

The plant pathogen implicated in the Irish potato famine, Phytophthora infestans, continues to reemerge globally. Understanding changes in the genome during emergence can provide insights useful for managing this pathogen. Previous work has relied on studying individuals from the United States, South America, Europe, and China reporting that these can occur as diploids, triploids, or tetraploids and are clonal. We studied variation in sexual populations at the pathogen’s center of origin, in Mexico, where it has been reported to reproduce sexually as well as within clonally reproducing, dominant clones from the United States and Europe. Our results newly show that sexual populations at the center of origin are diploid, whereas populations elsewhere are more variable and show genome-wide variation in gene copy number. We propose a model of evolution whereby new pathogen clones emerge predominantly by increasing the gene copy number genome-wide.
KEYWORDS: Irish famine, Phytophthora, copy number variation, oomycetes, plant pathogen, plant pathology, ploidy, population genomics, potato late blight
ABSTRACT
The plant pathogen that caused the Irish potato famine, Phytophthora infestans, continues to reemerge globally. These modern epidemics are caused by clonally reproducing lineages. In contrast, a sexual mode of reproduction is observed at its center of origin in Mexico. We conducted a comparative genomic analysis of 47 high-coverage genomes to infer changes in genic copy number. We included samples from sexual populations at the center of origin as well as several dominant clonal lineages sampled worldwide. We conclude that sexual populations at the center of origin are diploid, as was the lineage that caused the famine, while modern clonal lineages showed increased copy number (3×). Copy number variation (CNV) was found genome-wide and did not to adhere to the two-speed genome hypothesis. Although previously reported, tetraploidy was not found in any of the genomes evaluated. We propose a model of dominant clone emergence supported by the epidemiological record (e.g., EU_13_A2, US-11, US-23) whereby a higher copy number provides fitness, leading to replacement of prior clonal lineages.
INTRODUCTION
The Irish famine pathogen, Phytophthora infestans (Mont.) de Bary, notorious for destroying the potato crop in Ireland in the 19th century, continues to reemerge globally as one of the world’s costliest plant pathogens (1). This pathogen causes late blight on potato worldwide and is considered the most economically important pathogen of this crop. This organism is thought to have originated in central Mexico (2, 3), where it is found alongside two closely related, endemic sister-taxa defining Phytophthora clade 1c, namely, P. mirabilis and P. ipomoeae (4, 5, 51). Elsewhere in the world, it emerges as clonal lineages (6–9). These emergent clonal lineages are frequently ephemeral, disappearing after a season or two (8, 10). However, novel clones occasionally emerge and become dominant, replacing the formerly dominant lineages. While this pathogen continues to reemerge globally, we know very little about the mechanisms involved in pathogen emergence and the genomic features that are associated with these newly emerging, dominant clones.
P. infestans exhibits two distinct lifestyles worldwide. In central Mexico, the pathogen exists as a sexual, randomly mating population (1–3, 52). Throughout much of the remainder of the world, P. infestans is distributed as distinct clonal lineages that reproduce mitotically. Until the early 1990s, a single lineage, US-1, dominated the global populations (11). US-1 was thought to be the lineage that triggered the Great Famine. However, more recent work identified FAM-1 as the famine-causing lineage (12), a lineage that differs from but might be ancestral to US-1 (13, 14). During the mid-1990s, late blight reemerged in the United States as novel clonal genotypes that had not been previously observed (15, 16). The epidemiologically most notable genotypes included US-8 and US-11, which were characterized as having resistance to the fungicide metalaxyl. During the late 2000s, novel lineages emerged in the United States, including US-22, US-23, and US-24 (7, 8). Similar observations were made in Europe, where the 13_A2 clonal lineage became dominant in the late 2000s and where it displaced 6_A1 in the United Kingdom and other previously existing clonal lineages (9). While populations in most of Europe are clonal, sexual populations have been described in northern Europe (9, 17–22). The global population structure of P. infestans is therefore characterized as having a sexually reproducing population in Mexico as well as reemerging clonal epidemics in the United States and most of the rest of the world (except northern Europe), consisting of distinct clonal lineages that displace older clonal lineages.
The P. infestans genome has been characterized as being a two-speed genome. These two speeds refer to two compartments, gene-dense regions containing predominantly housekeeping genes, and gene-sparse regions enriched for effectors (proteins that are secreted from the pathogen and associated with infection), including RxLR genes (23, 24), genes containing an arginine, any amino acid, leucine, and an arginine motif. It is thought that dramatic changes to the gene-sparse, transposon, and effector-rich portion of the genome are responsible for most of the adaptation in clonal lineages. For example, Cooke et al. (9) studied the recent emergence of the 13_A2 clonal lineage in the United Kingdom that largely displaced clonal lineages existing in the United Kingdom by about 2008. This study documented that this lineage was more aggressive, thus outcompeting and displacing older lineages. They also reported large changes in copy number variation (CNV), gene loss, mutations, and gene expression patterns that distinguished 13_A2 from previous lineages. These genomic changes are thought to underlie its emergence.
In addition to the two-speed genome model, several studies have documented variation in ploidy. Phytophthora species are considered to be diploid (25). Extensive cytological work documented that P. infestans was primarily diploid yet indicated that some isolates might be of higher ploidy (26, 27). Several cytological studies indicated that individuals from sexual populations in Mexico were diploid, whereas individuals from clonal populations elsewhere frequently exhibited higher levels of ploidy (28, 29). More recently, Yoshida et al. (12) analyzed whole-genome sequences to show that the allele balance (e.g., the frequency of each allele sequenced at heterozygous positions) for some individuals was triploid or tetraploid. This observation of higher ploidy was further supported by work combining microsatellite analyses, flow cytometry, and high-throughput sequencing of 18 genomes (predominantly from the Netherlands) (30). This body of prior cytological and genomic work provides support for a model that clonal populations are often triploid or tetraploid, while some populations/strains might be diploid. However, these observations are based on individual samples, not allowing broader inferences about populations at large, and have not included a representative sample from sexual populations.
We resequenced genomes of P. infestans to explore variation in gene copy number and in a representative global sample that included a sexual population and select members of clonal lineages. We combined our genome data with recently published whole-genome data to obtain a population of 47 high-coverage samples (see Text Files S1 and S2 in reference 53) that provide power for testing the hypotheses of finding differences in ploidy, CNV, and genic content in P. infestans. For this study, we defined ploidy as a genome-wide change in copy number (i.e., whole-genome duplication), whereas copy number refers to a change observed at the subchromosomal level. We tested the hypotheses that sexual populations were diploid with little CNV, while clonal populations were predominantly triploid or tetraploid with high CNV. We also tested the hypothesis that CNV and the presence/absence polymorphism are enriched in gene-sparse, effector-rich portions of the genome (as expected from the two-speed genome hypothesis). We also expected to find that CNV and presence/absence polymorphisms differed in clonal versus sexual populations. Finally, we tested the hypothesis that similar changes in CNV might be observed in other heterothallic Phytophthora species for which genomic data for populations was available, such as P. parasitica and P. capsici. Our findings provide a new perspective on how plasticity in ploidy, copy number, and presence/absence polymorphisms contribute to the emergence of the Irish potato famine pathogen and other Phytophthora pathogens.
RESULTS
Resequencing populations of P. infestans.
To understand variation in CNV and gene content, we resequenced and used previously published populations of the potato late blight pathogen, P. infestans, from the center of origin in Mexico (n = 16) and dominant clonal lineages in the United States, Europe, and South America (Fig. 1). To allow for robust inference of gene copy number, we used only genomes with a genic average adjusted read depth (AARD) of 12× or greater (see Text S3 in reference 53). This resulted in a total of 47 high-quality P. infestans genomes (see Text S1 in reference 53).
FIG 1.
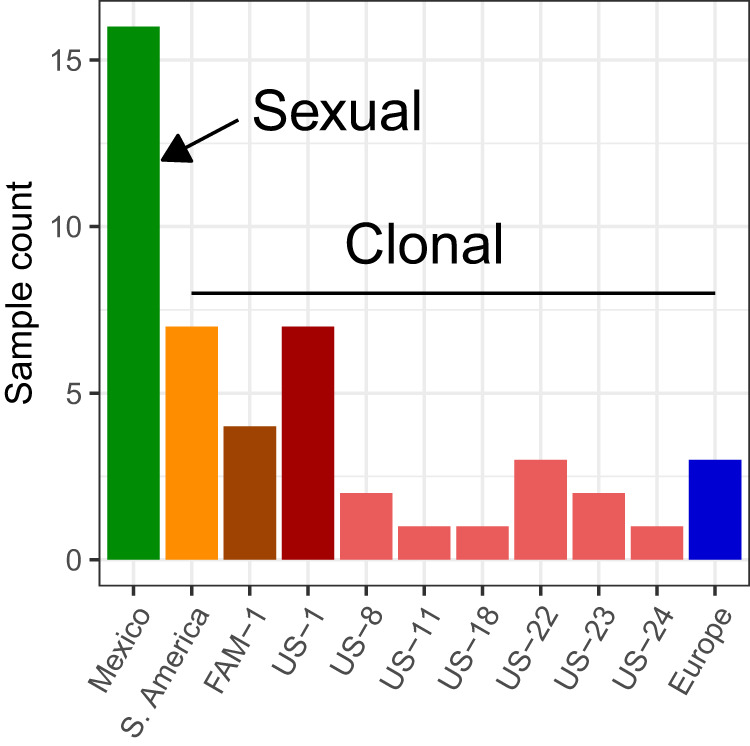
Samples included sexual and clonal populations selected for high sequence coverage. The samples were collected from throughout the world and included samples from Mexico that are sexually reproducing (green bar) as well as samples from the rest of the world that are clonally reproducing (all other bars) (2, 52). In order to attain high-quality samples from the literature and our own resequencing for the inference of copy number variation, only samples with at least 12× adjusted average read depth (AARD) were included (see Text S1 in reference 53).
Genic copy number varies continuously in P. infestans.
We observed genic CNV among populations (Fig. 2A) and a gradient of genic copy number ranging from predominantly 2× to predominantly 3× (Fig. 2B). We did not observe classes of individuals that would represent tetraploid individuals. Isolates from the United States belonging to clonal lineages have a gradient of gene copy number (Fig. 2C). Strains in U.S. lineages that were predominantly 2× were mostly found in the well-represented lineage US-22 (n = 3) and in US-18 (n = 1). Similarly, in Europe, isolates that were both predominantly 2× and 3× were observed. The exception to this balance of copy number appeared to be in South America, where almost the entire sample was predominantly 3× (Fig. 2C). Samples from Mexico had a low percentage of gene copy numbers assigned to 3× (<20%; Fig. 2A and C), and the majority of genes occurred in two copies. While previous studies focused on variation in ploidy (12, 26–30), our work supports variation in genome size in P. infestans occurring largely at a subgenomic level; Mexican, FAM-1, and US-22 samples were predominantly 2× with narrow variation that can be interpreted as diploidy, whereas samples from South America, US-1, other U.S. lineages, and those from Europe showed large variation (Fig. 2A).
FIG 2.

Strains of P. infestans show variation in gene copy number. (A) Shown are the percentage of triploid genes in sexual and clonal populations. Populations from Mexico, FAM-1, and US-22 are predominantly diploid, while populations from elsewhere in the world are predominantly triploid. (B) Bar plots of the proportion of inferred gene copy number based on allele balance over all genes for each individual. Ranking isolates from 2× to 3× show a continuous change in the proportion of gene copy number across all samples rather than discrete 2× or 3× clusters. (C) The second bar chart organizes the samples by geographic origin, starting on the left from Mexico and progressing to South America, FAM-1 (the Irish famine lineage [12]), US-1, US-8, US-11, US-18, US-22, US-23 (currently the most abundant in the United States; see Text S4 in reference 53), US-24, and finally, Europe.
The variation in CNV was also explored for samples where tissue was extracted from historical herbarium samples (FAM-1: M-0182896, Pi1889; US-1: Kew122, Kew126) (Fig. 2C). These samples were not cultured on medium and were not exposed to the modern fungicide metalaxyl and demonstrated variability in gene copy number as well (Fig. 2A and C), suggesting that CNV may have been a natural condition in clonal lineages of P. infestans. Note that two of the four samples that we determined to be of sufficient sequence depth to call copy number were from the 20th century (Kew122 and Kew126, both collected in 1955; see Text S1 in reference 53) and clustered with US-1 (14), while the other two were from the 19th century and clustered with FAM-1 (M-0182896 collected in 1877 and Pi1889 collected in 1889). This indicates that CNV was observed throughout the time series of the data and was not restricted to modern samples that were cultured on medium.
Gene loss occurs in both clonal and sexual populations.
We explored the hypothesis that gene loss (relative to the reference genome T30-4) had occurred collectively within a lineage or independently. The breadth of coverage (BOC) for a gene is the proportion of positions that were sequenced at least once in the reference genome (24). For example, a BOC of 0.75 would indicate that 75% of the positions in a gene were sequenced at least once. We used a BOC of 0 to define a gene loss event and presented samples for populations that included at least six individuals (groupings with more were randomly subset to a sample size of six) (Fig. 3; see Text S5 in reference 53). Gene loss was most pronounced in RxLR and crinkler (CRN) effectors but was found in all gene classes (average range of 0 to 1 for core, CAZy, necrosis inducing-like protein [NPP1], secreted small cysteine-rich protein [SCR], and elicitin) (see Text S5 in reference 53). Gene loss among the isolates from Mexico ranged from 38 to 112 gene deletions. However, we found only one shared deletion among all samples within the clonal lineage (Fig. 3, bottom panel). Clonally reproducing isolates from South America demonstrated a loss of 39 to 63 genes, with only 9 gene losses shared in common among these isolates. Among 6 individuals belonging to lineage US-1, we observed a range of loss of 21 to 68 genes but only 5 gene losses common among all of the sampled lineages (Fig. 3). Gene loss is a dominant feature in the gene-sparse regions harboring >95% of genes subject to gene loss in all samples of the genome. However, the specific gene lost within any particular sample is unique and random and apparently affects clonal and sexual populations equally.
FIG 3.

Gene loss in pathogenicity factors and pseudogenes occurs in individuals within populations regardless of clonal or sexual reproductive mode. The top three rows show results for individual samples, and the bottom row shows the summary for the populations sampled from Mexico (green), South America (orange), and the US-1 clonal lineage (red). Gene loss is common in individuals that may lose between 21 and 112 pathogenicity genes or pseudogenes. However, when comparing multiple samples within a population, few losses are shared. Gene loss is happening on an individual basis and is not dependent on ancestry. This pattern is observed in sexually reproducing populations (Mexico) as well as in clonally reproducing populations (South America and US-1). Gene loss is defined here as a gene from a sample with an adjusted average read depth of at least 12 and where 0 positions (base pairs) in the gene were sequenced (BOC = 0). The background for each panel is a heatmap indicating gene abundance in relation to their 5′ and 3′ intergenic spacing. Points indicate gene deletions and are colored by their functional annotation as defined in Haas et al. (23).
Genic copy number variation was not associated with specific classes of genes.
We found that in the sexually reproducing population from Mexico that was predominantly diploid, all gene categories had more 2× genes than 3× genes (Fig. 4, green). In contrast, for the populations from South America (orange) and US-1 (red), which were clonally reproducing, we found that all gene classes had more 3× genes than 2× genes regardless of gene family. CNV occurs throughout gene space without a preference for functional annotation (Fig. 4).
FIG 4.

Gene copy number variation is not restricted to a particular class of gene. The x axis shows the proportion of genes within each gene class (columns labeled at top) classified as being 2× or 3× copy number. The y axis shows the fraction of genes classified into 2× or 3× within a gene class. Columns show gene families, and rows show results by population. Isolates from Mexico (green), where P. infestans is sexually reproducing, had a gene copy number predominantly of two for all classes of genes. Isolates from South America and US-1, both considered clonally reproducing, had a gene copy number predominantly of three for all gene classes. Gene copy number varies throughout gene space and is not associated with function. Box and whisker plots summarize points that represent samples (n = 6) and the proportion of genes that were either 2× or 3× (based on the total number of 2× and 3× genes). A sample size of six was used (as in Fig. 3) to have equal samples sizes. Core, core orthologous genes; other gene families are defined as in Haas et al. (23).
Gene copy number variation occurred in core orthologous genes.
Core orthologous Phytophthora genes are reported to occur only once in P. infestans, P. ramorum, and P. sojae (23) and are thought to be highly conserved. Based on the two-speed genome hypothesis, one might expect higher copy number to preferentially occur in the gene-sparse region. We plotted all core orthologous genes present at 3× by their 5′ and 3′ intergenic distances (Fig. 5). We observed substantial numbers of genes inferred to have three copies (3×) among core orthologous genes in the gene-dense portion of each genome (Fig. 5). This indicates that this portion of the genome may be more dynamic than previously thought.
FIG 5.

Gene copy number does not follow the two-speed genome hypothesis. Core orthologous genes with a copy number of three are enriched in the gene-dense (rather than gene-sparse) regions of the genome. The background for each panel is a heatmap indicating gene abundance as in Fig. 3. Orange points (with transparency) are plotted over this background, where each point is a core orthologous gene that was determined to have three copies and was positioned based on their 3′ (y axis) and 5′ (x axis) intergenic distance as in Fig. 3. Background grid and axis ticks are identical to those in Fig. 3.
The phenomenon of genic CNV is shared with other members of the Phytophthora genus.
We explored if the variation in ploidy apparent in P. infestans is observed in other heterothallic Phytophthora taxa. We looked at species for which population-level genome data were available, including P. andina (clade 1c), P. parasitica (clade 1), and P. capsici (clade 2) (clades as assigned by Blair et al. [31]). The taxon P. andina appears to be diploid in our limited sample (Fig. 6). However, we observed more heterozygous positions than in the other taxa (Fig. 6). This is consistent with the interpretation that P. andina is a homoploid hybrid that arose from a cross between P. infestans and another undescribed Phytophthora species (32). The more distantly related P. parasitica appeared diploid as well. However, its relatively high sequence depth allowed resolution of minor peaks, indicating that a fraction of genes occur at three copies (particularly in the sample P1569). The taxon most distantly related to P. infestans included in our analysis was P. capsici. Three of the P. capsici samples appeared to be diploid, while one sample (Pc389) appeared to be triploid. These results suggest that our findings of variation in ploidy and CNV within P. infestans are also shared among other species of Phytophthora.
FIG 6.

Ploidy variation is observed in other heterothallic Phytophthora species. Histograms showing the major and minor allele frequency observed for individuals. Samples of P. infestans are diploid or triploid. The hybrid taxon P. andina appeared predominantly diploid. Note that the y axis indicates many more heterozygous positions in these samples relative to the other species. The clade 1 species P. parasitica is diploid. However, note the “shoulder” peaks at the 3× expectation for samples P10297and P1569. The clade 2 species P. capsici demonstrated a predominance of diploidy, with the single sample Pc389 being triploid. The x axis ticks mark 0, 1/5, 1/4, 1/3, 1/2, 2/3, 3/4, 4/5, and 1 mark our expectations for pentaploid, tetraploid, triploid, and diploid. The y axis ticks in the histograms are in units of 10,000 heterozygous sites. Phylogeny adapted from Martin et al. (49). Larger histograms are presented in Text Files S6 and S7 in the supplemental material (53).
DISCUSSION
To characterize the emergence of new clonal lineages of the Irish famine pathogen, Phytophthora infestans, we resequenced whole genomes of select populations. We focused on contrasting several dominant clonal lineages in the United States as well as sexual populations from the center of origin in Mexico for which we were able to obtain samples. Prior work (see below) focused primarily on individuals rather than populations and did not include sexual populations. The genomes were compared with previously sequenced, high-quality genomes to determine ploidy, CNV, and gene content. Recent epidemiological records indicated that new clonal lineages have emerged repeatedly in the United States and Europe (see Text S4 in reference 53). For example, the lineage US-1 was the first to establish itself in the U.S. but was eventually displaced by US-8, US-11, and more recently, by US-23 (1) (see Text S4 in reference 53). Similarly, populations in the United Kingdom were displaced by 13_A2 in the past decade and, more recently, by 6_A1 (9). While variation in ploidy has been described in individuals from clonal lineages of P. infestans, our work provides several new key insights based on population-level patterns, expanding on prior work focusing on single clonal strains.
Clonal lineages show higher copy numbers than sexual populations at the center of origin.
The populations studied show a gradient of CNV from 2× to 3× (Fig. 2B). Populations of P. infestans that are sexually reproducing at the species’ center of diversity in Mexico are predominantly diploid (Fig. 2C). This contrasts with dominant clonal populations from the rest of the world, which are predominantly triploid. This provides support for the hypothesis that there may be a connection between copy number, epidemic fitness, and mode of reproduction. Higher copy number might increase expression of advantageous genes. This hypothesis is, however, difficult or impossible to test experimentally and is not experimentally supported.
Isolates were predominantly diploid or triploid but not tetraploid.
We observed only diploid and triploid strains but no tetraploid individuals as reported previously (12). We reanalyzed some of the same samples and data, including the European lineage 13_A2, previously characterized as being tetraploid. In our analysis, 13_A2 had mostly three gene copies and would thus be classified as being triploid (Fig. 2), which is in agreement with a more recent report (30). Part of this discrepancy is due to changes in technology. Plotting histograms of allele balance has typically included all variants, including homozygous genotypes. Because homozygous sites are much more abundant than heterozygous sites, this tends to drive the scaling of the plot. To avoid this, previous work limited plots to a frequency range of 0.2 to 0.8. We subset our data to only the heterozygous genotypes, resulting in a plot of 0 to 1, and subset the data by omitting variants with unusually high or low sequence depth. This is a significant improvement in methodology for inferring ploidy or CNV based on allele balance (33).
Gene loss occurred within individuals in both sexual populations and clonal lineages.
We tested the hypothesis that gene loss was shared by ancestry. This would provide the expectation that members of a clonal lineage show fixed polymorphisms within that clonal lineage. We used breadth of coverage to identify the presence/absence of genes relative to the reference genome. Instead, we found that individuals within a clonal lineage (e.g., from South America or US-1; Fig. 3) showed gene loss within individuals at a rate similar to that of the sexual population (Mexico; Fig. 3). Furthermore, gene loss affected many gene families, including effectors, and was located throughout the genome. This is consistent with the hypothesis that pathogenicity factors are thought to be enriched in the gene-sparse portion of the genome (23, 34, 35).
CNV is found throughout the genome and affects all gene families, including core genes and effectors, equally.
Our expectation following the proposed two-speed genome hypothesis (23, 24) was to find CNV enriched in the gene-sparse, transposon, and effector-rich portion of the genome, where CNV could provide a means of creating novel paralogs. To our surprise, CNV affects housekeeping genes and effectors equally (Fig. 4) and is randomly dispersed throughout the whole genome. In the diploid genomes from Mexico, we found that core orthologous genes, pseudogenes, and several pathogenicity factors were all predominantly 2×. Genomes of clonally reproducing strains from South America and the lineage US-1 were found to have core orthologous genes, pseudogenes, and pathogenicity factors that were predominantly 3×. We also expected CNV to be higher in pathogenicity factors than in core orthologs, yet levels of CNV were not different, regardless of gene class.
Variation in copy number can be found in other Phytophthora species.
We also evaluated if changes in ploidy could be observed in other heterothallic Phytophthora species. We used genomes for moderate population sizes from P. andina, P. parasitica (= P. nicotianae), and P. capsici available at the Sequence Read Archive to address this question (Fig. 6). Within Phytophthora clade 1c, P. andina appeared predominantly diploid. P. andina has been recognized as a hybrid with two parental species, one of which is P. infestans, while the other hybrid parent is unknown (32, 36). The genomes of P. andina had one haplotype from each parental species as expected and were predominantly 2× copy number. P. parasitica, a distant relative of P. infestans basal to clade 1, was diploid. However, two strains (P10297 and P1569) had minor peaks at our expectation for three copies, indicating that fractions of these genomes may vary in copy number at 3×. Our ability to resolve these peaks was likely due to the high sequence depth of these samples relative to the other available taxa. In clade 2, the more distant P. capsici appeared predominantly diploid for 3 strains; however, one strain (Pc389) was triploid. These results suggest that variation in ploidy and/or copy number may be a common feature throughout the Phytophthora genus, consistent with other recent reports (37, 38).
We propose a model of emergence where triploid clones emerge and eventually displace prior clonal lineages.
Our work provides striking support for a model of predominantly diploid populations at the center of origin reinforced by sexuality and predominantly triploid clonal lineages elsewhere in the world (Fig. 7). In this model, novel clonal lineages emerging globally are predominantly triploid. These triploid lineages might be more fit and thus able to displace other extant lineages. A new lineage emerging from a sexual cross in Mexico is expected to be initially diploid and will gradually show an increase to three copies per gene. Older previously dominant lineages might thus be more triploid (e.g., US-1) than dominant younger lineages (e.g., US-23). Some lineages are ephemeral (e.g., US-18, US-22). The recently emerged diploid lineage US-22 was only observed between 2009 and 2011 and might be less fit (1, 10) and, curiously, shows predominantly 2× copies per gene. To the best of our knowledge, all lineages that became dominant in space and time are or were triploid, with the exception of FAM-1. It remains to be established if higher genic copy number confers higher epidemic fitness to a clonal lineage. Experimentally addressing this question might prove challenging given the fact that CNV is a whole-genome phenomenon. However, there are several studies supporting the idea that some clonal lineages (which are 3× in our analysis) displacing older lineages were indeed fitter. Kato and colleagues showed that US-8 strains have larger lesions and sporulate more than US-1 strains (39). Similarly, Cooke and colleagues, using mark-recapture methods in the field, showed that the 13_A2 strains were among the most aggressive clones compared to the strains evaluated and outcompeted previously dominant clonal lineages (9).
FIG 7.

Model of the genomic processes observed to date that are thought to underlie the patterns of emergence observed for clonal lineages of the Irish famine pathogen. This model proposes that sexual populations are diploid, while clonal populations are predominantly triploid. Some clones, likely rare and by chance, are clones that become dominant, displacing prior clonal lineages, as has been observed repeatedly in the United States. These dominant clones are predominantly triploid, including US-8, US-11, US-23, and the aggressive European lineage Blue13_A2 (represented in our sample by 06_3928A), while other lineages that are diploid, such as US-18 and US-22, are ephemeral or cryptic (see Text S4 in reference 53) (9). Sexual reproduction reinforces diploidy, while a triploid status might interfere with sexual reproduction but might confer fitness.
Conclusions.
The late blight pathogen P. infestans continues to reemerge, causing financial loss for farmers and threatening food security, particularly in developing countries (1). We report the observation that P. infestans isolates are diploid in central Mexico, where they reproduce sexually, and emerging dominant clonal lineages are predominantly triploid. These findings provide novel support for the hypotheses that a change in copy number might drive emergence of clonal lineages of the Irish famine pathogen.
MATERIALS AND METHODS
Sequence alignment and variant calling.
The sample came from previously published sources (9, 12–14, 23, 24) as well as 11 new Phytophthora infestans genomes we sequenced (see Text S1 in reference 53). Isolates US040009, FP-GCC, US100006, FL2009P4, and ND822Pi were sequenced at the UC Davis Genome Center. Isolates PIC97136, PIC97146, PIC97335, PIC97442, PIC97750, and PIC97785 were sequenced at Oregon State University’s Center for Genome Research and Biocomputing on an Illumina HiSeq 2000 platform. Additionally, five samples each of P. mirabilis and P. ipomoeae (see Text S2 in reference 53) were also sequenced at Oregon State University’s Center for Genome Research and Biocomputing on an Illumina HiSeq 2000 platform. All other samples were obtained from publicly available repositories (see Text S1 and S2 in reference 53). Newly sequenced genomes are publicly available at the Sequence Read Archive (BioProject number PRJNA542680; Text S3 in reference 53).
The FASTQ format files were aligned to the P. infestans T30-4 reference (23) using the Burrows-Wheeler Aligner MEM algorithm (BWA-MEM) 0.7.10 (40, 41). The resulting SAM format file was converted to BAM format, the mate information was fixed, and the MD and NM tags were added using SAMTools (41). PCR and optical duplicates were marked using Picard’s MarkDuplicates (42). The per gene sequence depth and coverage over all T30-4 genes was calculated using SAMtools mpileup (41). From the mpileup data, the number of positions that were sequenced at least once and a median of coverage were calculated. In order to correct our measure of coverage for GC bias, we calculated an adjusted average read depth (AARD) (24). A median was chosen as a robust alternative to an average; however, we refer to our measure here as AARD to be consistent with the existing literature. The genes were sorted into bins based on percentiles of GC content. The adjusted median read depth was then taken by multiplying the median read depth for each gene by the ratio of the median read depth of all genes divided by the median average read depth for all genes in the GC bin of the gene. The AARD for each genome was summarized using violin plots (43), and a threshold of mean AARD of at least 12 was used as a threshold for inclusion of a genome for further analysis.
Variants were called from the BAM files for diploid genotypes to create genomic variant call format (gVCF) files using the Genome Analysis Toolkit (GATK) HaplotypeCaller (44, 45). Diploid genotypes were called using the GATK’s GenotypeGVCFs. The samples P10127, P10650, P11633, P12204, P1362, P6096, and P7722 were flagged by the GATK’s HaplotyeCaller as having legacy quality encoding. These samples were run with the option fix_misencoded_quality_scores to accommodate this.
Gene copy number inference based on allele balance.
The inference of gene copy number was made based on the ratio of alleles observed at heterozygous positions (12, 30). The VCF specification (46) provides the option for variant callers to report the number of times each allele was sequenced at a variable position. In a diploid heterozygote, the expectation is that each allele will be observed at an equal frequency or a ratio of one half. A triploid heterozygote will be expected to have alleles observed at a ratio of one third. A tetraploid heterozygote will be expected to have alleles observed at a ratio of one quarter. Note that some combinations are indistinguishable and therefore uninformative. For example, a tetraploid heterozygote with only two alleles (e.g., A/A/C/C) will have each allele observed at a ratio of one half. This will be indistinguishable from our expectation from a diploid heterozygote. The ratio of alleles observed at each variable position has been used by other authors to make inferences about ploidy (12, 30). Shortcomings of the present use of the ratio of alleles are that it has been presented graphically as a histogram and that the data appear “noisy” in that they do not form a strong consensus at an expected CNV value. A problem with the graphical representation of data arises when a large number of samples are to be explored or when the genome is subset into a large number of fractions, such as in windowing analyses. A numerical summary table provides the ratio of alleles observed in any genome or in any fraction of a genome. The problem of noisy data may in part be due to variants of low quality (i.e., technical error) or potential variation in ploidy throughout a genome or subgenomic region (i.e., biological variation).
The challenge of identifying high-quality variants and numerically summarizing them was addressed by our method of allele balance analysis (33). The data were quality filtered using the sequence depth of the most abundant allele for all variants in a genome. An 80% confidence interval was created to eliminate variants with the lowest 10% and highest 10% sequence coverage. This confidence interval was then applied to the second-most-abundant allele as well. The VCF file was further subset to only heterozygous positions. The allele balance ratio for each heterozygous variant was calculated by dividing the number of times the most abundant allele was sequenced by the number of times the most abundant allele and the second-most-abundant allele were sequenced, resulting in a proportion. Finally, 200,000-bp windows were made using the allele ratio data. This window size was chosen for P. infestans because it was sufficiently large to include a population of heterozygous positions (we observed a heterozygous position every 1 to 2 kbp) but small enough to obtain fine-scale resolution. The data were then assigned to bins ranging from 0 to 1 that are 0.02 frequencies wide, and the bin with the greatest density was used as a summary for the window. This is analogous to the modal frequency. This summary was then categorized to a ploidy level by assigning it to the closest expected ratio (i.e., 1/2, 2/3, 3/4, 4/5). Each genome was now summarized into windows of ploidy. In order to assign copy numbers to genes, the coordinates of each gene were referenced in the windowed genome, and the copy number of the window where the gene was located was used to assign a copy number to the gene. This is critical because we do not expect most genes to contain enough heterozygous positions to infer an accurate estimate of copy number. Once a copy number was determined, a confidence in this estimate was made by subtracting the observed proportion from the determined proportion and dividing by the bin width so that the value ranges from 0 to 1. Calculations were performed in R (47) and using vcfR (33, 48).
Gene loss based on breadth of coverage.
In order to determine gene loss, we measured breadth of coverage (BOC) for each gene in each genome. We used SAMtools mpileup (41) to count the per position sequence coverage over all 18,179 genes in the P. infestans T30-4 genome (23). From these data, the number of positions that were sequenced at least once and a median of coverage were collected. Breadth of coverage was calculated by dividing the number of positions that were sequenced at least once by the gene length (i.e., the proportion of positions sequenced in a gene). We used a BOC of 0 to indicate the loss of a gene.
Gene class and density.
Published gene annotations (23) were used to assign genes to gene classes (core, pseudogene, RxLR, etc.). The flanking intergenic region (FIR) lengths (i.e., intergenic distances) were calculated using a previously available script (https://figshare.com/articles/Calculate_FIR_length_perl_script/707328). This information was used to create FIR plots for individuals and populations from Mexico, South America, and the lineage US-1 using R (47) and ggplot2 (43). In order to explore whether genes of a particular class from populations from Mexico, South America, and the lineage US-1 were enriched for a particular copy number, the genes were assigned a copy number (based on allele balance) and plotted as box and whisker plots using ggplot2 (43). In order to visualize whether genes determined to have three copies were more abundant in the gene-dense or gene-sparse portion of the genome, FIR plots were created as described above but using core orthologous genes that were determined to have three copies.
Copy number variation in other species of Phytophthora.
In order to address whether copy number variation occurred in other species of Phytophthora, we queried NCBI for samples that had Illumina sequence data as well as an assembled genome reference for the species. These data were processed as the P. infestans data were processed. In order to visualize these data in a phylogenetic context, a tree from Martin et al. (49) was obtained from TreeBase (50). The data were then plotted in R (47).
Data and code.
All R code and data necessary to reproduce the figures are available on GitHub (https://github.com/grunwaldlab/P_infestans_CNV).
ACKNOWLEDGMENTS
Val Fieland, Karan Fairchild, Meg Larsen, and Caroline Press provided much appreciated technical support. We greatly appreciate receiving data and cultures from Bill Fry and the USABlight community (https://usablight.org/), which were used for Text S4 in reference 53. We thank the Center for Genome Research and Biocomputing (CGRB) at Oregon State University for genome sequencing and support of our computational research on the CGRB cloud.
This research was supported in part by U.S. Department of Agriculture (USDA) Agricultural Research Service Grant 2072-22000-041-00-D and USDA National Institute of Food and Agriculture grants 2011-68004-30154 and 2018-67013-27823. Mention of trade names or commercial products in the manuscript are solely for the purpose of providing specific information and do not imply recommendation or endorsement.
We declare no competing interests.
Footnotes
Citation Knaus BJ, Tabima JF, Shakya SK, Judelson HS, Grünwald NJ. 2020. Genome-wide increased copy number is associated with emergence of dominant clones of the Irish potato famine pathogen Phytophthora infestans. mBio 11:e00326-20. https://doi.org/10.1128/mBio.00326-20.
REFERENCES
- 1.Fry WE, Birch PRJ, Judelson HS, Grünwald NJ, Danies G, Everts KL, Gevens AJ, Gugino BK, Johnson DA, Johnson SB, McGrath MT, Myers KL, Ristaino JB, Roberts PD, Secor G, Smart CD. 2015. Five reasons to consider Phytophthora infestans a reemerging pathogen. Phytopathology 105:966–981. doi: 10.1094/PHYTO-01-15-0005-FI. [DOI] [PubMed] [Google Scholar]
- 2.Goss EM, Tabima JF, Cooke DEL, Restrepo S, Fry WE, Forbes GA, Fieland VJ, Cardenas M, Grünwald NJ. 2014. The Irish potato famine pathogen Phytophthora infestans originated in central Mexico rather than the Andes. Proc Natl Acad Sci U S A 111:8791–8796. doi: 10.1073/pnas.1401884111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grünwald NJ, Flier WG. 2005. The biology of Phytophthora infestans at its center of origin. Annu Rev Phytopathol 43:171–190. doi: 10.1146/annurev.phyto.43.040204.135906. [DOI] [PubMed] [Google Scholar]
- 4.Galindo J, Hohl H. 1985. Phytophthora mirabilis, a new species of Phytophthora. Sydowia 38:87–96. [Google Scholar]
- 5.Flier WG, Grünwald NJ, Kroon LPNM, Van Den Bosch TBM, Garay-Serrano E, Lozoya-Saldaña H, Bonants PJM, Turkensteen LJ. 2002. Phytophthora ipomoeae sp. nov., a new homothallic species causing leaf blight on Ipomoea longipedunculata in the Toluca Valley of central Mexico. Mycol Res 106:848–856. doi: 10.1017/S0953756202006123. [DOI] [Google Scholar]
- 6.Fry WE, Goodwin SB, Dyer AT, Matuszak JM, Drenth A, Tooley PW, Sujkowski LS, Koh YJ, Cohen BA, Deahl KL, Inglis DA, Sandlan KP. 1993. Historical and recent migrations of Phytophthora infestans: chronology, pathways, and implications. Plant Dis 77:653–655. doi: 10.1094/PD-77-0653. [DOI] [Google Scholar]
- 7.Hu C-H, Perez FG, Donahoo R, McLeod A, Myers K, Ivors K, Secor G, Roberts PD, Deahl KL, Fry WE, Ristaino JB. 2012. Recent genotypes of Phytophthora infestans in the eastern United States reveal clonal populations and reappearance of mefenoxam sensitivity. Plant Dis 96:1323–1330. doi: 10.1094/PDIS-03-11-0156-RE. [DOI] [PubMed] [Google Scholar]
- 8.Fry WE, McGrath MT, Seaman A, Zitter TA, McLeod A, Danies G, Small IM, Myers K, Everts K, Gevens AJ, Gugino BK, Johnson SB, Judelson H, Ristaino J, Roberts P, Secor G, Seebold K, Snover-Clift K, Wyenandt A, Grünwald NJ, Smart CD. 2013. The 2009 late blight pandemic in the eastern United States: causes and results. Plant Dis 97:296–306. doi: 10.1094/PDIS-08-12-0791-FE. [DOI] [PubMed] [Google Scholar]
- 9.Cooke DEL, Cano LM, Raffaele S, Bain RA, Cooke LR, Etherington GJ, Deahl KL, Farrer RA, Gilroy EM, Goss EM, Grünwald NJ, Hein I, MacLean D, McNicol JW, Randall E, Oliva RF, Pel MA, Shaw DS, Squires JN, Taylor MC, Vleeshouwers VGAA, Birch PRJ, Lees AK, Kamoun S. 2012. Genome analyses of an aggressive and invasive lineage of the Irish potato famine pathogen. PLoS Pathog 8:e1002940. doi: 10.1371/journal.ppat.1002940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Danies G, Myers K, Mideros MF, Restrepo S, Martin FN, Cooke DEL, Smart CD, Ristaino JB, Seaman AJ, Gugino BK, Grünwald NJ, Fry WE. 2014. An ephemeral sexual population of Phytophthora infestans in the northeastern United States and Canada. PLoS One 9:e116354. doi: 10.1371/journal.pone.0116354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goodwin SB, Cohen BA, Fry WE. 1994. Panglobal distribution of a single clonal lineage of the Irish potato famine fungus. Proc Natl Acad Sci U S A 91:11591–11595. doi: 10.1073/pnas.91.24.11591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yoshida K, Schuenemann VJ, Cano LM, Pais M, Mishra B, Sharma R, Lanz C, Martin FN, Kamoun S, Krause J, Thines M, Weigel D, Burbano HA. 2013. The rise and fall of the Phytophthora infestans lineage that triggered the Irish potato famine. Elife 2:e00731. doi: 10.7554/eLife.00731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martin MD, Cappellini E, Samaniego JA, Zepeda ML, Campos PF, Seguin-Orlando A, Wales N, Orlando L, Ho SYW, Dietrich FS, Mieczkowski PA, Heitman J, Willerslev E, Krogh A, Ristaino JB, Gilbert MTP. 2013. Reconstructing genome evolution in historic samples of the Irish potato famine pathogen. Nat Commun 4:2172. doi: 10.1038/ncomms3172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martin MD, Vieira FG, Ho SY, Wales N, Schubert M, Seguin-Orlando A, Ristaino JB, Gilbert M. 2016. Genomic characterization of a South American Phytophthora hybrid mandates reassessment of the geographic origins of Phytophthora infestans. Mol Biol Evol 33:478–491. doi: 10.1093/molbev/msv241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fry WE, Goodwin SB. 1997. Re-emergence of potato and tomato late blight in the United States. Plant Dis 81:1349–1357. doi: 10.1094/PDIS.1997.81.12.1349. [DOI] [PubMed] [Google Scholar]
- 16.Fry WE, Goodwin SB. 1997. Resurgence of the Irish potato famine fungus. Bioscience 47:363–371. doi: 10.2307/1313151. [DOI] [Google Scholar]
- 17.Fry WE. 2016. Phytophthora infestans: new tools (and old ones) lead to new understanding and precision management. Annu Rev Phytopathol 54:529–547. doi: 10.1146/annurev-phyto-080615-095951. [DOI] [PubMed] [Google Scholar]
- 18.Yuen JE, Andersson B. 2013. What is the evidence for sexual reproduction of Phytophthora infestans in Europe? Plant Pathology 62:485–491. doi: 10.1111/j.1365-3059.2012.02685.x. [DOI] [Google Scholar]
- 19.Brurberg MB, Elameen A, Le VH, Naerstad R, Hermansen A, Lehtinen A, Hannukkala A, Nielsen B, Hansen J, Andersson B, Yuen J. 2011. Genetic analysis of Phytophthora infestans populations in the Nordic European countries reveals high genetic variability. Fungal Biol 115:335–342. doi: 10.1016/j.funbio.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 20.Montarry J, Andrivon D, Glais I, Corbiere R, Mialdea G, Delmotte F. 2010. Microsatellite markers reveal two admixed genetic groups and an ongoing displacement within the French population of the invasive plant pathogen Phytophthora infestans. Mol Ecol 19:1965–1977. doi: 10.1111/j.1365-294X.2010.04619.x. [DOI] [PubMed] [Google Scholar]
- 21.Chowdappa P, Kumar NBJ, Madhura S, Kumar MSP, Myers KL, Fry WE, Squires JN, Cooke D. 2013. Emergence of 13_A2 blue lineage of Phytophthora infestans was responsible for severe outbreaks of late blight on tomato in South-West India. J Phytopathol 161:49–58. doi: 10.1111/jph.12031. [DOI] [Google Scholar]
- 22.Li Y, van der Lee T, Zhu JH, Jin GH, Lan CZ, Zhu SX, Zhang RF, Liu BW, Zhao ZJ, Kessel G, Huang SW, Jacobsen E. 2013. Population structure of Phytophthora infestans in China: geographic clusters and presence of the EU genotype Blue_13. Plant Pathol 62:932–942. doi: 10.1111/j.1365-3059.2012.02687.x. [DOI] [Google Scholar]
- 23.Haas BJ, Kamoun S, Zody MC, Jiang RHY, Handsaker RE, Cano LM, Grabherr M, Kodira CD, Raffaele S, Torto-Alalibo T, Bozkurt TO, Ah-Fong AMV, Alvarado L, Anderson VL, Armstrong MR, Avrova A, Baxter L, Beynon J, Boevink PC, Bollmann SR, Bos JIB, Bulone V, Cai G, Cakir C, Carrington JC, Chawner M, Conti L, Costanzo S, Ewan R, Fahlgren N, Fischbach MA, Fugelstad J, Gilroy EM, Gnerre S, Green PJ, Grenville-Briggs LJ, Griffith J, Grünwald NJ, Horn K, Horner NR, Hu C-H, Huitema E, Jeong D-H, Jones AME, Jones JDG, Jones RW, Karlsson EK, Kunjeti SG, Lamour K, Liu Z, et al. 2009. Genome sequence and analysis of the Irish potato famine pathogen Phytophthora infestans. Nature 461:393–398. doi: 10.1038/nature08358. [DOI] [PubMed] [Google Scholar]
- 24.Raffaele S, Farrer RA, Cano LM, Studholme DJ, MacLean D, Thines M, Jiang RHY, Zody MC, Kunjeti SG, Donofrio NM, Meyers BC, Nusbaum C, Kamoun S. 2010. Genome evolution following host jumps in the Irish potato famine pathogen lineage. Science 330:1540–1543. doi: 10.1126/science.1193070. [DOI] [PubMed] [Google Scholar]
- 25.Brasier CM. 1992. Evolutionary biology of Phytophthora. I. Genetic system, sexuality and the generation of variation. Annu Rev Phytopathol 30:153–171. doi: 10.1146/annurev.py.30.090192.001101. [DOI] [Google Scholar]
- 26.Sansome E, Brasier CM. 1973. Diploidy and chromosomal structural hybridity in Phytophthora infestans. Nature 241:344–345. doi: 10.1038/241344a0. [DOI] [Google Scholar]
- 27.Sansome E. 1977. Polyploidy and induced gametangial formation in British isolates of Phytophthora infestans. Microbiology 99:311–316. doi: 10.1099/00221287-99-2-311. [DOI] [Google Scholar]
- 28.Tooley PW, Therrien CD. 1987. Cytophotometric determination of the nuclear DNA content of 23 Mexican and 18 non-Mexican isolates of Phytophthora infestans. Exp Mycol 11:19–26. doi: 10.1016/0147-5975(87)90032-6. [DOI] [Google Scholar]
- 29.Catal M, King L, Tumbalam P, Wiriyajitsomboon P, Kirk WW, Adams GC. 2010. Heterokaryotic nuclear conditions and a heterogeneous nuclear population are observed by flow cytometry in Phytophthora infestans. Cytometry A 77:769–775. doi: 10.1002/cyto.a.20888. [DOI] [PubMed] [Google Scholar]
- 30.Li Y, Shen H, Zhou Q, Qian K, van der Lee T, Huang S. 2017. Changing ploidy as a strategy: the Irish potato famine pathogen shifts ploidy in relation to its sexuality. Mol Plant Microbe Interact 30:45–52. doi: 10.1094/MPMI-08-16-0156-R. [DOI] [PubMed] [Google Scholar]
- 31.Blair JE, Coffey MD, Park S-Y, Geiser DM, Kang S. 2008. A multi-locus phylogeny for Phytophthora utilizing markers derived from complete genome sequences. Fungal Genet Biol 45:266–277. doi: 10.1016/j.fgb.2007.10.010. [DOI] [PubMed] [Google Scholar]
- 32.Goss EM, Cardenas ME, Myers K, Forbes GA, Fry WE, Restrepo S, Grünwald NJ. 2011. The plant pathogen Phytophthora andina emerged via hybridization of an unknown Phytophthora species and the Irish potato famine pathogen, P. infestans. PLoS One 6:e24543. doi: 10.1371/journal.pone.0024543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Knaus BJ, Grünwald NJ. 2018. Inferring variation in copy number using high throughput sequencing data in R. Front Genet 9:123. doi: 10.3389/fgene.2018.00123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Raffaele S, Kamoun S. 2012. Genome evolution in filamentous plant pathogens: why bigger can be better. Nat Rev Microbiol 10:417–430. doi: 10.1038/nrmicro2790. [DOI] [PubMed] [Google Scholar]
- 35.Dong S, Raffaele S, Kamoun S. 2015. The two-speed genomes of filamentous pathogens: waltz with plants. Curr Opin Genet Dev 35:57–65. doi: 10.1016/j.gde.2015.09.001. [DOI] [PubMed] [Google Scholar]
- 36.Oliva RF, Kroon L, Chacón G, Flier WG, Ristaino JB, Forbes GA. 2010. Phytophthora andina sp. nov., a newly identified heterothallic pathogen of solanaceous hosts in the Andean highlands. Plant Pathol 59:613–625. doi: 10.1111/j.1365-3059.2010.02287.x. [DOI] [Google Scholar]
- 37.Kasuga T, Bui M, Bernhardt E, Swiecki T, Aram K, Cano LM, Webber J, Brasier C, Press C, Grünwald NJ, Rizzo DM, Garbelotto M. 2016. Host-induced aneuploidy and phenotypic diversification in the sudden oak death pathogen Phytophthora ramorum. BMC Genomics 17:1–17. doi: 10.1186/s12864-016-2717-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hu J, Shrestha S, Zhou Y, Mudge J, Liu X, Lamour K. 2020. Dynamic extreme aneuploidy (DEA) in the vegetable pathogen Phytophthora capsici and the potential for rapid asexual evolution. PLoS One 15:e0227250. doi: 10.1371/journal.pone.0227250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kato M, Mizubuti ES, Goodwin SB, Fry WE. 1997. Sensitivity to protectant fungicides and pathogenic fitness of clonal lineages of Phytophthora infestans in the United States. Phytopathology 87:973–978. doi: 10.1094/PHYTO.1997.87.9.973. [DOI] [PubMed] [Google Scholar]
- 40.Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Broad Institute. Picard Tools. https://broadinstitute.github.io/picard/.
- 43.Wickham H. 2009. ggplot2: elegant graphics for data analysis. Springer, New York, NY. [Google Scholar]
- 44.Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, Banks E, Garimella KV, Altshuler D, Gabriel S, DePristo MA. 2013. From FastQ data to high-confidence variant calls: the genome analysis toolkit best practices pipeline. Curr Protoc Bioinformatics 43:11.10.1–11.10.33. doi: 10.1002/0471250953.bi1110s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ. 2011. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, McVean G, Durbin R, 1000 Genomes Project Analysis Group. 2011. The variant call format and VCFtools. Bioinformatics 27:2156–2158. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.R Core Team. 2018. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- 48.Knaus BJ, Grünwald NJ. 2017. VCFR: a package to manipulate and visualize variant call format data in R. Mol Ecol Resour 17:44–53. doi: 10.1111/1755-0998.12549. [DOI] [PubMed] [Google Scholar]
- 49.Martin FN, Blair JE, Coffey MD. 2014. A combined mitochondrial and nuclear multilocus phylogeny of the genus Phytophthora. Fungal Genet Biol 66:19–32. doi: 10.1016/j.fgb.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 50.Vos RA, Balhoff JP, Caravas JA, Holder MT, Lapp H, Maddison WP, Midford PE, Priyam A, Sukumaran J, Xia X, Stoltzfus A. 2012. NeXML: rich, extensible, and verifiable representation of comparative data and metadata. Syst Biol 61:675–689. doi: 10.1093/sysbio/sys025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kroon LP, Brouwer H, de Cock AW, Govers F. 2012. The genus Phytophthora anno 2012. Phytopathology 102:348–364. doi: 10.1094/PHYTO-01-11-0025. [DOI] [PubMed] [Google Scholar]
- 52.Flier WG, Grünwald NJ, Kroon LPNM, Sturbaum AK, van den Bosch TBM, Garay-Serrano E, Lozoya-Saldaña H, Fry WE, Turkensteen LJ. 2003. The population structure of Phytophthora infestans from the Toluca Valley of central Mexico suggests genetic differentiation between populations from cultivated potato and wild Solanum spp. Phytopathology 93:382–390. doi: 10.1094/PHYTO.2003.93.4.382. [DOI] [PubMed] [Google Scholar]
- 53.Knaus BJ, Tabima JF, Shakya SK, Judelson HS, Grünwald NJ. 2020. Supplements for Knaus et al. manuscript on changes variation in genic copy number in Phytophthora infestans and relatives. GitHub https://github.com/grunwaldlab/Supplements_Knaus_Pinfestans_CNV.