

The XAF1-E134* variant increases the cancer risk for carriers of the TP53-R337H allele.
Abstract
Cancer risk is highly variable in carriers of the common TP53-R337H founder allele, possibly due to the influence of modifier genes. Whole-genome sequencing identified a variant in the tumor suppressor XAF1 (E134*/Glu134Ter/rs146752602) in a subset of R337H carriers. Haplotype-defining variants were verified in 203 patients with cancer, 582 relatives, and 42,438 newborns. The compound mutant haplotype was enriched in patients with cancer, conferring risk for sarcoma (P = 0.003) and subsequent malignancies (P = 0.006). Functional analyses demonstrated that wild-type XAF1 enhances transactivation of wild-type and hypomorphic TP53 variants, whereas XAF1-E134* is markedly attenuated in this activity. We propose that cosegregation of XAF1-E134* and TP53-R337H mutations leads to a more aggressive cancer phenotype than TP53-R337H alone, with implications for genetic counseling and clinical management of hypomorphic TP53 mutant carriers.
INTRODUCTION
The cancer phenotype conferred by germline TP53 mutations is characterized by marked clinical heterogeneity, with no obvious pattern in age of onset, cancer type, and likelihood of subsequent malignancies (1). Carriers of pathogenic TP53 variants are susceptible to a wide spectrum of childhood and adult-onset malignancies, and phenotypic variability exists among carriers, suggesting that environmental and additional genetic factors contribute to cancer risk. Epidemiological and clinical studies revealed the presence of a unique TP53-R337H mutation in Southern and Southeastern Brazil (2–4). Carriers of the founder R337H allele have an increased risk of developing adrenocortical tumors (ACTs) (2–4) and other tumor types, such as breast carcinoma, soft-tissue sarcoma, osteosarcoma, choroid plexus carcinoma (CPC), and thyroid and lung cancers (5–11). Furthermore, family histories of cancer associated with the founder TP53-R337H allele range from isolated cancer cases to those fulfilling classic criteria for Li-Fraumeni syndrome (12).
Although R337H is the only reported widespread TP53 founder allele (4), the extended constitutive haplotype shared by carriers has not been established. We propose that additional variants and polymorphisms in the TP53-R337H haplotype contribute to phenotypic variation and cancer risk. Whole-genome sequencing (WGS) and whole-exome sequencing (WES) analyses of pediatric ACTs associated with the TP53-R337H allele (13) identified a subset of cases who share a recurrent long-range chromosome 17p13 haplotype, containing a nonsense variant (E134*) in the X-linked inhibitor of apoptosis (XIAP)–associated factor 1 (XAF1) gene a putative tumor suppressor functioning in a positive feedback loop with p53 (14). Given the heterogeneity of cancer phenotypes among TP53-R337H carriers and the existence of divergent haplotypes, one of them segregating both mutated alleles, we determined whether XAF1 acts as a modifier of p53 function and cancer susceptibility.
RESULTS
Delineating the common haplotype shared by TP53-R337H carriers
WGS and WES analyses of 12 unrelated Brazilian patients with ACT carrying the TP53-R337H allele (13) revealed an identical 29,971–base pair (bp) sequence (chr17: 7,570,956 to 7,600,926, GRCh37/hg19; corresponding to 0.046 cM based on HapMap) in all carriers. This region encompasses the TP53 and WRAP53 loci and the corresponding internal polymorphisms (hereafter referred to as R337H-only haplotype; Fig. 1). Of note, the TP53-R337H founder allele (4) bears one copy (nonduplicate) of the 16-bp polymorphism in intron 3 (rs17878362) and arginine at codon 72 (rs1042522). Furthermore, a long-range haplotype encompassing a 1,868,598-bp region (chr17: 6,140,570 to 8,009,167, GRCh37/hg19; corresponding to 4.44 cM based on HapMap) was identified in a subset of ACT cases (5 of 12; 42%) (hereafter referred to as extended haplotype). This extended haplotype harbors the “T” allele for single-nucleotide polymorphism (SNP) rs146752602 (chr17: 6,663,899, GRCh37/hg19), resulting in a stop-gain variant (E134*/Glu134Ter) in the putative tumor suppressor XAF1 (Fig. 1). Rare variants observed in the 2-Mb region of chromosome 17p13 were annotated. Only the XAF1-E134* variant showed consistent segregation with the TP53-R337H allele within this region (table S1).
Fig. 1. Schematic diagram of chromosome 17p13 spanning 2-Mb region encompassing TP53-R337H and XAF1-E134* variants.

Identification and location of genes in this region as well as the genotyped SNPs and microsatellite markers are shown. Positions are given relative to build GRCh37/hg19. R337H-only and extended (TP53-R337H + XAF1-E134*) haplotypes observed in the population cohort study are represented.
The R337H haplotypes were further verified by phasing, using genome-wide SNP array data from five unrelated multigenerational TP53-R337H families. Although array data lacked sufficient density to capture the full complement of SNPs obtained by WGS/WES, we inferred genotypes and concluded that TP53-R337H carriers in these families share the common extended haplotype (Fig. 2A) that cosegregates with affected family members. The extended haplotype containing TP53-R337H and XAF1-E134* alleles was also present in relatives not expressing a cancer phenotype, indicating that the combined mutations may not confer complete penetrance (Fig. 2, B and C) (15).
Fig. 2. Risk haplotype and representative pedigrees associated with the extended haplotype as determined by phasing.
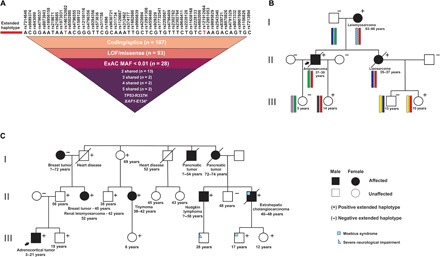
(A) Common SNPs from the Axiom array spanning 2-Mb region in the chromosome 17p13 (see Fig. 1). Of the rare variants falling within haplotype boundaries, only TP53-R337H and XAF1-E134* variants remained after filtering for pathogenicity. ExAC: Exome Aggregation Consortium; MAF: Minor Allele Frequency. (B) Brazilian family (proband no. 108; table S2) diagnosed with angiosarcoma and their relatives. SNPs were used to phase the haplotypes. The risk haplotype harboring both mutant alleles (TP53-R337H and XAF1-E134*) is represented in red bars. The remaining haplotype is represented by different colors as segregating in family members. (C) Spanish family (proband no. 50; table S2). Age at diagnosis and present age or age at death are indicated in each pedigree. ExAC, Exome Aggregation Consortium; MAF, Minor Allele Frequency.
The estimated age of the extended haplotype is 577 years [assuming a 28-year generation time; 95% confidence interval (CI), 208 to 1853] (16), bracketing with colonization of Brazil by Europeans and its previously suggested dispersal route into Southern Brazil (17). Consistently, all four R337H carriers from Europe (Spain, Portugal, and France) included in our study share the common TP53-R337H founder allele as the Brazilians. However, the extended haplotype was observed in the Spanish families and a subset of Brazilian carriers.
TP53-R337H and XAF1-E134* allele frequencies
The TP53-R337H variant (rs121912664) is rare and virtually absent in a worldwide meta-cohort, with an allele frequency of 0.000009151 (gnomAD V2.1.1 control dataset) (18). The TP53-R337H allele was not identified in cancer-free participants reported in the ABraOM (Arquivo Brasileiro Online de Mutações (19) and SELA (Serviço de Sequenciamento em Larga Escala) exome sequence databases (1348 Southeastern Brazilian individuals and 2696 chromosomes) and Global Biobank Engine dataset (337,199 British individuals) (20). However, a neonatal screening study of 171,649 newborns in Paraná, Southern Brazil detected this variant in 461 individuals (three homozygous carriers), corresponding to an estimated allele frequency of 0.001 (21). The TP53-R337H variant is reported in ClinVar (22) as pathogenic (allele ID: 12379).
The XAF1-E134* allele (rs146752602) is observed in non-Finnish Europeans (762 of 128,578 alleles) and worldwide (978 of 281,940 alleles) at an allele frequency of 0.006 and 0.004, respectively (https://gnomad.broadinstitute.org/variant/17-6663899-G-T) (18). This variant occurred at an allele frequency of 0.007 in the Global Biobank Engine database (20) and 0.004 in the ABraOM (19) and SELA Brazilian databases.
The XAF1-E134* variant was also identified in 38 adult survivors of childhood cancer (allelic frequency of 0.006) in the St. Jude Lifetime (SJLIFE) cohort (23). Notably, one patient who had developed rhabdomyosarcoma also harbored the hypomorphic TP53-G187S variant. Two additional potentially pathogenic germline XAF1 mutations were observed in neuroblastoma (XAF1-K235fs) and Hodgkin’s lymphoma (XAF1-Q283*) survivors, both with germline wild-type TP53 (wt-TP53).
TP53-R337H and XAF1-E134* frequencies in newborns from Southern Brazil
In a general screening of 42,438 newborns from Southern Brazil, we identified 147 TP53-R337H carriers. Notably, 101 (69%) also harbored the XAF1-E134* variant. Analysis of parental genomic DNA revealed that both mutated alleles segregated from the same parent. Family history of cancer was unknown for all positive cases.
The frequency of the single XAF1-E134* allele in Southern Brazil was verified by genotyping 3000 newborns negative for the TP53-R337H variant. We identified 23 XAF1-E134*–positive individuals, corresponding to an allele frequency of 0.004 (23 of 6000 chromosomes), consistent with the ABraOM (19) and SELA databases.
R337H-only and extended haplotype frequency in cancer populations
Genotyping of 203 patients with cancer carrying the TP53-R337H founder allele demonstrated that 161 also harbored the XAF1-E134* variant. Segregation analysis determined that both mutated alleles were within the same haplotype. Four homozygous R337H and E134* carriers (proband nos. 46, 61, 90, and 193; table S2) were excluded from further cancer risk analysis to mitigate potentially confounding effects. The relative frequency of the extended haplotype in the remaining 199 patients with cancer was higher (79%) [odds ratio (OR), 1.70; 95% CI, 1.10 to Inf (Infinity); P = 0.022] (Table 1) compared to the newborn group. Stratifying by tumor type, the extended haplotype (93%) was also enriched in patients with a first diagnosis of sarcoma (OR, 6.33; 95% CI, 1.76 to Inf; P = 0.003) (Table 1). Ten additional patients diagnosed with CPC (n = 3), prostate (n = 2), lymphoma (n = 2), thyroid (n = 1), lung (n = 1), and renal (n = 1) carcinoma (Table 1 and table S2) were collectively analyzed as a group because of limited sample size. All harbored the extended haplotype cosegregating the TP53-R337H and XAF1-E134* alleles (Table 1). The relative frequency of the extended haplotype in patients with ACT and breast cancer was comparable to the newborn group even when considering age at diagnosis (Table 1).
Table 1. Tumor distribution in probands harboring R337H-only and extended haplotype.
| Study participants | Total | TP53-R337H |
TP53- R337H + XAF1- E134* |
FDR | P | OR* | CI |
|
Control (newborn screening) |
147 | 46 | 101 | — | — | — | — |
| ACT [total cases (N)] | 102 | 26 | 76 | 0.314 | 0.198 | 1.33 | 0.799 to Inf |
| ACT (<5 years) | 77 | 20 | 57 | 0.314 | 0.251 | 1.297 | 0.742 to Inf |
| ACT (≥5 years) | 25 | 6 | 19 | 0.351 | 0.316 | 1.439 | 0.586 to Inf |
| Sarcomas | 30 | 2 | 28 | 0.03 | 0.003 | 6.329 | 1.756 to Inf |
|
Breast cancer [total cases (N)] |
57 | 14 | 43 | 0.314 | 0.22 | 1.397 | 0.743 to Inf |
| Breast cancer (≤45 years) |
31 | 7 | 24 | 0.314 | 0.23 | 1.558 | 0.680 to Inf |
| Breast cancer (>45 years) |
26 | 7 | 19 | 0.423 | 0.423 | 1.235 | 0.524 to Inf |
| Other cancers | 10 | 0 | 10 | 0.07 | 0.028 | 9.621 | 1.245 to Inf |
| Total | 199† | 42 | 157 | 0.07 | 0.022 | 1.7 | 1.097 to Inf |
|
Multiple tumors [total cases (N)] |
33 | 3 | 30 | 0.03 | 0.006 | 4.525 | 1.517 to Inf |
*For other cancers, group OR was estimated by adding 0.5 to the contingency table. Other ORs are conditional maximum likelihood estimator.
†Four probands (three ACTs and one CPC) homozygous for both variants were excluded from the analysis so all individuals in the analysis are heterozygous for TP53-R337H.
Multiple primary tumors across TP53-R337H carriers
Thirty-three probands (29 females and 4 males) developed multiple primary malignancies (Table 1 and table S2), 30 of which harbored the extended haplotype spanning both mutated alleles (91%) (OR, 4.53; 95% CI, 1.52 to Inf; P = 0.006) (Fig. 3). Patients with breast cancer constitute 52% of these cases (17 of 33), with 16 (including all contralateral breast cancers) harboring the extended haplotype (table S2). Notably, patients developing a third (n = 12) or fourth (n = 3) primary tumor were exclusively associated with the extended haplotype (Fig. 3 and table S2).
Fig. 3. Distribution of tumor types as a second, third, or fourth malignancy among patients with the R337H-only and extended haplotypes.
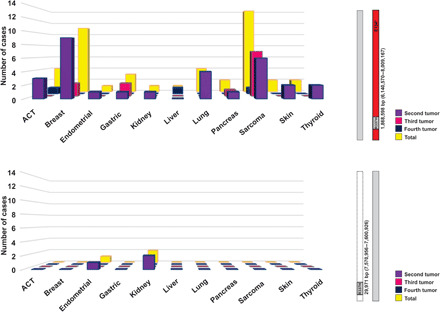
Thirty-three probands (29 females and 4 males) developed multiple primary malignancies. Multiple primary tumors in probands with the extended haplotype (n = 30) are visualized in the upper panel. Multiple primary tumors in probands with the R337H-only haplotype (n = 3) are visualized in the lower panel.
Considering all tumor types after first diagnosis (n = 48), sarcomas were most prevalent (n = 14; 29%) and included three cases of malignant phyllodes of the breast, followed by breast cancer (n = 11; 23%) and adrenocortical carcinoma (n = 4; 8%) (Fig. 3 and table S2).
Family cancer history
Inspection of the 203 pedigrees identified six families that fulfilled classic LFS (Li-Fraumeni syndrome) criteria, whereas the majority met the revised Chompret criteria (n = 141; table S2) (1). Most probands shared the R337H-only or extended haplotype with one parent. However, recombination events leading to the loss of XAF1-E134* (n = 3) or TP53-R337H (n = 1) alleles were observed in individuals from four independent families (nos. 79, 92, 131, and 149) (table S2). For instance, proband no. 149 diagnosed with breast cancer at age 27 was positive for the TP53-R337H founder allele but negative for the XAF1-E134* variant, whereas her sister who developed breast cancer at age 29 shared both derived alleles with their mother. Their father tested negative for both variants.
Exogenous XAF1 increases p53 transcriptional activity
To study the effect of XAF1 on p53 transactivation function, we transiently transfected Saos-2 cells with promoter-luciferase reporters with or without p53 and XAF1 expression vectors. wt-TP53 strongly induced the promoter containing intact p53-binding consensus sites (PG13) but not mutant sites (MG15). Full-length wild-type XAF1 (wt-XAF1) and XAF1-E134* had no effect individually or combined with either reporter (Fig. 4A and fig. S1A). However, wt-XAF1 stimulated wt-p53 transactivation, leading to increased luciferase expression. The nonsense XAF1-E134* was remarkably attenuated in activating p53 and interfered with wt-XAF1 by blocking its ability to increase p53 transactivation in a dominant-negative manner (Fig. 4A and fig. S1A). Comparable transactivation results were obtained using TP53-R337H (Fig. 4A and fig. S1A). Western blot analysis showed that changes in promoter-reporter activities in response to p53 due to coexpression with wt-XAF1 and/or XAF1-E134* were not a result of altered protein levels (Fig. 4B and fig. S1B). Studies with an independent p53-responsive promoter-reporter (p50-2) yielded essentially identical results (fig. S1, C and D). Of interest, wt-XAF1, but not XAF1-E134*, stimulated the transactivation function of other hypomorphic p53 variants such as T125M, R175L, R290H, and G334R (Fig. 4). The pathogenic DNA binding mutant TP53-R175H was transcriptionally inactive irrespective of XAF1. These findings support XAF1 as a functional modifier of wild-type, R337H, and other hypomorphic p53 variants.
Fig. 4. XAF1 increases the transcriptional activity of hypomorphic TP53 variants.
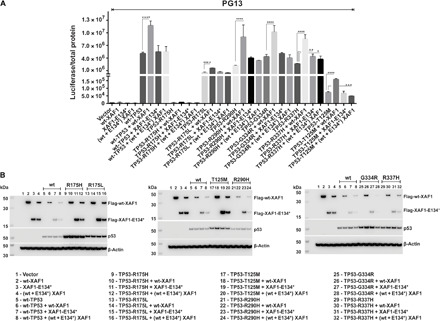
Saos-2 cells were transiently transfected with p53-responsive promoter-luciferase reporters with or without p53 and XAF1 expression vectors, as described in the Supplementary Materials. PG13 promoter-reporter luciferase assay. Wild-type XAF1, but not XAF1-E134*, stimulated the transactivation function of wild-type p53 and other hypomorphic p53 variants such as T125M, R175L, R290H, and G334R. (A) Western blot analysis of p53 and XAF1 in the transfected cell lysates. (B) The columns represent the mean of three independent experiments (±SD), each performed in duplicate. Error bars indicate SDs. Asterisks indicate statistical significance, as determined by one-way analysis of variance (ANOVA) *P < 0.005; **P = 0.0096; ***P = 0.0009; ****P = <0.0001.
Endogenous XAF1 modifies p53 function in primary human fibroblasts
To study the modifier effect of XAF1 upon p53 under more physiological conditions, we analyzed three low-passage primary human fibroblast lines derived from individuals with the following genotypes: (i) wt-XAF1 and wt-TP53 (HF001), (ii) heterozygous XAF1-E134* and TP53-R337H haplotype (HF003), and (iii) homozygous XAF1-E134* and TP53-R337H (HF004). As expected, untreated HF001 cells expressed a basal level of endogenous wild-type p53, which was markedly induced in parallel with its target genes p21CIP1, PUMA, and MDM2 in response to ionizing radiation (IR) [5 grays (Gy)] (Fig. 5A). Similarly, p21CIP1, PUMA, and MDM2 were induced upon DNA damage in the heterozygous HF003 fibroblasts. By contrast, p21CIP1, PUMA, and MDM2 basal levels are low in untreated HF004 fibroblasts and remain unaltered following DNA damage, despite expressing high levels of p53-R337H. These findings suggest that the p53 pathway is functionally compromised in these cells (Fig. 5A). The primary fibroblasts may be defective in the IR response due to the loss of functional XAF1. To address this hypothesis, we used a CRISPR-Cas9 approach to correct the endogenous XAF1-E134* allele and restore wt-XAF1 expression in these cells. Two successfully edited independent clones were identified (HF004-cl1 and HF004-cl2), each harboring wild-type and mutant XAF1 alleles. Re-establishing wt-XAF1 resulted in high expression of full-length XAF1, possibly due to copy-number gain of chromosome 17 in a subset of cells (fig. S2), and restoration of p21CIP1 basal expression in HF004-cl1 and HF004-cl2 cells (Fig. 5A). DNA damage moderately induced p21CIP1, PUMA, and MDM2 (Fig. 5A). These findings support a physiologic role for endogenous XAF1 as a positive regulator of p53 function.
Fig. 5. Correction of the XAF1-E134* mutation using CRISPR-Cas9 in human low-passage fibroblasts restores full-length XAF1 and partial p53 responsiveness.
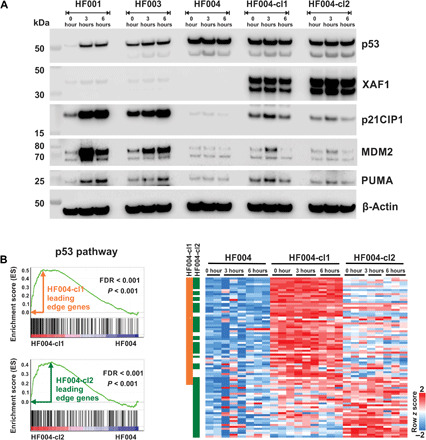
(A) Western blot analysis of MDM2, XAF1, p21CIP1, PUMA, and p53 expression in untreated and irradiated (5 Gy) low-passage fibroblasts. HF001 (wild-type XAF1 and TP53), HF003 (heterozygous XAF1- E134*; TP53-R337H), HF004 (homozygous XAF1-E134*; TP53-R337H), and derived HF004 clones with CRISPR-Cas9–corrected E134*(HF004cl1 and HF004cl2). (B) Gene set enrichment analysis (GSEA) showing the enrichment of p53 pathway signature in edited clones and heat map showing the expression levels in each cell line before and after IR treatment. Color scale represents SDs from the mean (z-score; range, −2 to 2). FDR, false discovery rate.
Gene set enrichment analysis (GSEA) was performed to compare transcriptional programs active in parental HF004 and derived edited clones (HF004-cl1 and HF004-cl2) cell lines before and after IR. Consistently, p53 was among the top significant pathways identified (Fig. 5B and tables S3 and S4). p53 activity was compromised in parental cells but restored in both clones (Fig. 5B). In addition, apoptosis and G2-M checkpoint pathways were also identified as the principal involved pathways in this analysis (fig. S3 and tables S3 and S4). Our results provide previously unidentified insights into cancer cell signaling by including XAF1 as modulator of p53 activity.
DISCUSSION
WGS and WES analyses of pediatric ACTs from Southern Brazil identified a common locus containing identical TP53 and WRAP53 sequences, including corresponding polymorphisms in all TP53-R337H carriers. Moreover, an extended chromosome 17p13 haplotype containing a nonsense mutation in the putative tumor-suppressor gene XAF1 (E134*) was observed in a subset of cases. These divergent haplotypes were verified in newborns from Southern Brazil, showing that 69% of TP53-R337H haplotypes also harbor the XAF1-E134* variant.
XAF1 is a zinc-finger, pro-apoptotic protein that was originally identified in a yeast two-hybrid screen using XIAP as the bait (24). Expression of XAF1 is frequently inactivated in human cancers, primarily due to aberrant promoter methylation and gene silencing, implicating XAF1 in tumor suppression (25). Gain- and loss-of-function studies using cell lines and mouse xenograft models further support XAF1 as a tumor suppressor (14, 25–29). Ectopic expression of XAF1 in parental HCT116 (TP53+/+) cells markedly repressed xenograft tumor growth. In contrast, genetically engineered HCT116 cells that lack endogenous TP53 (TP53−/−) were resistant to XAF1 overexpression and continued to grow in vivo (14). XAF1 reportedly functions in a positive feedback loop with p53, in part, by binding directly to the N-terminal proline-rich domain of p53, thereby interfering with MDM2 binding and ubiquitination of p53 (14). Together, these findings demonstrate that XAF1 can function as a tumor suppressor in a p53-dependent manner.
Consistent with these findings, our promoter-reporter studies demonstrate that full-length exogenous XAF1 enhances the transactivation function of wild-type, R337H, and other hypomorphic p53 variants, whereas XAF1-E134* is markedly attenuated in this regulatory activity. In addition, correction of endogenous of mutant XAF1 in low-passage human HF004 fibroblasts (XAF1E134*/E134*; TP53R337H/R337H) by CRISPR-Cas9 gene editing restored full-length XAF1 expression, reestablished the p21CIP1 basal expression, and the induction of p21CIP1, PUMA, and MDM2 levels upon DNA damage. Furthermore, GSEA identified p53, apoptosis, and G2-M checkpoint pathways among the highest ranked in the XAF1 reversed engineered cell clones (HF004-cl1 and HF004-cl2) compared to the HF004 parental cells. These data support XAF1 as a physiological modulator of p53 function.
Given that XAF1 can modify p53 function, it is reasonable to propose that the extended haplotype could affect tumor type, age of onset, and/or penetrance in TP53-R337H carriers. Genetic screening of 42,438 Southern Brazilian newborns revealed that 1 in 290 individuals harbored the TP53-R337H mutation, 69% of which was also positive for the XAF1-E134* mutation. We understand that positive carriers in this group are cancer prone but with unknown risk, which may reduce the power of detecting an association between haplotype and cancer susceptibility. Nevertheless, the extended haplotype harboring both mutated alleles was significantly enriched in patients who developed sarcoma at first diagnosis (93%) compared to the newborn group (69%). In addition, sarcoma was the most prevalent cancer in subsequent primary tumor diagnoses and one of the most frequently reported malignancies in family members of probands with the extended haplotype. These findings raise the intriguing possibility that XAF1 and p53 may have tissue-specific functions, consistent with other inherited mutations predisposing carriers to specific tumor types, such as BRCA1/BRCA2, RB, or WT1 (30, 31). The impact of XAF1-E134* on TP53-R337H is further evident by the significant excess of multiple primary cancers in carriers of the extended haplotype (91%).
Recombination events were observed whereby individuals from families associated with the extended haplotype have lost either the TP53-R337H or XAF1-E134* allele. These findings underscore the need to validate not only TP53 status but also the constitutive chromosome 17p13 haplotype in the proband and extended family members. The clinical significance and impact of the R337H variant and the corresponding haplotype will be particularly important for future studies assessing penetrance and cancer risk.
In conclusion, we identified XAF1-E134* as a linked variant that acts in concert with the TP53-R337H allele in modulating the cancer phenotype. These findings suggest that the R337H haplotype may affect cancer risk more than simply the R337H allele alone. We also propose that the modifier function of XAF1 is relevant not only to the TP53-R337H founder allele but also to other hypomorphic TP53 alleles. Further studies to advance our understanding of the impact of germline TP53 variants in the context of risk-modifying variants such as XAF1 will have important implications for genetic counseling, surveillance, and clinical management of carriers.
CONCLUSION
The heterogeneity of cancer phenotypes among carriers of the founder TP53-R337H mutation could be explained by the existence of divergent haplotypes, one of them cosegregating mutated alleles in two distinct tumor-suppressor genes: TP53 and XAF1. Screening for both variants and segregation analysis needs to be considered in this context.
MATERIALS AND METHODS
Study design
Physicians from different institutions in Brazil and Europe provided DNA samples from patients with cancer, ascertained as a proband harboring the TP53-R337H allele. Cases were retrospectively selected on the basis of availability of genomic DNA and independent of age at diagnosis, cancer predisposition syndrome, or family history of cancer. Genotyping was performed on genomic DNA from 203 patients with cancer from Brazil (n = 199), Spain (n = 2), Portugal (n = 1), and France (n = 1) (fig. S4). Genomic DNA from 582 family members was also included. Haplotypes were determined by segregation analysis of genomic DNA from parents and descendants or by loss of heterozygosity (LOH) in tumor samples or inferred by a population approach (Supplementary Materials). Clinical, demographic characteristics, and family history of cancer were available (Table 1 and table S2). Genomic DNA from 42,438 newborns undergoing routine genetic screening (Paraná State, Brazil, from January 2016 to July 2017) was analyzed using a polymerase chain reaction (PCR)–based assay (Supplementary Materials) to determine the frequencies of XAF1-E134* and TP53-R337H variants (newborn cohort). This study was approved by the local ethics committees and the institutional review board at St. Jude Children’s Research Hospital.
Haplotype reconstruction
A 2-Mb region on chromosome 17p13, including the TP53 locus (chr17: 6,000,000 to 8,000,000, GRCh37/hg19), was analyzed using WGS (n = 10) and WES (n = 2) data from 12 unrelated Brazilian patients with ACT harboring TP53-R337H allele (European Genome-phenome Archive: EGAS00001000257) (13). Because of chromosome 17 copy-neutral LOH in tumor tissue (13), haplotypes of the 2-Mb region could be inferred without prior phasing. The resulting haplotypes were verified in 25 individuals (21 TP53-R337H carriers) from five unrelated families, with available SNP data obtained by Axiom Genome-Wide CEU 1 Array (Affymetrix) analysis (Supplementary Materials).
Database query
TP53-R337H (rs121912664) and XAF1-E134* (rs146752602) variant frequencies were retrieved from the (i) Genome Aggregation Database (https://gnomad.broadinstitute.org/) (18); (ii) Global Biobank Engine with genome-wide association analysis of 337,199 white British individuals (20); and (iii) ABraOM (n = 609) (19) and SELA (n = 739) (www.premium.fm.usp.br/index.php?mpg=11.42.00&lab=SELA) databases, containing exome sequences of cancer-free individuals from Southeastern Brazil. TP53-R337H and XAF1-E134* variant frequencies were also derived from the SJLIFE cohort (23), with genomic information from 3006 adult survivors of childhood cancer.
Genetic, molecular, in vitro, and in vivo functional studies
TP53 and XAF1 variants were genotyped by PCR-restriction fragment-length polymorphism analysis and Sanger sequencing (fig. S5). The TP53 and XAF1 loci were mapped by SNPs and microsatellite marker analyses, as described in the Supplementary Materials. Functional promoter-reporter analyses and gene editing studies using CRISPR-Cas9 (correction of endogenous XAF1-E134* in primary human fibroblasts) and expression analysis by microarray and cytogenetic analysis are described in the Supplementary Materials.
Statistical analyses
Conditional on individuals harboring the germline TP53-R337H founder variant and a cancer diagnosis, a target cohort of 203 patients was analyzed. Within each tumor type observed, as total or stratified by age, we estimated the prevalence of the haplotype that segregates TP53-R337H and XAF1-E134* mutations, along with exact 95% CI. A lower bound of the 95% CI not including the prevalence rate of 70% (observed in the newborn cohort) would be suggestive of an increased prevalence of this haplotype within that tumor type. Statistical analysis was carried out using GraphPad Prism and Stata v.12 packages.
Supplementary Material
Acknowledgments
We especially thank the patients and their family members for participating in this study. We thank M. Loyd and S. Spencer from the Hartwell Center, V. Valentine and J. Wilbourne from Cytogenetics, and R. Holcomb from the Anatomic Pathology at St. Jude Children’s Research Hospital. We thank V. Shanker for scientific editorial assistance and D. Kumar Srivastava, PhD for statistical assistance. Funding: This work was supported by the Cancer Center Support grant CA21765 and the American Lebanese Syrian Associated Charities (ALSAC). We also recognize the support of the Speer Charitable Trust and the EXPOGEN-CANCER CNRS International Associated Laboratory. Author contributions: Conception and design: E.M.P. and G.P.Z. Provision of study materials or patients: E.M.P., B.C.F., H.C.R.G., M.N.F., M.C.B.V.F., P.A.-P., E.M.S.F.R., G.F., T.E.B.C., S.A.S., E.I.P., H.S., J.L.F.-S., C.L., G.C., D.V., V.O.-F., L.B., T.E., E.M.S., K.N.M., M.I.A., K.C.d.A., L.K., A.P., A.C.L., B.B.M., M.Q.A., V.B.B., C.M.B., E.W.S.S., C.M., C.R.N.R., M.M., W.Z., K.J., A.V., P.P.K., K.M.S., H.K., M.M.P., I.Z.S.P., C.R.-G., R.C.R., and G.P.Z. Data analysis and interpretation: E.M.P., B.C.F., W.C., M.Y., E.L., K.V.H., J.W., M.R.C., A.J.M., E.L., K.E.N., M.K., J.P.C., S.P.-M., M.G.T., E.R., Y.D., J.Z., G.N., G.W., and G.P.Z. Manuscript writing: E.M.P. and G.P.Z. Final approval of manuscript: All authors. Competing interests: H.C.R.G. reports personal fees from AstraZeneca do Brasil Ltda. and the Instituto Hermes Pardini S.A. A.P. reports personal fees from Bayer, EUSA, Array, and Roche. M.I.A. reports personal fees from AstraZeneca do Brasil Ltda. E.M.P., R.C.R., and G.P.Z. report a patent GENOTYPING ASSAYS TO IDENTIFY MUTATIONS IN XAF1 pending (PCT/IB2019/053202, filed 17 April 2019) to St. Jude Children’s Research Hospital. The other authors declare that they have no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/26/eaba3231/DC1
REFERENCES AND NOTES
- 1.McBride K. A., Ballinger M. L., Killick E., Kirk J., Tattersall M. H., Eeles R. A., Thomas D. M., Mitchell G., Li-Fraumeni syndrome: Cancer risk assessment and clinical management. Nat. Rev. Clin. Oncol. 11, 260–271 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Ribeiro R. C., Sandrini F., Figueiredo B., Zambetti G. P., Michalkiewicz E., Lafferty A. R., DeLacerda L., Rabin M., Cadwell C., Sampaio G., Cat I., Stratakis C. A., Sandrini R., An inherited p53 mutation that contributes in a tissue-specific manner to pediatric adrenal cortical carcinoma. Proc. Natl. Acad. Sci. U.S.A. 98, 9330–9335 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Latronico A. C., Pinto E. M., Domenice S., Fragoso M. C. B. V., Martin R. M., Zerbini M. C., Lucon A. M., Mendonca B. B., An inherited mutation outside the highly conserved DNA-binding domain of the p53 tumor suppressor protein in children and adults with sporadic adrenocortical tumors. J. Clin. Endocrinol. Metab. 86, 4970–4973 (2001). [DOI] [PubMed] [Google Scholar]
- 4.Pinto E. M., Billerbeck A. E. C., Fragoso M. C. B. V., Domenice S., Mendonca B. B., Latronico A. C., Founder effect for the highly prevalent R337H mutation of tumor suppressor p53 in Brazilian patients with adrenocortical tumors. Arq. Bras. Endocrinol. Metabol. 48, 647–650 (2004). [DOI] [PubMed] [Google Scholar]
- 5.Cury N. M., Ferraz V. E., Silva W. A. Jr., TP53 p.R337H prevalence in a series of Brazilian hereditary breast cancer families. Hered. Cancer Clin. Pract. 12, 8 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Achatz M. I. W., Olivier M., Le Calvez F., Martel-Planche G., Lopes A., Rossi B. M., Ashton-Prolla P., Giugliani R., Palmero E. I., Vargas F. R., Da Rocha J. C. C., Vettore A. L., Hainaut P., The TP53 mutation, R337H, is associated with Li-Fraumeni and Li-Fraumeni-like syndromes in Brazilian families. Cancer Lett. 245, 96–102 (2007). [DOI] [PubMed] [Google Scholar]
- 7.Marcel V., Palmero E. I., Falagan-Lotsch P., Martel-Planche G., Ashton-Prolla P., Olivier M., Brentani R. R., Hainaut P., Achatz M. I., TP53 PIN3 and MDM2 SNP309 polymorphisms as genetic modifiers in the Li-Fraumeni syndrome: Impact on age at first diagnosis. J. Med. Genet. 46, 766–772 (2009). [DOI] [PubMed] [Google Scholar]
- 8.Custodio G., Taques G. R., Figueiredo B. C., Gugelmin E. S., Figueiredo M. M. O., Watanabe F., Pontarolo R., Lalli E., Torres L. F., Increased incidence of choroid plexus carcinoma due to the germline TP53 R337H mutation in southern Brazil. PLOS ONE 6, e18015 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oliveira C. R., Mendonça B. B., de Camargo O. P., Pinto E. M., Nascimento S. A., Latorre-Mdo R., Zerbini M. C., Classical osteoblastoma, atypical osteoblastoma, and osteosarcoma: A comparative study based on clinical, histological, and biological parameters. Clinics 62, 167–174 (2007). [DOI] [PubMed] [Google Scholar]
- 10.Formiga M. N. C., de Andrade K. C., Kowalski L. P., Achatz M. I., Frequency of thyroid carcinoma in Brazilian TP53 p.R337H carriers with Li Fraumeni syndrome. JAMA Oncol. 3, 1400–1402 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Couto P. P., Bastos-Rodrigues L., Schayek H., Melo F. M., Lisboa R. G. C., Miranda D. M., Vilhena A., Bale A. E., Friedman E., De Marco L., Spectrum of germline mutations in smokers and non-smokers in Brazilian non-small-cell lung cancer (NSCLC) patients. Carcinogenesis 38, 1112–1118 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Achatz M. I., Zambetti G. P., The inherited p53 mutation in the Brazilian population. Cold Spring Harb. Perspect. Med. 6, a026195 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pinto E. M., Chen X., Easton J., Finkelstein D., Liu Z., Pounds S., Rodriguez-Galindo C., Lund T. C., Mardis E. R., Wilson R. K., Boggs K., Yergeau D., Cheng J., Murder H. L., Manne J., Jenkins J., Mastellaro M. J., Figueiredo B. C., Dyer M. A., Pappo A., Zhang J., Downing J. R., Ribeiro R. C., Zambetti G. P., Genomic landscape of paediatric adrenocortical tumours. Nat. Commun. 6, 6302 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee M.-G., Han J., Jeong S.-I., Her N.-G., Lee J.-H., Ha T.-K., Kang M.-J., Ryu B.-K., Chi S.-G., XAF1 directs apoptotic switch of p53 signaling through activation of HIPK2 and ZNF313. Proc. Natl. Acad. Sci. U.S.A. 111, 15532–15537 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nadeau J. H., Modifier genes in mice and humans. Nat. Rev. Genet. 2, 165–174 (2001). [DOI] [PubMed] [Google Scholar]
- 16.Pagani L., Diekmann Y., Sazzini M., De Fanti S., Rondinelli M., Farnetti E., Casali B., Caretto A., Novara F., Zuffardi O., Garagnani P., Mantero F., Thomas M. G., Luiselli D., Rossi E., Three reportedly unrelated families with Liddle syndrome inherited from a common ancestor. Hypertension 71, 273–279 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garritano S., Gemignani F., Palmero E. I., Olivier M., Martel-Planche G., Le Calvez-Kelm F., Brugiéres L., Vargas F. R., Brentani R. R., Ashton-Prolla P., Landi S., Tavtigian S. V., Hainaut P., Achatz M. I., Detailed haplotype analysis at the TP53 locus in p.R337H mutation carriers in the population of Southern Brazil: Evidence for a founder effect. Hum. Mutat. 31, 143–150 (2010). [DOI] [PubMed] [Google Scholar]
- 18.Lek M., Karczewski K. J., Minikel E. V., Samocha K. E., Banks E., Fennell T., O’Donnell-Luria A. H., Ware J. S., Hill A. J., Cummings B. B., Tukiainen T., Birnbaum D. P., Kosmicki J. A., Duncan L. E., Estrada K., Zhao F., Zou J., Pierce-Hoffman E., Berghout J., Cooper D. N., Deflaux N., DePristo M., Do R., Flannick J., Fromer M., Gauthier L., Goldstein J., Gupta N., Howrigan D., Kiezun A., Kurki M. I., Moonshine A. L., Natarajan P., Orozco L., Peloso G. M., Poplin R., Rivas M. A., Ruano-Rubio V., Rose S. A., Ruderfer D. M., Shakir K., Stenson P. D., Stevens C., Thomas B. P., Tiao G., Tusie-Luna M. T., Weisburd B., Won H.-H., Yu D., Altshuler D. M., Ardissino D., Boehnke M., Danesh J., Donnelly S., Elosua R., Florez J. C., Gabriel S. B., Getz G., Glatt S. J., Hultman C. M., Kathiresan S., Laakso M., McCarroll S., McCarthy M. I., McGovern D., McPherson R., Neale B. M., Palotie A., Purcell S. M., Saleheen D., Scharf J. M., Sklar P., Sullivan P. F., Tuomilehto J., Tsuang M. T., Watkins H. C., Wilson J. G., Daly M. J., MacArthur D. G.; Exome Aggregation Consortium , Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Naslavsky M. S., Yamamoto G. L., De Almeida T. F., Ezquina S. A. M., Sunaga D. Y., Pho N., Bozoklian D., Sandberg T. O. M., Brito L. A., Lazar M., Bernardo D. V., Amaro E. Jr., Duarte Y. A. O., Lebrão M. L., Passos-Bueno M. R., Achatz M. I., Exomic variants of an elderly cohort of Brazilians in the ABraOM database. Hum. Mutat. 38, 751–763 (2017). [DOI] [PubMed] [Google Scholar]
- 20.McInnes G., Tanigawa Y., DeBoever C., Lavertu A., Olivieri J. E., Aguirre M., Rivas M. A., Global Biobank Engine: Enabling genotype-phenotype browsing for biobank summary statistics. Bioinformatics 35, 2495–2497 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Custódio G., Parise G. A., Filho N. K., Komechen K., Sabbaga C. C., Rosati R., Grisa L., Parise I. Z. S., Pianovski M. A. D., Fiori C. M. C. M., Ledesma J. A., Barbosa J. R. S., Figueiredo F. R. O., Sade E. R., Ibañez H., Arram S. B. I., Stinghen S. T., Mengarelli L. R., Figueiredo M. M. O., Carvalho D. C., Avilla S. G. A., Woiski T. D., Poncio L. C., Lima G. F. R., Pontarolo R., Lalli E., Zhou Y., Zambetti G. P., Ribeiro R. C., Figueiredo B. C., Impact of neonatal screening and surveillance for the TP53 R337H mutation on early detection of childhood adrenocortical tumors. J. Clin. Oncol. 31, 2619–2626 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Landrum M. J., Lee J. M., Benson M., Brown G. R., Chao C., Chitipiralla S., Gu B., Hart J., Hoffman D., Jang W., Karapetyan K., Katz K., Liu C., Maddipatia Z., Malheiro A., McDaniel K., Ovetsky M., Riley G., Zhou G., Holmes J. B., Kattman B. L., Maglott D. R., ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 46, D1062–D1067 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Z., Wilson C. L., Easton J., Thrasher A., Mulder H., Liu Q., Hedges D. J., Wang S., Rusch M. C., Edmonson M. N., Levy S., Lanctot J. Q., Caron E., Shelton K., Currie K., Lear M., Patel A., Rosencrance C., Shao Y., Vadodaria B., Yergeau D., Sapkota Y., Brooke R. J., Moon W., Rampersaud E., Ma X., Chang T.-C., Rice S. V., Pepper C., Zhou X., Chen X., Chen W., Jones A., Boone B., Ehrhardt M. J., Krasin M. J., Howell R. M., Phillips N. S., Lewis C., Srivastava D., Pui C.-H., Kesserwan C. A., Wu G., Nichols K. E., Downing J. R., Hudson M. M., Yasui Y., Robinson L. L., Zhang J., Genetic risk for subsequent neoplasms among long-term survivors of childhood cancer. J. Clin. Oncol. 36, 2078–2090 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liston P., Fong W. G., Kelly N. L., Toji S., Miyazaki T., Conte D., Tamai K., Craig C. G., McBurney M. W., Korneluk R. G., Identification of XAF1 as an antagonist of XIAP anti-caspase activity. Nat. Cell Biol. 3, 128–133 (2001). [DOI] [PubMed] [Google Scholar]
- 25.Plenchette S., Cheung H. H., Fong W. G., LaCasse E. C., Korneluk R. G., The role of XAF1 in cancer. Curr. Opin. Investig. Drugs 8, 469–476 (2007). [PubMed] [Google Scholar]
- 26.Fong W. G., Liston P., Rajcan-Separovic E., St. Jean M., Craig C., Korneluk R. G., Expression and genetic analysis of XIAP-associated factor 1 (XAF1) in cancer cell lines. Genomics 70, 113–122 (2000). [DOI] [PubMed] [Google Scholar]
- 27.Victoria-Acosta G., Vazquez-Santillan K., Jimenez-Hernandez L., Muñoz-Galindo L., Maldonado V., Martinez-Ruiz G. U., Melendez-Zajgla J., Epigenetic silencing of the XAF1 gene is mediated by the loss of CTCF binding. Sci. Rep. 5, 14838 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jeong S.-I., Kim J.-W., Ko K.-P., Ryu B.-K., Lee M.-G., Kim H.-J., Chi S.-G., XAF1 forms a positive feedback loop with IRF-1 to drive apoptotic stress response and suppress tumorigenesis. Cell Death Dis. 9, 806 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shin C.-H., Lee M.-G., Han J., Jeong S.-I., Ryu B.-K., Chi S.-G., Identification of XAF1–MT2A mutual antagonism as a molecular switch in cell-fate decisions under stressful conditions. Proc. Natl. Acad. Sci. U.S.A. 114, 5683–5688 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schneider G., Schmidt-Supprian M., Rad R., Saur D., Tissue-specific tumorigenesis: Context matters. Nat. Rev. Cancer 17, 239–253 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haigis K. M., Cichowski K., Elledge S. J., Tissue-specificity in cancer: The rule, not the exception. Science 363, 1150–1151 (2019). [DOI] [PubMed] [Google Scholar]
- 32.Abecasis G. R., Cherny S. S., Cookson W. O., Cardon L. R., Merlin—Rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 30, 97–101 (2002). [DOI] [PubMed] [Google Scholar]
- 33.Delaneau O., Zagury J.-F., Marchini J., Improved whole-chromosome phasing for disease and population genetic studies. Nat. Methods 10, 5–6 (2013). [DOI] [PubMed] [Google Scholar]
- 34.Lander E. S., Green P., Construction of multilocus genetic linkage maps in humans. Proc. Natl. Acad. Sci. U.S.A. 84, 2363–2367 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.O’Connell J., Gurdasani D., Delaneau O., Pirastu N., Ulivi S., Cocca M., Traglia M., Huang J., Huffman J. E., Rudan I., McQuillan R., Fraser R. M., Campbell H., Polasek O., Asiki G., Ekoru K., Hayward C., Wright A. F., Vitart V., Navarro P., Zagury J.-F., Wilson J. F., Toniolo D., Gasparini P., Soranzo N., Sandhu M. S., Marchini J., A general approach for haplotype phasing across the full spectrum of relatedness. PLOS Genet. 10, e1004234 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pinto E. M., Ribeiro R. C., Li J., Taja-Chayeb L., Carrasco L. F., Peña-Torres M. L., Vidal-Millán S., Maldonado-Mtz H., Dueñas-Gonzáles A., McGregor L., Zambetti G. P., An identical, complex TP53 mutation arising independently in two unrelated families with diverse cancer profiles: The complexity of interpreting cancer risk in carriers. Oncogene 1, e1 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zambetti G. P., Bargonetti J., Walker K., Prives C., Levine A. J., Wild-type p53 mediates positive regulation of gene expression through a specific DNA sequence element. Genes Dev. 6, 1143–1152 (1992). [DOI] [PubMed] [Google Scholar]
- 38.Hinds P. W., Finlay C. A., Quartin R. S., Baker S. J., Fearon E. R., Vogelstein B., Levine A. J., Mutant p53 DNA clones from human colon carcinomas cooperate with ras in transforming primary rat cells: A comparison of the “hot spot” mutant phenotypes. Cell Growth Differ. 1, 571–580 (1990). [PubMed] [Google Scholar]
- 39.el-Deiry W. S., Tokino T., Velculescu V. E., Levy D. B., Parsons R., Trent J. M., Lin D., Mercer W. E., Kinzler K. W., Vogelstein B., WAF1, a potential mediator of p53 tumor suppression. Cell 75, 817–825 (1993). [DOI] [PubMed] [Google Scholar]
- 40.Pappas K., Xu J., Zairis S., Resnick-Silverman L., Abate F., Steinbach N., Ozturk S., Saal L. H., Su T., Cheung P., Schmidt H., Aaronson S., Hibshoosh H., Manfredi J., Rabadan R., Parsons R., p53 maintains baseline expression of multiple tumor suppressor genes. Mol. Cancer Res. 15, 1051–1062 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Connelly J. P., Pruett-Miller S. M., CRIS.py: A versatile and high-throughput analysis program for CRISPR-based genome editing. Sci. Rep. 9, 4194 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Irizarry R. A., Hobbs B., Collin F., Beazer-Barclay Y. D., Antonellis K. J., Scherf U., Speed T. P., Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4, 249–264 (2003). [DOI] [PubMed] [Google Scholar]
- 43.Subramanian A., Tamayo P., Mootha V. K., Mukherjee S., Ebert B. L., Gillette M. A., Paulovich A., Pomeroy S. L., Golub T. R., Lander E. S., Mesirov J. P., Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/26/eaba3231/DC1