

Abstract
ATAD2 is a cancer-associated protein whose bromodomain has been described as among the least druggable of that target class. Starting from a potent lead, permeability and selectivity were improved through a dual approach: 1) using CF2 as a sulfone bio-isostere to exploit the unique properties of fluorine, and 2) using 1,3-interactions to control the conformation of a piperidine ring. This resulted in the first reported low-nanomolar, selective and cell permeable chemical probe for ATAD2.
Keywords: bioisosteres, conformation analysis, epigenetics, fluorine, medicinal chemistry
High expression levels of ATAD2 (ATPase family, AAA domain containing 2), also called ANCCA (AAA + nuclear coregulator cancer-associated), correlate with poor outcomes in several cancers, and its knockdown modulates multiple tumor cell growth factors.[1–3] Efforts to target this protein have focused on competitive binding to the acetyl-lysine (KAc) site of its bromodomain, but the role of the bromodomain in the biology of ATAD2 is unclear. We have developed chemical tools to try to understand this, and recently reported the discovery of quinolinones and naphthyridones such as 1–3 and 5–7 that bind to the KAc site of ATAD2.[4, 5]
The profound biological effects associated with BET inhibition complicate interpretation of phenotypes observed with unselective inhibitors of other bromodomains.[6] While potent against ATAD2, naphthyridone 5 had only modest selectivity over the BETs, represented in Table 1 by the first bromodomain of BRD4 (BRD4 BD1). We recently reported selectivity improvements from occupying an electrostatically positive site near the KAc pocket, the RVF shelf, with negative polarity (compare 5 to 6 to see the effect of introducing cyclic sulfones at the C3’ position of the piperidine ring with (R,R) stereochemistry).[5] An X-ray structure of ATAD2 bound to 3 showed that its sulfone oxygen atoms displace a weakly-bound water molecule and accept two hydrogen bonds from the guanidinium group of Arg1077 (Figure 1a). The WPF shelf of the BET bromodomains (the analogous subsite to the ATAD2 RVF shelf) contains lipophilic amino acids Trp81 and Met149 in place of ATAD2 residues Arg1007 and Arg1077, so is less tolerant of the sulfone groupQs polarity. Compounds with hydrophobic C3’-substituents such as the cyclohexylmethylenes 2 and 5 are more potent against the BETs, which is consistent with the crystal structure of 2 bound to BRD4 BD1, where the C3’-cyclohexyl ring rests on the WPF shelf (Figure 1b). Unfortunately, while the sulfone C3’ group improved selectivity it also impacted passive permeability, raising doubts over the suitability of 6 and 7 as tool compounds for chromatin-bound ATAD2. Here, we describe the conclusion of our optimization of the series to overcome this.
Table 1. Micromolar lead to nanomolar ATAD2 inhibitors. For statistics see Table S1a, Supporting Information.
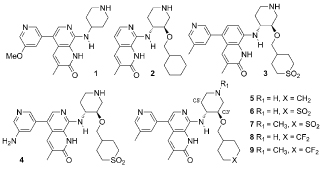 | ||||||||
|---|---|---|---|---|---|---|---|---|
| 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | |
| ATAD2 TR-FRET pIC50 | 5.6 | 7.2 | 6.9 | 6.7 | 6.9 | 6.5 | 7.4 | 7.1 |
| ATAD2 Bromosphere pIC50 | 5.4 | 7.4 | 7.3 | 6.5 | 7.5 | 7.0 | 7.2 | 7.1 |
| BRD4 BD1 TR-FRET pIC50 | 5.4 | 5.4 | 5.0 | 5.8 | 4.8 | 4.1 | 5.2 | 5.1 |
| TR-FRET selectivity (logs) | 0.2 | 1.8 | 1.9 | 0.9 | 2.1 | 2.4 | 2.2 | 2.0 |
| Chrom logD (pH 7.4) | 3.3 | 2.1 | 0.4 | 4.0 | 1.6 | 2.3 | 3.0 | 4.1 |
| Polar surface area [Å2] | 79 | 113 | 152 | 92 | 126 | 117 | 92 | 83 |
| Artificial membrane permeability (nms−1, pH 7.4) | 130 | <3 | <3 | 138 | <3 | <10 | 86 | 395 |
Figure 1.

A) 2 views of the X-ray structure of 3 bound to ATAD2 (PDB 5a83). B) Superimposed X-ray structures of BRD4 BD1 bound to 1 (orange, PDB 5a5s) and 2 (cyan, PDB 5a85). C) Superimposed X-ray structures of BRD4 BD1 bound to 1 (orange, PDB 5a5s) and 4 (green, PDB 5lj2) showing the di-axial conformation of the piperidine ring. D) ATAD2 crystallographic binding modes of 16 (cyan, PDB 5lj0) and 3 (green, PDB 5a83). E) Binding modes in BRD4 BD1 of 16 (cyan, PDB 5lj1) and 4 (green, PDB 5lj2). For refinement statistics see Table S3; for density maps Figure S6, Supporting Information.
Alternative C3’ functional groups of intermediate polarity had been found inferior to the sulfone in potency or selectivity.[5] We therefore considered the possibilities offered by fluorine substitution. The effect of fluorine on features of organic molecules such as conformation, pKa, permeability and metabolism has recently been reviewed.[7] Fluorine combines unique properties of polarity, lipophilicity and low polarisability.[8–10] Regarding noncovalent interactions, the ability of organofluorine to participate in hydrogen bonds has been debated.[10–15] While the lone pairs of organic fluorine seem too tightly held to hydrogen-bond, strong charge–dipole interactions can be formed with cations.[15]
The bi-lobal negative charge patches characteristic of the molecular surface of the SO2 group of 3 are well oriented to interact with the ATAD2 RVF shelf residue Arg1077 (Figure 1a). The CF2 group possesses geometrically similar electronegative patches (Figure S1, Supporting Information) and should be compatible with polar interactions with Arg1077. Indeed, the guanidinium group of arginine has been described as highly fluorophilic.[14] We reasoned that CF2 may provide an isosteric replacement for SO2, that its large dipole moment relative to CH2 (2.57 D in 1,1-difluorocyclohexane[16]) might disfavour interaction with the hydrophobic BRD4 BD1 WPF shelf, and that the expected increase in lipophilicity should have a positive effect on permeability.
Fluorinated derivatives 8 and 9 were made according to previously reported procedures[5] and their profile compared to the direct sulfone analogues 6 and 7 (Table 1). The higher logD and lower polar surface area of the difluoromethylene compounds relative to the sulfones resulted in dramatically improved artificial membrane permeability, comparable to the cyclohexyl analogues (compare 5, 6 and 8). The difluoromethylenes 8 and 9 had greater TR-FRET ATAD2 potency than the sulfones (6 and 7), which we rationalize by a decreased desolvation penalty. While the selectivity of 8 and 9 for ATAD2 over BRD4 BD1 was slightly lower than the analogous sulfones 6 and 7, it was significantly better than the cyclohexyl 5 and matched our probe critera of >2 logs. Overall, in this series the CF2 group proved to be an excellent SO2 isostere.
In parallel, we sought ways to increase the selectivity window by better understanding the binding mode of analogues of 6 to BRD4 BD1. We assumed that this would be similar to previous analogues such as 1 and 2 (Figure 1b). The naphthyridone binds in the KAc-pocket, with the 3’ substituent of 2 located on the WPF shelf as outlined above. We obtained a crystal structure of BRD4 BD1 bound to 4, the amino-pyridine analogue of 6. The bound position of the naphthyridone of 4 is very like that of 1. Unexpectedly, rather than the 3’ substituent lying on the WPF shelf as expected by analogy with 2, the piperidine ring of 4 adopts a trans di-axial conformation, positioning the SO2-containing 3’-substituent in a solvent-exposed position far from the WPF shelf (Figure 1c). This is in sharp contrast to the trans di-equatorial conformation seen in ATAD2 for sulfone-containing 3’-substituted compounds like 3 (Figure 1a). In this axial conformation, the piperidine nitrogen of 4 makes a salt-bridge interaction with the sidechain of BRD4 Asp144. However, as this was not seen with 1 or 2 this cannot be the main driver for the adoption of the di-axial conformation in BRD4 BD1. Rather, this conformation allows the polar sulfone group to evade the hydrophobic WPF shelf environment.
Interestingly, gas-phase ab initio calculations on model systems favor the di-axial conformation, which is stabilised by a 1,3 interaction between the alkoxy oxygen and the protonated piperidine nitrogen (Figure S1c, Supporting Information). In a continuum water model the strong electrostatic contribution was diminished and di-equatorial was slightly preferred. NMR experiments on 4 in water indicated an equilibrium, which was rapid on the NMR timescale between these two piperidine chair conformers, in an approximately 5:2 ratio in favour of the trans di-equatorial (see Supporting Information). Hence, both molecular modelling and experimental data support the trans di-axial conformer as a low energy form for this structure.
These observations suggested that selectivity for ATAD2 over BRD4 BD1 might be increased by stabilization of the equatorial piperidine conformation over axial. Introduction of a substituent cis to the ether at the C5’ position would create a 1,3 steric repulsion in the tri-axial conformation, favouring the tri-equatorial. We tested this idea by introducing a methoxy substituent in the C5’ position via the route shown in Scheme 1.
Scheme 1.

Conditions: a) m-CPBA, CH2Cl2, 0°C; b) NaH, MeI, DMF, 000B0;C, 83% (2 steps); c) LiClO4, NaN3, CH3CN, 8000B0;C, 35%; d) tBuOK, THF, 0°C then RCH2OTf, 70–72%; e) H2, Pd/C, MeOH, room temperature, 90–91%; f) 14, tBuONa, BrettPhos, Pd(OAc)2 or Pd(dba)2, THF, 60°C, 37–43%; g) NBS, CH2Cl2, –10 °C, 94–97%; h) ArB(OH)2, K2CO3, Pd(OAc)2, cataCXium A, dioxane/H2O, microwave, 100°C, 40–49 %; i) TFA, reflux, >93%.
Epoxide 11 was easily obtained as a single diastereoisomer from alcohol 10 and could be opened with sodium azide in the presence of LiClO4 to give a 1:1 mixture of regioisomers, which were separable.[17] Following alkylation and reduction of 12, amines 13a,b could be coupled with naphthyridone 14 in moderate yields.[5] Regioselective bromination, Suzuki coupling and deprotection in acidic conditions provided the inhibitors 15–17. Chiral purification could be performed at the final stage or following the Buchwald coupling.
Data for conformationally biased analogues are shown in Table 2. The C5’-OMe C3’-sulfone-containing piperidine 15 retained the ATAD2 activity of des-methoxy 6, but its BRD4 BD1 activity was significantly weaker, resulting in an increased selectivity window of >3 logs. We believe that this is due to the destabilization of the axial conformation favored for BRD4 BD1 binding relative to the equatorial favored by ATAD2. This conclusion was supported by NMR data for 15 in water which indicated a strong preference for the tri-equatorial conformation, with no evidence for significant presence of tri-axial (see Supporting Information).
Table 2. Comparison of di- and tri-substituted inhibitors. For statistics see Table S1a, Supporting Information.
| 6 | 15 | 8 | 16 | 17 | |
|---|---|---|---|---|---|
| ATAD2 TR-FRET pIC50 | 6.9 | 6.9 | 7.4 | 7.3 | 5.5 |
| ATAD2 Bromosphere pIC50 | 7.5 | 8.4 | 7.2 | 8.0 | |
| BRD4 BD1 TR-FRET pIC50 | 4.8 | 3.8 | 5.2 | ≤4.5 | ≤4.4 |
| TR-FRET selectivity (logs) | 2.1 | 3.1 | 2.2 | ≥2.8 | ≥1.1 |
| Chrom logD (pH 7.4) | 1.6 | 1.7 | 3.0 | 3.2 | 3.1 |
| Polar surface area (Å2) | 126 | 135 | 92 | 101 | 101 |
| Artificial membrane permeability (nms−1, pH 7.4) | <3 | <3 | 86 | 190 | 156 |
Encouraged by this, we also made 16, the equatorially-biased C5’-OMe analogue of the difluorocyclohexyl 8. By TR-FRET, this retained the enhanced selectivity of 15, being equipotent to 8 against ATAD2 with reduced BRD4 BD1 activity (Table 2). As before, replacing the sulfone by difluoromethyl resulted in improved permeability. Compound 17 (the opposite enantiomer of 16) is significantly weaker against ATAD2, so represents a useful negative control for cellular assays.
Crystal structures were obtained of 16 bound to both ATAD2 and to BRD4 BD1. As expected, in ATAD2 the binding mode of 16 is very similar to that of 3, with the entire molecules closely superimposable (Figure 1d). The piperidine binds in a tri-equatorial conformation with the C5’ methoxy group making no apparent interaction with the protein. The two fluorine atoms are close to the guanidinium terminal nitrogen of Arg1077 (3.1 Å, 3.3 Å) suggesting that while probably not hydrogen-bonding the group shows good electrostatic complementarity to the ATAD2 RVF shelf. In BRD4 BD1, 16 showed a very similar binding mode to 4 (Figure 1e). Surprisingly, even in the presence of the C5’-OMe group, the piperidine ring of 16 adopts a tri-axial conformation. 15 binds to BRD4 BD1 in a similar tri-axial conformation (data not shown). These results illustrate that ligands may adopt higher-energy conformers when their binding site requires.
Table 3 shows further characterization of the potency and selectivity of 16. By isothermal titration calorimetry a pKd of 8.1 (Kd 8 nm) was estimated for ATAD2, with no interaction detected with the tandem BRD4 bromodomains (Figure S2, Supporting Information). Whether measured by ITC or TR-FRET, the potency against the BET bromodomains was close to the lower limit, giving a selectivity window of over 2.8 logs. 16 has similar potency in the ATAD2 Bromosphere assay, confirming its activity against endogenous full-length ATAD2. The potency against ATAD2 and the closely related ATAD2B bromodomain were comparable by TR-FRET. The enantiomeric compound 17 has a similar profile, but is consistently around 2 logs weaker against ATAD2.
Table 3. Profile of 16 (GSK8814) and its enantiomer 17 (GSK8815). For statistics see Table S1b, Supporting Information.
| 16 | 17 | |
|---|---|---|
| ATAD2 pKd ITC | 8.1 | 5.5 |
| ATAD2 BROMOScan pKi | 8.9 | n.d.[a] |
| ATAD2 TR-FRET pIC50 | 7.3 | 5.5 |
| ATAD2B TR-FRET pIC50 | 7.7 | 5.5 |
| BRD4 tandem pKd ITC | <5.3 | <5.3 |
| BRD2 BD1/ BD2 TR-FRET pIC50 | ≤4.4/<4.3 | ≤4.9/≤4.6 |
| BRD3 BD1/ BD2 TR-FRET pIC50 | <4.3/<4.3 | ≤4.6/≤4.7 |
| BRD4 BD1/ BD2 TR-FRET pIC50 | ≤4.5/<3.3 | ≤4.4/≤4.7 |
| BRDT BD1/ BD2 TR-FRET pIC50 | ≤4.3/<4.3 | ≤4.5/≤4.4 |
| CLND solubility [μM] | >439 | 326 |
n.d.=not determined.
The selectivity of 7 and 16 against the wider bromodomain family was assessed in the BROMOscan panel (Table S2, Figure S3, Supporting Information). 16 is one of the most selective bromodomain inhibitors so far reported in this panel, with ATAD2 pKd of 8.9, displaying ≥3.0 logs selectivity over 30/33 bromodomains tested, the exceptions being ATAD2B, TAF1 BD2 (2.2 logs) and TAF1L BD2 (2.5 logs). As a further assessment of possible off-target activity, 16 was inactive against an internal GSK panel of 40 targets considered as potential liabilities (data not shown).
With compound 16 showing improved passive permeability over the sulfone analogue 7, we sought additional evidence for increased cellular permeability. For this, we used a Nano-BRET assay measuring displacement of labelled ATAD2 bromodomain from histone H3.3 (Promega). As expected, treatment with 16 but not 7 or the negative control enantiomer 17 resulted in dose-dependent displacement (Figure 2; Figure S4, Supporting Information). Interestingly, 16 was ineffective at displacing full-length ATAD2 from H3.3.
Figure 2.

Unlike the impermeable sulfone analog 7 (grey circles), 16 (black diamonds) displaces the ATAD2 bromodomain construct in a cellular nanoBRET displacement assay. 16 does not displace full-length ATAD2 (crosses). (Error bars = SD).
We attempted to reproduce some of the reported antiproliferative effects of ATAD2 knockdown using 16. This is the first time that this has been evaluated using a small molecule inhibitor targeting the bromodomain. Disappointingly, effects on colony formation and inhibition of genes involved in cell cycle and division were only seen at high compound concentrations (Figure S5, Supporting Information). Therefore, we cannot be sure that the effects were caused by ATAD2 inhibition, although they were not seen to such an extent with the negative control 17. Considering the in vitro potency and permeability of 16, these results suggest that the opportunity to regulate proliferation in a therapeutic context via the ATAD2 bromodomain may be limited. There is presently little data associating the anti-proliferative effects seen upon ATAD2 knockdown with its bromodomain, although an intact bromodomain is required for binding acetylated histones.[18, 19] Another group recently concluded that the ATPases of the SMARCA2/4 proteins provide better oncology opportunities than their bromodomains.[20] The RNA knockdown data may be consistent with reports that other ATAD2 domains are necessary for its function: the N-terminal region is required for oligomerization and binding of acetylated histone H4 and chromatin,[1, 21] and the ATPase for E2-stimulated gene expression.[18] Perhaps the oligomeric state of ATAD2 results in chromatin binding with such avidity that far more potent bromodomain inhibitors would be needed. However, it is possible that the bromodomain of ATAD2 exerts a more subtle influence and that systems other than those reported here may be more sensitive to inhibitors. For instance, it has recently been proposed that ATAD2 has a more critical function in differentiating embryonic stem cells.[22] Hence, we hope that reporting this probe, GSK8814 (16) and its less active control GSK8815 (17), to the scientific community will enable others to investigate the biology of ATAD2 further. We recommend not exceeding low-micromolar compound concentrations in cells to avoid any possible unknown off-target effects.
To conclude, we report the optimization of ATAD2 inhibitors for improved cell permeability and selectivity over the BET bromodomains. This was accomplished using what we believe to be a novel use of CF2 as a polar hydrophobic isostere of SO2. This modification retained the favourable ATAD2 interactions of the sulfone and selectivity over the BETs, yet gave a dramatic improvement in logD and passive permeability. In parallel, an unexpected difference in the bound piperidine conformation in ATAD2 and BRD4 BD1 was exploited to further improve selectivity. The highly unusual tri-axial conformation adopted by 16 in BRD4 BD1 highlights the difference between the conformational preferences of substituted piperidines and cyclohexanes. This effort culminated in the identification of the first reported nanomolar, selective and cell permeable ATAD2 bromodomain inhibitor.
Supplementary Material
Supporting information (X-ray, ITC, assay, molecular modelling, LNCaP biology, NMR and chemistry supplementary methods) and the ORCID identification numbers for the authors of this article can be found under http://dx.doi.org/10.1002/anie.201603928.
Acknowledgements
We thank Bhumika Karamshi and Laurie Gordon for TR-FRET data, Emma Jones for protein production, Abigail Lucas and Fiona Shilliday for crystallization support, and Jacqui M8ndez and Danette Daniels of Promega Corporation for facilitating NanoBRET assays. A.J.B., K.H.C., S.R. and T.K. acknowledge grants from Wellcome Trust (092096), Cancer Research UK (C6946/A14492, RG17001), and BBSRC (RG69031).
Contributor Information
Dr Paola Grandi, Cellzome GmbH, GlaxoSmithKline, Meyerhofstrasse 1, 69117, Heidelberg (Germany).
Dr Tony Kouzarides, Gurdon Institute, Tennis Court Road, Cambridge, CB2 1QN (UK).
Dr Anne-Marie Michon, Cellzome GmbH, GlaxoSmithKline, Meyerhofstrasse 1, 69117, Heidelberg (Germany).
Dr Christina Rau, Cellzome GmbH, GlaxoSmithKline, Meyerhofstrasse 1, 69117, Heidelberg (Germany).
Dr Samuel Robson, Gurdon Institute, Tennis Court Road, Cambridge, CB2 1QN (UK).
Robert J. Sheppard, GlaxoSmithKline, Gunnels Wood Road, Stevenage, SG1 2NY, (UK)
References
- [1].Caron C, et al. Oncogene. 2010;29:5171. doi: 10.1038/onc.2010.259. [DOI] [PubMed] [Google Scholar]
- [2].Boussouar F, Jamshidikia M, Morozumi Y, Rousseaux S, Khochbin S. Biochim Biophys Acta Gene Regul Mech. 2013;1829:1010. doi: 10.1016/j.bbagrm.2013.06.003. [DOI] [PubMed] [Google Scholar]
- [3].Zou JX, Guo L, Revenko AS, Tepper CG, Gemo AT, Kung HJ, Chen HW. Cancer Res. 2009;69:3339. doi: 10.1158/0008-5472.CAN-08-3440. [DOI] [PubMed] [Google Scholar]
- [4].Demont EH, et al. J Med Chem. 2015;58:5649. doi: 10.1021/acs.jmedchem.5b00772. [DOI] [PubMed] [Google Scholar]
- [5].Bamborough P, et al. J Med Chem. 2015;58:6151. doi: 10.1021/acs.jmedchem.5b00773. [DOI] [PubMed] [Google Scholar]
- [6].Müller S, Knapp S. MedChemComm. 2014;5:288. [Google Scholar]
- [7].Gillis EP, Eastman KJ, Hill MD, Donnelly DJ, Meanwell NA. J Med Chem. 2015;58:8315. doi: 10.1021/acs.jmedchem.5b00258. [DOI] [PubMed] [Google Scholar]
- [8].Biffinger JC, Kim HW, Dimagno SG. ChemBioChem. 2004;5:622. doi: 10.1002/cbic.200300910. [DOI] [PubMed] [Google Scholar]
- [9].Muller K, Faeh C, Diederich F. Science. 2007;317:1881. doi: 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]
- [10].Smart BE. J Fluorine Chem. 2001;109:3. [Google Scholar]
- [11].Howard JA, Hoy VJ, O’Hagan D, Smith GT. Tetrahedron. 1996;52:12613. [Google Scholar]
- [12].Brammer L, Bruton EA, Sherwood P. Cryst Growth Des. 2001;1:277. [Google Scholar]
- [13].Dunitz JD. ChemBioChem. 2004;5:614. doi: 10.1002/cbic.200300801. [DOI] [PubMed] [Google Scholar]
- [14].Zhou P, Zou J, Tian F, Shang Z. J Chem Inf Model. 2009;49:2344. doi: 10.1021/ci9002393. [DOI] [PubMed] [Google Scholar]
- [15].O’Hagan D. Chem Soc Rev. 2008;37:308. [Google Scholar]
- [16].Loudet M, Metras F, Petrissans J, Deschamps J, Pfister G. J Mol Struct. 1974;20:357. [Google Scholar]
- [17].Tokuda O, Aikawa T, Ikemoto T, Kurinoto I. Tetrahedron Lett. 2010;51:2832. [Google Scholar]
- [18].Zou JX, Revenko AS, Li LB, Gemo AT, Chen HW. Proc Natl Acad Sci USA. 2007;104:18067. doi: 10.1073/pnas.0705814104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ciro M, et al. Cancer Res. 2009;69:8491. doi: 10.1158/0008-5472.CAN-09-2131. [DOI] [PubMed] [Google Scholar]
- [20].Vangamudi B, et al. Cancer Res. 2015;75:3865. doi: 10.1158/0008-5472.CAN-14-3798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Zhan Y, et al. Epigenetics Chromatin. 2015;8:37. doi: 10.1186/s13072-015-0026-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Morozumi Y, et al. J Mol Cell Biol. 2016 doi: 10.1093/jmcb/mjv060. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.