

Room-temperature phosphorescence materials are manufactured by a facile click reaction between phosphor and polymer.
Abstract
To achieve efficient polymer-based room-temperature phosphorescence (RTP) materials, covalently embedding phosphors into the polymer matrix appeared as the most appealing approach. However, it is still highly challenging to fabricate RTP materials on a large scale because of the inefficient binding engineering and time-consuming covalent reactions. Here, we have proposed a scalable preparation approach for RTP materials by the facile B─O click reaction between boronic acid–modified phosphors and polyhydroxy polymer matrix. The ab initio molecular dynamics simulations demonstrated that the phosphors were effectively immobilized, resulting in the suppressed nonradiative transitions and activated RTP emission. In comparison to the reported covalent binding time of several hours, such a B─O click reaction can be accomplished within 20 s under ambient environment. The developed strategy simplified the construction of polymer-based RTP polymeric materials by the introduction of facile click chemistry. Our success provides inspirations and possibilities for the scale-up production of RTP materials.
INTRODUCTION
Polymer-based room-temperature phosphorescence (RTP) materials have attracted increasing attention in the past decades in the field of organic flexible electronics because of their fantastic advantages, including good flexibility, stretchability, and low cost (1–3). The past few decades have witnessed a great advance in the synthesis of polymer-based RTP materials (4, 5). There are two main categories for the polymer-based RTP materials (2). The first category, known as nondoped polymer materials, represented for the materials with phosphor in the backbone of polymer itself (6). The second category is achieved by embedding phosphors in polymer matrix (doped RTP polymers) (7, 8). The doped RTP materials showed prosperity in constructing efficient RTP polymeric materials because of the substantial role of polymer matrix in suppressing the nonradiative transitions of phosphors and activating RTP generation. Currently, most of the existing doped RTP materials are implemented via noncovalent interactions between phosphors and polymer matrices (9, 10). Disappointedly, noncovalent interaction (e.g., electrostatic interaction or van der Waals force) is a nondirectional weak linking, and phase separation is usually unavoidable (11, 12). Instead, covalent cross-linking might overcome such deficiencies (13, 14). Encouragingly, Zhao and coworkers successfully pioneered a long-lived heavy atom–free amorphous organic phosphorescence material through the formation of strong C─O─C covalent interactions. However, the preparation for RTP material is implemented under rigorous reaction conditions (11). In addition, a kind of catalyst or initiator is occasionally required to trigger a covalent reaction, and thus, this method is impractical for RTP material production because the catalysts are difficult to remove (15–17). Such drawbacks of the developed covalent cross-linking reaction would impede the large-scale applications of polymer-based RTP materials in flexible electronics manufacture. Accordingly, a reliable large-scale strategy for engineering highly efficient polymer-based RTP materials is highly desirable.
Click chemistry methodologies, especially the catalyst-free click reaction, have drawn numerous attention recently because of its merits with relatively mild reaction condition, high efficiency, and high yield (18, 19). These prominent features are essential benefits that could be extensively exploited in organic synthesis, polymerization, and bioengineering (20). Satisfactorily, the previous time-consuming procedures induced by multiple reaction steps and complex purification can be avoided by click reaction, leading to triumph in scale-up fabrication from laboratorial to industrial scale (21). To this end, functional polymers, hierarchical nanostructures, and even bioactive fragments can be synthesized in large scale (22, 23). These achievements make click chemistry methodology as an excellent candidate not only in the scientific studies but also for the large-scale manufacturing in engineering and industrial fields. Early studies revealed that the B─O click reaction could couple boronic species with organic sugars or polyol compounds to form a cyclic boronic ester with structural rigidity (24). The strong binding affinities between boronic group and hydroxyls have provided the versatility in molecular recognition and sensing (25). Importantly, the formation of B─O linkage is apt to finish under mild environments (26), inspiring us to take advantage of B─O covalent linkage to achieve highly efficient polymer-based RTP materials.
In this contribution, we have introduced a facile and catalyst-free click reaction into the synthesis of covalently linked phosphor-polymers RTP materials. Efficient RTP material can be constructed through strong B─O covalent bonds between tetraphenylethylene-diboronic acid (named as TPEDB) molecule and polyvinyl alcohol (PVA) matrix within 20 s under ambient environment (Fig. 1). This click reaction is energetically favorable, according to the Gibbs free energy change of −1.017 eV through the density functional theory (DFT) calculations. The number of B─O covalent bonds can be finely modulated through the click tailoring, contributing to the strong RTP intensity and long lifetime up to 768.6 ms for TPEDB-PVA polymeric material. The ab initio molecular dynamics (AIMD) simulations demonstrated that the efficient RTP was attributed to the suppressed molecular rotation and restricted nonradiative transition of TPEDB. The present strategy provides a platform for the large-scale manufacturing and industrialization of efficient polymer-based RTP materials, which may open up an avenue to realize the practical applications of RTP materials.
Fig. 1. Schematic representation for the polymer-based RTP.
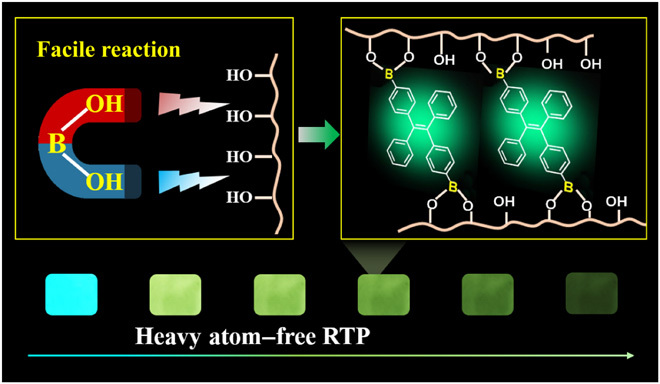
Facile and large-scale approach of RTP through B─O click reaction between phosphors with boronic acid and polymer with hydroxyl groups.
RESULTS
Fabrication of efficient RTP materials by a facile click reaction
To establish the covalent B─O bond with PVA matrix, two boronic acid units functionalized tetraphenylethylene (named as TPEDB; fig. S1) were used. The reaction between TPEDB and PVA can be facilely accomplished under ambient environment. The fluorescence of TPEDB was turned on within 20 s, and the pH of TPEDB was reduced from 10.88 to 10.76 upon the addition of PVA. These results demonstrated the occurrence of the covalent B─O click reaction. A flexible RTP polymeric material was successfully achieved after the solvent evaporation. The morphological studies of the TPEDB-PVA polymeric materials were implemented by scanning electronic microscopy and atomic force microscopy (AFM). A uniform and continuous surface can be obtained for the TPEDB-PVA, and the thickness was estimated to be ~27 μm by the side-view images (fig. S2). The elemental distribution in the TPEDB-PVA polymeric material was analyzed by the energy-dispersive x-ray spectroscopy (EDX) and mapping analysis. The results showed the homogeneously dispersed boron, oxygen, and carbon elements in both the horizontal and vertical directions throughout the film (fig. S2, B to D). The perfect matching from the merged picture of elements indicated the good combination between phosphors and polymer matrices.
The luminescent spectra of the TPEDB-PVA polymeric materials were recorded: fluorescent emission at 450 nm and green RTP emission at 535 nm (Fig. 2A). The Stokes shift of the phosphorescence spectra reached over 200 nm, which manifested the typical characteristic of RTP emission. A persistent lifetime up to 768.6 ms can be observed at 535 nm for the TPEDB-PVA polymeric material, with the fluorescent lifetime of 4.5 ns at 450 nm (fig. S3). Using an ultraviolet light-emitting diode (UV-LED) as light source (illustrated in fig. S4), bright cyan fluorescence can be observed. After the removal of light irradiation, the polymeric material exhibited intense green phosphorescence, which could be traced by the naked eyes with faded emission to 4 s (Fig. 2B). To study the origin of RTP, TPEDB, PVA, and TPEDB-x%PVA polymeric materials were drop-casted on quartz glass to investigate their phosphorescence spectra. It can be seen that the pristine TPEDB showed weak RTP emission, whereas the PVA matrix was non-emissive after UV irradiation (fig. S5A). With the addition of PVA into TPEDB molecules, RTP performances of TPEDB-PVA materials were promoted, indicating the role of PVA as a matrix to activate RTP of TPEDB. Moreover, with an increase in the PVA contents, the RTP intensities of TPEDB-PVA polymeric materials increased to a maximum and then decreased (fig. S5B). The strongest RTP intensity for the polymeric materials can be observed when PVA reached 60 mg. It can be speculated that the excess PVA could weaken the molecular interaction between TPEDB molecules (2).
Fig. 2. Luminescent behaviors of TPEDB-PVA polymeric materials.
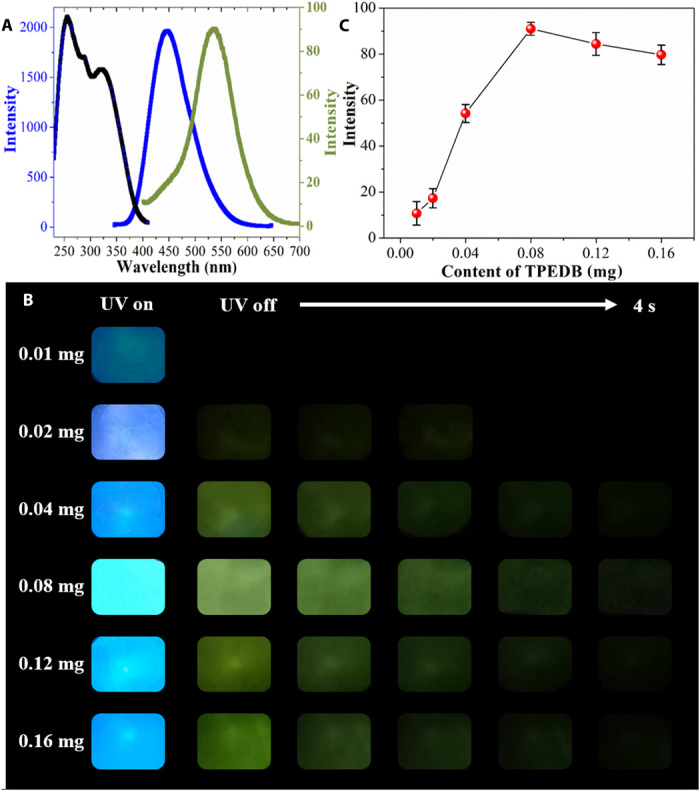
(A) Fluorescent excitation (black), emission (blue), and RTP emission (green) spectra of TPEDB-PVA polymeric material. (B) Photographs of TPEDB-PVA polymeric material under 365-nm ultraviolet (UV) irradiation and at different time intervals after removal of UV irradiation. Photo credit: Rui Tian (first author), Beijing University of Chemical Technology. (C) RTP intensities of TPEDB-PVA polymeric materials with different contents of TPEDB.
The effect of doping contents for TPEDB in TPEDB-PVA polymeric materials has been investigated. At lower content (0.01 to 0.04 mg of TPEDB), weak phosphorescence intensity can be recorded in the spectra, which can be hardly observed by the naked eyes. With the increased addition of TPEDB, the RTP intensities were promoted continuously (Fig. 2C). The optimum content of TPEDB was determined to be 0.08 mg in the TPEDB-PVA polymeric material to achieve the strongest RTP intensity. At excessive content of TPEDB, the hydroxyl groups in PVA matrices were inadequate to localize all the TPEDB molecules through B─O covalent bond, resulting in the energy dissipation through molecular motions of free TPEDB (2). Moreover, the UV-visible absorption spectra for TPEDB-PVA polymeric materials have been recorded. The strong absorbance around 350 nm indicated the promoted n → π* transition (fig. S6), which may be induced by the covalent binding between TPEDB and PVA. This may, in turn, facilitate the spin-orbit coupling and populate the triplet state through the enhanced intersystem crossing (27, 28). Therefore, an efficient RTP can be achieved through a facile and rapid cross-linking reaction between boronic acid modified phosphor and polymer matrix with hydroxyl groups.
Superiorities of the covalent bonds
To verify the importance of the B─O covalent interaction, controlled experiments of polymer in the absence of hydroxyl groups or boronic acid groups were implemented. Typically, TPEDB molecules were embedded into different hydroxyl-free polymers such as positively charged poly(diallyldimethyl-ammonium chloride) (PDDA), negatively charged poly(styrene-4-sulfonate) (PSS), and poly(vinylidene fluoride) (PVDF). As shown in fig. S7, the RTP intensities decreased distinctly for TPEDB-polymer materials in the absence of hydroxyl groups. This result was attributed to the lack of active sites and disability to form B─O bonds between phosphors and polymer matrices. Moreover, the weak noncovalent interaction contributed little to the immobilization and RTP activation of the TPEDB. The function of the boronic acid groups was further investigated by strategically positioning boronic acid groups on the TPE moiety. Neither covalent nor hydrogen bonding can form between bare TPE and PVA molecules, leading to faint RTP emissions for TPE-PVA material. In addition, TPE core functionalized with only one boronic acid (TPEMB) showed a weak RTP emission, as a result of the insufficient bonding with PVA matrices. These results demonstrated the necessity of both hydroxyl groups and boronic acid groups to form a stable covalent linkage for efficient RTP materials.
Regulation of the covalent bonds
The above results indicated that the covalent cross-linkage between TPEDB and PVA played substantial roles in the RTP performances. To verify the speculation, PVA with different alcoholysis degrees (72, 87, 92, and 98%, denoted as PVA72, PVA87, PVA92, and PVA98) has been applied. It can be realized that PVA98 had approximately 6, 11, and 26% more hydroxyl groups than PVA92, PVA87, and PVA72, respectively. As anticipated, the increasing alcoholysis degree of PVA from 72 to 98% led to the promoted RTP and fluorescent performances for the TPEDB-PVA polymeric materials (Fig. 3, A and B). The intense green phosphorescence with afterglow of 4 s for TPEDB-PVA98 was distinctly weakened for TPEDB-PVA92 and TPEDB-PVA87, and no RTP can be observed for TPEDB-PVA72 by the naked eye (fig. S8A). The phosphorescence decay data verified this phenomenon (Fig. 3C): The RTP lifetime of 768.6 ms for TPEDB-PVA98 was shortened to 443.3 and 140.1 ms for TPEDB-PVA92 and TPEDB-PVA87, respectively, and no phosphorescence lifetime above microsecond can be observed for the TPEDB-PVA72 polymeric material. This phenomenon is ascribed to the fact that the replacement of hydroxyl groups by acetate groups could lead to the smaller localization effect of TPEDB and the poorer RTP performances (fig. S8B).
Fig. 3. Luminescent performances of TPEDB-PVA polymeric materials with varied alcoholysis degree of PVA.
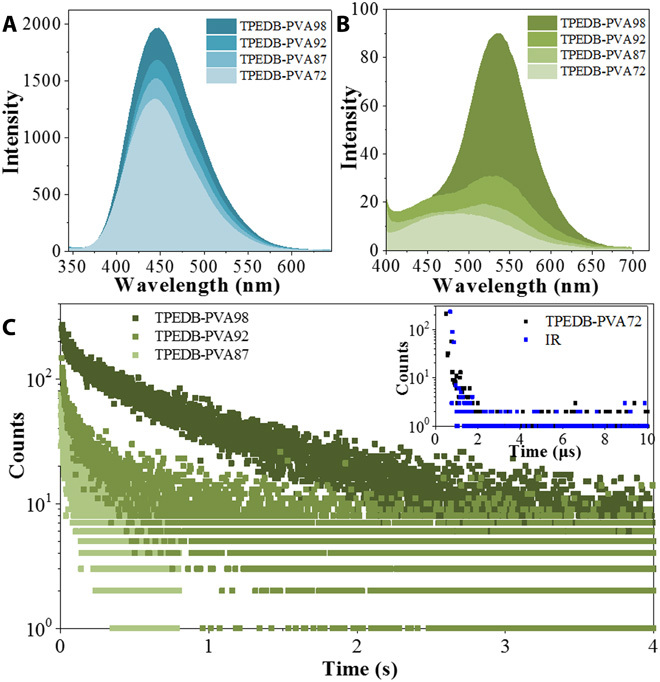
(A) Fluorescent emission, (B) phosphorescent emission spectra, and (C) RTP lifetime of TPEDB-PVA polymeric materials (TPEDB of 0.08 mg) with alcoholysis degree of PVA ranged from 87%, 92%, to 98%, and the inset shows the radiative curve of TPEDB-PVA72 polymeric material (black) and the instrumental reference (blue).
To verify the interactions between TPEDB and PVA with varied alcoholysis degree, x-ray diffraction (XRD) measurements were carried out for PVA and TPEDB-PVA polymeric materials. It can be seen that the semicrystalline peak at 19.6° for PVA showed a shift to the higher degree along with the decreased intensity upon the addition of TPEDB (Fig. 4A). Moreover, the peak at 22.6° increased obviously, demonstrating the formation of cross-linkage of PVA (11). In comparison with PVA72, PVA87, and PVA92, the biggest changes in PVA98 showed the greatest environmental variation by the reaction of TPEDB molecules with PVA. Furthermore, Fourier transform infrared (FTIR) measurements for TPEDB-PVA polymeric materials with varied alcoholysis degree in PVA were carried out. As shown in Fig. 4B, the characteristic peak attributed to the stretching of B─O bond can be observed in the range of 1310 to 1430 cm−1 for the TPEDB-PVA polymeric materials (29). The peaks were intensified with the varied PVA chains from PVA72 to PVA98, demonstrating that the more covalent bond could be built between TPEDB and PVA98 than TPEDB and PVA with less hydroxyl group. Moreover, a set of broad peaks assigned to the hydrogen bond appeared in the range of 3000 to 3500 cm−1, and these peaks were heightened along with the increasing alcoholysis degree of PVA. These results indicated that the hydrogen bonds were formed between the adjacent PVA molecules in constructing a rigid network. Therefore, the increased quantity of hydroxyl groups in PVA provided more linking sites for both covalent and hydrogen bonds in the system, establishing a much more favorable environment to confine the phosphors and activate their RTP.
Fig. 4. Structural studies for TPEDB-PVA polymeric materials.
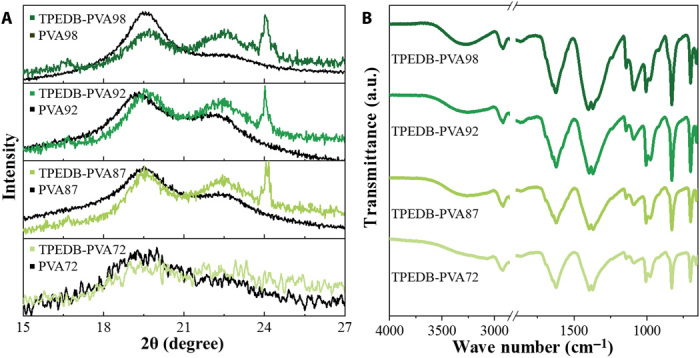
(A) XRD patterns and (B) FTIR spectra for TPEDB-PVA polymeric material (TPEDB of 0.08 mg) with alcoholysis degree of PVA ranging from 72%, 87%, 92%, to 98%. a.u., arbitrary units.
Mechanism study for the polymeric materials
To understand the B─O reaction, the origin of phosphorescence, and the enhancement of phosphorescence by the covalent localization of phosphor, DFT calculations were performed. First, the Gibbs free energy change of the click reaction (TPEDB + PVA → TPEDB-PVA + H2O) was calculated to be −1.017 eV (Fig. 5A), indicating that this reaction was quite favorable in the view of energy with the ultrafast reaction rate. Moreover, the energy levels of the ground state (S0), first singlet excited state (S1), and first triplet excited state (T1) for TPEDB (table S1) and PVA were calculated and displayed in Fig. 5B. For TPEDB, the energy levels of S0, S1, and T1 were −5.728, −3.266, and −3.757 eV versus the vacuum level, respectively. The results demonstrated that the electrons of TPEDB can be excited from S0 to S1 under irradiation, and then transfer from S1 to T1 and back to S0, resulting in the efficient radiation as RTP. In contrast, the energy levels of S0, S1, and T1 versus the vacuum level for PVA were −6.716, −2.751, and −2.318 eV, respectively. The T1 state of PVA lied higher than S1 state energetically, making the intersystem crossing forbidden. Therefore, the origin of phosphorescence was from TPEDB, and PVA was used as a non-emissive polymer matrix to stabilize TPEDB molecules.
Fig. 5. DFT calculations of TPEDB-PVA polymeric materials.

(A) Gibbs free energy change of the click reaction (TPEDB + PVA → TPEDB-PVA + H2O). (B) Molecular orbitals and energy levels of the ground state (S0), first singlet excited state (S1), and first triplet excited state (T1) for TPEDB molecule and PVA. (C) Definition of the dihedral angle (d) between the phenyl ring and C═C planes. (D) Distribution of dihedral angle d during AIMD for TPEDB-PVA (100, 67, and 50) and TPEDB with hydroxyl-free polymers (PDDA, PSS, and PVDF).
Furthermore, it has been reported that the nonradiative recombination for TPE-based molecules mainly derives from the rotation of phenyl ring (30, 31). To this end, the dihedral angle (abbreviated as d), defined as in Fig. 5C, demonstrated the rotation degree of phenyl ring in TPEDB under the confinement effect of covalent network. The AIMD simulations were performed on TPEDB-polymer materials (TPEDB-PVA100, TPEDB-PVA67, TPEDB-PVA50, TPEDB-PDDA, TPEDB-PSS, and TPEDB-PVDF). To explain and predict the molecular behaviors through the computational cost-effective model construction, the alcoholysis degree used in theoretical calculations varied in a larger scope (50, 67, and 100%) than that in experiments (72 to 98%). The geometry of each model after AIMD simulation was displayed in fig. S9. The distribution of dihedral angle d during the AIMD simulation for each model was shown in Fig. 5D. It can be observed that the d of TPEDB embedded in PVA (especially PVA100) distributed in smaller scope than that in PDDA, PSS, and PVDF (Fig. 5D). Moreover, the integral of d away from the equilibrium state was defined as to describe the molecular motion, where P is the possibility and is the averaged d. The integral values were calculated to be 1.449°, 3.352°, 4.053°, 4.384°, 5.631°, and 4.802° for TPEDB-PVA100, TPEDB-PVA67, TPEDB-PVA50, TPEDB-PDDA, TPEDB-PSS, and TPEDB-PVDF, respectively (fig. S9). The results indicated the better suppressed rotation of TPEDB in the covalent linkage provided by PVA than that in noncovalent binding by hydroxyl-free polymers. In addition, the effect of restraint increases along with the alcoholysis degree of PVA ranging from 50 to 100%. To reveal the intrinsic reason of this restraining effect, the binding energies between TPEDB and polymers were calculated according to eqs. S1 and S2. As expected, the binding energies for TPEDB-PVA polymeric materials (being −14.49, −8.31, and −6.12 eV for TPEDB-PVA100, TPEDB-PVA67, and TPEDB-PVA50, respectively) are much larger than that for TPEDB-PDDA (−3.77 eV), TPEDB-PSS (−0.44 eV), and TPEDB-PVDF (−0.71 eV). The enormous differences in binding energy further explained the stronger confinement of the covalent linkage, in comparison with the noncovalent bond network. Notably, the calculation results further explained the vital role of covalent linkage constructed by PVA in suppressing the nonradiative recombination and enhancing RTP performances for TPEDB molecules (32–34). Therefore, the RTP performances of TPEDB-PVA polymeric materials can be facilely manipulated by the quantity of hydroxyl groups in PVA.
Practicability of TPEDB-PVA polymeric materials
To explore the potential applications of TPEDB-PVA polymeric materials, the solubility and stability of the TPEDB-PVA polymeric materials were investigated. As seen in Fig. 6A, the TPEDB-PVA polymeric materials were completely dissolved in water after 2 min at 60°C. The fluorescence of the dissolved TPEDB-PVA polymeric material turned to weak blue fluorescence (Fig. 6A, inset). The good solubility of TPEDB-PVA polymeric RTP materials might be attributed to the fact that there are many hydroxyl groups available after the B─O click reaction. Moreover, the photostability of TPEDB-PVA polymeric materials was studied under UV irradiation. The RTP intensity at ~535 nm could maintain 80% of its original value after 80 min (Fig. 6B). The resistance toward UV irradiation of the TPEDB-PVA polymeric materials can be ascribed to the protection of TPEDB by PVA matrix. The solution-processing ability and decent photostability made this polymeric material as potential candidate as optoelectronic polymeric materials.
Fig. 6. Practicability of TPEDB-PVA RTP polymeric materials.
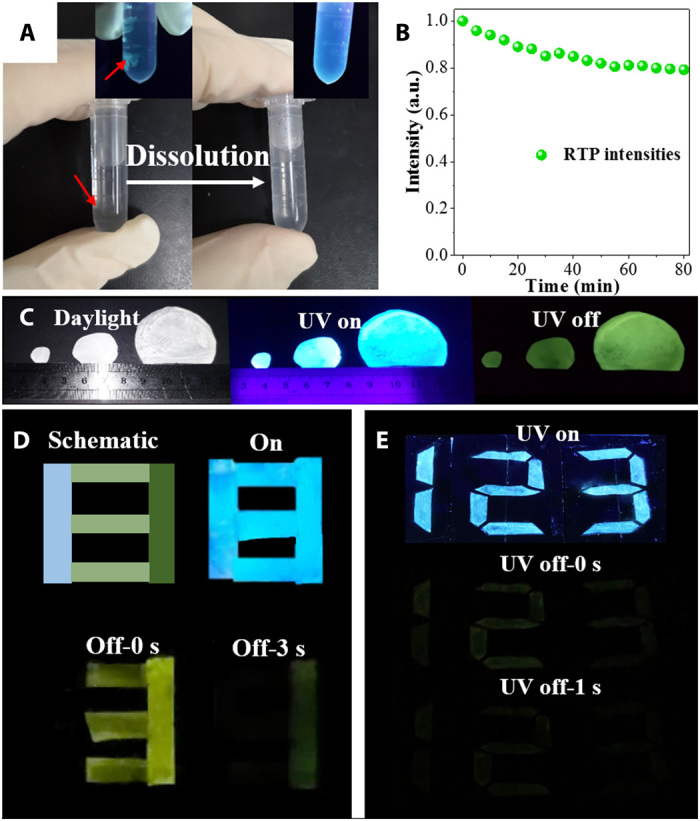
(A) Solubility in water (the inset shows the photos captured under UV irradiation) [photo credit: Rui Tian (first author), Beijing University of Chemical Technology], (B) photostability under UV irradiation, (C) photographs of scalable preparation of TPEDB-PVA polymeric materials (radii of 0.5, 1.0, and 2.5 cm) [photo credit: Qi Xu (coauthor), Beijing University of Chemical Technology], (D) lifetime-dependent data encryption, and (E) digital coding written by TPEDB ink on PVA under and after UV irradiation.
Scalable preparation of the TPEDB-PVA polymeric materials was realized, depending on the sizes of the petri dishes. TPEDB-PVA polymeric materials with radii of 0.5, 1.0, and 2.5 cm were successfully prepared (Fig. 6C). On the basis of the large-scale preparation, the data encryption of TPEDB-PVA polymeric materials was achieved. The number “8” was encoded by TPEDB-PVA with different alcoholysis degrees of PVA, which showed intense cyan fluorescence under UV excitation (Fig. 6D). Upon removing the light source, numbers “3” and “1” with green afterglow can be distinguished after different time delays. The different display of numbers was induced by the distinct RTP lifetimes of TPEDB-PVA polymeric materials with varied alcoholysis degrees in PVA chains. In addition, the specific interaction between TPEDB and PVA inspired us to explore the application of TPEDB as a kind of security ink for anti-counterfeiting on PVA matrix. The pure PVA matrix was prepared, and the TPEDB solution was used as ink for painting. As expected, numbers “123” with clear and definite boundary were patterned on PVA substrate instantly by TPEDB ink (Fig. 6E). Under UV irradiation, blue numbers can be observed. When the UV irradiation is removed, the green RTP can still be seen. Therefore, the facile click reaction between TPEDB and PVA can be regarded as a powerful and multifunctional tool in anti-counterfeiting and digital encoding.
DISCUSSION
To summarize, we have presented an efficient polymer-based RTP material through a facile one-step B─O click strategy. The RTP performances were finely regulated by the number of B─O covalent bonds. Because of the merits of simple operation, high efficiency, and scalable preparation, this developed engineering approach opens possibilities for method innovation in constructing polymeric RTP materials. This success facilitates the use of RTP materials in many applications, such as light-emitting devices and data security. It seems reasonable to anticipate that this strategy could be expanded to many other diverse RTP materials by tuning the reagents of click reaction.
MATERIALS AND METHODS
Materials
All reagents in experiments were of analytical grade and used without further purification. K2CO3, KHCO3, and dimethyl sulfoxide (DMSO) were purchased from Sinopharm Chemical Reagent Co. Ltd. PVA with different degrees of hydrolysis was purchased from Aladdin Chemical Co. Ltd.: PVA98 (hydrolysis degree of 98 to 99%), PVA92 (hydrolysis degree of 92 to 94%), PVA87 (hydrolysis degree of 87 to 89%), and PVA72 (hydrolysis degree of 72 to 74%). PDDA, PSS, and PVDF were purchased from Sigma-Aldrich. TPEDB acid was purchased from AIEgen Biotech Co. Ltd. and used without further treatment.
Preparation of TPEDB-PVA polymeric materials
To maintain the alkaline of the aqueous solution (26), a carbonate buffer containing K2CO3 and KHCO3 was prepared. Afterward, alkaline TPEDB solution was prepared in the buffer, and the concentration of TPEDB solution was 100 μM. PVA aqueous solution with the weight content of 3% was prepared in hot deionized water. The TPEDB-PVA suspensions were then prepared by dropwise addition of PVA (2.0 ml) into TPEDB solution with different concentrations under continuous shaking. The final contents of TPEDB in TPEDB-PVA polymeric materials were determined as 0.01, 0.02, 0.04, 0.08, 0.12, and 0.16 mg, respectively. The suspension of each TPEDB-PVA polymeric material was slowly poured into culture dishes. The dishes with TPEDB-PVA polymeric materials were then treated with ultrasound to drive out the bubbles. After drying at 60°C for 3 hours, thin and transparent films were obtained.
Similarly, the controlled experiments (TPEDB-polymer, TPEDB of 0.08 mg and polymer of 2 ml) based on PVA with different hydrolysis degrees (72, 87, 92, and 98%) and other hydroxyl-free polymers (PDDA, PSS, and PVDF) were carried out in the same way.
Digital coding and encryption of TPEDB-PVA polymeric materials
The TPEDB-PVA polymeric materials with different alcoholysis degrees of PVA have been used as different parts in letter “8.” The encryption was realized by UV light on, UV light off (instantly), and UV light off after 3 s.
The pure PVA films were fabricated as follows: PVA solution with a weight content of 3% was prepared, and the as-prepared solution was poured into culture dishes and dried at 60°C. TPEDB was written as security ink on the PVA substrate directly. The writing procedure was processed without additional treatment for transforming. The encryption was realized by UV light on, UV light off (instantly), and UV light off after 2 s. The photos were taken by mobile phone.
Computational model construction
The model of TPEDB was built according to its molecular formula (fig. S1). To study the effect of alcoholysis degree of PVA on the phosphorescence of TPEDB, PVA molecules with alcoholysis degree ranging from 50, 67, to 100% were constructed, which were named as PVA50, PVA67, and PVA100, respectively. On the other hand, the models for the controlled samples including PDDA, PSS, and PVDF were also built according to fig. S1 as comparisons. Each polymer contained five monomers.
Then, the models of TPEDB-PVA50, TPEDB-PVA67, TPEDB-PVA100, TPEDB-PDDA, TPEDB-PSS, and TPEDB-PVDF were built by placing one TPEDB molecule and eight polymer chains into a box with the space group of P1 (lattice parameters of α = β = γ = 90°). The other three lattice parameters a, b, and c were set according to the size of each polymer chain.
Computational methods
The geometries of models TPEDB, PVA50, PVA67, PVA100, PDDA, PSS, and PVDF were optimized with the Gaussian 09 software package (35). The geometries of the first singlet and triplet excited states for TPEDB were also calculated. DFT with the exchange-correlation functional of B3LYP (36) at the level of 6-311+G(d) was applied. The Gibbs free energy was obtained by calculating the frequency. To calculate the excited states of TPEDB, time-dependent DFT was performed.
The AIMD simulations for TPEDB-PVA50, TPEDB-PVA67, TPEDB-PVA100, TPEDB-PDDA, TPEDB-PSS, and TPEDB-PVDF were performed with the DMol3 code (37) with the exchange-correlation functional of generalized gradient approximation (GGA) Perdew-Burke-Ernzerhof (PBE) (38). All electron method was used for core treatment, and double numerical plus polarization basis set was applied. The self-consistent field tolerance was 2.0 × 10−6 hartree. The AIMD simulations were performed in isothermal-isobaric (NPT) ensemble, with a temperature of 298 K and a pressure of 0.1 MPa. Temperature and pressure were controlled using the Andersen method (39) and the Berendsen method (40), respectively. A total simulation time of 20 ps was performed for each model with a time step of 1 fs.
The covalent binding energy between TPEDB and PVA, EB, was calculated according to Eq. 1
| (1) |
where ETPEDB-PVA, Ewater, ETPEDB, and EPVA are the energy of TPEDB-PVA (50, 67, or 100), H2O, TPEDB, and PVA (50, 67, or 100), respectively.
The noncovalent binding energy between TPEDB and PDDA (or PSS, PVDF), EB, was calculated with Eq. 2
| (2) |
Sample characterizations
The fluorescence and phosphorescence spectra of all the samples were measured on an F-7000 spectrophotometer (Hitachi, Japan) with an excitation wavelength of 330 nm. The fluorescence emission spectra range from 350 to 650 nm with voltages of photomultiplier tube of 400 V. The phosphorescence spectra were collected in the range of 400 to 700 nm. The photostability measurements were implemented by capturing the phosphorescence spectra every 5 min under continuous UV irradiation. Fluorescence lifetime measurements were recorded with an Edinburgh Instruments FL 980 fluorimeter. Phosphorescence lifetimes of polymeric materials were measured on an Edinburgh Instruments FL 920 fluorimeter. FTIR spectra of the flexible films were recorded on a Nicolet 6700 spectrometer (Thermo Electron) with ZnSe crystal for the attenuated total reflection of the film. XRD patterns of the polymeric materials were measured on a Rigaku 2500 VB2+PC diffractometer with a count time of 10 s per step. The morphologies of films were investigated by a scanning electron microscope (Hitachi S-3500), and the accelerating voltage applied was 20 kV, in combination with EDX for the determination of elemental composition. The morphology data of AFM were implemented on a NanoScope 9.1 instrument (Bruker, Germany).
Supplementary Material
Acknowledgments
Funding: This work was supported by the National Natural Science Foundation of China (21838007, 21521005, 21575010, 21701004, and 21974008) and the Innovation and Promotion Project of Beijing University of Chemical Technology. Author contributions: R.T. and C.L. conceived the experiments. R.T. and Q.X. carried out the experiments. S.-M.X. implemented the simulation. R.T., S.-M.X., and C.L. contributed to data analysis and writing of this manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/21/eaaz6107/DC1
REFERENCES AND NOTES
- 1.Chen X., Xu C., Wang T., Zhou C., Du J., Wang Z., Xu H., Xie T., Bi G., Jiang J., Zhang X., Demas J. N., Trindle C. O., Luo Y., Zhang G., Versatile room-temperature-phosphorescent materials prepared from n-substituted naphthalimides: Emission enhancement and chemical conjugation. Angew. Chem. Int. Ed. Engl. 55, 9872–9876 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Gan N., Shi H., An Z., Huang W., Recent advances in polymer-based metal-free room-temperature phosphorescent materials. Adv. Funct. Mater. 28, 1802657 (2018). [Google Scholar]
- 3.Ma X., Wang J., Tian H., Assembling-induced emission: An efficient approach for amorphous metal-free organic emitting materials with room-temperature phosphorescence. Acc. Chem. Res. 52, 738–748 (2019). [DOI] [PubMed] [Google Scholar]
- 4.Lee D., Bolton O., Kim B. C., Youk J. H., Takayama S., Kim J., Room temperature phosphorescence of metal-free organic materials in amorphous polymer matrices. J. Am. Chem. Soc. 135, 6325–6329 (2013). [DOI] [PubMed] [Google Scholar]
- 5.Ogoshi T., Tsuchida H., Kakuta T., Yamagishi T.-a., Taema A., Ono T., Sugimoto M., Mizuno M., Ultralong room-temperature phosphorescence from amorphous polymer poly(styrene sulfonic acid) in air in the dry solid state. Adv. Funct. Mater. 28, 1707369 (2018). [Google Scholar]
- 6.Chengjian K., Liu B., Enhancing the performance of pure organic room-temperature phosphorescent luminophores. Nat. Commun. 10, 2111 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cai S., Ma H., Shi H., Wang H., Wang X., Xiao L., Ye W., Huang K., Cao X., Gan N., Ma C., Gu M., Song L., Xu H., Tao Y., Zhang C., Yao W., An Z., Huang W., Enabling long-lived organic room temperature phosphorescence in polymers by subunit interlocking. Nat. Commun. 10, 4247 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hirata S., Totani K., Zhang J., Yamashita T., Kaji H., Marder S. R., Watanabe T., Adachi C., Efficient persistent room temperature phosphorescence in organic amorphous materials under ambient conditions. Adv. Funct. Mater. 23, 3386–3397 (2013). [Google Scholar]
- 9.Jiang K., Zhang L., Lu J., Xu C., Cai C., Lin H., Triple-mode emission of carbon dots: Applications for advanced anti-counterfeiting. Angew. Chem. Int. Ed. Engl. 55, 7231–7235 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Kwon M. S., Lee D., Seo S., Jung J., Kim J., Tailoring intermolecular interactions for efficient room-temperature phosphorescence from purely organic materials in amorphous polymer matrices. Angew. Chem. Int. Ed. Engl. 53, 11177–11181 (2014). [DOI] [PubMed] [Google Scholar]
- 11.Su Y., Phua S. Z. F., Li Y., Zhou X., Jana D., Liu G., Lim W. Q., Ong W. K., Yang C., Zhao Y., Ultralong room temperature phosphorescence from amorphous organic materials toward confidential information encryption and decryption. Sci. Adv. 4, eaas9732 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kwon M. S., Yu Y., Coburn C., Phillips A. W., Chung K., Shanker A., Jung J., Kim G., Pipe K., Forrest S. R., Youk J. H., Gierschner J., Kim J., Suppressing molecular motions for enhanced room-temperature phosphorescence of metal-free organic materials. Nat. Commun. 6, 8947 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang G., Palmer G. M., Dewhirst M. W., Fraser C. L., A dual-emissive-materials design concept enables tumour hypoxia imaging. Nat. Mater. 8, 747–751 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ma X., Xu C., Wang J., Tian H., Amorphous pure organic polymers for heavy-atom-free efficient room-temperature phosphorescence emission. Angew. Chem. Int. Ed. Engl. 57, 10854–10858 (2018). [DOI] [PubMed] [Google Scholar]
- 15.Zhang G., Chen J., Payne S. J., Kooi S. E., Demas J. N., Fraser C. L., Multi-emissive difluoroboron dibenzoylmethane polylactide exhibiting intense fluorescence and oxygen-sensitive room-temperature phosphorescence. J. Am. Chem. Soc. 129, 8942–8943 (2007). [DOI] [PubMed] [Google Scholar]
- 16.Pfister A., Zhang G., Zareno J., Horwitz A. F., Fraser C. L., Boron polylactide nanoparticles exhibiting fluorescence and phosphorescence in aqueous medium. ACS Nano 2, 1252–1258 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen H., Xu L., Ma X., Tian H., Room temperature phosphorescence of 4-bromo-1,8-naphthalic anhydride derivative-based polyacrylamide copolymer with photo-stimulated responsiveness. Polym. Chem. 7, 3989–3992 (2016). [Google Scholar]
- 18.Liu B., Deng X., Xie Z., Cheng Z., Yang P., Lin J., Thiol-ene click reaction as a facile and general approach for surface functionalization of colloidal nanocrystals. Adv. Mater. 29, 1604878 (2017). [DOI] [PubMed] [Google Scholar]
- 19.Kukwikila M., Gale N., El-Sagheer A. H., Brown T., Tavassoli A., Assembly of a biocompatible triazole-linked gene by one-pot click-DNA ligation. Nat. Chem. 9, 1089–1098 (2017). [DOI] [PubMed] [Google Scholar]
- 20.Arslan M., Acik G., Tasdelen M. A., The emerging applications of click chemistry reactions in the modification of industrial polymers. Polym. Chem. 10, 3806–3821 (2019). [Google Scholar]
- 21.Hou K., Zeng Y., Zhou C., Chen J., Wen X., Xu S., Cheng J., Pi P., Facile generation of robust POSS-based superhydrophobic fabrics via thiol-ene click chemistry. Chem. Eng. J. 332, 150–159 (2018). [Google Scholar]
- 22.Xu Q., Huang T., Li S., Li K., Li C., Liu Y., Wang Y., Yu C., Zhou Y., Emulsion-assisted polymerization-induced hierarchical self-assembly of giant sea urchin-like aggregates on a large scale. Angew. Chem. Int. Ed. Engl. 57, 8043–8047 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Taemaitree L., Shivalingam A., El-Sagheer A. H., Brown T., An artificial triazole backbone linkage provides a split-and-click strategy to bioactive chemically modified CRISPR sgRNA. Nat. Commun. 10, 1610 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lorand J. P., Edwards J. O., Polyol complexes and structure of the benzeneboronate ion. J. Org. Chem. 24, 769–774 (1959). [Google Scholar]
- 25.Yang K.-A., Barbu M., Halim M., Pallavi P., Kim B., Kolpashchikov D. M., Pecic S., Taylor S., Worgall T. S., Stojanovic M. N., Recognition and sensing of low-epitope targets via ternary complexes with oligonucleotides and synthetic receptors. Nat. Chem. 6, 1003–1008 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu Y., Deng C., Tang L., Qin A., Hu R., Sun J. Z., Tang B. Z., Specific detection of D-glucose by a tetraphenylethene-based fluorescent sensor. J. Am. Chem. Soc. 133, 660–663 (2011). [DOI] [PubMed] [Google Scholar]
- 27.Jiang K., Wang Y., Gao X., Cai C., Lin H., Facile, quick, and gram-scale synthesis of ultralong-lifetime room-temperature-phosphorescent carbon dots by microwave irradiation. Angew. Chem. Int. Ed. Engl. 57, 6216–6220 (2018). [DOI] [PubMed] [Google Scholar]
- 28.An Z., Zheng C., Tao Y., Chen R., Shi H., Chen T., Wang Z., Li H., Deng R., Liu X., Huang W., Stabilizing triplet excited states for ultralong organic phosphorescence. Nat. Mater. 14, 685–690 (2015). [DOI] [PubMed] [Google Scholar]
- 29.Nishiyabu R., Shimizu A., Boronic acid as an efficient anchor group for surface modification of solid polyvinyl alcohol. Chem. Commun. 52, 9765–9768 (2016). [DOI] [PubMed] [Google Scholar]
- 30.Dong J., Li X., Peh S. B., Yuan Y. D., Wang Y., Ji D., Peng S., Liu G., Ying S., Yuan D., Jiang J., Ramakrishna S., Zhao D., Restriction of molecular rotors in ultrathin two-dimensional covalent organic framework nanosheets for sensing signal amplification. Chem. Mater. 31, 146–160 (2018). [Google Scholar]
- 31.Dong J., Li X., Zhang K., Di Yuan Y., Wang Y., Zhai L., Liu G., Yuan D., Jiang J., Zhao D., Confinement of aggregation-induced emission molecular rotors in ultrathin two-dimensional porous organic nanosheets for enhanced molecular recognition. J. Am. Chem. Soc. 140, 4035–4046 (2018). [DOI] [PubMed] [Google Scholar]
- 32.Yuan W. Z., Shen X. Y., Zhao H., Lam J. W. Y., Tang L., Lu P., Wang C., Liu Y., Wang Z., Zheng Q., Sun J. Z., Ma Y., Tang B. Z., Crystallization-induced phosphorescence of pure organic luminogens at room temperature. J. Phys. Chem. C 114, 6090–6099 (2010). [Google Scholar]
- 33.Xiong Y., Zhao Z., Zhao W., Ma H., Peng Q., He Z., Zhang X., Chen Y., He X., Lam J. W. Y., Tang B. Z., Designing efficient and ultralong pure organic room-temperature phosphorescent materials by structural isomerism. Angew. Chem. Int. Ed. Engl. 57, 7997–8001 (2018). [DOI] [PubMed] [Google Scholar]
- 34.Zhao W., Cheung T. S., Jiang N., Huang W., Lam J. W. Y., Zhang X., He Z., Tang B. Z., Boosting the efficiency of organic persistent room-temperature phosphorescence by intramolecular triplet-triplet energy transfer. Nat. Commun. 10, 1595 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, Gaussian 09, Revision D.01 (Gaussian Inc., 2009).
- 36.Beche A. D., Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 98, 5648–5652 (1993). [Google Scholar]
- 37.Delley B., An all-electron numerical method for solving the local density functional for polyatomic molecules. J. Chem. Phys. 92, 508–517 (1990). [Google Scholar]
- 38.Perdew J. P., Burke K., Ernzerhof M., Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996). [DOI] [PubMed] [Google Scholar]
- 39.Andersen H. C., Molecular dynamics simulations at constant pressure and/or temperature. J. Chem. Phys. 72, 2384–2393 (1980). [Google Scholar]
- 40.Berendsen H. J. C., Postma J. P. M., Van Gunsteren W. F., DiNola A., Haak J. R., Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690 (1984). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/21/eaaz6107/DC1