

Abstract
In silico T-cell epitope prediction plays an important role in immunization experimental design and vaccine preparation. Currently, most epitope prediction research focuses on peptide processing and presentation, e.g., proteasomal cleavage, transporter associated with antigen processing (TAP), and major histocompatibility complex (MHC) combination. To date, however, the mechanism for the immunogenicity of epitopes remains unclear. It is generally agreed upon that T-cell immunogenicity may be influenced by the foreignness, accessibility, molecular weight, molecular structure, molecular conformation, chemical properties, and physical properties of target peptides to different degrees. In this work, we tried to combine these factors. Firstly, we collected significant experimental HLA-I T-cell immunogenic peptide data, as well as the potential immunogenic amino acid properties. Several characteristics were extracted, including the amino acid physicochemical property of the epitope sequence, peptide entropy, eluted ligand likelihood percentile rank (EL rank(%)) score, and frequency score for an immunogenic peptide. Subsequently, a random forest classifier for T-cell immunogenic HLA-I presenting antigen epitopes and neoantigens was constructed. The classification results for the antigen epitopes outperformed the previous research (the optimal AUC = 0.81, external validation data set AUC = 0.77). As mutational epitopes generated by the coding region contain only the alterations of one or two amino acids, we assume that these characteristics might also be applied to the classification of the endogenic mutational neoepitopes also called “neoantigens.” Based on mutation information and sequence-related amino acid characteristics, a prediction model of a neoantigen was established as well (the optimal AUC = 0.78). Further, an easy-to-use web-based tool “INeo-Epp” was developed for the prediction of human immunogenic antigen epitopes and neoantigen epitopes.
1. Introduction
An antigen consists of several epitopes, which can be recognized either by B- or T-cells and/or molecules of the host immune system. However, usually only a small number of amino acid residues that comprise a specific epitope are necessary to elicit an immune response [1]. The properties of these amino acid residues causing immunogenicity are unknown. HLA-I antigen peptides are processed and presented as follows: (a) cytosolic and nuclear proteins are cleaved to short peptides by intracellular proteinases; (b) some are selectively transferred to the endoplasmic reticulum (ER) by the TAP transporter, and subsequently are treated by endoplasmic reticulum aminopeptidase; and (c) antigen-presenting cells (APCs) present peptides containing 8-11 AA (amino acid) residues on HLA class I molecules to CD8+ T-cells [2]. Researchers can now simulate antigen processing and presentation by computational methods to predict binding peptide-MHC complexes (p-MHC). Several types of software systems have been developed, including NetChop [3], NetCTL [4], NetMHCpan [5], and MHCflurry [6]. However, despite that the binding to MHC molecules of most peptides is predicted, only 10%~15% of those have been shown to be immunogenic [7–10]. For neoantigens, the result was approximately 5% (range: 1%-20%) due to central immunotolerance [11, 12]. As a result, the cycle for vaccine development and immunization research is extended. Here, we aim to develop a T-cell HLA class-I immunogenicity prediction method to further identify real epitopes/neoepitopes from p-MHC to shorten this cycle.
Many experimental human epitopes have been collected and summarized in the immune epitope database (IEDB) [13], which makes it feasible to mathematically predict human epitopes. However, there still exist two limitations: (i) a high level of MHC polymorphism produces a severe challenge for T-cell epitope prediction and (ii) there is an extremely unequal distribution of data to compare epitopes and nonepitopes. It is not conducive to analyze the potential deviation existing in TCR recognition owing to the presentation of different HLA peptides. A general analysis of all HLA-presented peptides, ignoring the specific pattern of TCR recognition of individual HLA-presented peptides, may result in a lower predictive accuracy.
With the advances in HLA research, Sette and Sidney [14] classified, for the first time, overlapping peptide binding repertoires into nine major functional HLA supertypes (A1, A2, A3, A24, B7, B27, B44, B58, and B62). In 2008, Sidney et al. [15] made a further refinement, in which over 80% of the 945 different HLA-A and B alleles can be assigned to the original nine supertypes. It has not been reported whether peptides presented by different HLA alleles influence TCR recognition. Hence, we collected experimental epitopes according to HLA alleles and assumed that epitopes belonging to the same HLA supertypes have similar properties.
Moreover, screening for endogenic mutational neoepitopes is one of the core steps in tumor immunotherapy. In 2017, Ott et al. [16] and Sahin et al. [17] confirmed that peptides and RNA vaccines made up of neoantigens in melanoma can stimulate and proliferate CD8+ and CD4+ T-cells. In addition, a recent research suggests that including neoantigen vaccination not only can expand the existing specific T-cells but also can induce a wide range of novel T-cell specificity in cancer patients and enhance tumor suppression [18]. Meanwhile, a tumor can be better controlled by the combination therapy of neoantigen vaccine and programmed cell death protein 1 (PD-1)/PD1 ligand 1 (PDL-1) therapy [19, 20]. Nevertheless, a considerable number of predicted candidate p-MHC from somatic cell mutations may be false positive, which would fail to stimulate TCR recognition and immune response. This is undoubtedly a challenge for designing vaccines against neoantigens.
In our study, based on HLA-I T-cell peptides collected from experimentally validated antigen epitopes and neoantigen epitopes, we aim to build a novel method to further reduce the range of immunogenic epitope screening based on predicted p-MHC. Finally, a simple web-based tool, INeo-Epp (immunogenic epitope/neoepitope prediction), was developed for prediction of human antigen and neoantigen epitopes.
2. Materials and Methods
The flow chart for “INeo-Epp” prediction is shown in Figure 1.
Figure 1.
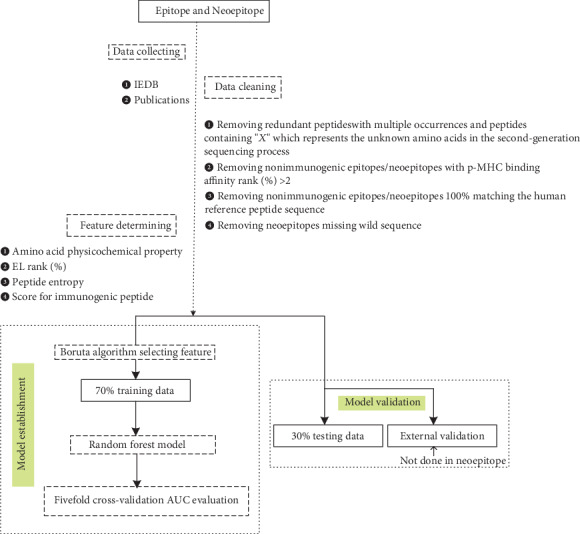
The flow chart for “INeo-Epp” prediction.
2.1. Construction of Immunogenic and Nonimmunogenic Epitopes
Peptides that can promote cytokine proliferation are considered to be immunogenic epitopes. However, nonimmunogenic epitopes may result from the following reasons: (a) p-MHC is truly unrecognized by TCR, (b) peptides are not presented by MHC (quantitatively expressed as rank(%) > 2, see Rank(%) Score (C24) for details), and (c) negative selection/clonal presentation is induced by excessive similarity to autologous peptides [21]. In this work, to further study the recognition preferences of T-cells, peptides with >2 rank(%) were regarded as not in contact with TCR, and sequences 100% matching the human reference peptides (ftp://ftp.ensembl.org/pub/release-97/fasta/homo_sapiens/pep/) were regarded as exhibiting immune tolerance. Hence, we removed these from the definition of nonimmunogenic peptides.
2.2. Construction of Data Sets: Epitopes, External Validation of Epitopes, and Neoepitopes
Antigen epitope data were collected from IEDB (linear epitope, human, T-cell assays, MHC class I, any disease was chosen). Data collection criteria accommodated for each HLA allele quantity > 50 and frequency > 0.5% (refer to allele frequency database [22]) (Table 1, check Table S1 for detailed information).
Table 1.
Summary of IEDB epitope data.
| HLA supertype | IEDB HLA data | Number | HLA allele frequency Asian/Black/Caucasian | Motif view | |
|---|---|---|---|---|---|
| Negative | Positive | ||||
| A1 | A01:01 | 811 | 103 | 0.154/0.046/0.164 | 1-2(ST)-3-4-5-6-7-8-9(Y) |
| A26:01 | 83 | 19 | 0.041/0.014/0.030 | 1(DE)-2(ITV)-3-4-5-6-7-8-9(FMY) | |
| A2 | A02:01 | 1883 | 1580 | 0.049/0.123/0.275 | 1-2(LM)-3-4-5-6-7-8-9(ILV)-10(V) |
| A3 | A11:01 | 196 | 174 | 0.139/0.014/0.060 | 1-2(IMSTV)-3-4-5-6-7-8-9(K)-10(K) |
| A03:01 | 1400 | 169 | 0.063/0.083/0.139 | 1-2(ILMTV)-3-4-5-6-7-8-9(K)-10(K) | |
| A24 | A24:02 | 207 | 219 | 0.136/0.024/0.084 | 1-2(WY)-3-4-5-6-7-8-9(FIW) |
| A23:01 | 1138 | 12 | 0.006/0.109/0.019 | 1-2(WY)-3-4-5-6-7-8-9-10(F) | |
| B7 | B35:01 | 63 | 248 | 0.062/0.068/0.055 | 1-2(P)-3-4-5-6-7-8-9(FMY) |
| B07:02 | 523 | 244 | 0.034/0.005/0.0143 | 1-2(p)-3-4-5-6-7-8-9(FLM) | |
| B51:01 | 13 | 51 | 0.074/0.021/0.047 | 1-2(P)-3-4-5-6-7-8-9(IV) | |
| B8 | B08:01 | 317 | 195 | 0.036/0.037/0.114 | 1-2-3-4-5(HKR)-6-7-8-9(FILMV) |
| B27 | B27:05 | 100 | 86 | 0.008/0.008/0.037 | 1(RY)-2(R)-3(FMLWY)-4-5-6-7-8-9 |
| B44 | B37:01 | 1036 | 10 | 0.034/0.005/0.014 | — |
| B40:01 | 67 | 65 | 0.022/0.012/0.052 | — | |
| B44:02 | 73 | 66 | 0.008/0.020/0.095 | 1-2(E)-3-4-5-6-7-8-9(FIWY) | |
| B58 | B58:01 | 11 | 62 | 0.041/0.037/0.007 | 1-2(AST)-3-4-5-6-7-8-9(W) |
| B62 | B15:01 | 3 | 70 | 0.016/0.010/0.060 | 1-2(LMQ)-3-4-5-6-7-8-9(FY) |
| Total | 7924 | 3373 | |||
| Remove negative rank(%) > 2 | 5123 | 3373 | |||
| Remove negative human 100% similar | 4943 | 3373 | |||
The external antigen epitope validation set was collected from seven published independent human antigen studies [23–29], consisting of 577 nonimmunogenic epitopes and 85 immunogenic epitopes (Table 2, S2 Table).
Table 2.
External data included in validation set.
| Publication time | PMID | Author | Nonepitopes | Epitopes |
|---|---|---|---|---|
| 2013 | 23580623 | Weiskopf et al. | 477 | 42 |
| 2018 | 29397015 | Luxenburger et al. | 100 | 26 |
| 2018 | 30260541 | Xia et al. | — | 1 |
| 2018 | 30487281 | Vahed et al. | — | 4 |
| 2018 | 30518652 | Khakpoor et al. | — | 2 |
| 2018 | 30587531 | Huth et al. | — | 4 |
| 2018 | 30815394 | Sekyere et al. | — | 6 |
| Total | 577 | 85 | ||
| Remove negative with rank(%) > 2 and HLA supertypes (not appeared in training set) | 321 | 69 | ||
Here, we removed peptides for which HLA supertypes do not appear in the training set, because we assume peptides belonging to the same HLA supertypes to have similar properties. In the external validation set, some peptides bind to rare HLA supertypes. Their characteristics were not included in the training set. Hence, these peptides in the external validation data might lead to a classification bias.
The neoantigen data were collected from 11 publications [19, 30–39] and IEDB mutational epitopes, and 13 published data sets collected by Bjerregaard et al. in one publication [40] in 2017 (see Table 3, S3 Table for details) were also included.
Table 3.
Neoepitope data included in this study.
| Publication time | PMID | Author | Tumor type | Nonimmunogenic neoepitopes | Immunogenic neoepitopes | T-cell assay |
|---|---|---|---|---|---|---|
| 2013-12 | 24323902 | D. A. Wick et al. | Ovarian cancer | — | 1 | ELISPOT |
| 2015-9 | 26359337 | E. M. Van Allen et al. | Melanoma | — | 18 | Clinical benefit |
| 2015-11 | 26752676 | T. Karasaki et al. | Lung adenocarcinoma | — | 4 | — |
| 2016-1 | 26901407 | A. Gros et al. | Melanoma | 12 | 14 | ELISPOT |
| 2016-5 | 27198675 | E. Strønen, et al. | Melanoma | 1134 | 16 | CTL clone |
| 2016-12 | 28405493 | A. Nelde et al. | Lymphoma | — | 2 | ELISPOT |
| 2017-6 | 28619968 | X. Zhang et al. | Breast cancer | — | 4 | Flow cytometry |
| 2017-10 | 29104575 | M. Markus et al. | Melanoma | 10 | 16 | — |
| 2017-11 | 29187854 | A.-M. Bjerregaard et al. | Polytype | 1874 | 42 | ELISPOT et al. |
| 2017-11 | 29132146 | V. P. Balachandran et al. | Pancreatic | — | 10 | Flow cytometry |
| 2018-5 | 29720506 | T. Matsuda et al. | Ovarian cancer | — | 3 | ELISPOT |
| 2018-12 | 29409514 | K. Sonntag et al. | Pancreatic ductal carcinoma | — | 3 | Flow cytometry |
| 2018-10 | 30357391 | V. Randi et al. | — | 6 | 35 | — |
| Total | 3030 | 168 | ||||
| Remove duplication | 2837 | 164 | ||||
| Remove negative rank(%) > 2 and human 100% similar | 1697 | 164 | ||||
2.3. Construction of Potential Immunogenicity Feature
2.3.1. Calculation of Peptide Characteristics Based on Amino Acid Sequences
The formula for calculating peptide characteristics is shown in (1). PN, P2, and PC (N-terminal, position 2, C-terminal as anchored sites by default) are considered to be embedded in HLA molecules and have no contact with TCRs; therefore, they were not evaluated.
| (1) |
where P is peptide, c is characteristic. Pc represents the characteristics of peptides, A represents amino acids, N represents the N-terminal in a peptide, C represents the C-terminal in a peptide, Pos represents the amino acid position in a peptide, and PAc represents characteristics of amino acids in peptides.
2.3.2. Frequency Score for Immunogenic Peptide (C22)
Amino acid distribution frequency differences between immunogenic and nonimmunogenic peptides at TCR contact sites (excluding anchor sites) were considered as a feature:
| (2) |
where Pie+ represents immunogenic peptides, Pie− represents nonimmunogenic peptides. fA′ represents amino acid frequency in the TCR contact position. Pie+(fA′) represents the frequency of amino acids in immunogenic peptides at TCR contact sites.
2.3.3. Calculating Peptide Entropy (C23)
Peptide entropy [41] was used as a feature:
| (3) |
where PH represents peptide entropy. fA represents amino acid frequency in the human reference peptide sequence. PfA represents the frequency in the human reference peptide sequence of amino acids in epitope peptides.
2.3.4. Rank(%) Score (C24)
HLA binding prediction was performed using NetMHCpan 4.0. Rank(%) provides a robust filter for the identification of MHC-binding peptides, in which rank(%) was recommended as an evaluation standard, rank(%) < 0.5 as strong binders, 0.5 < rank(%) < 2 as weak binders, and rank(%) > 2 as no binders.
2.4. Fivefold Cross-Validation, Feature Selection, Random Forests, and ROC Generation
The 5-fold cross-validation was implemented in R using the caret package [42] (method = “repeatedcv,” number = 5, repeats = 3). The feature screening results were generated in R using the package Boruta [43] (a novel random forest-based feature selection algorithm for finding all relevant variables, which provides unbiased and stable selection of important and nonimportant attributes from an information system). It iteratively removes the features which are proven by a statistical test to be less relevant than random probes. It uses Z score (computed by dividing the average loss by its standard deviation) as the importance measure, and it takes into account the fluctuations of the mean accuracy loss among trees in the forest. R package randomForest [44] was used for training data (the R language machine learning package caret provides automatic iteration selection of optimal parameters: mtry = 15 for antigen epitope and mtry = 14 for neoantigen epitope; the remaining parameters use default values). R package ROCR [45] was used for drawing ROC.
2.5. Web Tool Implementation
The front end of Ineo-Epp was constructed via HTML/JavaScript/CSS. The back end was written in PHP, connecting the web interface and Apache web server. A python script was used for calculating peptide characteristics and extracting mutation information. Models were built using R.
3. Results
Ultimately, 11,297 validated epitopes and nonepitopes with lengths of 8-11 amino acids were collected from IEDB. T-cell responses included activation, cytotoxicity, proliferation, IFN-γ release, TNF release, granzyme B release, IL-2 release, and IL-10 release. Seventeen different HLA alleles were collected (Figure 2(a)), and the detailed antigen length distribution is shown in Figure 2(b). Additionally, we collected the neoantigen data from 12 publications, including 2837 nonneoepitopes and 164 neoepitopes (Figure 2(c)), and the detailed neoantigen length distribution is shown in Figure 2(d).
Figure 2.
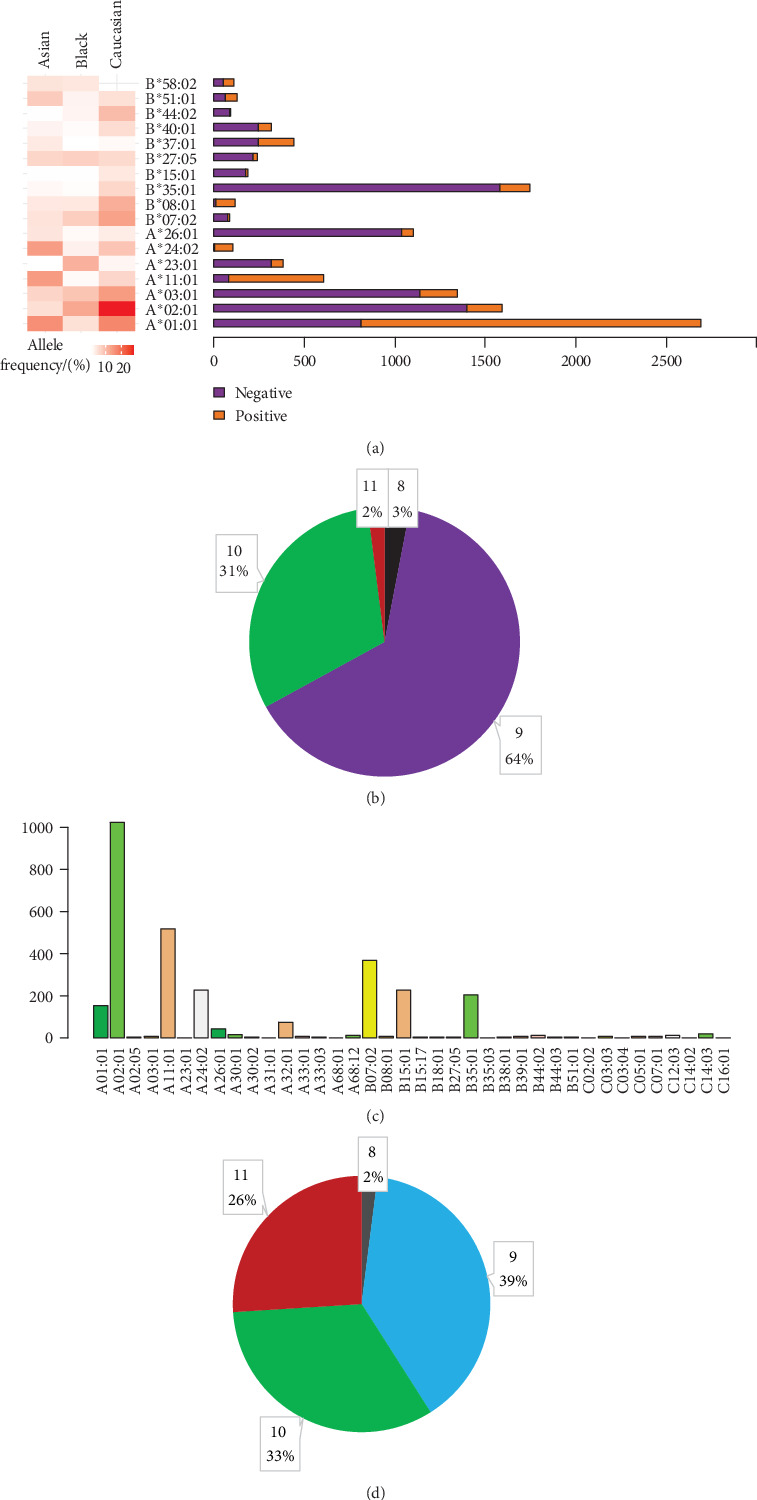
Epitope/neoepitope peptide composition and amino acid length distribution. (a) Detailed data distribution of seventeen HLA alleles of antigen peptides, the proportion of each HLA allele (positive and negative) epitopes, and the corresponding HLA frequency in Asians, Blacks, Caucasians. (b) Proportion of antigen peptides with lengths of 8-11 AA. (c) Data distribution of HLA alleles of neoantigen peptides. (d) Proportion of neoantigen peptides with lengths of 8-11 AA.
The TCR contact position plays a crucial role in the analysis of immunogenicity, as TCRs might be more sensitive to some amino acids; the amino acid preference in the antigen epitope peptide and the antigen nonepitope peptide was further analyzed after excluding anchor sites (N-terminal, position 2, and C-terminal) (Figure 3). We found that TCRs tend to identify hydrophobic amino acids. For example, 3/4 hydrophobic amino acids (L, W, P, A, V, and M) occur more frequently in immunogenicity epitopes. Charged amino acids (e.g., D and K) are enriched in nonepitopes, whereas the rest of the charged amino acids (R, H, and E) show no difference. Based on the result in Figure 3, the amino acid distribution difference at the TCR contact sites was regarded by us as one of the immunogenicity features (i.e., Frequency Score for Immunogenic Peptide (C22)).
Figure 3.
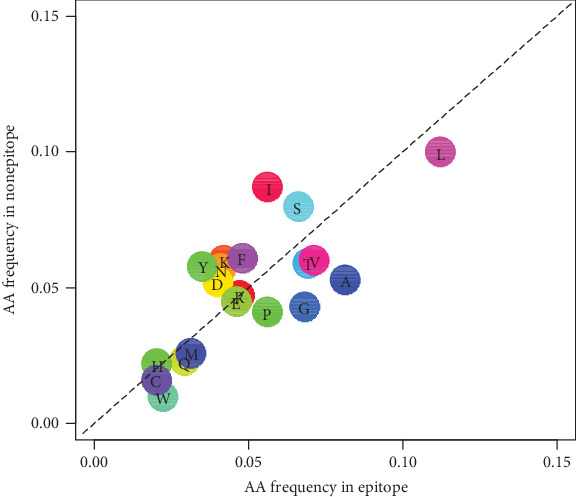
Antigen epitope amino acid distribution frequency in the TCR contact site of epitopes and nonepitopes. Frequency distribution of amino acids at TCR contact sites in antigen epitope and nonepitope peptides, and the amino acids below the dotted line are preferred by the epitope.
3.1. Classification Prediction Model for Antigen Epitopes
We constructed the features of peptides on the basis of the characteristics of amino acids (see Calculation of Peptide Characterstics Based on Amino Acid Sequences). All amino acid characteristics were selected from ProtScale [46] in ExPASy (SIB Bioinformatics Resource Portal). The 21 involved features are as follows: Kyte-Doolittle numeric hydrophobicity scale (C1) [47], molecular weight (C2), bulkiness (C3) [48], polarity (C4) [49], recognition factors (C5) [50], hydrophobicity (C6) [51], retention coefficient in HPLC (C7) [52], ratio hetero end/side (C8) [49], average flexibility (C9) [53], beta-sheet (C10) [54], alpha-helix (C11) [55], beta-turn (C12) [55], relative mutability (C13) [56], number of codon(s) (C14), refractivity (C15) [57], transmembrane tendency (C16) [58], accessible residues (%) (C17) [59], average area buried (C18) [60], conformational parameter for coil (C19) [55], total beta-strand (C20) [60], and parallel beta-strand (C21) [61] (see Table S4 for details). Also, Frequency Score for Immunogenic Peptide (C22), Calculating Peptide Entropy (C23), and Rank(%) Score (C24) were also taken into consideration. Together, 24 immunogenic features were collected, and all features were retained for antigen epitope prediction after screening using the R package Boruta. Compared with other characteristics, the frequency score for immunogenic peptide and rank(%) have higher impact, suggesting that they have more significant influence on antigen epitope classification (Figure 4(a)).
Figure 4.
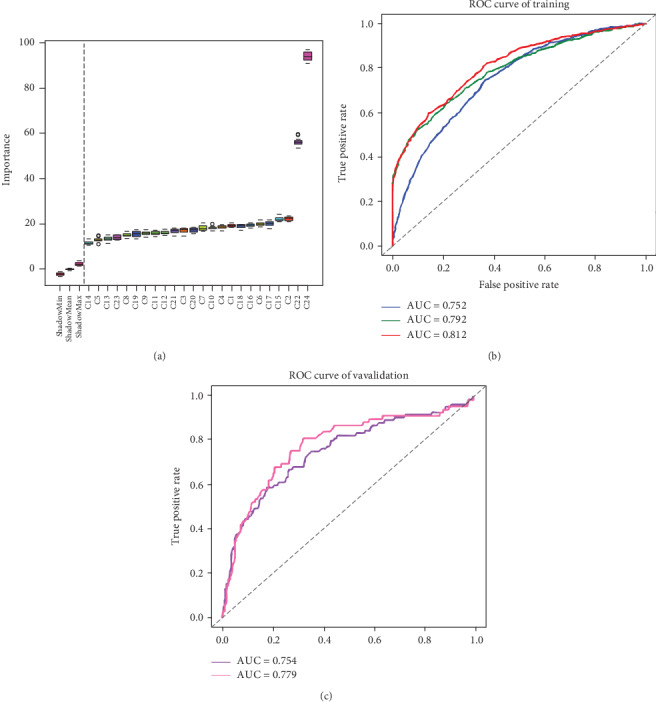
Feature selection in antigen epitopes and ROC curves of antigen epitope classification. (a) Peptide features: twenty-four features were screened, and we defined the features on the right of the dotted line as being effective. (b) Trained model: the line in blue represents antigen epitopes without screening; the line in green represents the selection with the deletion of the rank(%) > 2 nonepitope; the line in red represents the selection with the deletion of the nonepitopes 100% matching the human reference peptide sequence. (c) External validation: the ROC curves for the external verification set. The line in purple represents modeling using antigen epitopes without filtering, and the line in pink represents modeling using antigen epitopes removing nonepitopes with rank(%) > 2 and HLA for which supertypes did not appear in the training set.
The receiver operator characteristic (ROC) curve of models are shown in Figure 4. The fivefold cross-validation AUC was 0.81 in the prediction model for the antigen epitope (line in red, Figure 4(b)), and the externally validated (see Table 2) AUC was 0.75 (line in purple, Figure 4(c)). Here, we tried to remove peptides for which HLA supertypes did not appear in the training set from the externally validated antigen data, and the AUC, specificity, and sensitivity were increased to 0.78, 0.71, and 0.72, respectively (line in pink, Figure 4(c)). This, to some extent, verifies our conjecture about TCR specific recognition of different HLA alleles presenting peptides.
3.2. Classification Prediction Model for Neoantigen Epitopes
Neoantigens derived from somatic mutations are different from the wild peptide sequences. Therefore, some mutation-related characteristics were also taken into account. For instance, difference in hydrophobility before and after mutation (C25), differential agretopicity index (DAI, C26) [62], and whether the mutation position was anchored (C27). Finally, 27 features were selected for the neoantigen epitope prediction model. However, only 25 neoantigen-related features were retained after running Boruta, because C25 and C27 were removed. Also, rank(%) showed a marked effect (Figure 5(a)). In the fivefold cross-validation of the prediction model for neoantigen epitopes, AUC was 0.78 (Figure 5(b)).
Figure 5.
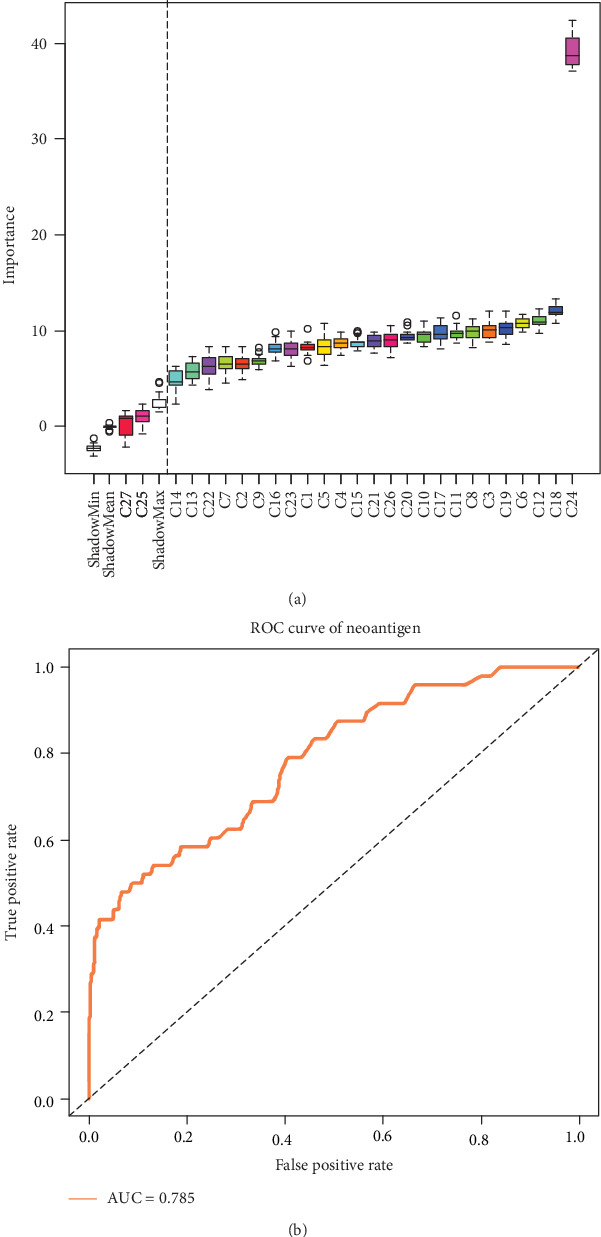
Feature selection in neoantigen epitopes and ROC curves of neoantigen epitope classification. (a) Twenty-seven features were screened, and the 25 features on the right of the dotted line were reserved for modeling using a random forest algorithm. (b) ROC curves of neoantigen epitope classification.
3.3. Web Server for TCR Epitope Prediction
Based on the abovementioned validated features, we established a web server for TCR epitope prediction, named “INeo-Epp.” This tool can be used to predict both immunogenic antigen and neoantigen epitopes. For antigens, the nine main HLA supertypes can be used. We recommend the peptides with the lengths of 8-12 residues, but not less than 8. N-terminal, position 2, and C-terminal were treated as anchored sites by default. A predictive score value greater than 0.5 is considered as immunogenicity (positive-high), a score between 0.4 and 0.5 is considered as positive-low, and a score less than 0.4 is considered as negative-high. It is critical to make sure that the HLA-subtype must match your peptides (rank(%) < 2). Where HLA-subtypes mismatch, a large deviation of the rank(%) value may strongly influence the results. Additionally, the neoantigen model requires providing wild type and mutated sequences at the same time to extract mutation-associated characteristics, and currently only immunogenicity prediction for neoantigens of single amino acid mutations are supported. Users can choose example options to test the INeo-Epp (http://www.biostatistics.online/ineo-epp/neoantigen.php).
4. Discussion
Due to the complexity of antigen presenting and TCR binding, the mechanism of TCR recognition has not been clearly revealed. In 2013, Calis et al. [63] developed a tool for epitope identification for mice and humans (AUC = 0.68). Although mice and human beings are highly homologous, the murine epitopes may very likely cause limitations in identifying human epitopes. Inspired by J. A. Calis, our research here focused on human beings' epitopes and has been conducted in a larger data set.
By analyzing epitope immunogenicity from the perspective of amino acid molecular composition, we observed that TCRs do have a preference for hydrophobic amino acid recognition. For short peptides presented by different HLA supertypes, TCRs may have different identification patterns. The immunogenicity prediction based on all HLA-presenting peptides may affect the accuracy of the prediction results. That is, if the prediction could focus on specified HLA-presenting peptides, the results may improve. Therefore, in our work we used HLA supertypes to improve the prediction of HLA-presenting epitopes, including antigen epitopes and neoantigen epitopes, for a better recognition by TCRs. At present, neoantigen epitopes that can be collected in accordance with the standard for experimental verification are too few, the data of positive and negative neoantigens are unbalanced, and there is not enough data to be used for an external verification set. In the future, we will continue to refine and expand our training and verification datasets. Recently, Laumont et al. [64] demonstrated that noncoding regions aberrantly expressing tumor-specific antigens (aeTSAs) may represent ideal targets for cancer immunotherapy. These epitopes can also be studied in the future. Increased epitope data may also help empower the prediction of potentially immunogenic peptides or neopeptides.
5. Conclusions
Neoantigen prediction is the most important step at the start of preparation of a neoantigen vaccine. Bioinformatics methods can be used to extract tumor mutant peptides and predict neoantigens. Most current strategies aimed at and ended in presenting peptide predictions, and among the results of these predictions, probably only fewer than 10 neoantigens might be clinically immunogenic and produce effective immune response. It is time-consuming and costly to experimentally eliminate the false positively predicted peptides. Our methods as developed in this study and the INeo-Epp tool may help eliminate false positive antigen/neoantigen peptides and greatly reduce the amount of candidates to be verified by experiments. We believe that in the age of biological system data explosion, computational approaches are a good way to enhance research efficiency and direct biological experiments. With the development of machine learning and deep learning, we expect that the prediction of epitope immunogenicity will be continually improved.
In summary, this study provides a novel T-cell HLA class-I immunogenicity prediction method from epitopes to neoantigens, and the INeo-Epp can be applied not only to identify putative antigens, but also to identify putative neoantigens.
It needs to be stated here that we published the preprint [65] of this article in July 2019. This is a modified version.
Acknowledgments
We sincerely thank Drs. Menghuan Zhang, Hong Li, and Qibing Leng for our valuable discussion. We also acknowledge Dr. Michael Liebman for his critical reading and editing. This work was funded by the National Natural Science Foundation of China (No. 31870829), the Shanghai Municipal Health Commission, and the Collaborative Innovation Cluster Project (No. 2019CXJQ02).
Contributor Information
Yong Zhao, Email: yzhao@shou.edu.cn.
Yong Lin, Email: yong_lynn@163.com.
Lu Xie, Email: luxiex2017@outlook.com.
Data Availability
The data used to support the findings of this study are included within the supplementary information file(s).
Disclosure
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflicts of Interest
The authors declare that there is no conflict of interest regarding the publication of this paper.
Supplementary Materials
S1 Table IEDB antigen epitopes summary. Detailed description of 17 HLA molecules collected from IEDB. (XLSX) S2 Table External validation antigen epitopes summary. Epitope details of 7 publications. (XLSX) S3 Table Neoantigen epitopes summary. Epitope details of 13 publications. (XLSX) S4 Table Summary of amino acid characteristics. For all amino acid characteristics (n=21) that are described in the ExPASy. (XLSX).
References
- 1.Desai D. V., Kulkarni-Kale U. T-cell epitope prediction methods: an overview. Methods in Molecular Biology. 2014;1184:333–364. doi: 10.1007/978-1-4939-1115-8_19. [DOI] [PubMed] [Google Scholar]
- 2.Goldberg A. L., Rock K. L. Proteolysis, proteasomes and antigen presentation. Nature. 1992;357(6377):375–379. doi: 10.1038/357375a0. [DOI] [PubMed] [Google Scholar]
- 3.Keşmir C., Nussbaum A. K., Schild H., Detours V., Brunak S. Prediction of proteasome cleavage motifs by neural networks. Protein Engineering, Design and Selection. 2002;15(4):287–296. doi: 10.1093/protein/15.4.287. [DOI] [PubMed] [Google Scholar]
- 4.Larsen M. V., Lundegaard C., Lamberth K., et al. An integrative approach to CTL epitope prediction: a combined algorithm integrating MHC class I binding, TAP transport efficiency, and proteasomal cleavage predictions. European Journal of Immunology. 2005;35(8):2295–2303. doi: 10.1002/eji.200425811. [DOI] [PubMed] [Google Scholar]
- 5.Jurtz V., Paul S., Andreatta M., Marcatili P., Peters B., Nielsen M. NetMHCpan-4.0: improved peptide-MHC class I interaction predictions integrating eluted ligand and peptide binding affinity data. Journal of Immunology. 2017;199(9):3360–3368. doi: 10.4049/jimmunol.1700893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O'Donnell T. J., Rubinsteyn A., Bonsack M., Riemer A. B., Laserson U., Hammerbacher J. MHCflurry: open-source class I MHC binding affinity prediction. Cell Systems. 2018;7(1):129–132.e4. doi: 10.1016/j.cels.2018.05.014. [DOI] [PubMed] [Google Scholar]
- 7.Wang M., Lamberth K., Harndahl M., et al. CTL epitopes for influenza A including the H5N1 bird flu; genome-, pathogen-, and HLA-wide screening. Vaccine. 2007;25(15):2823–2831. doi: 10.1016/j.vaccine.2006.12.038. [DOI] [PubMed] [Google Scholar]
- 8.Pérez C. L., Larsen M. V., Gustafsson R., et al. Broadly immunogenic HLA class I supertype-restricted elite CTL epitopes recognized in a diverse population infected with different HIV-1 subtypes. Journal of Immunology. 2008;180(7):5092–5100. doi: 10.4049/jimmunol.180.7.5092. [DOI] [PubMed] [Google Scholar]
- 9.Lundegaard C., Hoof I., Lund O., Nielsen M. State of the art and challenges in sequence based T-cell epitope prediction. Immunome Research. 2010;6(Suppl 2):p. S3. doi: 10.1186/1745-7580-6-S2-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sanchez-Trincado J. L., Gomez-Perosanz M., Reche P. A. Fundamentals and Methods for T- and B-Cell Epitope Prediction. Journal of Immunology Research. 2017;2017:14. doi: 10.1155/2017/2680160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kristensen V. N. The Antigenicity of the Tumor Cell — Context Matters. New England Journal of Medicine. 2017;376(5):491–493. doi: 10.1056/nejmcibr1613793. [DOI] [PubMed] [Google Scholar]
- 12.Kiyotani K., Chan H. T., Nakamura Y. Immunopharmacogenomics towards personalized cancer immunotherapy targeting neoantigens. Cancer Science. 2018;109(3):542–549. doi: 10.1111/cas.13498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ponomarenko J., Papangelopoulos N., Zajonc D. M., Peters B., Sette A., Bourne P. E. IEDB-3D: structural data within the immune epitope database. Nucleic Acids Research. 2010;39(Database):D1164–D1170. doi: 10.1093/nar/gkq888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sette A., Sidney J. Nine major HLA class I supertypes account for the vast preponderance of HLA-A and -B polymorphism. Immunogenetics. 1999;50(3-4):201–212. doi: 10.1007/s002510050594. [DOI] [PubMed] [Google Scholar]
- 15.Sidney J., Peters B., Frahm N., Brander C., Sette A. HLA class I supertypes: a revised and updated classification. BMC Immunology. 2008;9(1):p. 1. doi: 10.1186/1471-2172-9-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ott P. A., Hu Z., Keskin D. B., et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. 2017;547(7662):217–221. doi: 10.1038/nature22991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sahin U., Derhovanessian E., Miller M., et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature. 2017;547(7662):222–226. doi: 10.1038/nature23003. [DOI] [PubMed] [Google Scholar]
- 18.Hu Z., Ott P. A., Wu C. J. Towards personalized, tumour-specific, therapeutic vaccines for cancer. Nature Reviews Immunology. 2018;18(3):168–182. doi: 10.1038/nri.2017.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Van Allen E. M., Miao D., Schilling B., et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. 2015;350(6257):207–211. doi: 10.1126/science.aad0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Efremova M., Finotello F., Rieder D., Trajanoski Z. Neoantigens generated by individual mutations and their role in cancer immunity and immunotherapy. Frontiers in Immunology. 2017;8:p. 1679. doi: 10.3389/fimmu.2017.01679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Klein L., Hinterberger M., Wirnsberger G., Kyewski B. Antigen presentation in the thymus for positive selection and central tolerance induction. Nature Reviews Immunology. 2009;9(12):833–844. doi: 10.1038/nri2669. [DOI] [PubMed] [Google Scholar]
- 22.Gonzalez-Galarza F. F., McCabe A., Melo dos Santos E. J., et al. Allele frequency net database. Methods in Molecular Biology. 2018;1802:49–62. doi: 10.1007/978-1-4939-8546-3_4. [DOI] [PubMed] [Google Scholar]
- 23.Weiskopf D., Angelo M. A., de Azeredo E. L., et al. Comprehensive analysis of dengue virus-specific responses supports an HLA-linked protective role for CD8+ T cells. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(22):E2046–E2053. doi: 10.1073/pnas.1305227110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luxenburger H., Graß F., Baermann J., et al. Differential virus-specific CD8+T-cell epitope repertoire in hepatitis C virus genotype 1 versus 4. Journal of Viral Hepatitis. 2018;25(7):779–790. doi: 10.1111/jvh.12874. [DOI] [PubMed] [Google Scholar]
- 25.Xia Y., Pan W., Ke X., et al. Differential escape of HCV from CD8+T cell selection pressure between China and Germany depends on the presenting HLA class I molecule. Journal of Viral Hepatitis. 2019;26(1):73–82. doi: 10.1111/jvh.13011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vahed H., Agrawal A., Srivastava R., et al. Unique Type I Interferon, Expansion/Survival Cytokines, and JAK/STAT Gene Signatures of Multifunctional Herpes Simplex Virus-Specific Effector Memory CD8+TEMCells Are Associated with Asymptomatic Herpes in Humans. Journal of Virology. 2019;93(4):p. e01882. doi: 10.1128/jvi.01882-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khakpoor A., Ni Y., Chen A., et al. Spatiotemporal Differences in Presentation of CD8 T Cell Epitopes during Hepatitis B Virus Infection. Journal of Virology. 2019;93(4) doi: 10.1128/jvi.01457-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huth A., Liang X., Krebs S., Blum H., Moosmann A. Antigen-specific TCR signatures of cytomegalovirus infection. The Journal of Immunology. 2019;202(3):979–990. doi: 10.4049/jimmunol.1801401. [DOI] [PubMed] [Google Scholar]
- 29.Sekyere S. O., Schlevogt B., Mettke F., et al. HCC immune surveillance and antiviral therapy of hepatitis C virus infection. Liver Cancer. 2019;8(1):41–65. doi: 10.1159/000490360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wick D. A., Webb J. R., Nielsen J. S., et al. Surveillance of the tumor mutanome by T cells during progression from primary to recurrent ovarian cancer. Clinical Cancer Research. 2014;20(5):1125–1134. doi: 10.1158/1078-0432.ccr-13-2147. [DOI] [PubMed] [Google Scholar]
- 31.Karasaki T., Nagayama K., Kawashima M., et al. Identification of Individual Cancer-Specific Somatic Mutations for Neoantigen- Based Immunotherapy of Lung Cancer. Journal of Thoracic Oncology. 2016;11(3):324–333. doi: 10.1016/j.jtho.2015.11.006. [DOI] [PubMed] [Google Scholar]
- 32.Gros A., Parkhurst M. R., Tran E., et al. Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients. Nature Medicine. 2016;22(4):433–438. doi: 10.1038/nm.4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stronen E., Toebes M., Kelderman S., et al. Targeting of cancer neoantigens with donor-derived T cell receptor repertoires. Science. 2016;352(6291):1337–1341. doi: 10.1126/science.aaf2288. [DOI] [PubMed] [Google Scholar]
- 34.Nelde A., Walz J. S., Kowalewski D. J., et al. HLA class I-restrictedMYD88L265P-derived peptides as specific targets for lymphoma immunotherapy. OncoImmunology. 2017;6(3):p. e1219825. doi: 10.1080/2162402X.2016.1219825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang X., Kim S., Hundal J., et al. Breast cancer neoantigens can induce CD8+T-cell responses and antitumor immunity. Cancer Immunology Research. 2017;5(7):516–523. doi: 10.1158/2326-6066.CIR-16-0264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Müller M., Gfeller D., Coukos G., Bassani-Sternberg M. ‘Hotspots’ of antigen presentation revealed by human leukocyte antigen ligandomics for neoantigen prioritization. Frontiers in Immunology. 2017;8:p. 1367. doi: 10.3389/fimmu.2017.01367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Balachandran V. P., Initiative A. P. C. G., Łuksza M., et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature. 2017;551(7681):512–516. doi: 10.1038/nature24462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Matsuda T., Leisegang M., Park J.-H., et al. Induction of neoantigen-specific cytotoxic T cells and construction of T-cell receptor-engineered T cells for ovarian cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2018;24(21):5357–5367. doi: 10.1158/1078-0432.CCR-18-0142. [DOI] [PubMed] [Google Scholar]
- 39.Sonntag K., Hashimoto H., Eyrich M., et al. Immune monitoring and TCR sequencing of CD4 T cells in a long term responsive patient with metastasized pancreatic ductal carcinoma treated with individualized, neoepitope-derived multipeptide vaccines: a case report. Journal of translational medicine. 2018;16(1) doi: 10.1186/s12967-018-1382-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bjerregaard A.-M., Nielsen M., Jurtz V., et al. An analysis of natural T cell responses to predicted tumor neoepitopes. Frontiers in Immunology. 2017;8 doi: 10.3389/fimmu.2017.01566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shannon C. E. A mathematical theory of communication. Bell System Technical Journal. 1948;27(3):379–423. doi: 10.1002/j.1538-7305.1948.tb01338.x. [DOI] [Google Scholar]
- 42.Kuhn M. Building predictive models in R using the caret package. Journal of Statistical Software. 2008;28(5):1–26. [Google Scholar]
- 43.Kursa M. B., Rudnicki W. R. Feature selection with theBorutaPackage. Journal of Statistical Software. 2010;36(11) doi: 10.18637/jss.v036.i11. [DOI] [Google Scholar]
- 44.Law I., Wiener M. Classification and regression by randomForest. 3. Vol. 2. R News; 2002. [Google Scholar]
- 45.Sing T., Sander O., Beerenwinkel N., Lengauer T. ROCR: visualizing classifier performance in R. Bioinformatics. 2005;21(20):3940–3941. doi: 10.1093/bioinformatics/bti623. [DOI] [PubMed] [Google Scholar]
- 46.Walker J. M. The proteomics protocols handbook. Biochemistry. 2006;71(6):696–696. [Google Scholar]
- 47.Kyte J., Doolittle R. F. A simple method for displaying the hydropathic character of a protein. Journal of Molecular Biology. 1982;157(1):105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 48.Zimmerman J. M., Eliezer N., Simha R. The characterization of amino acid sequences in proteins by statistical methods. Journal of Theoretical Biology. 1968;21(2):170–201. doi: 10.1016/0022-5193(68)90069-6. [DOI] [PubMed] [Google Scholar]
- 49.Grantham R. Amino acid difference formula to help explain protein evolution. Science. 1974;185(4154):862–864. doi: 10.1126/science.185.4154.862. [DOI] [PubMed] [Google Scholar]
- 50.Fraga S. Theoretical prediction of protein antigenic determinants from amino acid sequences. Canadian Journal of Chemistry. 1982;60(20):2606–2610. doi: 10.1139/v82-374. [DOI] [Google Scholar]
- 51.Sweet R. M., Eisenberg D. Correlation of sequence hydrophobicities measures similarity in three- dimensional protein structure. Journal of Molecular Biology. 1983;171(4):479–488. doi: 10.1016/0022-2836(83)90041-4. [DOI] [PubMed] [Google Scholar]
- 52.Meek J. L. Prediction of peptide retention times in high-pressure liquid chromatography on the basis of amino acid composition. Proceedings of the National Academy of Sciences of the United States of America. 1980;77(3):1632–1636. doi: 10.1073/pnas.77.3.1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rose G., Geselowitz A., Lesser G., Lee R., Zehfus M. Hydrophobicity of amino acid residues in globular proteins. Science. 1985;229(4716):834–838. doi: 10.1126/science.4023714. [DOI] [PubMed] [Google Scholar]
- 54.Chou P. Y., Fasman G. D. Prediction of the secondary structure of proteins from their amino acid sequence. Advances in enzymology and related areas of molecular biology. 1978;47:45–148. doi: 10.1002/9780470122921.ch2. [DOI] [PubMed] [Google Scholar]
- 55.Deléage G., Roux B. An algorithm for protein secondary structure prediction based on class prediction. Protein Engineering. 1987;1(4):289–294. doi: 10.1093/protein/1.4.289. [DOI] [PubMed] [Google Scholar]
- 56.Burger A. Atlas of protein sequence and structure 1969. Journal of Medicinal Chemistry. 1970;13(2):337–337. doi: 10.1021/jm00296a903. [DOI] [Google Scholar]
- 57.Jones D. D. Amino acid properties and side-chain orientation in proteins: a cross correlation approach. Journal of Theoretical Biology. 1975;50(1):167–183. doi: 10.1016/0022-5193(75)90031-4. [DOI] [PubMed] [Google Scholar]
- 58.Zhao G., London E. Strong correlation between statistical transmembrane tendency and experimental hydrophobicity scales for identification of transmembrane helices. Journal of Membrane Biology. 2009;229(3):165–168. doi: 10.1007/s00232-009-9178-0. [DOI] [PubMed] [Google Scholar]
- 59.Janin J. Surface and inside volumes in globular proteins. Nature. 1979;277(5696):491–492. doi: 10.1038/277491a0. [DOI] [PubMed] [Google Scholar]
- 60.Green J. R., Korenberg M. J., David R., Hunter I. W. Recognition of adenosine triphosphate binding sites using parallel cascade system identification. Annals of Biomedical Engineering. 2003;31(4):462–470. doi: 10.1114/1.1561293. [DOI] [PubMed] [Google Scholar]
- 61.Lifson S., Sander C. Antiparallel and parallel β-strands differ in amino acid residue preferences. Nature. 1979;282(5734):109–111. doi: 10.1038/282109a0. [DOI] [PubMed] [Google Scholar]
- 62.Duan F., Duitama J., al Seesi S., et al. Genomic and bioinformatic profiling of mutational neoepitopes reveals new rules to predict anticancer immunogenicity. Journal of Experimental Medicine. 2014;211(11):2231–2248. doi: 10.1084/jem.20141308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Calis J. J. A., Maybeno M., Greenbaum J. A., et al. Properties of MHC class I presented peptides that enhance immunogenicity. PLoS Computational Biology. 2013;9(10, article e1003266) doi: 10.1371/journal.pcbi.1003266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Laumont C. M., Vincent K., Hesnard L., et al. Noncoding regions are the main source of targetable tumor-specific antigens. Science Translational Medicine. 2018;10(470):p. eaau5516. doi: 10.1126/scitranslmed.aau5516. [DOI] [PubMed] [Google Scholar]
- 65.Wang G., Wan H., Jian X., et al. INeo-Epp: T-cell HLA class I immunogenic or neoantigenic epitope prediction via random forest algorithm based on sequence related amino acid features. bioRxiv; 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
S1 Table IEDB antigen epitopes summary. Detailed description of 17 HLA molecules collected from IEDB. (XLSX) S2 Table External validation antigen epitopes summary. Epitope details of 7 publications. (XLSX) S3 Table Neoantigen epitopes summary. Epitope details of 13 publications. (XLSX) S4 Table Summary of amino acid characteristics. For all amino acid characteristics (n=21) that are described in the ExPASy. (XLSX).
Data Availability Statement
The data used to support the findings of this study are included within the supplementary information file(s).