.
The recommended SDS concentration and treatment time have been optimized to balance the prevention of leakage of nuclear contents and sufficient enzyme penetration to the nuclei. The degree of nuclear leakage can also be quantified by the ratio of supernatant RNA (sRNA) to nuclear-retained RNA (nRNA) and the ratio of supernatant DNA (sDNA) to nuclear-retained DNA (nDNA) (checkpoint at Step 23) (Fig. 3d–f). Sufficient enzyme penetration is reflected by obtaining the desired lengths of the fragmented DNA and RNA (checkpoint in Step 25). The desired lengths of fragmented DNA range from 200 to 1,500 bp (Fig. 4a). The desired lengths of fragmented RNA range from 180 to 500 nt (Fig. 4b).
Figure 4.
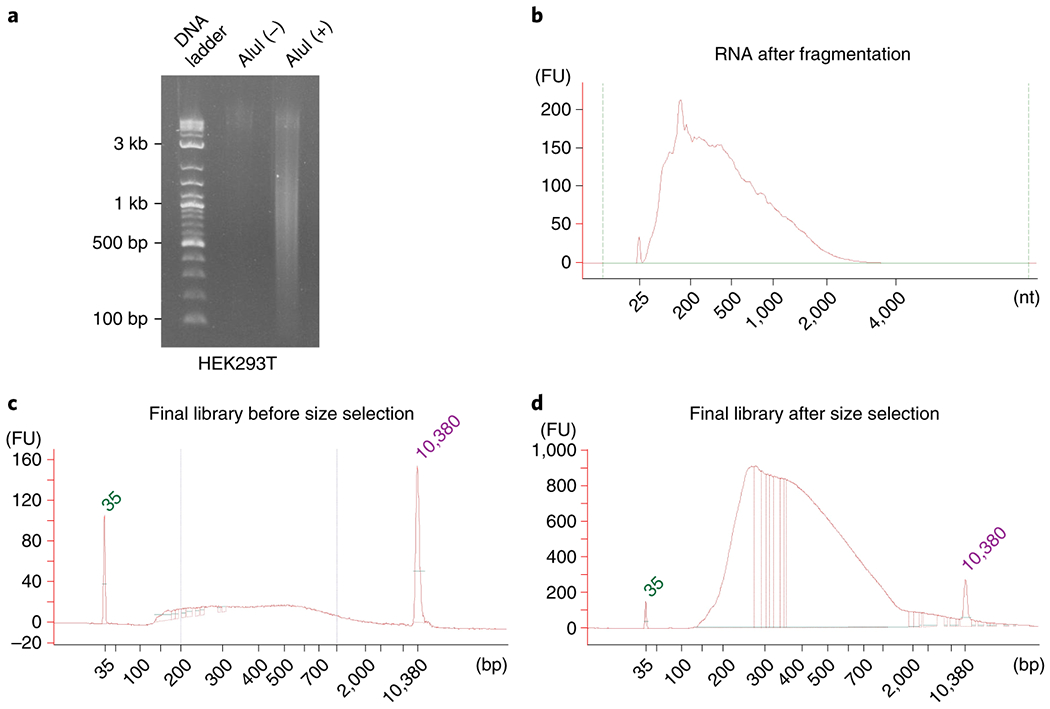
Size distributions of intermediate and final products from HEK293T cells. (a) Size distributions of un-fragmented (Alul (−)) and fragmented DNA (Alul (+)) resolved on a precast 1 % (wt/vol) agarose E-gel. 2-log DNA ladder was used in as ladder, (b) Size distribution of fragmented RNA resolved on an Agilent Bioanalyzer, (c-d) Size distributions of iMARGI sequencing library before (c) and after (d) size selection. The peaks at 35 and 10,380 bp are size markers which are not included in the sequencing library. The two vertical bars indicate 200 bp and 1,000 bp, respectively.
RNA-linker and linker-DNA ligation in nuclei
The top strand and the bottom strand of the linker are annealed to create suitable linkers for ligations. To prepare the fragmented RNA for ligation, T4 polynucleotide kinase is applied to convert any 3’ phosphate groups on the fragmented RNA into 3’ hydroxyl (-OH) groups. RNA with 3’ -OH is subsequently ligated with the top strand of the linker which has a adenylated 5’ end using T4 RNA ligase 2-truncated KQ. Unligated linkers are removed by extensive washes. To prepare the fragmented DNA for ligation, an “A” base is added to the DNA strand with an exposed 3’ end using Klenow fragment which lacks 3’ to 5’exonuclease activity. The A-tailed DNA is subsequently ligated with the linker through sticky end ligation.
Collecting ligation products
After the ligation steps, nuclei are collected, washed, and incubated with extraction buffer to extract nucleic acids and reverse crosslinks. The extracted nucleic acids include the desired ligation products in the form of RNA-linker-DNA, as well as incomplete products in the forms of RNA, DNA, and RNA-linker. The biotin on the linkers of the RNA-linker products is cleaved by exonucleases. After the cleavage, the desired ligation products are harvested by streptavidin beads, followed by stringent wash steps to remove any nucleic acids attached due to non-specific binding.
Constructing the sequencing library
Reverse transcription is performed to produce cDNA, resulting in a complementary strand of cDNA-linker-DNA sequence (Fig. 1b). This complementary strand is released from the streptavidin beads by using denaturing buffer which contains 0.1 M NaOH and 0.1 mM EDTA. The released strand is circularized using CircLigase™ (Fig. 1c). This single-stranded circular DNA is annealed with a hybridization oligo to create a double-stranded region containing a BamHI restriction site. A subsequent BamH1 cut linearizes the DNA, which splits the linker into two halves. The two halves of the linker at the two ends of the re-linearized DNA are compatible with sequencing primers (Fig. 1d). The linearized DNA is amplified with NEBNext PCR primers for Illumina, size-selected (Fig. 4c–d), and subjected to paired-end sequencing.
Checkpoints
We designed checkpoints to gauge the success of several key experimental steps. These checkpoints examine the success of annealing the two strands of the linkers (Step 46), the integrity of the extracted nuclei (Step 9 and Step 23, Fig. 3), the efficiency of DNA and RNA fragmentation (Step 25) (Fig. 4a–b), and the size distribution of the sequencing library (Steps 105 and 113) (Fig. 4c–d). Figure 5 shows results from testing different conditions to optimize DNA fragmentation. Empirical thresholds for quantitative evaluation of broken nuclei are given in Table 3. [AU: Please check if text is OK or edit if needed. Figure 5 needs to be cited before figure 6 but after figure 4 and Table 3 needs to be cited in main text.] Moreover, we provide checkpoints to ensure sequencing quality (Step 115) and appropriate data processing (Steps 116, 117, 119, and 123).
Figure 5.
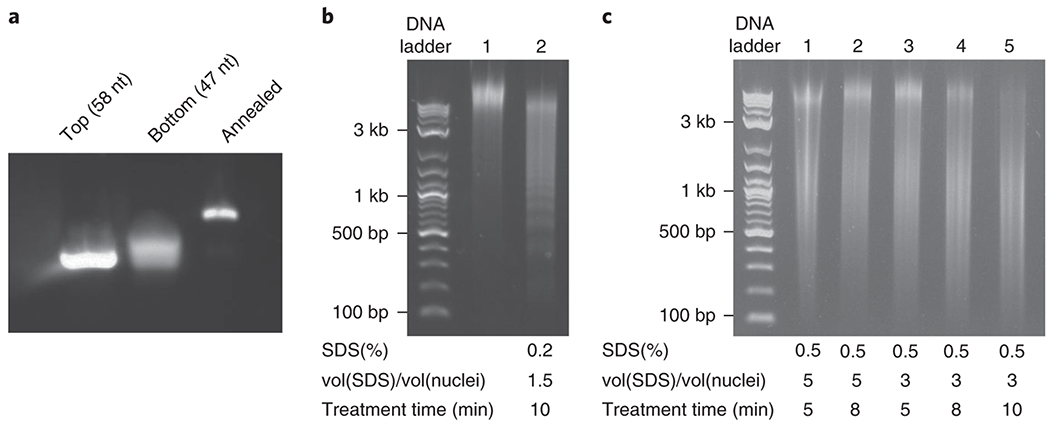
Checkpoints and troubleshooting, (a) Checkpoint for annealing the two strands of the linker sequence. Sizes of the top strand (58 nt), bottom strand (47 nt) and the annealed product are resolved on a precast 2% agarose E-gel. (b-c) Troubleshooting for insufficient DNA fragmentation. Size distributions of un-fragmented (control, Lane 1 in b) and fragmented DNA (Lane 2 in b, Lanes 1 – 5 in c) under different parameters of nuclear permeabilization. 2-log DNA ladder was used in the ladder lane. The corresponding parameters including SDS concentration (SDS%), volume ratios [v(SDS)/v(nuclei)], and treatment time are marked below each lane. Lane 5 of Panel c corresponds to the default parameters of the iMARGI protocol. The parameters resulting in optimal DNA fragmentation without disrupting nuclear integrity should be chosen.
Table 3.
Empirical thresholds for quantitative evaluation of broken nuclei. These thresholds should be applied to the ratios of the measured amounts of sRNA and nRNA (sRNA/nDNA column) and the amounts of sDNA and nDNA (sDNA/nDNA column). The sRNA and sDNA should be obtained from the supernatant in Steps 23, 28, 32, 50, and 56 (rows). nRNA and nDNA should be obtained from Step 72. Any obtained ratio at any of these steps beyond the listed threshold of that step is a sign of having a non-negligible fraction of broken nuclei.
| Step # for retrieving sRNA and sDNA | sRNA/nRNA | sDNA/nDNA |
|---|---|---|
| Step 23 | 10/100 | 1/100 |
| Step 28 | 1/100 | 1/100 |
| Step 32 | 1/100 | 1/100 |
| Step 50 | 1/100 | 1/100 |
| Step 56 | 1/100 | 1/100 |
Figure 6.
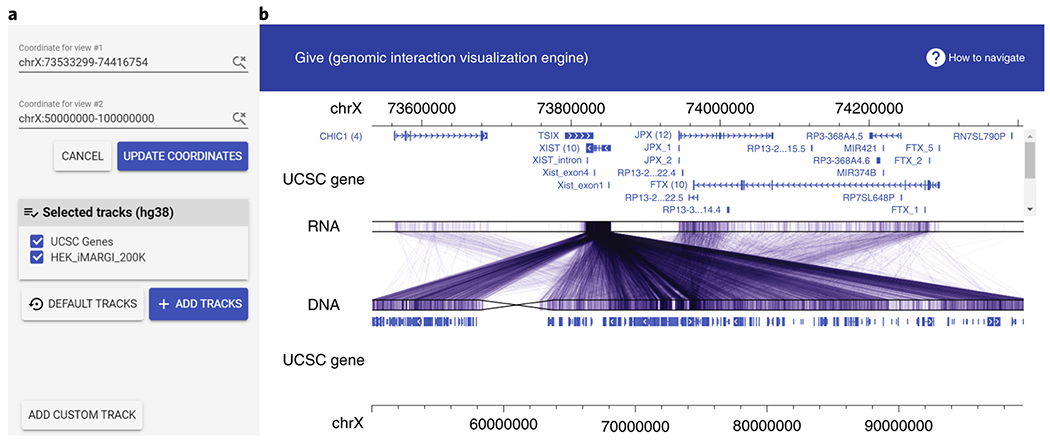
Visualization of iMARGI data in GIVE, (a) User defined genome coordinates and data tracks, (b) A two-layer presentation of the genome. Every line corresponds to a mapped read pair, where the RNA end is mapped to the top layer (RNA lane) and the DNA end is mapped to the bottom layer (DNA lane). Users can adjust the coordinates of the two layers separately using the input box in the left panel. The current setting shows human Chromosome X: 73,533,299 – 74,416754 in the top layer, covering the XIST gene and its genomic neighborhood, and Chromosome X: 50,000,000 – 100,000,000 in the bottom layer, covering 50 Mb of the X chromosome.
Data processing
We have automated the data processing steps with iMARGI-Docker. The key steps include quality control (QC) of the sequencing data (Step 115), checking the hardware and software requirements (Step 116), QC of the mapped data (Step 123), and conversion of the mapped data to other data formats that are compatible with other popular data analysis and visualization tools (Steps 127-129).
Materials
Biological materials
-
Cell lines of interest. We have used a variety of cell types successfully, such as human embryonic kidney (HEK) cells (ATTC, ATCC® CRL-1573™), human foreskin fibroblasts (HFF), human umbilical vein endothelial cells (HUVEC) and human embryonic stem cells (hESC) H1. All data shown in this paper were generated from HEK293T cells (ATTC, ATCC® CRL-1573™).
! CAUTION The cell lines used in your research should be regularly checked to ensure they are authentic and are not infected with mycoplasma.
Reagents
Penicillin-Streptomycin (Thermo Fisher Scientific, cat.no. 15140122)
Fetal Bovine Serum (Thermo Fisher Scientific, cat.no. A2720803)
DMEM (Genesee Scientific, cat.no. 25-500)
mTeSR™1 Basal Medium (400 mL; STEMCELL Technologies, cat.no. 85851)
mTeSR™1 5× Supplement (100 mL; STEMCELL Technologies, cat.no. 85852)
ReLeSR™ (STEMCELL Technologies, cat.no. 05872/05873)
Corning® Matrigel® hESC-qualified Matrix (Corning, cat.no. 354277)
DMEM/F-12 (STEMCELL Technologies, cat.no. 36254)
16% (wt/vol) Formaldehyde (Thermo Fisher Scientific, cat.no. 28908) ! CAUTION Formaldehyde is toxic upon ingestion, inhalation, or contact with skin. Handle formaldehyde in a chemical fume hood with caution. Protective gloves, lab coat and eye protection should be worn. Formaldehyde waste should be discarded in an appropriate container according to local regulation.
UltraPure Distilled Water (DNase, RNase and Protease-Free; Invitrogen, cat.no. 10977-015)
Glycine (Fisher Scientific, cat.no. BP381-1)
Protease Inhibitor Tablet (Roche, cat.no. 04693132001)
Tris-HCI (1 M, pH 7.5; Invitrogen, cat.no. 15567027)
Tris-HCI (1 M, pH 6.5; Teknova, cat.no. T1065)
Nonidet P 40 (Sigma-Aldrich, cat.no. 74385-1L)
10× Cutsmart Buffer (New England Biolabs (NEB), cat.no. B7204S)
20% (wt/vol) SDS (Invitrogen, cat.no. AM9820)
Triton X-100 (Acros Organics, cat.no. 215682500)
Alul (10 U/μL; NEB, cat.no. R0137L)
Ribonuclease Inhibitor RNasinPlus (40 U/μL; Promega, cat.no. N2615)
10× PBS (Invitrogen, cat.no. AM9625)
RNase I (50 U/μL; NEB, cat.no. M0243S)
RNase A (10 mg/mL; Thermo Scientific, cat.no. EN0531)
TURBO DNase (2 U/μL; Invitrogen, cat. no. AM2239)
MgCI2 (1 M; Invitrogen, cat.no. AM9530G)
DTT (1 M in H2O; Sigma-Aldrich, cat.no. 43816-10ML)
T4 Polynucleotide Kinase (10 U/μL; NEB, cat.no. M0201L)
10× NEBuffer 2 (NEB, cat.no. B7002S)
Klenow Fragment (3’−5’ exo-) (5 U/μL; NEB, cat.no. M0212L)
dNTP Solution Set (100 mM, 0.25 mL of each dNTP; NEB, cat.no. N0446S)
5’DNA Adenylation Kit (NEB, cat.no. E2610L)
Mth RNA Ligase (NEB, cat.no. M2611A, included in 5’DNA Adenylation Kit)
Dynabeads MyOne SILANE (40 mg/mL; Invitrogen, cat.no. 37002D)
Isopropanol (Sigma-Aldrich, cat.no. I9516-500ML) ! CAUTION Isopropanol is highly flammable, both as a liquid and as vapor. It can cause irritation to eyes and skin. Avoid inhalation of vapors, mist and gas. Handle with caution and ensure adequate ventilation.
10× T4 RNA ligase reaction buffer (NEB, cat.no. B0216S)
-
T4 RNA Ligase 2, Truncated KQ (200 U/μL; NEB, cat.no. M0373L)
CRITICAL: T4 RNA ligase 2, truncated KQ must not be replaced by other ligases, such as T4 RNA ligase 1 or T4 RNA ligase 2. T4 RNA ligase 2, truncated KQ is specifically used in this protocol to ligate the pre-adenylated 5’ DNA linker end to the 3’ OH end of the RNA in the absence of ATP to prevent RNA self-ligation. Other ligases are not compatible with this protocol.
50% (wt/vol) PEG8000 (NEB, cat.no. B1004S)
10× DNA Ligase Reaction Buffer (NEB, cat.no. B0202S)
T4 DNA Ligase (2000 U/μL; NEB, cat.no. M0202M)
BSA (20 mg/mL; NEB, cat.no. B9000S)
EDTA (0.5 M, pH 8.0; Ambion, cat.no. AM9261)
Proteinase K (20 mg/mL; NEB, cat.no. P8107S)
Phenol:Chloroform:Isoamyl Alcohol (Phenol:ChCI3:IAA) (25:24:1) (pH 7.9; Ambion, cat.no. AM9730) ! CAUTION Phenol:ChCI3:IAA (25:24:1) is toxic upon ingestion, inhalation or contact with skin. Handle Phenol:ChCI3:IAA (25:24:1) in chemical fume hood with caution. Protective gloves, lab coat and eye protection should be worn. Phenol:ChCI3:IAA (25:24:1) waste should be discarded in an appropriate container according to local regulation. CRITICAL: This reagent must not be replaced by acidic extraction reagent (pH 4.5), which will remove DNA during extraction. The reagent used in this protocol ensures the extraction of both DNA and RNA
NaCI (5 M; Invitrogen, cat.no. AM9759)
Sodium Acetate (3 M, pH 5.2; VWR, cat.no. E521-100ML) ! CAUTION Sodium acetate can cause skin irritation and serious eye damage. Avoid contact with eyes. Wear protective lab coat and gloves.
Ethyl alcohol (Pure, 200 proof, for molecular biology; Sigma-Aldrich, cat.no. E7023-500ML) ! CAUTION Ethyl alcohol is highly flammable, both as liquid and vapor. It can cause eye irritation. Avoid breathing vapors, mist and gas. Handle with caution and ensure adequate ventilation.
Phase Lock Gel (Heavy 2 mL; 5 Prime, cat.no. 2302830)
Qubit dsDNA BR kit (Invitrogen, cat.no. Q32850)
Qubit dsDNA HS kit (Invitrogen, cat.no. Q32854)
Qubit RNA BR kit (Invitrogen, cat.no. Q10211)
Qubit RNA HS kit (Invitrogen, cat.no. Q32855)
2-log DNA ladder (NEB, cat.no. N3200S)
Exonuclease I (NEB, cat.no. M0293L)
T4 DNA Polymerase (3 U/μL; NEB, cat.no. M0203L)
Tween 20 (Fisher Scientific, cat.no. BP337-500)
Dynabeads MyOne Streptavidin C1 (Invitrogen, cat.no. 65002)
SupersScript III Reverse Transcriptase (200 U/μL; kit supplied with 5× first-strand buffer and 0.1 M DTT; Invitrogen, cat.no. 18080044)
10× T4 Polynucleotide Kinase Reaction Buffer (NEB, cat.no. B0201S)
Hydrochloric acid (HCI) (11.6-12 M; Sigma, cat.no. H1758-100ML) ! CAUTION HCI can cause severe skin burns and eye damage. Wear protective lab coat, gloves, eye protection and face protection. Use in a well-ventilated area.
Sodium Hydroxide (NaOH) Solution (10 M in H2O; Sigma, cat.no. 72068-100ML) ! CAUTION 10 M NaOH can cause severe skin burns and eye damage. Wear protective lab coat, gloves, eye protection and face protection. Handle with caution.
ATP (10 mM; NEB, cat.no. P0756L)
CircLigase ssDNA Ligase (100 U/μL; kit supplied with CircLigase 10× reaction buffer, 1 mM ATP and 50 mM MnCI2; Epicentre, cat.no. CL4115K)
BamHI (20 U/μL; NEB, cat.no. R3136S)
NEBNext High-fidelity 2× PCR Master Mix (NEB, cat.no. M0541S)
MinElute PCR Purification Kit (Qiagen, cat.no. 28006)
MinElute Gel Extraction Kit (Qiagen, cat.no. 28604)
RNA 6000 Pico Reagents Part I (Agilent Technologies, cat.no. 5067-1513)
RNA 6000 Pico Ladder (Agilent, cat.no. 5067-1535)
High Sensitivity DNA Reagents (Agilent Technologies, cat.no. 5067-4626)
NucBlue Fixed Cell ReadyProbes Reagent (ThermoFisher, cat.no. R37606)
iMARGI linkers and library PCR primers
-
Linker top strand:
5’/Phos/NNAGATCGGAAGAGCGTCGTGTAGGGAGGATCCGTTCAGACGTGTGCTCTTCC*GA/iBiodT/CT3’ (“iBiodT” stands for internal biotin modification on dT) (Integrated DNA Technologies) ▲ CRITICAL The C* indicates a phosphorothioated C base. This linker top strand should be purified by RNase Free HPLC and lyophilized. Dissolve linker in UltraPure distilled H2O and measure the concentration. Store at −20 °C for up to two years. The concentration of linker top strand used in our lab is 380 μM.
-
Linker bottom strand:
5’/Phos/GATCGGAAGAGCACACGTCTGAACGGATCCTCCCTACACGACGCTCT3’ (Integrated DNA Technologies) ▲ CRITICAL This linker bottom strand should be purified by RNase Free HPLC and lyophilized. Dissolve linker in UltraPure distilled H2O and measure the concentration. Store at −20 °C for up to two years. The concentration of linker bottom strand used in our lab is 216 μM.
-
Cut_oligo sequence:
5’TCGTGTAGGGAGGATCCGTTCAGACGTGTGCTCT/3InvdT/3’ (“3InvdT” stands for inverted dT at the 3’ end) (Integrated DNA Technologies) ▲ CRITICAL This Cut_oligo strand should be purified by RNase Free HPLC and lyophilized. Dissolve Cut_oligo strand in UltraPure distilled H2O and measure the concentration. Prepare a final concentration of 10 μM Cut_oligo working stock using UltraPure distilled H2O. Store at −20 °C for up to two years.
-
NEBNext Universal PCR primerfor Iliumina:
5’AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATC*T3’ (New England Biolabs, cat.no. E7335S). Store at −20 °C for up to one year.
-
NEBNext Index primer for Illumina (Index Primers Set 1):
5’CAAGCAGAAGACGGCATACGAGAT[6 bp barcode]
GTGACTGGAGTTCAGACGTGTGCTCTTCCGATC-S-T3’
(New England Biolabs, cat.no. E7335S). Store at −20 °C for up to one year.
Equipment
Cell and Tissue Culture Dishes (100×20 mm, Genesee Scientific, cat.no. 25-202; 150×20 mm, Genesee Scientific, cat.no. 25-203)
BioLite Cell Culture Treated Flasks (75 cm2, Thermo Scientific, cat.no. 12556010)
6-well Cell Culture Plates (VWR, cat.no. 10062892)
Cell Scrapers (Fisher Scientific, cat.no. 087711A)
Incubator (Thermo Scientific, cat.no. 13-998-086, set incubator to 37 °C, 5% CO2)
15 mL Conical Tube (VWR, cat.no. 5250606)
50 mL Conical Tube (VWR, cat. no. 5250608)
1.5 mL DNA LoBind Tubes (Eppendorf, cat.no. 022431021)
2 mL DNA LoBind Tubes (Eppendorf, cat.no. 022431048)
5 mL Protein LoBind Tubes (Eppendorf, cat.no. 0030108302)
Dounce Homogenizer (2 mL; Sigma-Aldrich, cat.no. D8938)
Thermomixer (Thermomixer C; Eppendorf, cat.no. 5382000023)
Microcentrifuge (Centrifuge 5424 R; Eppendorf, cat.no. 5404000138)
Centrifuge (Centrifuge 5810 R; Eppendorf, cat.no. 022625501)
Thermocycler(Mastercycler nexus gradient; Eppendorf, cat.no. 6331000025)
E-gel EX Agarose Gels (1%, Invitrogen, cat.no. G402001; 2%, Invitrogen, cat.no. G402002)
E-gel iBase Power System (Invitrogen, cat.no. 10001123)
Qubit Fluorometer (Invitrogen, cat.no. Q32866)
NanoDrop Lite Spectrophotometer (Thermo Scientific, cat.no. ND-LITE)
0.2 mL PCR Tubes (Applied Biosystem, cat.no. N8010540)
0.2 mL PCR 8-Well Tube Strips (VWR, cat.no. 89401-570)
DynaMag Magnet (Thermo Fisher Scientific, cat.no. 12301D)
Magnetic Separation Stand (Promega, cat.no. Z5342)
2100 Bioanalyzer (Agilent, cat.no. G2939BA)
Blades (We use stainless steel disposable scalpels for gel-cutting, Integra Miltex, cat.no. 4-410)
Gel Imager (GelDoc-lt TS2 310 Imager, UVP, cat. no. 71004-578)
Rocker (UltraCruz 2D rocker, Santa Cruz Biotechnology, cat.no. sc-358757)
Pipettes
Filter tips
Vortex, any benchtop model
Rotator (Mini Labroller Rotator; Labnet, cat.no. H5500)
Sequencer (Illumina HiSeq4000)
A computer or server with a minimum of 16 GB memory running on any of the recommended Linux distributions, including Ubuntu, Debian, Fedora and CentOS. CRITICAL Multi-core CPU is advantage for accelerating the computing time of data processing. 16 or more cores are recommended. Enough hard disk space is necessary. 200 GB free disk space is required for the example procedure of data processing.
Docker Community Edition (CE) software (https://docs.docker.com/install/). Docker CE version 18.09.5 is used here.
iMARGI-Docker software (https://github.com/Zhong-Lab-UCSD/iMARGI-Docker). In this paper, the current version v1.1.1 is pulled from Docker-Hub (https://hub.docker.eom/r/zhonglab/imargi).
An example iMARGI dataset (SRR8206679 in the NCBI Sequence Read Archive database, 361.2 million sequencing read pairs).
Reagent setup
1 M Glycine
Add 3.74 g of glycine into a 50 mL tube and adjust the volume to 50 mL with UltraPure distilled H2O. Dissolve glycine by inverting the tube. Use the solution immediately.
0.5 M Glycine
Add 1.87 g of glycine into 50 mL tube and adjust the volume to 50 mL with UltraPure distilled H2O. Dissolve glycine by inverting the tube. Use the solution immediately.
50× protease inhibitor cocktail
Dissolve one tablet of protease inhibitor cocktail in 1 mL UltraPure distilled H2O. Prepare 50 μL aliquots and store at −20 °C for at least 12 weeks.
1× PBS
Dilute 1 mL of 10× PBS with 9 mL of UltraPure distilled H2O. Store at room temperature (RT) (22-25 °C) for up to 6 months.
0.1 M DTT
Dilute 0.5 mL of 1 M DTT with 4.5 mL of UltraPure distilled H2O. Store the solution at −20 °C for up to 12 months.
10 mM dATP
Dilute 50 μL of 100 mM ATP with 450 μL of UltraPure distilled H2O. Store the solution at −20 °C for up to 12 months.
1 mM NaOH
Dilute 1 mL of 10 mM NaOH solution with 9 mL of UltraPure distilled H2O. Store at RT for up to 12 months.
1 M HCl
Dilute 1 mL of ~12 M HCl with 11 mL of UltraPure distilled H2O. Store at RT for up to 12 months.
10% (vol/vol) Tween 20
Mix 10 μL of Tween 20 with 90 μL of UltraPure distilled H2O. Store at RT for up to 12 months.
70% (vol/vol) Ethanol
Mix 35 mL of 100%(vol/vol) ethanol with 15 mL of UltraPure distilled H2O. Store at RT for up to 6 months.
20% (vol/vol) Nonidet P 40
Mix 20 μL of Nonidet P 40 with 80 μL of UltraPure distilled H2O. Store at RT for up to 12 months.
1 M NaCl
Dilute 2 mL of 5 M NaCl solution with 8 mL of UltraPure distilled H2O. Store at RT for up to 12 months.
1× Cell lysis buffer
Mix 10 μL of 1 M Tris-HCl, pH 7.5, 10 μL of 1 M NaCl, 10 μL of 20% (vol/vol) Nonidet P 40, 20 μL of 50× protease inhibitor cocktail and 950 μL of UltraPure distilled H2O. Use lysis buffer immediately. The final composition for 1× Cell lysis buffer is 10 mM Tris-HCl, pH 7.5, 10 mM NaCl, 0.2% (vol/vol) Nonidet P 40, 1× protease inhibitor cocktail.
1× Cutsmart buffer
Dilute 1 mL of 10× Cutsmart buffer with 9 mL UltraPure distilled H2O. Store the buffer at −20 °C for up to 6 months.
0.5% (wt/vol) SDS
Mix 5 μL of 20% (wt/vol) SDS, 100 μL of 1 × Cutsmart buffer and 95 μL of UltraPure distilled H2O. Use the buffer immediately.
10% (vol/vol) Triton X-100
Mix 20 μL of Triton X-100, 100 μL of 1× Cutsmart buffer and 80 μL of UltraPure distilled H2O. Use the buffer immediately.
5× PNK phosphatase buffer, pH 6.5
Mix 350 μL of 1 M Tris-HCl, pH 6.5, 50 μL of 1 M MgCI2, 100 μL of 0.1 M DTT and 500 μL of UltraPure distilled H2O. Store the buffer at −20 °C for up to 6 months.
PNK wash buffer
Mix 1 mL of 1 M Tris-HCl, pH 7.5, 0.5 mL of 1 M MgCl2, 0.1 mL 100% (vol/vol) Tween-20 and 48.4 mL of UltraPure distilled H2O. Store at RT for up to 6 months. The final composition for PNK wash buffer is 20 mM Tris-HCl, pH 7.5, 10 mM MgCl2 and 0.2% (vol/vol) Tween-20.
Extraction buffer
Mix 25 μL of 1 M Tris-HCl, pH 7.5, 25 μL of 20% (wt/vol) SDS, 1 μL of 0.5 M EDTA, 10 μL of 5 M NaCl, 25 μL of Proteinase K and 414 μL of UltraPure distilled H2O to make a final volume of 500 μL. Freshly prepare before extraction and reverse crosslinking step. The final composition of the vextraction buffer is 50 mM Tris-HCl, pH 7.5, 1% (wt/vol) SDS, 1 mM EDTA, 100 mM NaCl and 1 mg/mL Proteinase K.
2× B&W buffer
Mix 300 μL of 1 M Tris-HCl, pH 7.5, 60 μL of 0.5 M EDTA, 12 mL of 5 M NaCl, 60 μL 10% (vol/vol) Tween-20 and 55.8 mL of UltraPure distilled H2O. Store at RT for up to 6 months. The final composition of the 2× B&W buffer is 10 mM Tris-HCl, pH 7.5, 1 mM EDTA, 2 M NaCl and 0.02% (vol/vol) Tween-20.
1× B&W buffer
Dilute 10 mL of 2× B&W buffer with 10 mL of UltraPure distilled H2O. Store at RT for up to 6 months.
Denaturing buffer
Mix 100 μL of 1 M NaOH, 1 μL of 100 mM EDTA and 899 μL of UltraPure distilled H2O. Store at RT for up to 12 months. The final composition of the denaturing buffer is 0.1 M NaOH and 0.1 mM EDTA.
High salt biotin wash buffer
Mix 3 mL of 1 M Tris-HCl, pH 7.5, 240 mL of 5 M NaCl, 600 μL of 0.5 M EDTA, 600 μL of Tween-20 and 55.8 mL of UltraPure distilled H2O. Store at 4 °C for up to 1 months. The final composition of the high salt biotin wash buffer is 10 mM Tris-HCl, pH 7.5, 4 M NaCl, 1 mM EDTA and 0.2% (vol/vol) Tween-20.
Cell culture medium
For HEK293T cells, DMEM supplemented with a final concentration of 10% (vol/vol) FBS and 1% Penicillin/Streptomycin. Store at 4 °C for up to one month.
Procedure
Crosslinking cells in solution. ● Timing 2 h
Grow cells in the appropriate culture medium to 90% confluency. Three to five million cells are enough to generate one iMARGI library. However, we recommend starting with more cells (i.e., ~ 2 × 107) and crosslinking and aliquoting cells 1 × 107 cells per each tube to perform technical replicates if necessary. For cells cultured in suspension or loosely adherent cells (e.g. HEK293T cells), we recommend crosslinking in tube to minimize the loss of cells [see option (A)]. For adherent cells that attach well, we recommend crosslinking on plate [see option (B)].
(A). Crosslinking cells in tube
For adherent cells only: aspirate the medium from tissue culture plates. Rinse cells grown on cell culture plates with 7 mL 1× PBS and put the plates on ice. Scrape cells into the PBS already in the plates and transfer to a 50 mL tube. Wash plates with 7 mL 1× PBS once and combine the residual cells with the cells in the tube. Cell counting is recommended at this step. For suspension cells, directly start from Step 1A(ii).
Spin down detached adherent cells or cells in suspension at 500g for 3 minutes (min) at 4 °C. Discard supernatant. Resuspend cells in 15 mL 1× PBS.
Add 1 mL of fresh 16% (wt/vol) formaldehyde to a final concentration of 1% (wt/vol) and fix cells at RT for 10 min with rotation.
Quench crosslinking reaction by adding 4 mL of 1 M glycine to a final concentration of 0.2 M.
Incubate at RT for 10 min with rotation, and then on ice for 10 min.
Spin crosslinked cells at 2000g for 5 min at 4°C.
Discard supernatant into an appropriate collection container by pipetting.
Rinse cells once with 15 mL ice cold 1× PBS without resuspending the cell pellet.
Spin at 2000g for 5 min at 4°C to pellet cells. Carefully discard supernatant by aspirating.
■ PAUSE POINT Cells can be flash-frozen in liquid nitrogen and stored at −80 °C for at least one year. If frozen, gently thaw cells on ice before proceeding to the rest of the protocol.
(B). Crosslinking cells on plate
Aspirate medium from tissue culture plates. Rinse cells grown on cell culture plates with 5 mL 1× PBS.
Add 7 mL freshly made 1% (wt/vol) formaldehyde to plates and incubate cells at RT for 10 min with gentle rocking on a rocker.
Quench crosslinking reaction by adding 4.6 mL of 0.5 M glycine to a final of 0.2 M.
Incubate cells at RT for 10 min with rotation, and then on ice for another 10 min.
Remove solution by pipetting into an appropriate liquid waste container.
Transfer plate on ice and rinse cells twice with 5 mL cold 1× PBS.
Add 5 mL 1× PBS to the plate. Scrape cells from the plate and transfer into a 15 mL conical tube.
Add 5 mL 1× PBS to plate and transfer the cells left in the plate to the same 15 mL conical tube.
Spin at 2000g for 5 min at 4°C to pellet cells. Carefully discard supernatant by aspirating.
■ PAUSE POINT cells can be flash-frozen in liquid nitrogen and stored at −80 °C for at least one year. If frozen, gently thaw cells on ice before proceeding to the rest of the protocol.
Cell lysis and DNA fragmentation. ● Timing 3 h plus overnight reaction
-
2
Resuspend one aliquot of crosslinked cells (~ 1 × 107 cells) from Step 1Aix or 1Bix in 1 mL of freshly prepared cold cell lysis buffer and incubate on ice for 15 min.
-
3
Transfer the mixture into 2 mL Dounce homogenizer.
-
4
Homogenize the cells on ice with pestle A. Slowly move pestle up and down for 10 times, incubate on ice for 1 min and then do 10 more strokes. Further incubate cells on ice for 10 min.
▲ CRITICAL STEP Pestle A is used in this step to homogenize cells. Pestle A has a looser fitting which can help the lysis of cells but keep the nuclei intact.
-
5
Weigh two empty 1.5 mL Eppendorf tubes.
-
6
Transfer the cell lysis mixture into one of the 1.5 mL tubes weighed in Step 5.
-
7
Centrifuge at 2500g at 4 °C for 2 min to pellet nuclei and discard supernatant carefully.
-
8
Carefully resuspend pelleted nuclei in 500 μL of 1× Cutsmart buffer. Split 250 μL of mixture with nuclei into the other 1.5 mL tube weighed in Step 5.
-
9
Centrifuge both tubes of nuclei from Step 8 at 2500g at 4 °C for 1 min to pellet nuclei and discard supernatant carefully. After splitting the nuclei mixture in Step 8, the nuclei pelleted in Step 9 is from approximately 5 × 106 cells, which will be used in the rest of the protocol. The other half of nuclei pellet can be processed at the same time as a replicate if necessary or used to examine nuclei integrity as described in Box 1.
-
10
Weigh the 1.5 mL tube containing nuclei pellet from Step 9 after discarding the supernatant.
-
11
Estimate the weight of nuclei pellet by subtracting the weight of empty tube in Step 5 from the total weight of tube and pellet in Step 10.
-
12
Gently resuspend nuclei from Step 10 in 0.5% (wt/vol) SDS following this formula: vol(SDS in μL)/wt(nuclei in mg)=3. For example, if the measured weight of nuclei pellet is 15 mg, add 45 μL of 0.5% (wt/vol) SDS to the nuclei pellet.
-
13
Incubate SDS-nuclei mixture at 62 °C for 10 min in a thermomixer shaking at 800 rpm.
-
14
Quench the SDS reaction by adding 1× Cutsmart buffer and 10% (vol/vol) Triton X-100. The final SDS concentration will be 0.1% (wt/vol) and final Triton X-100 concentration will be 1% (vol/vol).
-
15
Incubate at 37 °C for 15 min in a thermomixer shaking at 800 rpm.
-
16
Pellet nuclei by centrifuging at 2500g at 4 °C for 1 min. Discard supernatant carefully.
? TROUBLE SHOOTING
-
17
Wash nuclei by adding 500 μL of 1× Cutsmart buffer to gently resuspend nuclei, centrifuge at 2500g at 4 °C for 1 min and discard the supernatant. Repeat the wash once.
? TROUBLE SHOOTING
-
18Resuspend nuclei in 300 μL of the following chromatin digestion mix:
Reagent Volume (μL) Final concentration H2O 198 10× Cutsmart buffer 30 1× Alul (10 U/μL) 70 2.3 U/μL RNasinPlus (40 U/μL) 2 0.3 U/μL Total volume 300 19 Digest chromatin overnight at 37 °C in a thermomixer shaking at 850 rpm for 30 second (s) per 1 min.
Box 1: Checkpoint 1 for imaging nuclei integrity.
This checkpoint is recommended for researchers performing an iMARGI experiment for the first time. In this checkpoint, the integrity of nuclei will be examined under a microscope.
Resuspend the remaining aliquot of pelleted nuclei (~5 × 106 cells after splitting in Step 8) in 500 μL cold 1x PBS in 1.5 mL tube.
Add one drop of NucBlue fixed cell stain and incubate at RT in the dark for 10 min. After the incubation, pellet nuclei at 2000g for 5 min at 4 °C by centrifugation.
Resuspend nuclei in 500 μL cold 1× PBS, and then dispense 10 μL of nuclei mixture onto a microscope slide and cover with cover glass.
Image slide with 40x air or 60x oil objective with standard DAPI emission/excitation filter on a standard inverted epifluorescence microscope. Specifically, we used an Olympus IX83 inverted microscope with a 60x oil objective (NA=1.42) with 50 ms exposure at 1x gain. This DAPI stained aliquot of nuclei can be subjected to Steps 10–19 in parallel with the other aliquot of nuclei in Step 9.
Repeat this checkpoint after permeabilization of the nuclear membrane (after Step 17) and DNA fragmentation (after Step19) for the DAPI stained aliquot of nuclei. The nuclei are considered intact if the DAPI signal is confined to the nuclei and overlaps with the differential interference contrast (DIC) images of nuclei (Fig. 3a–c).
RNA fragmentation, preparation of RNA and DNA ends for ligation, and ligation of linker to RNA. ● Timing 4 h plus overnight reaction
-
20
Dilute RNase I 10-fold in 1× PBS.
-
21
Add 1 μL of diluted RNase I to the chromatin digestion mix from Step 19 and incubate at 37 °C for 3 min.
-
22
Transfer tube on ice for at least 5 min.
-
23
Pellet nuclei at 2500g for 1 min at 4 °C. Discard supernatant carefully or transfer supernatant to a new 1.5 mL tube for nuclei integrity examination (described in Box 2).
-
24
Wash nuclei with 300 μL PNK wash buffer twice as described in Step 17.
-
25
Checkpoint. This step examines the efficiencies of chromatin fragmentation and RNA fragmentation. Transfer 10 μL of nuclei pellet from Step 23 to a new tube. Add 100 μL of extraction buffer and then incubate the mixture at 65 °C for 2 h to reverse crosslinking. Purify nucleic acids extracted from nuclei using 50 μL SILANE beads. Elute nucleic acids using 50 μL UltraPure distilled H2O but do not remove beads. Split the nucleic acids/beads mixture in half: add 1 μL of RNase A to one half; add 2.5 μL of 10× TURBO DNase buffer and 1 μL of TURBO DNase into the other half. Incubate at 37 °C for 30 min. Purify nucleic acids using SILANE beads that are already in the mix. Elute DNA or RNA, respectively. Incubate beads with 15 μL of UltraPure distilled H2O and elute as described in Step 46. Check the chromatin fragmentation pattern by loading eluted DNA on a precast 1% agarose E-gel. A smear of DNA fragments between ~100 bp to ~ 2kb is considered as efficient chromatin fragmentation (Fig. 4a). Check RNA fragmentation efficiency by loading eluted RNA on a Bioanalyzer. The degree of RNA fragmentation is deemed appropriate when most fragments range from 50 nt to 1,000 nt (Fig. 4b).
? TROUBLE SHOOTING
-
26Prepare RNA 3’ end dephosphorylation reaction mix as follows:
Reagents Volume (μL) Final concentration H2O 148 5× PNK phosphatase buffer, pH 6.5 40 1× T4 PNK (10 U/μL) 10 0.5 U/μL RNasinPlus (40 U/μL) 2 0.4 U/μL Total volume 200 -
27
Resuspend nuclei from Step 24 in the reaction mix and incubate at 37 °C for 30 min shaking at 800 rpm in a thermomixer.
-
28
Pellet nuclei at 2500g for 1 min at 4 °C. Discard supernatant or transfer supernatant to a new 1.5 mL tube for nuclei integrity examination as described in Box 2.
-
29
Wash nuclei twice with 300 μL PNK wash buffer on ice.
-
30Prepare DNA dA-tailing reaction mix as follows:
Reagents Volume (μL) Final concentration H2O 164 10× NEBuffer 2 20 1× Klenow fragment (3’−5’ exo-) (5 U/μL) 12 0.3 U/μL 10 mM dATP 2 0.1 U/μL 10% (vol/vol) Triton X-100 2 0.1 % (vol/vol) Total volume 200 -
31
Resuspend nuclei pellet in the reaction mix and incubate at 37 °C for 30 min shaking at 800 rpm in a thermomixer.
-
32
Pellet nuclei at 2500g for 1 min at 4 °C. Discard supernatant or transfer supernatant to a new 1.5 mL tube for nuclei integrity examination as described in Box 2.
-
33
Wash twice with 300 μL PNK wash buffer, discard the supernatant from second wash and keep the pelleted nuclei on ice.
-
34Prepare the following adenylation reaction mix in a 0.2 mL PCR tube for the linker top strand:
Reagents Volume (μL) Final concentration H2O 3.5 10× 5’ DNA adenylation reaction buffer 3 1× 1 mM ATP 3 0.1 mM linker top strand oligo (380 μM) 2.5 30 μM 50 μM Mth RNA ligase 18 30 μM Total volume 30 -
35
Incubate the reaction mixture in a thermocycler at 65 °C for 1 h and then at 85 °C for 5 min to inactivate the enzyme.
-
36
Add 4.2 μL (900 pmol) of the linker bottom strand (216 μM) to the adenylated linker top stand and incubate samples in the thermocycler using the following program to anneal top and bottom strands: 95°C for 2 min; then 71 cycles of 20 s, starting from 95°C and decrease the temperature by 1°C each cycle down to 25°C; and hold at 25°C.
▲ CRITICAL STEP Freshly prepare adenylated linker top strand and anneal to the bottom strand in each experiment. The volume of linker top strand and bottom strand needed in Step 34 depend on the actual concentration of the linker oligos.
-
37
Purify the annealed linker using 200 μL SILANE beads. To do so, firstly, transfer 200 μL SILANE beads mixture into a 1.5 mL tube. Place on a magnetic stand, wait for beads to attach to the magnet, and then discard the stock solution.
-
38
Resuspend SILANE beads with 300 μL buffer RLT, place tube on a magnetic stand and wait for beads to attach to the magnet. Discard supernatant carefully.
-
39
Remove the tube from the magnetic stand.
-
40
Resuspend beads in 3.5× original sample volume of buffer RLT. The sample volume is 30 μL reaction volume from Step 34 plus the 4.2 μL of bottom linker, which is 34.2 μL in total. Add the 34.2 μL of annealed linker sample from Step 36 to the bead suspension.
-
41
Add 4.5× original sample volume of isopropanol to the bead/sample mixture. Mix well by inverting the tube for 5 times.
-
42
Incubate the sample with beads for 10 min at RT with rotation.
-
43
Place the tube on a magnetic stand and wait for beads attach to the magnet. Discard supernatant carefully.
-
44
Wash beads with 300 μL of 70% (vol/vol) ethanol, wait for beads to attach to the magnet and discard supernatant carefully.
-
45
Repeat the washing in Step 44 once. Discard supernatant and let the beads air dry for 5 to 10 min.
-
46
Elute sample in 50 μL of UltraPure distilled H2O. Place tube on magnet, wait for beads attach to the magnet and transfer eluate to a new tube. All the later SILANE beads purifications will follow the procedure outlined in Steps 37-46 unless mentioned. The beads and sample volume may vary and will be specified at each subsequent step.
▲ CRITICAL STEP The final eluate collected after magnetic separation could be less than 50 μL. All the eluted linker will be added to the linker-RNA ligation reaction in Step 47.
Checkpoint. To check the annealing efficiency of linker top and bottom strand, dilute 1 μL each of linker top, linker bottom and annealed double-stranded (ds) linker sample from Step 46 with 19 μL of UltraPure distilled H2O separately. Load 20 μL each of diluted linker top, linker bottom and ds linker onto a precast 2% agarose E-gel. The successfully annealed ds linker will shift to a larger size and run at a higher molecular weight than the single-stranded top and bottom strand (Fig. 5a).
-
47Prepare the following linker-RNA ligation mix in a 2 mL tube:
Reagents Volume (μL) Final concentration H2O make up to 200 μL Linker (Step 46) entire eluate (≤ 50 μL) 10× T4 RNA ligase reaction buffer 20 1× 50% (vol/vol) PEG 8000 80 20%(vol/vol) 10% (vol/vol) Triton X-100 2 0.1% (vol/vol) RNasinPlus (40 U/μL) 2 0.4 U/μL T4 RNA ligase 2, truncated KQ (200 U/μL) 10 10 U/μL Total volume 200 -
48
Resuspend nuclei from Step 33 in the linker-RNA ligation mix and incubate at 22 °C for 6h with intermittent mixing (900 rpm; mix for 30 s, stop for 15 s and start mixing again) and then 16 °C overnight shaking at 850 rpm in a thermomixer.
▲ CRITICAL STEP The reaction mix with PEG is very viscous. Pipet carefully to avoid residual reaction mix on the tip. We recommend using a 2 mL tube to facilitate mixing. Therefore, nuclei need to be transferred from a 1.5 mL tube to a 2 mL tube at this step. Take 100 μL of reaction mix from Step 47 and add to the nuclei pellet from Step 33. Resuspend and transfer all the nuclei to the 2 mL tube for reaction.
Box 2: Checkpoint 2 for quantitative evaluation of nuclei integrity.
This optional checkpoint is recommended for researchers performing an iMARGI experiment for the first time. This checkpoint is complementary to the checkpoint performed at Step 9. In this checkpoint, the concentrations of RNA and DNA in nuclei and in the supernatant will be measured, which will be used to quantitatively evaluate nuclei integrity.
Remove the supernatant from Steps 23, 28, 32, 50 and 56 after the reactions in those steps have been completed (Fig. 3d) and transfer to new tubes, add 10 μL proteinase K, and incubate tubes at 65°C for 2 h to reverse crosslinking.
Purify nucleic acids using 50 μL of SILANE beads and elute with 50 μL of UltraPure distilled H2O but do not remove from beads. Please refer to Step 36 to 46 for SILANE beads purification. The sample volume will be the volume of the supernatant taken out after Steps 23, 28, 32, 50 and 56, which will be approximately the same as the reaction volume in Steps 18, 26, 30, 47 and 52.
Split sample/beads mixture in half into two tubes: treat one tube with 1 μL of RNase A to remove RNA in the sample, and the other tube with 1 μL TURBO DNase + 2.5 μL TURBO DNA Buffer to remove DNA in the sample.
Incubate each tube at 37°C for 30 min.
Purify nucleic acids using SILANE beads already in the mixture. Elute with 20 μL UltraPure distilled H2O and transfer purified DNA and RNA into new tubes.
Measure the concentration of DNA or RNA, respectively, in each tube using Qubit dsDNA HS assay kit or Qubit RNA HS assay kit. Calculate the amount of purified supernatant DNA (sDNA) and supernatant RNA (sRNA). Since the nucleic acids were split in two tubes after the first elution, one should multiply the amount of sDNA or sRNA by two to obtain the total amount of sDNA or sRNA leaked from nuclei.
Compare the amounts of sDNA and sRNA after each step with the amounts of the final nuclei extracted DNA (nDNA) and RNA (nRNA) from Step 72, by taking the ratios of these amounts (Fig. 3, e and f). Small ratios indicate non-leaking nuclei, whereas large ratios indicate broken nuclei (see Table 3 for thresholds).
Proximity ligation. ● Timing 45 min plus overnight reaction
-
49
Stop the linker-RNA ligation reaction by adding 20 μL 0.5 M EDTA and incubate at 16 °C for 5 min.
-
50
Pellet nuclei and discard supernatant or transfer supernatant to a new tube for examination of nuclei integrity as described in Box 2.
-
51
Wash five times with 500 μL PNK wash buffer to remove free linkers as described in Step 17.
▲ CRITICAL STEP Nuclei may not pellet at the bottom of the 2 mL tube very securely. Carefully remove the supernatant after each wash to avoid losing nuclei.
-
52Prepare the following proximity ligation mixture without T4 DNA ligase:
Reagents Volume (μL) Final concentration H2O 1660 10× DNA ligase reaction buffer 200 1× 10 % (vol/vol) Triton X-100 20 0.1% (vol/vol) BSA (20 mg/mL) 100 1 mg/mL T4 DNA ligase (2000 U/μL) 4 4 U/μL RNasinPlus (40 U/μL) 16 0.5 U/μL Total volume 2000 -
53
Before adding the T4 DNA ligase, resuspend nuclei in the reaction mix, and, afterwards add T4 DNA ligase.
▲ CRITICAL STEP Nuclei need to be transferred from 2 mL tube to a 5 mL tube at this step. Take 500 μL of the reaction mix excluding the T4 DNA ligase from Step 52 and add to the 2 mL tube with nuclei pellet. Resuspend the nuclei and transfer all the nuclei to the 5 mL tube. Add T4 DNA ligase to the 5 mL tube after transferring all the nuclei to the 5 mL tube.
-
54
Incubate the reaction mixture at 16 °C overnight with intermittent mixing in thermomixer (650 rpm; 30 s per min)
▲ CRITICAL STEP The proximity ligation is performed in a diluted condition in a 5 mL tube to minimize random ligation.
Reverse crosslinking and DNA/RNA extraction. ● Timing 5 h
-
55
Stop the proximity ligation reaction by adding 200 μL of 0.5 M EDTA and incubate at 16 °C for 15 min.
-
56
Pellet nuclei at 2500g at 4 °C for 1 min. Discard the supernatant or transfer it to a new tube for nuclei integrity examination as described in Box 2.
-
57
Wash nuclei pellet twice with 500 μL 1× PBS as described in Step 17.
-
58
Resuspend nuclei in 250 μL of extraction buffer to extract nucleic acid and to reverse crosslinks.
▲ CRITICAL STEP The extraction buffer can form a white precipitate if put on ice. We recommend using the extraction buffer immediately after preparation.
-
59
Incubate the mixture at 65 °C on a thermomixer for 3 h shaking at 800 rpm.
-
60
Add an equal volume of Phenol:CHCl3:IAA (25:24:1) (pH 7.9) to the reverse crosslinking mixture. Vortex for 1 min until the mixture turns white.
-
61
Pre-spin a Phase Lock Gel tube at 1500g for 1 min at RT. Add the mixture from Step 60 to the Phase Lock Gel tube.
▲ CRITICAL STEP Phase lock gel could stick to the wall of the tube. Spin Phase Lock Gel tube before use.
-
62
Centrifuge Phase Lock Gel tube at 12,000g for 5 min at RT, then transfer the top aqueous phase to a new 1.5 mL tube.
▲ CRITICAL STEP Avoid touching the phase lock gel with the pipet tip when transferring the top aqueous phase.
-
63
Add 250 μL of Phenol:CHCl3:IAA (25:24:1) (pH 7.9) to the 1.5 mL tube from Step 62. Vortex for 1 min until the mixture turns white.
-
64
Add the mixture to a new Phase Lock Gel tube. Centrifuge at 12,000g for 5 min at RT, and then transfer the aqueous phase to a new 1.5 mL tube.
-
65
Perform ethanol precipitation by adding 1/10th volume of 3 M sodium acetate (pH 5.2) and 3× volume of ice cold 100% (vol/vol) ethanol to the 1.5 mL tube from Step 64. Mix well by inverting the tube three times.
-
66
Incubate at −80 °C for 30 min.
■ PAUSE POINT The ethanol precipitation mix can also be incubated at −20 °C overnight.
-
67
Centrifuge the tube at 14000 rpm for 30 min at 4 °C.
-
68
Remove and discard the supernatant and gently add 1 mL of cold 70% (vol/vol) ethanol to the precipitated nucleic acid pellet. Do not disturb the pellet.
-
69
Centrifuge again at 14000 rpm for 10 min in 4 °C.
-
70
Remove and discard the supernatant and air-dry pellet for 5 min to 10 min.
-
71
Carefully resuspend the pellet in 50 μL of UltraPure distilled H2O.
▲ CRITICAL STEP The dried nucleid acid pellet may become transparent and invisible in the tube. Add UltraPure distilled H2O into the tube gently. Completely dissolve nucleic acid pellet by pipetting.
-
72
Measure the DNA and RNA concentration using a Qubit dsDNA BR kit and a Qubit RNA BR kit, respectively. A typical yield of DNA is ~10-15 μg for ~5 × 106 cells, the amount of RNA depends on the cell type.
■ PAUSE POINT Purified nucleic acids can be stored at −20 °C for up to 6 months.
Removal of biotin from un-ligated linkers. ● Timing 3 h
-
73Separate the purified nucleic acid into multiple 0.2 mL PCR tubes (PCR strips) containing a maximum of 8 μg nucleic acid in each tube. Prepare the reaction mixture in each tube as follows:
Reagents Volume (1×) (μL) Final concentration H2O to 144 μL 10× NEB buffer 2 15 1× BSA (20 mg/mL) 1 0.1 mg/mL Exo I (20 U/μL) 5 0.7 U/μL RNasinPlus (40 U/μL) 2 0.6 U/μL Nuclei Acid (DNA+RNA; Step 72) variable 8 μg maximum each tube Total volume 144 -
74
Incubate reactions in PCR strips at 37 °C for 30 min in a thermocycler.
-
75Add the following mixture to each PCR tube to a final volume of 150 μL.
Reagents Volume (1×) (μL) Final concentration 10 mM dATP 1.5 0.1 mM 10 mM dGTP 1.5 0.1 mM T4 DNA polymerase (3 U/ μL) 3 0.06 U/ μL Total volume 6 -
76
Incubate at 12 °C for 2 h in a thermocycler.
-
77
Combine all the tubes in the PCR strip into a single 1.5 mL tube and add a final concentration of 50 mM EDTA to stop the reaction.
■ PAUSE POINT Sample mixture can be stored at −20 °C overnight.
Biotin pull-down of RNA-DNA chimeric sequences, reverse transcription, and ssDNA circularization ■ Timing 5-6 h plus overnight reaction
-
78
Add 200 μL of Streptavidin C1 beads to a 15 mL tube.
-
79
Wash beads with 300 μL 1× B&W buffer, place the tube on a DynaMag magnet for 1 min, and discard supernatant. Repeat the wash for three times.
-
80
Resuspend beads with a volume of 2× B&W buffer equal to the volume of the sample from Step 77. Add the sample from Step 77 to the beads and incubate at RT for 30 min with gentle rotation.
-
81
Pull down the beads with a magnet and extensively wash beads seven times, using 7 mL high salt biotin wash buffer for each wash.
▲ CRITICAL STEP Washing the streptavidin beads with a high salt biotin wash buffer is important for removing non-specific nucleic acids that bind to the beads. Incubate the high salt biotin wash buffer with the beads for 2 min on a rotator during each wash. Then, place the tubes on magnet, wait for the solution to turn clear, and discard supernatant.
-
82
Resuspend the beads with 1mL of PNK wash buffer and transfer the beads mixture to a new 2 mL tube.
-
83
Put the 2 mL tube from Step 82 on magnet and remove and discard PNK wash buffer.
▲ CRITICAL STEP High salt biotin wash buffer left with beads may affect later enzymatic reactions. Therefore, washing beads with PNK wash buffer is essential to remove the residual high salt biotin wash buffer completely before enzymatic reactions.
-
84Prepare reverse the transcription reaction mix as follows:
Reagents Volume (μL) Final concentration H2O 22 5× First-strand buffer 8 1× 10 mM dNTP 2 0.5 mM 100 mM DTT 2 5 mM RNasinPlus (40 U/μL) 2 2 U/μL Superscript III RT (200 U/μL) 4 20 U/μL Total volume 40 -
85
Resuspend the beads in the reverse transcription mix and incubate at 50 °C for 1 h shaking at 800 rpm in a thermomixer.
-
86
Pull down beads with magnet and discard the supernatant.
-
87
Wash beads twice with 300 μL 1× B&W buffer.
-
88
Wash beads once with 300 μL PNK wash buffer.
-
89Prepare DNA 5’ end phosphorylation mixture as follows:
Reagents Volume (μL) Final concentration H2O 74 10× T4 PNK reaction buffer, pH 7.6 10 1× T4 PNK (10 U/μL) 5 0.5 U/μL 10% (vol/vol) Triton X-100 1 0.1% (vol/vol) 10 mM ATP 10 1 mM Total 100 -
90
Resuspend beads in the phosphorylation mixture and incubate at 37 °C for 1 h.
-
91
Wash beads with 300 μL 1× B&W buffer twice. Remove and discard the 1× B&W buffer.
-
92
Resuspend the beads in 100 μL of denaturing buffer to dissociate single-stranded cDNA-DNA (ssDNA) chimeric sequences from the beads.
-
93
Incubate tube at RT for 15 min, pull down beads using a magnet and transfer the supernatant to a new tube.
▲ RITICAL STEP After incubation in the denaturing buffer, ssDNA is dissociated from its biotinylated complementary strand and is released into the supernatant. Therefore, after magnetic separation, the supernatant should be collected.
-
94
Incubate the collected supernatant at 98 °C for 20 min to completely hydrolyze the complementary RNA strand and then neutralize th supernatant with 10 μL of 1 M HCl and 10 μL 1 M Tris-HCl, pH 7.5.
-
95
Purify the resulting 120 μL of ssDNA using 100 μL SILANE beads.
-
96
Elute ssDNA from the beads with 20μL UltraPure distilled H2O.
-
97Prepare the ssDNA circularization mix in a 0.2 mL PCR tube as follows:
Reagents Volume (μL) Final concentration ssDNA (Step 96) 15 10× CircLigase Buffer 2 1× 1mM ATP 1 0.05 mM 50 mM MnCl2 1 2.5 mM CircLigase (100 U/μL) 1 5 U/μL Total volume 20 ▲ CRITICAL STEP All the ssDNA will be added into the mixture in Step 97. Based on our experience, 15 μL of ssDNA can be collected after eluting with 20 μL of UltraPure distilled H2O.
-
98
Incubate the reaction mixture at 60 °C for 4 h in thermocycler, then heat inactivate the enzyme by incubating at 80 °C for 10 min. Hold at 4 °C.
■ PAUSE POINT The reaction can be held at 4 °C in thermocycler overnight.
Cut_Oligo annealing, BamHI digestion and library amplification ● Timing 4 h
-
99Prepare the following oligo annealing mix:
Reagents Volume (μL) Final concentration H2O 23 10× Cutsmart buffer 3 1× 10μM Cut_oligo 1 0.2 μM Total volume 27 -
100
Add the annealing mix to the 20 μL of circularized ssDNA from Step 98.
-
101
Anneal the oligos using the same annealing program as described in Step 36 in a thermocycler.
-
102
Add 3 μL of BamHI to the oligo annealing mixture and incubate for 1 h at 37 °C in a thermocycler to linearize DNA
-
103
Purify 50 μL of linearized DNA using 50 μL SILANE beads and elute with 25 μL of UltraPure distilled H2O.
-
104
Optimization of the number of PCR cycles used for library amplification is important for maximizing the complexity of iMARGI libraries and minimizing the PCR or amplification bias. We recommend optimizing the PCR cycle number by setting up a 50 μL test PCR reaction mix using 5 μL of linearized DNA, 25 μL of 2× NEBNext HC PCR Master Mix, 1 μL of Universal Primer (10 μM), 1 μL of Index Primer (10 μM) and 18 μL of H2O. Aliquot the PCR reaction mix into five 10 μL reaction for PCR cycle screening. Five different cycle numbers (e.g.10, 13, 15, 18, 22 cycles) can be tested. Perform PCR as described in Step 107, but vary the cycle numbers.
? TROUBLE SHOOTING
-
105
Determine the appropriate PCR cycle number by running PCR products on agarose gel or a Bioanalyzer.
Checkpoint: To visualize size distribution of PCR products on a gel, load 1 μL of each PCR product onto a precast 1% agarose E-gel. PCR cycle numbers resulting in fragments much larger than 1 kb should be avoided. Use the lowest PCR cycle number possible to avoid over-amplification. To check the size distribution of the products on a Bioanalyzer, dilute each PCR product to 50 μL using UltraPure distilled H2O. Purify the diluted products using GIAquick PCR Purification kit according to the manufacturer’s instruction and elute the QIAquick spin column with 50 μL of UltraPure distilled H2O. Check the size distribution of the library by loading 1 μL of purified PCR product onto a Bioanalyzer using the High Sensitivity DNA kit. The majority of library after PCR is expected to range from 150 bp to 1,000 bp (Fig. 4c).
? TROUBLE SHOOTING
-
106After the PCR cycle number is determined in Steps 104 and 105, prepare the library amplification mixture as follows:
Reagents Volume (μL) Final concentration H2O 40 linearized DNA (Step 103) 5 2× NEBNext HF PCR Master Mix 50 1× Universal primer (10 μM) 2.5 0.25 μM Index primer (10 μM) 2.5 0.25 μM Total volume 100 -
107Since the library amplification mixture contains more DNA template than the pilot PCR in Step 104, one can choose the optimized cycle number determined in Step 105 minus one cycle number in the final library generation PCR. Use the following PCR program:
Cycle number Denature Anneal Extend 1 98 °C, 30 s 2-12a 98 °C, 10 s 65 °C, 30 s 72 °C, 30 s 13 72 °C, 5 min aUse optimized cycle number. In our iMARGI experiments, we used 12 cycles for library generation.■ PAUSE POINT Samples can be kept at 4 °C overnight. In our lab, we usually finish Step 99-113 in one day. Therefore, we consider Steps 99-113 as Day 8.
Library size selection ● Timing 2 h
-
108
Purify the final PCR product using the MinElute PCR Purification kit according to the manufacturer’s instruction and elute the MinElute column twice, each time with 12 μL UltraPure distilled H2O, thus obtaining ~24 μL in total.
-
109
Load the resulting ~24μL of library onto a precast 2% agarose E-gel to perform size selection. Mix 1 μL of 2-log DNA ladder with 19 μL of UltraPure distilled H2O and run on the 2% agarose E-gel with the library using E-gel system
-
110
Take the agarose gel out from the E-gel cast and visualize the bands under UV light in the gel imager. Cut out the gel piece corresponding to a size ranging from 250 bp to 1000 bp, collect the gel pieces, and transfer them into a 1.5 mL tube.
! CAUTION Wear a protective lab coat, gloves and an eye shield to protect yourself from UV irradiation. Dispose the rest of the gel in an appropriate container.
-
111
Extract DNA from the gel pieces using a MinElute gel extraction kit according to the manufacturer’s instruction and elute the MinElute column twice (12 μL UltraPure distilled H2O each time; ~24 μL in total).
-
112
Check the concentration of final library by Qubit dsDNA HS kit.
-
113
Checkpoint. Run 1 μL of library on a Bioanalyzer using the High Sensitivity DNA kit and check the quality and size distribution of the library (Fig. 4d). The final library concentration should be at least 1 ng/μL if the library average size is within the range from 380 bp to 410 bp. Based on our experience, with the correct number of cells, 12 PCR cycles in Step 107 will be sufficient to generate enough material for sequencing. To determine the minimum number of cells needed to generate an iMARGI library for sequencing, vary the cell number in Step 2 and repeat from Step 2 to 113. If the final library concentration is at least 1 ng/μL measured in Step 112, and the library average size is within the range of 380 bp to 410 bp measured in Step 113 using Bioanalyzer, the amount of input cells is sufficient.
Sequencing ● Timing variable, depending on sequencing facility [AU: Editor has added text. Please check if OK.]
-
114
Sequence the library with 100 cycles pair-end sequencing with an Illumina sequencer. We used HiSeq4000 to sequence our libraries. Use sufficient Illumina sequencing lanes of runs to produce 300 million or more read pairs.
-
115
Checkpoint. Check sequencing quality with the FastQC software28. The following minimum requirements must be reached: (1) In the Per Base Sequence Quality module, the lower quartile of base quality score must be greater than 10 in every position, and the median quality score must be greater than 25; (2) In the Per Base Sequence Content module, both the proportion of “C”s at the first position and that of “T”s at the second position of Read 2 must be greater than 50%. (3) In the Per Sequence Quality module, the average quality per read must be greater than 27; (4) In the Per Base N Content module, the proportion of Ns at any position should be smaller than 5%; (5) In the Sequence Length Distribution module, read lengths of all reads must be identical. Do not rely the other quality metrics in FastQC, because they are not compatible with iMARGI’s experimental design. Please resort to general troubleshooting strategies for DNA sequencing if criteria 1, 3, 4, 5 were not reached. Please resort to Troubleshooting (Table 4) if criterion 2 was not reached.
? TROUBLE SHOOTING
Table 4.
Troubleshooting table.
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| 16 | (i) Obvious loss of nuclei pellets after SDS treatment (ii) Disappearance of nuclei pellets and transparent gel-like precipitate at the top of the tube |
Nuclei are lysed during SDS treatment in Step 12 | Reduce the SDS concentration, volume to weight ratio [v(SDS)/wt(nuclei)] or SDS treatment time. The recommended range for SDS concentration is 0.2-0.5% (wt/vol). The recommended range for v(SDS)/wt(nuclei) is 1-5. The recommended treatment time is between 5 to 10 minutes at 62 °C. Carry out the protocol through Step 16 and observe nuclei pellet at the top of the tube. |
| 17 | Difficult to resuspend nuclei in wash buffer after SDS treatment | Nuclei become sticky after SDS treatment | Gently resuspend nuclei and break nuclei clumps by pipetting. Pipet gently to avoid bursting nuclei. |
| 25 | (i) Insufficient DNA fragmentation. Most DNA fragments are larger than 1,000 bp (Lane 2, Fig. 5b). (ii) Insufficient or over digestion of RNA. If RNA fragments are mostly larger than 1000 nt and contain obvious ribosomal RNA peaks in the bioanalyzer results, they are not fragmented very well; if RNA fragments are mostly between 20 to 100 nt, they are overly digested. |
(i) Inefficient SDS treatment in Step 12 or insufficient amount of restriction enzyme AluI used in Step 18 (ii) The amount of RNase I used to fragment RNA is not optimal in Step 20 and 21. |
(i) Test chromatin fragmentation efficiencies under different volume ratios [v(SDS)/wt(nuclei)], SDS treatment time (Fig. 5b and c) or AluI concentrations. Carry out the procedure up to Step 25 and check the efficiency of DNA fragmentation. DNA in the size range of 100 bp to 3,000 bp indicates good fragmentation (Fig. 4a). (ii) Try a different fold dilution of RNase I in 1× PBS; the recommended RNase I fold dilution is from 100-fold to 10000-fold depending on the digestion results. Carry out the procedure up to Step 25 and check the efficiency of RNA fragmentation. A good example of RNA fragmentation is shown in Fig. 4b. |
| 104 | Too many PCR cycles are required to generate enough material for sequencing (>14 cycles) or the number of cycles varies too much between technical replicates. | Too little material after Step 103, which could be caused by starting with an insufficient number of cells or by low efficiency of DNA-linker proximity ligation. Based on our experience, the quality of annealed linker is critical for the efficiency of proximity ligation. If the single-stranded top linker is in excess after annealing, it will lead to lots of RNA ligated to the single-stranded top linker, thus preventing DNA from being ligated to the linker. | (i) Perform the procedure from the start with more cells. (ii) Redo Steps 34 to 46 and Checkpoint. If the amount of excess top linker is comparable to the amount of annealed ds linker, lower the amount of top linker or increase the amount the top linker and perform Steps 34 to 46 to make sure a complete annealing of the linker by checking that there is no excess top linker left in-annealed. A little excess top strand will not affect the DNA-linker ligation step. |
| 105 | Library fragment sizes are too small (< 500 bp) when visualized on an agarose gel. | DNA-linker proximity ligation efficiency is too low. | Perform Steps 34 to 46 again and optimize linker annealing as mentioned above. |
| 115 | Criterion 2 in this checkpoint was not reached. | Insufficient DNA fragmentation. | Test chromatin fragmentation efficiencies under different volume ratios [v(SDS)/wt(nuclei)], SDS treatment time (Fig. 5b and c) or AluI concentrations (see Step 25). |
Data processing ● Timing 14 h
▲CRITICAL Processing of iMARGI data will be carried out with a standard data processing pipeline using iMARGI-Docker (https://sysbio.ucsd.edu/imargi_pipeline). The iMARGI sequencing dataset at the NCBI Sequence Read Archive (SRA) with accession number SRR820667915 was used as the placeholder dataset in all commands of this section. Please replace this placeholder dataset with your custom dataset.
-
116
Checkpoint. Ensure the computer has 16GB or more memory and is running one of the following Linux distributions: Ubuntu, Debian, Fedora, or CentOS. Make sure that there is enough free space on hard disk. Each 300 million sequencing read pairs require 200 GB free space on hard disk.
-
117
Checkpoint. Check whether Docker has been installed by typing the following command in a Terminal window:
docker -v
If a Docker version is returned, for example “Docker version 18.09.5, build e8ff056”, Docker has been installed. Otherwise, install Docker with root privilege using the command:
sudo curl -fsSL https://get.docker.com |sh -
-
118
Add non-root user to the Docker group with the following command and replace the placeholder demo_user with the Linux username of the user. Then you need to login again with the added user’s account.
sudo usermod -aG docker demo_user
-
119
Checkpoint. Check whether Docker service has been started with the following command:
docker info
If a set of system information is returned, Docker service has been started, continue to Step 120. If the output shows “Cannot connect to the Docker daemon”, start Docker service with the command: sudo service docker start on Ubuntu, Debian, or Fedora, or with the command: sudo systemctl start docker on CentOS.
-
120
Install iMARGI-Docker with the command:
docker pull zhonglab/imargi
-
121
Create a working directory. Download the example dataset (SRR8206679) from NCBI SRA and convert this dataset into FASTQ format. Move the reference genome (GRCh38/hg38) into the working directory.
mkdir ~/imargi_example
cd ~/imargi_example
mkdir data ref
docker run --rm -t -v ~/imargi_example:/imargi zhonglab/imargi f astq-dump --gzip --split-3 SRR8206679
mv SRR8206679_*.fastq.gz ./data
gunzip -d GRCh38 no alt analysis_set_GCA_000001405.15.fasta.gz mv GRCh38_no_alt_analysis_set_GCA_000001405.15.fasta ./ref
-
122
Run the iMARGI data processing pipeline using iMARGI-Docker container by specifying SRR8206679 as the input sequencing data (with the −1 and −2 flags) and GRCh38 as the reference genome (with the –g flag).
cd ~/imargi_example
mkdir ./output
docker run --rm -t -v ~/imargi_example:/imargi zhonglab/imargi i margi_wrapper.sh -r hg38 -N HEK_iMARGI -t 16 -g ./ref/GRCh38_no_al_t_analysis_set_GCA_000001405.15.fasta −1 ./data/SRR8206679_1.fastq gz −2 ./data/SRR8206679_2.fastq.gz -o ./output
The output files include the mapped RNA-DNA read pairs (final_HEK_iMARGI.pairs.gz). This is a compressed .pairs file. The .pairs file format is a standard file format for storing pairs of genomics locations, defined by the NIH 4D Nucleome network29.
-
123
Checkpoint. Open the summary output text file (pipelineStats.log) from the previous step. The first line should read “Sequence mapping QC passed” or “Sequence mapping QC failed”. Only proceed if sequence mapping QC has been passed. The sequence mapping QC is passed when both of the following two criteria are satisfied: (1) At least 50% of the read pairs had at least one end uniquely mapped to the genome (GRCh38/hg38); (2) At least 50% of the uniquely mapped non-duplicate read pairs were retained after the filtering steps. Please note that the deduplication and filtering steps are encapsulated within the previous step, and the user does not have to execute specific commands for these operations.
Further analysis (Optional) ● Timing 3 h
-
124
Count the numbers of intra- and inter-chromosomal interaction read pairs.
docker run --rm -t -v ~/imargi_example:/imargi zhonglab/imargi i margi_stats.sh -i ./output/final_HEK_iMARGI.pairs.gz -o ./output/report_final_HEK_iMARGI.txt
-
125
Remove the proximal read pairs. The proximal read pairs are defined as the read pairs where the RNA end and the DNA end are mapped to the same chromosome and within 200,000 bp. Don’t do this step if you want to analyze nascent transcripts.
docker run --rm -t -v ~/imargi_example:/imargi zhonglab/imargi i margi_distfilter.sh -d 200000 -i ./output/final_HEK_iMARGI.pairs.g z -o ./output/filter200k_final_HEK_iMARGI.pairs.gz
-
126
Annotate the interaction file (.pairs) with gene information from a GTF annotation file, such as GTF annotations built by GENCODE. The gene annotations corresponding to the mapped RNA end and the mapped DNA end of a read pair will be added to the output file as column “gene1” and column “gene2”, respectively.
cd ~/imargi_example
wget ftp://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_24/gencode.v24.annotation.gtf.gz
gunzip -d gencode.v24.annotation.gtf.gz
mv gencode.v24.annotation.gtf ./ref
docker run --rm -t -v ~/imargi_example:/imargi zhonglab/imargi i margi_annotate.sh -A gtf -a ./ref/gencode.v24.annotation.gtf -i ./output/filter200k_final_HEK_iMARGI.pairs.gz -o ./output/annot_filter200k_final_HEK_iMARGI.pairs.gz
Converting .pairs file to other file types (Optional) ● Timing 1 h
-
127
Convert the .pairs file into a .bedpe file30 with the following command. Files in .bedpe format can be further analyzed by other software including bedtools30.
docker run --rm -t -v ~/imargi_example:/imargi zhonglab/imargi i margi_convert.sh -i ./output/ filter200k_final_HEK_iMARGI.pairs.gz -f bedpe -o ./output/filter200k_final_HEK_iMARGI.bedpe.gz
-
128
Convert the .pairs file into .cool and .mcool files31 with the following command. The two dimensional contact matrix in a .mcool file can be visualized by HiGlass32 as a heatmap.
docker run --rm -t -v ~/imargi example:/imargi zhonglab/imargi i margi_convert.sh -i ./output/ filter200k_final_HEK_iMARGI.pairs.gz -f cool -o ./output/filter200k_final_HEK_iMARGI.cool
-
129
Convert the .pairs file into a GIVE interaction (.gin) file33 with the following command. Every read pair in a .gin file can be visualized by GIVE33 as a line connecting the mapped location of the RNA end (RNA lane, Fig. 6) and the DNA end (DNA lane, Fig. 6).
docker run --rm -t -v ~/imargi_example:/imargi zhonglab/imargi i margi_convert.sh -i ./output/ filter200k_final_HEK_iMARGI.pairs.gz -f give -o ./output/filter200k_final_HEK_iMARGI.gin
Troubleshooting
Troubleshooting advice can be found in Table 4 and Fig. 5, a–c.
Timing
Day 1: Cell crosslinking (Step 1). Allocate 2 hours for this step.
Day 2: Cell lysis and DNA fragmentation (Steps 2-19). Allocate 3 hours of hands-on time and an overnight reaction.
Day 3: RNA fragmentation, preparation of RNA and DNA ends for ligation, linker adenylation, and RNA-linker ligation (Steps 20-48). Allocate 4 hours of work and an overnight reaction.
Day 4: Linker-DNA ligation (Steps 49-54): Allocate 45 minutes of work and an overnight reaction.
Day 5: Crosslinking reversal and DNA/RNA extraction (Steps 55-72). Allocate 5 hours.
Day 6: Removal of biotin from unligated linkers (Steps 73-77). Allocate 3 hours.
Day 7: Biotin pull-down, reverse transcription, and ssDNA circularization (Steps 78-98). Allocate 5-6 hours of work and overnight reaction.
Day 8: Cut_oligo annealing, BamHI digestion, library amplification and size selection, and quality check (Steps 99-113). Allocate 6 hours in total.
Day 9 and beyond: Sequencing (Step 114). Allocate appropriate time for sequencing.
Anticipated results
The DNA sequences of a fully constructed iMARGI library are expected to be between 200 bp and 1,000 bp in length (Fig. 4d). The desired read depths depend on the research question. Due to the diverse types of chromatin-associated RNAs and diverse modes of RNA-chromatin interactions, we are not able to recommend a uniform sequencing depth for all future studies. However, the XIST-X chromosome interaction and the interaction between Malat1 lincRNA and NEAT1 gene locus could be reliably identified in HEK293T cells with approximately 30 million total read pairs 14. In general, we recommend 300 million or more read pairs from paired-end sequencing. The first two bases of Read 2 are expected to be enriched with the CT dinucleotide corresponding to the AluI restriction sites. While the minimum requirement in quality control (QC) requires 50% or more C and T at each of the first two positions (see Checkpoint at Step 115), high quality libraries can reach 90% or more C and T at their respective positions. While the minimum QC requirement at the mapping step requires 50% of the read pairs to have at least one end to be uniquely mapped, and 50% of the uniquely mapped non-duplicate read pairs to be retained after the filtering steps (see Checkpoint at Step 123), a high quality library can reach 80% in both metrics.
Although RNA-chromatin interactions vary by cell type and cellular environment, the following observations are expected from a successful experiment. The number of uniquely mapped non-redundant iMARGI read pairs decreases as the distance between the two mapped ends increases, in an approximately linear manner in log-log scale (see Figure 2A in Reference15). Approximately 40% of the read pairs come from nascent transcripts and their nearby genomic sequences, as determined by the distance between the two mapped ends of a read pair. Prominent interactions between XIST and the X chromosome should be observed from differentiated female cells (see Figure 3C of Reference14). The genome-wide distribution of small nuclear RNAs is expected to be non-uniform (see Figure 2 of Reference18), and the genomic regions enriched with small nuclear RNAs are expected to correlate with the A compartment, the nuclear compartment that is more active in transcription (see Figure 5 of Reference18).
Code availability
The iMARGI-Docker software and its documentation are available at: https://sysbio.ucsd.edu/imargi_pipeline. The software is completely open source, under the BSD 2 license. The source code is available at https://github.com/Zhong-Lab-UCSD/iMARGI-Docker. The pre-built Docker image can be pulled from the Docker Hub. The version used in this paper is v1.1.1.
Data availability
An iMARGI dataset is deposited to NCBI Sequence Read Archive (SRA) under accession number: SRR8206679.
Supplementary Material
Acknowledgements
We thank Xingzhao Wen for help with graphics, 4DN DCIC (http://dcic.4dnucleome.org) for discussions on the data processing pipeline, Alvin Zheng, Arya Kaul, Kian Faizi, Niema Moshiri, Jingyao Chen, Xuerui Huang, Ziyang Zhang for testing the iMARGI-Docker software, and Kiran Sriram and Aleysha Chen for proofreading. This work is funded by DP1HD087990 (to S.Z.), NIH 4D Nucleome U01CA200147 (to S.C. and S.Z.), and NIH R00HL122368 (to Z.C.).
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Competing interests
S.Z. is a cofounder of Genemo Inc.
Related links
Key references using this protocol
Sridhar, B. et al. Current Biology 27(4), 602–609 (2017) https://doi.org/10.1016/j.cub.2017.01011
Yan,Z. et al. PNAS116 (8),3328-3337 (2019) https://doi.org/10.1073/pnas.1819788116
Chen, W. et al. iScience 4:204-215 (2018) https://doi.org/10.1016/j.isci.2018.06.005
References
- 1.Nguyen TC,Zaleta-Rivera K, Huang X, Dai X &Zhong S. RNA, Action through Interactions. Trends in genetics: TIG 34, 867–882 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Penny GD, Kay GF, Sheardown SA, Rastan S & Brockdorff N RequirementforXist in X chromosome inactivation. Nature 379,131–137 (1996). [DOI] [PubMed] [Google Scholar]
- 3.Yang F et al. The IncRNA Firre anchorsthe inactiveXchromosometothe nucleolus by binding CTCF and maintains H3K27me3methylation. Genome biology 16,52 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Graf M et al. Telomere Length Determines TERRA and R-Loop Regulation through the Cell Cycle. Cell 170, 72–85 e14 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Yu R, Wang X & Moazed D Epigenetic inheritance mediated by coupling of RNAi and histone H3K9 methylation. Nature 558, 615–619 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Watanabe T et al. Role for piRN As and noncoding RNA in de novo DNA methylation of the imprinted mouse Rasgrfl locus. Science 332, 848–852 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miao Y et al. Enhancer-associated longnon-codingRNALEENEregulatesendothelial nitric oxide synthase and endothelialfunction. Nature communications 9,292 ( 2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Place RF, Li LC, Pookot D, Noonan EJ & Dahiya R MicroRNA-373 inducesexpression of genes with complementary promotersequences. Proceedings of the National Academy of Sciences of the United States ofAmerica 105, 1608–1613 ( 2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morris KV, Chan SW, Jacobsen SE & Looney DJ Small interfering RNA-induced transcriptional gene silencing in human cells. Science 305, 1289–1292 ( 2004). [DOI] [PubMed] [Google Scholar]
- 10.Rinn JL et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs.Cell 129, 1311–1323 ( 2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Simon MD et al. Thegenomicbindingsitesofa noncoding RNA. Proceedings of the National Academy of Sciences of the United States of America 108, 20497–20502 ( 2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chu C, Qu K, Zhong FL, Artandi SE & Chang HY Genomic Maps of Long Noncoding RNA Occupancy Reveal Principlesof RNA-Chromatin Interactions. MOLECULAR CELL 44, 667–678 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Engreitz JM et al. The Xist IncRNA ExploitsThree-DimensionalGenome Architecture to Spread Acrossthe X Chromosome. Science 341,1237973 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sridhar B et al. Systematic Mapping of RNA-Chromatin Interactions In Vivo. Current Biology 27, 602–609 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yan Z et al. Genome-wideco-localization of RNA-DNAinteractionsandfusion RNA pairs. Proceedings of the National Academy of Sciences of the United States of America 116, 3328–3337 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bell JC et al. Chromatin-associated RNAsequencing(ChAR-seq) maps genome-wide RNA-to-DN A contacts. eLife 7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li X et al. GRID-seq revealsthe global RNA-chromatin interactome. Nature Biotechnology 35, 940–940 ( 2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen W et al. RNAsas Proximity-LabelingMediaforldentifyingNuclearSpeckle Positions Relativetothe Genome. iScienceA, 204–215 (2018). [Google Scholar]
- 19.Yin Y et al. U1 snRNP regulates chromatin retention of noncodingRN As. PreprintatbioRixv https://www.biorxiv.org/content/10.1101/310433vl (2018). [Google Scholar]
- 20.Sun G et al. A bias-reducingstrategyin profilingsmall RNAsusingSolexa. RNA 17,2256–2262 (2011).22016383 [Google Scholar]
- 21.Jayaprakash AD, Jabado O, Brown BD & Sachidanandam R Identification and remediation of biases inthe activity of RNA ligases in small-RNAdeep sequencing. N ucieic Acids Res 39, el41 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sorefan K et al. Reducing ligation bias of small RNAs in libraries for next generation sequencing. Silence 3, 4 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li W, Freudenberg J & Miramontes P Diminishing return forincreased Mappability with longersequencingreads: implicationsofthe k-merdistributionsinthe humangenome. BMC bioinformatics 15, 2 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao J et al. Genome-wide identification of polycomb-associated RNAs by RIP-seq. MolCellAQ, 939–953 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hadjiolov AA,Tencheva ZS &Bojadjieva-Mikhailova AG. Isolation and some characteristics of cell nucleifrom brain cortex of adult cat. The Journalof cell bioiogylS, 383–393 (1965). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Suzuki K, Bose P, Leong-Quong RY, Fujita DJ & Riabowol K REAP: Atwo minute cell fractionation method. BMC research notes 3,294 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nabbi A & Riabowol K Isolation of Nuclei. Cold Spring Harbor protocols 2015, 731–734 (2015). [DOI] [PubMed] [Google Scholar]
- 28.Andrews S FastQC: a quality controltool forhighthroughput sequence data. http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc (2010). [Google Scholar]
- 29.Dekker J et al. The4D nucleome project. Nature 549, 219–226 ( 2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Quinlan AR & Hall IM J.B. BEDTools: a flexiblesuite of utilitiesforcomparinggenomic features. 26, 841–842 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Abdennur N&Mirny L Cooler:scalablestorageforHi-Cdataand othergenomically-labeled arrays. 557660 (2019). [Google Scholar]
- 32.Kerpedjiev P et al. HiGlass: web-based visual exploration and analysis of genome interaction maps. Genome biology 19, 125 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cao X,Yan Z, Wu Q, Zheng A & Zhong S GIVE: portable genome browsers for persona I websites. Genome biology 19,92 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
An iMARGI dataset is deposited to NCBI Sequence Read Archive (SRA) under accession number: SRR8206679.