

Abstract
Runt-related transcription factor-1 (RUNX1), also known as acute myeloid leukaemia 1 protein (AML1), is a member of the core-binding factor family of transcription factors which modulate cell proliferation, differentiation, and survival in multiple systems. It is a master-regulator transcription factor, which has been implicated in diverse signalling pathways and cellular mechanisms during normal development and disease. RUNX1 is best characterized for its indispensable role for definitive haematopoiesis and its involvement in haematological malignancies. However, more recently RUNX1 has been identified as a key regulator of adverse cardiac remodelling following myocardial infarction. This review discusses the role RUNX1 plays in the heart and highlights its therapeutic potential as a target to limit the progression of adverse cardiac remodelling and heart failure.
Keywords: Runx1, Myocardial infarction, Adverse cardiac remodelling, Cardiovascular diseases, Heart failure, Excitation–contraction coupling, Calcium
1. Introduction
The healthcare burden and socioeconomic impact of heart failure is increasing on a global scale leading to significant levels of disability, reduced quality of life, and mortality.1,2 Adverse changes in the architecture and function of the heart, collectively referred to as cardiac remodelling, are critical to the development of heart failure. Adverse cardiac remodelling occurs in response to many cardiovascular diseases including acute myocardial infarction (MI), hypertension, arrhythmias, and valve disease. For example, following an MI, adverse cardiac remodelling clinically manifests as left ventricular (LV) wall thinning, impaired contractility, and dilation; progression of which is linked to increased deaths or hospitalizations due to heart failure (Figure 1).3
Figure 1.

Pathological ventricular remodelling. Schematic showing remodelling of the left ventricle (LV) following myocardial infarction (MI). Following MI, a fibrosous scar forms in the infarcted tissue accompanied by myocyte loss. Adjacent to the infarcted tissue in the region bordering the remote LV, the border-zone region, myocytes thicken and elongate and excitation–contraction coupling becomes impaired. In terms of gross architecture of the LV, the early phase of remodelling is characterized by thinning and elongation of the infarcted zone followed by ventricular dilation, where the LV transitions from an elliptical shape to a spherical shape. Schematic was prepared using Servier Medical Art by Servier under a Creative Commons Attribution 3.0 Unported License.
At the cellular and molecular level, adverse cardiac remodelling is the result of a complex series of alterations to transcriptional, structural (e.g. hypertrophy), and electrophysiological signalling pathways (Figure 1) in cardiomyocytes.4 These changes are accompanied by fibroblast proliferation and collagen deposition (fibrosis), vascular smooth muscle cell proliferation, endothelial dysfunction, and inflammation.4
Current long-term therapy to prevent or reverse adverse remodelling remains inadequate and therefore novel strategies to preserve LV function and limit adverse cardiac remodelling are needed to treat patients with cardiovascular disease and improve prognosis.5 This review discusses the structure and function of the master-regulator transcription factor RUNX1. In particular, we discuss its novel and far-reaching role within the cardiovascular system, highlighting it as a target of interest for adverse cardiac remodelling that merits further experimental and clinical investigation.
2. RUNX1
The RUNX (runt-related) family of genes encode the α-subunits of a family of transcription factors that orchestrate proliferation, differentiation, and cell survival in multiple lineages. In mammalian species, three α-subunits exist, known as RUNX1, RUNX2, and RUNX3, each with its own distinct spatial-temporal and tissue-specific pattern of expression.6,7 All the RUNX proteins partner with a constitutively expressed β subunit (core-binding factor β; CBFβ), to form a transcriptionally active heterodimer that can either activate or repress target gene expression. Although each RUNX protein interacts with the same target consensus sequence, they display distinct and non-redundant biological functions.7
RUNX1 is best characterized for its role as a key transcriptional regulator of haematopoiesis and its involvement in blood malignancies.8 Mice with a homozygous knockout of Runx1 lack definitive haematopoiesis and are unable to survive past an early embryonic stage (days 11.5–12.5) due to severe haemorrhage within the central nervous system (CNS), peritoneum, and pericardium.9 In humans, the RUNX1 gene is one of the most common targets of chromosomal and genetic alterations in acute leukaemia.10 Whilst the focus of RUNX1 research has therefore predominately been in the cancer field, accumulating evidence suggests that RUNX1 has more widespread functions in a range of organs and pathologies than previously considered.
3. RUNX1 structure and regulation
Like the other members of the RUNX family, the RUNX1 protein contains the highly conserved 128 aa region known as the Runt homology (runt) domain which mediates binding to DNA and facilitates interaction with CBFβ.11 In its free form, RUNX1 has a relatively poor capacity to bind DNA due to negative regulation from regions at the N- and C-terminal limits of the Runx domain.12,13 On binding to CBFβ, structural rearrangements of the runt domain take place which unmask and stabilize the DNA-binding site resulting in a markedly increased affinity for DNA.12,14 Moreover, whilst the RUNX1 protein itself has a short half-life of approximately 60 min, formation of the RUNX1–CBFβ heterodimer protects RUNX1 from degradation by the ubiquitin ligase complex, increasing its half-life three-fold.15 Transcriptional ability is further modulated via protein-interacting domains within the RUNX1 C-terminus including the PY motif (proline-rich peptide that interacts with proteins with a WW domain), and the VWRPY motif [which acts as a docking site for the assembly of the Groucho/Transducin-like enhancer of split (TLE) family of co-repressor proteins].16,17 Via these domains, RUNX1 can form a platform for the assembly of other transcription factors, co-activators, and chromatin modulators to form large transcriptional complexes16 (Figure 2A). It is likely that the particular combination of mediators recruited to these complexes dictate the profile of the genes targeted and whether they are activated or repressed, leading to both indirect regulation and context-dependent function.16
Figure 2.

RUNX1 structure and regulation. Illustration of RUNX1 transcriptional regulatory complexes for activation and repression of gene expression (A). Schematic depicting RUNX1 gene structure (B). Expression of RUNX1 is initiated by two promoters: distal P1 and proximal P2. (C) Alternative promoters and gene-splicing results in different 5ʹ-UTRs. The P1-5ʹ-UTR contains four exons and the P2-5ʹ-UTR has a single exon and an internal ribosome entry site (IRES). This results in three major isoforms of RUNX1; 1A and 1B transcribed from P2 and isoform 1C transcribed from P1. (D) Schematic of the proteins encoded by the RUNX1 isoforms with the major functional domains marked: runt-homology domain (RHD) and transactivation domain (TAD). Figure prepared using information from various sources.186–188
Expression of RUNX1 itself is tightly controlled at the transcriptional, translational, and post-translational level, which also contributes to the strong context specificity of RUNX1 function. The RUNX1 gene is under the control of a dual promoter system comprising a distal P1 and a proximal P2 promotor and alternative usage of these promoters leads to the generation of mRNA isoforms that differ in their 5ʹ-untranslated region (UTR) and N-terminal coding sequences18,19 (Figure 2B, C). The biological significance of the dual promoter system has not been fully elucidated; however, their differential expression during developmental haematopoiesis appears important for cell specification.20 Expression of the P2 transcript predominates during haematopoietic stem and progenitor cell (HSC) emergence from the haemogenic endothelium during embryonic development, whereas P1 transcript expression increases as HSCs colonize the liver and becomes the dominant isoform in both liver HSCs and in adult HSCs in the bone marrow.20,21 The differing lengths of the 5ʹ-UTR of the P1 and P2 transcripts may also facilitate their differential expression in distinct cellular contexts by influencing their translation efficiency. The human RUNX1 P1 transcript has a 5ʹUTR that is 452 base pairs (bp) in length and is translated efficiently; however, the 5ʹ-UTR of the P2 transcript is substantially longer at 1631 bp and forms a secondary structure which appears to hinder its translation in vitro.22 Whilst the P1-5ʹ-UTR is translated by a cap-dependent mechanism (the most common means of translation direction under basal conditions), translation of P2 transcripts is directed by the presence of an internal ribosomal entry site (IRES), which recruits the ribosome to directly initiate translation thus allowing the canonical, cap-dependent mode of translation initiation to be bypassed.22 IRES-mediated translation has been suggested to predominate under conditions of stress, such as hypoxia or apoptosis when cap-dependent translation is diminished.23 Messenger RNA isoforms with different translational regulation may allow a shift from the predominant expressed RUNX1 protein isoform and may tailor RUNX function as part of an adaptive response to stress.7
Further diversity in the RUNX1 gene products is augmented by splicing and exon skipping of the P1 and P2 transcripts to yield further isoforms with different functions and expression patterns.7,18 For example, in humans, P1-driven transcription produces the RUNX1c isoform, whereas P2 activity yields the RUNX1a and RUNX1b isoforms.18 This gives rise to a range of protein isoforms, ranging in size from 20 to 52 kDa18,24 (Figure 2D). RUNX1a is truncated at exon 7 and so has DNA-binding capacity but lacks the C-terminal transcriptional regulatory domain seen in RUNX1b and RUNX1c. It has therefore been suggested that RUNX1a may act as an antagonist to transcriptional activation driven by RUNX1b and RUNX1c.18,24 Indeed, in a murine myeloid cell line, ectopic expression of AML1a (RUNX1a) only or a combination of AML1a and AML1b (RUNX1b) was found to have an opposing effect on the response of the cells to granulocyte colony-stimulating factor (G-CSF). Overexpression of AML1a alone was found to block granulocytic differentiation induced by G-CSF, whereas additional expression of AML1b was found to rescue granulocytic differentiation.25
Additional control over RUNX1 stability and function is achieved by a range of post-translational mechanisms including phosphorylation, acetylation, and ubiquitination (Figure 3). Acting as scaffolds for multiple protein complexes the outcome of RUNX binding to DNA is heavily influenced by interaction with different activating or repressive co-factors. Post-translational modifications can influence RUNX1 activity, affinity for DNA, and its stability at least in part by directing its binding to other co-factors and in this way provides a means to fine-tune RUNX1 activity in response to external stimuli or cellular events.26 For example, phosphorylation was shown to enhance the transactivation activity of RUNX1.27 Subsequently, phosphorylation has been shown to reduce interaction with histone deacetylases28 and other repressors such as sin3a.29 Conversely, phosphorylation (albeit on specific residues) has also been shown to reduce activity.30 RUNX1 can be phosphorylated in response to cytokine/growth factor-induced signalling, underlining how transcription factor activity can be modulated by external factors.30 Given the importance of post-translational modifications it is not surprising that aberrant phosphorylation of RUNX1 has been linked to disease. FMS-like tyrosine kinase 3 (FLT3) mutations resulting in constitutive activation are common in AML and have been correlated with high levels of RUNX1. A study by Behrens et al.31 revealed that constitutive FLT3 activation was associated with phosphorylation of RUNX1 residues in the inhibitory domain of RUNX1, resulting in increased expression of downstream oncogenic targets. Thus, it is possible that FLT3 activation may recruit RUNX1 as its oncogenic partner at least in part through modifying its activity.
Figure 3.
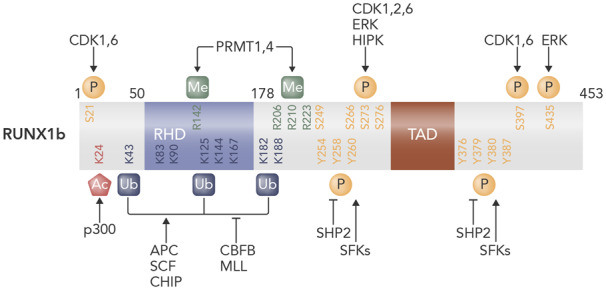
Post-translational modifications of human RUNX1. Schematic depicting post-translational modifications of RUNX1b. Runt-homology domain (RHD) is the DNA- and CBFβ-binding domain. Transactivation domain (TAD) is important for transcriptional activation. Numbers refer to amino acid residues from N terminus. Ac, acetylation; APC, anaphase-promoting complex; K, lysine; Me, methylation; P, phosphorylation; R, arginine; S, serine; SCF, Skp1/Cullin/F-box protein complex; T, threonine; Ub, ubiquitination; Y, tyrosine. Figure prepared using information from various sources.189,190
Other modifications to RUNX1 affect its interaction with regulatory co-factors, for example, methylation can affect RUNX1 partner interactions to both promote32 and repress33 transcriptional activity. Similarly, p300-mediated acetylation of RUNX1 has been associated with increased DNA binding and transcriptional activity.34 Ubiquitination frequently marks proteins, including RUNX1, for degradation by the proteasome but its effects can be more subtle and, in some cases, can result in protein stabilization.35
The highly complex regulation of RUNX1 expression is perhaps reflective of a need to tightly control and direct the cellular processes of cellular differentiation, proliferation, and lineage commitment in which RUNX1 is critical. Consequently, dysregulation or altered expression of RUNX1, which is well documented in cancer patients, leads to disrupted cellular function and disease.10
4. RUNX1 in the developing heart
In the developing embryo, RUNX1 is the most broadly expressed of all the RUNX proteins and is expressed in a range of tissues including the mesenchymal tissue of the heart and in vascular tissue.6 The importance of RUNX1 in the development of the vasculature is highlighted in the phenotype observed in Runx1 KO mice. These mice show a complete lack of definitive haematopoiesis and abnormal vasculature development in many organs.9 In the heart, these mice have an underdeveloped coronary plexus and smaller ventricular free wall vessels.36 This coincides with changes in heart structure including ventricular septal defects and the development of thin myocardium. In ex vivo preparations of explanted ventricle from embryonic stage 11.5, Runx1 null preparations showed less endothelial sprouting.36 The effects on the developing vasculature may be linked to a haematopoietic function; however, a contributing non-haemogenic role of RUNX1, perhaps directly affecting vessel and heart structure cannot be ruled out based on this study alone.36,37
Expression of RUNX1 in the neonatal heart is higher compared with adult heart tissue.38 Although the reasons for this are unexplored, it is interesting to note that genes with RUNX1-binding sites within their promoter region are overrepresented in the collection of genes that become methylated during the first week of life.39 Promoter methylation is an epigenetic modification which usually leads to gene silencing by blocking the access of transcriptional machinery.40 In this setting, increased gene methylation may be important in the maturation process by switching off genes necessary for heart development to support transition to a more adult phenotype.39 Of note, methylation of RUNX1 target genes during the 7 days after birth coincides with a loss of the hearts proliferative and regenerative capacity.39 This is an intriguing finding given the fact that there is a known link between RUNX1 and the transcriptional co-activators Yap (Yes-associated protein) and TAZ (transcriptional co-activator with PDZ binding motif), which have a regenerative role in the neonatal and adult heart.41–43
As RUNX1 has been shown to be involved in female sex development and is a mediator of female hormone signalling, with RUNX1 dysregulation involved in breast, ovarian, and uterine cancers,44,45 it is important to consider that RUNX1 may have differing roles within the female vs. male cardiovascular system.
5. RUNX1 expression in cardiac injury
Although expression of RUNX1 in the adult heart is reported to be low, several studies have demonstrated that RUNX1 is increased in the context of cardiac pathology.46–49 This was first demonstrated by Gattenlohner et al.48 in 2003 using human heart tissue autopsies from patients with diagnosed ischaemic cardiomyopathy. Up-regulation of RUNX1 mRNA in the ischaemic tissue was accompanied by overexpression of the 52 kDa RUNX1 isoform and down-regulation of a 38 kDa isoform predicted to be the RUNX1 AML-1 delta N isoform which has a known dominant-negative function.48,50 In a separate study, RUNX1 was also up-regulated in heart tissue from patients with chronic dilated cardiomyopathy compared with control myocardium.47 These findings have been reinforced by rodent models of MI which have shown induction of RUNX1 in the nuclei of cardiomyocytes within the myocardial region bordering the infarcted tissue (border zone) as early as 24 h post-MI.46,47 Whilst in the early stages after MI the up-regulation of RUNX1 seems specific to the border zone, by 8 weeks post-injury Runx1 mRNA appears up-regulated in the LV remote from the infarct.46 Interestingly, cardiac up-regulation of RUNX1 does not appear to be specific to ischaemic pathology as RUNX1 was also found to be increased experimentally in animal models of: (i) diabetic cardiomyopathy; (ii) pressure overload; and (iii) dilated cardiomyopathy.51–53 More recently, RUNX1 has been shown to be involved in zebrafish heart regeneration following cardiac injury.54 Cryo-injury resulted in increased RUNX1 expression within various cell types of the myocardium including cardiomyocytes, myofibroblasts, endocardial/endothelial, epicardial, and thrombocytes, where it inhibits heart repair.
6. Triggers of RUNX1 expression
Up-regulation of RUNX1 in damaged tissue is reported in non-cardiac tissues including the CNS, lungs, and skeletal muscle.55–58 In the nervous system, RUNX1 is up-regulated in response to sciatic nerve crush injury and controlled cortical impact injury.56,59 Comparable with observations made in the heart where RUNX1 was found to be increased in the infarct border zone at 24 h post-MI,46 Logan et al.56 also identified increased RUNX1 expression at the injury site as early as 1-day post-cortical injury. RUNX1 induction at early time points in a range of tissues following different insults may indicate that RUNX1 is up-regulated as part of a common response to injury independent of the precise nature of the original insult.56 An interesting comparison can be made between RUNX1 expression in cardiac and skeletal myocytes. Like heart tissue, RUNX1 is increased in skeletal myocytes in a range of pathologies including ischaemic muscle, cardiotoxin treated muscle, and mouse models of Duchenne musclar dystrophy and amylotrophic lateral sclerosis.57,60,61 Denervation of skeletal muscle is associated with a rapid and marked induction in RUNX1 expression; for example, in rat hindlimb muscle a two-fold induction of Runx1 mRNA was detected at 1-day post-denervation, rising to 50- to 100-fold by day 5 post-denervation.58,62 Significantly, the up-regulation of RUNX1 in response to denervation can be attenuated by the daily application of either electrical stimulation or stretch to the denervated muscle.63 RUNX1 expression is increased in aged muscle alongside denervation and sarcopenia and can similarly be reduced by physical intervention. In a small number of older obese men and women and in aged female mice with sarcopenia resistance training, which is known to preserve the neuromuscular junction and improve innervation,64 was found to reduce RUNX1 expression.65,66 Interestingly, in skeletal muscle and in ischaemic cardiomyopathy RUNX1 up-regulation coincides with the up-regulation of the neural cell adhesion molecule (NCAM, CD56).48 Whereas, NCAM up-regulation in rhabdomyosarcoma occurred where myocytes were separated by infiltrating tumour, NCAM up-regulation in ischaemic heart tissue arose where cardiomyocytes were separated by scar tissue. If NCAM expression is up-regulated downstream of RUNX1 this may suggest that physical disruption of cell–cell contact or adhesion may contribute to RUNX1 regulation,48 possibly by affecting cell communication.
Changes in RUNX1 expression in response to alterations in electrical activity, cell–cell communication or as a result of physical strain or tension may explain why RUNX1 up-regulation is first observed in the border zone following MI. After MI, cardiomyocytes within the border zone exhibit abnormal cell–cell communication due to altered gap-junction distribution and physical disruption of cell contact by interstitial fibrosis and necrotic myocytes, leading to electrical heterogeneity and the precipitation of arrhythmias.67 The MI border zone is also subjected to high physical stress and abnormal stretch created at the boundaries between viable cardiomyocytes alongside dead cells and non-contractile scar tissue.68–70 As the heart attempts to compensate for the hypocontractile infarct, the remote myocardium also becomes subject to abnormal wall stress which may lead to the up-regulation of RUNX1 in this region at a later stage post-MI.71
The apposition of the border zone alongside the infarct means that viable border zone cardiomyocytes are also directly exposed to the inflammatory milieu of the infarcted tissue.72 Post-infarction, there is a rapid immune response to cellular damage, resulting in the release of damage signals, chemokines, and cytokines and the recruitment of leucocytes into the damaged area.73 Consequently, there is an intense surge of immune signals which could act as a local trigger for RUNX1 expression. Oncostatin-M, which is a member of the IL-6 cytokine family and is secreted by inflammatory cells in response to injury,74,75 has already been identified as a potent inducer of RUNX1 in cultured cardiomyocytes.47,51 In addition, transgenic mice with a heart-specific overexpression of monocyte chemotactic protein-1 (MCP-1) exhibit pathological macrophage infiltration in the myocardium, which occurs alongside enhanced oncostatin-M and RUNX1 expression, providing evidence that an association between inflammation, oncostatin-M, and RUNX1 up-regulation may exist.47,76 Other inflammatory mediators with known links to RUNX1 include the pro-inflammatory cytokine IL-1β and the transcription factor TGF-β. Treatment of a human glioblastoma cell line with IL-1β was shown to up-regulate RUNX1 expression via the p38 MAPK cascade77 and TGF-β has been shown to increase RUNX1 expression in a range of cell types including adult hippocampal precursor cells,78 mesenchymal stem cells, CD4 T cells, and a mouse hepatocyte cell line.79,80 Currently, the effects of these inflammatory mediators on RUNX1 expression in the myocardium remain unknown.
7. RUNX1 and adverse cardiac remodelling
Work conducted using mice with an inducible cardiomyocyte-specific Runx1 deficiency was the first to provide evidence that the up-regulation of RUNX1 post-MI drives adverse cardiac remodelling.46 At baseline, Runx1-deficient mice had equivalent echocardiographic contractile parameters to their littermate controls. However, following MI stark differences in contractile function and myocardial remodelling emerged. Whilst classic adverse cardiac remodelling occurred in control mice post-MI including declining systolic function, thinning of the left ventricular free wall, and dilation of the left ventricular chamber, the same parameters were remarkably absent in Runx1-deficient mice.46 At 8 weeks post-MI, control mice had developed eccentric hypertrophy, characterized by cardiomyocyte elongation and thinning. In contrast, this was notably undetectable in the Runx1-deficient mice. The link between RUNX1 and adverse cardiac remodelling has now been corroborated by others in mice81 and also in cryo-injured zebrafish hearts, where Runx1 knockout prevented adverse cardiac remodelling by promoting faster scar degradation.54 The cellular composition of the scar was differentially regulated so that smooth muscle and collagen gene expression was significantly reduced, subsequently reducing the amount of collagen, and fibrin deposition. Myofibroblast formation was also reduced whilst fibrinolysis increased, thus allowing invasion of proliferative cardiomyocytes and hence enhancing muscle regeneration.54
The improvement in function and myocardial architecture of mouse hearts with Runx1-deficiency post-MI may be explained by an effect of RUNX1 on calcium homeostasis in cardiomyocytes. At 2 weeks post-MI, cardiomyocytes isolated from Runx1-deficient hearts had increased amplitude of electrically stimulated sarcoplasmic reticulum (SR)-mediated Ca2+ release, a faster rate of calcium removal from the cytosol by the SR Ca2+-ATPase (SERCA) and a higher SR content.46 Although the absolute protein levels of the endogenous SERCA inhibitor phospholamban (PLN) were similar between Runx1-deficient and control cardiomyocytes, the proportion of PLN phosphorylated on Ser16 and Thr17 residues was increased in Runx1-deficient cardiomyocytes. This was accompanied by a parallel decrease in protein phosphatase 1, the phosphatase responsible for the dephosphorylation of PLN.46 In its phosphorylated state, the inhibitory actions of PLN on SERCA is relieved, allowing enhanced Ca2+ uptake into the SR which lowers end-diastolic cytosolic Ca2+ concentration and improves the relaxation of the heart during diastole.82,83 SR Ca2+ content is also increased, leading to augmented release of Ca2+ with each electrically stimulated transient and therefore improved contraction.84,85
The insights offered by McCarroll et al., into the protection afforded by Runx1-deficiency fit with other studies in the field where improvements in cardiomyocyte calcium handling can have a profound beneficial effect on cardiac structure and function.84,86,87 Interestingly, the improved contractility noted in Runx1-deficient mice occurred in the absence of an effect on infarct size, another key determinant of systolic function after MI.46 This finding additionally implies that the protective influence of Runx1-deficiency likely relates to the ability of viable cardiomyocytes to functionally compensate for cardiomyocyte death via preserving calcium handling rather than a direct effect of Runx1-deficiency on the salvage of ischaemic cardiomyocytes. Importantly and in agreement with mouse studies, several calcium-dependent genes were up-regulated in Runx1 knockout zebrafish hearts.54
Thus far, the direct mechanisms linking RUNX1 to changes in cardiomyocyte Ca2+ handling are unknown and require further study. Given the preponderance of Ca2+ dysregulation across cardiac pathologies, including diabetic and hypertensive heart disease,88,89 and its contribution to heart failure progression,90 it will be useful to establish whether manipulation of RUNX1 in other cardiac disease contexts improve cardiac function.
8. Interaction of RUNX1 with signalling pathways involved in cardioprotection and adverse cardiac remodelling
Improved Ca2+ homeostasis may not be the exclusive mechanism underlying protective influence of RUNX1 deficiency on the heart. As a master transcription factor, RUNX1 has the potential to simultaneously influence several downstream signalling pathways which may also dictate cardiomyocyte phenotype and the global response of the heart to injury. Although the direct targets of RUNX1 in the heart have not been elucidated, RUNX1 has been demonstrated to interact with signalling pathways in other tissues that are implicated in cardioprotection and adverse cardiac remodelling.
RUNX1 is known to interact with signalling mediated by hypoxia-inducible factor 1-alpha (HIF-1α).91 HIF-1α belongs to the HIF family of transcription factors which are key orchestrators of the cellular response to ischaemia.92 Many studies have demonstrated that augmented HIF-1α signalling is protective in the context of MI and leads to improved heart contractility, angiogenesis and reduced infarct size93,94; however, chronic activation of HIF-1α signalling in the long term may exacerbate adverse cardiac remodelling and advance progression to heart failure.95–97 In haematopoietic cells with forced expression of RUNX1 and HIF-1α, RUNX1 has been shown to physically interact with HIF-1α and reduce transcription of HIF-1α targets including vascular endothelial growth factor (VEGF) and glucose transporter 1 (GLUT 1). Conversely, forced expression of HIF-1α was found to enhance RUNX1-mediated transcription.98 In a separate study, RUNX1 overexpression in a glioblastoma cell line down-regulated genes involved in the hypoxic response including the known HIF-1α targets hexokinase 2 (HK 2), caveolin 1 (CAV1), adenosine A2B receptor, and protein phosphatase 1 regulatory subunit 3C gene (PPP1R3C).99–101 RUNX1 itself may be up-regulated by HIF-1α, as chemical stabilizers of HIF-1α including dimethyloxalylglycine (DMOG) and cobalt chloride (CoCl2) have been shown to increase RUNX1 expression102–104 and a correlation between RUNX1 and HIF-1α transcripts was identified in hippocampal transcriptomic data from in-bred mouse strains.78 Whether RUNX1 interacts with HIF-1α in the heart has not been elucidated.
TGF-β-mediated signalling is also linked to RUNX1 and implicated in cardiac remodelling. RUNX1 expression is activated by TGF-β and several of the biological effects of TGF-β stimulation have been shown to involve RUNX1 including myofibroblast differentiation and fibrosis.78,105,106 RUNX1 can direct TGF-β signalling through physical interactions with SMAD proteins, the intracellular transducers of TGF-β receptor activation.78,107 Shortly after MI, TGF-β activation is considered protective and fundamental for infarct healing108; however, prolonged and excessive TGF-β signalling promotes adverse cardiac remodelling including interstitial fibrosis and hypertrophy.108,109 Work is needed to determine whether RUNX1 is linked to TGF-β signalling in the heart and whether this interaction is involved in cardiac pathology.
Another means by which RUNX1 can alter cellular function and response to injury is via interaction with microRNAs (miRNAs). miRNAs are small non-coding RNAs which post-transcriptionally modify gene expression by interacting with the 3ʹUTR of target mRNAs. As microRNAs can target hundreds of mRNA sequences and thus simultaneously affect several targets within the one pathway, small changes in their expression has the potential to amount to a significant biological effect.110,111 ChIP-seq data from studies in haematopoietic cells have shown that RUNX1 physically binds over 200 miRNA genes suggesting a potential for RUNX1 to influence a remarkable number of miRNA networks.112 An increasing number of studies are providing experimental support for this observation and as a result, the number of validated RUNX1 miRNA targets reported is continuously increasing, including the identification of RUNX1 miRNA targets which have known links to cardiovascular disease.113,114 For example, miR-24 which is down-regulated by RUNX1, decreases in the ischaemic border zone after experimental MI.114–117 Moreover, miR-24 appears to have a cardioprotective role within this setting, as local delivery of miR-24 into the border zone after infarction reduced cardiomyocyte apoptosis, decreased infarct size, and improved cardiac contractility.115 It is possible that RUNX1 may contribute to reduced miR-24 in the aftermath of MI, and in doing so may adversely affect the cardiac outcome; however, this is yet to be investigated.
The miR-17-92 cluster is a further example of a group of miRNAs that are linked to both RUNX1 and cardiac pathology. The miRNAs in this cluster target the 3ʹUTR of RUNX1 to down-regulate its protein expression and are also themselves targeted by RUNX1 as part of a negative feedback loop.117,118 Of note, the miR-17-92 cluster has been associated with cardiomyocyte differentiation and proliferation, and furthermore, in a mouse model of MI, miR-17-92 overexpression in cardiomyocytes was reported to improve cardiac function and increase the number of proliferating cardiomyocytes within the infarct border zone.118,119
9. RUNX1 targets in skeletal muscle
An interesting comparison can be made between the role of RUNX1 in skeletal and cardiac muscle post-injury and may shed light on potential RUNX1 targets in the heart. Both skeletal and cardiac myocytes have a primary contractile function and are thus equipped with highly organized arrangements of myofilaments and complex calcium signalling mechanisms to facilitate this.120 Notably, both cell types respond to a range of insults via the up-regulation of RUNX1.120 However, in denervated skeletal muscle inactivation of RUNX1 by mutation severely affects the response of muscle fibres to denervation, leading to worsened fibre atrophy, excessive autophagy and structural abnormalities including misalignment and fragmentation of the z discs, dilation of the SR, and an altered myofilament composition.62 Thus, in skeletal muscle fibres, RUNX1 appears to protect the structural and functional integrity of skeletal muscle fibres in the face of injury or insult. This contrasts with its role in the heart post-MI, where RUNX1 appears to reduce cardiomyocyte function and promotes adverse cardiac remodelling. The differences between RUNX1 function in cardiac and skeletal muscle are fascinating yet are not easy to reconcile. They perhaps stem from the highly context dependency of RUNX1 function and may arise either because RUNX1 affects distinct processes in the two cell types or may exist because RUNX1 has opposing effects on the same target pathways. Interestingly, several of the 29 genes found to be dysregulated in RUNX1 mutated denervated skeletal muscle have previously been linked to cardiac contractility and cardiac remodelling. For example, RUNX1 was found to maintain or induce the expression of osteopontin, PLN, thrombospondin 1 in the face of denervation.62,121,122 Following MI, osteopontin up-regulation in cardiomyocytes has been found to negatively correlate with left and right ventricular ejection fraction and positively correlate with cardiomyocyte hypertrophy.123 In contrast, thrombospondin 1 up-regulation in the extracellular matrix post-MI appears beneficial by preventing excessive infiltration of macrophages and myofibroblasts into the peri-infarct zone.124 Further investigation is now necessary to explore whether RUNX1 acts on similar targets in cardiac muscle and to understand the consequence of these RUNX1 interactions in the context of cardiac disease.
10. RUNX1 in differentiation and proliferation
The role of RUNX1 in proliferation and differentiation is well documented. In tissues such as the skin, intestine, mammary gland, and in the haematopoietic system RUNX1 participates in the regulation of stem cell quiescence by directing entry and exit from the cell cycle.125,126 In general, RUNX1 appears to support or maintain proliferation, in part by promoting G1 to S progression through the cell cycle.126–128 The control of proliferation appears intimately linked with cellular differentiation, the balance of which may depend on the level RUNX1 expression. Inhibition of RUNX1 in mouse neurosphere cultures with the inhibitor Ro5-3335, a benzodiazepine which may reduce RUNX1 activity by disrupting RUNX1–CBFβ interactions129 was shown to inhibit proliferation of neural stem or progenitor cell populations without affecting cell viability or differentiation.130 In contrast, lentiviral-mediated RUNX1 overexpression did not affect the proliferative capacity of the cells but instead up-regulated markers of neuronal differentiation and the development of a neuronal-like morphology.130
In the heart, RUNX1 may be similarly linked to cell differentiation. This may become important after cardiac injury because the cellular differentiation status of cardiomyocytes in the injured or stressed myocardium131 is believed to be closely linked to the remodelling that takes place. Cardiomyocyte dedifferentiation, a phenotype characterized by the loss of the mature sarcomeric structure and the activation of a foetal gene expression profile, occurs in response to injury and has been reported in the ischaemic heart and the MI border zone, in dilated cardiomyopathy, and the pressure overloaded heart.47,51,52,131–133 Cells with a dedifferentiated phenotype also stain positive for RUNX1; however, whether up-regulation of RUNX1 plays a direct role in orchestrating dedifferentiation in the heart has not been explored.47,51 Of note, in one study which looked at the ability of adult murine cardiomyocytes to dedifferentiate in an in vitro co-culture model with neonatal rat ventricular cardiomyocytes, dedifferentiation, and proliferation of the adult cardiomyocytes coincided with an increase in RUNX1 expression, which was lost when the cardiomyocytes re-differentiated.132 Cardiomyocyte dedifferentiation may confer stress resistance to hypoxic conditions and/or metabolic strain which may allow performance to be sustained during pathophysiological stress, providing the damage imposed on the myocardium is self-limiting in nature.131 However, in the long term, chronic expression of foetal genes involved in metabolism, calcium homeostasis, and contractility may lead to mitochondrial dysfunction, impaired contractility contributing to adverse cardiac remodelling, and thus promote progression to heart failure.76,134 Improved understanding of the transcription factors driving cardiomyocyte dedifferentiation in response to injury may therefore be a crucial step towards the development of therapies which attenuate adverse cardiac remodelling.
Transcription factors that regulate the balance between proliferation and differentiation are also of interest because of the centrality of these processes to tissue repair and regeneration. In many tissues, resident stem or progenitor cells can proliferate and differentiate to replace injured cells.135 RUNX1 has already been identified as a key regulator of stem cell behaviour in the haematopoietic system, skeletal muscle, hair follicles, and the nervous system.125,126,136 RUNX1 appears to have a role in skeletal muscle regeneration in mice that have lost skeletal muscle dystrophin function (mdx mice).137
In the absence of dystrophin, the mice repeatedly undergo cycles of myonecrosis followed by regeneration which preserves muscle mass. However, when the mdx mice were developed to have a skeletal muscle Runx1-deficiency in addition to the loss of dystrophin, muscle mass was not maintained beyond 2 weeks, suggesting impaired regeneration of muscle fibres.137 The Runx1-deficient mdx mice were found to have severe muscle deterioration and fibrosis, accompanied by an impaired exercise capacity and reduced evidence of regenerative fibres. In the same study, primary myoblasts with a RUNX1 deficiency showed reduced proliferation, G1 arrest and premature differentiation whereas, ectopic expression of RUNX1 in myoblasts was found to reduce differentiation.137 RUNX1 may facilitate regeneration in this context by preventing premature differentiation of proliferating myoblasts until enough cells have accumulated to permit effective repair.
In contrast to skeletal muscle, the heart has a very limited capacity to regenerate and this is evidenced by the permanent damage to the myocardium caused by infarction.138,139 Although there is evidence suggesting that cardiomyocytes can proliferate, the rate of renewal is very low, estimated at 0.5–2% of cardiomyocytes each year.140,141 Cardiomyocyte proliferation may increase following injury; however, any contributions to tissue repair are considered small.140,142 This contrasts with findings in the neonatal mouse heart as well as zebrafish and newts where a robust proliferative regenerative response is mounted following cardiac resection.143–145 Understanding the developmental mechanisms of cardiomyocyte proliferation and regeneration and how these are altered in the adult heart may therefore be a crucial step towards the identification of reparative pathways that could be harnessed to improve cardiac regeneration in the future.146 Indeed, a recent study has provided promising evidence that stimulation of endogenous cardiomyocyte proliferation programmes in large mammals is a valid approach to facilitate cardiac repair by increasing myocardial mass and contractility after MI.147
Whilst RUNX1 has been identified to be a key player in zebrafish heart regeneration following cardiac injury54 its role in cardiomyocyte proliferation programmes in mammals remains unknown. Investigation of this gap in our knowledge is warranted given that RUNX1 regulates proliferation, differentiation, and tissue regeneration in other settings.
RUNX1 has also been shown to interact with the Hippo pathway, a key pathway linked to cardiac regeneration in neonatal as well as adult hearts.148,149 Like RUNX1, the Hippo pathway and its downstream effectors YAP and TAZ are involved in proliferation, differentiation, and expansion of stem cell populations.150 Adult mice with a heterozygous deletion of YAP show increased scarring and depressed cardiac contractility following MI and conversely, experimental induction of YAP in cardiomyocytes post-MI has the opposite effect.151,152 RUNX1 has been shown to interact via its PY motif to YAP WW domain to co-regulate target genes.43,153 Co-expression of RUNX1 and YAP leads to the repression of the YAP gene signature and a functional attenuation of the effects of YAP activation in mammary epithelial cell lines.43 Although little is known about the interaction of YAP and RUNX1 in the heart, it is interesting to note that oncostatin-M, a potent inducer of RUNX1, is activated downstream of YAP in cardiomyocytes, suggesting that an indirect link between RUNX1 and YAP may exist within the heart.52
11. RUNX1 in non-myocytes
Whilst studies have focused on the function of RUNX1 within cardiomyocytes to date, a role for RUNX1 in other non-cardiomyocyte cells types may also exist. Other cell types within the myocardium include fibroblasts, endothelial cells, resident immune cells, and neural cells, each with their own role in cardiac physiology and disease. Interestingly, increased Runx1 mRNA expression is reported in non-cardiomyocyte cells following MI, with 14% of non-cardiomyocytes within the infarct zone expressing RUNX1 at day 1 post-MI and up to 26% and 35% of non-cardiomyocytes expressing RUNX1 within the infarct zone and border zone, respectively, by 2 weeks post-MI.46 Similarly, in a mouse model of DCM, RUNX1 expression was increased 4.8-fold in DCM non-myocytes compared to non-myocytes in control hearts.53 The exact non-myocyte cell types in which RUNX1 is up-regulated is unknown, and whether this affects cardiac remodelling remains unexplored. However, in a recent study which employed a combination of computational transcription factor-binding analysis and RNA-seq data, the RUNX1 binding motif was identified as a main motif in cardiac fibroblast-specific active enhancers. This suggests that RUNX1 may have an important role in cardiac fibroblasts.154 Certainly, RUNX1 has been shown to regulate proliferation in stromal fibroblasts from human prostate-derived mesenchymal stem cells and is involved in the activation of fibroblasts to myofibroblasts.105 As the activation and proliferation of fibroblasts in the heart after injury is an integral part of the cardiac repair process and yet in the longer term contributes to cardiac pathology, it will be interesting to see if RUNX1 is implicated within this process in the future.155
Many studies document a role for RUNX1 in the development, polarization, and function of the mammalian immune system. A full discussion of RUNX1 in inflammation and immunity is beyond the remit of this review and can be found elsewhere.156–158 However, it is important to consider that the ability of RUNX1 to modulate the inflammatory phenotype in response to damage may have profound implications in the injured heart, where immune cell function impacts upon remodelling and repair.159,160 Generally, a loss of RUNX1 function is associated with heightened inflammatory responses.161–163 Mice with a Runx1 deletion in alveolar epithelial cells had up-regulated NF-κB signalling, augmented pulmonary inflammation and an earlier onset of death after pulmonary LPS exposure.55 Further studies have suggested that RUNX1 attenuates NF-κB signalling by directly binding IκB kinase (IKK) in the cytoplasm and preventing it from targeting IκBα, an endogenous inhibitor of NF-κB, for degradation.55,162 However, common to other RUNX1 functions, the effect of RUNX1 on inflammation appears context dependent, as in some settings RUNX1 appears pro-inflammatory in its action. For example, in myometrial cells silencing of Runx1 with siRNA led to reduced up-regulation of IL-1β, IL-6, and the adhesion molecules VCAM1 and ICAM1 mRNA in response to treatment with TNF-α.164 Similarly, in mouse peritoneal macrophages cultured in vitro the C-terminus of RUNX1 interacts with the p50 subunit of NF-κB to co-transcriptionally activate the production of IL-6 in response to stimulation with LPS.165 The findings that RUNX1 can act as both a positive and negative regulator of NF-κB signalling is particularly intriguing, as NF-κB is a major orchestrator of the inflammatory actions of cytokines in heart disease.166 Importantly, NF-κB has been identified as both a cardioprotective mediator and an active participant in the progression of cardiac disease depending on the spatial-temporal resolution of its activation.166,167 Therefore, any factor capable of modulating its activity or directing its transcriptional profile in either a positive or negative manner may be expected to influence the outcomes of cardiac pathology.
Like the other non-myocyte cell types resident in the heart, endothelial cells are gaining increasing attention for playing an active role in cardiomyocyte function and disease progression.168–170 Whilst a comprehensive review of the functions of RUNX1 in endothelial cells is beyond the scope of this review, the link between RUNX1 and angiogenesis will be considered further due to the significance of angiogenesis in tissue salvage post-MI and in cardiac remodelling.171 The importance of RUNX1 in the developing vasculature is highlighted by the phenotype of Runx1-deficient embryos which exhibit defective vascular formation and suffer fatal haemorrhages in the CNS, pericardium, and peritoneum.9,37 Generally, RUNX1 has a pro-angiogenic effect on endothelial cells. In endothelial precursor cells derived from mouse embryos, RUNX1 was shown to direct angiogenesis by inducing endothelial cell differentiation and vascular network formation through the induction of VE-cadherin and repression of the anti-angiogenic factor insulin-like growth factor-binding protein 3.172 Similarly, siRNA knockdown of Runx1 in cultured human retinal microvasculature endothelial cells (HRMECs) decreased endothelial migration and proliferation.173 In endothelial tube formation assays, an in vitro methodology used to study the angiogenic capacity of cultured cells, treatment of HRMECs with the RUNX1 inhibitor Ro5-3335 was associated with reduced tubular structure formation, indicating depressed angiogenic function.173 Interestingly, in another study also using tube forming assays, treatment of human umbilical vein endothelial cells (HUVECs) with conditioned media prepared from RUNX1-silenced glioblastoma cells was able to have a similar suppressive effect on angiogenesis.77 This suggests that RUNX1 expression in non-vascular cell types may be able to direct endothelial function and angiogenic capacity via the release of paracrine signals.
Paradoxically, RUNX1 has been shown to both directly and indirectly limit the expression of the potent pro-angiogenic factor VEGF.98,174,175 This would suggest that RUNX1 may limit angiogenesis in some circumstances, and in support, silencing of RUNX1 in acute myeloid leukaemia cells increased VEGFA promoter activity and conditioned media from these cells led to a significant improvement in the in vitro angiogenic capacity of HUVECs.175 Like its other functions, it may be that RUNX1 acts in a context-dependent manner and possess the capacity to both stimulate or restrain angiogenic potential depending on the cellular milieu.
In the injured heart, expansion of the vascular network through angiogenesis is a key response which protects against adverse remodelling and progression to heart failure.176,177 In view of the evidence linking RUNX1 to angiogenesis, it may be valuable to explore whether the up-regulation of RUNX1 in cardiomyocytes or in non-myocytes in the border zone following MI influences angiogenesis within this region.
12. RUNX1 as a novel target in heart disease
Heart failure remains a major global health and economic burden.178 There is a persistent unmet need for better treatments as patients with heart failure with reduced ejection fraction continue to have poor long-term outcomes. Importantly, there appears to be a lack of translation between basic science discovery and drug development with most therapies failing phase III clinical trials despite promising phase II testing.179 Critically, there are currently no approved treatments for heart failure patients with preserved ejection fraction, although phase III trials are on-going (Novartis PARAGON-HF trial and AstraZeneca DETERMINE trial).180
Novel therapies aimed at targeting a master-regulator transcription factor such as RUNX1 may achieve a more efficacious therapeutic response by impacting on multiple downstream signalling pathways to mitigate the adverse cardiac remodelling that initiates heart failure. Given its involvement in systolic dysfunction46 inflammation,164,165 fibrosis and angiogenesis,172,173 RUNX1 has been highlighted as an attractive therapeutic target.
Transcription factors have historically been viewed as unlikely and challenging ‘druggable’ targets due to their pleiotropic actions in multiple cell types and systems during normal development and disease.181 More recently and thanks to advances in our knowledge of transcription factor structure and function, transcription factor drivers of cancer have been directly targeted using small molecule protein–protein interaction inhibitors.182
Importantly, the small molecule benzodiazepine Ro5-3335 was identified as a disruptor of the CBFβ–RUNX interaction.129 Ro5-3335 was shown to repress CBFβ/RUNX-dependent transactivation in reporter assays and repress RUNX1-dependent haematopoiesis in zebrafish by altering the conformation of the CBFβ–RUNX1 complex. Subsequently, Ro5-3335 has been used to inhibit RUNX1 during a variety of pathophysiological processes including, but not exclusively in, macrophages during septic shock,165 in retinal angiogenesis173 and, acute myeloid leukaemia.183 However, direct binding of Ro5-3335 to CBFβ or RUNX has not been well documented, thus whether Ro5-3335 is a direct inhibitor of Runx remains contentious.184
Illendula et al.184 developed a selective small-molecule inhibitor of RUNX activity. Further optimization of this compound led to a new small-molecule inhibitor, AI-14-91, which binds selectively to CBFβ and prevents it from binding RUNX proteins via an allosteric mechanism.185 AI-14-91 has been optimized and shown to inhibit RUNX1 function in leukaemia and basal-like breast cancer cells.185 As mentioned, adverse cardiac remodelling is a complex response to injury involving cardiomyocyte loss, hypertrophy, fibrosis, extracellular matrix remodelling, and angiogenesis. As RUNX1 has the potential to impact all these processes, it is pertinent to determine whether AI-14-91 can prevent adverse remodelling and progression to heart failure following cardiac injury and is the focus of on-going studies.
13. Conclusions
Accumulating evidence supports that RUNX1 is up-regulated at early time points in the heart in response to a range of cardiac pathologies. New developments in the field have demonstrated that RUNX1 activation in cardiomyocytes after MI is detrimental to systolic function and cardiac structure and have thus unveiled RUNX1 as an attractive novel target to mitigate adverse cardiac remodelling. Understanding of the role of RUNX1 in the heart is in its infancy and there are many pertinent questions to be asked about the identity of the downstream targets of RUNX1 in the heart. Crossing discipline boundaries to understand RUNX1 biology across different organs has offered insight into the potential functions of RUNX1, however, due to the strong context-dependent nature of RUNX1 further work is now needed to evaluate whether the effects of RUNX1 observed in other tissues translate into the cardiac context. This exploration should extend beyond cardiomyocytes to study the role of RUNX1 in other cardiac cell types in heart physiology and pathology. As a master transcription factor, RUNX1 has the potential to act across an entire network of signalling pathways. Unravelling the functional consequences of this network represents an exciting challenge with a translational potential.
Conflict of interest: none declared.
Funding
Funding was received from the British Heart Foundation, PhD studentship FS/15/64/32035 and Project grant PG/18/9/33548.
References
- 1. Conrad N, Judge A, Tran J, Mohseni H, Hedgecott D, Crespillo AP, Allison M, Hemingway H, Cleland JG, McMurray JJV, Rahimi K.. Temporal trends and patterns in heart failure incidence: a population-based study of 4 million individuals. Lancet 2018;391:572–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dewan P, Rørth R, Jhund PS, Ferreira JP, Zannad F, Shen L, Køber L, Abraham WT, Desai AS, Dickstein K, Packer M, Rouleau JL, Solomon SD, Swedberg K, Zile MR, McMurray JJV.. Income inequality and outcomes in heart failure: a global between-country analysis. JACC Heart Fail 2019;7:336–346. [DOI] [PubMed] [Google Scholar]
- 3. Konstam MA, Kramer DG, Patel AR, Maron MS, Udelson JE.. Left ventricular remodeling in heart failure: current concepts in clinical significance and assessment. JACC Cardiovasc Imaging 2011;4:98–108. [DOI] [PubMed] [Google Scholar]
- 4. Burchfield JS, Xie M, Hill JA.. Pathological ventricular remodeling: mechanisms: part 1 of 2. Circulation 2013;128:388–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cokkinos DV, Belogianneas C.. Left ventricular remodelling: a problem in search of solutions. Eur Cardiol 2016;11:29–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Levanon D, Brenner O, Negreanu V, Bettoun D, Woolf E, Eilam R, Lotem J, Gat U, Otto F, Speck N, Groner Y.. Spatial and temporal expression pattern of Runx3 (Aml2) and Runx1 (Aml1) indicates non-redundant functions during mouse embryogenesis. Mech Dev 2001;109:413–417. [DOI] [PubMed] [Google Scholar]
- 7. Levanon D, Groner Y.. Structure and regulated expression of mammalian RUNX genes. Oncogene 2004;23:4211–4219. [DOI] [PubMed] [Google Scholar]
- 8. Cameron ER, Blyth K, Hanlon L, Kilbey A, Mackay N, Stewart M, Terry A, Vaillant F, Wotton S, Neil JC.. The Runx genes as dominant oncogenes. Blood Cells Mol Dis 2003;30:194–200. [DOI] [PubMed] [Google Scholar]
- 9. Wang Q, Stacy T, Binder M, Marin-Padilla M, Sharpe AH, Speck NA.. Disruption of the Cbfa2 gene causes necrosis and hemorrhaging in the central nervous system and blocks definitive hematopoiesis. Proc Natl Acad Sci USA 1996;93:3444–3449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Blyth K, Cameron ER, Neil JC.. The RUNX genes: gain or loss of function in cancer. Nat Rev Cancer 2005;5:376–387. [DOI] [PubMed] [Google Scholar]
- 11. Ogawa E, Maruyama M, Kagoshima H, Inuzuka M, Lu J, Satake M, Shigesada K, Ito Y.. PEBP2/PEA2 represents a family of transcription factors homologous to the products of the Drosophila runt gene and the human AML1 gene. Proc Natl Acad Sci U S A 1993;90:6859–6863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kanno T, Kanno Y, Chen LF, Ogawa E, Kim WY, Ito Y.. Intrinsic transcriptional activation-inhibition domains of the polyomavirus enhancer binding protein 2/core binding factor alpha subunit revealed in the presence of the beta subunit. Mol Cell Biol 1998;18:2444–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wolf-Watz M, Xie XQ, Holm M, Grundstrom T, Hard T.. Solution properties of the free and DNA-bound Runt domain of AML1. Eur J Biochem 1999;261:251–260. [DOI] [PubMed] [Google Scholar]
- 14. Backstrom S, Wolf-Watz M, Grundstrom C, Hard T, Grundstrom T, Sauer UH.. The RUNX1 Runt domain at 1.25A resolution: a structural switch and specifically bound chloride ions modulate DNA binding. J Mol Biol 2002;322:259–272. [DOI] [PubMed] [Google Scholar]
- 15. Huang G, Shigesada K, Ito K, Wee HJ, Yokomizo T, Ito Y.. Dimerization with PEBP2beta protects RUNX1/AML1 from ubiquitin-proteasome-mediated degradation. EMBO J 2001;20:723–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chuang LS, Ito K, Ito Y.. RUNX family: regulation and diversification of roles through interacting proteins. Int J Cancer 2013;132:1260–1271. [DOI] [PubMed] [Google Scholar]
- 17. Seo W, Tanaka H, Miyamoto C, Levanon D, Groner Y, Taniuchi I.. Roles of VWRPY motif-mediated gene repression by Runx proteins during T-cell development. Immunol Cell Biol 2012;90:827–830. [DOI] [PubMed] [Google Scholar]
- 18. Miyoshi H, Ohira M, Shimizu K, Mitani K, Hirai H, Imai T, Yokoyama K, Soceda E, Ohkl M.. Alternative splicing and genomic structure of the AML1 gene involved in acute myeloid leukemia. Nucl Acids Res 1995;23:2762–2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ghozi MC, Bernstein Y, Negreanu V, Levanon D, Groner Y.. Expression of the human acute myeloid leukemia gene AML1 is regulated by two promoter regions. Proc Natl Acad Sci U S A 1996;93:1935–1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bee T, Liddiard K, Swiers G, Bickley SRB, Vink CS, Jarratt A, Hughes JR, Medvinsky A, de Bruijn MFTR.. Alternative Runx1 promoter usage in mouse developmental hematopoiesis. Blood Cells Mol Dis 2009;43:35–42. [DOI] [PubMed] [Google Scholar]
- 21. Telfer JC, Rothenberg EV.. Expression and function of a stem cell promoter for the murine CBFalpha2 gene: distinct roles and regulation in natural killer and T cell development. Dev Biol 2001;229:363–382. [DOI] [PubMed] [Google Scholar]
- 22. Pozner A, Goldenberg D, Negreanu V, Le S-Y, Elroy-Stein O, Levanon D, Groner Y.. Transcription-coupled translation control of AML1/RUNX1 is mediated by cap- and internal ribosome entry site-dependent mechanisms. Mol Cell Biol 2000;20:2297–2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Spriggs KA, Stoneley M, Bushell M, Willis AE.. Re-programming of translation following cell stress allows IRES-mediated translation to predominate. Biol Cell 2008;100:27–38. [DOI] [PubMed] [Google Scholar]
- 24. Levanon D, Glusman G, Bangsow T, Ben-Asher E, Male DA, Avidan N, Bangsow C, Hattori M, Taylor TD, Taudien S, Blechschmidt K, Shimizu N, Rosenthal A, Sakaki Y, Lancet D, Groner Y.. Architecture and anatomy of the genomic locus encoding the human leukemia-associated transcription factor RUNX1/AML1. Gene 2001;262:23–33. [DOI] [PubMed] [Google Scholar]
- 25. Tanaka T, Tanaka K, Ogawa S, Kurokawa M, Mitani K, Yazaki Y, Shibata Y, Hirai H.. An acute myeloid leukemia gene, AML1, regulates transcriptional activation and hemopoietic myeloid cell differentiation antagonistically by two alternative spliced forms. Leukemia 1997;11(Suppl 3):299–302. [PubMed] [Google Scholar]
- 26. Wang L, Huang G, Zhao X, Hatlen MA, Vu L, Liu F, Nimer SD.. Post-translational modifications of Runx1 regulate its activity in the cell. Blood Cells Mol Dis 2009;43:30–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tanaka T, Kurokawa M, Ueki K, Tanaka K, Imai Y, Mitani K, Okazaki K, Sagata N, Yazaki Y, Shibata Y, Kadowaki T, Hirai H.. The extracellular signal-regulated kinase pathway phosphorylates AML1, an acute myeloid leukemia gene product, and potentially regulates its transactivation ability. Mol Cell Biol 1996;16:3967–3979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Guo H, Friedman AD.. Phosphorylation of RUNX1 by cyclin-dependent kinase reduces direct interaction with HDAC1 and HDAC3. J Biol Chem 2011;286:208–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Imai Y, Kurokawa M, Yamaguchi Y, Izutsu K, Nitta E, Mitani K, Satake M, Noda T, Ito Y, Hirai H.. The corepressor mSin3A regulates phosphorylation-induced activation, intranuclear location, and stability of AML1. Mol Cell Biol 2004;24:1033–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Huang H, Woo AJ, Waldon Z, Schindler Y, Moran TB, Zhu HH, Feng G-S, Steen H, Cantor AB.. A Src family kinase-Shp2 axis controls RUNX1 activity in megakaryocyte and T-lymphocyte differentiation. Genes Dev 2012;26:1587–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Behrens K, Maul K, Tekin N, Kriebitzsch N, Indenbirken D, Prassolov V, Müller U, Serve H, Cammenga J, Stocking C.. RUNX1 cooperates with FLT3-ITD to induce leukemia. J Exp Med 2017;214:737–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhao X, Jankovic V, Gural A, Huang G, Pardanani A, Menendez S, Zhang J, Dunne R, Xiao A, Erdjument-Bromage H, Allis CD, Tempst P, Nimer SD.. Methylation of RUNX1 by PRMT1 abrogates SIN3A binding and potentiates its transcriptional activity. Genes Dev 2008;22:640–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Vu LP, Perna F, Wang L, Voza F, Figueroa ME, Tempst P, Erdjument-Bromage H, Gao R, Chen S, Paietta E, Deblasio T, Melnick A, Liu Y, Zhao X, Nimer SD.. PRMT4 blocks myeloid differentiation by assembling a methyl-RUNX1-dependent repressor complex. Cell Rep 2013;5:1625–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yamaguchi Y, Kurokawa M, Imai Y, Izutsu K, Asai T, Ichikawa M, Yamamoto G, Nitta E, Yamagata T, Sasaki K, Mitani K, Ogawa S, Chiba S, Hirai H.. AML1 is functionally regulated through p300-mediated acetylation on specific lysine residues. J Biol Chem 2004;279:15630–15638. [DOI] [PubMed] [Google Scholar]
- 35. Yonezawa T, Takahashi H, Shikata S, Sawasaki T, Kitamura T, Goyama S.. The ubiquitin ligase RNF38 promotes RUNX1 ubiquitination and enhances RUNX1-mediated suppression of erythroid transcription program. Biochem Biophys Res Commun 2018;505:905–909. [DOI] [PubMed] [Google Scholar]
- 36. Lluri G, Huang V, Touma M, Liu X, Harmon AW, Nakano A.. Hematopoietic progenitors are required for proper development of coronary vasculature. J Mol Cell Cardiol 2015;86:199–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Takakura N, Watanabe T, Suenobu S, Yamada Y, Noda T, Ito Y, Satake M, Suda T.. A role for hematopoietic stem cells in promoting angiogenesis. Cell 2000;102:199–209. [DOI] [PubMed] [Google Scholar]
- 38. Eulalio A, Mano M, Ferro MD, Zentilin L, Sinagra G, Zacchigna S, Giacca M.. Functional screening identifies miRNAs inducing cardiac regeneration. Nature 2012;492:376–381. [DOI] [PubMed] [Google Scholar]
- 39. Gornikiewicz B, Ronowicz A, Krzeminski M, Sachadyn P.. Changes in gene methylation patterns in neonatal murine hearts: implications for the regenerative potential. BMC Genomics 2016;17:231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Portela A, Esteller M.. Epigenetic modifications and human disease. Nat Biotechnol 2010;28:1057–1068. [DOI] [PubMed] [Google Scholar]
- 41. Xin M, Kim Y, Sutherland LB, Murakami M, Qi X, McAnally J, Porrello ER, Mahmoud AI, Tan W, Shelton JM, Richardson JA, Sadek HA, Bassel-Duby R, Olson EN.. Hippo pathway effector Yap promotes cardiac regeneration. Proc Natl Acad Sci USA 2013;110:13839–13844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Flinn MA, Jeffery BE, O’Meara CC, Link BA.. Yap is required for scar formation but not myocyte proliferation during heart regeneration in zebrafish. Cardiovasc Res 2019;115:570–577. [DOI] [PubMed] [Google Scholar]
- 43. Kulkarni M, Tan TZ, Sulaiman NBS, Lamar JM, Bansal P, Cui J, Qiao Y, Ito Y.. RUNX1 and RUNX3 protect against YAP-mediated EMT, stem-ness and shorter survival outcomes in breast cancer. Oncotarget 2018;9:14175–14192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chimge NO, Frenkel B.. The RUNX family in breast cancer: relationships with estrogen signaling. Oncogene 2013;32:2121–2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Riggio AI, Blyth K.. The enigmatic role of RUNX1 in female-related cancers—current knowledge & future perspectives. FEBS J 2017;284:2345–2362. [DOI] [PubMed] [Google Scholar]
- 46. McCarroll CS, He W, Foote K, Bradley A, Mcglynn K, Vidler F, Nixon C, Nather K, Fattah C, Riddell A, Bowman P, Elliott EB, Bell M, Hawksby C, MacKenzie SM, Morrison LJ, Terry A, Blyth K, Smith GL, McBride MW, Kubin T, Braun T, Nicklin SA, Cameron ER, Loughrey CM.. Runx1 deficiency protects against adverse cardiac remodeling after myocardial infarction. Circulation 2018;137:57–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kubin T, Pöling J, Kostin S, Gajawada P, Hein S, Rees W, Wietelmann A, Tanaka M, Lörchner H, Schimanski S, Szibor M, Warnecke H, Braun T.. Oncostatin M is a major mediator of cardiomyocyte dedifferentiation and remodeling. Cell Stem Cell 2011;9:420–432. [DOI] [PubMed] [Google Scholar]
- 48. Gattenlohner S, Waller C, Ertl G, Bultmann BD, Muller-Hermelink HK, Marx A.. NCAM(CD56) and RUNX1(AML1) are up-regulated in human ischemic cardiomyopathy and a rat model of chronic cardiac ischemia. Am J Pathol 2003;163:1081–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang Y, Ma S, Wang Q, Hu W, Wang D, Li X, Su T, Qin X, Zhang X, Ma K, Chen J, Xiong LZe, Cao F.. Effects of cannabinoid receptor type 2 on endogenous myocardial regeneration by activating cardiac progenitor cells in mouse infarcted heart. Sci China Life Sci 2014;57:201–208. [DOI] [PubMed] [Google Scholar]
- 50. Zhang YW, Bae SC, Huang G, Fu YX, Lu J, Ahn MY, Kanno Y, Kanno T, Ito Y.. A novel transcript encoding an N-terminally truncated AML1/PEBP2 alphaB protein interferes with transactivation and blocks granulocytic differentiation of 32Dcl3 myeloid cells. Mol Cell Biol 1997;17:4133–4145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhang X, Ma S, Zhang R, Li S, Zhu D, Han D, Li X, Li C, Yan W, Sun D, Xu B, Wang Y, Cao F.. Oncostatin M-induced cardiomyocyte dedifferentiation regulates the progression of diabetic cardiomyopathy through B-Raf/Mek/Erk signaling pathway. Acta Biochim Biophys Sin 2016;48:257–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ikeda S, Mizushima W, Sciarretta S, Abdellatif M, Zhai P, Mukai R, Fefelova N, Oka S-I, Nakamura M, Del Re DP, Farrance I, Park JY, Tian B, Xie L-H, Kumar M, Hsu C-P, Sadayappan S, Shimokawa H, Lim D-S, Sadoshima J.. Hippo deficiency leads to cardiac dysfunction accompanied by cardiomyocyte dedifferentiation during pressure overload. Circ Res 2019;124:292–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Burke MA, Chang S, Wakimoto H, Gorham JM, Conner DA, Christodoulou DC, Parfenov MG, DePalma SR, Eminaga S, Konno T, Seidman JG, Seidman CE.. Molecular profiling of dilated cardiomyopathy that progresses to heart failure. JCI Insight 2016;1:e86898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Koth J, Wang X, Killen AC, Stockdale WT, Potts HG, Jefferson A, Bonkhofer F, Riley PR, Patient R, Gottgens B, Mommersteeg M TM. Runx1 promotes scar deposition and inhibits myocardial proliferation and survival during zebrafish heart regeneration. bioRxiv 2019:799163. [DOI] [PMC free article] [PubMed]
- 55. Tang X, Sun L, Jin X, Chen Y, Zhu H, Liang Y, Wu Q, Han X, Liang J, Liu X, Liang Z, Wang G, Luo F.. Runt-related transcription factor 1 regulates LPS-induced acute lung injury via NF-kappaB signaling. Am J Respir Cell Mol Biol 2017;57:174–183. [DOI] [PubMed] [Google Scholar]
- 56. Logan TT, Villapol S, Symes AJ.. TGF-beta superfamily gene expression and induction of the Runx1 transcription factor in adult neurogenic regions after brain injury. PLoS One 2013;8:e59250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gonzalez de Aguilar J-L, Niederhauser-Wiederkehr C, Halter B, De Tapia M, Di Scala F, Demougin P, Dupuis L, Primig M, Meininger V, Loeffler J-P.. Gene profiling of skeletal muscle in an amyotrophic lateral sclerosis mouse model. Physiol Genomics 2008;32:207–218. [DOI] [PubMed] [Google Scholar]
- 58. Zhu X, Yeadon JE, Burden SJ.. AML1 is expressed in skeletal muscle and is regulated by innervation. Mol Cell Biol 1994;14:8051–8057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zusso M, Methot L, Lo R, Greenhalgh AD, David S, Stifani S.. Regulation of postnatal forebrain amoeboid microglial cell proliferation and development by the transcription factor Runx1. J Neurosci 2012;32:11285–11298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Porter JD, Merriam AP, Leahy P, Gong B, Khanna S.. Dissection of temporal gene expression signatures of affected and spared muscle groups in dystrophin-deficient (mdx) mice. Hum Mol Genet 2003;12:1813–1821. [DOI] [PubMed] [Google Scholar]
- 61. Bao M, Liu S, Yu XY, Wu C, Chen Q, Ding H, Shen C, Wang B, Wang S, Song YH, Li Y. Runx1 promotes satellite cell proliferation during ischemia—induced muscle regeneration. Biochem Biophys Res Commun 2018;503:2993–2997. [DOI] [PubMed] [Google Scholar]
- 62. Wang X, Blagden C, Fan J, Nowak SJ, Taniuchi I, Littman DR, Burden SJ. Runx1 prevents wasting, myofibrillar disorganization, and autophagy of skeletal muscle. Genes Dev 2005;19:1715–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Russo TL, Peviani SM, Durigan JL, Gigo-Benato D, Delfino GB, Salvini TF.. Stretching and electrical stimulation reduce the accumulation of MyoD, myostatin and atrogin-1 in denervated rat skeletal muscle. J Muscle Res Cell Motil 2010;31:45–57. [DOI] [PubMed] [Google Scholar]
- 64. Valdez G, Tapia JC, Kang H, Clemenson GD, Gage FH, Lichtman JW, Sanes JR.. Attenuation of age-related changes in mouse neuromuscular synapses by caloric restriction and exercise. Proc Natl Acad Sci USA 2010;107:14863–14868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. White Z, Terrill J, White RB, McMahon C, Sheard P, Grounds MD, Shavlakadze T.. Voluntary resistance wheel exercise from mid-life prevents sarcopenia and increases markers of mitochondrial function and autophagy in muscles of old male and female C57BL/6J mice. Skelet Muscle 2016;6:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Messi ML, Li T, Wang ZM, Marsh AP, Nicklas B, Delbono O.. Resistance training enhances skeletal muscle innervation without modifying the number of satellite cells or their myofiber association in obese older adults. Gerona 2016;71:1273–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Dillon SM, Allessie MA, Ursell PC, Wit AL.. Influences of anisotropic tissue structure on reentrant circuits in the epicardial border zone of subacute canine infarcts. Circ Res 1988;63:182–206. [DOI] [PubMed] [Google Scholar]
- 68. Ashikaga H, Mickelsen SR, Ennis DB, Rodriguez I, Kellman P, Wen H, McVeigh ER.. Electromechanical analysis of infarct border zone in chronic myocardial infarction. Am J Physiol Heart Circ Physiol 2005;289:H1099–H1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Sakai K, Watanabe K, Millard RW.. Defining the mechanical border zone: a study in the pig heart. Am J Physiol 1985;249:H88–H94. [DOI] [PubMed] [Google Scholar]
- 70. Mazhari R, Omens JH, Covell JW, McCulloch AD.. Structural basis of regional dysfunction in acutely ischemic myocardium. Cardiovasc Res 2000;47:284–293. [DOI] [PubMed] [Google Scholar]
- 71. D’Elia N, D’Hooge J, Marwick TH.. Association Between Myocardial Mechanics and Ischemic LV Remodeling. JACC Cardiovasc Imaging 2015;8:1430–1443. [DOI] [PubMed] [Google Scholar]
- 72. Frangogiannis NG. The reparative function of cardiomyocytes in the infarcted myocardium. Cell Metab 2015;21:797–798. [DOI] [PubMed] [Google Scholar]
- 73. Frangogiannis NG. The inflammatory response in myocardial injury, repair, and remodelling. Nat Rev Cardiol 2014;11:255–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Lorchner H, Poling J, Gajawada P, Hou Y, Polyakova V, Kostin S, Adrian-Segarra JM, Boettger T, Wietelmann A, Warnecke H, Richter M, Kubin T, Braun T. Myocardial healing requires Reg3beta-dependent accumulation of macrophages in the ischemic heart. Nat Med 2015;21:353–362. [DOI] [PubMed] [Google Scholar]
- 75. Gwechenberger M, Moertl D, Pacher R, Huelsmann M.. Oncostatin-M in myocardial ischemia/reperfusion injury may regulate tissue repair. Croat Med J 2004;45:149–157. [PubMed] [Google Scholar]
- 76. Poling J, Gajawada P, Lorchner H, Polyakova V, Szibor M, Bottger T, Warnecke H, Kubin T, Braun T. The Janus face of OSM-mediated cardiomyocyte dedifferentiation during cardiac repair and disease. Cell Cycle 2012;11:439–445. [DOI] [PubMed] [Google Scholar]
- 77. Sangpairoj K, Vivithanaporn P, Apisawetakan S, Chongthammakun S, Sobhon P, Chaithirayanon K.. RUNX1 regulates migration, invasion, and angiogenesis via p38 MAPK pathway in human glioblastoma. Cell Mol Neurobiol 2017;37:1243–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Fukui H, Runker A, Fabel K, Buchholz F, Kempermann G.. Transcription factor Runx1 is pro-neurogenic in adult hippocampal precursor cells. PLoS One 2018;13:e0190789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Kim T, Zhelyabovska O, Liu J, Yang Q.. Generation of an inducible, cardiomyocyte-specific transgenic mouse model with PPAR beta/delta overexpression. Methods Mol Biol 2013;952:57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Klunker S, Chong MMW, Mantel P-Y, Palomares O, Bassin C, Ziegler M, Rückert B, Meiler F, Akdis M, Littman DR, Akdis CA.. Transcription factors RUNX1 and RUNX3 in the induction and suppressive function of Foxp3+ inducible regulatory T cells. J Exp Med 2009;206:2701–2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Li X, Zhang S, Wa M, Liu Z, Hu S.. MicroRNA-101 protects against cardiac remodeling following myocardial infarction via downregulation of runt-related transcription factor 1. J Am Heart Assoc 2019;8:e013112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Carr AN, Schmidt AG, Suzuki Y, del Monte F, Sato Y, Lanner C, Breeden K, Jing S-L, Allen PB, Greengard P, Yatani A, Hoit BD, Grupp IL, Hajjar RJ, DePaoli-Roach AA, Kranias EG.. Type 1 phosphatase, a negative regulator of cardiac function. Mol Cell Biol 2002;22:4124–4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Elliott EB, Kelly A, Smith GL, Loughrey CM.. Isolated rabbit working heart function during progressive inhibition of myocardial SERCA activity. Circ Res 2012;110:1618–1627. [DOI] [PubMed] [Google Scholar]
- 84. Haghighi K, Bidwell P, Kranias EG.. Phospholamban interactome in cardiac contractility and survival: a new vision of an old friend. J Mol Cell Cardiol 2014;77:160–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Eisner D, Bode E, Venetucci L, Trafford A.. Calcium flux balance in the heart. J Mol Cell Cardiol 2013;58:110–117. [DOI] [PubMed] [Google Scholar]
- 86. Fattah C, Nather K, McCarroll CS, Hortigon-Vinagre MP, Zamora V, Flores-Munoz M, McArthur L, Zentilin L, Giacca M, Touyz RM, Smith GL, Loughrey CM, Nicklin SA.. Gene therapy with angiotensin-(1-9) preserves left ventricular systolic function after myocardial infarction. J Am Coll Cardiol 2016;68:2652–2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Miller S, Currie S, Loughrey C, Kettlewell S, Seidler T, Reynolds D, Hasenfuss G, Smith G.. Effects of calsequestrin over-expression on excitation-contraction coupling in isolated rabbit cardiomyocytes. Cardiovasc Res 2005;67:667–677. [DOI] [PubMed] [Google Scholar]
- 88. Mandavia CH, Aroor AR, Demarco VG, Sowers JR.. Molecular and metabolic mechanisms of cardiac dysfunction in diabetes. Life Sci 2013;92:601–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Lalande S, Johnson BD.. Diastolic dysfunction: a link between hypertension and heart failure. Drugs Today 2008;44:503–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Peana D, Domeier TL.. Cardiomyocyte Ca(2+) homeostasis as a therapeutic target in heart failure with reduced and preserved ejection fraction. Curr Opin Pharmacol 2017;33:17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Lee SH, Manandhar S, Lee YM.. Roles of RUNX in hypoxia-induced responses and angiogenesis. Adv Exp Med Biol 2017;962:449–469. [DOI] [PubMed] [Google Scholar]
- 92. Semenza GL. Hypoxia-inducible factor 1 and cardiovascular disease. Annu Rev Physiol 2014;76:39–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Kido M, Du L, Sullivan CC, Li X, Deutsch R, Jamieson SW, Thistlethwaite PA.. Hypoxia-inducible factor 1-alpha reduces infarction and attenuates progression of cardiac dysfunction after myocardial infarction in the mouse. J Am Coll Cardiol 2005;46:2116–2124. [DOI] [PubMed] [Google Scholar]
- 94. Ong SG, Hausenloy DJ.. Hypoxia-inducible factor as a therapeutic target for cardioprotection. Pharmacol Ther 2012;136:69–81. [DOI] [PubMed] [Google Scholar]
- 95. Bekeredjian R, Walton CB, MacCannell KA, Ecker J, Kruse F, Outten JT, Sutcliffe D, Gerard RD, Bruick RK, Shohet RV.. Conditional HIF-1alpha expression produces a reversible cardiomyopathy. PLoS One 2010;5:e11693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Holscher M, Schafer K, Krull S, Farhat K, Hesse A, Silter M, et al. Unfavourable consequences of chronic cardiac HIF-1alpha stabilization. Cardiovasc Res 2012;94:77–86. [DOI] [PubMed] [Google Scholar]
- 97. Moslehi J, Minamishima YA, Shi J, Neuberg D, Charytan DM, Padera RF, Signoretti S, Liao R, Kaelin WG.. Loss of hypoxia-inducible factor prolyl hydroxylase activity in cardiomyocytes phenocopies ischemic cardiomyopathy. Circulation 2010;122:1004–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Peng ZG, Zhou MY, Huang Y, Qiu JH, Wang LS, Liao SH, Dong S, Chen GQ.. Physical and functional interaction of Runt-related protein 1 with hypoxia-inducible factor-1 alpha. Oncogene 2008;27:839–847. [DOI] [PubMed] [Google Scholar]
- 99. Bogoch Y, Friedlander-Malik G, Lupu L, Bondar E, Zohar N, Langier S, Ram Z, Nachmany I, Klausner JM, Pencovich N.. Augmented expression of RUNX1 deregulates the global gene expression of U87 glioblastoma multiforme cells and inhibits tumor growth in mice. Tumour Biol 2017;39:1010428317698357. [DOI] [PubMed] [Google Scholar]
- 100. Shen GM, Zhang FL, Liu XL, Zhang JW.. Hypoxia-inducible factor 1-mediated regulation of PPP1R3C promotes glycogen accumulation in human MCF-7 cells under hypoxia. FEBS Lett 2010;584:4366–4372. [DOI] [PubMed] [Google Scholar]
- 101. Ong S-G, Lee WH, Theodorou L, Kodo K, Lim SY, Shukla DH, Briston T, Kiriakidis S, Ashcroft M, Davidson SM, Maxwell PH, Yellon DM, Hausenloy DJ.. HIF-1 reduces ischaemia-reperfusion injury in the heart by targeting the mitochondrial permeability transition pore. Cardiovasc Res 2014;104:24–36. [DOI] [PubMed] [Google Scholar]
- 102. Tokumoto Y, Tamaki S, Kabe Y, Takubo K, Suematsu M.. Quiescence of adult oligodendrocyte precursor cells requires thyroid hormone and hypoxia to activate Runx1. Sci Rep 2017;7:1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Lim S-E, Esain V, Kwan W, Theodore LN, Cortes M, Frost IM, Liu SY, North TE.. HIF1alpha-induced PDGFRbeta signaling promotes developmental HSC production via IL-6 activation. Exp Hematol 2017;46:83–95.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Harris JM, Esain V, Frechette GM, Harris LJ, Cox AG, Cortes M, Garnaas MK, Carroll KJ, Cutting CC, Khan T, Elks PM, Renshaw SA, Dickinson BC, Chang CJ, Murphy MP, Paw BH, Vander Heiden MG, Goessling W, North TE.. Glucose metabolism impacts the spatiotemporal onset and magnitude of HSC induction in vivo. Blood 2013;121:2483–2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Kim W, Barron DA, San Martin R, Chan KS, Tran LL, Yang F, Ressler SJ, Rowley DR.. RUNX1 is essential for mesenchymal stem cell proliferation and myofibroblast differentiation. Proc Natl Acad Sci USA 2014;111:16389–16394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Zhou T, Luo M, Cai W, Zhou S, Feng D, Xu C, Wang H.. Runt-related transcription factor 1 (RUNX1) promotes TGF-beta-induced renal tubular epithelial-to-mesenchymal transition (EMT) and renal fibrosis through the PI3K subunit p110delta. EBioMedicine 2018;31:217–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Hanai J-I, Chen LF, Kanno T, Ohtani-Fujita N, Kim WY, Guo W-H, Imamura T, Ishidou Y, Fukuchi M, Shi M-J, Stavnezer J, Kawabata M, Miyazono K, Ito Y.. Interaction and functional cooperation of PEBP2/CBF with Smads. Synergistic induction of the immunoglobulin germline Calpha promoter. J Biol Chem 1999;274:31577–31582. [DOI] [PubMed] [Google Scholar]
- 108. Bujak M, Frangogiannis NG.. The role of TGF-beta signaling in myocardial infarction and cardiac remodeling. Cardiovasc Res 2007;74:184–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Dobaczewski M, Chen W, Frangogiannis NG.. Transforming growth factor (TGF)-beta signaling in cardiac remodeling. J Mol Cell Cardiol 2011;51:600–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. van Rooij E, Olson EN.. MicroRNAs: powerful new regulators of heart disease and provocative therapeutic targets. J Clin Invest 2007;117:2369–2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Selbach M, Schwanhausser B, Thierfelder N, Fang Z, Khanin R, Rajewsky N.. Widespread changes in protein synthesis induced by microRNAs. Nature 2008;455:58–63. [DOI] [PubMed] [Google Scholar]
- 112. Rossetti S, Sacchi N.. RUNX1: a microRNA hub in normal and malignant hematopoiesis. IJMS 2013;14:1566–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Neil JC, Hay J, Borland G.. RUNX oncoproteins and miRNA networks. Oncotarget 2017;8:62818–62819. United States [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Zaidi SK, Dowdy CR, van Wijnen AJ, Lian JB, Raza A, Stein JL, Croce CM, Stein GS.. Altered Runx1 subnuclear targeting enhances myeloid cell proliferation and blocks differentiation by activating a miR-24/MKP-7/MAPK network. Cancer Res 2009;69:8249–8255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Qian L, Van Laake LW, Huang Y, Liu S, Wendland MF, Srivastava D.. miR-24 inhibits apoptosis and represses Bim in mouse cardiomyocytes. J Exp Med 2011;208:549–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. van Rooij E, Sutherland LB, Thatcher JE, DiMaio JM, Naseem RH, Marshall WS, Hill JA, Olson EN.. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc Natl Acad Sci USA 2008;105:13027–13032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Fontana L, Pelosi E, Greco P, Racanicchi S, Testa U, Liuzzi F, Croce CM, Brunetti E, Grignani F, Peschle C.. MicroRNAs 17-5p-20a-106a control monocytopoiesis through AML1 targeting and M-CSF receptor upregulation. Nat Cell Biol 2007;9:775–787. [DOI] [PubMed] [Google Scholar]
- 118. Chen J, Huang Z-P, Seok HY, Ding J, Kataoka M, Zhang Z, Hu X, Wang G, Lin Z, Wang S, Pu WT, Liao R, Wang D-Z.. mir-17-92 cluster is required for and sufficient to induce cardiomyocyte proliferation in postnatal and adult hearts. Circ Res 2013;112:1557–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Wang J, Greene SB, Bonilla-Claudio M, Tao Y, Zhang J, Bai Y, Huang Z, Black BL, Wang F, Martin JF.. Bmp signaling regulates myocardial differentiation from cardiac progenitors through a MicroRNA-mediated mechanism. Dev Cell 2010;19:903–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Shadrin IY, Khodabukus A, Bursac N.. Striated muscle function, regeneration, and repair. Cell Mol Life Sci 2016;73:4175–4202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Singh M, Dalal S, Singh K.. Osteopontin: at the cross-roads of myocyte survival and myocardial function. Life Sci 2014;118:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Mustonen E, Ruskoaho H, Rysa J.. Thrombospondins, potential drug targets for cardiovascular diseases. Basic Clin Pharmacol Toxicol 2013;112:4–12. [DOI] [PubMed] [Google Scholar]
- 123. Stawowy P, Blaschke F, Pfautsch P, Goetze S, Lippek F, Wollert-Wulf B, Fleck E, Graf K.. Increased myocardial expression of osteopontin in patients with advanced heart failure. Eur J Heart Fail 2002;4:139–146. [DOI] [PubMed] [Google Scholar]
- 124. Frangogiannis NG, Ren G, Dewald O, Zymek P, Haudek S, Koerting A, Winkelmann K, Michael LH, Lawler J, Entman ML.. Critical role of endogenous thrombospondin-1 in preventing expansion of healing myocardial infarcts. Circulation 2005;111:2935–2942. [DOI] [PubMed] [Google Scholar]
- 125. Deltcheva E, Nimmo R.. RUNX transcription factors at the interface of stem cells and cancer. Biochem J 2017;474:1755–1768. [DOI] [PubMed] [Google Scholar]
- 126. Wang CQ, Jacob B, Nah GS, Osato M.. Runx family genes, niche, and stem cell quiescence. Blood Cells Mol Dis 2010;44:275–286. [DOI] [PubMed] [Google Scholar]
- 127. Bernardin-Fried F, Kummalue T, Leijen S, Collector MI, Ravid K, Friedman AD.. AML1/RUNX1 increases during G1 to S cell cycle progression independent of cytokine-dependent phosphorylation and induces cyclin D3 gene expression. J Biol Chem 2004;279:15678–15687. [DOI] [PubMed] [Google Scholar]
- 128. Hoi CSL, Lee SE, Lu SY, McDermitt DJ, Osorio KM, Piskun CM, Peters RM, Paus R, Tumbar T.. Runx1 directly promotes proliferation of hair follicle stem cells and epithelial tumor formation in mouse skin. Mol Cell Biol 2010;30:2518–2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Cunningham L, Finckbeiner S, Hyde RK, Southall N, Marugan J, Yedavalli VRK, Dehdashti SJ, Reinhold WC, Alemu L, Zhao L, Yeh J-RJ, Sood R, Pommier Y, Austin CP, Jeang K-T, Zheng W, Liu P.. Identification of benzodiazepine Ro5-3335 as an inhibitor of CBF leukemia through quantitative high throughput screen against RUNX1-CBFbeta interaction. Proc Natl Acad Sci USA 2012;109:14592–14597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Logan TT, Rusnak M, Symes AJ.. Runx1 promotes proliferation and neuronal differentiation in adult mouse neurosphere cultures. Stem Cell Res 2015;15:554–564. [DOI] [PubMed] [Google Scholar]
- 131. Szibor M, Poling J, Warnecke H, Kubin T, Braun T.. Remodeling and dedifferentiation of adult cardiomyocytes during disease and regeneration. Cell Mol Life Sci 2014;71:1907–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Wang WE, Li L, Xia X, Fu W, Liao Q, Lan C, Yang D, Chen H, Yue R, Zeng C, Zhou L, Zhou B, Duan DD, Chen X, Houser SR, Zeng C.. Dedifferentiation, proliferation, and redifferentiation of adult mammalian cardiomyocytes after ischemic injury. Circulation 2017;136:834–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Ausma J, Schaart G, Thoné F, Shivalkar B, Flameng W, Depré C, Vanoverschelde J-L, Ramaekers F, Borgers M.. Chronic ischemic viable myocardium in man: aspects of dedifferentiation. Cardiovasc Pathol 1995;4:29–37. [DOI] [PubMed] [Google Scholar]
- 134. Dirkx E, da Costa Martins PA, De Windt LJ.. Regulation of fetal gene expression in heart failure. Biochim Biophys Acta 2013;1832:2414–2424. [DOI] [PubMed] [Google Scholar]
- 135. Wells JM, Watt FM.. Diverse mechanisms for endogenous regeneration and repair in mammalian organs. Nature 2018;557:322–328. [DOI] [PubMed] [Google Scholar]
- 136. Appleford PJ, Woollard A.. RUNX genes find a niche in stem cell biology. J Cell Biochem 2009;108:14–21. [DOI] [PubMed] [Google Scholar]
- 137. Umansky KB, Gruenbaum-Cohen Y, Tsoory M, Feldmesser E, Goldenberg D, Brenner O, Groner Y.. Runx1 transcription factor is required for myoblasts proliferation during muscle regeneration. PLoS Genet 2015;11:e1005457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Tzahor E, Poss KD.. Cardiac regeneration strategies: staying young at heart. Science 2017;356:1035–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Sutton MG, Sharpe N.. Left ventricular remodeling after myocardial infarction: pathophysiology and therapy. Circulation 2000;101:2981–2988. [DOI] [PubMed] [Google Scholar]
- 140. Eschenhagen T, Bolli R, Braun T, Field LJ, Fleischmann BK, Frisén J, Giacca M, Hare JM, Houser S, Lee RT, Marbán E, Martin JF, Molkentin JD, Murry CE, Riley PR, Ruiz-Lozano P, Sadek HA, Sussman MA, Hill JA.. Cardiomyocyte regeneration: a consensus statement. Circulation 2017;136:680–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Cai CL, Molkentin JD.. The elusive progenitor cell in cardiac regeneration: slip slidin’ away. Circ Res 2017;120:400–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Hsieh PCH, Segers VFM, Davis ME, MacGillivray C, Gannon J, Molkentin JD, Robbins J, Lee RT.. Evidence from a genetic fate-mapping study that stem cells refresh adult mammalian cardiomyocytes after injury. Nat Med 2007;13:970–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Poss KD, Wilson LG, Keating MT.. Heart regeneration in zebrafish. Science 2002;298:2188–2190. [DOI] [PubMed] [Google Scholar]
- 144. Witman N, Murtuza B, Davis B, Arner A, Morrison JI.. Recapitulation of developmental cardiogenesis governs the morphological and functional regeneration of adult newt hearts following injury. Dev Biol 2011;354:67–76. [DOI] [PubMed] [Google Scholar]
- 145. Porrello ER, Mahmoud AI, Simpson E, Hill JA, Richardson JA, Olson EN, Sadek HA.. Transient regenerative potential of the neonatal mouse heart. Science 2011;331:1078–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Payan SM, Hubert F, Rochais F.. Cardiomyocyte proliferation, a target for cardiac regeneration. Biochim Biophys Acta Mol Cell Res 2019;1867:11.8461. [DOI] [PubMed] [Google Scholar]
- 147. Gabisonia K, Prosdocimo G, Aquaro GD, Carlucci L, Zentilin L, Secco I, Ali H, Braga L, Gorgodze N, Bernini F, Burchielli S, Collesi C, Zandonà L, Sinagra G, Piacenti M, Zacchigna S, Bussani R, Recchia FA, Giacca M.. MicroRNA therapy stimulates uncontrolled cardiac repair after myocardial infarction in pigs. Nature 2019;569:418–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Liu S, Martin JF.. The regulation and function of the Hippo pathway in heart regeneration. Wires Dev Biol 2019;8:e335. [DOI] [PubMed] [Google Scholar]
- 149. Heallen T, Morikawa Y, Leach J, Tao G, Willerson JT, Johnson RL, Martin JF.. Hippo signaling impedes adult heart regeneration. Development 2013;140:4683–4690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Moya IM, Halder G.. Hippo-YAP/TAZ signalling in organ regeneration and regenerative medicine. Nat Rev Mol Cell Biol 2019;20:211-226. [DOI] [PubMed] [Google Scholar]
- 151. Lin Z, von Gise A, Zhou P, Gu F, Ma Q, Jiang J, Yau AL, Buck JN, Gouin KA, van Gorp PRR, Zhou B, Chen J, Seidman JG, Wang D-Z, Pu WT.. Cardiac-specific YAP activation improves cardiac function and survival in an experimental murine MI model. Circ Res 2014;115:354–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Del Re DP, Yang Y, Nakano N, Cho J, Zhai P, Yamamoto T, Zhang N, Yabuta N, Nojima H, Pan D, Sadoshima J.. Yes-associated protein isoform 1 (Yap1) promotes cardiomyocyte survival and growth to protect against myocardial ischemic injury. J Biol Chem 2013;288:3977–3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Yagi R, Chen LF, Shigesada K, Murakami Y, Ito Y.. A WW domain-containing yes-associated protein (YAP) is a novel transcriptional co-activator. EMBO J 1999;18:2551–2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Golan-Lagziel T, Lewis YE, Shkedi O, Douvdevany G, Caspi LH, Kehat I.. Analysis of rat cardiac myocytes and fibroblasts identifies combinatorial enhancer organization and transcription factor families. J Mol Cell Cardiol 2018;116:91–105. [DOI] [PubMed] [Google Scholar]
- 155. Porter KE, Turner NA.. Cardiac fibroblasts: at the heart of myocardial remodeling. Pharmacol Ther 2009;123:255–278. [DOI] [PubMed] [Google Scholar]
- 156. Wong WF, Kohu K, Chiba T, Sato T, Satake M.. Interplay of transcription factors in T-cell differentiation and function: the role of Runx. Immunology 2011;132:157–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Voon DC, Hor YT, Ito Y.. The RUNX complex: reaching beyond haematopoiesis into immunity. Immunology 2015;146:523–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Tang X, Sun L, Wang G, Chen B, Luo F.. RUNX1: a regulator of NF-kB signaling in pulmonary diseases. Curr Protein Pept Sci 2018;19:172–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Santos-Zas I, Lemarie J, Tedgui A, Ait-Oufella H.. Adaptive immune responses contribute to post-ischemic cardiac remodeling. Front Cardiovasc Med 2018;5:198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Hulsmans M, Sam F, Nahrendorf M.. Monocyte and macrophage contributions to cardiac remodeling. J Mol Cell Cardiol 2016;93:149–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Wong WF, Kohu K, Nakamura A, Ebina M, Kikuchi T, Tazawa R, Tanaka K, Kon S, Funaki T, Sugahara-Tobinai A, Looi CY, Endo S, Funayama R, Kurokawa M, Habu S, Ishii N, Fukumoto M, Nakata K, Takai T, Satake M.. Runx1 deficiency in CD4+ T cells causes fatal autoimmune inflammatory lung disease due to spontaneous hyperactivation of cells. J Immunol 2012;188:5408–5420. [DOI] [PubMed] [Google Scholar]
- 162. Nakagawa M, Shimabe M, Watanabe-Okochi N, Arai S, Yoshimi A, Shinohara A, Nishimoto N, Kataoka K, Sato T, Kumano K, Nannya Y, Ichikawa M, Imai Y, Kurokawa M.. AML1/RUNX1 functions as a cytoplasmic attenuator of NF-kappaB signaling in the repression of myeloid tumors. Blood 2011;118:6626–6637. [DOI] [PubMed] [Google Scholar]
- 163. Komine O, Hayashi K, Natsume W, Watanabe T, Seki Y, Seki N, Yagi R, Sukzuki W, Tamauchi H, Hozumi K, Habu S, Kubo M, Satake M.. The Runx1 transcription factor inhibits the differentiation of naive CD4+ T cells into the Th2 lineage by repressing GATA3 expression. J Exp Med 2003;198:51–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Lappas M. Runt-related transcription factor 1 (RUNX1) deficiency attenuates inflammation-induced pro-inflammatory and pro-labour mediators in myometrium. Mol Cell Endocrinol 2018;473:61–71. [DOI] [PubMed] [Google Scholar]
- 165. Luo M-C, Zhou S-Y, Feng D-Y, Xiao J, Li W-Y, Xu C-D, Wang H-Y, Zhou T.. Runt-related transcription factor 1 (RUNX1) binds to p50 in macrophages and enhances TLR4-triggered Inflammation and septic shock. J Biol Chem 2016;291:22011–22020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Bartekova M, Radosinska J, Jelemensky M, Dhalla NS.. Role of cytokines and inflammation in heart function during health and disease. Heart Fail Rev 2018;23:733–758. [DOI] [PubMed] [Google Scholar]
- 167. Jones WK, Brown M, Wilhide M, He S, Ren X.. NF-kappaB in cardiovascular disease: diverse and specific effects of a “general” transcription factor? Cardiovasc Toxicol 2005;5:183–202. [DOI] [PubMed] [Google Scholar]
- 168. Leucker TM, Jones SP.. Endothelial dysfunction as a nexus for endothelial cell-cardiomyocyte miscommunication. Front Physiol 2014;5:328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Talman V, Kivela R.. Cardiomyocyte-endothelial cell interactions in cardiac remodeling and regeneration. Front Cardiovasc Med 2018;5:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Perbellini F, Watson SA, Bardi I, Terracciano CM.. Heterocellularity and cellular cross-talk in the cardiovascular system. Front Cardiovasc Med 2018;5:143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Kobayashi K, Maeda K, Takefuji M, Kikuchi R, Morishita Y, Hirashima M, Murohara T.. Dynamics of angiogenesis in ischemic areas of the infarcted heart. Sci Rep 2017;7:7156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Iwatsuki K, Tanaka K, Kaneko T, Kazama R, Okamoto S, Nakayama Y, Ito Y, Satake M, Takahashi S-I, Miyajima A, Watanabe T, Hara T.. Runx1 promotes angiogenesis by downregulation of insulin-like growth factor-binding protein-3. Oncogene 2005;24:1129–1137. [DOI] [PubMed] [Google Scholar]
- 173. Lam JD, Oh DJ, Wong LL, Amarnani D, Park-Windhol C, Sanchez AV, Cardona-Velez J, McGuone D, Stemmer-Rachamimov AO, Eliott D, Bielenberg DR, van Zyl T, Shen L, Gai X, D’Amore PA, Kim LA, Arboleda-Velasquez JF.. Identification of RUNX1 as a mediator of aberrant retinal angiogenesis. Diabetes 2017;66:1950–1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Liu C, Xu D, Xue B, Liu B, Li J, Huang J.. Upregulation of RUNX1 suppresses proliferation and migration through repressing VEGFA expression in hepatocellular carcinoma. Pathol Oncol Res 2019;1-11. [DOI] [PubMed] [Google Scholar]
- 175. ter Elst A, Ma B, Scherpen FJG, de Jonge HJM, Douwes J, Wierenga ATJ, Schuringa JJ, Kamps WA, de Bont ESJM.. Repression of vascular endothelial growth factor expression by the runt-related transcription factor 1 in acute myeloid leukemia. Cancer Res 2011;71:2761–2771. [DOI] [PubMed] [Google Scholar]
- 176. Oka T, Akazawa H, Naito AT, Komuro I.. Angiogenesis and cardiac hypertrophy: maintenance of cardiac function and causative roles in heart failure. Circ Res 2014;114:565–571. [DOI] [PubMed] [Google Scholar]
- 177. Cochain C, Channon KM, Silvestre JS.. Angiogenesis in the infarcted myocardium. Antioxid Redox Signal 2013;18:1100–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Benjamin EJ, Muntner P, Alonso A, Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Chang AR, Cheng S, Das SR, Delling FN, Djousse L, Elkind MSV, Ferguson JF, Fornage M, Jordan LC, Khan SS, Kissela BM, Knutson KL, Kwan TW, Lackland DT, Lewis TT, Lichtman JH, Longenecker CT, Loop MS, Lutsey PL, Martin SS, Matsushita K, Moran AE, Mussolino ME, O’Flaherty M, Pandey A, Perak AM, Rosamond WD, Roth GA, Sampson UKA, Satou GM, Schroeder EB, Shah SH, Spartano NL, Stokes A, Tirschwell DL, Tsao CW, Turakhia MP, VanWagner LB, Wilkins JT, Wong SS, Virani SS; , On behalf of the American Heart Association Council on Epidemiology and Prevention Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics-2019 update: a report from the American Heart Association. Circulation 2019;139:e56–e528. [DOI] [PubMed] [Google Scholar]
- 179. Gheorghiade M, Larson CJ, Shah SJ, Greene SJ, Cleland JGF, Colucci WS, Dunnmon P, Epstein SE, Kim RJ, Parsey RV, Stockbridge N, Carr J, Dinh W, Krahn T, Kramer F, Wahlander K, Deckelbaum LI, Crandall D, Okada S, Senni M, Sikora S, Sabbah HN, Butler J.. Developing new treatments for heart failure: focus on the heart. Circ Heart Fail 2016;9:e002727. [DOI] [PubMed] [Google Scholar]
- 180. Solomon SD, Rizkala AR, Lefkowitz MP, Shi VC, Gong J, Anavekar N, Anker SD, Arango JL, Arenas JL, Atar D, Ben-Gal T, Boytsov SA, Chen C-H, Chopra VK, Cleland J, Comin-Colet J, Duengen H-D, Echeverría Correa LE, Filippatos G, Flammer AJ, Galinier M, Godoy A, Goncalvesova E, Janssens S, Katova T, Køber L, Lelonek M, Linssen G, Lund LH, O’Meara E, Merkely B, Milicic D, Oh B-H, Perrone SV, Ranjith N, Saito Y, Saraiva JF, Shah S, Seferovic PM, Senni M, Sibulo AS, Sim D, Sweitzer NK, Taurio J, Vinereanu D, Vrtovec B, Widimský J, Yilmaz MB, Zhou J, Zweiker R, Anand IS, Ge J, Lam CSP, Maggioni AP, Martinez F, Packer M, Pfeffer MA, Pieske B, Redfield MM, Rouleau JL, Van Veldhuisen DJ, Zannad F, Zile MR, McMurray JJV.. Baseline characteristics of patients with heart failure and preserved ejection fraction in the PARAGON-HF trial. Circ Heart Fail 2018;11:e004962. [DOI] [PubMed] [Google Scholar]
- 181. Bushweller JH. Targeting transcription factors in cancer—from undruggable to reality. Nat Rev Cancer 2019;19:611–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Lambert M, Jambon S, Depauw S, David-Cordonnier MH.. Targeting transcription factors for cancer treatment. Molecules 2018;23:1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Richter LE, Wang Y, Becker ME, Coburn RA, Williams JT, Amador C, Hyde RK.. HDAC1 is a required cofactor of CBFbeta-SMMHC and a potential therapeutic target in inversion 16 acute myeloid leukemia. Mol Cancer Res 2019;17:1241–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Illendula A, Pulikkan JA, Zong H, Grembecka J, Xue L, Sen S, Zhou Y, Boulton A, Kuntimaddi A, Gao Y, Rajewski RA, Guzman ML, Castilla LH, Bushweller JH.. Chemical biology. A small-molecule inhibitor of the aberrant transcription factor CBFbeta-SMMHC delays leukemia in mice. Science 2015;347:779–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Illendula A, Gilmour J, Grembecka J, Tirumala VSS, Boulton A, Kuntimaddi A, Schmidt C, Wang L, Pulikkan JA, Zong H, Parlak M, Kuscu C, Pickin A, Zhou Y, Gao Y, Mishra L, Adli M, Castilla LH, Rajewski RA, Janes KA, Guzman ML, Bonifer C, Bushweller JH.. Small molecule inhibitor of CBFbeta-RUNX binding for RUNX transcription factor driven cancers. EBioMedicine 2016;8:117–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Real PJ, Navarro-Montero O, Ramos-Mejía V, Ayllón V, Bueno C, Menéndez P.. The role of RUNX1 isoforms in hematopoietic commitment of human pluripotent stem cells. Blood 2013;121:5250–5252. United States [DOI] [PubMed] [Google Scholar]
- 187. Sood R, Kamikubo Y, Liu P.. Role of RUNX1 in hematological malignancies. Blood 2017;129:2070–2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Foote K. Characterisation of Cardiac Function and RUNX Expression in Two Separate Models of Cardiac Disease. University of Glasgow; 2012. http://theses.gla.ac.uk/3746/. [Google Scholar]
- 189. Goyama S, Huang G, Kurokawa M, Mulloy JC.. Posttranslational modifications of RUNX1 as potential anticancer targets. Oncogene 2015;34:3483–3492. [DOI] [PubMed] [Google Scholar]
- 190. Blumenthal E, Greenblatt S, Huang G, Ando K, Xu Y, Nimer SD.. Covalent modifications of RUNX proteins: structure affects function. Adv Exp Med Biol 2017;962:33–44. [DOI] [PubMed] [Google Scholar]