

Abstract
Brain glycogen is stored mainly in astrocytes, although neurons also have an active glycogen metabolism. Glycogen has gained relevance as a key player in brain function. In this regard, genetically modified animals have allowed researchers to unravel new roles of this polysaccharide in the brain. Remarkably, mice in which glycogen synthase is abolished in the brain, and thus devoid of brain glycogen, are viable, thereby indicating that the polysaccharide in this organ is not a requirement for survival. While there was growing evidence supporting a role of glycogen in learning and memory, these animals have now confirmed that glycogen participates in these two processes.
The association of epilepsy with brain glycogen has also attracted attention. Analysis of genetically modified mice indicates that the relation between brain glycogen and epilepsy is complex. While the formation of glycogen aggregates clearly underlies epilepsy, as in Lafora Disease (LD), the absence of glycogen also favors the occurrence of seizures.
LD is a rare genetic condition that affects children. It is characterized by epileptic seizures and neurodegeneration, and it develops rapidly until finally causing death. Research into this disease has unveiled new aspects of glycogen metabolism. Animal models of LD accumulate polyglucosan bodies formed by aberrant glycogen aggregates, called Lafora bodies (LBs). The abolition of glycogen synthase (GS) prevents the formation of LBs and the development of LD, thereby indicating that glycogen accumulation underlies this disease and the associated symptoms, and thus establishing a clear relation between the accumulation of glycogen aggregates and the incidence of seizures.
Although it was initially accepted that LBs were essentially neuronal, it is now evident that astrocytes also accumulate polyglucosan aggregates in LD. However, the appearance and composition of these deposits differs from that observed in neurons. Of note, the astrocytic aggregates in LD models show remarkable similarities with corpora amylacea (CA), a type of polyglucosan aggregate observed in the brains of aged mice and humans. The abolition of GS in mice also impedes the formation of CA with age and at the same time prevents the formation of a number of protein aggregates associated with aging. Therefore CA may play a role in age-related neurological decline.
Keywords: Learning, Memory, Long-term potentiation, Epilepsy, Lafora disease, Corpora amylacea, Hypoxia
1. Brain Glycogen in Learning and Epilepsy
Glycogen is produced by GS—the only enzyme able to synthesize glucose polymers in mammals—and degraded by glycogen phosphorylase (GP) in the cytosol and by alpha-glucosidase in the lysosome. Mammals express two isoforms of GS encoded by GYS1 and GYS2. The latter encodes the hepatic isoform (LGS), whose expression is restricted to the liver, while the former encodes the muscle isoform (MGS), which is widely expressed in all organs except the liver (Kaslow et al. 1985). MGS is regulated by phosphorylation at multiple serine residues located in the amino- and carboxy-terminal domains of the enzyme. Phosphorylation by several kinases, including GSK3, induces the inactivation of the enzyme (Roach et al. 1998). Dephosphorylation is facilitated by scaffolding proteins, such as Protein Targeting to Glycogen (PTG), which bring the catalytic subunit of protein phosphatase 1 (PP1) into contact with GS on the glycogen particle, thus causing its dephosphorylation and consequent activation (Vilchez et al. 2007). GS is also allosterically activated by glucose-6-phosphate (G6P) in the brain (Goldberg and O’Toole 1969) and in other tissues (Bouskila et al. 2010; von Wilamowitz-Moellendorff et al. 2013). Glycogen is a branched molecule, which is a crucial property that confers solubility in water. Therefore, the synthesis and degradation of this polysaccharide requires two additional enzymes, namely glycogen branching enzyme, which generates the branching points during the synthesis, and glycogen debranching enzyme (GBE), which removes them during degradation.
Glycogen concentration in the brain is much lower than in the liver or muscle, thus explaining why its function in brain has been largely overlooked. However, a growing body of evidence has accumulated in the last decade indicating that brain glycogen plays an important role in memory formation and learning, in susceptibility to epilepsy, and in many other brain functions. These roles have been elucidated mainly by experiments in which glycogen usage has been impeded by means of specific metabolic inhibitors of GP and more recently confirmed with the use of genetically modified animal models.
The role of glycogen in memory formation was first demonstrated in young chicks with bead discrimination experiments, in which chicks are trained to avoid beads of a specific color (Gibbs et al. 2006). Intracranial injection of 1,4-dideoxy-1,4-imino-d-arabinitol (DAB), a GP inhibitor, or 2-deoxyglucose, which blocks glycolysis, impaired learning of this task. The authors of that study thus concluded that glycogenolysis and aerobic glycolysis are necessary for memory formation (reviewed in (Gibbs 2015)). Other studies performed in rats using DAB injection or knocking down lactate transporters in astrocytes or neurons are consistent with the idea that the mobilization of astrocytic glycogen to generate lactate to feed neurons is critical for memory formation (ANLS hypothesis, see below) (reviewed in (Alberini et al. 2018)).
Epilepsy results from a sudden synchronization of the activity of a group of neurons. During epileptic seizures, neurons and astrocytes dramatically increase their energy consumption. Therefore, the availability of glycogen in this context could be of great importance. In this regard, high glycogen content has been found in biopsies of the brains of epileptic patients (Dalsgaard et al. 2007). Moreover, in the methionine sulfoximine epileptic model, an increase in brain glycogen content has been reported (Phelps 1975; Hevor et al. 1985), and susceptibility of two inbred mouse strains to methionine sulfoximine inversely correlates with their capacity to accumulate glycogen in the brain (Bernard-Helary et al. 2000). Furthermore, the amount of glycogen in epileptic foci is reduced after seizures induced by kainate (Walls et al. 2014) and other epileptogenic agents or situations (Lopez-Ramos et al. 2015). These series of studies have shown a correlation between glycogen content and epilepsy. However, they do not categorically prove that glycogen is directly involved in the disorder.
The use of genetically modified animals in the study of the physiological and pathological aspects of brain glycogen presents clear advantages over methods based on drug treatments, which are hampered by non-specificity and uncontrolled side effects. Furthermore, deletion of specific genes only in the brain of these animals ensures that the results reflect the importance of glycogen exclusively in this organ.. Furthermore and in contrast to pharmacological approaches, genetic manipulation allows researchers to increase the amount of glycogen in specific cell types.
In this regard, the generation of a mouse model that lacks GS specifically in the nervous system (GYS1Nestin-KO), and thus depleted of glycogen in the brain, has shed light on the long-standing question regarding the role of glycogen in this organ (Duran et al. 2013; Lopez-Ramos et al. 2015). The first conclusions drawn from this model is that the animals are viable and that they have a normal lifespan. The same is true for mice lacking GYS1, which are depleted of glycogen in the whole body except the liver (Raben et al. 2001). These results support the notion that brain glycogen is not an absolute requirement for life and that the brain can survive on free glucose. However, GYS1Nestin-KO animals are not normal, as they present deficiencies in learning and memory and an increased susceptibility to epilepsy (Duran et al. 2013; Lopez-Ramos et al. 2015).
GYS1Nestin-KO mice show a significant deficit in capacity to learn an instrumental conditioning task (Fig. 1a). They also show deficits in concomitant activity-dependent changes in synaptic strength, the most striking of which is the almost complete absence of Long-Term Potentiation (LTP), the long-lasting increase in the strength of excitatory synapses after a high-frequency stimulation protocol (Duran et al. 2013) (Fig. 1b). LTP is believed to be the cellular process that underlies information storage within neural systems (Martinez and Derrick 1996; Gruart et al. 2006). These observations clearly demonstrate the key role of brain glycogen in the proper and timed acquisition of relatively difficult associative learning tasks. The characterization of GYS1Nestin-KO mice has also contributed to shedding light on the relation between brain glycogen and epilepsy. Animals lacking brain glycogen are more susceptible to hippocampal seizures after the administration of kainate, a drug widely used to induce epilepsy (Lopez-Ramos et al. 2015) (Fig. 2). This observation thus indicates that glycogen availability contributes to the maintenance of the proper equilibrium between excitatory and inhibitory neurotransmission.
Fig. 1.
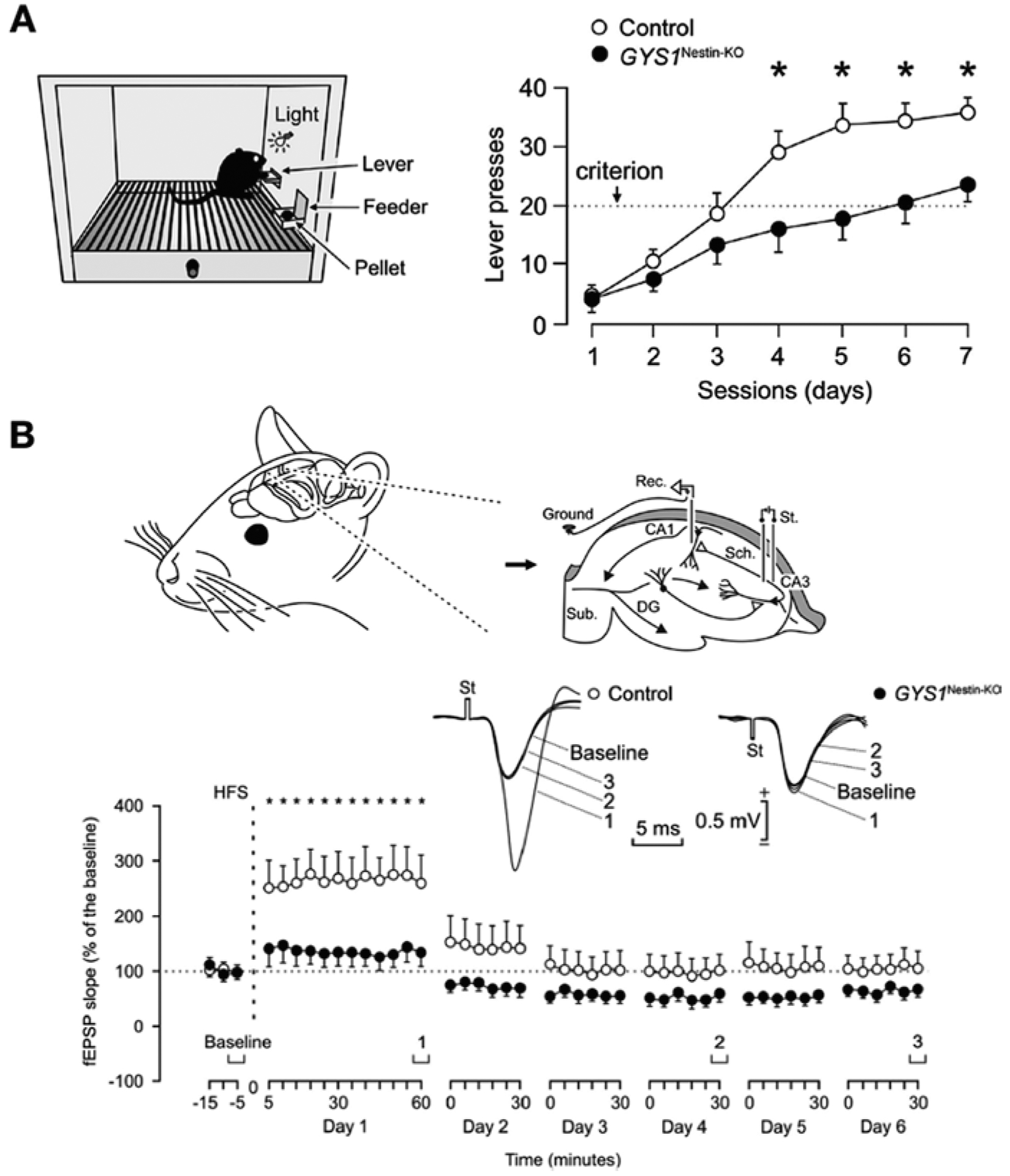
Adapted from (Duran et al. 2013). (a) Impaired performance of GYS1Nestin-KO mice in an operant conditioning task. Mice were trained in a Skinner box to press a lever to obtain a food pellet (left). Lever presses in the first 7 days of training of task (right). Dotted line corresponds to criterion. (b) Animals were chronically implanted with stimulating electrodes in the hippocampal Schaffer collaterals and with a recording electrode in the ipsilateral pyramidal CA1 area. An extra wire was attached to the bone as ground (Top) (DG, dentate gyrus; Sub., subiculum). Time course of long-term potentiation (LTP) evoked in the CA3-CA1 synapse after a high-frequency stimulation (HFS) session (bottom) (mean ± s.e.m. fEPSP slopes given as a percentage of values collected during baseline recordings (100%)). Representative examples of fEPSPs collected at the indicated times are plotted at the top. ∗Statistically significant (P < 0.05) differences between the two groups
Fig. 2.
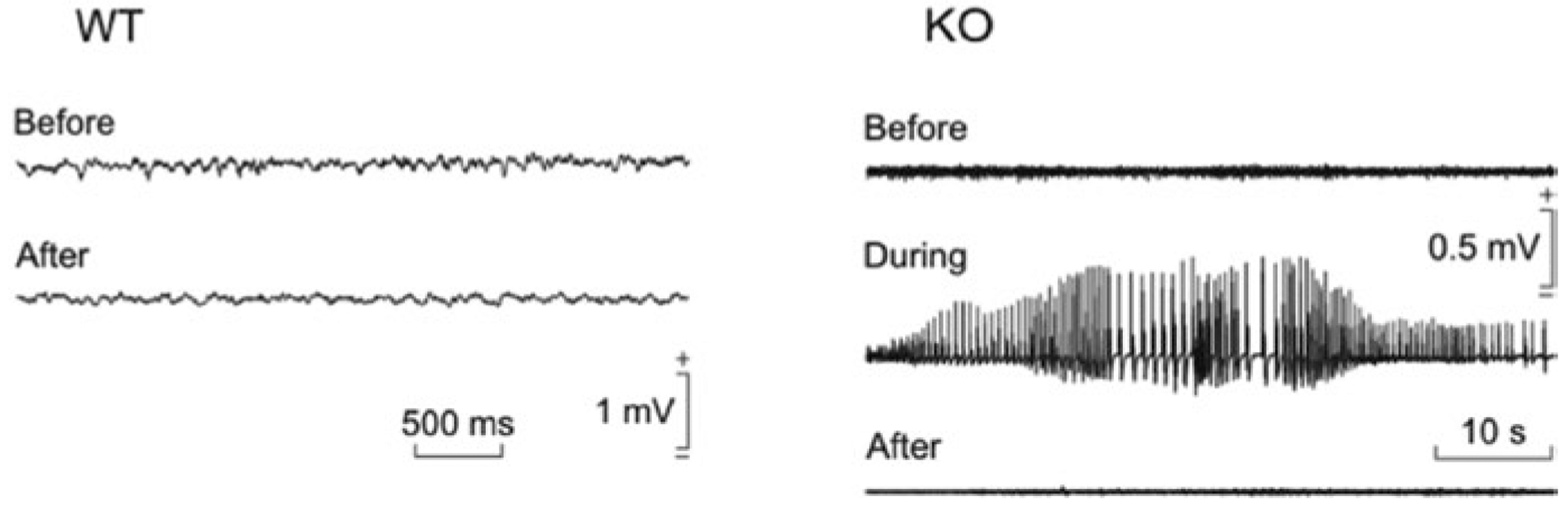
Adapted from (Lopez-Ramos et al. 2015). Effects of kainate injection on spontaneous electric potential generated by neurons (Local Field Potentials, LFPs) recorded in the hippocampus of behaving mice. Representative examples of LFPs recorded from a wild-type (WT) and a GYS1Nestin-KO (KO) mouse before and 30 min after a kainate injection (8 mg/kg,i.p.)
2. Astrocytes Vs. Neurons
A further complication in the study of brain glycogen arises from the fact that the polysaccharide is present mainly in astrocytes. Therefore, in brain function, all the physiological roles of glycogen as an energy reserve or contributing to the consolidation of memory have been essentially attributed to astrocytic glycogen. It has been hypothesized that astrocytes accumulate glycogen for the benefit of neurons. However, since astrocytes do not have glucose-6-phosphatase they cannot release free glucose to be taken up by neurons. Therefore, the question as to how neurons can benefit from glycogen stored in astrocytes is puzzling. A simple explanation is that astrocytes use their own glycogen in times of high energy demand, sparing interstitial glucose for neurons. A more sophisticated mechanism implies that astrocytes degrade glycogen to lactate, which is released, and taken up and consumed by neurons—a hypothesis known as the astrocyte-neuron lactate shuttle (ANLS). This hypothesis continues to be debated (reviewed in (Waitt et al. 2017)).
Another difficulty encountered when studying brain glycogen arises from its rapid degradation in post-mortem conditions. This explains why glycogen is difficult to detect in cell types with a low glycogen content, such as neurons, and why its role in these cells has been overlooked (Hertz and Chen 2018). Localization studies performed in microwave-fixed brains (a procedure that stops enzymatic activity immediately and thus preserves the metabolic state) indicate that neurons also contain glycogen, although at much lower concentrations than astrocytes (Oe et al. 2016). Furthermore, the presence of glycogen deposition in neurons in certain conditions such as LD (see below) demonstrates that these cells do synthesize glycogen. In this regard, it has been shown that primary cultured neurons accumulate glycogen, although in small amounts (Fig. 3a), and express GS and GP, thus indicating that these cells have the capacity to synthesize and degrade this polysaccharide (Vilchez et al. 2007; Saez et al. 2014). The availability of glycogen in these neurons is relevant for their tolerance to hypoxia (Fig. 3b). Therefore, a pressing question was to dissect the relative roles of neuronal and astrocyte glycogen in learning and epilepsy. The characterization of a mouse model depleted of glycogen specifically in neurons of the forebrain (GYS1Camk2a-KO) involved in memory and learning has shed light on this long-standing issue (Duran et al. 2019). As in the case of GYS1Nestin-KO animals, in which GS is depleted from both neurons and astrocytes, GYS1Camk2a-KO mice showed decreased LTP evoked in the hippocampal CA3-CA1 synapse and a significant deficiency in the acquisition of an instrumental learning task (Fig. 3c). In contrast, they did not present the greater susceptibility to hippocampal seizures and myoclonus observed in the GYS1Nestin-KO model. These results unequivocally demonstrate the presence of an active glycogen metabolism in neurons in vivo and its fundamental role in the proper acquisition of new motor and cognitive abilities and in the changes in synaptic strength underlying such acquisition.
Fig. 3.

(a) Adapted from (Saez et al. 2014). Neurons contain glycogen, which is mobilized in hypoxia. Glycogen content was determined in control (Normoxia, exposed to environmental 21% O2) and treated neurons (Hypoxia, exposed to 1% O2) and represents the mean ± s.e.m. (n.7). ∗∗∗P < 0.001 versus Normoxia. (b) Adapted from (Saez et al. 2014). Glycogen synthase (GS) has a protective role in neurons under hypoxia. Death fold change after increasing exposure to hypoxia in GS wild-type (WT) (black) and knockout (KO) (white) neurons. (c) Adapted from (Duran et al. 2019). Performance of control and GYS1Camk2a-KO mice was studied in an operant conditioning task. Mice were placed in a Skinner box and trained to press a lever to obtain a pellet only when a light bulb was switched on. Control mice outperformed GYS1Camk2a-KO mice [F(9,225) = 2.82; P = 0.01]
A conclusion drawn from these studies is that neuronal glycogen is responsible for some of the roles previously attributed exclusively to astrocytic glycogen (Hertz and Chen 2018). Therefore the relevance of neuronal glycogen for brain function should be reconsidered.
3. Lafora Disease
A rare genetic disease, Progressive Myoclonic Epilepsy 2 (EPM2), also known as Lafora disease (LD), has greatly contributed to enhancing our knowledge of glycogen metabolism in the brain and its relation to epilepsy. This disease is an invariably fatal epilepsy that affects both genders equally. Its onset occurs during adolescence, in apparently healthy children, causing absence seizures and/or visual auras. Patients then typically experience generalized tonic-clonic seizures and insidious decline in cognitive function. LD at onset is difficult to distinguish from idiopathic generalized epilepsies. Myoclonic seizures, staring spells, and generalized convulsions follow and escalate over time. LD patients also develop epileptic and non-epileptic visual hallucinations (Gentry et al. 2018; Nitschke et al. 2018).
LD is caused by mutations in either NHLRC1, which encodes malin, an E3-ubiquitin ligase, or EPM2A, which encodes laforin, a serine-threonine phosphatase. Patients carrying loss-of-function mutations in either of these two genes are indistinguishable. Genetically modified animal models, i.e. the malin knockout (malinKO) (DePaoli-Roach et al. 2010; Valles-Ortega et al. 2011) and the laforin knockout mice (Ganesh et al. 2002), are ideal systems in which to study the disease.
The hallmark of LD is the presence of large inclusions of glycogen aggregates known as polyglucosan bodies (PGBs) or, more specifically, Lafora bodies (LBs), in the brain (Fig. 4) and in other tissues such as muscle and heart (Cavanagh 1999). LBs were traditionally considered to be neuronal inclusions that occur concomitantly with neurodegeneration and epilepsy and that are related to the inexorable worsening of the condition until death in early adulthood (Machado-Salas et al. 2012). However, studies with LD mouse models have demonstrated the presence of PGB also in astrocytes (Valles-Ortega et al. 2011; Auge et al. 2018; Rubio-Villena et al. 2018). Thus, malinKO mice present two types of PGB in the brain, one affecting neurons and the other astrocytes. These two types differ in size, distribution and presence of specific neo-epitopes that are recognized by natural antibodies present in the sera of mammals (Auge et al. 2018). Interestingly, astrocytic, but not neuronal, PGBs also appear in aged control animals, in animal models of accelerated aging and in animals with enhanced glycogen synthesis in the brain (Auge et al. 2018). This observation indicates that the absence of malin triggers the formation of LBs in neurons and enhances the formation of PGBs in astrocytes. Therefore, only the PGBs present in the neurons are specific to LD, while those in astrocytes can be found in other conditions. Given their similarity to corpora amylacea (see below), these astrocytic PGBs could be referred to as corpora amylacea-like granules (CAL) (Fig. 4a).
Fig. 4.

Lafora bodies in malinKO brain. (a) GS immunostaining of control and malinKO brains. Arrows indicate clusters of CAL inclusions, arrowheads indicate nLBs. (b) Adapted from (Valles-Ortega et al. 2011). Electron microscopy images of CA1 region. Micrographs depict the presence of LBs and glycogen granules in dendrites (a1, a2) and in astrocytes (c1). ∗, Lafora Body; black arrowhead: postsynaptic density; B: synaptic bouton; DS: dendritic spine. Scale bars are 5 μm in c1 and 0.5 μm in a1 and a2
The mechanism by which these aggregates form remains unclear. However, given that laforin is able to remove phosphate from glycogen, it has been proposed that the accumulation of phosphorylated glycogen is the main underlying factor involved. This increased phosphate would underlie a change in glycogen structure, decreasing its degree of branching and making it less soluble (Gentry et al. 2009). However, this does not explain the similar accumulation of LBs in the absence of malin, a situation marked by an increase in laforin (Duran et al. 2014). Furthermore, the phosphatase activity of laforin does not appear to be required to prevent LD (Gayarre et al. 2014), and glycogen hyperphosphorylation does not cause the formation of LBs (Nitschke et al. 2017). As mentioned above, while CAL granules are also formed in aging and other conditions in which glycogen synthesis is increased, neuronal LBs appear only in models lacking malin or laforin. However, the presence of high numbers of CAL granules in the malinKO mouse model suggests that the lack of malin alters glycogen metabolism not only in neurons but also in astrocytes. Nevertheless, there is an important difference. While the absence of malin (or laforin) is a requirement to trigger the formation of neuronal LBs, it is not a prerequisite to generate astrocytic CAL granules. However, the absence of malin clearly enhances the formation of these aggregates.
Since the malin–laforin complex had been described to have functions other than that of regulating glycogen synthesis, such as the control of autophagy, the role of glycogen accumulation in the etiopathology of LD was a matter of debate. In this regard, autophagy impairment was hypothesized to underlie neurodegeneration in LD (Criado et al. 2012; Knecht et al. 2012). A first indication that glycogen accumulation is the direct cause of the neurodegeneration and functional impairments seen in LD was that the deletion of PTG prevents the manifestation of LD in the malinKO model (Turnbull et al. 2011). These animals showed reduced glycogen accumulation and the resolution of neurodegeneration and myoclonic epilepsy. However, since PTG is a regulatory protein of PP1, which has many targets, it could not be ruled out that the observed effects were due to changes in the phosphorylation of other targets.
A direct demonstration that glycogen accumulation induces neurodegeneration came from experiments in which a mutant form of MGS in which 9 regulatory serine residues were mutated to alanine, rendering MGS that is not unactivatable by phosphorylation (MGS-9A), was specifically expressed in Purkinje neurons of mice (Duran et al. 2012). Glycogen accumulation in these neurons resulted in a time-dependent loss of these cells by apoptosis and the associated motor impairment (Fig. 5a). Similarly, overexpression of MGS-9A in fly (Drosophila) neurons leads to the accumulation of glycogen, reduced lifespan and locomotion defects (Fig. 5b).
Fig. 5.

Adapted from (Duran et al. 2012). (a) Progressive loss of Purkinje neurons in animals overexpressing MGS-9A in these cells (mMGS-9APcp2). Cerebellar sections of p15-p75 mMGS-9APcp2 animals labelled to visualize Purkinje cells by Calbindin protein expression (brown). Sections were also labelled with haematoxylin (blue). Scale bars, 500 μm (left) and 30 μm (right). (b) Survival assay of control (GFAP) or flies overexpressing wild type MGS (hMGS-wt), MGS-9A (hMGS-9A) or a catalytically inactive form of MGS-9A (hMGS-9A(D)) specifically in neurons
However, final evidence of the causal role of glycogen accumulation in the etiopathology of LD was provided by experiments that showed that not only the double laforin-MGS KO (Pederson et al. 2013) and double malin-MGS KO animals (Duran et al. 2014) do not show the accumulation of brain glycogen (Fig. 6a, b) but also that the neurological alterations inherent to LD are rescued. In this regard, a malin-deficient mouse that cannot synthesize glycogen in the brain (malinKO + MGSKO) does not show the increase in markers of neurodegeneration seen in the malinKO model (Fig. 6c). Furthermore, the partial reduction of MGS expression by deleting only one of the GYS1 alleles (malinKO + MGShet) is sufficient to decrease the levels of glycogen (Fig. 6b) and the neurodegeneration markers (Fig. 6c) almost to the levels of control animals (Duran et al. 2014). The increase in LTP of hippocampal synapses and the susceptibility to kainate-induced epilepsy seen in the malinKO model are also reversed in these malinKO + MGShet animals (Fig. 6d). These observations thus indicate that the malinKO mice are rescued from their neurological dysfunctions when GS is diminished. This finding gains relevance in the context of the treatment of LD, as it implies that partial inhibition of GS activity may be sufficient to prevent the progression of the disease. This notion has fueled interest in identifying inhibitors of GS or oligonucleotides able to block its expression in the CNS, as potential treatments for the disease.
Fig. 6.
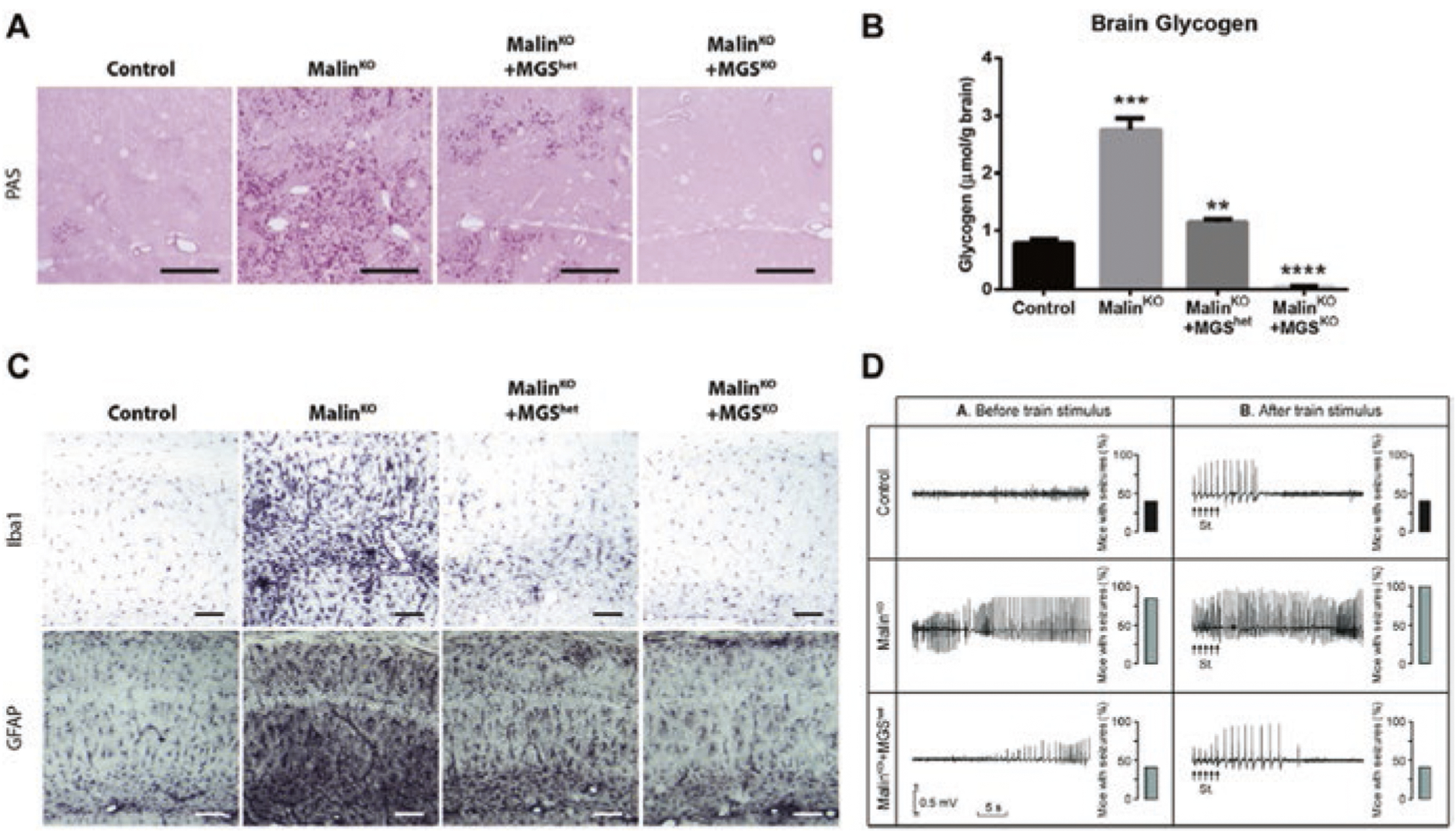
Adapted from (Duran et al. 2014). (a) MGS is indispensable for the formation of LBs. PAS staining shows that LBs accumulated in malinKO brains are absent in malinKO + MGSKO and greatly reduced in malinKO + MGShet brains. (b) Graph represents biochemical determination of brain glycogen concentration. MalinKO + MGSKO brains are devoid of glycogen, and the concentration of the polysaccharide is clearly reduced in malinKO + MGShet brains. (c) Neurodegeneration is rescued in malinKO mice that cannot synthesize glycogen in the brain. GFAP and Iba1 stainings, as markers of neurodegeneration, show increased staining in the malinKO mice with respect to the controls, thus indicating neurodegeneration. This staining is normalized in the malinKO + MGSKO animals and partially normalized in the malinKO + MGShet animals. (d) Increased susceptibility to kainate-induced epilepsy is also dependent on MGS. Representative hippocampal local field potential (LFP) recordings were carried out 30 min after kainic acid injection before and after train stimulation. The percentage (%) of mice presenting seizures within each group is shown to the right of the LFP recordings. MalinKO animals show enhanced susceptibility, which is normalized in malinKO + MGShet animals
At this point, it is interesting to draw attention to a paradox, namely that both the absence and the accumulation of brain glycogen (MGSNestin-KO and malinKO, respectively) induce increased susceptibility to epilepsy (Duran et al. 2014; Lopez-Ramos et al. 2015). This paradox could be attributed to non-degradable nature of the glycogen accumulated in LD, thus bringing about a similar situation to that found when there is a lack of glycogen. In this regard, it has been proposed that abnormal glycogen structure contributes to susceptibility to epilepsy (DiNuzzo et al. 2015). In contrast, while the accumulation of brain glycogen in malinKO brains generates an increase in LTP in the CA3-CA1 synapse, the absence of brain glycogen in the MGSNestin-KO model results in the loss of LTP in the same synapse (Duran et al. 2013, 2014). Thus, in this case, the increase in LTP in malinKO mice is not a consequence of an incapacity to mobilize glycogen.
As mentioned before, autophagy impairment had been hypothesized to underlie neurodegeneration in LD. Interestingly, autophagy impairment is rescued in malinKO + MGSKO animals, and partially rescued in the malinKO + MGShet animals (Fig. 7a). These observations thus demonstrate that the accumulation of glycogen is not a consequence of autophagy impairment, but rather the cause of it, and show that glycogen accumulation precedes autophagy and not vice versa. To reinforce this idea, other models of glycogen accumulation in the CNS in which malin levels have not been modified (namely animals overexpressing 9A-MGS or PTG) also show impaired autophagy (Duran et al. 2014) (Fig. 7b).
Fig. 7.

Adapted from (Duran et al. 2014). (a) Analysis of autophagy markers. Accumulation of p62 and NBR1 in malinKO brains was dependent on the expression of MGS. Brain extracts from 11-month-old mice were analyzed by western blot with antibodies against p62 and NBR1. Actin was used as loading control. (b) Brain extracts from 3-week-old 9A-MGSOE and littermate controls were analyzed as in a
The extensive neuronal loss and severe neuropathological phenotypes observed in patients and animal models of LD suggest that neurons are particularly vulnerable to excess glycogen accumulation (Delgado-Escueta 2007; Valles-Ortega et al. 2011; Duran et al. 2014; Lopez-Gonzalez et al. 2017). This evidences the need for tight control of glycogen synthesis in neurons since an excess of the polysaccharide results in the death of this cell population by apoptosis (Vilchez et al. 2007; Duran et al. 2012). However, the aforementioned accumulation of glycogen in astrocytes may also contribute to the physiopathology of LD.
Nevertheless, LD is not the only condition in which glycogen is deposited in an abnormally manner in nervous tissue. In other diseases like adult polyglucosan body disease and diabetic neuropathy, but also in normal aging, glycogen accumulates in the form of PGBs in neural tissue.
4. Adult Polyglucosan Body Disease
Adult Polyglucosan Body Disease (APBD) is a rare progressive neurodegenerative disorder caused by mutations in (GBE). The lack of GBE produces abnormal glycogen with low solubility due to the lack of branching, thereby leading to the accumulation of PGBs. Loss-of-function mutations of the enzyme cause glycogen storage disease type IV, also known as Andersen disease, a condition that affects neonates and children. In this condition, glycogen accumulates most severely in liver, cardiac and muscle cells, resulting in cirrhosis and death within 5 years. APBD, the adult-onset form of the disease, results from the partial loss of activity of the enzyme (Bruno et al. 1993). It is characterized by the deposition of PGBs in nervous tissue, which induces severe leukodystrophy and atrophy of the spine and medulla (Lossos et al. 1998). The accumulation of glycogen in astrocytes is sufficient to cause the disease (Dainese et al. 2013).
5. Diabetic Neuropathy and Diabetic Retinopathy
Diabetic neuropathy and diabetic retinopathy are two common complications of diabetes mellitus, affecting approximately 60% and 30% of diabetic patients respectively (Vincent and Feldman 2004; Simo et al. 2014). Peripheral nerves are surrounded by the perineurium, which, although acting as a diffusion barrier, is not as efficient as the blood–brain barrier. Consequently, peripheral nerves are in a less well-regulated microenvironment compared to the CNS. In normal conditions, nerve glycogen is present mainly in Schwann cells. However, glycogen accumulation has been described in the axons of peripheral nerves in diabetic patients (Yagihashi and Matsunaga 1979; Mancardi et al. 1985) and in several animal models of diabetes (Moore et al. 1981; Zotova et al. 2008). In fact, intra-axonal glycogen deposition is one of the parameters used to assess diabetic neuropathy (Orloff and Greenleaf 1990). This glycogen is accumulated in large spherical deposits similar to LBs (Powell et al. 1977). In the retina, glycogen is present in Müller cells and in various types of neuron, especially those of the inner retina (Rungger-Brandle et al. 1996). Although the retina has one of the highest metabolic demands of any tissue, retinal oxygen tension is relatively low, especially in the inner layers. This conditioning might explain why retinal neurons accumulate more glycogen than brain neurons in normal conditions. Interestingly, several reports describe an increased deposition of glycogen in the retina of animal models of diabetic retinopathy (Sosula et al. 1974; Sanchez-Chavez et al. 2008; Osorio-Paz et al. 2012). In the light of the discovery of the toxic role of glycogen accumulation in LD, it is reasonable to hypothesize that glycogen accumulation in the nerves and retinas of diabetic patients could contribute to the physiopathology of diabetic neuropathy and diabetic retinopathy.
6. Aging
The accumulation of PGBs, known as corpora amylacea (CA), has also been observed in the aged human brain (Cavanagh 1999). CA share multiple histological and biochemical characteristics with LBs, including a composition of insoluble, poorly branched polysaccharide, resistance to digestion by amylase, and minor protein content.
Remarkably, the significance of PGB accumulation with regard to aging has been largely overlooked, and only a few authors have proposed that CA have a relevant role in neurodegeneration (Singhrao et al. 1993). Although human brain CA are formed mainly by polyglucosan, the presence of waste elements is a recurrent feature of these structures. This observation suggests that that they are involved in trapping and sequestering potentially hazardous products (Cavanagh 1999; Pirici et al. 2014; Rohn 2015). CA contain a number of neo-epitopes—specific epitopes that are not present in healthy brain structures but appear in situations of cellular stress and tissue damage (Binder 2010; Auge et al. 2017). The neo-epitopes in CA are recognized by natural IgM antibodies, thus revealing the potential role of the natural immune system in the removal of these aggregates (Auge et al. 2017).
In the same way in which CA accumulate with age in the human brain, CAL granules progressively appear in the aging mouse brain. These granules are present in a wide range of mouse strains but are particularly abundant in the senescence-accelerated mouse prone 8 (SAMP8) model (Manich et al. 2016). This model is a non-genetically modified strain of mice with an accelerated aging process and it shares characteristics with aged humans, such as a reduced lifespan, lordosis, hair loss, and reduced physical activity (Takeda 2009). In these animals, CAL granules appear in various regions of the brain as early as 3 months of age, and their number increases faster than in other strains (Jucker et al. 1994a, b; Del Valle et al. 2010). Remarkably, glycogen also accumulates in the brain of aged flies (Drosophila). This has relevant consequences for neurological function in vivo. If endogenous glycogen synthase (dGS) is knocked down by RNAi using neuron-specific drivers, glycogen deposition in the brain is reduced. Adult males expressing dGS-RNAi have a significantly longer median (log-rank test) and maximum lifespan than driver-only-expressing controls (Fig. 8a). Young males expressing dGS-RNAi show a normal climbing response, with no significant change in climbing speed relative to control flies. In contrast, aged males showed a higher average and maximum climbing speed than controls (Fig. 8b). Interestingly, these changes are not observed in female flies.
Fig. 8.
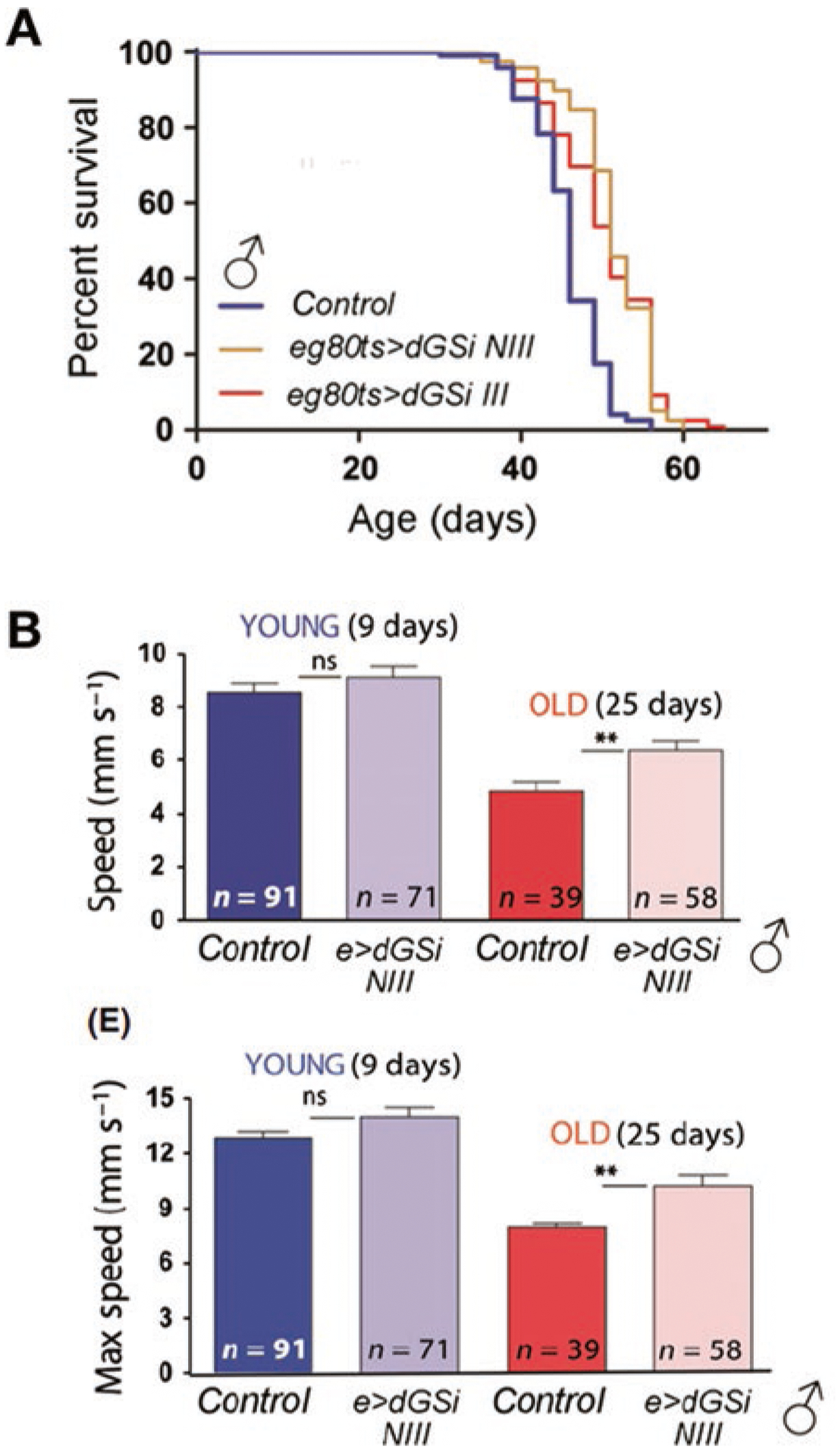
Adapted from (Sinadinos et al. 2014). Functional consequences of reduced GS in the nervous system of aging Drosophila. (a) Survival curves of control and flies with reduced expression of GS (eg80ts > dGSi NIII and eg80ts > dGSi III). (b) Average (top graph) maximum (bottom graph) climbing speed of young (9 d, blue) and old (25 d, red) male control flies and flies with reduced expression of GS (e > dGSi NIII)
As indicated earlier, neo-epitopes are present on CAL granules in mouse brains and on CA in human brains (Manich et al. 2016; Auge et al. 2017). Although the composition of CA must be re-examined due to the possible false positive staining in immunohistochemical studies (Auge et al. 2017), there is wide consensus that CA contain waste products (Cavanagh 1999; Pirici et al. 2014) and ubiquitin (Cisse and Schipper 1995; Wilhelmus et al. 2011; Pirici et al. 2014). It has been proposed that the presence of neo-epitopes on CA is related to the removal of these bodies via the natural immune system after their extrusion (Auge et al. 2017). In this regard, CAL granules contain p62 protein, which has a ubiquitin-binding domain and is involved in the sequestration of ubiquitinated proteins and organelles (Liu et al. 2016). As expected, CAL granules are not present in any brain region of MGSKO mice, thereby confirming that glycogen is an essential component of the aggregates. Interestingly, aggregates of alpha-synuclein, hsp70, and ubiquitin, which normally can all be found in the brains of aged controls, are also absent in MGSKO mouse brains (Sinadinos et al. 2014). This observation indicates that glycogen is involved in the formation of protein-based aggregates, such as age-dependent accumulations of aggregation-prone or stress-response proteins. The increased accumulation of these markers in the brains of young malinKO mice is consistent with this proposal.
Collectively, these findings suggest that the progressive accumulation of glycogen aggregates in aged brains is a widespread phenomenon that contributes to neurological decline and that mutations in malin and laforin in LD dramatically increase the rate of this process. These observations thus point to glycogen synthesis as a promising target for reducing the age-related deterioration of the nervous system.
In summary, new evidence, mainly from genetically modified animals, has shed light on the role of glycogen in both astrocytes and neurons. It is now acknowledged that glycogen is required for normal functioning of the brain, but that its overaccumulation induces neurodegeneration, as demonstrated in Lafora disease.
Abbreviations
- ANLS
Astrocyte-neuron lactate shuttle
- APBD
Adult polyglucosan body disease
- CAL
Corpora amylacea-like
- DAB
1,4-dideoxy-1,4-imino-d-arabinitol
- EPM2
Progressive myoclonic epilepsy 2
- GBE
Glycogen branching enzyme
- GP
Glycogen phosphorylase
- GS
Glycogen synthase
- HFS
High-frequency stimulus
- LBs
Lafora bodies
- LD
Lafora disease
- LFP
Local field potential
- LGS
Liver glycogen synthase
- LTP
Long-term potentiation
- MGS
Muscle glycogen synthase
- PGBs
Polyglucosan bodies
- PP1
Protein phosphatase 1
- PTG
Protein targeting to glycogen
- SAMP8
Senescence accelerated mouse prone 8
Contributor Information
Jordi Duran, Institute for Research in Biomedicine (IRB Barcelona), Barcelona Institute of Science and Technology, Barcelona, Spain; Centro de Investigación Biomédica en Red de Diabetes y Enfermedades Metabólicas Asociadas (CIBERDEM), Madrid, Spain.
Agnès Gruart, Division of Neurosciences, Pablo de Olavide University, Seville, Spain.
Juan Carlos López-Ramos, Division of Neurosciences, Pablo de Olavide University, Seville, Spain.
José M. Delgado-García, Division of Neurosciences, Pablo de Olavide University, Seville, Spain
Joan J. Guinovart, Institute for Research in Biomedicine (IRB Barcelona), Barcelona Institute of Science and Technology, Barcelona, Spain Centro de Investigación Biomédica en Red de Diabetes y Enfermedades Metabólicas Asociadas (CIBERDEM), Madrid, Spain; Department of Biochemistry and Molecular Biomedicine, University of Barcelona, Barcelona, Spain.
References
- Alberini CM, Cruz E et al. (2018) Astrocyte glycogen and lactate: new insights into learning and memory mechanisms. Glia 66(6):1244–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auge E, Cabezon I et al. (2017) New perspectives on corpora amylacea in the human brain. Sci Rep 7:41807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auge E, Pelegri C et al. (2018) Astrocytes and neurons produce distinct types of polyglucosan bodies in Lafora disease. Glia 66:2094–2107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard-Helary K, Lapouble E et al. (2000) Correlation between brain glycogen and convulsive state in mice submitted to methionine sulfoximine. Life Sci 67(14):1773–1781 [DOI] [PubMed] [Google Scholar]
- Binder CJ (2010) Natural IgM antibodies against oxidation-specific epitopes. J Clin Immunol 30(Suppl 1):S56–S60 [DOI] [PubMed] [Google Scholar]
- Bouskila M, Hunter RW et al. (2010) Allosteric regulation of glycogen synthase controls glycogen synthesis in muscle. Cell Metab 12(5):456–466 [DOI] [PubMed] [Google Scholar]
- Bruno C, Servidei S et al. (1993) Glycogen branching enzyme deficiency in adult polyglucosan body disease. Ann Neurol 33(1):88–93 [DOI] [PubMed] [Google Scholar]
- Cavanagh JB (1999) Corpora-amylacea and the family of polyglucosan diseases. Brain Res Brain Res Rev 29(2–3):265–295 [DOI] [PubMed] [Google Scholar]
- Cisse S, Schipper HM (1995) Experimental induction of corpora amylacea-like inclusions in rat astroglia. Neuropathol Appl Neurobiol 21(5):423–431 [DOI] [PubMed] [Google Scholar]
- Criado O, Aguado C et al. (2012) Lafora bodies and neurological defects in Malin-deficient mice correlate with impaired autophagy. Hum Mol Genet 21(7):1521–1533 [DOI] [PubMed] [Google Scholar]
- Dainese L, Monin ML et al. (2013) Abnormal glycogen in astrocytes is sufficient to cause adult polyglucosan body disease. Gene 515(2):376–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalsgaard MK, Madsen FF et al. (2007) High glycogen levels in the hippocampus of patients with epilepsy. J Cereb Blood Flow Metab 27(6):1137–1141 [DOI] [PubMed] [Google Scholar]
- Del Valle J, Duran-Vilaregut J et al. (2010) Early amyloid accumulation in the hippocampus of SAMP8 mice. J Alzheimers Dis 19(4):1303–1315 [DOI] [PubMed] [Google Scholar]
- Delgado-Escueta AV (2007) Advances in lafora progressive myoclonus epilepsy. Curr Neurol Neurosci Rep 7(5):428–433 [DOI] [PubMed] [Google Scholar]
- DePaoli-Roach AA, Tagliabracci VS et al. (2010) Genetic depletion of the malin E3 ubiquitin ligase in mice leads to lafora bodies and the accumulation of insoluble laforin. J Biol Chem 285(33):25372–25381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiNuzzo M, Mangia S et al. (2015) Does abnormal glycogen structure contribute to increased susceptibility to seizures in epilepsy? Metab Brain Dis 30(1):307–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran J, Tevy MF et al. (2012) Deleterious effects of neuronal accumulation of glycogen in flies and mice. EMBO Mol Med 4(8):719–729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran J, Saez I et al. (2013) Impairment in long-term memory formation and learning-dependent synaptic plasticity in mice lacking glycogen synthase in the brain. J Cereb Blood Flow Metab 33(4):550–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran J, Gruart A et al. (2014) Glycogen accumulation underlies neurodegeneration and autophagy impairment in Lafora disease. Hum Mol Genet 23(12):3147–3156 [DOI] [PubMed] [Google Scholar]
- Duran J, Gruart A et al. (2019) Lack of Neuronal Glycogen Impairs Memory Formation and Learning-Dependent Synaptic Plasticity in Mice. Frontiers in Cellular Neuroscience 13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganesh S, Delgado-Escueta AV et al. (2002) Targeted disruption of the Epm2a gene causes formation of Lafora inclusion bodies, neurodegeneration, ataxia, myoclonus epilepsy and impaired behavioral response in mice. Hum Mol Genet 11(11):1251–1262 [DOI] [PubMed] [Google Scholar]
- Gayarre J, Duran-Trio L et al. (2014) The phosphatase activity of laforin is dispensable to rescue Epm2a−/− mice from Lafora disease. Brain 137(Pt 3):806–818 [DOI] [PubMed] [Google Scholar]
- Gentry MS, Dixon JE et al. (2009) Lafora disease: insights into neurodegeneration from plant metabolism. Trends Biochem Sci 34(12):628–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentry MS, Guinovart JJ et al. (2018) Lafora disease offers a unique window into neuronal glycogen metabolism. J Biol Chem 293(19):7117–7125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs ME (2015) Role of Glycogenolysis in memory and learning: regulation by noradrenaline, Serotonin and ATP. Front Integr Neurosci 9:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs ME, Anderson DG et al. (2006) Inhibition of glycogenolysis in astrocytes interrupts memory consolidation in young chickens. Glia 54(3):214–222 [DOI] [PubMed] [Google Scholar]
- Goldberg ND, O’Toole AG (1969) The properties of glycogen synthetase and regulation of glycogen biosynthesis in rat brain. J Biol Chem 244(11):3053–3061 [PubMed] [Google Scholar]
- Gruart A, Munoz MD et al. (2006) Involvement of the CA3-CA1 synapse in the acquisition of associative learning in behaving mice. J Neurosci 26(4):1077–1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertz L, Chen Y (2018) Glycogenolysis, an astrocyte-specific reaction, is essential for both astrocytic and neuronal activities involved in learning. Neuroscience 370:27–36 [DOI] [PubMed] [Google Scholar]
- Hevor TK, Delorme P et al. (1985) Glycogen content and fructose-1, 6-biphosphatase activity in methionine sulfoximine epileptogenic mouse brain and liver after protein synthesis inhibition. Neuropathol Appl Neurobiol 11(2):129–139 [DOI] [PubMed] [Google Scholar]
- Jucker M, Walker LC et al. (1994a) Age-related fibrillar deposits in brains of C57BL/6 mice. A review of localization, staining characteristics, and strain specificity. Mol Neurobiol 9(1–3):125–133 [DOI] [PubMed] [Google Scholar]
- Jucker M, Walker LC et al. (1994b) Age-related deposition of glia-associated fibrillar material in brains of C57BL/6 mice. Neuroscience 60(4):875–889 [DOI] [PubMed] [Google Scholar]
- Kaslow HR, Lesikar DD et al. (1985) L-type glycogen synthase. Tissue distribution and electrophoretic mobility. J Biol Chem 260(18):9953–9956 [PubMed] [Google Scholar]
- Knecht E, Criado-Garcia O et al. (2012) Malin knockout mice support a primary role of autophagy in the pathogenesis of Lafora disease. Autophagy 8(4):701–703 [DOI] [PubMed] [Google Scholar]
- Liu WJ, Ye L et al. (2016) p62 links the autophagy pathway and the ubiqutin-proteasome system upon ubiquitinated protein degradation. Cell Mol Biol Lett 21:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Gonzalez I, Viana R et al. (2017) Inflammation in Lafora disease: evolution with disease progression in Laforin and Malin Knock-out mouse models. Mol Neurobiol 54(5):3119–3130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Ramos JC, Duran J et al. (2015) Role of brain glycogen in the response to hypoxia and in susceptibility to epilepsy. Front Cell Neurosci 9:431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lossos A, Meiner Z et al. (1998) Adult polyglucosan body disease in Ashkenazi Jewish patients carrying the Tyr329Ser mutation in the glycogen-branching enzyme gene. Ann Neurol 44(6):867–872 [DOI] [PubMed] [Google Scholar]
- Machado-Salas J, Avila-Costa MR et al. (2012) Ontogeny of Lafora bodies and neurocytoskeleton changes in Laforin-deficient mice. Exp Neurol 236(1):131–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancardi GL, Schenone A et al. (1985) Polyglucosan bodies in the sural nerve of a diabetic patient with polyneuropathy. Acta Neuropathol 66(1):83–86 [DOI] [PubMed] [Google Scholar]
- Manich G, Cabezon I et al. (2016) Periodic acid-Schiff granules in the brain of aged mice: from amyloid aggregates to degenerative structures containing neo-epitopes. Ageing Res Rev 27:42–55 [DOI] [PubMed] [Google Scholar]
- Martinez JL Jr, Derrick BE (1996) Long-term potentiation and learning. Annu Rev Psychol 47:173–203 [DOI] [PubMed] [Google Scholar]
- Moore SA, Peterson RG et al. (1981) Glycogen accumulation in tibial nerves of experimentally diabetic and aging control rats. J Neurol Sci 52(2–3):289–303 [DOI] [PubMed] [Google Scholar]
- Nitschke F, Sullivan MA et al. (2017) Abnormal glycogen chain length pattern, not hyperphosphorylation, is critical in Lafora disease. EMBO Mol Med 9(7):906–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitschke F, Ahonen SJ et al. (2018) Lafora disease - from pathogenesis to treatment strategies. Nat Rev Neurol 14(10):606–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oe Y, Baba O et al. (2016) Glycogen distribution in the microwave-fixed mouse brain reveals heterogeneous astrocytic patterns. Glia 64(9):1532–1545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orloff MJ, Greenleaf G et al. (1990) Reversal of diabetic somatic neuropathy by whole-pancreas transplantation. Surgery 108(2):179–189; discussion 189–190 [PubMed] [Google Scholar]
- Osorio-Paz I, Sanchez-Chavez G et al. (2012) Control of glycogen content in retina: allosteric regulation of glycogen synthase. PLoS One 7(2):e30822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pederson BA, Turnbull J et al. (2013) Inhibiting glycogen synthesis prevents lafora disease in a mouse model. In: Ann Neurol, vol 74, pp 297–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelps CH (1975) An ultrastructural study of methionine sulphoximine-induced glycogen accumulation in astrocytes of the mouse cerebral cortex. J Neurocytol 4(4):479–490 [DOI] [PubMed] [Google Scholar]
- Pirici I, Margaritescu C et al. (2014) Corpora amylacea in the brain form highly branched three-dimensional lattices. Romanian J Morphol Embryol 55(3 Suppl):1071–1077 [PubMed] [Google Scholar]
- Powell H, Knox D et al. (1977) Alloxan diabetic neuropathy: electron microscopic studies. Neurology 27(1):60–66 [DOI] [PubMed] [Google Scholar]
- Raben N, Danon M et al. (2001) Surprises of genetic engineering: a possible model of polyglucosan body disease. Neurology 56(12):1739–1745 [DOI] [PubMed] [Google Scholar]
- Roach PJ, Cheng C et al. (1998) Novel aspects of the regulation of glycogen storage. J Basic Clin Physiol Pharmacol 9(2–4):139–151 [DOI] [PubMed] [Google Scholar]
- Rohn TT (2015) Corpora amylacea in neurodegenerative diseases: cause or effect? Int J Neurol Neurother 2(3):031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio-Villena C, Viana R et al. (2018) Astrocytes: new players in progressive myoclonus epilepsy of Lafora type. Hum Mol Genet 27(7):1290–1300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rungger-Brandle E, Kolb H et al. (1996) Histochemical demonstration of glycogen in neurons of the cat retina. Invest Ophthalmol Vis Sci 37(5):702–715 [PubMed] [Google Scholar]
- Saez I, Duran J et al. (2014) Neurons have an active glycogen metabolism that contributes to tolerance to hypoxia. J Cereb Blood Flow Metab 34:945–955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Chavez G, Hernandez-Berrones J et al. (2008) Effect of diabetes on glycogen metabolism in rat retina. Neurochem Res 33(7):1301–1308 [DOI] [PubMed] [Google Scholar]
- Simo R, Hernandez C et al. (2014) Neurodegeneration in the diabetic eye: new insights and therapeutic perspectives. Trends Endocrinol Metab 25(1):23–33 [DOI] [PubMed] [Google Scholar]
- Sinadinos C, Valles-Ortega J et al. (2014) Neuronal glycogen synthesis contributes to physiological aging. Aging Cell 13(5):935–945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singhrao SK, Neal JW et al. (1993) Corpora amylacea could be an indicator of neurodegeneration. Neuropathol Appl Neurobiol 19(3):269–276 [DOI] [PubMed] [Google Scholar]
- Sosula L, Beaumont P et al. (1974) Glycogen accumulation in retinal neurons and glial cells of streptozotocin-diabetic rats. Quantitative electron microscopy. Diabetes 23(3):221–231 [DOI] [PubMed] [Google Scholar]
- Takeda T (2009) Senescence-accelerated mouse (SAM) with special references to neurodegeneration models, SAMP8 and SAMP10 mice. Neurochem Res 34(4):639–659 [DOI] [PubMed] [Google Scholar]
- Turnbull J, DePaoli-Roach AA et al. (2011) PTG depletion removes Lafora bodies and rescues the fatal epilepsy of Lafora disease. PLoS Genet 7(4):e1002037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valles-Ortega J, Duran J et al. (2011) Neurodegeneration and functional impairments associated with glycogen synthase accumulation in a mouse model of Lafora disease. EMBO Mol Med 3(11):667–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilchez D, Ros S et al. (2007) Mechanism suppressing glycogen synthesis in neurons and its demise in progressive myoclonus epilepsy. Nat Neurosci 10(11):1407–1413 [DOI] [PubMed] [Google Scholar]
- Vincent AM, Feldman EL (2004) New insights into the mechanisms of diabetic neuropathy. Rev Endocr Metab Disord 5(3):227–236 [DOI] [PubMed] [Google Scholar]
- Waitt AE, Reed L et al. (2017) Emerging roles for glycogen in the CNS. Front Mol Neurosci 10:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walls AB, Eyjolfsson EM et al. (2014) A subconvulsive dose of kainate selectively compromises astrocytic metabolism in the mouse brain in vivo. J Cereb Blood Flow Metab 34(8):1340–1346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Wilamowitz-Moellendorff A, Hunter RW et al. (2013) Glucose-6-phosphate-mediated activation of liver glycogen synthase plays a key role in hepatic glycogen synthesis. Diabetes 62(12):4070–4082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelmus MM, Verhaar R et al. (2011) Novel role of transglutaminase 1 in corpora amylacea formation? Neurobiol Aging 32(5):845–856 [DOI] [PubMed] [Google Scholar]
- Yagihashi S, Matsunaga M (1979) Ultrastructural pathology of peripheral nerves in patients with diabetic neuropathy. Tohoku J Exp Med 129(4):357–366 [DOI] [PubMed] [Google Scholar]
- Zotova EG, Schaumburg HH et al. (2008) Effects of hyperglycemia on rat cavernous nerve axons: a functional and ultrastructural study. Exp Neurol 213(2):439–447 [DOI] [PMC free article] [PubMed] [Google Scholar]