

Goat domestication selection mainly focused on immune and neural genes and involved both standing and introgressed variation.
Abstract
Goat domestication was critical for agriculture and civilization, but its underlying genetic changes and selection regimes remain unclear. Here, we analyze the genomes of worldwide domestic goats, wild caprid species, and historical remains, providing evidence of an ancient introgression event from a West Caucasian tur-like species to the ancestor of domestic goats. One introgressed locus with a strong signature of selection harbors the MUC6 gene, which encodes a gastrointestinally secreted mucin. Experiments revealed that the nearly fixed introgressed haplotype confers enhanced immune resistance to gastrointestinal pathogens. Another locus with a strong signal of selection may be related to behavior. The selected alleles at these two loci emerged in domestic goats at least 7200 and 8100 years ago, respectively, and increased to high frequencies concurrent with the expansion of the ubiquitous modern mitochondrial haplogroup A. Tracking these archaeologically cryptic evolutionary transformations provides new insights into the mechanisms of animal domestication.
INTRODUCTION
Domestication presents an extreme shift of physiological and behavioral stress for free-living animals (1) to a high-density and disease-prone anthropogenic environment, especially for herbivores. The goat (Capra hircus) was one of the first domesticated livestock species, demonstrating remarkable adaptability and versatility (2, 3), and it has been intimately associated with human dispersal (4). Recent studies have identified candidate targets of selection during goat domestication, including loci related to pigmentation, xenobiotic metabolism, and milk production (5, 6); however, the evolutionary dynamics of key genes involved in adaptation during the early phase of domestication remain unclear.
Goat domestication is believed to have begun ~11,000 years ago from a mosaic of wild bezoar populations (Capra aegagrus) (2, 6). However, other wild Capra species are widely distributed and many of them can hybridize with domestic goats (7–9). Their contribution to the goat domestication process remains unexplored. Introgression of adaptive variants has been widely recognized as a notable evolutionary phenomenon in humans (10), sheep (11), and cattle (12), and it may be particularly effective for increasing fitness without negative pleiotropic effects as demonstrated in other species (13). Here, we conducted a comprehensive population genomic survey of modern goat populations distributed across the world, six wild Capra species, and previously published ancient goat genomes to investigate temporal shifts in key genetic variants under selection during goat domestication.
RESULTS
Population structure and origin of domestic goats
We sequenced 101 Capra genomes (coverage, 3 to 47 times; average, 12 times), including 88 domestic goats collected from three different continents, one bezoar, one Alpine ibex (Capra ibex), three Siberian ibex (Capra sibirica), three Markhors (Capra falconeri), one West Caucasian tur (Capra caucasica), and four Nubian ibex × domestic goat hybrids (Capra nubiana × C. hircus) (Fig. 1A and text S1). We also sequenced five ancient goat samples at ~0.04 to 13.44 times coverage (Fig. 1A and text S1), including the earliest known Chinese archaeological samples from the Neolithic time period (14). Together with the publicly available genomic sequences for modern goat and bezoar (table S1) (5, 15), as well as ancient genomes from (table S4) (6), we compiled a worldwide collection of 164 modern domestic goats, 52 ancient goats, 24 modern bezoars, and 4 ancient bezoars.
Fig. 1. Geographic distribution and genetic affinities of wild and domestic goats used in this study.
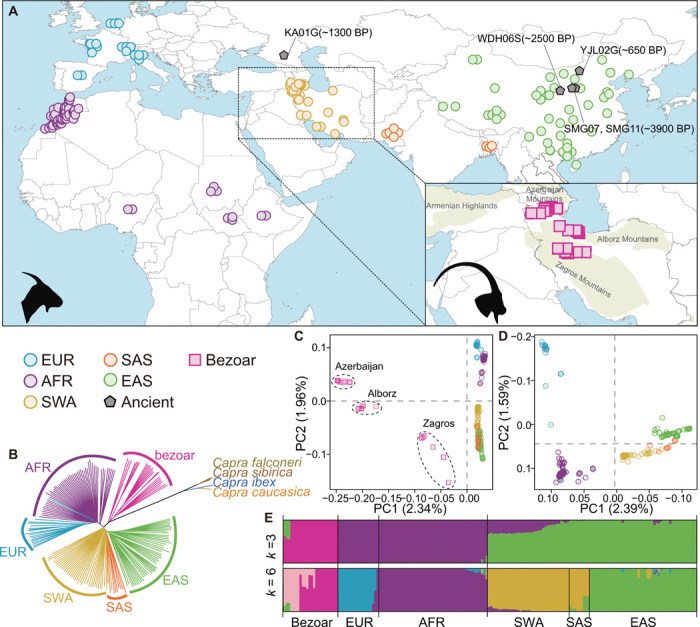
(A) Locations of wild (squares), domestic (circles), and ancient (pentagon) goats for each major geographic group. Domestic goats are colored to mirror their geographic origin. BP, before the present. (B) A neighbor-joining tree from the genome sequences used in this study. The branches are colored following the same color code used in (A). (C and D) PCA of bezoars and domestic goats (C) and domestic goats only (D). (E) ADMIXTURE results for k = 3 and k = 6, which had the low cross-validation error.
A whole-genome neighbor-joining phylogenetic tree reveals that all domestic goats form a monophyletic sister lineage to bezoars (Fig. 1B and fig. S4), confirming that modern domestic goats are the descendants of bezoar-like ancestors (16). The other four wild Capra species (C. ibex, C. sibirica, C. falconeri, and C. caucasica), which are referred to as the ibex-like species (17), fall exclusively in another branch divergent from bezoar-goat (Fig. 1B and fig. S4). Principal components analysis (PCA) shows that the three bezoar populations, which were collected in the Middle East (Fig. 1A) near the domestication center (2, 18), are structured and cluster corresponding to their geographic origins (Fig. 1C). Within domestic goats, PCA and model-based clustering (k = 3) show that Asian goats are genetically distinct from European (EUR) and African (AFR) samples (Fig. 1, D and E). At k = 6, Asian goats further split into two geographic subgroups: Southwest Asia–South Asia (SWA-SAS) and East Asia (EAS) (Fig. 1E). This geographic structure is also supported by TreeMix (fig. S8) and haplotype-based statistics (fig. S9), in agreement with the scenario that the ancestors of present-day domestic goat populations followed distinct dispersal routes along the east-west axis of Afro-Eurasia (Fig. 2A and table S6) (4). This dispersal scenario was further supported by the observation that populations far from the domestication center (non-SWA) had elevated linkage disequilibrium and reduced genetic diversity (fig. S10).
Fig. 2. Gene flow during the early stage of goat domestication and diffusion.
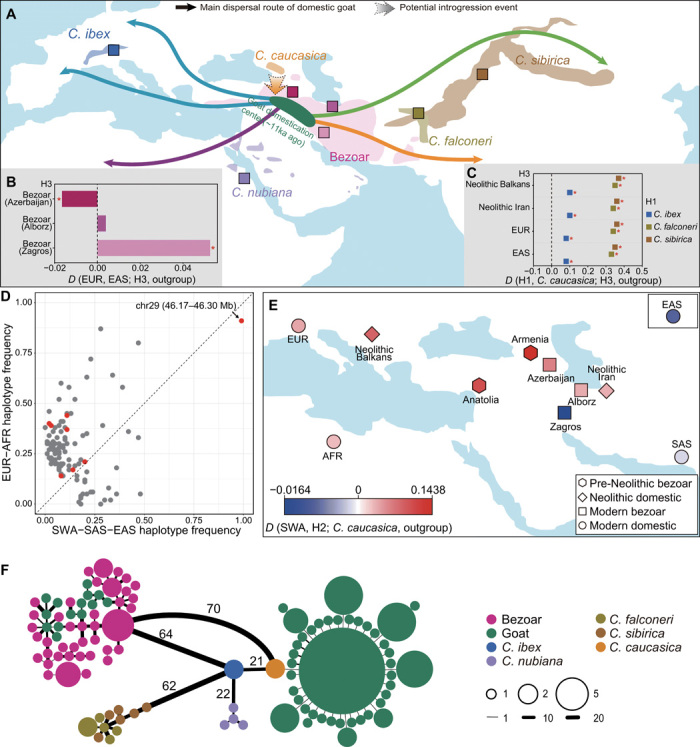
(A) Geographic distribution of the wild Capra species and the dispersal routes of domestic goats out of their domestication areas (3, 7). Sampling locations for ibex-like species are shown by squares. (B) Allele sharing between modern bezoars (H3) and domestic goats. (C) Allele sharing between domestic goats (H3) and ibex-like species (H1). See also table S8 for other populations (H3) investigated. Statistically significant results, defined by |Z scores| ≥ 3, are marked with a red asterisk for (B) and (C). (D) A scatter plot of introgressed haplotype frequency in EUR-AFR and Asian goat populations. The red dots represent immune-related loci. (E) A heat map of D statistic testing for the differential affinity between SWA (blue) and H2 (red), where H2 represents the individual or population of bezoars and domestic goats located in the map. The East Asian goats are depicted in the box in the upper right corner. (F) Haplotype network from the number of pairwise differences at the MUC6 nonrepeat region. All domestic goats are colored in green, and wild Capra species are divided into five subgroups, including the haplotypes from hybrids of Nubian ibex × domestic goat. The radius of the pie chart and the width of edges were log2-transformed.
Our demographic analyses using multiple sequentially Markovian coalescent (MSMC), SMC++, and ∂a∂i indicate that the divergence times between modern Asian and European goat populations predated the archaeologically estimated domestication time (~11,000 years ago) (fig. S12 and text S2). In addition, D statistics revealed that the bezoars from Zagros display higher genetic affinity to eastern domestic populations, whereas the bezoars in Azerbaijan show higher affinity to western domestic populations (Fig. 2B and fig. S15A). Therefore, the deep coalescence times found between east-west population pairs can be explained by their origin in structured ancestral bezoar populations (6) or by postdomestication recruitment from different local bezoar populations.
Gene flow from ibex-like species to predomestication bezoars and modern goats
D statistics reveal that all four ibex-like species have significant signals of allele sharing with ancient and modern goats, indicative of admixture (Fig. 2C and table S8). We then examined this genome-wide pattern of admixture between ibex-like species and domestic goats using D statistics and identity by state in 20-kb sliding windows. We further verified candidate introgressed regions using Sprime and maximum likelihood (ML) phylogenetic trees. Using a conservative criterion (namely, only keeping putative introgressed haplotypes with a frequency higher than 0.1 in goats), we identified 112 genomic segments overlapping with 81 protein-coding genes with signatures of introgression from ibex-like species (Fig. 2D, fig. S16, and data file S1). A Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis for these genes shows that the most significantly enriched category is amoebiasis (hypergeometric test, adjusted P < 5.28 × 10−3: table S10), which is related to parasite invasion and immunosuppression, including four genes (SERPINB3, SERPINB4, CD1B, and COL4A4). Three additional genes (BPI, MAN2A1, and CD2AP) are also involved in immune function (19–21). In these segments, we observed a pronounced signature of putatively introgressed alleles from the West Caucasian tur (fig. S17), consistent with this species showing the greatest genome-wide allele sharing with domestic goats (Fig. 2C).
While investigating the history of West Caucasian tur admixture, we found that the West Caucasian tur shared more alleles with all four predomesticated bezoars (an Armenian bezoar dating to >47,000 years ago and three Anatolian bezoars dating to ~13,000 years ago) and especially Armenian bezoar, compared to present-day bezoars and domesticates (Fig. 2E). Further f3 statistics in the form of f3 (modern bezoar, West Caucasian tur; target) support gene flow from the West Caucasian tur to ancient Armenian bezoar (table S9). Together with the fact that three predomestication Anatolian bezoars had a tur-like mitochondrial haplotype (6), our results provide evidence for a predomestication admixture event of bezoar with a tur-like species. This ancient admixture event is further supported by TreeMix analyses (fig. S8C) and the close geographical proximity between the West Caucasian tur and the ancient Armenian bezoar, with both being, in turn, closer to the goat domestication center than any other living ibex-like species (Fig. 2A). Although all modern goat genomes suggest admixture with tur, Neolithic Balkan and modern EUR and AFR goats show more allele sharing with tur compared to Asian goats (fig. S15B), probably because of their higher genetic affinity with the ancient Armenian and Anatolian bezoars (Fig. 2E) (6). Therefore, predomestication introgression from a tur-like species may have contributed alleles to ancient bezoars and thereby early domesticated goats and their derived modern populations.
Notably, one introgressed region on chromosome 29 has a nearly fixed introgressed haplotype (frequency = 95.7%) in the modern worldwide domestic goats (Fig. 2F and fig. S22A). This region contains one intact protein-coding gene, MUC6, which encodes a gastrointestinally secreted mucin that forms a protective glycoprotein coat involved in host innate immune responses to the invasion of multiple gastrointestinal pathogens (22–24). We searched for potential donor populations in our sequenced wild caprid genomes. The haplotype network constructed with the nonrepeated region of MUC6 shows that the most common domestic haplotype (MUC6D) is similar to West Caucasian tur, differing by only one mutation, while being different from the minor frequency haplotypes of domestic goats and the common haplotype in bezoar (MUC6B) (Fig. 2G). To further validate this introgression signal, we estimated the most recent common ancestor of the divergent haplotypes and investigated whether selection acting on either a de novo mutation or on standing variation could possibly lead to the pattern in this region within domestic goats. Coalescence time estimates (fig. S18A) and neutral simulations (fig. S18B) suggest that the observed pattern of haplotype differentiation is highly unlikely in the absence of interspecific gene flow. Therefore, the nearly fixed MUC6D in goats was most likely introgressed from a lineage close to the West Caucasian tur, consistent with the genome-wide signal.
Domestication-mediated selection on immune and neural genes
To identify key selective sweeps in goat domestication, we compared the worldwide domestic goat populations with all 24 bezoars by estimating pairwise genetic differentiation (FST), differences in nucleotide diversity (π ln-ratio bezoar/domestic), and cross-population extended haplotype homozygosity (XP-EHH) in 50-kb sliding windows along the genome (figs. S19 and S20). We defined the windows with significant values (Z test, P < 0.005) in all three statistics (FST > 0.195, π ln-ratio > 0.395, and XP-EHH > 2.10) as putative selective sweeps. After merging consecutive outlier windows, 105 unique regions containing 403 protein-coding genes were identified (Fig. 3A and data file S2). Eighteen of these regions have been identified in previous domestication scans, including those associated to phenotypic effects related to immunity, neural pathways or processes, pigmentation, and productivity traits associated with milk composition and hair characteristics (data file S2). The modest overlap with previous studies is mainly due to differences in sample sets, selection scan methodology, and different versions of the reference genome (text S3).
Fig. 3. Genomic regions with selection signals in domestic goats.
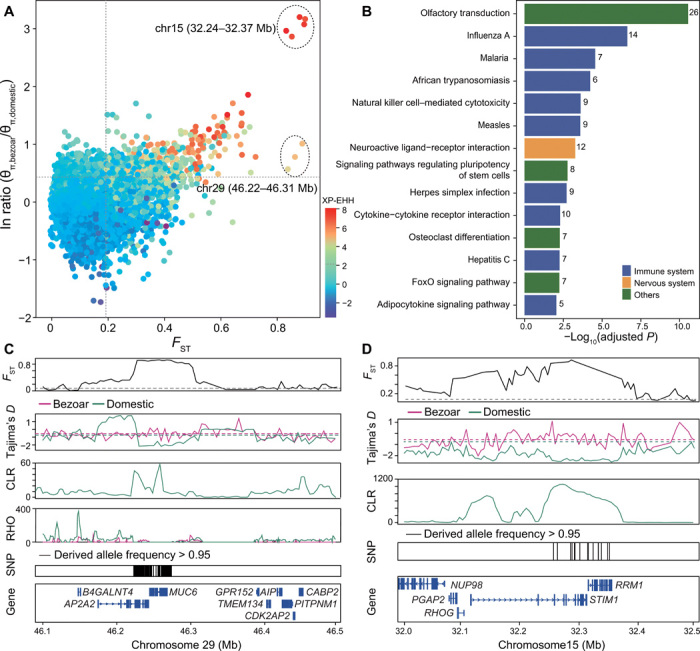
(A) Distribution of the pairwise fixation index (FST) (x axis), π ln ratio (y axis) and value of XP-EHH (color) between bezoars and domestic goats. The dashed vertical and horizontal lines indicate the significance threshold (corresponding to Z test, P < 0.005, where FST > 0.195, π ln ratio > 0.395, and XP-EHH > 2.1) used for extracting outliers. (B) KEGG pathways identified as significantly overrepresented (hypergeometric test, adjusted P < 0.01). (C and D) Selective sweep and distribution of the recombination (RHO) on chromosome 29 (46.22 to 46.31 Mb) (C) and selective sweep on chromosome 15 (32.24 to 32.37 Mb) (D). The putative sweep region is additionally validated by FST, Tajima’s D, and CLR test. Horizontal dashed lines represent the whole-genome mean for the corresponding parameters. Gene annotations in the sweep region and SNPs nearly fixed for derived alleles in domestic goats (C and D) are indicated at the bottom.
A KEGG analysis showed that 9 of the 14 significantly enriched pathways (hypergeometric test, adjusted P < 0.01) are immune related (hypergeometric test, adjusted P = 5 × 10−3 to 2.35 × 10−7) (Fig. 3B and table S12), including influenza A, malaria, African trypanosomiasis, natural killer cell–mediated cytotoxicity, measles, herpes simplex infection, cytokine–cytokine receptor interaction, hepatitis C, and adipocytokine signaling pathway. We surveyed the literature and identified 40 additional genes with immune function (table S13), obtaining a total of 41 regions that contain 77 immune-related genes. Among these, FST is particularly high in the MUC6 region, and we detected 16 missense single-nucleotide polymorphism (SNP) mutations showing FST > 0.88 in this gene (windowed FST = 0.89, π ln-ratio = 1.01, and XP-EHH = 5.25; Fig. 3C and table S14). Two of the 16 missense mutations were predicted to be deleterious, and these deleterious alleles occur at very low frequency (mean = 0.038) in the domestic goat population (fig. S21). Overall, this region contains 228 SNPs nearly fixed for the derived allele in modern goats (frequency > 95%, absent in bezoars) accounting for 93.8% of such variants in the whole genome (a total of 243, data file S3), illustrating the singular nature of this selection signal.
The selection enrichment analysis furthermore pinpoints 12 genes involved in neuroactive ligand–receptor interaction (hypergeometric test, adjusted P = 3.70 × 10−5). We observed an additional 37 genes linked to other functions in the nervous system that were under selection during goat domestication (table S15). One genomic region located on chromosome 15 shows the strongest combined signal of selection (FST = 0.90, π ln-ratio = 3.20, and XP-EHH = 8.09) (Fig. 3A). Evidence for large negative Tajima’s D values, high composite likelihood ratio (CLR) scores, and extensive haplotype sharing in domestic goats all suggest strong positive selection at this locus (Fig. 3D and fig. S22B). This locus contains 15 SNPs almost fixed for the derived allele in domestic goats and harbors two protein-coding genes, STIM1 and RRM1 (data file S3). STIM1 is an endoplasmic reticulum calcium sensor involved in regulating Ca2+ and metabotropic glutamate receptor signaling in the neural system (25, 26). It is known for its role in modulating the excitability of Purkinje neurons in the cerebellum, which play a key role in motor learning and the integration of sensory-motor information (27). RRM1 encodes ribonucleoside-diphosphate reductase large subunit, an enzyme essential for the production of deoxyribonucleotides, and influences sensitivity to valproic acid–induced neural tube defects in mice (28). Hence, we hypothesize that this strongly selected locus may be related to neural function and/or behavior.
Introgressed MUC6 plays a role in pathogen resistance
The MUC6 region constitutes the only intersection between the selection and introgression scans. In sheep and cattle, MUC6 is associated with gastrointestinal parasite resistance (29, 30). The results of transcriptome sequencing, quantitative real-time polymerase chain reaction (qPCR), and immunohistochemistry revealed that MUC6 is specifically expressed in the abomasum and duodenum of goat (Fig. 4A and fig. S23). Structurally, MUC6 has a high and variable number of tandem repeats (VNTRs) rich in Pro, Thr, and Ser residues, which can influence the covalent attachment of O-glycans (31). We therefore used PacBio SMRT transcriptome sequencing to investigate the difference between the MUC6D and MUC6B at the transcriptional level. Besides a number of small indels, an 82–amino acid deletion containing three copies of VNTR after the 2789th amino acid was uniquely identified in MUC6D compared to MUC6B (figs. S24 and S25). The different number of VNTRs in these two haplotypes may influence the function of MUC6, which is key to the generation of gastrointestinal mucous gel (23, 32). To examine the potential difference of pathogen resistance of the MUC6D and MUC6B variants, we carried out an epidemiological survey in a polymorphic goat population. By evaluating fecal egg counts (FEC) for gastrointestinal nematodes in 143 MUC6D/MUC6D, 111 MUC6D/MUC6B, and 14 MUC6B/MUC6B ewes at 13 months of age, we found that the goats carrying MUC6D/MUC6D exhibited lower FEC than those carrying the other two genotypes (Fig. 4B and data file S4), implying that the goats carrying MUC6D might be more resistant to gastrointestinal nematodes. To control for the genetic background, we also sequenced a subset of 117 animals to 5 average depth (data file S4). A genome-wide association analysis for FEC by incorporation of principal component covariates and pairwise relatedness found a strong association signal that contains MUC6 locus on chromosome 29 (42.77 to 51.33 Mb) (Fig. 4C). These results support that the introgressed MUC6D in domestic goats is most likely under selection due to its advantage in the host innate immune response toward potential gastrointestinal pathogens (23).
Fig. 4. The association of MUC6 with gastrointestinal nematodes resistance.
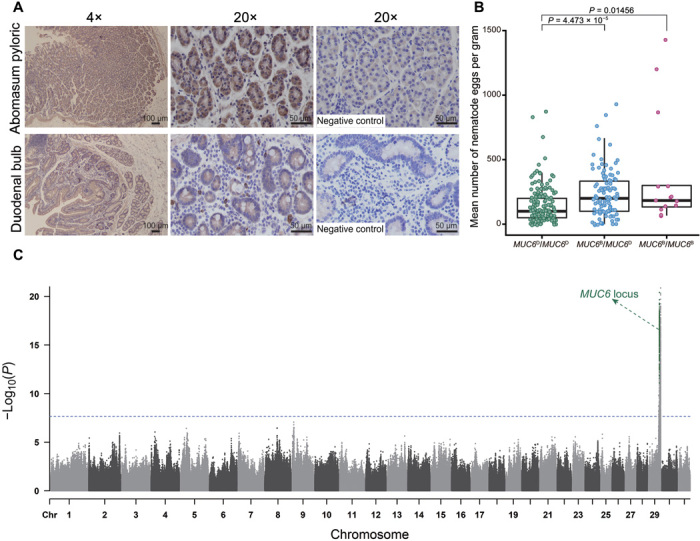
(A) Immunohistochemistry for MUC6 in abomasum pyloric and duodenal bulb of a goat. Photomicrographs at 4× and 20× are shown on the left and in the middle. The negative controls (20×) are shown on the right. (B) The statistical association between MUC6 genotype and the FECs for gastrointestinal nematodes. Wilcoxon rank sum test was used to compute the P values. (C) Manhattan plot of genome-wide association results for FECs. The significant SNPs within MUC6 locus are highlighted in dark green. The dotted horizontal line indicates the threshold (−Log10(P) = 7.65).
The origin and diffusion of domestic STIM1-RRM1 and MUC6 alleles
An in-depth genetic survey of ancient genomes throughout the Near East revealed that two ancient Balkan goats dating to ~8100 years ago carried STIM1-RRM1D (fig. S28 and data file S5). MUC6D appeared later at ~7200 years ago in Southwest Iran (fig. S28 and data file S6), a period characterized by an increased density of settlements in the Fertile Crescent (33). Herding goats at higher densities in a crowded and disease-prone anthropogenic environment would likely have exerted an increased selective pressure for livestock pathogen resistance (34). The first detected ancient animals having STIM1-RRM1D or MUC6D both carry mitochondrial haplogroup A, although this mitochondrial haplogroup had a low frequency and narrow distribution before 7500 years ago (6). By ~6500 years ago, the frequency of the STIM1-RRM1D and MUC6D increased to ~60% in the Near East (Fig. 5A) and spread to China ~3900 years ago (Fig. 5B), concomitant with the consolidation and diffusion of livestock-based economies throughout Eurasia (35, 36). The expansion of these two selected loci was also contemporaneous with the overwhelming spread of mitochondrial haplogroup A. In contrast, the frequencies of the two major Y-chromosome haplogroups remained relatively unchanged over time (Fig. 5B).
Fig. 5. The emergence and diffusion of domestic STIM1-RRM1 and MUC6 haplotypes are concurrent with the spread of mtDNA haplogroup A.
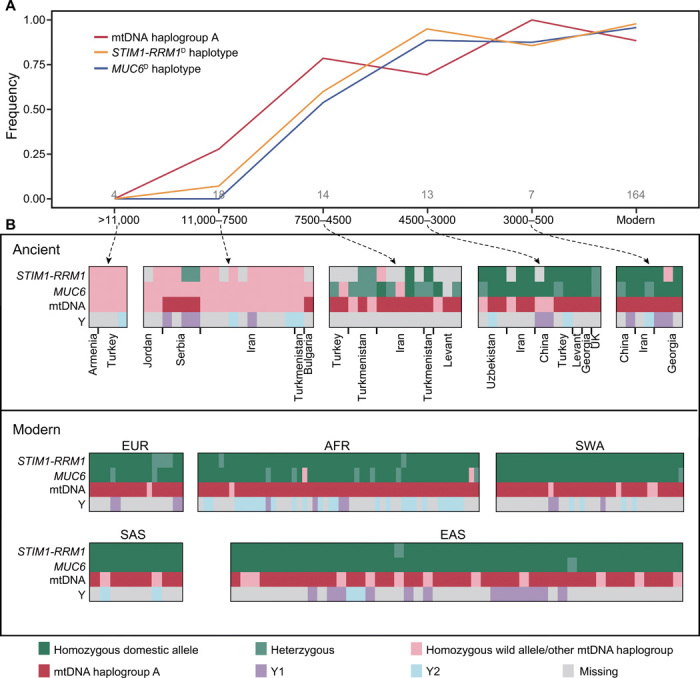
(A) The temporal changes in the frequency of the STIM1-RRM1D, MUC6D, and mtDNA haplogroup A from predomestication bezoars to modern domestic goats. The dates are expressed as cal. BP. (B) Genotypes of STIM1-RRM1 and MUC6, and mtDNA and Y-chromosome haplogroups. The presences in homozygous or heterozygous states are shown in green and light green, respectively. The absence of the domestic allele is depicted in light pink. The light gray color symbolizes missing information.
DISCUSSION
The present study generated genomic data from a substantial number of domestic and wild Capra species to characterize adaptive introgression and genetic changes during goat domestication. Collectively, both the selective sweep enrichment analyses and the two outstanding selection signals found in MUC6 and STIM1-RRM1 in domestic goats are consistent with the hypothesis that genes related to pathogen resistance (37) and behavior have been particularity targeted during goat domestication.
Several studies have shown that adaptive introgression can provide beneficial alleles that allow the recipient populations to adapt to new environments (12, 38, 39). In humans, immune-related loci acquired substantially advantageous alleles by immigrant modern humans admixing with archaic humans who were probably well adapted to local environments and pathogens through their long exposure to them (40–42). We here report numerous genomic segments putatively introgressed from an ibex-like species. These introgressed segments are enriched in genes with an immune function, suggesting that historic gene flow from wild species played an important role in shaping the diversity of immune system phenotypes in domestic goat and possibly increased its adaptive potential. When comparing the set of putatively introgressed alleles in each segment to our sampled ibex-like genomes, we find that most match rates range from 0.5 to 0.8 (fig. S17), indicating the degree of similarity between the introgressing and our sequenced ibex-like individuals. Additional wild Capra genome sequences in the future may clarify the origin of these alleles, although it is also possible that the introgression came from a now extinct branch on the Capra tree.
In particular, a number of lines of evidence support that gene flow between West Caucasian tur and goats occurred before the onset of domestication. The West Caucasian tur is distributed around the Black Sea coast (Fig. 2A), geographically close to the goat domestication center. It inhabits a humid, subtropical environment where the expected pathogen exposure and parasite load may be considerably higher than in more arid regions of Southwest Asia. The rigorous identification of introgression segments in domestic goats shows that the nearly fixed MUC6D was introgressed from a West Caucasian tur-like species. Although the earliest sign of introgression from West Caucasian tur is old (>47,000 years ago), we did not find the MUC6D in ancient and modern sampled bezoars. Two possibilities can explain this observation. First, given the pervasive gene flow between different Capra species (8, 9, 43), we cannot rule out the possibility of multiple introgression from ibex-like species before and after goat domestication. Second, because of the scarcity of ancient and modern bezoars, it may be that the introgressed variants were segregating at low frequencies in these populations. Nonetheless, our results indicate that the introgression of advantageous MUC6D might enhance innate immune reactivity against potential gastrointestinal pathogens under a grazing agro-ecological niche (44) with previously unknown infectious disease pressures.
Our dataset including several ancient goat samples allowed us to tentatively track the emergence and spread of the advantageous variants in these two important domestication loci (Fig. 5). The first occurrences of the STIM1-RRM1D and MUC6D locus coincided with the otherwise rare mitochondrial DNA (mtDNA) haplogroup A, and the three increased in frequency roughly concurrently (Fig. 5A), becoming nearly fixed in modern goat populations. These results suggest that the global diffusion of variations central to the domestication process was possibly mediated by a subset of female goats carrying the mtDNA haplogroup A. Despite this, modern goat populations remain differentiated from each other. The most likely explanation we can think of is that even Neolithic goat husbandry entailed some kind of breeding strategy under which immigrant matrilines containing globally advantageous alleles were deliberately crossbred with local populations, which probably carried local genetic adaptations, rather than simply replacing local populations.
Overall, our results provide evidence for adaptive introgression and the genetic basis of selected traits during the domestication of goats. We highlight that livestock domestication is a dynamic evolutionary process, with adaptive leaps driven by introgression and selection. Our results indicate that domestication may have profound impacts on neural traits and pathogen resistance, which helped manage herds to adapt to an anthropogenic environment.
MATERIALS AND METHODS
Sample collection
We collected 106 samples including 88 domestic goats (C. hircus), one bezoar (C. aegagrus), three Markhors (C. falconeri), three Siberian ibex (C. sibirica), one Alpine ibex (C. ibex), one West Caucasian tur (C. caucasica), five ancient goats spanning from the Neolithic to the Middle Ages, and four Nubian ibex hybrids (C. nubiana × C. hircus) for whole-genome sequencing. Among them, the Nubian ibex hybrids offer a potential source for determining whether the introgressed haplotypes could originate from the Nubian ibex or closely related ibex. All procedures involving sample collection were approved by the Northwest A&F University Animal Care Committee (permit number: NWAFAC1019), making all efforts to minimize invasiveness. The C. caucasica (tooth) sample was obtained from a zoo specimen donated to Ménagerie du Jardin des Plantes in 1976 of unknown provenance (wild or captive-born) but with no evidence of recent admixture (text S1). The individual died in 1982 and was subsequently sampled by the Muséum National d’Histoire Naturelle (MNHN, sample ID MNHN ZM AC 1982-1092). In addition, we also obtained whole-genome data and their geographical coordinates of the sampling sites from previous studies (5, 15). Details of the samples used in this study are given in tables S1 to S4.
DNA extraction and sequencing
For modern samples, genomic DNA was extracted from whole blood using the standard phenol-chloroform method and checked for quality and quantity on the Qubit 2.0 fluorometer (Invitrogen). Library construction for resequencing was performed with 1 to 3 μg of genomic DNA using standard library preparation protocols and insert sizes from 300 to 500 base pair (bp). All libraries were sequenced on either the Illumina HiSeq 2500 or X Ten platforms.
For the historical sample (C. caucasica), tooth pulverization, DNA extraction, and Illumina sequencing library preparation were performed in Trinity College Dublin, Ireland as described in (6). The double-stranded DNA sequencing library was constructed without uracil-DNA glycosylase treatment. The sample library was then sequenced using a HiSeq 2500 platform (single end, 1 × 101 bp). Details on the processing of five ancient samples are described in the Supplementary Materials.
Read alignment and variant calling
Filtered reads from all individuals were aligned to the latest goat reference genome by the Burrows-Wheeler Aligner (BWA v0.7.15). To obtain high-quality SNPs, we carried out SNP calling and genotyping at two separate stages. Variants were found on a population scale (without outgroup argali Ovis ammon) using a SAMtools model implemented in the analysis of next-generation sequencing data (ANGSD) (45) with “-only_proper_pairs 1 -uniqueOnly 1 -remove_bads 1 -minQ 20 -minMapQ 30 -C 50 -doMaf 1 -doMajorMinor 1 -GL 1 -setMinDepth [Total_read_depth/3] -setMaxDepth [Total_read_depth*3] -doCounts 1 -dosnpstat 1 -SNP_pval 1.” The sites that could be called in at least 90% of the samples and had a strand bias score below 90% were retained. A P value threshold of 1 × 10−6 and minor allele frequency value > 1/2n were used as SNP discovery criteria, where n was the sample size. We also kept the sites at which the sampled individuals were homozygous for the alternative alleles. To obtain the hard-called genotypes in all Capra species and outgroups, we used a workflow adapted from GATK v3.7.0 HaplotypeCaller (46) best practice. Briefly, the genome variants, in genomic variant call format (gVCF) for each sample, were identified with the HaplotypeCaller. After all the gVCF files were merged, a raw population genotype file with the SNPs and indels was created. Optional variant hard filter values were applied for SNPs using GATK VariantFiltration as Quality by Depth (QD) < 2.0, root mean square of Mapping Quality (MQ) < 40.0, Fisher Strand (FS) > 60.0, HaplotypeScore >13.0, and MQRankSum ≤ 12.5. Last, only biallelic SNPs identified by both GATK and ANGSD that all show no more than 10% missing data were extracted using VCFtools v0.1.15 (47) with “--min-alleles 2 --max-alleles 2 --max-missing 0.9.” Then, all sample sets of filtered variant calls were used for imputation and phasing using Beagle v4.1 (48, 49) with default parameters, except for “niterations,” which was set to 10.
To retrieve the nucleotides that were accessible to variant discovery (used in the demographic estimation), hard filtering of the invariant sites was carried out with thresholds based on sequencing coverage (one-third– to threefold of corresponding mean depth) and missing data rate (no more than 10%) following the same strategies adopted for variant sites.
Population structure and phylogenetic analysis
Neighbor-joining tree was constructed for the whole-genome SNP set (table S5) using MEGA v6.0 based on pairwise genetic distances matrix calculated by PLINK v1.9. We also made use of the 5,043,096 fourfold degenerate sites to build an ML tree using RAxML v8.2.9. PCA was performed using smartpca, part of the EIGENSOFT v6.1. A Tracy-Widom test was used to determine the significance level of the eigenvectors. ADMIXTURE was used to perform an unsupervised clustering analysis. We increased the number of predefined genetic clusters from k = 2 to k = 7 and ran the analysis 20 times for each k. We further compared the individual-level haplotype similarity using fineSTRUCTURE v2.1.3. TreeMix v1.12 was also used to infer a population-level phylogeny using the ML approach.
Demographic reconstruction
To infer patterns of effective population size and population separations over historical time, we used MSMC2, using the statistically phased autosomal genotypes. We also used the SMC++, which does not rely on haplotype phase information. In addition, we calculated the likelihood of the observed site frequency spectrum conditioned on several demographic models using ∂a∂i (fig. S13). For the best model, nonparametric bootstrapping (100 replicates) was performed to determine the variance of each parameter (fig. S14 and table S7). To obtain reliable time estimates, we used a phylogenetic comparison (50) of goat and sheep (Ovis aries) to calibrate the mutation rate for goats. Using an average generation time of 2 years for goats (51), we estimated a mutation rate of 4.32 × 10−9 per site per generation, which is similar to that obtained in (52).
Gene flow analysis
We computed f3 statistics and D statistics to formally test hypotheses of gene flow. To identify regions introgressed into modern domestic goats from ibex-like species, we calculated D statistics using nonoverlapping 20-kb sliding windows across the genome. Windows within top 5% of the D statistics were considered as outliers and were further examined. An introgressed segment should have low sequence divergence to the putative donor species, whereas sequence divergence between the donor species and bezoar should be higher. Therefore, we also calculated the identity by state distance matrix of the outlier regions using ANGSD. Differences in sequence divergence were determined by a t test–based approach using a P value of 0.01 as cutoff. The windows which passed the above two filtering steps were merged as the significantly diverged regions.
We then applied Sprime (53) for investigating whether introgression can explain the significantly diverged regions. The first step of Sprime is to read the phased genotypes of the target and outgroup individuals. The outgroup is a population that is closely related to the ancestor(s) of the target population but that is not expected to have experienced admixture from ibex-like species that contributed to the target population. The 24 modern bezoars are used as the outgroup population, although they may themselves contain introgressed sequence from ibex-like species. This may lead to the occurrence of false negatives but will minimize the risk of false positives. We consider this an acceptable trade-off given our objective. Sprime first groups variants into three classes: those common in the outgroup, those not seen in the outgroup, and those uncommon but present in the outgroup. The threshold frequency used to distinguish common versus uncommon variants is 0.01 (53) for the analyses presented here. The common alleles (those with frequency > 0.01 in 24 modern bezoars) are excluded from further consideration because these variants in domestic goats are more likely inherited from their wild progenitor (bezoar). We assumed a score threshold of 150,000 (53) and a mutation rate of 4.32 × 10−9 per site per generation to identify introgressed alleles in domestic goat genomes. The next step is to find putatively introgressed segments (i.e., sets of alleles) for each significantly diverged region. We process one region at a time and find the set of alleles with the highest score. These comprise the putative introgressed segment. The segment boundaries were defined by the position of first and last introgressed alleles. We consider only segments with at least 100 putatively introgressed alleles that are of nonbezoar origin. To determine whether a target individual carries the introgressed haplotype, at least 50% of introgressed alleles must be found in that haplotype. We primarily focus on the most common introgressed regions (i.e., introgressed haplotype frequency > 0.1) from ibex-like species to domestic goats.
We further checked the introgression status by constructing ML trees with MEGA v6.0 and searching for domestic goats located within the ibex-like clade (fig. S16). For each region, we also reported a match if the genotype of ibex like species included the putative introgressed allele and a mismatch otherwise. The match rate was calculated as the number of matches divided by the number of compared sites (matches and mismatches) (fig. S17). Note that the match rate should not imply that the sampled ibex-like genomes represent the direct source population(s), but the degree of similarity between the introgressing and sampled ibex-like species.
Selective sweep analysis
We screened for sweeps selected during goat domestication by parsing specific 50-kb windows that showed low diversity in domestic goats and had high divergence (a high fixation index, FST, and haplotype differences, XP-EHH) between domestic goats and wild goats. After calculating all tests, the windows with P values less than 0.005 (Z test) were considered significant signals. Candidate genes under selection were defined as those overlapped by sweep regions or within 20 kb of the signals. Two additional statistics, the Tajima’s D and CLR test were applied to confirm the top signals.
Functional enrichment analyses
We characterized the most relevant functions of the protein-coding genes overlapping with the introgressed regions and selective sweeps by searching for overrepresented KEGG pathways. Goat protein sequences were used to conduct functional enrichment tests on the target genes using the KOBAS 3.0 server (54). The P value was calculated using a hypergeometric distribution. False discovery rate (FDR) correction was performed to adjust for multiple testing. Pathways with an FDR-corrected P value of <0.01 were considered statistically significantly enriched (tables S10 and S12).
Genotyping loci underlying domestication in ancient genomes
To investigate the genotypes of the STIM1-RRM1 and MUC6 loci in ancient samples, we used a total of 243 SNPs (15 SNPs within STIM1-RRM1 locus and 228 SNPs within MUC6 locus, respectively), which showed derived allele frequency > 0.95 in 164 modern domestic goats (defined as domestic haplotypes: STIM1-RRM1D and MUC6D) and was absent in 24 modern bezoars and 4 ancient bezoars (Hovk1, Direkli1-2, Direkli6, and Direkli5) (defined as bezoar haplotypes: STIM1-RRM1B and MUC6B). Because of the low coverage for some ancient genomes, we used allele reads count at SNP positions to determine the genotypes of ancient goats at STIM1-RRM1 locus and MUC6 locus. For each ancient goat and SNP, we determined the numbers of reads corresponding to the ancestral and derived allele using GATK v3.7.0 UnifiedGenotyper (--min_base_quality_score 15--output_mode EMIT_ALL_SITES --standard_min_confidence_threshold_for_calling 20) (46). For each locus in each ancient goat, we calculated the proportion of the summed derived allele reads count versus total allele reads count. If the proportion was lower than 10%, the genotype of ancient goat was considered to be homozygous for ancestral allele (bezoar like). The genotype was set to heterozygous if the proportion was between 10 and 90%. If the proportion was more than 90%, it was classified as homozygous for derived allele (domestic like). We set the genotype to be missing when the ancient goats only had one single informative read (mapping to the predefined SNPs) mapped to the locus.
Expression and epidemiologic survey related to MUC6 locus
qPCR analysis
Total RNA was extracted using goat tissues from gastrointestinal tract (including abomasum, duodenum, jejunum, ileum, and caecum) using Eastep Super Total RNA Extraction Kit (Promega, Shanghai, China). Complementary DNA was generated via PrimerScript RT Reagent Kit with genomic DNA Eraser (Perfect Real Time) (TaKaRa, Beijing, China). qPCR analysis was performed with FastStart Universal SYBR Green Master (Roche, Shanghai, China) on a Bio-Rad instrument. MUC6 primers (forward primer: 5′-CAGCCAGGACAAAATCATGA-3′ and reverse primer: 5′-CTCTGGTCTGGCCTCTGAAC-3′) were designed using National Center for Biotechnology Information Primer-BLAST (http://ncbi.nlm.nih.gov/tools/primer-blast/). GAPDH (forward primer: 5′-ACACCCTCAAGATTGTCAGC-3′ and reverse primer: 5′-ATAAGTCCCTCCACGATGC-3′) was used as internal reference. Gene expression results were calculated using the delta-delta cycle threshold (2−ΔΔCT) method.
Immunohistochemistry analysis
For histological analysis, dissected goat tissues (abomasum pyloric and duodenal bulb) were fixed in 4% paraformaldehyde and embedded in paraffin for sectioning (5 μm thick). The tissues sections were deparaffinized, and rehydration was followed by antigen retrieval using a citrate-buffered solution in a microwave at 100°C for 15 min. After cooling down to room temperature, quenching of endogenous peroxidase, and protein block, the sections were treated with a primary antibody (cat. no. D161001, Sangon Biotech, Shanghai, China) overnight at 4°C. Negative control sections were obtained by incubating with the primary antibody diluted buffer. Subsequently, the secondary antibody (cat. no. D110058, Sangon Biotech, Shanghai, China) was used to detect primary antibody. Specific protein immunoreactivity was visualized with the substrate chromogen 3,3′- diaminobenzidine. Last, the slides were rinsed in water, counterstained with hematoxylin, and mounted with coverslips.
Full-length transcript sequencing
To determine the sequence differences between the domestic (MUC6D) and bezoar (MUC6B) MUC6 haplotypes, total RNA was isolated from the abomasum pyloric with high expression of MUC6 from two heterozygous goats and sequenced by PacBio Sequel. Pacbio SMRTbell libraries were sequenced on two separate SMRT cells (Annoroad Gene Technology Co. Ltd., Beijing, China). A total of 23,338,976 subreads were generated with a mean accuracy of 80% and an average length of 1755 nt. High-quality circular consensus sequences (CCSs) were obtained using the Iso-Seq 3 application in the PacBio SMRT Analysis v6.0.0 (https://github.com/PacificBiosciences/IsoSeq), with parameters “--noPolish --minPasses 1.” Last, we got a total of 960,271 CCSs. These CCSs were aligned to the latest goat reference genome using Minimap2 (55), and we picked out 251 CCSs that mapped to the MUC6 mRNA sequence. We used the CCS that belonged to the bezoar haplotype to manually assemble the mRNA sequence of the MUC6B. Then, we realigned the MUC6B mRNA sequence to MUC6D mRNA sequence (XM_018042766.1) by MEGA v6.0 and carefully checked the indels. The abundance of tandem repeats in MUC6 (XP_017898255.1) was examined by using the rapid automatic detection and alignment of repeat finding program (https://www.ebi.ac.uk/Tools/pfa/radar/) (56). Three major types of repeat units with distinct sequence features were observed. We detected a 246-bp insertion in the MUC6B mRNA sequence located at the 32nd exon, containing three copies of type III units between the 2789th amino acid and the 2871th amino acid (fig. S24). We further validated this insertion by aligning short reads from West Caucasian tur to the MUC6B mRNA sequences (fig. S25).
Test population
To further test the genotype-phenotype associations, we first examined the genotypes at the MUC6 locus using blood samples from breeding rams from a company located in Inner Mongolia, China. This company runs more than 9000 cashmere goats, mainly for superfine cashmere (fiber diameter < 14 μm). The animals are dewormed routinely with oxfendazole, ivermectin, avermectin, and levamisole three times annually. Of the 20 tested breeding rams, we identified 4 animals with MUC6D/MUC6B genotype. We sampled their offspring partly on the basis of the breeding records. Blood samples of 495 animals were collected in January 2019.
Genotypic and phenotypic analysis
DNA was extracted from the blood samples. The MUC6 locus was genotyped by PCR (forward primer: 5′-CAGCACTATCTCCCATACATC-3′ and reverse primer: 5′-GTGGAGCTGAGCTGACACTT-3′) and Sanger sequencing. The frequency of MUC6B was 17.3% (n = 338 MUC6D/MUC6D, n = 143 MUC6D/MUC6B, and n = 14 MUC6B/MUC6B).
After genotyping, we collected fresh fecal samples from a subset of 268 animals from the rectum in April 2019, a time when gastrointestinal nematodes numbers were anticipated to be elevated. The gastrointestinal parasitic infestations were examined using the McMaster’s technique as described in (57). Briefly, 2 g of fecal samples were transferred into a container, and 60 ml of saturated sodium chloride was added. The suspension was thoroughly stirred with a glass stick and poured through a strainer (80-mesh screen) into the new container. The container with the suspension was closed tightly and carefully inverted several times. Then, the suspension was taken up from the container to fill the glass plate with two McMaster counting chambers. The size of the counting chamber was 10 mm by 10 mm by 1.5 mm. The nematode eggs were then counted under microscopic observation at ×100 magnification. Observed nematodes in the feces included the common abomasal and intestinal goat nematodes: Hemonchus contortus and Nematodirus sp. (fig. S26).
The number of nematodes eggs per gram (EPG) was calculated according to the following formula: EPG = (n1 + n2)/2 ÷ 0.15 × 60 ÷ 2, where (n1 + n2)/2 is the average number of eggs per chamber, 0.15 is the effective volume (milliliters) for counting chamber, 60 is the total volume (milliliters) of suspension, 2 is the weight (grams) of feces examined. EPG was measured on three replicates of each fecal sample, and the average of the three replicates was used for analysis.
Data analysis
The average fecal nematode egg counts were not normally distributed; therefore, we used Wilcoxon rank sum test to test the null hypothesis that there was no association between genotype and phenotype.
Genome-wide association study
To have more even representation of sampling of the three genotypes, we used 10 MUC6B/MUC6B samples and randomly chose 46 MUC6B/MUC6D and 61 MUC6D/MUC6D for whole-genome sequencing (data file S4). The 117 individuals were sequenced at approximately fivefold genome coverage using BGISEQ-500 platform. SNP calling and estimation of genotype likelihoods were performed in ANGSD using the following settings: “-baq 1 -C 50 -only_proper_pairs 1 -uniqueOnly 1 -remove_bads 1 -minQ 20 -minMapQ 25 -doMaf 1 -minInd 106 -skipTriallelic 1 -doMajorMinor 1 -GL 1 -setMinDepth 250 -setMaxDepth 1,000 -doCounts 1 -doGlf 2 -SNP_pval 1e-6.” To correct for population stratification and cryptic relatedness, we adopted genotype likelihood–based approaches implemented in PCAngsd v0.973 (58) to perform PCA and estimate pairwise relatedness by supplying the following options: “-kinship -inbreed 2 -minMaf 0.05.” Beagle v3.3.2 (48) was used to convert the genotype likelihoods to actual genotypes. Only sites for which the allelic R2 (larger values of allelic R2 indicate more accurate genotype imputation) was >0.95 were retained. The imputed genotype dataset was converted to tped/tfam format using PLINK v1.9. A total of 12.47 million high-quality SNPs (minor allele frequency > 0.05, using whole-genome sequencing data alone) were used to perform genome-wide association study in EMMAX (59) software by incorporation of the first three principal component covariates (fig. S27, A and B) and kinship matrix. The phenotype was rank-transformed to normality (Shapiro-Wilk test, P = 0.02228) to counteract departures from normality (fig. S27C).
The genome-wide critical value was determined by permutation: The phenotype data were permuted 200 times; for each permutated phenotype, a genome-wide association analysis was conducted and the genome-wide lowest P value was recorded. We then took the 5% lowest tail from the 200 recoded minimal P values as the threshold for genome-wide significance (P = 2.257−8). The Manhattan and quantile-quantile plots (fig. S27D) were generated using the R package CMplot (https://github.com/YinLiLin/R-CMplot).
Supplementary Material
Acknowledgments
We thank members of the NextGen project for sharing data. We thank L. Mohammed for providing Nubian ibex hybrid blood samples. We thank High-Performance Computing (HPC) of NWAFU for providing computing resources. Funding: This project was supported by grants from the National Natural Science Foundation of China (31822052 and 31572381 to Y.J. and 31572369 to Y.Ch.), the Talents Team Construction Fund of Northwestern Polytechnical University (NWPU) and Strategic Priority Research Program of CAS (XDB13000000) to W.W., a DFF Sapere Aude grant to R.H., and the Tang Scholar at Northwest A&F University (NWAFU) to Xiaolong Wang. The Chinese Government contribution to CAAS-ILRI Joint Laboratory on Livestock and Forage Genetic Resources in Beijing (2018-GJHZ-01) is appreciated. Author contributions: Y.J. and Y.Ch. conceived the project and designed the research. Z.Z., M.L., and Xihong Wang performed most of the analysis with contributions from Z.Y., Y.L., Y.Ca., Q.C., J.Li., K.W., X.P., Y.W., S.H., M.G., T.Z., Y.Z., Y.G., Y.X., N.X., and Y.Y. Xiaolong Wang, W.Z., J.H., L.Ch., A.E., and M.O. prepared the modern DNA samples. J.Le. provided the historical sample of West Caucasian tur. D.C. prepared the ancient samples. Xihong Wang, Z.Z., Y.J., and M.L. drafted the manuscripts with input from all authors. Y.J., W.W., R.H., G.Z., K.G.D., D.G.B., L.Co., and T.S.S. revised the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. Individual genome sequence data are available at the Sequence Read Archive (https://ncbi.nlm.nih.gov/sra) under accession codes PRJNA387635 and PRJNA361447.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/21/eaaz5216/DC1
REFERENCES AND NOTES
- 1.Larson G., Fuller D. Q., The evolution of animal domestication. Annu. Rev. Ecol. Evol. Syst. 45, 115–136 (2014). [Google Scholar]
- 2.Zeder M. A., Domestication and early agriculture in the Mediterranean Basin: Origins, diffusion, and impact. Proc. Natl. Acad. Sci. U.S.A. 105, 11597–11604 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.F. Pereira, A. Amorim, in Origin and Spread of Goat Pastoralism (John Wiley & Sons, 2010). [Google Scholar]
- 4.Colli L., Milanesi M., Talenti A., Bertolini F., Chen M., Crisà A., Daly K. G., Del Corvo M., Guldbrandtsen B., Lenstra J. A., Rosen B. D., Vajana E., Catillo G., Joost S., Nicolazzi E. L., Rochat E., Rothschild M. F., Servin B., Sonstegard T. S., Steri R., Van Tassell C. P., Ajmone-Marsan P., Crepaldi P., Stella A.; AdaptMap Consortium , Genome-wide SNP profiling of worldwide goat populations reveals strong partitioning of diversity and highlights post-domestication migration routes. Genet. Sel. Evol. 50, 58 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alberto F. J., Boyer F., Orozco-terWengel P., Streeter I., Servin B., de Villemereuil P., Benjelloun B., Librado P., Biscarini F., Colli L., Barbato M., Zamani W., Alberti A., Engelen S., Stella A., Joost S., Ajmone-Marsan P., Negrini R., Orlando L., Rezaei H. R., Naderi S., Clarke L., Flicek P., Wincker P., Coissac E., Kijas J., Tosser-Klopp G., Chikhi A., Bruford M. W., Taberlet P., Pompanon F., Convergent genomic signatures of domestication in sheep and goats. Nat. Commun. 9, 813 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Daly K. G., Delser P. M., Mullin V. E., Scheu A., Mattiangeli V., Teasdale M. D., Hare A. J., Burger J., Verdugo M. P., Collins M. J., Kehati R., Erek C. M., Bar-Oz G., Pompanon F., Cumer T., Çakirlar C., Mohaseb A. F., Decruyenaere D., Davoudi H., Çevik O., Rollefson G., Vigne J.-D., Khazaeli R., Fathi H., Doost S. B., Sorkhani R. R., Vahdati A. A., Sauer E. W., Kharanaghi H. A., Maziar S., Gasparian B., Pinhasi R., Martin L., Orton D., Arbuckle B. S., Benecke N., Manica A., Horwitz L. K., Mashkour M., Bradley D. G., Ancient goat genomes reveal mosaic domestication in the Fertile Crescent. Science 361, 85–88 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Pidancier N., Jordan S., Luikart G., Taberlet P., Evolutionary history of the genus Capra (Mammalia, Artiodactyla): Discordance between mitochondrial DNA and Y-chromosome phylogenies. Mol. Phylogenet. Evol. 40, 739–749 (2006). [DOI] [PubMed] [Google Scholar]
- 8.Hammer S. E., Schwammer H. M., Suchentrunk F., Evidence for introgressive hybridization of captive markhor (Capra falconeri) with domestic goat: Cautions for reintroduction. Biochem. Genet. 46, 216–226 (2008). [DOI] [PubMed] [Google Scholar]
- 9.Grossen C., Keller L., Biebach I.; International Goat Genome Consortium, Croll D., Introgression from domestic goat generated variation at the major histocompatibility complex of alpine ibex. PLOS Genet. 10, e1004438 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Racimo F., Sankararaman S., Nielsen R., Huerta-Sánchez E., Evidence for archaic adaptive introgression in humans. Nat. Rev. Genet. 16, 359–371 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hu X.-J., Yang J., Xie X.-L., Lv F.-H., Cao Y.-H., Li W.-R., Liu M.-J., Wang Y.-T., Li J.-Q., Liu Y.-G., Ren Y.-L., Shen Z.-Q., Wang F., Hehua E., Han J.-L., Li M. H., The genome landscape of tibetan sheep reveals adaptive introgression from argali and the history of early human settlements on the qinghai-tibetan plateau. Mol. Biol. Evol. 36, 283–303 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu D.-D., Ding X.-D., Wang S., Wójcik J. M., Zhang Y., Tokarska M., Li Y., Wang M.-S., Faruque O., Nielsen R., Zhang Q., Zhang Y.-P., Pervasive introgression facilitated domestication and adaptation in the Bos species complex. Nat. Ecol. Evol. 2, 1139–1145 (2018). [DOI] [PubMed] [Google Scholar]
- 13.Hedrick P. W., Adaptive introgression in animals: Examples and comparison to new mutation and standing variation as sources of adaptive variation. Mol. Ecol. 22, 4606–4618 (2013). [DOI] [PubMed] [Google Scholar]
- 14.Sun Z., Shao J., Liu L., Cui J., Bonomo M. F., Guo Q., Wu X., Wang J., The first Neolithic urban center on China's north Loess Plateau: The rise and fall of Shimao. Archaeol. Res. Asia 14, 33–45 (2018). [Google Scholar]
- 15.Dong Y., Zhang X., Xie M., Arefnezhad B., Wang Z., Wang W., Feng S., Huang G., Guan R., Shen W., Bunch R., Culloch R. M., Li Q., Li B., Zhang G., Xu X., Kijas J. W., Salekdeh G. H., Wang W., Jiang Y., Reference genome of wild goat (Capra aegagrus) and sequencing of goat breeds provide insight into genic basis of goat domestication. BMC Genomics 16, 431–431 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Manceau V., Després L., Bouvet J., Taberlet P., Systematics of the genus Capra inferred from mitochondrial DNA sequence data. Mol. Phylogenet. Evol. 13, 504–510 (1999). [DOI] [PubMed] [Google Scholar]
- 17.D. M. Shackleton, Wild Sheep and Goats and their Relatives: Status Survey and Conservation Action Plan for Caprinae (International Union for Conservation of Nature and Natural Resources, 1997). [Google Scholar]
- 18.Naderi S., Rezaei H.-R., Pompanon F., Blum M. G. B., Negrini R., Naghash H.-R., Balkiz Ö., Mashkour M., Gaggiotti O. E., Ajmone-Marsan P., Kence A., Vigne J.-D., Taberlet P., The goat domestication process inferred from large-scale mitochondrial DNA analysis of wild and domestic individuals. Proc. Natl. Acad. Sci. U.S.A. 105, 17659–17664 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miguel M. A., Mingala C. N., Screening of pig (Sus scrofa) bactericidal permeability-increasing protein (BPI) gene as marker for disease resistance. Anim. Biotechnol. 30, 146–150 (2019). [DOI] [PubMed] [Google Scholar]
- 20.Yan W., Zheng C., He J., Zhang W., Huang X.-A., Li X., Wang Y., Wang X., Eleutheroside B1 mediates its anti-influenza activity through POLR2A and N-glycosylation. Int. J. Mol. Med. 42, 2776–2792 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Omoumi A., Fok A., Hsiung G.-Y., P4-246: Analysis of two immune function genes (CR1 and CD2AP) in Alzheimer's disease in two large Canadian cohorts. Alzheimers Dement. 8, P722 (2012). [Google Scholar]
- 22.Babu S. D., Jayanthi V., Devaraj N., Reis C. A., Devaraj H., Expression profile of mucins (MUC2, MUC5AC and MUC6) in Helicobacter pylori infected pre-neoplastic and neoplastic human gastric epithelium. Mol. Cancer 5, 10 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McGuckin M. A., Lindén S. K., Sutton P., Florin T. H., Mucin dynamics and enteric pathogens. Nat. Rev. Microbiol. 9, 265–278 (2011). [DOI] [PubMed] [Google Scholar]
- 24.Benavides M. V., Sonstegard T. S., Van Tassell C., Genomic regions associated with sheep resistance to gastrointestinal nematodes. Trends Parasitol. 32, 470–480 (2016). [DOI] [PubMed] [Google Scholar]
- 25.Hartmann J., Karl R. M., Alexander R. P. D., Adelsberger H., Brill M. S., Rühlmann C., Ansel A., Sakimura K., Baba Y., Kurosaki T., Misgeld T., Konnerth A., STIM1 controls neuronal Ca2+ signaling, mGluR1-dependent synaptic transmission, and cerebellar motor behavior. Neuron 82, 635–644 (2014). [DOI] [PubMed] [Google Scholar]
- 26.Majewski Ł., Maciag F., Boguszewski P. M., Wasilewska I., Wiera G., Wójtowicz T., Mozrzymas J., Kuznicki J., Overexpression of STIM1 in neurons in mouse brain improves contextual learning and impairs long-term depression. Biochim. Biophys. Acta. Mol. Cell Res. 1864, 1071–1087 (2017). [DOI] [PubMed] [Google Scholar]
- 27.Ryu C., Jang D. C., Jung D., Kim Y. G., Shim H. G., Ryu H.-H., Lee Y.-S., Linden D. J., Worley P. F., Kim S. J., STIM1 regulates somatic Ca2+ Signals and intrinsic firing properties of cerebellar purkinje neurons. J. Neurosci. 37, 8876–8894 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Craig J. C., Bennett G. D., Miranda R. C., Mackler S. A., Finnell R. H., Ribonucleotide reductase subunit r1: A gene conferring sensitivity to valproic acid-induced neural tube defects in mice. Teratology 61, 305–313 (2000). [DOI] [PubMed] [Google Scholar]
- 29.Rinaldi M., Dreesen L., Hoorens P. R., Li R. W., Claerebout E., Goddeeris B., Vercruysse J., Van Den Broek W., Geldhof P., Infection with the gastrointestinal nematode Ostertagia ostertagi in cattle affects mucus biosynthesis in the abomasum. Vet. Res. 42, 61 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Simpson H. V., Umair S., Hoang V. C., Savoian M. S., Histochemical study of the effects on abomasal mucins of Haemonchus contortus or Teladorsagia circumcincta infection in lambs. Vet. Parasitol. 226, 210–221 (2016). [DOI] [PubMed] [Google Scholar]
- 31.Moniaux N., Escande F., Porchet N., Aubert J.-P., Batra S. K., Structural organization and classification of the human mucin genes. Front. Biosci. 6, D1192–D1206 (2001). [DOI] [PubMed] [Google Scholar]
- 32.Hasnain S. Z., Gallagher A. L., Grencis R. K., Thornton D. J., A new role for mucins in immunity: Insights from gastrointestinal nematode infection. Int. J. Biochem. Cell Biol. 45, 364–374 (2013). [DOI] [PubMed] [Google Scholar]
- 33.Lawrence D., Philip G., Hunt H., Snape-Kennedy L., Wilkinson T. J., Long term population, city size and climate trends in the fertile crescent: A first approximation. PLOS ONE 11, e0152563 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Harper K. N., Armelagos G. J., Genomics, the origins of agriculture, and our changing microbe-scape: Time to revisit some old tales and tell some new ones. Am. J. Phys. Anthropol. 152, 135–152 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pereira F., Davis S. J. M., Pereira L., McEvoy B., Bradley D. G., Amorim A., Genetic signatures of a Mediterranean influence in Iberian Peninsula sheep husbandry. Mol. Biol. Evol. 23, 1420–1426 (2006). [DOI] [PubMed] [Google Scholar]
- 36.Arbuckle B. S., Pace and process in the origins of animal husbandry in Neolithic Southwest Asia. Bioarchaeol. Near East 8, 53–81 (2014). [Google Scholar]
- 37.Vilà C., Seddon J., Ellegren H., Genes of domestic mammals augmented by backcrossing with wild ancestors. Trends Genet. 21, 214–218 (2005). [DOI] [PubMed] [Google Scholar]
- 38.Huerta-Sánchez E., Jin X., Asan, Bianba Z., Peter B. M., Vinckenbosch N., Liang Y., Yi X., He M., Somel M., Ni P., Wang B., Ou X., Huasang, Luosang J., Cuo Z. X. P., Li K., Gao G., Yin Y., Wang W., Zhang X., Xu X., Yang H., Li Y., Wang J., Wang J., Nielsen R., Altitude adaptation in Tibetans caused by introgression of Denisovan-like DNA. Nature 512, 194–197 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jones M. R., Mills L. S., Alves P. C., Callahan C. M., Alves J. M., Lafferty D. J. R., Jiggins F. M., Jensen J. D., Melo-Ferreira J., Good J. M., Adaptive introgression underlies polymorphic seasonal camouflage in snowshoe hares. Science 360, 1355–1358 (2018). [DOI] [PubMed] [Google Scholar]
- 40.Dannemann M., Andrés A. M., Kelso J., Introgression of neandertal- and denisovan-like haplotypes contributes to adaptive variation in human toll-like receptors. Am. J. Hum. Genet. 98, 22–33 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abi-Rached L., Jobin M. J., Kulkarni S., McWhinnie A., Dalva K., Gragert L., Babrzadeh F., Gharizadeh B., Luo M., Plummer F. A., Kimani J., Carrington M., Middleton D., Rajalingam R., Beksac M., Marsh S. G. E., Maiers M., Guethlein L. A., Tavoularis S., Little A.-M., Green R. E., Norman P. J., Parham P., The shaping of modern human immune systems by multiregional admixture with archaic humans. Science 334, 89–94 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mendez F. L., Watkins J. C., Hammer M. F., Neandertal origin of genetic variation at the cluster of OAS immunity genes. Mol. Biol. Evol. 30, 798–801 (2013). [DOI] [PubMed] [Google Scholar]
- 43.Giacometti M., Roganti R., Tann D. D., Stahlberger-Saitbekova N., Obexer-Ruff G., Alpine ibex Capra ibex ibex x domestic goat C. aegagrus domesticahybrids in a restricted area of southern Switzerland. Wildl. Biol. 10, 137–143 (2004). [Google Scholar]
- 44.Junker K., Horak I. G., Penzhorn B., History and development of research on wildlife parasites in southern Africa, with emphasis on terrestrial mammals, especially ungulates. Int. J. Parasitol. Par. 4, 50–70 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Korneliussen T. S., Albrechtsen A., Nielsen R., ANGSD: Analysis of next generation sequencing data. BMC Bioinform. 15, 356 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M. A., The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Danecek P., Auton A., Abecasis G., Albers C. A., Banks E., Depristo M. A., Handsaker R. E., Lunter G., Marth G. T., Sherry S. T., McVean G., Durbin R.; 1000 Genomes Project Analysis Group , The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Browning S. R., Browning B. L., Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am. J. Hum. Genet. 81, 1084–1097 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Browning B. L., Browning S. R., Genotype imputation with millions of reference samples. Am. J. Hum. Genet. 98, 116–126 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhao S., Zheng P., Dong S., Zhan X., Wu Q., Guo X., Hu Y., He W., Zhang S., Fan W., Zhu L., Li D., Zhang X., Chen Q., Zhang H., Zhang Z., Jin X., Zhang J., Yang H., Wang J., Wang J., Wei F., Whole-genome sequencing of giant pandas provides insights into demographic history and local adaptation. Nat. Genet. 45, 67–71 (2013). [DOI] [PubMed] [Google Scholar]
- 51.de Magalhães J. P., Costa J., A database of vertebrate longevity records and their relation to other life-history traits. J. Evol. Biol. 22, 1770–1774 (2009). [DOI] [PubMed] [Google Scholar]
- 52.Chen L., Qiu Q., Jiang Y., Wang K., Lin Z., Li Z., Bibi F., Yang Y., Wang J., Nie W., Su W., Liu G., Li Q., Fu W., Pan X., Liu C., Yang J., Zhang C., Yin Y., Wang Y., Zhao Y., Zhang C., Wang Z., Qin Y., Liu W., Wang B., Ren Y., Zhang R., Zeng Y., da Fonseca R. R., Wei B., Li R., Wan W., Zhao R., Zhu W., Wang Y., Duan S., Gao Y., Zhang Y. E., Chen C., Hvilsom C., Epps C. W., Chemnick L. G., Dong Y., Mirarab S., Siegismund H. R., Ryder O. A., Gilbert M. T. P., Lewin H. A., Zhang G., Heller R., Wang W., Large-scale ruminant genome sequencing provides insights into their evolution and distinct traits. Science 364, eaav6202 (2019). [DOI] [PubMed] [Google Scholar]
- 53.Browning S. R., Browning B. L., Zhou Y., Tucci S., Akey J. M., Analysis of human sequence data reveals two pulses of archaic denisovan admixture. Cell 173, 53–61.e59 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xie C., Mao X., Huang J., Ding Y., Wu J., Dong S., Kong L., Gao G., Li C.-Y., Wei L., KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 39, W316–W322 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li H., Minimap and miniasm: Fast mapping and de novo assembly for noisy long sequences. Bioinformatics 32, 2103–2110 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Heger A., Holm L., Rapid automatic detection and alignment of repeats in protein sequences. Proteins 41, 224–237 (2000). [DOI] [PubMed] [Google Scholar]
- 57.Whitlock H. V., Some modifications of the McMaster helminth egg-counting technique and apparatus. J. Council Sci. Ind. Res. Au. 21, 177–180 (1948). [Google Scholar]
- 58.Meisner J., Albrechtsen A., Inferring population structure and admixture proportions in low depth NGS Data. Genetics 210, 719–731 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kang H. M., Sul J. H., Service S. K., Zaitlen N. A., Kong S.-Y., Freimer N. B., Sabatti C., Eskin E., Variance component model to account for sample structure in genome-wide association studies. Nat. Genet. 42, 348–354 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bolger A. M., Lohse M., Usadel B., Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bickhart D. M., Rosen B. D., Koren S., Sayre B. L., Hastie A. R., Chan S., Lee J., Lam E. T., Liachko I., Sullivan S. T., Burton J. N., Huson H. J., Nystrom J. C., Kelley C. M., Hutchison J. L., Zhou Y., Sun J., Crisà A., de León F. A. P., Schwartz J. C., Hammond J. A., Waldbieser G. C., Schroeder S. G., Liu G. E., Dunham M. J., Shendure J., Sonstegard T. S., Phillippy A. M., Van Tassell C. P., Smith T. P. L., Single-molecule sequencing and chromatin conformation capture enable de novo reference assembly of the domestic goat genome. Nat. Genet. 49, 643–650 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.H. Li, Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv:1303.3997v2 [q-bio.GN], (26 May 2013).
- 63.Okonechnikov K., Conesa A., García-Alcalde F., Qualimap 2: Advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 32, 292–294 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen N., Cai Y., Chen Q., Li R., Wang K., Huang Y., Hu S., Huang S., Zhang H., Zheng Z., Song W., Ma Z., Ma Y., Dang R., Zhang Z., Xu L., Jia Y., Liu S., Yue X., Deng W., Zhang X., Sun Z., Lan X., Han J., Chen H., Bradley D. G., Jiang Y., Lei C., Whole-genome resequencing reveals world-wide ancestry and adaptive introgression events of domesticated cattle in East Asia. Nat. Commun. 9, 2337 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Reimer P. J., Bard E., Bayliss A., Beck J. W., Blackwell P. G., Ramsey C. B., Buck C. E., Cheng H., Edwards R. L., Friedrich M., Grootes P. M., Guilderson T. P., Haflidison H., Hajdas I., Hatté C., Heaton T. J., Hoffmann D. L., Hogg A. G., Hughen K. A., Kaiser K. F., Kromer B., Manning S. W., Niu M., Reimer R. W., Richards D. A., Scott E. M., Southon J. R., Staff R. A., Turney C. S. M., van der Plicht J., IntCal13 and Marine13 radiocarbon age calibration curves 0–50,000 Years cal BP. Radiocarbon 55, 1869–1887 (2013). [Google Scholar]
- 66.Yang D. Y., Eng B., Waye J. S., Dudar J. C., Saunders S. R., Technical note: Improved DNA extraction from ancient bones using silica-based spin columns. Am. J. Phys. Anthropol. 105, 539–543 (1998). [DOI] [PubMed] [Google Scholar]
- 67.Schubert M., Lindgreen S., Orlando L., AdapterRemoval v2: Rapid adapter trimming, identification, and read merging. BMC. Res. Notes 9, 88 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li H., Durbin R., Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.; 1000 Genome Project Data Processing Subgroup , The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jónsson H., Ginolhac A., Schubert M., Johnson P. L. F., Orlando L., mapDamage2. 0: Fast approximate Bayesian estimates of ancient DNA damage parameters. Bioinformatics 29, 1682–1684 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pedersen B. S., Quinlan A. R., Mosdepth: Quick coverage calculation for genomes and exomes. Bioinformatics 34, 867–868 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Peltzer A., Jäger G., Herbig A., Seitz A., Kniep C., Krause J., Nieselt K., EAGER: Efficient ancient genome reconstruction. Genome Biol. 17, 60 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Martin M., Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 17, 10–12 (2011). [Google Scholar]
- 74.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M. A. R., Bender D., Maller J., Sklar P., de Bakker P. I. W., Daly M. J., Sham P. C., PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tamura K., Stecher G., Peterson D., Filipski A., Kumar S., MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Letunic I., Bork P., Interactive tree of life (iTOL) v3: An online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 44, W242–W245 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cornish-Bowden A., Nomenclature for incompletely specified bases in nucleic acid sequences: rcommendations 1984. Nucleic Acids Res. 13, 3021–3030 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Stamatakis A., RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Patterson N., Price A. L., Reich D., Population structure and eigenanalysis. PLOS Genet. 2, e190 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Alexander D. H., Novembre J., Lange K., Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Team R. D. C., R: A language and environment for statistical computing. R foundation for statistical computing. Computing 1, 12–21 (2016). [Google Scholar]
- 82.Puechmaille S. J., The program structure does not reliably recover the correct population structure when sampling is uneven: Subsampling and new estimators alleviate the problem. Mol. Ecol. Resour. 16, 608–627 (2016). [DOI] [PubMed] [Google Scholar]
- 83.Excoffier L., Dupanloup I., Huerta-Sánchez E., Sousa V. C., Foll M., Robust demographic inference from genomic and SNP data. PLOS Genet. 9, e1003905 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pickrell J. K., Pritchard J. K., Inference of population splits and mixtures from genome-wide allele frequency data. PLOS Genet. 8, e1002967 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lawson D. J., Hellenthal G., Myers S., Falush D., Inference of population structure using dense haplotype data. PLOS Genet. 8, e1002453 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Barrett J. C., Fry B., Maller J., Daly M. J., Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics 21, 263–265 (2005). [DOI] [PubMed] [Google Scholar]
- 87.Schiffels S., Durbin R., Inferring human population size and separation history from multiple genome sequences. Nat. Genet. 46, 919–925 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Handsaker R. E., Van Doren V., Berman J. R., Genovese G., Kashin S., Boettger L. M., McCarroll S. A., Large multiallelic copy number variations in humans. Nat. Genet. 47, 296–303 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Terhorst J., Kamm J. A., Song Y. S., Robust and scalable inference of population history from hundreds of unphased whole genomes. Nat. Genet. 49, 303–309 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gutenkunst R. N., Hernandez R. D., Williamson S. H., Bustamante C. D., Inferring the joint demographic history of multiple populations from multidimensional SNP frequency data. PLOS Genet. 5, e1000695 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nielsen R., Korneliussen T., Albrechtsen A., Li Y., Wang J., SNP calling, genotype calling, and sample allele frequency estimation from New-Generation Sequencing data. PLOS ONE 7, e37558 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Johnson J. B., Omland K. S., Model selection in ecology and evolution. Trends Ecol. Evol. 19, 101–108 (2004). [DOI] [PubMed] [Google Scholar]
- 93.Briggs A. W., Good J. M., Green R. E., Krause J., Maricic T., Stenzel U., Lalueza-Fox C., Rudan P., Brajković D., Kućan Ž., Gušić I., Schmitz R., Doronichev V. B., Golovanova L. V., de la Rasilla M., Fortea J., Rosas A., Pääbo S., Targeted retrieval and analysis of five Neandertal mtDNA genomes. Science 325, 318–321 (2009). [DOI] [PubMed] [Google Scholar]
- 94.Katoh K., Standley D. M., MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wang X., Zheng Z., Cai Y., Chen T., Li C., Fu W., Jiang Y., CNVcaller: Highly efficient and widely applicable software for detecting copy number variations in large populations. GigaScience 6, 1–12 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Paradis E., pegas: An R package for population genetics with an integrated–modular approach. Bioinformatics 26, 419–420 (2010). [DOI] [PubMed] [Google Scholar]
- 97.J. A. Lenstra, Evolutionary and demographic history of sheep and goats suggested by nuclear, mtDNA and Y chromosome markers, in The Role of Biotechnology for the Characterization and Conservation of Crop, Forestry, Animal and Fishery Geneic Resources (International Workshop, Turin, Italy, 5 to 7 March 2005), pp. 1–4. [Google Scholar]
- 98.Bouckaert R., Heled J., Kühnert D., Vaughan T., Wu C.-H., Xie D., Suchard M. A., Rambaut A., Drummond A. J., BEAST 2: A software platform for Bayesian evolutionary analysis. PLOS Comput. Biol. 10, e1003537 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Darriba D., Taboada G. L., Doallo R., Posada D., jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 9, 772 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Patterson N., Moorjani P., Luo Y., Mallick S., Rohland N., Zhan Y., Genschoreck T., Webster T., Reich D., Ancient admixture in human history. Genetics 192, 1065–1093 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gaunitz C., Fages A., Hanghøj K., Albrechtsen A., Khan N., Schubert M., Seguin-Orlando A., Owens I. J., Felkel S., Bignon-Lau O., de Barros Damgaard P., Mittnik A., Mohaseb A. F., Davoudi H., Alquraishi S., Alfarhan A. H., Al-Rasheid K. A. S., Crubézy E., Benecke N., Olsen S., Brown D., Anthony D., Massy K., Pitulko V., Kasparov A., Brem G., Hofreiter M., Mukhtarova G., Baimukhanov N., Lõugas L., Onar V., Stockhammer P. W., Krause J., Boldgiv B., Undrakhbold S., Erdenebaatar D., Lepetz S., Mashkour M., Ludwig A., Wallner B., Merz V., Merz I., Zaibert V., Willerslev E., Librado P., Outram A. K., Orlando L., Ancient genomes revisit the ancestry of domestic and Przewalski’s horses. Science 360, 111–114 (2018). [DOI] [PubMed] [Google Scholar]
- 102.Soraggi S., Wiuf C., Albrechtsen A., Powerful inference with the D-statistic on low-coverage whole-genome data. G3 8, 551–566 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tang H., Siegmund D. O., Shen P., Oefner P. J., Feldman M. W., Frequentist estimation of coalescence times from nucleotide sequence data using a tree-based partition. Genetics 161, 447–459 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ewing G., Hermisson J., MSMS: A coalescent simulation program including recombination, demographic structure and selection at a single locus. Bioinformatics 26, 2064–2065 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gao F., Ming C., Hu W., Li H., New software for the fast estimation of population recombination rates (FastEPRR) in the genomic era. G3 6, 1563–1571 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Korneliussen T. S., Moltke I., Albrechtsen A., Nielsen R., Calculation of Tajima’s D and other neutrality test statistics from low depth next-generation sequencing data. Bmc Bioinformatics 14, 289 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Szpiech Z. A., Hernandez R. D., selscan: An efficient multithreaded program to perform EHH-based scans for positive selection. Mol. Biol. Evol. 31, 2824–2827 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Degiorgio M., Huber C. D., Hubisz M. J., Hellmann I., Nielsen R., S weep F inder 2: Increased sensitivity, robustness and flexibility. Bioinformatics 32, 1895–1897 (2015). [DOI] [PubMed] [Google Scholar]
- 109.Heinlein C. A., Chang C., The roles of androgen receptors and androgen-binding proteins in nongenomic androgen actions. Mol. Endocrinol. 16, 2181–2187 (2002). [DOI] [PubMed] [Google Scholar]
- 110.Veinberg P., Analysis of horn shape and coat coloration in Capra (Artiodactyla). Byulleten Moskovskogo Obshchestva Ispytatelei Prirody, Otdel biologicheskii 98, 3–14 (1993). [Google Scholar]
- 111.Jordan S., Miquel C., Taberlet P., Luikart G., Sequencing primers and SNPs for rapidly evolving reproductive loci in endangered ibex and their kin (Bovidae, Capra spp.). Mol. Ecol. Notes 6, 776–779 (2006). [Google Scholar]
- 112.Vieira F. G., Lassalle F., Korneliussen T. S., Fumagalli M., Improving the estimation of genetic distances from next-generation sequencing data. Biol. J. Linn. Soc. 117, 139–149 (2015). [Google Scholar]
- 113.Lefort V., Desper R., Gascuel O., FastME 2.0: A comprehensive, accurate, and fast distance-based phylogeny inference program. Mol. Biol. Evol. 32, 2798–2800 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Browning B. L., Browning S. R., Improving the accuracy and efficiency of identity-by-descent detection in population data. Genetics 194, 459–471 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hu H., Petousi N., Glusman G., Yu Y., Bohlender R., Tashi T., Downie J. M., Roach J. C., Cole A. M., Lorenzo F. R., Rogers A. R., Brunkow M. E., Cavalleri G., Hood L., Alpatty S. M., Prchal J. T., Jorde L. B., Robbins P. A., Simonson T. S., Huff C. D., Evolutionary history of Tibetans inferred from whole-genome sequencing. PLOS Genet. 13, e1006675 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Dong Y., Xie M., Jiang Y., Xiao N., Du X., Zhang W., Tosser-Klopp G., Wang J., Yang S., Liang J., Chen W., Chen J., Zeng P., Hou Y., Bian C., Pan S., Li Y., Liu X., Wang W., Servin B., Sayre B., Zhu B., Sweeney D., Moore R., Nie W., Shen Y., Zhao R., Zhang G., Li J., Faraut T., Womack J., Zhang Y., Kijas J., Cockett N., Xu X., Zhao S., Wang J., Wang W., Sequencing and automated whole-genome optical mapping of the genome of a domestic goat (Capra hircus). Nat. Biotechnol. 31, 135–141 (2013). [DOI] [PubMed] [Google Scholar]
- 117.Letunic I., Doerks T., Bork P., SMART 7: Recent updates to the protein domain annotation resource. Nucleic Acids Res. 40, D302–D305 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Clark E. L., Bush S. J., McCulloch M. E. B., Farquhar I. L., Young R., Lefevre L., Pridans C., Tsang H. G., Wu C., Afrasiabi C., Watson M., Whitelaw C. B., Freeman T. C., Summers K. M., Archibald A. L., Hume D. A., A high resolution atlas of gene expression in the domestic sheep (Ovis aries). PLOS Genet. 13, e1006997 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/21/eaaz5216/DC1