

Visual Abstract

Key Words: diastolic dysfunction, heart failure, pressure overload, sympathetic overdrive, t-tubule
Abbreviations and Acronyms: AAV9, adeno-associated virus 9; ANOVA, analysis of variance; AR, adrenergic receptor; ATPase, adenosine triphosphatase; BW, body weight; CAMKII, Ca2+/calmodulin-dependent protein kinase; cBIN1, cardiac bridging integrator 1; CMV, cytomegalovirus; Di-8-ANNEPs, 4-[2-[6-(Dioctylamino)-2-naphthalenyl]ethenyl]-1-(3-sulfopropyl)-pyridinium, inner salt; EC, excitation contraction; EDV, end diastolic volume; EF, ejection fraction; GFP, green fluorescent protein; HF, heart failure; HW, heart weight; HR, heart rate; HT, heterozygote; ISO, isoproterenol; jSR, junctional sarcoplasmic reticulum; LSD, least significant difference; LTCC, voltage-dependent L-type calcium channel; LV, left ventricular; LW, lung weight; PBS, phosphate-buffered saline; PKA, protein kinase A; PLN, phospholamban; RyR, ryanodine receptor; RWT, relative wall thickness; SD, standard deviation; SEM, standard error of the mean; SERCA2a, sarcoplasmic reticulum calcium ATPase pump 2a; SR, sarcoplasmic reticulum; STORM, stochastic optical reconstruction microscopy; TAC, transverse aortic constriction; TEM, transmission electron microscopy; t-tubule, transverse-tubule; vg, vector genome; WT, wild type
Highlights
-
•
T-tubule cBIN1-microdomains are disrupted in hearts with concentric hypertrophy.
-
•
cBIN1 replacement therapy rescues t-tubule microdomains and reduces concentric hypertrophy in post-ISO hearts inducing a hyper-efficient phenotype similar to athletic hearts.
-
•
cBIN1-microdomains organize t-tubule Cav1.2 and SERCA2a distribution for improved contractility and lusitropy.
-
•
Exogenous cBIN1 is also effective in protecting cardiac contractility and lusitropy in mouse hearts subjected to pressure overload.
Summary
Heart failure is an important, and growing, cause of morbidity and mortality. Half of patients with heart failure have preserved ejection fraction, for whom therapeutic options are limited. Here we report that cardiac bridging integrator 1 gene therapy to maintain subcellular membrane compartments within cardiomyocytes can stabilize intracellular distribution of calcium-handling machinery, preserving diastolic function in hearts stressed by chronic beta agonist stimulation and pressure overload. This study identifies that maintenance of intracellular architecture and, in particular, membrane microdomains at t-tubules, is important in the setting of sympathetic stress. Stabilization of membrane microdomains may be a pathway for future therapeutic development.
Heart failure (HF) is a global health concern, with an estimated 6.2 million people affected in the United States (1). Approximately 50% of patients with HF have preserved ejection fraction (EF) with diastolic failure, for which there is lack of effective treatment. Diastolic failure can result from ventricular remodeling and diastolic dysfunction, which can occur secondary to chronic sympathetic activation (2). Preventing remodeling within individual ventricular myocytes may improve overall cardiac remodeling and have therapeutic benefits for failing hearts.
Cardiac transverse tubules (t-tubule) are critical for the initiation of calcium transients and maintenance of efficient excitation-contraction (EC) coupling. Pathological t-tubule remodeling is a consequence of β-adrenergic stimulation in HF (3, 4, 5). Furthermore, impaired t-tubule microdomains have been implicated in HF progression (6, 7, 8, 9). In fact, t-tubule remodeling can be the tipping point from hypertrophy to failure (10). Normal calcium transients (11), which require L-type calcium channels (LTCCs) to be at t-tubule microdomains, are crucial to cardiac contraction and relaxation. The t-tubule membrane scaffolding protein cardiac bridging integrator 1 (cBIN1) (12), which facilitates LTCC trafficking (13) and clustering for dyad organization, is also under the regulation of β-adrenergic receptor (AR) signaling (14). Furthermore, cBIN1 is reduced in HF (14, 15, 16) and the resultant cBIN1-microdomain disruption impairs normal stress response, limiting contractility and promoting arrhythmias. Therapeutic approaches that preserve cBIN1-microdomains may benefit stressed hearts by protecting the calcium-handling machinery, slowing HF progression.
In the present study, we explored whether in vivo over-expression of exogenous cBIN1 can limit myocardial remodeling and dysfunction. Continuous isoproterenol infusion, which causes reduced myocardial cBIN1 expression and disorganized intracellular distribution of calcium-handling proteins, also induces pathological concentric hypertrophy with diastolic dysfunction. We find that normalization of cBIN1 through adeno-associated virus 9 (AAV9) mediated gene transfer both increases inotropy and preserves lusitropy, reducing pathological hypertrophy. Within cardiomyocytes, we find that exogenous cBIN1 preserves the intracellular distribution of LTCCs at t-tubules and the localization of the sarcoplasmic reticulum (SR) calcium-ATPase 2a (SERCA2a). The protective effects of cBIN1 are both isoform-specific and confirmed effective in a second model of transverse aortic constriction (TAC)–induced cardiac hypertrophy and HF, indicating that exogenous cBIN1-mediated preservation of t-tubule microdomains is a possible therapeutic approach to improve myocardial function in hearts under chronic stress.
Methods
An expanded Methods section is provided in the Supplemental Material.
Animal procedures
All mouse procedures were reviewed and approved by the Institutional Animal Care and Use Committee of Cedars-Sinai Medical Center. Adult male C57Bl/6 mice (Jackson Laboratory, Sacramento, California) were administered with 3 × 1010 vector genome (vg) of AAV9 transducing green fluorescent protein (GFP) or BIN1 isoforms (Welgen, Inc., Worcester, Massachusetts) via retro-orbital injection (17). Three weeks later, mice were implanted subcutaneously with osmotic mini-pumps releasing phosphate-buffered saline (PBS) or isoproterenol (ISO) (30 mg/kg/day). Fifty-six mice were randomized into GFP+PBS, GFP+ISO, cBIN1+PBS, or cBIN1+ISO group (N = 14/group). Another 50 mice were randomized into receiving AAV9-GFP, cBIN1, BIN1, BIN1+17, or BIN1+13 (N = 10/group) before ISO. AAV9 was used because it is a promising gene therapy vehicle and exhibits the highest cardiac tropism (18). The cytomegalovirus (CMV) promoter was used given its efficiency and safety in cardiac gene transfer (19). AAV9-CMV-GFP was used as the negative control virus because it does not induce cardiomyocyte toxicity and has been successfully used as a negative control virus in numerous gene therapy studies with animal models of cardiovascular diseases (20). For TAC study, either adult male cardiac-specific Bin1 heterozygotes (HT) (Bin1flox/+, Myh6-cre+) with their wild type (WT) (Bin1flox/+, Myh6-cre -) littermates (12) or adult male C57BL/6 mice (Jackson Laboratory, Sacramento, California) were used. All mice were anesthetized at the age of 8 to 10 weeks and subjected to open-chest TAC or mock surgery (Sham). TAC was performed by tying a 7-0 silk suture against a 27-gauge needle between the first and second branch off the aortic arch. For gene therapy, same as the ISO study, mice received retro-orbital injection of 3 × 1010 vg of AAV9 virus transducing cBIN1-V5 or GFP-V5 at 3 weeks prior to the onset of TAC.
Echocardiograms were recorded using a Vevo-3100 ultrasound system (Visual Sonics, Toronto, Ontario, Canada) equipped with a 70-MHz transducer. Protein interaction was analyzed using immunofluorescent imaging and biochemical coimmunoprecipitation. Peak intensity of Cav1.2 at t-tubules is quantified using Image J as previously reported (13). Power spectrum analysis was analyzed in Matlab using fast Fourier transform conversion (10,13). Intracellular protein distribution was analyzed using sucrose gradient fractionation using a previously established method (21). For calcium transient measurement, Cal-520-AM (AAT Bioquest, Sunnyvale, California) was used as previously described (14). Three-dimensional super-resolution stochastic optical reconstruction microscopy (STORM) images were obtained (14) for nearest neighbor analysis between LTCC–ryanodine receptor 2 (RyR) and SERCA2a-cBIN1 molecules.
Statistical analysis
Data were analyzed using GraphPad Prism version 7.0 (GraphPad Software, La Jolla, California). All data are presented as mean ± SEM or SD as specified. Normality was assessed using the Shapiro-Wilk test. Continuous variables were compared using Student's t-test/Mann-Whitney U and 1-way analysis of variance (ANOVA)/Kruskal-Wallis tests. Two-way ANOVA was used to determine differences between 2 AAV9 groups with different drug infusion, which was then followed by Fisher least significant difference (LSD) post-hoc adjustment for multiple pairwise comparisons. Categorical variables were analyzed using Fisher exact or chi-square tests. For survival comparison, log-rank test was used to compare Kaplan-Meier survival curves between groups. Two-sided p values were used and p < 0.05 was considered statistically significant.
Results
Exogenous CBIN1 reduces concentric hypertrophy in mouse hearts after ISO infusion
We investigated the effect of cBIN1 on myocardial function in animals subjected to 4 weeks of ISO infusion (Figure 1A). AAV9 was used to introduce myocardial expression of exogenous V5-tagged GFP or cBIN1 (22) 3 weeks prior to the onset of ISO. Anti-V5 labeling identified a similar percent of myocardial area with detectable V5 signal at 7 weeks after AAV9 injection (GFP, 62.4 ± 10.5%; cBIN1, 57.9 ± 8.0%), indicating successful transduction of exogenous protein in over half of cardiomyocytes (Supplemental Figure 1). It is possible that the remaining almost 40% of negatively stained cardiomyocytes may express exogenous proteins at a low level below the detection threshold of immunofluorescence. In all mice, ISO significantly increased heart weight to body weight ratio (HW/BW), indicating cardiac hypertrophy (Figure 1B). Cardiac geometry and function were assessed using echocardiography (Figures 1C to 1H). In AAV9-GFP–pretreated animals, ISO induced a significant increase in left ventricular (LV) mass and relative wall thickness (RWT), without altering end diastolic volume (EDV), consistent with echocardiography-based classification of concentric hypertrophy (23). In AAV9-cBIN1–pretreated animals, the ISO increase of LV mass was attenuated with a normal RWT and increased EDV, similar to echocardiography-classified “physiological hypertrophy-like LV remodeling” using a previous reported method (23) (Supplemental Figure 2A). Furthermore, α-smooth muscle actin is increased in GFP+ISO hearts but not in cBIN1+ISO hearts (Supplemental Figure 2B), indicating AAV9-cBIN1 limits ISO-induced LV hypertrophy. In AAV9-GFP–pretreated mice, ISO resulted in a small increase in EF (p = 0.050 vs. GFP+PBS and p = 0.007 vs. cBIN1+PBS group), yet E/e’ strongly increased, indicating the onset of diastolic dysfunction. In mice pretreated with AAV9-cBIN1, ISO still increased systolic function and importantly maintained a normal E/e’, indicating positive inotropy with preserved lusitropy. Furthermore, although without blood pressure measurement, which remains a limitation of the current study, ISO significantly increased heart rate (HR) in all animals, indicating that ISO is effective in causing hemodynamic stress. Yet, post-ISO HR was not different between GFP+ISO and cBIN1+ISO mice (Supplemental Table 1), confirming that further improved post-ISO cardiac output in AAV9-cBIN1 mice was due to muscle efficiency and not increased rate.
Figure 1.

Exogenous cBIN1 Reduces Concentric Hypertrophy in Post-ISO Mouse Hearts
(A) Experimental protocol: 56 mice were randomized into 4 experimental groups: AAV9-GFP+PBS, AAV9-GFP+ISO, AAV9-cBIN1+PBS, and AAV9-cBIN1+ISO (n = 14/group). (B) Mouse HW/BW in the 4 groups. (C) Representative images of longitudinal axis view of left ventricles at both end diastolic and end systolic phase at 4 weeks post-PBS or ISO infusion. Echocardiography analysis of end diastolic volume, LV mass, and relative wall thickness (D), ejection fraction (E), E/e’ (F), stroke volume (G), and cardiac output (H) is also included. Data are presented as mean ± SEM. Two-way ANOVA was used followed by Fisher LSD test for multiple comparison. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001, for PBS vs. ISO comparison within each AAV9 treatment group ##p <0.01 and ###p <0.001 for GFP vs. cBIN1 comparison within each drug infusion group. AAV9 = adeno-associated virus 9; ANOVA = analysis of variance; BW = body weight; cBIN1 = cardiac bridging integrator 1; GFP = green fluorescent protein; HW = heart weight; ISO = isoproterenol; LSD = least significant difference; LV = left ventricular; PBS = phosphate-buffered saline; SEM = standard error of the mean.
Chronic ISO-disrupted CBIN1-microdomains can be normalized by AAV9-CBIN1
It is well known that both myocardial inotropy and lusitropy are related to cardiomyocyte calcium cycling (24). cBIN1, the structural organizer for dyad microdomains (14), creates t-tubule microfolds to limit extracellular Ca2+ diffusion (12), facilitates microtubule-dependent forward trafficking of LTCCs (13), and clusters LTCCs that are already delivered to t-tubule membranes. We, therefore, explored how cBIN1-microdomains may remodel in hypertrophic hearts after chronic ISO infusion. Western blots of heart lysates indicate that ISO induces a significant reduction in cBIN1 protein, which is normalized by AAV9-cBIN1 (Figure 2A). Note that immunoprecipitation with anti-BIN1-exon 17 antibody followed by Western blot detection with anti-BIN1-exon 13 antibody confirms that the examined protein band is the cBIN1 (BIN1+13+17) isoform. Gross t-tubule network architecture was also examined in isolated cardiomyocytes labeled with a membrane dye, 4-[2-[6-(Dioctylamino)-2-naphthalenyl]ethenyl]-1-(3-sulfopropyl)-pyridinium, inner salt (Di-8-ANNEPs) (Figure 2B). Live-cell imaging followed by power spectrum analysis indicates that overall t-tubule organization (normalized peak power density) remains similar in GFP+ISO mice, and is increased by AAV9-cBIN1. Even though the gross t-tubule network remains organized, using transmission electron microscopy imaging, we noted in GFP+ISO hearts that there was a reduction in t-tubule microfolds, which were preserved in cBIN1+ISO hearts (Figure 2C). Quantitation of the degree of contoured t-tubules using a modified scoring system established previously (12) (1, round or dilated t-tubule lumen without folds; 2, non-circular contoured t-tubule lumen without folds; 3, t-tubules with 2 to 3 layer of folds; or 4, t-tubules with >3 layer of folds) identifies a significant reduction in t-tubule contour in GFP+ISO hearts, which is normalized in cBIN1+ISO hearts (p < 0.001, chi-square test). Note the exaggerated microfolds (more than 3 layers of folds, score of 4) are found in cBIN1+PBS hearts, a result of greater than physiological levels of cBIN1. These data indicate that cBIN1 is critical for the formation of t-tubule microfolds, the folds are down-regulated under chronic sympathetic overdrive, and the folds can be restored using cBIN1 exogenous therapy.
Figure 2.

ISO Reduces cBIN1 and Disrupts cBIN1-Microfolds, Which Is Normalized by AAV9-cBIN1
(A) Western blots of cBIN1 and GAPDH from heart lysates and immunoprecipitated heart lysates from GFP+PBS, GFP+ISO, cBIN1+PBS, and cBIN1+ISO hearts. Quantification in the bar graph to the right (N = 6–7 hearts per group). (B) Representative cardiomyocyte images with Di-8-ANNEPs labeling (top panel) (Scale bar, 10 μm) and power spectrum (bottom panel) of the corresponding boxed region of interest above. Quantification of peak power density is included in the bar graph to the left (N = 26 to 31 cells from 3 to 4 hearts per group). Data are presented as mean ± SEM. Two-way ANOVA was used followed by Fisher LSD test for multiple comparison. ∗p < 0.05 and ∗∗p < 0.01 for PBS vs. ISO comparison within each AAV9 treatment group; #p < 0.05 and ##p < 0.01 for GFP vs. cBIN1 comparison within each drug infusion group. (C) Transmission electron microscopy imaging of t-tubule (TT) microfolds from myocardial tissue from all 4 groups (scale bar, 1 μm). Quantitation of the degree of contour of TTs from each group is included in the bar graphs to the left (N = 232 to 305 TTs from 60 to 100 images of 5 to 6 myocardial sections and 2 to 3 hearts from each group). Chi-square test was used to compare TT contour between groups, p < 0.001 for comparison of GFP+PBS vs. GFP+ISO, GFP+ISO vs. cBIN1+ISO, and cBIN1+PBS vs. other groups. Di-8-ANNEPs = 4-[2-[6-(Dioctylamino)-2-naphthalenyl]ethenyl]-1-(3-sulfopropyl)-pyridinium, inner salt; GAPDH = glyceraldehyde 3-phosphate dehydrogenase; TTs = t-tubules; other abbreviations as in Figure 1.
We then explored Cav1.2 expression and intracellular distribution in cardiomyocytes. In post-ISO hearts, the net myocardial protein expression of Cav1.2 is similar (Figure 3A). However, myocardial tissue immunofluorescent labeling of Cav1.2 reveals that channel density along t-tubules was significantly reduced in GFP+ISO cardiomyocytes, which was normalized by AAV9-cBIN1 (Figures 3B and 3C, power spectrum and fluorescent profile analysis). These data are consistent with previous observations of altered Cav1.2 protein distribution despite similar total protein levels (15). With reduced Cav1.2 localization to t-tubules, the peak amplitude of calcium transient (ΔF/F0) was significantly reduced in GFP+ISO cardiomyocytes when compared with that from control GFP+PBS myocytes (Figure 3D), which was normalized by exogenous cBIN1. Ryanodine receptor 2 (RyR) total protein expression and intracellular distribution were not different across all groups (Supplemental Figure 3). However, RyRs became hyper-phosphorylated at both protein kinase A (PKA)-dependent S2808 and Ca2+/calmodulin-dependent protein kinase II (CAMKII)-dependent S2814, consistent with a previous report (25). Together with increased phosphorylation at T287 in CAMKIIδ, these data indicate that PKA and CAMKII activation-induced RyR hyperphosphorylation occurs after chronic ISO infusion. Importantly, AAV9-cBIN1 pretreatment successfully blunts these pathways and reduces RyR hyperphosphorylation (Supplemental Figure 4).
Figure 3.

cBIN1 Increases Cav1.2 Localization to t-Tubules
(A) Western blot of Cav1.2 in heart lysates from GFP+PBS, GFP+ISO, cBIN1+PBS, and cBIN1+ISO hearts. Quantification (Cav1.2/GAPDH and Cav1.2/Troponin) is included in the bar graphs to the right (n = 5 to 7 hearts per group). (B) Representative confocal images (100×) of anti-Cav1.2 labeling in mouse myocardium from each group (top 2 panels) (scale bar, 10 μm). The third panel includes power spectrum and the fourth panel includes fluorescence intensity profiles within the boxed areas along the cardiomyocyte longitudinal axis. Quantification of Cav1.2 peak power density and immunofluorescent intensities at t-tubules in each group is summarized in the bar graph in C (n = 15 to 32 cell images from 3 to 4 hearts per group). Scale bar: 10 m. (D) Representative calcium transient tracing from each group and quantification of peak amplitudes (ΔF/F0) (n = 61 to 88 cells from 6 hearts per group). Data are presented as mean ± SEM. Two-way ANOVA was used followed by Fisher LSD test for multiple comparison. ∗∗∗p < 0.001 for PBS vs. ISO comparison within each AAV9 treatment group; ###p < 0.001 for GFP vs. cBIN1 comparison within each drug infusion group. Abbreviations as in Figure 1 and Figure 2.
Exogenous CBIN1 improves SERCA2A distribution along SR
Cardiac lusitropy is most directly related to calcium reuptake via SERCA2a. To our surprise, despite impaired diastolic dysfunction in GFP+ISO hearts, total protein expression of SERCA2a was significantly increased after ISO infusion (Figures 4A and 4B). Total protein levels of phospholamban (PLN) and its phosphorylated forms (pS16 and pT17) are not altered (Supplemental Figure 4). Previous studies indicate that acute ISO-induced PLN phosphorylation can be normalized after chronic ISO infusion and even PLN dephosphorylation can occur due to activation of serine/threonine phosphatases PP1 and PP2A (26,27). Consistent with these reports, our results indicate that unchanged PLN phosphorylation after 4 weeks of ISO infusion is a possible net result of balanced local activation of both kinases and phosphatases. These data indicate that both SERCA2a protein and activity are not decreased in post-ISO hearts. Given the effect of cBIN1 on Cav1.2 localization, we then examined SERCA2a localization. Myocardial tissue sections with SERCA2a labeling were imaged with spinning-disc confocal microscopy and compared across groups (Figure 4C). In GFP+PBS hearts, a subpopulation of SERCA2a was concentrated to the t-tubule/ junctional sarcoplasmic reticulum (jSR) regions, giving rise to an organized distribution with a major power spectrum peak at 1.8 to 2 μm, corresponding to the full length of a sarcomere. Over-expression of cBIN1 in the cBIN1+PBS hearts further increased SERCA2a signals near t-tubule/jSR. In GFP+ISO hearts, intracellular distribution of SERCA2a was disorganized with a significant reduction in peak power density, which was normalized in cBIN1+ISO hearts (quantification in Figure 4D).
Figure 4.

cBIN1 Organizes Intracellular Distribution of SERCA2a in Post-ISO Hearts
(A) Western blot of SERCA2a in heart lysates from GFP+PBS, GFP+ISO, cBIN1+PBS, and cBIN1+ISO hearts. (B) Quantification (SERCA2a/Actin) is included in the bar graph (n = 6 to 8 hearts per group). (C) Representative confocal images of anti-SERCA2a labeling in mouse myocardium from each group (top 2 panels). Scale bar: 10 μm. The third panel includes the power spectrum of SERCA2a of the boxed area above. (D) Quantification of peak power density of SERCA2a (n = 11 to 15 cell images from 3–4 hearts per group). Data are expressed as mean ± SEM. Two-way ANOVA was used followed by Fisher LSD test for multiple comparison. ∗∗p < 0.01 and ∗∗∗p, 0.001 for PBS vs. ISO comparison within each AAV9 treatment group; ### p < 0.001 for GFP vs. cBIN1 comparison within each drug infusion group. Abbreviations as in Figure 1.
Intracellular distribution of Cav1.2 and SERCA2a was further explored using biochemical sucrose gradient-based fractionation of cardiac microsomes (21). As indicated in Supplemental Figure 5A, fraction 4 (F4) has the lowest recovery yield when compared with other fractions. However, even with a low yield, Cav1.2 and cBIN1 are detectable in F4 with limited Na+/K+-ATPase and depleted SERCA2a, indicating that F4 is enriched with t-tubule origin microsomes (Supplemental Figure 5B). When normalizing t-tubule protein concentration for F4 across all samples (2.5 μg protein loaded per lane), GFP+ISO hearts have a significant reduction in both cBIN1 and Cav1.2 protein per unit t-tubule when compared with control GFP+PBS hearts, which was normalized using AAV9-cBIN1 pretreatment (Figure 5A). These data are consistent with immunofluorescent imaging, which identified less t-tubule localization of Cav1.2 channels following ISO infusion and restoration with AAV9-cBIN1. On the other hand, SR proteins are detected only in fractions F2 and F3. When normalizing SR protein concentration for F2 and F3 (25 μg protein loaded per lane), F3 has relatively more RyR and less PLN than F2 (Figure 5B), indicating more enrichment of jSR toward the heavier F3 fraction. Quantification of SERCA2a expression in F2 and F3 identifies that when compared with AAV9-GFP, AAV9-cBIN1 causes a significant increase in SERCA2a distribution into the heavier and more jSR-enriched F3, but not the longitudinal SR-enriched F2 fraction (Figure 5B). Note that ISO alone does increase SERCA2a expression in F3 in AAV9-GFP mouse hearts, likely due to an overall increase in total protein expression of SERCA2a in post-ISO hearts (Figure 4A). These data indicate that, in ISO-infused hearts, exogenous cBIN1 can maintain t-tubule microdomains to localize Cav1.2 and SERCA2a to their functional sites.
Figure 5.

Sucrose Gradient Fractionation of Cardiac Microsomes
(A) Representative Western blots of Cav1.2 and cBIN1 in the F4 (TT) fraction of cardiac microsome from GFP+PBS, GFP+ISO, cBIN1+PBS, and cBIN1+ISO hearts (2.5 μg proteins loaded per lane). Quantification is included in the bar graphs (n = 3 hearts per group). (B) Representative Western blots of RyR, PLN, and SERCA2a in the F2 (longitudinal SR enriched) and F3 (jSR enriched) fractions of cardiac microsome from GFP+PBS, GFP+ISO, cBIN1+PBS, and cBIN1+ISO hearts (25 μg protein loaded per lane). Quantification of SERCA2a in F2 and F3 is included in the bar graphs to the right (n = 3 hearts per group). Data are expressed as mean ± SD. Two-way ANOVA was used followed by Fisher LSD test for multiple comparison. ∗p < 0.05, ∗∗p <0.01, and ∗∗∗p < 0.001 for PBS vs. ISO comparison within each AAV9 treatment group; #p < 0.05 and ##p < 0.01 for GFP vs. cBIN1 comparison within each drug infusion group. jSR = junctional sarcoplasmic reticulum; PLN = phospholamban; RyR = ryanodine receptor; SD = standard deviation; SERCA2a = sarcoplasmic reticulum calcium ATPase pump 2a; other abbreviations as in Figure 1.
Given the reduced Cav1.2 and SERCA2a at the t-tubule/jSR region, we next used STORM imaging to analyze nanoscale protein-protein co-localization for Cav1.2-RyR and SERCA2a-cBIN1 (Figure 6, Videos 1A, 1B, 1C, 1D, 2A, and 2B). Using nearest neighbor analysis, we quantified the distance between the individual Cav1.2 molecule and its closest RyR molecule. Histogram distribution of distances between Cav1.2-RyR molecules from whole cell images identifies a first peak near 40 nm in GFP+PBS, GFP+ISO, and cBIN1+ISO cardiomyocytes, corresponding to dyad couplons. In GFP+ISO hearts with preserved systolic function, the distribution histogram tends to shift to the right, yet with a still preserved first peak position (Figure 6B). Interestingly, cBIN1+PBS myocytes have a left-shifted histogram distribution and a significantly reduced Cav1.2-RyR peak distance, indicating tightened couplons likely brought closer by exaggerated cBIN1-microfolds as observed in transmission electron microscopy imaging. On the other hand, the distance between SERCA2a and its nearest neighbor cBIN1 at t-tubules has a trend of increase after ISO in AAV9-GFP–pretreated animals (p = 0.063; GFP+PBS vs. GFP+ISO), which is significantly decreased in AAV9-cBIN1–pretreated animals (p < 0.001; GFP+ISO vs. cBIN1+ISO) (Figures 6C to 6D). These data indicate that cBIN1-microfolds can regulate co-localization and interaction between EC coupling and calcium-handling proteins.
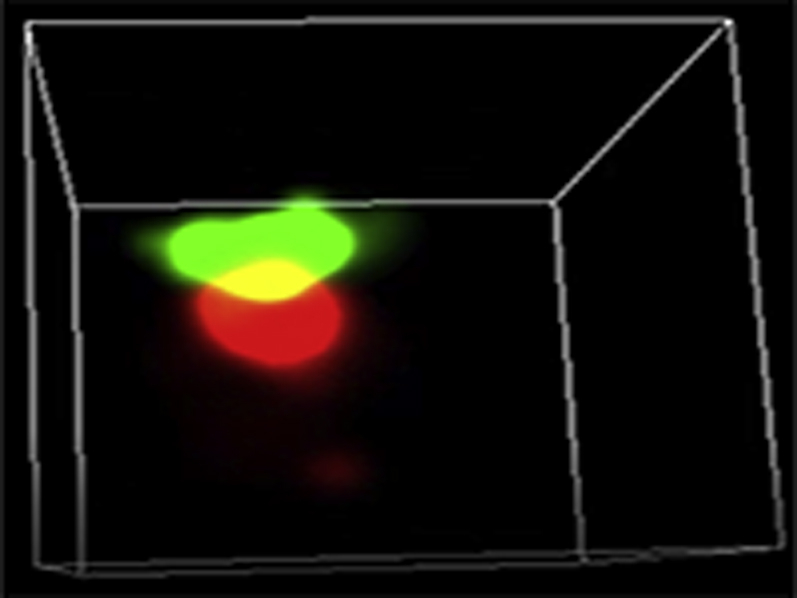
Representative three-dimensional (3D) STORM images of Cav1.2 (red) / RyR (green) couplons in cardiomyocytes isolated from GFP+PBS (A), cBIN1+PBS (B),GFP+ISO (C), or cBIN1+ISO (D) hearts.
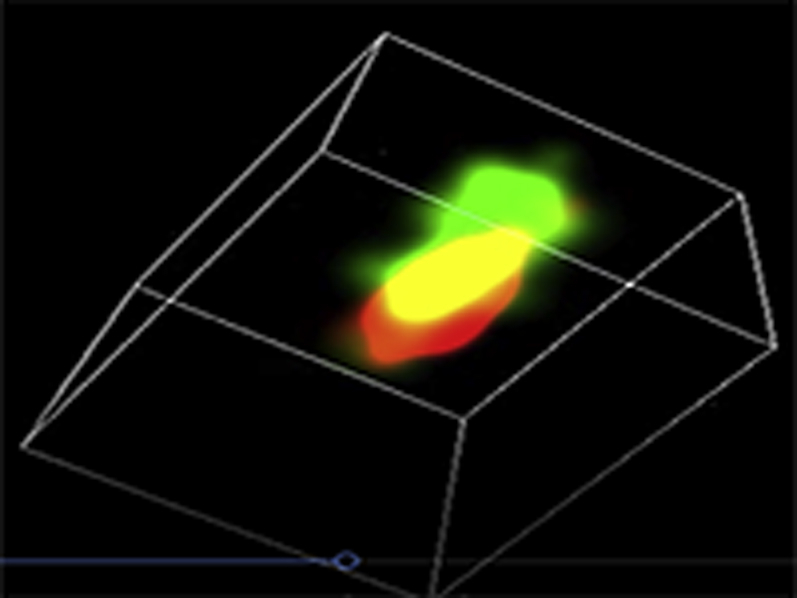
Representative three-dimensional (3D) STORM images of Cav1.2 (red) / RyR (green) couplons in cardiomyocytes isolated from GFP+PBS (A), cBIN1+PBS (B),GFP+ISO (C), or cBIN1+ISO (D) hearts.
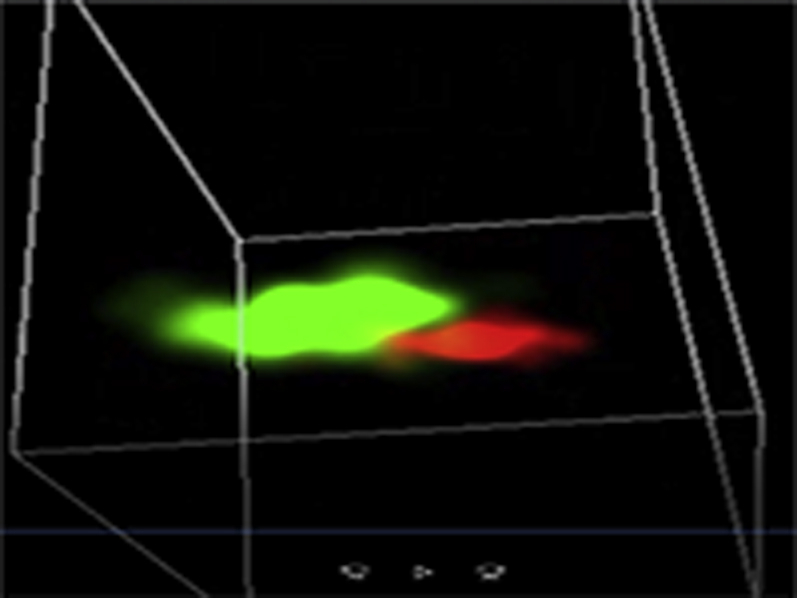
Representative three-dimensional (3D) STORM images of Cav1.2 (red) / RyR (green) couplons in cardiomyocytes isolated from GFP+PBS (A), cBIN1+PBS (B),GFP+ISO (C), or cBIN1+ISO (D) hearts.

Representative three-dimensional (3D) STORM images of Cav1.2 (red) / RyR (green) couplons in cardiomyocytes isolated from GFP+PBS (A), cBIN1+PBS (B),GFP+ISO (C), or cBIN1+ISO (D) hearts.
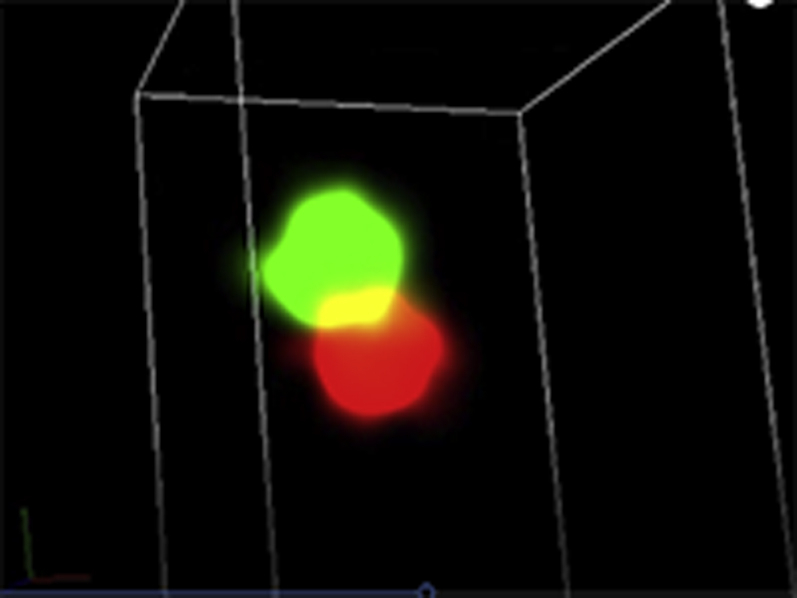
Representative three-dimensional (3D) STORM images of SERCA2a (green) / cBIN1 (red) couplons in cardiomyocytes isolated from GFP+PBS (A), cBIN1+PBS (B), GFP+ISO (C), or cBIN1+ISO (D) hearts.

Representative three-dimensional (3D) STORM images of SERCA2a (green) / cBIN1 (red) couplons in cardiomyocytes isolated from GFP+PBS (A), cBIN1+PBS (B), GFP+ISO (C), or cBIN1+ISO (D) hearts.
Figure 6.

Exogenous cBIN1 Brings Together Cav1.2-RyR and SERCA2a-cBIN1 Molecules in Cardiomyocytes
Super-resolution STORM imaging and nearest neighbor analysis of Cav1.2-RyR (A and B,Videos 1A, 1B, 1C, and 1D) and SERCA2a-cBIN1 (C and D,Videos 2A, and 2B) molecules in cardiomyocytes isolated from GFP+PBS, cBIN1+PBS, GFP+ISO, and cBIN1+ISO hearts. (A, C) Top-to-bottom, representative 2-D STORM cell images, representative 3-D STORM images of couplons, and histogram of nearest neighbor distance distribution obtained from full-cell 3-D STORM images. (B, D) Quantification of the first peak of nearest neighbor distance distribution histogram using full-cell image analysis (N = 7 to 17 cells from 2–3 animals per group). Data are expressed as mean ± SEM. Two-way ANOVA was used followed by Fisher LSD test for multiple comparison. ∗p < 0.05 for PBS vs. ISO comparison within each AAV9 treatment group; #p < 0.05 and ##p < 0.001 for GFP vs. cBIN1 comparison within each drug infusion group. STORM = stochastic optical reconstruction microscopy; other abbreviations as in Figures 1 and 5.
The phenotype of CBIN1+ISO hearts is isoform specific and unique to CBIN1
To further explore whether the observed phenotype of cBIN1+ISO hearts is an isoform-specific effect, we repeated the ISO protocol in 50 more mice, which were randomized to receive AAV9 transducing GFP and cBIN1, as well as the other 3 mouse cardiomyocyte expressing BIN1 isoforms including the small BIN1, BIN1+17, and BIN1+13. Similarly, 3 weeks after viral administration, mice were subjected to continuous subcutaneous ISO infusion at 30 mg/kg/day for 4 weeks. The protein expression of Cav1.2 and SERCA2a in post-ISO hearts was not significantly different when compared across 5 groups of mice transduced with GFP or BIN1 isoforms (Supplemental Figure 6A). Myocardial tissue immunofluorescent labeling of Cav1.2 channels reveals that channel density along t-tubules is significantly increased only in cBIN1-expressing hearts, but not the other BIN1 isoforms (Supplemental Figure 6B, quantification in 6D). Immunofluorescent imaging reveals that exogenous cBIN1 introduced by AAV9 organizes SERCA2a distribution (Supplemental Figure 6C, quantification in Supplemental Figure 6E), consistent with the data from Figure 4.
We next explored the functional consequence of different AAV9-BIN1 isoform pretreatment using echocardiography. cBIN1-expressing mice, when compared with the GFP group, lessened the ISO-induced increase in LV wall thickness, LV mass, and RWT (Figures 7A to 7D, Supplemental Table 2). In all animals, post-ISO cardiac output is significantly increased from its level at baseline, a result from the ISO-induced increase in HR (Supplemental Table 2). However, only cBIN1 hearts also have an improved systolic function, normalized E/e’, increased stroke volume, and further increased cardiac output when compared with post-ISO GFP hearts (Figures 7E to 7H). Of note, a partial cBIN1-like effect occurs in mice pretreated with BIN1+17, which significantly reduces LV mass, reduces E/e’, and attenuates ISO-increased RWT. The observed partial diastolic functional rescue from BIN1+17 is consistent with the partial rescue of intracellular distribution of SERCA2a using immunofluorescent imaging (Supplemental Figure 6C). However, due to the inability of BIN1+17 to increase Cav1.2 distribution at t-tubules, there is not a positive inotropic effect in AAV9-BIN1+17–pretreated hearts following ISO infusion.
Figure 7.

Echocardiography of Post-ISO Hearts Receiving AAV9-GFP, cBIN1, BIN1, BIN1+17, and BIN1+13
(A) Representative LV short axis M-mode images from each group at baseline (top) and 4 weeks after ISO treatment (bottom). In all of the 4-week post-ISO images, papillary muscles are marked by arrows (B to D). Quantitative analysis of LV mass (B), relative wall thickness (C), and ejection fraction (D) from each group (N = 10 mice per group). (E) Representative mitral valve inflow pulsed wave Doppler images (top) and tissue Doppler images of septal mitral valve annulus 4 weeks after ISO treatment (bottom). (F to H) Quantitative analysis of E/e’ (F), stroke volume (G), and cardiac output (H) from each group (N = 10 mice per group). Data are expressed as mean ± SEM. Two-way ANOVA was used followed by Fisher LSD test for multiple comparison. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 vs. baseline; #p < 0.05, ##p < 0.01, and ###p < 0.001 vs. the GFP group at 4 weeks post-ISO. Abbreviations as in Figure 1.
The cardiac protective effect of AAV9-CBIN1 is confirmed in TAC-induced HF
Next, we further explored the myocardial protective effect of cBIN1 in a separate mouse model of pressure overload induced using TAC. Mice with either genetic deficiency of cBIN1 or AAV9-transuced cBIN1 over-expression were tested in this study (Figure 8A). The deficiency study involved cardiac specific Bin1 HT mice and WT littermate controls (12), both subjected to TAC for 8 weeks. The over-expression study involved mice subjected to 8 weeks of TAC with prior injection of AAV9 transducing cBIN1-V5 or AAV9-GFP-V5, and mice subjected to an open-chest mock surgery (Sham). Mice were monitored and humanely killed at 8 weeks post-surgery. The viruses (AAV9-GFP/cBIN1-V5), dosage (3 x 1010 vg), administration time (3 weeks prior to surgery), and route (retro-orbital injection) are the same as those used in the ISO study. Aortic constriction in all TAC mice was confirmed by elevated trans-aortic pressure gradient (Figure 8B), establishing a similar increase in hemodynamic afterload in all mice receiving TAC. Kaplan-Meier curves summarizing severe HF-free (EF ≥35%) survival rates were included in Figure 8C. The rate of survival in Bin1 HT mice is 20.0% (nonsurvival 8 of 10, 2 deaths and 6 EF <35%), which is decreased from 71.4% (non-survival 4 of 14, 1 death and 3 EF <35%) in WT mice (p = 0.038 using log-rank test). As expected, all Sham mice survived through the entire experimental protocol (10 of 10, dotted black line). The rate of survival is decreased to 64.3% in the AAV9-GFP mice (non-survival 5 of 14, 5 EF <35%), which is significantly improved to 93.7% in the AAV9-cBIN1 group (non-survival 1 of 16, 1 EF <35%) (p = 0.020 using log-rank test). These data indicate that higher cBIN1 protein content in the heart is associated with better survival without development of systolic HF following pressure overload.
Figure 8.

cBIN1 Gene Transfer Improves Heart Failure–Free Survival in Post-TAC Mice
(A) Schematic protocol for the TAC study. (B) Trans-aortic pressure gradient measurement in all mice 5 days post-surgery. (C) Kaplan-Meier survival curves for heart failure–free survival (non-survival is death or EF <35%) in WT vs. Bin1 HT mice (left), and AAV9-GFP or cBIN1 pretreated mice (right). Log-rank test was used for survival comparison. HW/BW (D) and LW/BW (E) in all mice at 8 weeks post-TAC. (F) Representative M-mode echocardiography images of all mice 8 weeks after surgery. Echocardiography-measured left ventricular ejection fraction (G), end diastolic volume (H), LV mass (I), and E/e’ (J) for all mice at 8 weeks post-TAC. Data are expressed as mean ± SEM. Representative E and e’ images in the AAV9 treatment groups are included (right panel of J). Unpaired Student's t-test (or nonparametric Mann-Whitney test used for comparison between WT and Bin1 HT. One-way ANOVA or Kruskal-Wallis test followed by Fisher LSD test for multiple comparison was used for comparison among Sham, AAV9-GFP, and AAV9-cBIN1. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 for comparison vs. WT or Sham; #p < 0.05 and ##p < 0.01 for comparison of AAV9-GFP vs. AAV9-cBIN1. EF = ejection fraction; HT = heterozygote; LW = lung weight; TAC = transverse aortic constriction; WT = wild type; other abbreviations as in Figure 1.
At 8 weeks after TAC, surviving mice were humanely killed and evaluated for ratios of HW/BW and lung weight (LW)/BW (Supplemental Table 3, Figures 8D and 8E). Both HW/BW and LW/BW are significantly higher in Bin1 HT mice verses WT mice, indicating worsening of LV hypertrophy and lung edema with BIN1 deficiency. Regarding gene therapy, AAV9-cBIN1 significantly reduces LW/BW from that of the control GFP-TAC group to the level of Sham hearts, representing a striking reduction in TAC-induced pulmonary edema. Hypertrophy still occurs in the AAV9-cBIN1 hearts, although to a lesser extent. These data indicate that exogenous cBIN1 reduces TAC-induced hypertrophy and prevents deterioration into HF. Echocardiography analysis (Figures 8F to 8J, Supplemental Table 4) further indicates that when compared with WT-TAC mice, there is a significant decrease in EF and an increase in EDV in Bin1 HT-TAC hearts, indicating worsening of dilated cardiomyopathy with BIN1 deficiency. On the other hand, AAV9-cBIN1 significantly reduces TAC-induced LV dilation (EDV) and contractile dysfunction (EF), limiting the development of dilated cardiomyopathy. As a result, AAV9-cBIN1 pretreatment maintains stroke volume and cardiac output in post-TAC hearts without hearts being dilated. Furthermore, tissue doppler identified that the diastolic parameter E/e’ values of both lateral and septal walls are significantly improved in AAV9-cBIN1–pretreated hearts, indicating better diastolic function in mice with exogenous cBIN1. These data indicate that cBin1 gene therapy preserves myocardial systolic and diastolic function in stressed hearts and effectively prevents the development of dilated cardiomyopathy in mouse hearts subjected to pressure overload.
Discussion
This study indicates a beneficial effect of exogenous cBIN1 in preventing LV hypertrophy and cardiac dysfunction in stressed hearts. In mice subjected to continuous ISO infusion, exogenous cBIN1 offers an isoform-specific improvement in cardiac inotropy and lusitropy, limiting the development of LV hypertrophy. The cardiac protective effect of exogenous cBIN1 is further confirmed in mouse hearts subjected to pressure overload–induced HF.
Chronically elevated catecholamine levels and activation of cardiac β-ARs have a critical role in the pathogenesis of HF. Impaired myocardial structure and function have been observed in animals subjected to sustained sympathetic activation (28,29). ISO, which is a synthetic catecholamine and non-selective β-AR agonist, has been used in research to induce the model of LV hypertrophy and dysfunction (30). A high dose of ISO was used here to induce LV concentric hypertrophy with preserved systolic function. Chronic excessive cardiac workload–induced LV hypertrophy is associated with elevated risk of cardiovascular events (31), and preventing or reversing ventricular hypertrophy with preserved cardiac diastolic function is crucial to preventing the progression of stressed hearts to failing hearts. Here we found that cBIN1 attenuates chronic ISO-induced hypertrophy and at the same time conveys an isoform-specific improvement in stroke volume and cardiac output in hypertrophic hearts with preserved systolic function. The increase of LV volume in the cBIN1 hearts is not secondary to pump failure and dilated cardiomyopathy, but rather it reflects improvement in myocardial lusitropy (E/e’) with a parallel increase of intrinsic myocardial contractility (inotropy). This phenotype of cBIN1 hearts (Figure 1) is typical of athletic hearts in adaptation endurance training as characterized by chamber enlargement and increases of LV volume, stroke volume, and cardiac output (32, 33, 34). Aerobic exercise training has been reported to improve myocardial function and inotropic and lusitropic responses in both animal models (35,36) and patients with hypertension (37) and diastolic failure (38). Thus, exogenous cBIN1 may provide additional exercise-like benefit to patients with HF, improving exercise capacity and quality of life.
These post-ISO hearts are at a stage of hypertrophy with preserved systolic function, in which exogenous cBIN1 can effectively translate the increased demands on the heart into a functional effect. As a result, these functional and efficient cBIN1 hearts have limited hypertrophy development, which will likely prevent the next step of disease progression and HF development as occurs in the clinical setting. Next, the functional protective effect of exogenous cBIN1 in already decompensated hearts is also observed in a mouse model of TAC-induced hypertrophy and HF. Under pressure overload, compensated hypertrophy is an adaptive response. Over time, the adaptive response concedes to cardiac dilatation and the ensuing remodeling process becomes maladaptive, leading to worsening HF. We found that the fate of dilated cardiomyopathy development in pressure overload–stressed hearts is causally determined by myocardial content of cBIN1 protein. Following pressure overload, less cardiac BIN1 in genetically deleted Bin1 HT-TAC hearts is associated with more severe dilated cardiomyopathy, whereas greater cBIN1 with gene transfer improves cardiac systolic and diastolic function, limits HF, and improves HF-free survival (Figure 8). It remains unclear whether exogenous cBIN1 reduces myocyte death, which also contributes to LV dilation in post-TAC hearts. Future studies will be necessary to explore the effect of cBIN1 on myocyte survival in stressed hearts. Nevertheless, our data indicate that exogenous cBIN1 not only limits hypertrophy development in stressed hearts but also prevents myocardial transition from hypertrophy to dilated cardiomyopathy and HF in TAC mice.
The mechanism of improvement in cardiac inotropic function by cBIN1 is linked to its known effect in organizing t-tubule microdomains required for dyad organization and efficient EC coupling. cBIN1 creates t-tubule microfolds to organize a slow diffusion zone trapping extracellular t-tubule lumen ions, attracts LTCCs forward trafficking to t-tubules (13), clusters LTCCs that are already delivered to cell surface (39), and recruits RyRs to couple with LTCCs at dyads (14). Here we confirm in vivo that exogenous cBIN1, rather than any other BIN1 isoform, increases Cav1.2 localization to t-tubules (Figure 3, Supplemental Figure 6). These results support that preserved cBIN1-microdomain with organized LTCC distribution is responsible for the observed positive inotropic effect in sympathetically overdriven cBIN1 hearts. Whether cBIN1-microdomain regulates LTCC phosphorylation and its functional response to sympathetic stress including a well-established β-subunit-modulated Cav1.2 channel response (40,41) awaits future experimental explorations. Furthermore, RyR is critical to inotropy and hyper-phosphorylated leaky RyR plays a role in HF progression (42). Consistent with previous reports in an ISO model and human HF (25,42), we found that chronic ISO activates PKA and CAMKII-induced RyR hyperphosphorylation. AAV9-cBIN1 blunts these pathways, normalizing RyR phosphorylation following chronic sympathetic activation and preventing SR leak.
An additional novel finding from the current study is that exogenous cBIN1 increases SERCA2a function through organizing its intracellular distribution (Figures 4, 5, and 6). Chronic ISO-induced concentric hypertrophy with preserved systolic function is associated with disorganized intracellular distribution of SERCA2a yet increased overall protein expression. It is well accepted that SERCA2a activity is decreased in end-stage HF. Our data indicate that, in addition to reduced expression and impaired regulation by PLN, intracellular distribution of SERCA2a may also contribute to abnormal SR calcium reuptake activity in HF. Furthermore, as reported in adult rat ventricular cardiomyocytes with α-receptor agonist phenylephrine-induced hypertrophy, an adaptive increase in SERCA2a protein expression can occur due to elevated diastolic calcium-induced calcineurin/nuclear factor of activated T-cells activation (43). Thus, increased SERCA2a protein expression here is a possible adaptive response induced by elevated diastolic calcium concentration as indicated in elevated calcium-dependent phosphorylation at T287 of CAMKII. Thus, a transient increase in SERCA2a may occur at an early stage of all LV hypertrophy with preserved function. During disease progression, this adaptive increase in total SERCA2a protein expression will level off and even decrease as occurs in end-stage HF, resulting in severe diastolic and systolic failure. In cBIN1 hearts, organized SERCA2a along the SR indicates better calcium reuptake, therefore, less diastolic calcium overload for hearts still at a compensated stage. These results are consistent with a previous study in a rat model of HF that identified that increased BIN1 expression is associated with SERCA2a expression (44). Future studies exploring cBIN1 regulation of diastolic calcium concentration and calcineurin/nuclear factor of activated T-cells pathways will be needed to further understand its role in regulating SERCA2a expression and activity during disease progression. Note the effect on SERCA2a organization is not cBIN1-specific and can be partially induced by other BIN1 isoforms, particularly BIN1+17 (Supplemental Figure 6). This is consistent with the partial in vivo protective effects from BIN1+17 on cardiac hypertrophy and diastolic function (Figure 7). Whether and how BIN1 isoforms cooperate to organize SERCA2a distribution in normal and diseased cardiomyocytes require further exploration in future studies. Furthermore, through regulation of calcium-handling machineries at the SR, including SERCA2a distribution and RyR phosphorylation, cBIN1 may help maintain normal SR calcium load. As a limitation of the current study, future experiments are needed to quantify the effect of cBIN1 on SR calcium load, calcium release and reuptake kinetics, and arrhythmogenic spontaneous calcium release in chronically stressed hearts.
Nevertheless, the most robust protection of both inotropy and lusitropy in sympathetic overdriven hearts is only observed in the cBIN1 group (Figure 7), indicating possible further beneficial effect on lusitropy from cBIN1-dependent improvement in LTCC localization and dyad organization. With isoform-specific improvement in dyad organization, less orphaned leaky RyRs accumulate outside of dyads (14), limiting calcium leak from the SR and decreasing cytosolic calcium concentration during the diastolic phase. Together with the newly identified role on SERCA2a organization, our data indicate that a cBIN1-microdomain–related regulation offers a unique benefit in protecting cardiac lusitropy in addition to its inotropy effect. On the other hand, cBIN1 over-expression may also suppress the pathological effects of ISO stimulation by enhancing the control of β-AR signaling and the compartmentalization of secondary messengers and calcium-handling channels and pumps. Thus, by stabilizing t-tubule microdomains to regulate all aspects of calcium handling, cBIN1 produces a positive feedforward mechanism for efficient intracellular beat-to-beat calcium cycling. In future studies it will be interesting to identify whether exogenous cBIN1 alters β-AR expression, intracellular distribution, and functional regulation following chronic sympathetic activation.
Conclusions
We found that over-expression of exogenous cBIN1 is protective in mouse hearts subjected to chronic β-AR activation–induced concentric hypertrophy as well as pressure overload–induced hypertrophy and HF. As the first proof-of-concept study, there are a few limitations to our studies that need to be taken into consideration. First, the models used are limited to mouse models subjected to exaggerated stresses including a high dose of ISO infusion and severe pressure overload induced by aortic constriction. Future experiments in large mammals with common natural HF comorbidities, such as hypertension and diabetes, will be needed. Second, we used an AAV9 vector driven by the CMV promoter for gene delivery due to its consistent transduction efficiency and established cardiac tropism. Note the protective effect of exogenous cBIN1 in the current study occurred when only 60% of cardiomyocytes were transduced by AAV9 with detectable expression of exogenous protein. Improving the viral infectivity in cardiomyocytes can additionally help limit or prevent ISO-induced membrane disruption in all cardiomyocytes, increasing the protective effect on the entire heart. Further experiments using cBin1 packaged in AAV9 with an efficient and cardiac-specific promoter to induce sufficient exogenous protein expression in all cardiomyocytes will be needed before clinical trials testing the efficacy and efficiency of cBin1 gene therapy. Third, the current study lacks a full evaluation of hemodynamic parameters such as blood pressure. Although a similar HR increase in all post-ISO mice indicates effective and similar hemodynamic stress in animals with or without cBIN1, future studies are needed to establish whether cBIN1 will impact systemic hemodynamics and blood pressure. Finally, future studies are needed to explore how the cBIN1-microdomain regulates the organization of intracellular calcium-handling machineries, EC coupling, SR calcium load and release, diastolic calcium concentration and its downstream calcium-signaling pathways, interplay between signaling pathways of pathological and physiological hypertrophic remodeling, and molecular transition from compensated hypertrophy to decompensated cardiomyopathy.
Perspectives.
COMPETENCY IN MEDICAL KNOWLEDGE: The implications describe the effects of exogenous cBIN1 expression on cardiac remodeling and function in mouse hearts subjected to chronic sympathetic overdrive.
TRANSLATIONAL OUTLOOK: cBIN1 replacement therapy with a positive inotropy and lusitropy effect can be used as a potential new treatment option for HF.
Acknowledgments
The authors thank the Electron Imaging Center at California NanoSystems Institute of University of California-Los Angeles for transmission electron microscopy, the Cedars-Sinai Medical Center Histology Core for preparing cryosections, and Sarcotein Diagnostics for the recombinant anti-BIN1 exon 13 antibody.
Footnotes
This study was supported by grants from National Institute of Health, Bethesda, Maryland (HL133286 to Dr. Hong; HL152691 to Dr. Shaw, and HL138577 to Dr. Shaw); the American Heart Association, Dallas, Texas (Dr. Hong and Dr. Shaw), and the Department of Defense, Washington, DC (Dr. Hong and Dr. Shaw, PR160592). All authors have reported that they have no relationships relevant to the contents of this paper to disclose. All the work was done at Cedars-Sinai Medical Center and the University of Utah.
The authors attest they are in compliance with human studies committees and animal welfare regulations of the authors’ institutions and Food and Drug Administration guidelines, including patient consent where appropriate. For more information, visit the JACC: Basic to Translational Scienceauthor instructions page.
Appendix
For an expanded methods section as well as supplemental figures, videos, and tables, please see the online version of this paper.
Appendix
References
- 1.Virani S.S., Alonso A., Benjamin E.J. Heart Disease and Stroke Statistics-2020 Update: A Report From the American Heart Association. Circulation. 2020;141 doi: 10.1161/CIR.0000000000000757. e139–596. [DOI] [PubMed] [Google Scholar]
- 2.Verloop W.L., Beeftink M.M., Santema B.T. A systematic review concerning the relation between the sympathetic nervous system and heart failure with preserved left ventricular ejection fraction. PLoS One. 2015;10 doi: 10.1371/journal.pone.0117332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nikolaev V.O., Moshkov A., Lyon A.R. Beta2-adrenergic receptor redistribution in heart failure changes cAMP compartmentation. Science. 2010;327:1653–1657. doi: 10.1126/science.1185988. [DOI] [PubMed] [Google Scholar]
- 4.Ibrahim M., Gorelik J., Yacoub M.H., Terracciano C.M. The structure and function of cardiac t-tubules in health and disease. Proc Biol Sci. 2011;278:2714–2723. doi: 10.1098/rspb.2011.0624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schobesberger S., Wright P., Tokar S. T-tubule remodelling disturbs localized beta2-adrenergic signalling in rat ventricular myocytes during the progression of heart failure. Cardiovasc Res. 2017;113:770–782. doi: 10.1093/cvr/cvx074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Balycheva M., Faggian G., Glukhov A.V., Gorelik J. Microdomain-specific localization of functional ion channels in cardiomyocytes: an emerging concept of local regulation and remodelling. Biophys Rev. 2015;7:43–62. doi: 10.1007/s12551-014-0159-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hong T., Shaw R.M. Cardiac t-tubule microanatomy and function. Physiol Rev. 2017;97:227–252. doi: 10.1152/physrev.00037.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Glukhov A.V., Balycheva M., Sanchez-Alonso J.L. Direct evidence for microdomain-specific localization and remodeling of functional L-Type calcium channels in rat and human atrial myocytes. Circulation. 2015;132:2372–2384. doi: 10.1161/CIRCULATIONAHA.115.018131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sanchez-Alonso J.L., Bhargava A., O'Hara T. Microdomain-specific modulation of L-type calcium channels leads to triggered ventricular arrhythmia in heart failure. Circ Res. 2016;119:944–955. doi: 10.1161/CIRCRESAHA.116.308698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wei S., Guo A., Chen B. T-tubule remodeling during transition from hypertrophy to heart failure. Circ Res. 2010;107:520–531. doi: 10.1161/CIRCRESAHA.109.212324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bers D.M. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- 12.Hong T., Yang H., Zhang S.S. Cardiac BIN1 folds T-tubule membrane, controlling ion flux and limiting arrhythmia. Nat Med. 2014;20:624–632. doi: 10.1038/nm.3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hong T.T., Smyth J.W., Gao D. BIN1 localizes the L-type calcium channel to cardiac T-tubules. PLoS Biol. 2010;8 doi: 10.1371/journal.pbio.1000312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fu Y., Shaw S.A., Naami R. Isoproterenol promotes rapid ryanodine receptor movement to Bridging Integrator 1 (BIN1)-organized dyads. Circulation. 2016;133:388–397. doi: 10.1161/CIRCULATIONAHA.115.018535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hong T.T., Smyth J.W., Chu K.Y. BIN1 is reduced and Cav1.2 trafficking is impaired in human failing cardiomyocytes. Heart Rhythm. 2012;9:812–820. doi: 10.1016/j.hrthm.2011.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Caldwell J.L., Smith C.E., Taylor R.F. Dependence of cardiac transverse tubules on the BAR domain protein amphiphysin II (BIN-1) Circ Res. 2014;115:986–996. doi: 10.1161/CIRCRESAHA.116.303448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Basheer W.A., Xiao S., Epifantseva I. GJA1-20k arranges actin to guide Cx43 delivery to cardiac intercalated discs. Circ Res. 2017;121:1069–1080. doi: 10.1161/CIRCRESAHA.117.311955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zincarelli C., Soltys S., Rengo G., Rabinowitz J.E. Analysis of AAV serotypes 1-9 mediated gene expression and tropism in mice after systemic injection. Mol Ther. 2008;16:1073–1080. doi: 10.1038/mt.2008.76. [DOI] [PubMed] [Google Scholar]
- 19.Chen B.D., He C.H., Chen X.C. Targeting transgene to the heart and liver with AAV9 by different promoters. Clin Exp Pharmacol Physiol. 2015;42:1108–1117. doi: 10.1111/1440-1681.12453. [DOI] [PubMed] [Google Scholar]
- 20.Denegri M., Bongianino R., Lodola F. Single delivery of an adeno-associated viral construct to transfer the CASQ2 gene to knock-in mice affected by catecholaminergic polymorphic ventricular tachycardia is able to cure the disease from birth to advanced age. Circulation. 2014;129:2673–2681. doi: 10.1161/CIRCULATIONAHA.113.006901. [DOI] [PubMed] [Google Scholar]
- 21.Amoasii L., Hnia K., Chicanne G. Myotubularin and PtdIns3P remodel the sarcoplasmic reticulum in muscle in vivo. J Cell Sci. 2013;126:1806–1819. doi: 10.1242/jcs.118505. [DOI] [PubMed] [Google Scholar]
- 22.Fu Y., Shaw R.M. CASAAV technology to examine regulators of heart failure: cause or effect. Circ Res. 2017;120:1846–1848. doi: 10.1161/CIRCRESAHA.117.311074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gaasch W.H., Zile M.R. Left ventricular structural remodeling in health and disease: with special emphasis on volume, mass, and geometry. J Am Coll Cardiol. 2011;58:1733–1740. doi: 10.1016/j.jacc.2011.07.022. [DOI] [PubMed] [Google Scholar]
- 24.Periasamy M., Janssen P.M. Molecular basis of diastolic dysfunction. Heart Fail Clin. 2008;4:13–21. doi: 10.1016/j.hfc.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shan J., Kushnir A., Betzenhauser M.J. Phosphorylation of the ryanodine receptor mediates the cardiac fight or flight response in mice. J Clin Invest. 2010;120:4388–4398. doi: 10.1172/JCI32726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boknik P., Fockenbrock M., Herzig S. Protein phosphatase activity is increased in a rat model of long-term beta-adrenergic stimulation. Naunyn Schmiedebergs Arch Pharmacol. 2000;362:222–231. doi: 10.1007/s002100000283. [DOI] [PubMed] [Google Scholar]
- 27.Kirchhefer U., Hammer E., Heinick A. Chronic beta-adrenergic stimulation reverses depressed Ca handling in mice overexpressing inhibitor-2 of protein phosphatase 1. J Mol Cell Cardiol. 2018;125:195–204. doi: 10.1016/j.yjmcc.2018.10.022. [DOI] [PubMed] [Google Scholar]
- 28.Osadchii O.E. Cardiac hypertrophy induced by sustained beta-adrenoreceptor activation: pathophysiological aspects. Heart Fail Rev. 2007;12:66–86. doi: 10.1007/s10741-007-9007-4. [DOI] [PubMed] [Google Scholar]
- 29.Soltysinska E., Olesen S.P., Osadchii O.E. Myocardial structural, contractile and electrophysiological changes in the guinea-pig heart failure model induced by chronic sympathetic activation. Exp Physiol. 2011;96:647–663. doi: 10.1113/expphysiol.2011.058503. [DOI] [PubMed] [Google Scholar]
- 30.Balakumar P., Singh A.P., Singh M. Rodent models of heart failure. J Pharmacol Toxicol Methods. 2007;56:1–10. doi: 10.1016/j.vascn.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 31.Frey N., Katus H.A., Olson E.N., Hill J.A. Hypertrophy of the heart: a new therapeutic target? Circulation. 2004;109:1580–1589. doi: 10.1161/01.CIR.0000120390.68287.BB. [DOI] [PubMed] [Google Scholar]
- 32.Oliveira R.S., Ferreira J.C., Gomes E.R. Cardiac anti-remodelling effect of aerobic training is associated with a reduction in the calcineurin/NFAT signalling pathway in heart failure mice. J Physiol. 2009;587:3899–3910. doi: 10.1113/jphysiol.2009.173948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Neves V.J., Fernandes T., Roque F.R., Soci U.P., Melo S.F., de Oliveira E.M. Exercise training in hypertension: role of microRNAs. World J Cardiol. 2014;6:713–727. doi: 10.4330/wjc.v6.i8.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ooi J.Y., Bernardo B.C., McMullen J.R. The therapeutic potential of miRNAs regulated in settings of physiological cardiac hypertrophy. Future Med Chem. 2014;6:205–222. doi: 10.4155/fmc.13.196. [DOI] [PubMed] [Google Scholar]
- 35.MacDonnell S.M., Kubo H., Crabbe D.L. Improved myocardial beta-adrenergic responsiveness and signaling with exercise training in hypertension. Circulation. 2005;111:3420–3428. doi: 10.1161/CIRCULATIONAHA.104.505784. [DOI] [PubMed] [Google Scholar]
- 36.Hoydal M.A., Stolen T.O., Kettlewell S. Exercise training reverses myocardial dysfunction induced by CaMKIIdeltaC overexpression by restoring Ca2+ homeostasis. J Appl Physiol. 2016;121:212–220. doi: 10.1152/japplphysiol.00188.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pitsavos C., Chrysohoou C., Koutroumbi M. The impact of moderate aerobic physical training on left ventricular mass, exercise capacity and blood pressure response during treadmill testing in borderline and mildly hypertensive males. Hellenic J Cardiol. 2011;52:6–14. [PubMed] [Google Scholar]
- 38.Dieberg G., Ismail H., Giallauria F., Smart N.A. Clinical outcomes and cardiovascular responses to exercise training in heart failure patients with preserved ejection fraction: a systematic review and meta-analysis. J Appl Physiol. 2015;119:726–733. doi: 10.1152/japplphysiol.00904.2014. [DOI] [PubMed] [Google Scholar]
- 39.Fu Y., Hong T. BIN1 regulates dynamic t-tubule membrane. Biochim Biophys Acta. 2016;1863:1839–1847. doi: 10.1016/j.bbamcr.2015.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang L., Katchman A., Kushner J. Cardiac CaV1.2 channels require beta subunits for beta-adrenergic-mediated modulation but not trafficking. J Clin Invest. 2019;129:647–658. doi: 10.1172/JCI123878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miriyala J., Nguyen T., Yue D.T., Colecraft H.M. Role of CaVbeta subunits, and lack of functional reserve, in protein kinase A modulation of cardiac CaV1.2 channels. Circ Res. 2008;102 doi: 10.1161/CIRCRESAHA.108.171736. e54–64. [DOI] [PubMed] [Google Scholar]
- 42.Marx S.O., Reiken S., Hisamatsu Y. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101:365–376. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- 43.Anwar A., Taimor G., Korkusuz H. PKC-independent signal transduction pathways increase SERCA2 expression in adult rat cardiomyocytes. J Mol Cell Cardiol. 2005;39:911–919. doi: 10.1016/j.yjmcc.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 44.Lyon A.R., Nikolaev V.O., Miragoli M. Plasticity of surface structures and beta(2)-adrenergic receptor localization in failing ventricular cardiomyocytes during recovery from heart failure. Circ Heart Fail. 2012;5:357–365. doi: 10.1161/CIRCHEARTFAILURE.111.964692. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.