

Abstract
The major pathways of DNA double strand break (DSB) repair have key roles in suppressing genomic instability. However, if deployed in an inappropriate cellular context, these same repair functions can mediate chromosome rearrangements that underlie various human diseases, ranging from developmental disorders to cancer. Two major mechanisms of DSB repair predominate in mammalian cells, namely homologous recombination and non-homologous end joining. In this Review, we outline a ‘decision tree’ of DSB repair pathway choice in somatic mammalian cells, and consider how DSB repair dysfunction can lead to genomic instability. Stalled or broken replication forks present a distinctive challenge to the DSB repair system. Emerging evidence suggests that the ‘rules’ governing stalled fork repair pathway choice differ from those that operate at a conventional DSB.
Introduction
DNA double strand breaks (DSBs) and solitary DNA ends—whether products of chromosome breakage, replication fork stalling or telomere deprotection — pose an immediate threat to the stability of the genome, potentially provoking chromosome rearrangements and disrupting gene structure and function. Indeed, germline mutations in DSB repair genes cause genomic instability in numerous hereditary human disease syndromes that are associated with cancer predisposition, developmental disorders and premature aging1. Genetic disruption of any one of the major pathways of DSB repair causes genomic instability in mammalian primary cells, suggesting that the different DSB repair pathways normally work in harmony to minimize genome errors. However, not all breaks are created equal. A series of control mechanisms have evolved to ensure that the DSB repair pathway that is engaged is matched to the cellular context — including cell cycle phase and the local chromatin environment. This Review focuses on how these control mechanisms operate in normal cells and how their dysfunction can promote genomic instability. We first outline the pathways that are available for the repair of a conventional two-ended DSB and discuss the special challenge to the DSB repair system posed by one-ended breaks. We then consider the critical points at which commitment to each pathway occurs, and outline a ‘decision tree’ of DSB repair. Lastly, we address our emerging understanding of the ‘rules’ governing repair at stalled forks. Recent work shows that these rules differ substantially from those that operate at a conventional DSB. We suggest that at least one DSB repair pathway that has traditionally been considered error-prone, single strand annealing, may have a conservative function at stalled forks by suppressing tandem duplications at sites of aberrant replication fork restart.
Overview of DSB repair pathways
Two major pathways are predominantly involved in the repair of a two-ended DSB: non-homologous end joining (NHEJ) and homologous recombination (HR)2–8 (Figure 1). ‘Classical’ NHEJ (cNHEJ)—so called to distinguish it from alternative end-joining (aEJ), the rejoining of DNA ends in the absence of cNHEJ genes—is a rapid, high capacity pathway in mammalian cells that joins two DNA ends with minimal reference to DNA sequence. cNHEJ can, however, accommodate very limited base-pairing between the two processed DNA ends, potentially forming repair joints with up to 4 base pairs of ‘microhomology’7. By contrast, homologous recombination requires extensive sequence homology between the broken DNA and a donor DNA molecule, and entails templated DNA synthesis as a key step in the repair process.
Figure 1. Two major pathways of DNA double strand break repair.
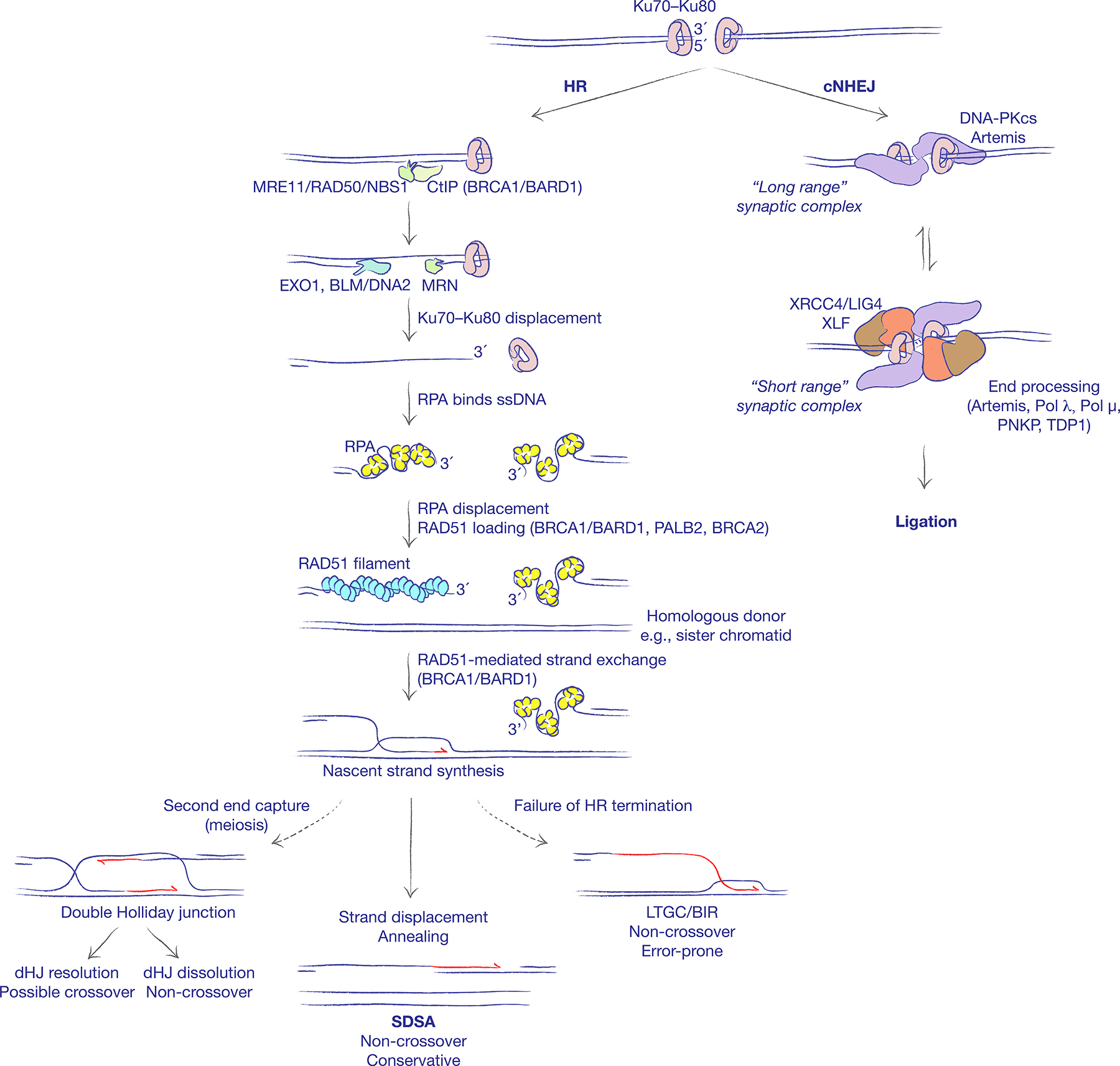
The binding of the Ku70–Ku80 heterodimer to DNA ends schedules repair of DNA double strand breaks (DSBs) by classical non-homologous end joining (cNHEJ). cNHEJ entails formation of a ‘long range’ synaptic complex, which is in equilibrium with a ‘short range’ synaptic complex. End processing by cNHEJ enzymes (as shown) and ligation are restricted to the short range complex. PNKP: Polynucleotide kinase-phosphatase. TDP1: Tyrosyl-DNA phosphodiesterase 1. The default engagement of cNHEJ can be disrupted by DNA end resection. The nuclease activity of MRE11 converts the blunt end into a 3ʹ single-stranded DNA (ssDNA) tail, displacing Ku70–Ku80 from the DNA end and establishing the possibility of repair by homologous recombination (HR). The replication protein A (RPA) complex avidly binds to ssDNA and must be displaced by recombination mediators to enable the formation of a RAD51 nucleoprotein filament. BRCA2 is the major recombination mediator in mammalian cells, likely acting in concert with PALB2 and the BRCA1–BARD1 heterodimer. Interactions between the two DNA ends at the recombination synapse, and operations on the D-loop formed following synapsis, influence which HR sub-pathway is engaged. The non-crossover synthesis-dependent strand annealing (SDSA) pathway is the predominant repair pathway in somatic cells. In meiotic cells, formation of a double Holliday junction (dHJ) intermediate can lead to crossing over. A failure to engage the second end of the break, or failure to displace the nascent strand leads to aberrant replicative HR responses of long tract gene conversion (LTGC) and break-induced replication (BIR). Established roles for BRCA gene products in HR are indicated in parentheses.
Classical non-homologous end joining
cNHEJ is initiated by the binding of the Ku70–Ku80 (also known as XRCC6–XRCC5) heterodimer to DSB ends. Although several molecules of Ku can be loaded onto a DNA end in vitro, direct imaging of Ku at DSBs in living mammalian cells suggests that one dimer of Ku normally binds to each DNA end of a chromosomal DSB9. Ku nucleates the recruitment of other cNHEJ factors including the DNA-dependent protein kinase catalytic subunit (DNA-PKcs), DNA ligase IV (LIG4) and the associated scaffolding factors XRCC4, XRCC4-like factor (XLF) and paralogue of XRCC4 and XLF (PAXX)10–14. XRCC4 is essential for LIG4 stability and function, while XLF and PAXX have partially redundant scaffolding roles, as revealed by studies of the cNHEJ-mediated process of V(D)J recombination in lymphocyte development15,16. Single molecule imaging of the cNHEJ reaction has revealed a two-stage mechanism of synapsis of the two ends of the DSB17. First, Ku70–Ku80 and DNA-PKcs establish long-range synapsis; second, the two ends become closely aligned in a process requiring XLF, non-catalytic functions of XRCC4–LIG4 and DNA-PKcs kinase activity18. A synaptic reaction can alternate between long-range and short-range states, suggesting that sampling of DNA end-binding partners is a dynamic process that is reversible until the process of ligation is completed. End processing by the nuclease Artemis, by specialized DNA polymerases λ and μ and other enzymes ensures compatibility of the ligated ends and is restricted to the short-range synaptic complex19. A number of accessory factors, some of which likely remain undiscovered, support or otherwise regulate C-NHEJ. These include the multifunctional Mre11/Rad50/Nbs1 (MRN) end recognition complex, which may assist in end bridging20–22 and apratxin and PNK-like factor (APLF), which interacts with Ku80 and with poly(ADP ribose)-modified proteins in the vicinity of the DSB23–25. Several additional positive and negative regulators of Ku70–Ku80 have been identified26–28. For a ‘Ku-binding motif” in a number of Ku70–Ku80-interacting proteins is thought to mediate their cNHEJ-regulatory functions27.
Ku70–Ku80 is an abundant nuclear complex, which has high affinity for DNA ends that are either blunt or possess limited single-stranded DNA (ssDNA) overhangs. Long ssDNA tails have reduced affinity for Ku70–Ku80 and are channeled towards cNHEJ less efficiently29. Nucleolytic processing of DNA overhangs or chemically modified ends by the cNHEJ nuclease Artemis can re-establish Ku70–Ku80 access to the DNA end30. The reversible nature of cNHEJ synapsis prior to ligation suggests that later steps of the pathway may also be subject to regulation. Indeed, DNA-PKcs autophosphorylation is an important regulator of cNHEJ, affecting both Ku70–Ku80 binding and complex disassembly during ligation18,31. Further, interspecies variations in the efficiency of cNHEJ may reflect differences in the abundance or regulation of DNA-PKcs.
Homologous recombination
The second major pathway of DSB repair, homologous recombination, is a multi-step process that differs from cNHEJ in key aspects. Unlike cNHEJ, which operates throughout the vertebrate cell cycle to repair DSBs, HR is largely restricted to the S and G2 phases of the cell cycle32. The major conservative HR pathway in somatic cells involves recombination between sister chromatids — the identical copies of a post-replicative chromosome32,33. Sequence identity, alignment and physical cohesion of the two sister chromatids are thought to favour sister chromatid recombination over other potential recombination partners. HR entails the loading of the recombinase RAD51 (the eukaryotic homologue of RecA) onto ssDNA, either at DNA ends that have undergone DNA end resection to generate extended 3´ ssDNA tails, or at post-replicative ssDNA gaps, where no DSB is present2,34. Eukaryotic DNA end resection is initiated by the MRE11–RAD50–NBS1 (MRN) complex (Mre11–Rad50–Xrs2 in Saccharomyces cerevisiae), which also serves as a scaffold for activation of the multi-functional DNA damage signalling kinase ATM35–37. Mre11 endonuclease activity nicks the DNA strand of the DSB that possesses a free 5´ terminus, up to 300 nt internal to the DNA end, and Mre11 3´–5´ exonuclease activity extends the nick towards the DNA end. Efficient initiation of ‘short-range’ resection by MRE11 endonuclease activity requires interaction with CtBP-interacting protein (CtIP; also known as RBBP8; Sae2 in S. cerevisiae and Ctp1 in Schizosaccharomyces pombe) and is stimulated by protein blocks at the DNA end, such as Ku70–Ku80, replication protein A (RPA) or nucleosomes38–45. This initial processing step is thought to displace Ku70–Ku80 from DNA ends (Figure 1) and also provides an entry point for ‘long range’ resection, in which Exonuclease 1 (EXO1), in parallel with the helicase Bloom syndrome protein (BLM; Sgs1 in S. cerevisiae) and the endonuclease DNA2, mediate unwinding and/or nucleolytic digestion of the 5´ strand of the DNA end to form a long 3´ ssDNA tail46–49. Several additional DNA end resection regulators, both positive and negative, have been described. BRCA1, the product of one of the two major hereditary breast and ovarian cancer predisposition genes, in complex with its heterodimeric partner BARD1, interacts with CtIP and MRN and has been implicated in DNA end resection as well as in later stages of HR (discussed below).
Single-stranded DNA rapidly becomes coated with the abundant ssDNA binding heterotrimeric complex replication protein A (RPA). ssDNA bound to RPA cannot pair with other ssDNA sequences. Thus, RPA ‘melts’ secondary structure in ssDNA and limits spurious interactions with ssDNA intermediates of other nuclear processes. RPA also forms a barrier to the loading of the RAD51 recombinase and must be displaced by recombination mediators if HR is to proceed34 (Figure 1). In budding yeast, Rad52 (Rad22 in S. pombe) is the key recombination mediator, while BRCA2, the product of the second major hereditary breast and ovarian cancer predisposition gene, serves that function in vertebrates and in some fungal species50–52. BRCA2, constitutively bound to the proteasomal component DSS1, interacts with both ssDNA and RAD51 monomers, as well as with BRCA1–BARD1 via the PALB2 protein5. BRCA2–DSS1 can act as a recombination mediator in vitro. The extent to which BRCA1–BARD1–PALB2 modulates this activity remains to be determined. The association of BRCA1 with proteins involved in DNA end resection and RAD51 loading suggests that BRCA1 might couple these two HR steps, perhaps analogous to how DNA end resection is coupled to RecA filament formation in Escherichia coli53. In E. coli, direct interactions between the RecB subunit of the DNA end resection complex RecBCD and RecA ensure timely loading of RecA at χ recombination hotspot sequences54.
The RAD51 filament is a dynamic structure, subject to competing activities that promote its stability or disassembly. In S. cerevisiae, Rad51 paralogues promote the stability of the Rad51 filament and restrain its disassembly by the antirecombinase Srs255–57. Loss of Srs2 alone promotes unrestrained, ‘toxic’ recombination and genomic instability58. These relationships indicate that RAD51 filament stability in normal physiology is regulated to optimize the efficiency of HR and, at the same time, to restrict RAD51 function to appropriate DNA substrates. RAD51-bound ssDNA mediates the homology search that defines HR, by invading duplex DNA molecules and facilitating base-pairing with complementary homologous DNA sequences in the invaded molecule. BRCA1–BARD1 has recently been implicated in facilitating RAD51-mediated homologous pairing, indicating that BRCA1 promotes multiple HR steps59. RecA or RAD51 nucleoprotein filaments form synaptic complexes containing a three-stranded DNA helix that supports heteroduplex formation — base-pairing between the invading strand and the complementary strand of the invaded molecule60. If a match of sufficient homology is made, the synapse is stabilized and the non-base-paired strand of the invaded molecule is displaced to form a displacement loop (D-loop)—a process driven by RAD51-mediated ATP hydrolysis and RAD51 filament disassembly61. The free 3´ end of the invading strand engages a DNA polymerase, extending the invading (nascent) strand using the invaded donor DNA molecule as a template for gene conversion (Figure 1). DNA polymerase δ (Pol δ) plays a major role in nascent strand synthesis, but translesion DNA polymerases have also been implicated in competition with Pol δ62–64. Gene conversion in yeasts and flies can entail multiple rounds of RAD51-mediated invasion, nascent strand extension, displacement and reinvasion65,66. The same process might also occur in vertebrates. Various motor proteins, including FANCM (Mph1 in S. cerevisiae and Fml1 in S. pombe), BLM and RTEL1 can disassemble D-loops, implicating these proteins in displacement of the nascent strand to limit the extent of gene conversion44,67–70.
Subpathways of HR
The major pathway of somatic HR, synthesis-dependent strand annealing (SDSA), invokes RAD51-mediated invasion by only one end of the two-ended DSB, whereas the second end is resected but remains passive2. How this asymmetry is established is not well understood. Interestingly, a capacity for asymmetric resection of ionizing radiation-induced two-ended breaks was revealed in S. cerevisiae mutants lacking MRX or Sae2 function, suggesting that MRX or MRN may control this process71. For a two-ended DSB, the non-invading second end of the break enables HR termination by annealing with the displaced nascent strand. Because it does not involve formation of a Holliday junction, SDSA is a non-crossover pathway (Figure 1). By contrast, the classical HR pathway, DSB repair (DSBR), which is prominent in meiotic recombination, entails formation of a double Holliday junction (dHJ)2. Depending on the polarity of dHJ resolution, this can result in crossing over between the recombining molecules, detected in somatic cells as a sister chromatid exchange72 (Figure 1). BLM, in complex with TopIIIα/RMI1/RMI2 (the BTR complex), promotes an alternative non-crossover dHJ dissolution mechanism73. Replicative responses following strand invasion, of which break-induced replication (BIR) is a prominent example, are discussed below.
Single strand annealing
Several additional pathways of DSB repair are recognized. Single strand annealing (SSA, Figure 2A) is a Rad51-independent mechanism that enables two homologous 3´-ssDNA ends (for example, at tandem repeats) to be joined by annealing, at the cost of a deletion between the repeats2. In repair of a conventional DSB, SSA is therefore an error-prone pathway. SSA requires extensive DNA end resection and RPA displacement to reveal complementary homologous sequences. Rad52 has a key role in RPA displacement and Rad51 loading in yeast, while its role in this process in mammalian cells appears less critical.
Figure 2. Alternative DSB repair pathways.
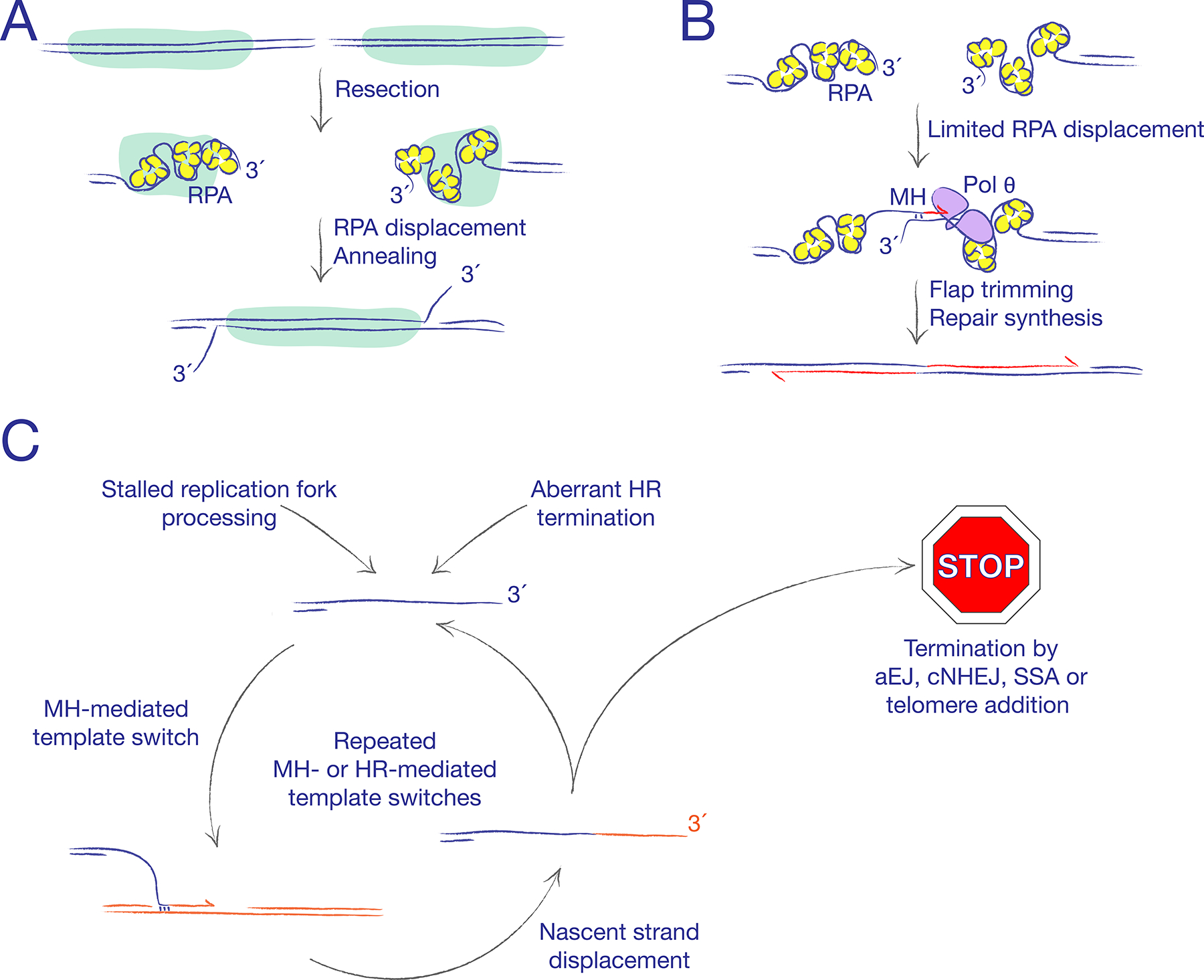
A. Single strand annealing (SSA) converts homologous repeats (marked in green) to a single copy, by annealing complementary single-stranded DNA (ssDNA) ends within each repeat. Replication protein A (RPA) must be displaced to expose complementary ssDNA for annealing. B. Alternative end joining (aEJ) rejoins DNA ends without use of classical non-homologous end joining (cNHEJ) proteins. MH: microhomology. Frequent use of microhomology-mediated end joining (MMEJ) is typical but not a defining feature of aEJ. The figure depicts the action of DNA polymerase θ (Pol θ), an important aEJ mediator in mammalian cells. C. Microhomology-mediated template switching can arise when a free 3ʹ ssDNA end lacks an immediately available partner for recombination or rejoining. The persistent ssDNA end is thought to interact with ssDNA gaps in neighbouring DNA molecules, leading to the synthesis of a few hundred base pairs templated on the ectopic donor strand. Multiple rounds of microhomology-mediated or HR-mediated template switching can give rise to complex breakpoints in cancer and in developmental disorders.
Alternative end joining
A related rejoining mechanism that operates on 3´-ssDNA ends is alternative end joining (aEJ, Figure 2B)—defined as non-homologous end joining that does not use the cNHEJ pathway74). Use of microhomology at the breakpoint is a prominent feature of aEJ and the term ‘microhomology-mediated end joining’ (MMEJ) is sometimes used synonymously with aEJ. However, this elision can be confusing, since limited microhomology use is also associated with cNHEJ. We will therefore use the term ‘MMEJ’ as a descriptive term, to note the presence of microhomology at a breakpoint, whether mediated by cNHEJ or aEJ. In metazoans, Pol θ (encoded by the POLQ gene) has been implicated in aEJ75,76. A Pol θ-associated helicase function can displace RPA from ssDNA, revealing internal microhomologies on ssDNA, while its polymerase function can stabilize the joint between the two DNA ends77,78. Additional DNA polymerases may be required to complete fill-in synthesis during aEJ62. Polq null mice reveal spontaneous genomic instability, implicating Pol θ (and possibly aEJ) in physiological functions79. These functions are probably executed at sites of stalled replication; in Caenorhabditis elegans, pol-q1 (encoding Pol θ) suppresses large chromosomal deletions at sites of fork stalling, at the expense of allowing small deletions to form80,81. Combined deletion of Ku70 and Polq in primary mouse cells induces a severe growth defect, while HR-defective cells reveal elevated expression of POLQ82–84. Taken together, these data suggest that Pol θ evolved to repair certain replication-associated DNA lesions that are poor substrates for cNHEJ. Pol θ has also been implicated as a mediator of pathological chromosome rearrangements84.
Recent work suggests that not all mammalian aEJ is mediated by Pol θ. Class switch recombination (CSR) is an end joining process that is involved in the rearrangement of immunoglobulin heavy chain (IgH) loci in cytokine-stimulated B cells, in which both cNHEJ and aEJ pathways repair DSBs induced at CSR ‘switch’ regions, triggered by activation-induced cytidine deaminase (AID)-mediated cytosine deamination74,85. Polq–/– mouse B cells have normal CSR frequencies and normal spectra of microhomology use at CSR breakpoints, indicating that POLQ is not required for MMEJ during CSR86,87. However, Polq is required for formation of CSR junctions that contain nucleotide insertions86. A recent study reported that Rad52–/– mouse B cells have increased CSR frequencies in comparison to wild type cells, but fail to form CSR products with microhomology >4 bp87. Since C-NHEJ is not associated with microhomology >4 bp, this finding raises the interesting possibility that RAD52 contributes to mammalian aEJ during CSR and may compete with cNHEJ in this context. RAD52 has not been implicated in MMEJ in other settings88. It remains to be determined what specific features of CSR might enable RAD52 to contribute to aEJ.
Repair responses to one-ended breaks
A distinctive challenge for the DSB repair system arises at sites of broken or collapsed replication forks, since solitary DNA ends or one-ended breaks can arise in this context89–92. In this case, there is no immediate partner for end joining, and the absence of a second DNA end denies the possibility of engaging error-free SDSA.
Break-induced replication
In S. cerevisiae, ‘break-induced replication’ (BIR, Figure 1), a product of RAD51-dependent gene conversion, extends the nascent strand of a one-ended invasion to the end of the chromosome, frequently copying more than 100 kb from the donor chromosome, unless a disruptive event such as a collision with a replication fork prematurely terminates the process91,93. BIR that engages a heterologous chromosome donor results in a non-reciprocal translocation94,95. In S. pombe, a Rad22Rad52-mediated form of BIR can occur at stalled forks93. BIR entails conservative DNA synthesis via a migrating bubble mechanism, generating extensive ssDNA tracts that are vulnerable to mutation and secondary chromosome rearrangements58,96–99. As a result, BIR is highly mutagenic. BIR in S. cerevisiae requires the gene PIF1, encoding the Pif1 helicase and POL32, encoding a non-essential subunit of Pol δ, whereas neither of these genes are required for conventional short tract SDSA96,100,101. However, SDSA-mediated long tract gene conversions (LTGCs) of only a few kilobases also require POL32102. BIR can be preceded by repeated rounds of LTGC and homologous template switching during the first ~10 kb of gene conversion66. These findings evince both similarities and distinctions between LTGC and BIR.
Other than the presence or absence of a second DNA end, the stability of D-loop/migrating bubble structures formed during nascent strand synthesis influences copying mechanism ‘choice’ during HR. Activities that mediate D-loop disassembly/nascent strand displacement channel HR towards SDSA, while factors that stabilize the D-loop favor LTGC or BIR and crossover outcomes in S. cerevisiae103. This interplay is supported by data implicating several motor proteins in D-loop metabolism. A recent study in S. cerevisiae, in which D-loop formation was directly quantified in response to a site-specific DSB, identified two parallel pathways of D-loop disruption, mediated by Mph1 and the Sgs1/Top3/Rmi1 (STR) complex or, in parallel, by the Srs2 helicase104. Similarly, the mammalian homologues of Mph1 (FANCM) and STR (the BLM/TopIIIα/RMI1/RMI2 complex), as well as several candidate homologues of Srs2, can disrupt D-loops in vitro103, and both BLM and FANCM suppress LTGC during mammalian HR92. Interestingly, SGS1 and MPH1 impose a delay in the onset of BIR at a one-ended break102,105,106. These data suggest that the fate of the D-loop is intimately related to the balance between conservative and error-prone outcomes of HR.
In mammalian cells, the longest DSB-induced Rad51-mediated gene conversions reported to date are LTGC products of <10 kb, substantially less than the >100kb BIR tracts observed in yeast. Cells lacking BRCA1, CtIP, BRCA2, or paralogues of RAD51 reveal a bias in favor of LTGC107–110. This bias could reflect a failure to engage the second end of the break during SDSA termination or a specific bias in favour of BIR-type copying mechanisms111. An emerging literature suggests that some BIR-like processes in mammalian cells are RAD51-independent. First, RAD51-independent mitotic DNA synthesis (‘MiDAS’) occurs at common fragile sites (regions of the genome that exit S phase with incompletely replicated DNA)112,113. MiDAS is mediated by RAD52, DNA polymerase δ subunit 3 (POLD3; the mammalian homologue of S. cerevisiae Pol32) and the structure-specific nuclease MUS81–EME1 — possibly implicating the processing of stalled replication forks as an initiating event. Second, RAD51-independent and Pol δ-mediated BIR tracts of up to ~70 kb are provoked by DSBs on telomeres maintained by the recombination-mediated ‘alternative lengthening of telomeres’ (ALT) pathway114. Third, LTGC triggered at stalled mammalian replication forks is RAD51-independent110.
Microhomology-mediated template switching
A distinct replicative response associated with a solitary 3´-ssDNA end is microhomology-mediated template switching (Figure 2C). This process entails microhomology-mediated synapsis of a free 3´-ssDNA tail with ssDNA donor sequences (possibly daughter strand gaps in postreplicative chromatin), followed by limited DNA synthesis of up to a few hundred base pairs, and completed by strand displacement. In S. cerevisiae, translesional DNA polymerases are implicated in the synthesis step99. Unlike BIR, the end product of microhomology-mediated template switching is not a full-blown chromosome translocation but the liberation of a 3´-ssDNA tail derived from the displaced nascent strand, similar in structure to the initiating 3´-ssDNA tail. Thus, microhomology-mediated template switching does not resolve the problem of the one-ended break; instead, it ‘kicks the can down the road’. Microhomology-mediated template switching has been invoked to explain complex chromosome rearrangement breakpoints in cancer and other diseases, in which multiple short (i.e., a few hundred bp) ectopic sequences derived from distinct remote chromosomal loci are present within the breakpoint115–117. Such complex breakpoints may be products of ‘futile cycles’ of repeated microhomology-mediated template switching between different donor loci. Work in E. coli and S. cerevisiae has emphasized an association of microhomology-mediated template switching with BIR (“microhomology-mediated BIR”)118–121. Multiple RAD51-mediated strand invasions can also generate complex breakpoints in yeast122. In contrast, experimental models of microhomology-mediated template switching in mammalian cells indicate an association with end joining92,116,123,124. Perhaps these species differences reflect the relative efficiency of end joining mechanisms in mammalian cells (where they have a major role) and yeast (where their role is minimal). In summary, the phenomenon of microhomology-mediated template switching suggests that solitary 3´-ssDNA tails are highly reactive DNA lesions that, if not channeled into a conservative repair pathway, can interact avidly with neighboring DNA molecules in uncontrolled and dangerous ways.
The chromatin response to DSBs
DSBs provoke an extensive chromatin response that has an important role in DSB repair. The PIKK-family DNA damage response kinases ATM (activated by interactions with MRN at the break36), ATR (activated by ssDNA and RPA sensors AtrIP and ETAA1125–129) and DNA-PKcs (activated by Ku70–Ku8018) have each been implicated in the phosphorylation of serine 139 in the carboxy-terminal tail of the histone variant H2A.X, forming “γ-H2A.X” chromatin domains130. The H2A.X signalling chromatin response is conserved from yeasts to mammals130. In vertebrates, γ-H2A.X directly recruits the adaptor protein MDC1, forming a specialized chromatin structure that can extend hundreds of kilobases away from the DSB131. γ-H2A.X/MDC1 decorated chromatin is multifunctional, supporting class switch recombination in activated B cells (an end-joining process), HR between sister chromatids and ATM signal amplification (mediated by interaction of MDC1 with the MRN complex), and also suppressing spurious end resection during V(D)J recombination in lymphocytes130,132–135. The MDC1-binding E3 ubiquitin ligase RNF8 catalyzes K63-linked polyubiquitylation of histone H2A, recruiting BRCA1–Abraxas–Rap80-containing complexes and a second E3 ubiquitin ligase, RNF168136,137. The BRCA1–Rap80 complex contains deubiquitylating enzymes, further editing the ubiquitin landscape of chromatin near the DSB, and has a role in antagonizing DNA end resection138–141. In parallel, 53BP1 — a homologue of the S. cerevisiae Rad9 and S. pombe Crb2 damage response proteins—is recruited to chromatin by binding to dual chromatin marks of histone H4 monomethylated on lysine 20 (H4K20me1) or H4K20me2, which are constitutive marks in mature chromatin and histone H2A monoubiquitylated on lysine 15 (H2AK15Ub; a target of RNF168)142. Both H2A.X-dependent and H2A.X-independent pathways of 53BP1 and BRCA1 recruitment to DSB sites have been described143. These complexes execute distinct functions in DSB repair, conforming quite well to a ‘histone code’ of DSB repair144. In chicken DT40 cells, 53BP1 null reveals epistasis with null alleles of cNHEJ genes and 53BP1 similarly regulates cNHEJ in mammalian cells, primarily in an H2A.X-independent manner144,145. Numerous additional biochemical modifications of chromatin occur in the vicinity of the break, including recruitment of complexes that remodel chromatin and execute histone replacement146–150. An emerging literature points to a role for RNA in the DSB response151,152. These modifications may contribute to repair, signalling and epigenetic reprogramming in the vicinity of the break.
A ‘decision tree’ of DSB repair
Given such a bewildering array of DSB repair pathways, how does the cell select the pathway most appropriate for each DSB? In principle, all DSB repair pathways might compete for access to all free DNA ends. However, for a conventional two-ended DSB, the two major conservative DSB repair pathways, cNHEJ and HR, are dominant. In contrast, error prone pathways (such as SSA, aEJ, microhomology-mediated template switching and BIR) may act more opportunistically, scavenging on the products of aborted or incomplete cNHEJ or HR, or on problematic lesions such as one-ended breaks. In this way, the DSB repair system could be understood as a decision tree (Figure 3, 4), the branch-points (‘nodes’) of which represent points of commitment to cNHEJ or HR, points of physiological sub-pathway divergence (for example, during late stages of HR), or points at which repair intermediates are vulnerable to hijack by error-prone repair pathways. Presumably, in a well-regulated cell, each decision node is tuned to maximize the probability of conservative repair and minimize error-prone outcomes. Pathological conditions perturb this regulatory balance by disrupting DSB repair regulatory genes, by flooding the cell with levels of DNA damage that exceed the capacity of physiological pathways, or by allowing the formation of complex DNA lesions for which there are no good outcomes. For example, some of the recently identified examples of catastrophic chromosome rearrangement—chromothripsis and chromoplexy—reflect the action of end joining on an overwhelming burden of simultaneously arising DSBs116,117.
Figure 3. A decision tree of DNA double strand break repair.
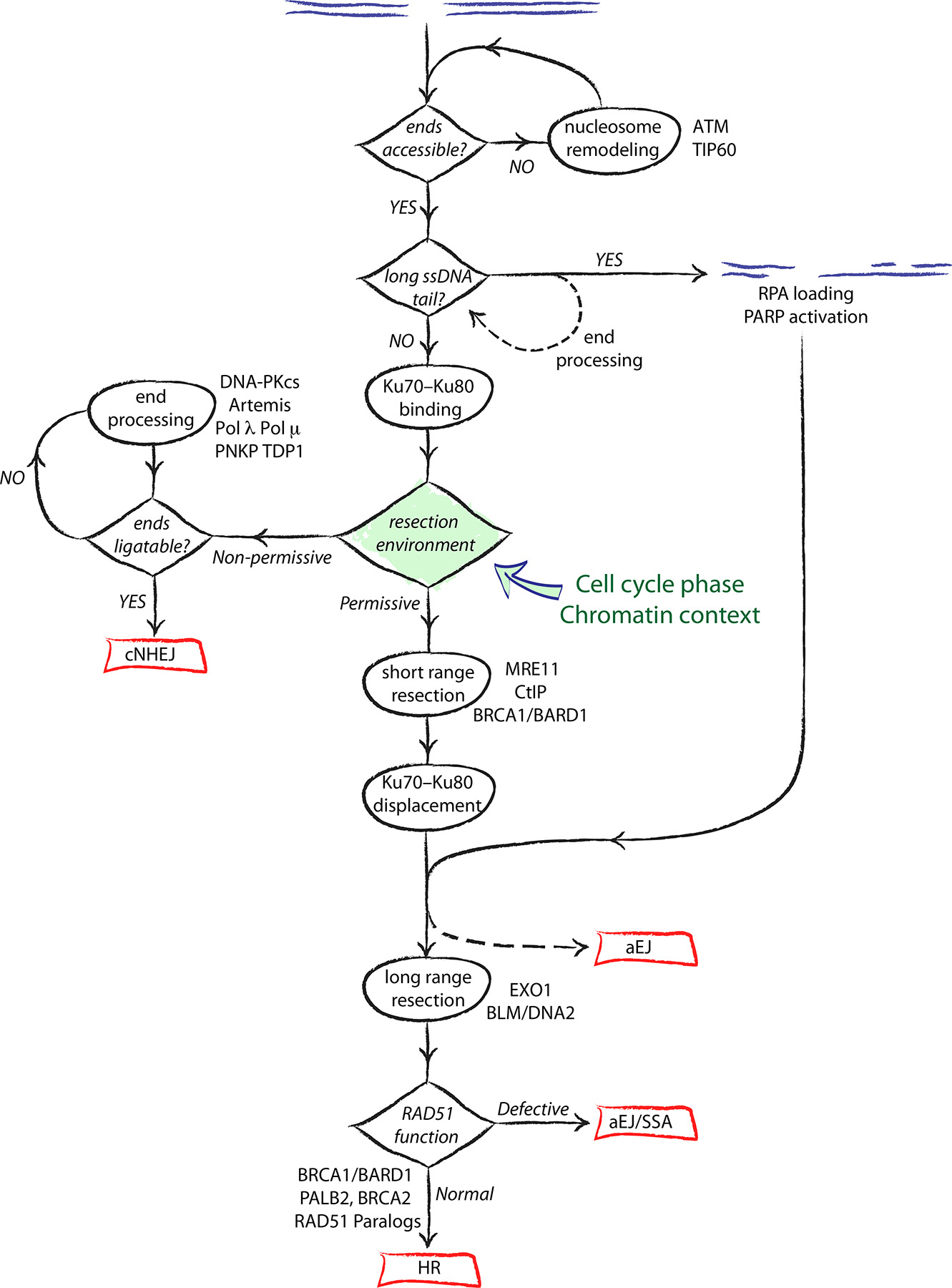
DNA end resection has a crucial role in determining repair pathway choice. Cellular environments that disfavor resection enable Ku70–Ku80 retention at the DNA end, leading to classical non-homologous end joining (cNHEJ). A pro-resection environment favours Ku70–Ku80 displacement and the engagement of homologous recombination (HR). Error-prone pathways such as alternative end joining (aEJ) and single strand annealing (SSA) can act opportunistically on complex DNA ends or on recombination intermediates, hijacking the conservative HR process and leading to chromosome rearrangements.
Figure 4. A decision tree of homologous recombination.
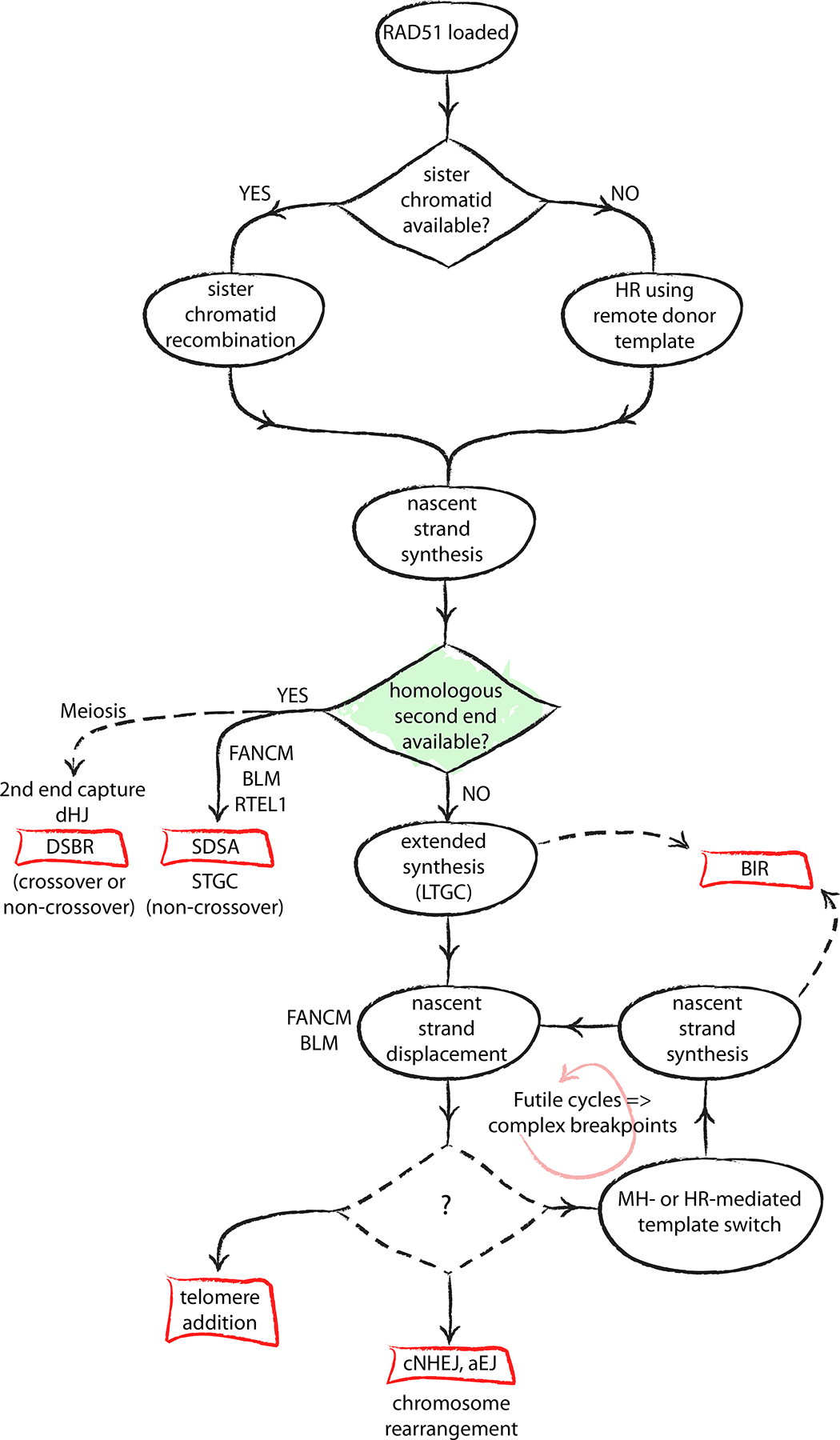
The schematic depicts the key role of the second end of the DNA double-stranded break (DSB) in determining the outcome of the homologous recombination (HR) process. Conservative outcomes are possible only if the second end is engaged for HR termination. The absence of a second end, or a failure to engage it in a timely fashion, leads to error-prone replicative HR outcomes — namely, long tract gene conversion (LTGC) and break-induced replication (BIR). Displacement of the nascent strand following LTGC places the solitary 3ʹ single-stranded DNA (ssDNA) end at risk of template switching and other spurious interactions, leading to complex breakpoints and chromosome rearrangements. The mechanisms that govern pathway selection for the displaced one-ended ssDNA end are unknown.
“The wand chooses the wizard”
An important determinant of DSB repair pathway choice is the initiating DNA lesion itself: “The wand chooses the wizard”153. As discussed above, attempts to repair a one-ended break or solitary DNA end are necessarily error-prone. For a two-ended break, whether the DNA ends contain single stranded tails can affect repair pathway choice, since Ku70–Ku80 binds weakly to long ssDNA tails29. Similarly, a DNA end that is chemically blocked or that forms within compacted chromatin may require processing or extensive chromatin remodeling as an accompaniment to DSB repair. Complex patterns of single stranded gaps close to the free DNA end might also affect DSB repair because of intense activation of poly(ADP-ribose) polymerase (PARP) at ssDNA gaps. The spatial relationship between DSBs that form at heterologous loci affects the probability of their interaction; more closely positioned DSBs are more likely to interact, increasing the likelihood of rearrangement between the spatially proximate but genomically remote loci154,155.
The time factor
A second important determinant of DSB repair pathway choice is the time factor. In mammalian cells, the bulk of radiation-induced DSBs (which form in a genome-wide fashion across all cell cycle phases) are repaired with rapid kinetics, with a half-life in the order of a few minutes156. This rapid phase of repair requires cNHEJ genes, while HR and contributes to slower phases of repair17,157,158. A slower phase of Artemis-dependent cNHEJ has also been described, likely reflecting the involvement of DNA end processing prior to ligation159. In yeast mating-type switching, the interval between RAD51 loading at a DSB and its association with the intrachromosomal homologous donor is ~15 minutes, while the initiation of nascent strand synthesis requires an additional ~15 minutes160,161. Imaging of RAD51-mediated synapsis during mammalian interchromosomal HR suggests similar kinetics162. Although HR between sister chromatids might occur more rapidly than this, the kinetics of even the most efficient forms of HR are likely delayed in comparison to cNHEJ. The kinetics of error-prone mammalian repair pathways such as aEJ, microhomology-mediated template switching and BIR—arguably, repair pathways of last resort—are unknown. However, it is reasonable to assume that these processes are executed more slowly than cNHEJ.
The efficient nature of mammalian cNHEJ, combined with the role of Ku70-Ku80 as an ‘early responder’ at DNA ends lacking an extensive ssDNA tail, suggests that cNHEJ is a default repair pathway163. Consistent with this idea, mammalian cNHEJ competes with HR for repair of a site-specific chromosomal DSB164. Similarly, at an HO endonuclease-induced DSB in S. cerevisiae, cNHEJ acts on unresected DNA ends in precedence to and without reference to the status of the HR system165.
Pathway choice and DNA end resection
At a molecular level, override of cNHEJ requires displacement of the Ku70-Ku80 complex from the DNA end. This may be accomplished by several mechanisms, including targeted degradation of Ku70-Ku80 by specific E3 ubiquitin ligases166. A major evolutionarily conserved mechanism for displacement of Ku70-Ku80 is the process of DNA end resection itself (Figure 1). Indeed, the engagement of DNA end resection is a key commitment step in HR, and its regulation through the cell cycle in part explains how HR is restricted to the S and G2 phases of the cell cycle.
Regulation of DNA end resection is one of the most important determinants of DSB repair pathway choice. Cell cycle-dependent kinase (CDK) activity, which increases as cells enter S phase, provides activating signals to the resection machinery and also to proteins that act later in HR167–172. Phosphorylation of CtIP on threonine 847 or S. cerevisiae Sae2 on serine 267 is essential for efficient activation of the MRE11 nuclease170,173. Thus, CtIP both senses cell cycle phase and transduces this information to initiate DNA end resection. A second significant connection made by CtIP in vertebrates is its binding to BRCA1 — an interaction that is regulated by phosphorylation of serine 327 on CtIP174. A role for BRCA1 in regulating DNA end resection during HR was suggested by the finding that BRCA1, like MRN and CtIP, is required for both HR-mediated and SSA-mediated repair of a site-specific DSB175. CtIP functions in HR, at least in part, independently of BRCA1; studies of the effect of BRCA1 loss on bulk DNA end resection have yielded variable results107,176–178.
Remarkably, deletion of the DNA damage response gene 53BP1 suppresses the severe genomic instability of BRCA1 mutants, as well as the sensitivity of some BRCA1 mutant cells to PARP inhibitors179. This phenotypic suppression is especially prominent in those BRCA1 hypomorphs for which the expressed BRCA1 gene product retains the ability to bind to PALB2–BRCA2–Rad51 and hence, presumably, retains RAD51-loading mediator functions180–182. How does loss of 53BP1 lead to this striking phenotypic reversal? In the repair of a conventional DSB, 53BP1 mediates cNHEJ and also suppresses DNA end resection144,179,183. The latter activity led to the proposal that 53BP1 deletion suppresses genomic instability in BRCA1 mutants by reversing a defect in DNA end resection. 53BP1 effectors include RIF1, PTIP and Rev7 (also called MAD2L2 — a subunit of the translesion DNA polymerase Pol ζ)184–190. Recent studies identified CTC-534A2.2, FAM35A and C20ORF19 as components of a Rev7-interacting ‘shieldin’ complex that functions downstream of 53BP1–RIF1–Rev7.191–195. Like 53BP1, the shieldin complex mediates cNHEJ, suppresses DNA end resection and mediates sensitivity of BRCA1-deficient cells to PARP inhibitors. FAM35A (also called SHLD2) contains ssDNA-binding oligonucleotide/oligosaccharide-binding (OB) fold domains, which are required for its antagonism of BRCA1. Interestingly, the results of a recent study suggested that shieldin promotes fill-in synthesis on ssDNA, potentially helping to blunt ssDNA tails194. The insight that shieldin antagonizes BRCA1 via interactions with ssDNA broadens the potential scope of 53BP1-shieldin complex function and raises some tantalizing questions: what are the key ssDNA structures over which BRCA1 and 53BP1 compete? Does 53BP1–shieldin execute functions on ssDNA additional to its known functions of resection suppression and cNHEJ? Which function of 53BP1–shieldin explains its role in conferring PARP inhibitor sensitivity to BRCA1 mutants?
The competition between BRCA1 and 53BP1 can also be visualized in the context of the γ-H2A.X chromatin domain130,196. The balance between BRCA1 and 53BP1 on chromatin is affected by TIP60-mediated acetylation of residues close to H4K20, which can disrupt 53BP1 binding to H4K20me1 or H4K20me2 marks197,198. An E3 ubiquitin ligase activity of BRCA1–BARD1 can ubiquitylate histone H2A on lysine 27, recruiting the chromatin remodeller SMARCAD1 and facilitating 53BP1 repositioning199. 53BP1 is also subject to direct regulation by TIRR, a protein that blocks the H4K20me-binding domain of 53BP1200.
Several mechanisms in addition to CDK-mediated phosphorylation of HR targets communicate cell cycle status to the DSB repair machinery. HR gene expression is up-regulated as cells transition from G1 into S phase. In mammalian cells in G1, DNA end resection is suppressed by the negative regulator HELB, which is inactivated as cells enter S phase201. The assembly of the BRCA1–PALB2–BRCA2–Rad51 recombinase complex is suppressed in G1 by proteasome-mediated degradation of PALB2, following its ubiquitylation by the E3 ubiquitin ligase cullin-3 (CUL3)–RBX1 and the adaptor protein Kelch-like ECH-associated protein 1 KEAP1202. A study of postreplicative chromatin provides an intriguing example of how S phase chromatin structure can favor HR. The TONSL–MMS22L heterodimer assists RAD51 loading and activity at stalled replication forks203–206. The ankyrin repeat domain of TONSL was shown to bind to unmethylated lysine 20 of histone H4 (H4K20me0), an unmodified state that is restricted to newly incorporated histones207. By this mechanism, immature postreplicative chromatin provides a docking site for TONSL–MMS22L, while the dearth of H4K20me1 and H4K20me2 marks might also deny 53BP1’s anti-BRCA1 activity stable access to chromatin to exert its anti-BRCA1 activity.
Entry into mitosis requires chromatin condensation and presents a unique challenge to DSB repair. Between late G2 and mid prophase the cell commits to mitotic entry even in the presence of DNA damage208. This transition is accompanied by an attenuation of DSB signalling. Although MRN is recruited to breaks in mitotic cells and ATM is activated, the chromatin response is restricted to γ-H2A.X modification and MDC1 recruitment, without activation of RNF8 and RNF168 E3 ubiquitin ligases or accumulation of BRCA1 or 53BP1 on chromatin209. This restraint of the DNA damage response is mediated by inhibitory phosphorylation of 53BP1 and RNF8 by mitotic kinases210,211. Indeed, unregulated reactivation of 53BP1 in mitosis provokes chromosome rearrangement and telomere fusions, reflecting inappropriate activation of cNHEJ.
Pathway choice and chromatin context
The chromatin context in which a DSB arises may have a broad influence on repair pathway choice. A study that measured the time-course of γ-H2A.X focus resolution (as a surrogate for DSB repair) in irradiated cells in G2 phase suggested that DSBs in heterochromatin are preferentially repaired by an HR mechanism requiring ATM212. Another study used chromatin-immunoprecipitation (ChIP) to evaluate the accumulation of HR and cNHEJ proteins at defined site-specific DSBs induced by the rare-cutting restriction endonuclease AsiSI213. In this setting, XRCC4 consistently accumulated in close proximity to each AsiSI target site, whereas RAD51 accumulation was more widely distributed around the break site, was maximal in G2 and showed a high level of variation in signal intensity between different AsiSI target sites. Of note, AsiSI target sites that revealed high levels of RAD51 accumulation were enriched in transcriptionally active genes, marked by the chromatin modification H3K36me3. The H3K36me3-binding factor LEDGF was implicated in RAD51 accumulation at transcriptionally active genes, consistent with previous work that linked this protein to CtIP function214. Interestingly, a ChIP study in undamaged cells revealed preferential accumulation of BRCA1 and PALB2 within the bodies of transcribed genes, suggesting that the accumulation of these proteins may be a scheduled accompaniment of transcription of some genes215. The mechanisms underlying these gene body-specific localization patterns are unclear. Possibly, collisions between replication and transcription, including the genome destabilizing properties of RNA-DNA hybrids (R-loops), might concentrate these factors at transcribed genes216–218. Alternatively, specific interactions with transcription complexes or epigenetic marks might be involved150.
In yeast, DSBs are mobilized to the nuclear periphery as part of a nucleus-wide choreography of repair219. The question of whether DSBs are mobilized within the mammalian nucleus has been addressed by a number of studies. Mammalian DSBs do not seem to undergo mobilization that is precisely equivalent to that observed in yeast220. However, γ-H2A.X foci were found to coalesce following DSB induction by alpha particles, which led to the suggestion that DSBs might undergo clustering during repair221. 53BP1 promotes mobility of deprotected telomeres, thereby facilitating long range rejoining222,223. Furthermore, endonuclease-induced DSBs that form in transcribed genes were observed to coalesce in G1, pending repair by HR at later stages of the cell cycle213. This observation has intriguing parallels with a cancer-associated chromosome rearrangement termed ‘chromoplexy’, in which chains of linked translocations between multiple transcribed loci mediate chromosome rearrangements in prostate cancer224. In both flies and mammalian cells, DSBs arising in constitutive heterochromatin are relocated to the periphery of heterchromatin before RAD51 becomes associated with the break sites225–227. Collectively, these studies suggest that regulated mobility of DNA ends occurs in specific chromatin contexts, contributing to a higher order level of DSB repair regulation.
Repair at stalled replication forks
Stalled replication forks differ from conventional DSBs in several important ways. The presence of branched DNA replication intermediates, ssDNA ‘daughter strand gaps’ (DSGs) and unresolved hemicatenanes in immature postreplicative chromatin, together with scaffolding by PCNA and other replisome components creates a unique environment for repair228–232. Attempted replication across a ssDNA nick in the parental template will convert the nick to a one-ended DSB, with the potential for misrepair of the broken fork, as discussed above. Forks that are stalled but not broken trigger a cascade of cellular responses that, when properly coordinated, are thought to minimize the risk of chromosome rearrangement at the site of stalling (Figure 5). These responses include: the activation of DNA damage signalling, controlled primarily by the Atr signaling kinase233,234; replisome disassembly (also termed ‘fork collapse’), in which the CMG replicative helicase is extracted by the valosin-containing protein (VCP)/p97 ATPase following ubiquitylation of MCM helicase subunits235; remodeling of DNA structure (‘fork remodeling’)90; and the activation of repair responses, of which HR is a major component236–238. Reinitiation of replication (‘replication restart’) can also occur at the site of stalling, which can be an error-prone process92,93. Knowledge of the different fork remodeling steps in mammalian cells is almost certainly incomplete, as is the ability to quantify each step satisfactorily. Currently, fork remodeling is recognized to include resection of nascent lagging strands239, fork reversal240 and endonucleolytic processing of the stalled and reversed fork241,242. In vertebrates, timely, organized cleavage of stalled forks is a key step of conservative repair by HR241,242. In contrast, pathological states allow unscheduled processing of the stalled fork, leading to misrepair and genomic instability.
Figure 5. Rad51 is an ‘early responder’ at stalled forks.
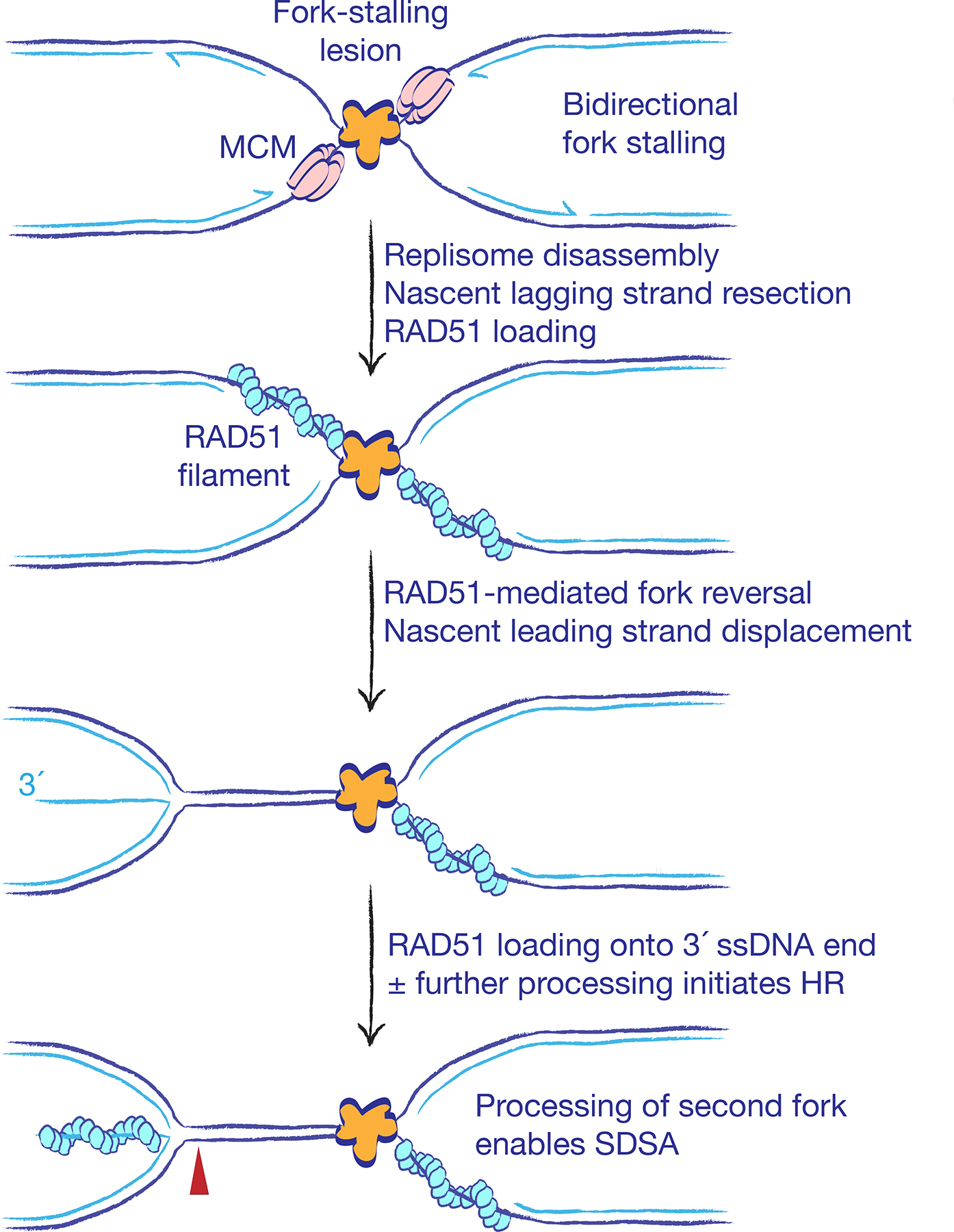
The early steps of stalled fork processing for conservative homologous recombination (HR) entail bidirectional fork stalling, nascent lagging strand resection, replisome disassembly (also termed fork collapse) and asymmetric fork reversal. RAD51 acts early in stalled fork processing to facilitate fork reversal, which remodels lagging strand ‘daughter strand gaps’ into long 3´ single-stranded DNA (ssDNA) tails formed from the displaced leading daughter strand. The combination of ssDNA structural intermediates and avid BRCA-mediated RAD51-loading activity block Ku70–Ku80 access to DNA ends at stalled forks, making HR the default repair pathway in this context. Red arrowhead indicates possible site of nuclease-mediated cleavage that could liberate a RAD51-coated DNA end for HR. Processing of the opposing fork arrested at the site of stalling generates a second DNA end and enables conservative repair by SDSA.
Replisome disassembly exposes the fork to topological stresses and remodelling activities that promote fork reversal90,231,243 (Figure 5). Fork reversal generates a cruciate ‘chicken foot’ structure with a solitary DNA end that is formed by the annealing of the leading and lagging nascent strands. Several key mediators of fork reversal in mammalian cells have been identified, including RAD51, PARP, the translocase HLTF and the annealing helicases SMARCAL1 and ZRANB389,244–246. The activity of RAD51 in fork reversal is counteracted by negative regulators such as PARI and RADX, which presumably restrict fork reversal to appropriate contexts247,248. In addition to its role in fork reversal, RAD51, in a Fanconi anaemia/BRCA pathway-dependent process, protects nascent daughter strands at stalled and reversed forks from degradation by MRE11249–253.
Conservative repair at stalled forks
Major insights into the steps of stalled fork processing and repair in vertebrates have come from in vitro studies of replication-coupled interstrand DNA cross-link (ICL) repair in Xenopus laevis egg extracts239,254. An ICL covalently links the two parental DNA strands and an absolute block to replication, unless it can be ‘traversed’ with the assistance of FANCM255 or directly ‘unhooked’ by the NEIL3 glycosylase256. In X. laevis, ICL repair is initiated following bidirectional replication fork stalling at the ICL242. The arrival of both opposing forks is required for replisome disassembly, asymmetric fork reversal and subsequent nucleolytic processing of the stalled fork for HR240,242,257. Nucleases regulated by the FANCD2–FANCI heterodimer introduce dual incisions in the stalled leading and lagging strands of one sister chromatid. This mechanism ensures that forks stalled at an ICL are processed to two-ended DSBs, thereby favouring conservative SDSA over LTGC or BIR. In contrast to the X. laevis ICL repair system, HR products at the replication termination switch (RTS1) replication fork barrier (RFB) in S. pombe occur without the formation of a DSB intermediate. Similarly, in S. cerevisiae, a site-specific RFB, derived from the E. coli replication termination system, comprising DNA replication terminus site-binding protein (Tus) bound to an array of seven ter sites, provokes no detectable incisions of the arrested fork258. Rearrangements at Tus–ter in S. cerevisiae arise from the processing of post-replicative ssDNA gaps259. Of note, the Fanconi anaemia pathway in yeasts is limited to orthologs of SLX4 (also known as FANCP) and FANCM; the stalled fork endonuclease-enabling FANCD2–FANCI heterodimer and its activator, the Fanconi core complex, are absent68,260. The presence of these later evolutionarily additions to the Fanconi anaemia pathway in higher eukaryotes might explain why scheduled incisions of the stalled fork have more prominent roles in stalled fork repair in vertebrates than in yeasts.
Rad51 is an ‘early responder’
In mammalian cells, HR responses to site-specific fork stalling have been studied in replicating ICL-containing episomal plasmids and by use of a chromosomal Tus–ter RFB92,110,261. In contrast to the polar fork arrest observed in E. coli and S. cerevisiae, a Tus–ter RFB composed of six ter repeats mediates bidirectional fork stalling in mammalian cells110. HR induced at a Tus–ter RFB in wild type mammalian cells is a non-crossover pathway, generating predominantly STGC products of two-ended recombination, mediated by the canonical Fanconi anaemia–BRCA–RAD51 pathway92,110. These properties suggest that conservative HR at stalled forks is mediated by SDSA. The two DNA ends that participate in Tus–ter RFB-induced HR are presumably derived from the two opposing forks arrested at the Tus–ter RFB (Figure 5). However, in contrast to HR triggered by a conventional DSB, where cNHEJ avidly competes with HR, Tus–ter RFB-induced HR is unaffected by the status of the cNHEJ genes Ku70 or XRCC4237. This suggests that the mechanism of SDSA at Tus/Ter RFBs differs significantly from SDSA at a conventional DSB. To understand this difference, it is helpful to review the known interactions between HR and cNHEJ at stalled forks.
Studies of genetic interactions between HR and cNHEJ have provided important insights into the mechanisms of stalled fork repair. In S. cerevisiae, the Ku70–Ku80 complex, acting independently of Lig4, mediates lethality of mre11 and sae2 resection mutants exposed to the topoisomerase I inhibitor camptothecin (CPT)—a drug that generates one-ended breaks at broken forks262,263. This suggests that Mre11/Sae2-mediated resection at CPT-induced breaks is required to overcome the barrier formed by Ku70–Ku80 binding to the DNA end. Similarly, in human cells, Ku70–Ku80 (inferred by associated DNA-PKcs activity) accumulates transiently at CPT lesions and is rapidly displaced by MRE11 and CtIP, whereas delayed resection at CPT lesions allows misrepair by cNHEJ264,265. A recent study showed that HR induced by a DNA nicking enzyme (“nickase”) is not affected by deletion of Ku70 or Xrcc4266. One possible explanation for this finding is that the absence of a second DNA end at sites of nickase-induced fork breakage denies cNHEJ a productive outcome, although this hypothesis remains to be tested.
Given that Tus–ter RFB-induced HR is a product of two-ended recombination, why is cNHEJ denied access to these HR intermediates?237. A clue as to the underlying mechanism came from ChIP analysis of RAD51 recruitment to the Tus–ter RFB. In contrast to the DSB response, in which the Rad51 ChIP signal extends for several kilobases on either side of the DSB213, RAD51 recruited to Tus–ter RFBs is localized to within 1 kb of the stall site and the ChIP signal is both more intense and more sustained than in the DSB response237. This distinctive pattern suggests that the principal DNA structures that recruit Rad51 to the stalled fork are not conventional DSBs — a conclusion corroborated by work in the X. laevis model of ICL repair236. Of note, lagging strand gaps normally arise, albeit transiently, during replication. Fork stalling renders these daughter strand gaps (DSGs) abnormally persistent, and nascent lagging strand resection at the site of fork stalling would further extend the size of the DSG239. Lagging strand DSGs could thus provide a platform for RAD51 recruitment as a very early step of stalled fork processing, prior to the formation of either a DNA end or a DSB236,245. An abundance of BRCA1 and BRCA2 at the stalled fork would ensure efficient RAD51 loading. In this model, RAD51 is an ‘early responder’ at stalled forks267, acting as a sentinel repair factor in the stalled fork response analogous to Ku’s pivotal role at a DSB (Figure 5). If leading and lagging strand synthesis were uncoupled at a leading strand DNA lesion, RAD51 might be loaded onto the resulting leading strand DSG prior to fork reversal267.
Early and sustained RAD51 recruitment to the stalled fork could explain the exclusion of cNHEJ during stalled fork HR. If stalled fork HR in mammalian cells is initiated by RAD51-mediated fork reversal240,245, the length of the 3´ ssDNA tail formed by fork reversal would be a reflection of the size of its precursor lesion, the lagging strand DSG (Figure 5). The first DNA end generated during stalled fork repair — an extended 3´ ssDNA tail produced by fork reversal — might therefore be incapable of binding Ku70-Ku80. In this way, the initial steps of stalled fork HR would remain ‘invisible’ to the cNHEJ pathway. Subsequent processing steps—more extensive fork reversal and nucleolytic incision of the reversed fork—could mobilize the RAD51-loaded DNA ends for SDSA. This model of stalled fork HR invokes two consecutive and distinct RAD51 loading steps: the first onto the lagging strand DSG, as a prelude to fork reversal; the second onto the 3´ ssDNA tail of the reversed fork, as a pre-requisite for SDSA.
There is still much to be learned about the asymmetries associated with stalled fork processing for SDSA. How does the Fanconi anaemia pathway select one sister chromatid for incision, while leaving the other intact? During asymmetrical fork reversal, how is one fork selected to undergo reversal, and how does this asymmetry relate to the asymmetrical processing of DNA ends that is innate to SDSA?
Error-prone fork repair and restart
Error-prone fork repair may involve several distinct types of fork processing errors, including: pathological fork processing by opportunistic nucleases; aberrant interactions of solitary DNA ends formed by fork reversal; and aberrant fork restart. In our study of rearrangements at a chromosomally targeted Tus–ter RFB, we used high throughput genome-wide translocation sequencing (HTGTS) to identify DNA ends at Tus–ter that form translocations92,268. The major translocation-competent DNA lesions detected at Tus–ter by HTGTS were solitary DNA ends/one-ended breaks. This finding appears paradoxical, given the two-ended model of stalled fork HR discussed above, and in light of other methods that have revealed DNA ends of both polarities at stalled forks269,270. A possible explanation of this discrepancy is that two-ended intermediates of conservative repair are protected from translocation, whereas solitary DNA ends produced by aberrant fork processing are relatively translocation-prone.
The time factor influences whether stalled forks are processed in a conservative or error-prone manner. In S. pombe, HR at the RTS1 RFB is detectable within ~10 minutes of a fork stalling event, whereas aberrant fork restart is initiated only after ~60 minutes93. In mammalian cells, RAD51 supports the restart of hydroxyurea (HU)-stalled forks at which fork collapse has not yet occurred271. Presumably, in the context of transient HU-mediated nucleotide pool depletion, RAD51 protects stalled fork structures for a limited time period, allowing the replisome to be reactivated once the nucleotide pool is restored. However, more extensive exposure to HU leads to localized DNA damage responses and possibly fork breakage271. Pathological MRE11-mediated degradation of nascent strands in BRCA mutants is first detectable after ~30 minutes’ incubation in HU, but is fully manifest only after ~5 hours249. Limited as the data is on this topic, it appears that pathological stalled fork repair pathways become operative only if physiological systems fail.
Replication restart and cancer
In bacteria, RecA-mediated invasion of the sister chromatid by a one-ended break at a broken fork is coupled to reassembly of a normal replisome by PriA, with resumption of conventional semi-conservative DNA synthesis272. To date, a PriA-like replisome re-loading activity has not been identified in eukaryotes. Consequently, once the replisome has been disassembled, fork restart in eukaryotes may be obligatorily error-prone. In S. pombe, forks stalled at an ectopically located RTS1 RFB engage both conservative and error-prone HR, including RAD51-dependent and RAD51-independent pathways of fork restart273–275. Stalled forks in S. pombe can be restarted by a Rad22Rad52-mediated mechanism, but the restarted fork is unstable, being prone to rearrangement up to 75 kb downstream of the RFB93,276. The restarted fork in this setting is probably extended by BIR. In both S. pombe and S. cerevisiae, the mutagenic impact of BIR at stalled or broken forks is limited by the arrival of an opposing normal fork derived from the neighboring replicon91,93. A role for RAD52 in restarting collapsed mammalian replication forks has also been proposed277.
RAD51-independent fork restart can occur at a mammalian Tus–ter RFB during the formation of tandem duplications in BRCA1 mutant cells, recapitulating a highly specific ~10 kb TD ‘rearrangement signature’ that is observed in human BRCA1-linked breast and ovarian cancers92,110,278,279. BRCA1, BARD1 and CtIP suppress tandem duplications, whereas BRCA2, Rad51 and other HR mediators have no effect on tandem duplication formation. Tandem duplication formation in BRCA1 mutants is seen in the stalled fork response but not in response to a conventional DSB92. This shows that aberrant stalled fork processing, not defective DSB repair, is the crucial trigger of tandem duplication formation in BRCA1-linked tumorigenesis. These findings directly implicate aberrant stalled fork restart in the formation of human cancer-associated chromosome rearrangements. Interestingly, FANCM and BLM — the same motor proteins that impose a delay on the onset of BIR in S. cerevisiae102,105,106 — specifically suppress the formation of tandem duplications in BRCA1 mutant cells92. These observations suggest that fork restart during tandem duplication formation may be mediated by BIR (Figure 6), and invite comparisons with aberrant fork restart in S. pombe93,276.
Figure 6. Single strand annealing may be a conservative repair pathway at stalled replication forks.
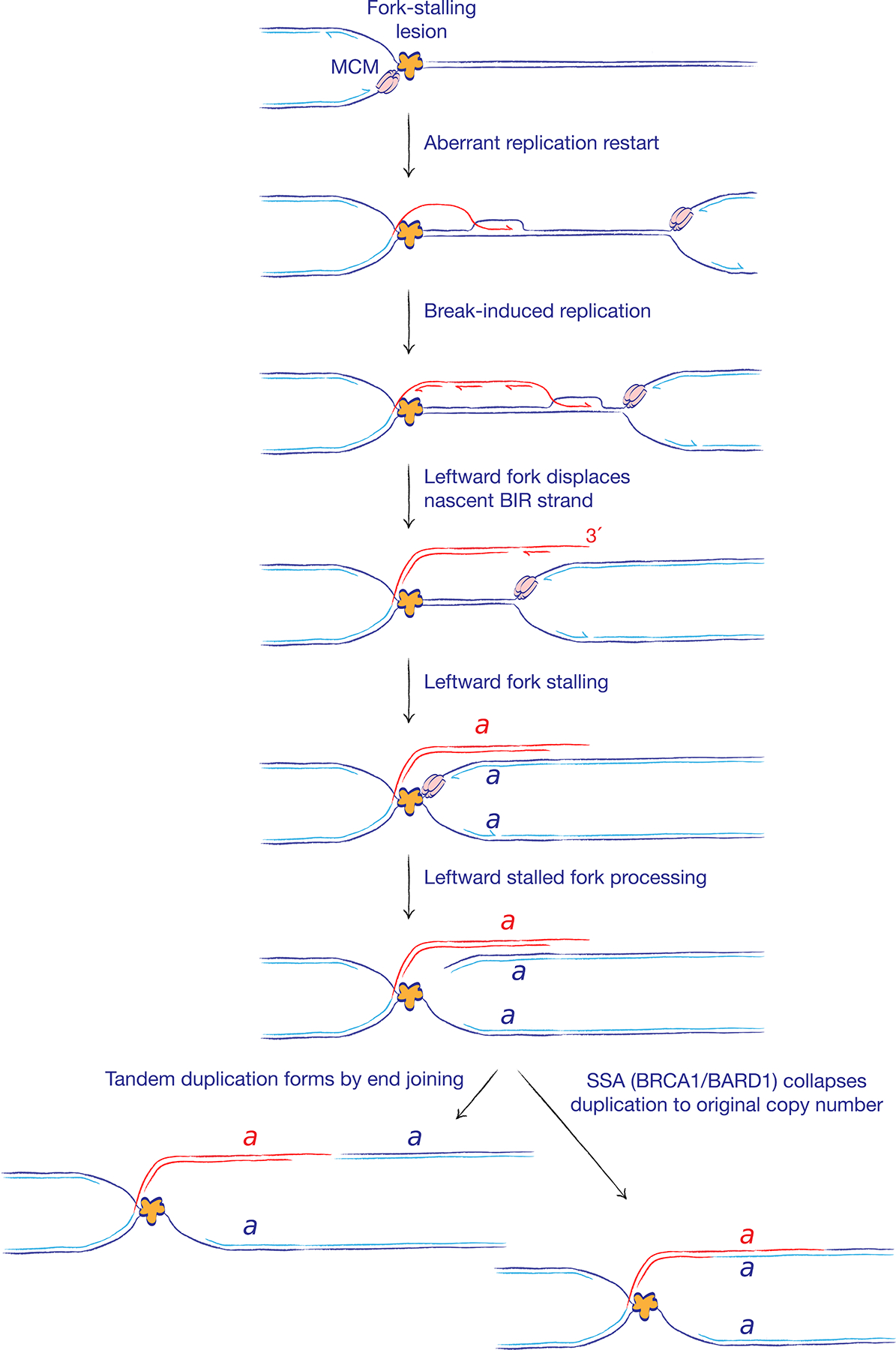
A solitary stalled fork may undergo aberrant fork restart, with the engagement of ‘break-induced replication’ (BIR)-type copying (red). Of note, BIR in this context might not entail a DNA break intermediate. Displacement of the BIR nascent strand by the converging opposing fork results in duplication of genomic segment a, bounded, at one end, by the site of fork stalling and, at the other end, by the site at which the BIR nascent strand was displaced. A non-homologous tandem duplication forms if these two DNA ends are repaired by end joining. By contrast, repair by single strand annealing (SSA; promoted by BRCA1–BARD1) would collapse the two copies of segment a back to a single copy, thereby suppressing tandem duplication formation and maintaining normal chromosome structure.
The final step of tandem duplication formation in BRCA1 mutant cells is mediated by cNHEJ, with formation of a non-homologous tandem duplication breakpoint (Figure 6)92. It is interesting to consider the possibility that this end joining step might be in competition with the error-prone SSA mechanism of DSB repair (Figure 2A). If the two DNA ends of the tandem duplication were repaired by SSA instead of cNHEJ, the duplicated segments would be collapsed to their original single copy status (Figure 6). In this setting, SSA, which is a BRCA1-dependent process, would perform a conservative function at stalled forks by counteracting the tendency of aberrantly restarted forks to form tandem duplications. Thus, a repair pathway that is considered to be error-prone in conventional DSB repair might mediate error-free repair at stalled forks.
Conclusion
Our understanding of the decision tree of mammalian DSB repair has reached quite sophisticated levels, enabling researchers to focus attention on higher order cellular processes that affect pathway choice, such as cell cycle status and the local chromatin environment. In contrast, some of the key ‘rules’ of repair pathway choice at stalled mammalian replication forks are only beginning to become clear — for example, the role of fork reversal and the exclusion of cNHEJ during conservative HR. The mechanisms that regulate remodelling of the stalled fork remain to be fully revealed and quantified in mammalian cells. We expect that ongoing research into repair pathway choice at stalled forks will yield additional insights into the origins of cancer and will reveal new therapeutic targets in diseases that are characterized by genomic instability.
Acknowledgements
The authors thank Drs. Johannes Walter, Joe Loparo, Andre Nussenzweig, Stephen Jackson, Edison Liu, David Cortez, and Agata Smogorzewska, as well as Scully lab members, for helpful discussions and for sharing unpublished research findings. This work was supported by awards R01CA095175, R01CA217991, OC160440, BC160172P1 and R21ES027776 (to R.S.) and P50CA168504 (to N.A.W.).
Footnotes
Competing interests
The authors declare no competing interests.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Ciccia A & Elledge SJ The DNA damage response: making it safe to play with knives. Mol Cell 40, 179–204, doi:S1097–2765(10)00747–1 [pii] 10.1016/j.molcel.2010.09.019 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Paques F & Haber JE Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiology and molecular biology reviews : MMBR 63, 349–404. (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sung P & Klein H Mechanism of homologous recombination: mediators and helicases take on regulatory functions. Nat Rev Mol Cell Biol 7, 739–750 (2006). [DOI] [PubMed] [Google Scholar]
- 4.Symington LS Role of RAD52 epistasis group genes in homologous recombination and double-strand break repair. Microbiology & Molecular Biology Reviews 66, 630–670 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Prakash R, Zhang Y, Feng W & Jasin M Homologous recombination and human health: the roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harb Perspect Biol 7, a016600, doi: 10.1101/cshperspect.a016600 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hartlerode AJ & Scully R Mechanisms of double-strand break repair in somatic mammalian cells. Biochem J 423, 157–168 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pannunzio NR, Watanabe G & Lieber MR Nonhomologous DNA End Joining for Repair of DNA Double-Strand Breaks. J Biol Chem, doi: 10.1074/jbc.TM117.000374 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ferguson DO et al. The nonhomologous end-joining pathway of DNA repair is required for genomic stability and the suppression of translocations. Proc Natl Acad Sci U S A 97, 6630–6633 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Britton S, Coates J & Jackson SP A new method for high-resolution imaging of Ku foci to decipher mechanisms of DNA double-strand break repair. The Journal of cell biology 202, 579–595, doi: 10.1083/jcb.201303073 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gottlieb TM & Jackson SP The DNA-dependent protein kinase: requirement for DNA ends and association with Ku antigen. Cell 72, 131–142 (1993). [DOI] [PubMed] [Google Scholar]
- 11.Nick McElhinny SA, Snowden CM, McCarville J & Ramsden DA Ku recruits the XRCC4-ligase IV complex to DNA ends. Mol Cell Biol 20, 2996–3003 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ahnesorg P, Smith P & Jackson SP XLF interacts with the XRCC4-DNA ligase IV complex to promote DNA nonhomologous end-joining. Cell 124, 301–313, doi: 10.1016/j.cell.2005.12.031 (2006). [DOI] [PubMed] [Google Scholar]
- 13.Buck D et al. Cernunnos, a novel nonhomologous end-joining factor, is mutated in human immunodeficiency with microcephaly. Cell 124, 287–299, doi: 10.1016/j.cell.2005.12.030 (2006). [DOI] [PubMed] [Google Scholar]
- 14.Ochi T et al. DNA repair. PAXX, a paralog of XRCC4 and XLF, interacts with Ku to promote DNA double-strand break repair. Science 347, 185–188, doi: 10.1126/science.1261971 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zha S et al. ATM damage response and XLF repair factor are functionally redundant in joining DNA breaks. Nature 469, 250–254, doi: 10.1038/nature09604 nature09604 [pii] (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kumar V, Alt FW & Frock RL PAXX and XLF DNA repair factors are functionally redundant in joining DNA breaks in a G1-arrested progenitor B-cell line. Proc Natl Acad Sci U S A 113, 10619–10624, doi: 10.1073/pnas.1611882113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Graham TG, Walter JC & Loparo JJ Two-Stage Synapsis of DNA Ends during Non-homologous End Joining. Mol Cell 61, 850–858, doi: 10.1016/j.molcel.2016.02.010 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Blackford AN & Jackson SP ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol Cell 66, 801–817, doi: 10.1016/j.molcel.2017.05.015 (2017). [DOI] [PubMed] [Google Scholar]
- 19.Stinson BM, Moreno AT, Walter JC & Loparo JJ Non-homologous end joining minimizes errors by coordinating DNA processing with ligation. bioRxiv, doi: 10.1101/563197. (2019). [DOI] [Google Scholar]
- 20.Xie A, Kwok A & Scully R Role of mammalian Mre11 in classical and alternative nonhomologous end joining. Nature structural & molecular biology 16, 814–818 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dinkelmann M et al. Multiple functions of MRN in end-joining pathways during isotype class switching. Nature structural & molecular biology 16, 808–813 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Williams RS et al. Mre11 dimers coordinate DNA end bridging and nuclease processing in double-strand-break repair. Cell 135, 97–109 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grundy GJ et al. APLF promotes the assembly and activity of non-homologous end joining protein complexes. EMBO J 32, 112–125, doi: 10.1038/emboj.2012.304 emboj2012304 [pii] (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Macrae CJ, McCulloch RD, Ylanko J, Durocher D & Koch CA APLF (C2orf13) facilitates nonhomologous end-joining and undergoes ATM-dependent hyperphosphorylation following ionizing radiation. DNA Repair (Amst) 7, 292–302, doi:S1568–7864(07)00359-X [pii] 10.1016/j.dnarep.2007.10.008 (2008). [DOI] [PubMed] [Google Scholar]
- 25.Rulten SL et al. PARP-3 and APLF function together to accelerate nonhomologous end-joining. Mol Cell 41, 33–45, doi:S1097–2765(10)00962–7 [pii] 10.1016/j.molcel.2010.12.006 (2011). [DOI] [PubMed] [Google Scholar]
- 26.Arnoult N et al. Regulation of DNA repair pathway choice in S and G2 phases by the NHEJ inhibitor CYREN. Nature 549, 548–552, doi: 10.1038/nature24023 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grundy GJ et al. The Ku-binding motif is a conserved module for recruitment and stimulation of non-homologous end-joining proteins. Nat Commun 7, 11242, doi: 10.1038/ncomms11242 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu XS et al. LRF maintains genome integrity by regulating the non-homologous end joining pathway of DNA repair. Nat Commun 6, 8325, doi: 10.1038/ncomms9325 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mimori T & Hardin JA Mechanism of interaction between Ku protein and DNA. J Biol Chem 261, 10375–10379 (1986). [PubMed] [Google Scholar]
- 30.Chang HH, Watanabe G & Lieber MR Unifying the DNA end-processing roles of the artemis nuclease: Ku-dependent artemis resection at blunt DNA ends. J Biol Chem 290, 24036–24050, doi: 10.1074/jbc.M115.680900 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chang HHY, Pannunzio NR, Adachi N & Lieber MR Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat Rev Mol Cell Biol 18, 495–506, doi: 10.1038/nrm.2017.48 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Takata M et al. Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. Embo J 17, 5497–5508 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kadyk LC & Hartwell LH Sister chromatids are preferred over homologs as substrates for recombinational repair in Saccharomyces cerevisiae. Genetics 132, 387–402 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.San Filippo J, Sung P & Klein H Mechanism of eukaryotic homologous recombination. Annu Rev Biochem 77, 229–257 (2008). [DOI] [PubMed] [Google Scholar]
- 35.Symington LS & Gautier J Double-strand break end resection and repair pathway choice. Annual review of genetics 45, 247–271, doi: 10.1146/annurev-genet-110410-132435 (2011). [DOI] [PubMed] [Google Scholar]
- 36.Lee JH & Paull TT Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science 304, 93–96 (2004). [DOI] [PubMed] [Google Scholar]
- 37.Lee JH & Paull TT ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science 308, 551–554 (2005). [DOI] [PubMed] [Google Scholar]
- 38.Limbo O et al. Ctp1 is a cell-cycle-regulated protein that functions with Mre11 complex to control double-strand break repair by homologous recombination. Mol Cell 28, 134–146 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sartori AA et al. Human CtIP promotes DNA end resection. Nature 450, 509–514 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lengsfeld BM, Rattray AJ, Bhaskara V, Ghirlando R & Paull TT Sae2 is an endonuclease that processes hairpin DNA cooperatively with the Mre11/Rad50/Xrs2 complex. Mol Cell 28, 638–651, doi:S1097–2765(07)00733–2 [pii] 10.1016/j.molcel.2007.11.001 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cannavo E & Cejka P Sae2 promotes dsDNA endonuclease activity within Mre11-Rad50-Xrs2 to resect DNA breaks. Nature 514, 122–125, doi: 10.1038/nature13771 (2014). [DOI] [PubMed] [Google Scholar]
- 42.Reginato G, Cannavo E & Cejka P Physiological protein blocks direct the Mre11-Rad50-Xrs2 and Sae2 nuclease complex to initiate DNA end resection. Genes Dev 31, 2325–2330, doi: 10.1101/gad.308254.117 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang W, Daley JM, Kwon Y, Krasner DS & Sung P Plasticity of the Mre11-Rad50-Xrs2-Sae2 nuclease ensemble in the processing of DNA-bound obstacles. Genes Dev 31, 2331–2336, doi: 10.1101/gad.307900.117 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stafa A, Donnianni RA, Timashev LA, Lam AF & Symington LS Template switching during break-induced replication is promoted by the Mph1 helicase in Saccharomyces cerevisiae. Genetics 196, 1017–1028, doi: 10.1534/genetics.114.162297 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Garcia V, Phelps SE, Gray S & Neale MJ Bidirectional resection of DNA double-strand breaks by Mre11 and Exo1. Nature 479, 241–244, doi: 10.1038/nature10515 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mimitou EP & Symington LS Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature 455, 770–774 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mimitou EP & Symington LS Ku prevents Exo1 and Sgs1-dependent resection of DNA ends in the absence of a functional MRX complex or Sae2. EMBO J 29, 3358–3369, doi: 10.1038/emboj.2010.193 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nimonkar AV et al. BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev 25, 350–362, doi: 10.1101/gad.2003811 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Daley JM et al. Enhancement of BLM-DNA2-Mediated Long-Range DNA End Resection by CtIP. Cell reports 21, 324–332, doi: 10.1016/j.celrep.2017.09.048 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang H, Li Q, Fan J, Holloman WK & Pavletich NP The BRCA2 homologue Brh2 nucleates RAD51 filament formation at a dsDNA-ssDNA junction. Nature 433, 653–657 (2005). [DOI] [PubMed] [Google Scholar]
- 51.Jensen RB, Carreira A & Kowalczykowski SC Purified human BRCA2 stimulates RAD51-mediated recombination. Nature 467, 678–683, doi:nature09399 [pii] 10.1038/nature09399 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thorslund T et al. The breast cancer tumor suppressor BRCA2 promotes the specific targeting of RAD51 to single-stranded DNA. Nature structural & molecular biology 17, 1263–1265, doi:nsmb.1905 [pii] 10.1038/nsmb.1905 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Anderson DG & Kowalczykowski SC The translocating RecBCD enzyme stimulates recombination by directing RecA protein onto ssDNA in a chi-regulated manner. Cell 90, 77–86 (1997). [DOI] [PubMed] [Google Scholar]
- 54.Spies M & Kowalczykowski SC The RecA binding locus of RecBCD is a general domain for recruitment of DNA strand exchange proteins. Mol Cell 21, 573–580 (2006). [DOI] [PubMed] [Google Scholar]
- 55.Krejci L et al. DNA helicase Srs2 disrupts the Rad51 presynaptic filament. Nature 423, 305–309 (2003). [DOI] [PubMed] [Google Scholar]
- 56.Liu J et al. Rad51 paralogues Rad55-Rad57 balance the antirecombinase Srs2 in Rad51 filament formation. Nature 479, 245–248, doi: 10.1038/nature10522 nature10522 [pii] (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Heyer WD, Ehmsen KT & Liu J Regulation of homologous recombination in eukaryotes. Annual review of genetics 44, 113–139, doi: 10.1146/annurev-genet-051710-150955 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Elango R et al. Break-induced replication promotes formation of lethal joint molecules dissolved by Srs2. Nat Commun 8, 1790, doi: 10.1038/s41467-017-01987-2 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhao W et al. BRCA1-BARD1 promotes RAD51-mediated homologous DNA pairing. Nature, doi: 10.1038/nature24060 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen Z, Yang H & Pavletich NP Mechanism of homologous recombination from the RecA-ssDNA/dsDNA structures. Nature 453, 489–484, doi: 10.1038/nature06971 (2008). [DOI] [PubMed] [Google Scholar]
- 61.van der Heijden T et al. Homologous recombination in real time: DNA strand exchange by RecA. Mol Cell 30, 530–538, doi: 10.1016/j.molcel.2008.03.010 (2008). [DOI] [PubMed] [Google Scholar]
- 62.McVey M, Khodaverdian VY, Meyer D, Cerqueira PG & Heyer WD Eukaryotic DNA Polymerases in Homologous Recombination. Annual review of genetics 50, 393–421, doi: 10.1146/annurev-genet-120215-035243 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kane DP, Shusterman M, Rong Y & McVey M Competition between replicative and translesion polymerases during homologous recombination repair in Drosophila. PLoS genetics 8, e1002659, doi: 10.1371/journal.pgen.1002659PGENETICS-D-11-01925 [pii] (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hicks WM, Kim M & Haber JE Increased mutagenesis and unique mutation signature associated with mitotic gene conversion. Science 329, 82–85, doi: 10.1126/science.1191125329/5987/82 [pii] (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.McVey M, Adams M, Staeva-Vieira E & Sekelsky JJ Evidence for multiple cycles of strand invasion during repair of double-strand gaps in Drosophila. Genetics 167, 699–705 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Smith CE, Llorente B & Symington LS Template switching during break-induced replication. Nature 447, 102–105 (2007). [DOI] [PubMed] [Google Scholar]
- 67.Barber LJ et al. RTEL1 maintains genomic stability by suppressing homologous recombination. Cell 135, 261–271, doi: 10.1016/j.cell.2008.08.016 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Whitby MC The FANCM family of DNA helicases/translocases. DNA Repair (Amst) 9, 224–236, doi: 10.1016/j.dnarep.2009.12.012 (2010). [DOI] [PubMed] [Google Scholar]
- 69.Xue X, Sung P & Zhao X Functions and regulation of the multitasking FANCM family of DNA motor proteins. Genes Dev 29, 1777–1788, doi: 10.1101/gad.266593.115 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vindigni A & Hickson ID RecQ helicases: multiple structures for multiple functions? HFSP J 3, 153–164, doi: 10.2976/1.3079540 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Westmoreland JW & Resnick MA Coincident resection at both ends of random, gamma-induced double-strand breaks requires MRX (MRN), Sae2 (Ctp1), and Mre11-nuclease. PLoS genetics 9, e1003420, doi: 10.1371/journal.pgen.1003420PGENETICS-D-12-02301 [pii] (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sarbajna S & West SC Holliday junction processing enzymes as guardians of genome stability. Trends Biochem Sci 39, 409–419, doi: 10.1016/j.tibs.2014.07.003 (2014). [DOI] [PubMed] [Google Scholar]
- 73.Bizard AH & Hickson ID The dissolution of double Holliday junctions. Cold Spring Harb Perspect Biol 6, a016477, doi: 10.1101/cshperspect.a016477 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yan CT et al. IgH class switching and translocations use a robust non-classical end-joining pathway. Nature 449, 478–482 (2007). [DOI] [PubMed] [Google Scholar]
- 75.Chan SH, Yu AM & McVey M Dual roles for DNA polymerase theta in alternative end-joining repair of double-strand breaks in Drosophila. PLoS genetics 6, e1001005, doi: 10.1371/journal.pgen.1001005 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yu AM & McVey M Synthesis-dependent microhomology-mediated end joining accounts for multiple types of repair junctions. Nucleic Acids Res 38, 5706–5717, doi: 10.1093/nar/gkq379 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mateos-Gomez PA et al. The helicase domain of Poltheta counteracts RPA to promote alt-NHEJ. Nature structural & molecular biology 24, 1116–1123, doi: 10.1038/nsmb.3494 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kent T, Chandramouly G, McDevitt SM, Ozdemir AY & Pomerantz RT Mechanism of microhomology-mediated end-joining promoted by human DNA polymerase theta. Nature structural & molecular biology 22, 230–237, doi: 10.1038/nsmb.2961 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shima N, Munroe RJ & Schimenti JC The mouse genomic instability mutation chaos1 is an allele of Polq that exhibits genetic interaction with Atm. Mol Cell Biol 24, 10381–10389, doi: 10.1128/MCB.24.23.10381-10389.2004 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Koole W et al. A Polymerase Theta-dependent repair pathway suppresses extensive genomic instability at endogenous G4 DNA sites. Nat Commun 5, 3216, doi: 10.1038/ncomms4216 (2014). [DOI] [PubMed] [Google Scholar]
- 81.Roerink SF, van Schendel R & Tijsterman M Polymerase theta-mediated end joining of replication-associated DNA breaks in C. elegans. Genome research 24, 954–962, doi: 10.1101/gr.170431.113 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wyatt DW et al. Essential Roles for Polymerase theta-Mediated End Joining in the Repair of Chromosome Breaks. Mol Cell, doi: 10.1016/j.molcel.2016.06.020 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ceccaldi R et al. Homologous-recombination-deficient tumours are dependent on Poltheta-mediated repair. Nature 518, 258–262, doi: 10.1038/nature14184 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mateos-Gomez PA et al. Mammalian polymerase theta promotes alternative NHEJ and suppresses recombination. Nature 518, 254–257, doi: 10.1038/nature14157 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Boboila C, Alt FW & Schwer B Classical and alternative end-joining pathways for repair of lymphocyte-specific and general DNA double-strand breaks. Advances in immunology 116, 1–49, doi: 10.1016/B978-0-12-394300-2.00001-6 (2012). [DOI] [PubMed] [Google Scholar]
- 86.Yousefzadeh MJ et al. Mechanism of suppression of chromosomal instability by DNA polymerase POLQ. PLoS genetics 10, e1004654, doi: 10.1371/journal.pgen.1004654 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zan H et al. Rad52 competes with Ku70/Ku86 for binding to S-region DSB ends to modulate antibody class-switch DNA recombination. Nat Commun 8, 14244, doi: 10.1038/ncomms14244 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bennardo N, Cheng A, Huang N & Stark JM Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS genetics 4, e1000110(2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Quinet A, Lemacon D & Vindigni A Replication Fork Reversal: Players and Guardians. Mol Cell 68, 830–833, doi: 10.1016/j.molcel.2017.11.022 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Neelsen KJ & Lopes M Replication fork reversal in eukaryotes: from dead end to dynamic response. Nat Rev Mol Cell Biol 16, 207–220, doi: 10.1038/nrm3935 (2015). [DOI] [PubMed] [Google Scholar]
- 91.Mayle R et al. Mus81 and converging forks limit the mutagenicity of replication fork breakage. Science 349, 742–747, doi: 10.1126/science.aaa8391 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Willis NA et al. Mechanism of tandem duplication formation in BRCA1-mutant cells. Nature 551, 590–595, doi: 10.1038/nature24477 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nguyen MO, Jalan M, Morrow CA, Osman F & Whitby MC Recombination occurs within minutes of replication blockage by RTS1 producing restarted forks that are prone to collapse. eLife 4, e04539, doi: 10.7554/eLife.04539 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Anand RP, Lovett ST & Haber JE Break-induced DNA replication. Cold Spring Harb Perspect Biol 5, doi: 10.1101/cshperspect.a010397a010397 [pii] cshperspect.a010397 [pii] (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Llorente B, Smith CE & Symington LS Break-induced replication: what is it and what is it for? Cell Cycle 7, 859–864 (2008). [DOI] [PubMed] [Google Scholar]
- 96.Saini N et al. Migrating bubble during break-induced replication drives conservative DNA synthesis. Nature 502, 389–392, doi: 10.1038/nature12584 nature12584 [pii] (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Roberts SA et al. Clustered mutations in yeast and in human cancers can arise from damaged long single-strand DNA regions. Mol Cell 46, 424–435, doi: 10.1016/j.molcel.2012.03.030 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Deem A et al. Break-induced replication is highly inaccurate. PLoS Biol 9, e1000594, doi: 10.1371/journal.pbio.1000594 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sakofsky CJ et al. Translesion Polymerases Drive Microhomology-Mediated Break-Induced Replication Leading to Complex Chromosomal Rearrangements. Mol Cell 60, 860–872, doi: 10.1016/j.molcel.2015.10.041 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wilson MA et al. Pif1 helicase and Poldelta promote recombination-coupled DNA synthesis via bubble migration. Nature 502, 393–396, doi: 10.1038/nature12585 nature12585 [pii] (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lydeard JR, Jain S, Yamaguchi M & Haber JE Break-induced replication and telomerase-independent telomere maintenance require Pol32. Nature 448, 820–823 (2007). [DOI] [PubMed] [Google Scholar]
- 102.Jain S et al. A recombination execution checkpoint regulates the choice of homologous recombination pathway during DNA double-strand break repair. Genes Dev 23, 291–303 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Heyer WD Regulation of recombination and genomic maintenance. Cold Spring Harb Perspect Biol 7, a016501, doi: 10.1101/cshperspect.a016501 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Piazza A et al. Dynamic Processing of Displacement Loops during Recombinational DNA Repair. Mol Cell, doi: 10.1016/j.molcel.2019.01.005 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Jain S, Sugawara N, Mehta A, Ryu T & Haber JE Sgs1 and Mph1 Helicases Enforce the Recombination Execution Checkpoint During DNA Double-Strand Break Repair in Saccharomyces cerevisiae. Genetics 203, 667–675, doi: 10.1534/genetics.115.184317 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mehta A, Beach A & Haber JE Homology Requirements and Competition between Gene Conversion and Break-Induced Replication during Double-Strand Break Repair. Mol Cell 65, 515–526 e513, doi: 10.1016/j.molcel.2016.12.003 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chandramouly G et al. BRCA1 and CtIP suppress long-tract gene conversion between sister chromatids. Nat Commun 4, 2404, doi: 10.1038/ncomms3404 ncomms3404 [pii] (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nagaraju G, Hartlerode A, Kwok A, Chandramouly G & Scully R XRCC2 and XRCC3 regulate the balance between short- and long-tract gene conversion between sister chromatids. Mol Cell Biol (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Nagaraju G, Odate S, Xie A & Scully R Differential regulation of short- and long-tract gene conversion between sister chromatids by Rad51C. Mol Cell Biol 26, 8075–8086 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Willis NA et al. BRCA1 controls homologous recombination at Tus/Ter-stalled mammalian replication forks. Nature 510, 556–559, doi: 10.1038/nature13295 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Costantino L et al. Break-induced replication repair of damaged forks induces genomic duplications in human cells. Science 343, 88–91, doi: 10.1126/science.1243211 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bhowmick R, Minocherhomji S & Hickson ID RAD52 Facilitates Mitotic DNA Synthesis Following Replication Stress. Mol Cell 64, 1117–1126, doi: 10.1016/j.molcel.2016.10.037 (2016). [DOI] [PubMed] [Google Scholar]
- 113.Minocherhomji S et al. Replication stress activates DNA repair synthesis in mitosis. Nature 528, 286–290, doi: 10.1038/nature16139 (2015). [DOI] [PubMed] [Google Scholar]
- 114.Dilley RL et al. Break-induced telomere synthesis underlies alternative telomere maintenance. Nature 539, 54–58, doi: 10.1038/nature20099 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lee JA, Carvalho CM & Lupski JR A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell 131, 1235–1247, doi: 10.1016/j.cell.2007.11.037 (2007). [DOI] [PubMed] [Google Scholar]
- 116.Zhang CZ, Leibowitz ML & Pellman D Chromothripsis and beyond: rapid genome evolution from complex chromosomal rearrangements. Genes Dev 27, 2513–2530, doi: 10.1101/gad.229559.113 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Willis NA, Rass E & Scully R Deciphering the Code of the Cancer Genome: Mechanisms of Chromosome Rearrangement. Trends Cancer 1, 217–230, doi: 10.1016/j.trecan.2015.10.007 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ira G & Haber JE Characterization of RAD51-independent break-induced replication that acts preferentially with short homologous sequences. Mol Cell Biol 22, 6384–6392 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Payen C, Koszul R, Dujon B & Fischer G Segmental duplications arise from Pol32-dependent repair of broken forks through two alternative replication-based mechanisms. PLoS genetics 4, e1000175, doi: 10.1371/journal.pgen.1000175 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Slack A, Thornton PC, Magner DB, Rosenberg SM & Hastings PJ On the mechanism of gene amplification induced under stress in Escherichia coli. PLoS genetics 2, e48, doi: 10.1371/journal.pgen.0020048 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hastings PJ, Ira G & Lupski JR A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS genetics 5, e1000327, doi: 10.1371/journal.pgen.1000327 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Piazza A, Wright WD & Heyer WD Multi-invasions Are Recombination Byproducts that Induce Chromosomal Rearrangements. Cell 170, 760–773 e715, doi: 10.1016/j.cell.2017.06.052 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Simsek D & Jasin M Alternative end-joining is suppressed by the canonical NHEJ component Xrcc4-ligase IV during chromosomal translocation formation. Nature structural & molecular biology 17, 410–416, doi: 10.1038/nsmb.1773 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Guirouilh-Barbat J et al. 53BP1 Protects against CtIP-Dependent Capture of Ectopic Chromosomal Sequences at the Junction of Distant Double-Strand Breaks. PLoS genetics 12, e1006230, doi: 10.1371/journal.pgen.1006230 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Bass TE et al. ETAA1 acts at stalled replication forks to maintain genome integrity. Nat Cell Biol 18, 1185–1195, doi: 10.1038/ncb3415 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Feng S et al. Ewing Tumor-associated Antigen 1 Interacts with Replication Protein A to Promote Restart of Stalled Replication Forks. J Biol Chem 291, 21956–21962, doi: 10.1074/jbc.C116.747758 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Haahr P et al. Activation of the ATR kinase by the RPA-binding protein ETAA1. Nat Cell Biol 18, 1196–1207, doi: 10.1038/ncb3422 (2016). [DOI] [PubMed] [Google Scholar]
- 128.Cortez D, Guntuku S, Qin J & Elledge SJ ATR and ATRIP: partners in checkpoint signaling. Science 294, 1713–1716, doi: 10.1126/science.1065521 (2001). [DOI] [PubMed] [Google Scholar]
- 129.Zou L & Elledge SJ Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 300, 1542–1548 (2003). [DOI] [PubMed] [Google Scholar]
- 130.Scully R & Xie A Double strand break repair functions of histone H2AX. Mutation research 750, 5–14, doi: 10.1016/j.mrfmmm.2013.07.007 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Stucki M & Jackson SP gammaH2AX and MDC1: anchoring the DNA-damage-response machinery to broken chromosomes. DNA Repair (Amst) 5, 534–543 (2006). [DOI] [PubMed] [Google Scholar]
- 132.Helmink BA et al. H2AX prevents CtIP-mediated DNA end resection and aberrant repair in G1-phase lymphocytes. Nature 469, 245–249, doi: 10.1038/nature09585 nature09585 [pii] (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Messick TE & Greenberg RA The ubiquitin landscape at DNA double-strand breaks. The Journal of cell biology 187, 319–326, doi: 10.1083/jcb.200908074 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Jackson SP & Durocher D Regulation of DNA damage responses by ubiquitin and SUMO. Mol Cell 49, 795–807, doi: 10.1016/j.molcel.2013.01.017 (2013). [DOI] [PubMed] [Google Scholar]
- 135.Lukas J, Lukas C & Bartek J More than just a focus: The chromatin response to DNA damage and its role in genome integrity maintenance. Nat Cell Biol 13, 1161–1169, doi: 10.1038/ncb2344 (2011). [DOI] [PubMed] [Google Scholar]
- 136.Doil C et al. RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell 136, 435–446 (2009). [DOI] [PubMed] [Google Scholar]
- 137.Stewart GS et al. The RIDDLE syndrome protein mediates a ubiquitin-dependent signaling cascade at sites of DNA damage. Cell 136, 420–434 (2009). [DOI] [PubMed] [Google Scholar]
- 138.Feng L, Huang J & Chen J MERIT40 facilitates BRCA1 localization and DNA damage repair. Genes Dev 23, 719–728 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Shao G et al. MERIT40 controls BRCA1-Rap80 complex integrity and recruitment to DNA double-strand breaks. Genes Dev 23, 740–754 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Hu Y et al. RAP80-directed tuning of BRCA1 homologous recombination function at ionizing radiation-induced nuclear foci. Genes Dev 25, 685–700, doi:gad.2011011 [pii] 10.1101/gad.2011011 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Coleman KA & Greenberg RA The BRCA1-RAP80 complex regulates DNA repair mechanism utilization by restricting end resection. J Biol Chem 286, 13669–13680, doi:M110.213728 [pii] 10.1074/jbc.M110.213728 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Fradet-Turcotte A et al. 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature 499, 50–54, doi: 10.1038/nature12318 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Celeste A et al. Histone H2AX phosphorylation is dispensable for the initial recognition of DNA breaks. Nat Cell Biol 5, 675–679 (2003). [DOI] [PubMed] [Google Scholar]
- 144.Xie A et al. Distinct roles of chromatin-associated proteins MDC1 and 53BP1 in mammalian double-strand break repair. Mol Cell 28, 1045–1057 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Nakamura K et al. Genetic dissection of vertebrate 53BP1: a major role in non-homologous end joining of DNA double strand breaks. DNA Repair (Amst) 5, 741–749 (2006). [DOI] [PubMed] [Google Scholar]
- 146.Polo SE & Jackson SP Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev 25, 409–433, doi:25/5/409 [pii] 10.1101/gad.2021311 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Price BD & D’Andrea AD Chromatin remodeling at DNA double-strand breaks. Cell 152, 1344–1354, doi: 10.1016/j.cell.2013.02.011 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Nakamura K et al. Regulation of homologous recombination by RNF20-dependent H2B ubiquitination. Mol Cell 41, 515–528, doi: 10.1016/j.molcel.2011.02.002 (2011). [DOI] [PubMed] [Google Scholar]
- 149.Moyal L et al. Requirement of ATM-dependent monoubiquitylation of histone H2B for timely repair of DNA double-strand breaks. Mol Cell 41, 529–542, doi: 10.1016/j.molcel.2011.02.015 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Kim J et al. Replication Stress Shapes a Protective Chromatin Environment across Fragile Genomic Regions. Mol Cell 69, 36–47 e37, doi: 10.1016/j.molcel.2017.11.021 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.d’Adda di Fagagna F A direct role for small non-coding RNAs in DNA damage response. Trends in cell biology 24, 171–178, doi: 10.1016/j.tcb.2013.09.008 (2014). [DOI] [PubMed] [Google Scholar]
- 152.Meers C, Keskin H & Storici F DNA repair by RNA: Templated, or not templated, that is the question. DNA Repair (Amst) 44, 17–21, doi: 10.1016/j.dnarep.2016.05.002 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Rowling JK Harry Potter and the Philosopher’s Stone. (Bloomsbury, 1997). [Google Scholar]
- 154.Zhang Y et al. Spatial organization of the mouse genome and its role in recurrent chromosomal translocations. Cell 148, 908–921, doi: 10.1016/j.cell.2012.02.002 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Roukos V et al. Spatial dynamics of chromosome translocations in living cells. Science 341, 660–664, doi: 10.1126/science.1237150 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Okayasu R & Iliakis G Ionizing radiation induces two forms of interphase chromosome breaks in Chinese hamster ovary cells that rejoin with different kinetics and show different sensitivity to treatment in hypertonic medium or beta-araA. Radiation research 136, 262–270 (1993). [PubMed] [Google Scholar]
- 157.Okayasu R & Iliakis G Evidence that the product of the xrs gene is predominantly involved in the repair of a subset of radiation-induced interphase chromosome breaks rejoining with fast kinetics. Radiation research 138, 34–43 (1994). [PubMed] [Google Scholar]
- 158.Scully R et al. Genetic analysis of BRCA1 function in a defined tumor cell line. Mol Cell 4, 1093–1099 (1999). [DOI] [PubMed] [Google Scholar]
- 159.Lobrich M & Jeggo P A Process of Resection-Dependent Nonhomologous End Joining Involving the Goddess Artemis. Trends Biochem Sci 42, 690–701, doi: 10.1016/j.tibs.2017.06.011 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Sugawara N, Wang X & Haber JE In vivo roles of Rad52, Rad54, and Rad55 proteins in Rad51-mediated recombination. Molecular Cell 12, 209–219 (2003). [DOI] [PubMed] [Google Scholar]
- 161.Haber JE A Life Investigating Pathways That Repair Broken Chromosomes. Annual review of genetics 50, 1–28, doi: 10.1146/annurev-genet-120215-035043 (2016). [DOI] [PubMed] [Google Scholar]
- 162.Cho NW, Dilley RL, Lampson MA & Greenberg RA Interchromosomal homology searches drive directional ALT telomere movement and synapsis. Cell 159, 108–121, doi: 10.1016/j.cell.2014.08.030 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Shibata A et al. Factors determining DNA double-strand break repair pathway choice in G2 phase. EMBO J 30, 1079–1092, doi: 10.1038/emboj.2011.27 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Pierce AJ, Hu P, Han M, Ellis N & Jasin M Ku DNA end-binding protein modulates homologous repair of double-strand breaks in mammalian cells. Genes & Development 15, 3237–3242 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Frank-Vaillant M & Marcand S Transient stability of DNA ends allows nonhomologous end joining to precede homologous recombination. Mol Cell 10, 1189–1199 (2002). [DOI] [PubMed] [Google Scholar]
- 166.Postow L Destroying the ring: Freeing DNA from Ku with ubiquitin. FEBS Lett 585, 2876–2882, doi:S0014–5793(11)00412–1 [pii] 10.1016/j.febslet.2011.05.046 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Aylon Y, Liefshitz B & Kupiec M The CDK regulates repair of double-strand breaks by homologous recombination during the cell cycle. EMBO J 23, 4868–4875, doi: 10.1038/sj.emboj.7600469 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Ira G et al. DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature 431, 1011–1017 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Caspari T, Murray JM & Carr AM Cdc2-cyclin B kinase activity links Crb2 and Rqh1-topoisomerase III. Genes Dev 16, 1195–1208 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Huertas P, Cortes-Ledesma F, Sartori AA, Aguilera A & Jackson SP CDK targets Sae2 to control DNA-end resection and homologous recombination. Nature 455, 689–692, doi:nature07215 [pii] 10.1038/nature07215 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Tomimatsu N et al. Phosphorylation of EXO1 by CDKs 1 and 2 regulates DNA end resection and repair pathway choice. Nat Commun 5, 3561, doi: 10.1038/ncomms4561 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Makharashvili N & Paull TT CtIP: A DNA damage response protein at the intersection of DNA metabolism. DNA Repair (Amst) 32, 75–81, doi: 10.1016/j.dnarep.2015.04.016 (2015). [DOI] [PubMed] [Google Scholar]
- 173.Anand R, Ranjha L, Cannavo E & Cejka P Phosphorylated CtIP Functions as a Co-factor of the MRE11-RAD50-NBS1 Endonuclease in DNA End Resection. Mol Cell 64, 940–950, doi: 10.1016/j.molcel.2016.10.017 (2016). [DOI] [PubMed] [Google Scholar]
- 174.Yu X & Chen J DNA damage-induced cell cycle checkpoint control requires CtIP, a phosphorylation-dependent binding partner of BRCA1 C-terminal domains. Mol Cell Biol 24, 9478–9486 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Stark JM, Pierce AJ, Oh J, Pastink A & Jasin M Genetic steps of mammalian homologous repair with distinct mutagenic consequences. Mol Cell Biol 24, 9305–9316 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Polato F et al. CtIP-mediated resection is essential for viability and can operate independently of BRCA1. The Journal of experimental medicine 211, 1027–1036, doi: 10.1084/jem.20131939 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Zhou Y, Caron P, Legube G & Paull TT Quantitation of DNA double-strand break resection intermediates in human cells. Nucleic Acids Res 42, e19, doi: 10.1093/nar/gkt1309 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Cruz-Garcia A, Lopez-Saavedra A & Huertas P BRCA1 accelerates CtIP-mediated DNA-end resection. Cell reports 9, 451–459, doi: 10.1016/j.celrep.2014.08.076 (2014). [DOI] [PubMed] [Google Scholar]
- 179.Bunting SF et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 141, 243–254 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Cao L et al. A selective requirement for 53BP1 in the biological response to genomic instability induced by Brca1 deficiency. Mol Cell 35, 534–541, doi: 10.1016/j.molcel.2009.06.037 S1097–2765(09)00548–6 [pii] (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Bouwman P et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nature structural & molecular biology 17, 688–695, doi:nsmb.1831 [pii] 10.1038/nsmb.1831 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Nacson J et al. BRCA1 Mutation-Specific Responses to 53BP1 Loss-Induced Homologous Recombination and PARP Inhibitor Resistance. Cell reports 24, 3513–3527 e3517, doi: 10.1016/j.celrep.2018.08.086 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Zimmermann M & de Lange T 53BP1: pro choice in DNA repair. Trends in cell biology 24, 108–117, doi: 10.1016/j.tcb.2013.09.003 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Chapman JR et al. RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Mol Cell 49, 858–871, doi: 10.1016/j.molcel.2013.01.002 S1097–2765(13)00003–8 [pii] (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Di Virgilio M et al. Rif1 prevents resection of DNA breaks and promotes immunoglobulin class switching. Science 339, 711–715, doi: 10.1126/science.1230624 science.1230624 [pii] (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Escribano-Diaz C et al. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol Cell 49, 872–883, doi: 10.1016/j.molcel.2013.01.001 (2013). [DOI] [PubMed] [Google Scholar]
- 187.Zimmermann M, Lottersberger F, Buonomo SB, Sfeir A & de Lange T 53BP1 regulates DSB repair using Rif1 to control 5’ end resection. Science 339, 700–704, doi: 10.1126/science.1231573 science.1231573 [pii] (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Boersma V et al. MAD2L2 controls DNA repair at telomeres and DNA breaks by inhibiting 5’ end resection. Nature 521, 537–540, doi: 10.1038/nature14216 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Xu G et al. REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature 521, 541–544, doi: 10.1038/nature14328 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Callen E et al. 53BP1 mediates productive and mutagenic DNA repair through distinct phosphoprotein interactions. Cell 153, 1266–1280, doi: 10.1016/j.cell.2013.05.023 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Gupta R et al. DNA Repair Network Analysis Reveals Shieldin as a Key Regulator of NHEJ and PARP Inhibitor Sensitivity. Cell 173, 972–988 e923, doi: 10.1016/j.cell.2018.03.050 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Dev H et al. Shieldin complex promotes DNA end-joining and counters homologous recombination in BRCA1-null cells. Nat Cell Biol 20, 954–965, doi: 10.1038/s41556-018-0140-1 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Ghezraoui H et al. 53BP1 cooperation with the REV7-shieldin complex underpins DNA structure-specific NHEJ. Nature 560, 122–127, doi: 10.1038/s41586-018-0362-1 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Mirman Z et al. 53BP1-RIF1-shieldin counteracts DSB resection through CST- and Polalpha-dependent fill-in. Nature 560, 112–116, doi: 10.1038/s41586-018-0324-7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Noordermeer SM et al. The shieldin complex mediates 53BP1-dependent DNA repair. Nature 560, 117–121, doi: 10.1038/s41586-018-0340-7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Chapman JR, Sossick AJ, Boulton SJ & Jackson SP BRCA1-associated exclusion of 53BP1 from DNA damage sites underlies temporal control of DNA repair. J Cell Sci 125, 3529–3534, doi:jcs.105353 [pii] 10.1242/jcs.105353 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Tang J et al. Acetylation limits 53BP1 association with damaged chromatin to promote homologous recombination. Nature structural & molecular biology 20, 317–325, doi: 10.1038/nsmb.2499 nsmb.2499 [pii] (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Jacquet K et al. The TIP60 Complex Regulates Bivalent Chromatin Recognition by 53BP1 through Direct H4K20me Binding and H2AK15 Acetylation. Mol Cell 62, 409–421, doi: 10.1016/j.molcel.2016.03.031 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Densham RM et al. Human BRCA1-BARD1 ubiquitin ligase activity counteracts chromatin barriers to DNA resection. Nature structural & molecular biology 23, 647–655, doi: 10.1038/nsmb.3236 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Drane P et al. TIRR regulates 53BP1 by masking its histone methyl-lysine binding function. Nature 543, 211–216, doi: 10.1038/nature21358 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Tkac J et al. HELB Is a Feedback Inhibitor of DNA End Resection. Mol Cell 61, 405–418, doi: 10.1016/j.molcel.2015.12.013 (2016). [DOI] [PubMed] [Google Scholar]
- 202.Orthwein A et al. A mechanism for the suppression of homologous recombination in G1 cells. Nature 528, 422–426, doi: 10.1038/nature16142 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 203.Duro E et al. Identification of the MMS22L-TONSL complex that promotes homologous recombination. Mol Cell 40, 632–644, doi: 10.1016/j.molcel.2010.10.023 (2010). [DOI] [PubMed] [Google Scholar]
- 204.O’Donnell L et al. The MMS22L-TONSL complex mediates recovery from replication stress and homologous recombination. Mol Cell 40, 619–631, doi: 10.1016/j.molcel.2010.10.024 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Piwko W et al. RNAi-based screening identifies the Mms22L-Nfkbil2 complex as a novel regulator of DNA replication in human cells. EMBO J 29, 4210–4222, doi: 10.1038/emboj.2010.304 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Piwko W et al. The MMS22L-TONSL heterodimer directly promotes RAD51-dependent recombination upon replication stress. EMBO J 35, 2584–2601, doi: 10.15252/embj.201593132 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Saredi G et al. H4K20me0 marks post-replicative chromatin and recruits the TONSL-MMS22L DNA repair complex. Nature 534, 714–718, doi: 10.1038/nature18312 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Rieder CL & Cole RW Entry into mitosis in vertebrate somatic cells is guarded by a chromosome damage checkpoint that reverses the cell cycle when triggered during early but not late prophase. The Journal of cell biology 142, 1013–1022 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Giunta S, Belotserkovskaya R & Jackson SP DNA damage signaling in response to double-strand breaks during mitosis. The Journal of cell biology 190, 197–207, doi: 10.1083/jcb.200911156 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Lee DH et al. Dephosphorylation enables the recruitment of 53BP1 to double-strand DNA breaks. Mol Cell 54, 512–525, doi: 10.1016/j.molcel.2014.03.020 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Orthwein A et al. Mitosis inhibits DNA double-strand break repair to guard against telomere fusions. Science 344, 189–193, doi: 10.1126/science.1248024 (2014). [DOI] [PubMed] [Google Scholar]
- 212.Goodarzi AA et al. ATM signaling facilitates repair of DNA double-strand breaks associated with heterochromatin. Mol Cell 31, 167–177, doi: 10.1016/j.molcel.2008.05.017 S1097–2765(08)00395-X [pii] (2008). [DOI] [PubMed] [Google Scholar]
- 213.Aymard F et al. Transcriptionally active chromatin recruits homologous recombination at DNA double-strand breaks. Nature structural & molecular biology 21, 366–374, doi: 10.1038/nsmb.2796 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Daugaard M et al. LEDGF (p75) promotes DNA-end resection and homologous recombination. Nature structural & molecular biology 19, 803–810, doi: 10.1038/nsmb.2314 (2012). [DOI] [PubMed] [Google Scholar]
- 215.Gardini A, Baillat D, Cesaroni M & Shiekhattar R Genome-wide analysis reveals a role for BRCA1 and PALB2 in transcriptional co-activation. EMBO J 33, 890–905, doi: 10.1002/embj.201385567 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Garcia-Muse T & Aguilera A Transcription-replication conflicts: how they occur and how they are resolved. Nat Rev Mol Cell Biol, doi: 10.1038/nrm.2016.88 (2016). [DOI] [PubMed] [Google Scholar]
- 217.Hill SJ et al. Systematic screening reveals a role for BRCA1 in the response to transcription-associated DNA damage. Genes Dev 28, 1957–1975, doi: 10.1101/gad.241620.114 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Hatchi E et al. BRCA1 Recruitment to Transcriptional Pause Sites Is Required for R-Loop-Driven DNA Damage Repair. Mol Cell 57, 636–647, doi: 10.1016/j.molcel.2015.01.011 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Seeber A & Gasser SM Chromatin organization and dynamics in double-strand break repair. Curr Opin Genet Dev 43, 9–16, doi: 10.1016/j.gde.2016.10.005 (2017). [DOI] [PubMed] [Google Scholar]
- 220.Soutoglou E et al. Positional stability of single double-strand breaks in mammalian cells. Nat Cell Biol 9, 675–682 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Aten JA et al. Dynamics of DNA double-strand breaks revealed by clustering of damaged chromosome domains. Science 303, 92–95 (2004). [DOI] [PubMed] [Google Scholar]
- 222.Dimitrova N, Chen YC, Spector DL & de Lange T 53BP1 promotes non-homologous end joining of telomeres by increasing chromatin mobility. Nature 456, 524–528, doi:nature07433 [pii] 10.1038/nature07433 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Lottersberger F, Karssemeijer RA, Dimitrova N & de Lange T 53BP1 and the LINC Complex Promote Microtubule-Dependent DSB Mobility and DNA Repair. Cell 163, 880–893, doi: 10.1016/j.cell.2015.09.057 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Baca SC et al. Punctuated evolution of prostate cancer genomes. Cell 153, 666–677, doi: 10.1016/j.cell.2013.03.021 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Amaral N, Ryu T, Li X & Chiolo I Nuclear Dynamics of Heterochromatin Repair. Trends in genetics : TIG 33, 86–100, doi: 10.1016/j.tig.2016.12.004 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Chiolo I et al. Double-strand breaks in heterochromatin move outside of a dynamic HP1a domain to complete recombinational repair. Cell 144, 732–744, doi: 10.1016/j.cell.2011.02.012 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Tsouroula K et al. Temporal and Spatial Uncoupling of DNA Double Strand Break Repair Pathways within Mammalian Heterochromatin. Mol Cell 63, 293–305, doi: 10.1016/j.molcel.2016.06.002 (2016). [DOI] [PubMed] [Google Scholar]
- 228.Lopes M, Foiani M & Sogo JM Multiple mechanisms control chromosome integrity after replication fork uncoupling and restart at irreparable UV lesions. Mol Cell 21, 15–27 (2006). [DOI] [PubMed] [Google Scholar]
- 229.Zeman MK & Cimprich KA Causes and consequences of replication stress. Nat Cell Biol 16, 2–9, doi: 10.1038/ncb2897 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Berti M & Vindigni A Replication stress: getting back on track. Nature structural & molecular biology 23, 103–109, doi: 10.1038/nsmb.3163 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Marians KJ Lesion Bypass and the Reactivation of Stalled Replication Forks. Annu Rev Biochem, doi: 10.1146/annurev-biochem-062917-011921 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Giannattasio M et al. Visualization of recombination-mediated damage bypass by template switching. Nature structural & molecular biology 21, 884–892, doi: 10.1038/nsmb.2888 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Costanzo V et al. An ATR- and Cdc7-dependent DNA damage checkpoint that inhibits initiation of DNA replication. Mol Cell 11, 203–213 (2003). [DOI] [PubMed] [Google Scholar]
- 234.Cimprich KA & Cortez D ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol 9, 616–627, doi:nrm2450 [pii] 10.1038/nrm2450 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Fullbright G, Rycenga HB, Gruber JD & Long DT p97 Promotes a Conserved Mechanism of Helicase Unloading during DNA Cross-Link Repair. Mol Cell Biol 36, 2983–2994, doi: 10.1128/MCB.00434-16 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Long DT, Raschle M, Joukov V & Walter JC Mechanism of RAD51-dependent DNA interstrand cross-link repair. Science 333, 84–87, doi: 10.1126/science.1204258 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Willis NA, Panday A, Duffey EE & Scully R Rad51 recruitment and exclusion of non-homologous end joining during homologous recombination at a Tus/Ter mammalian replication fork barrier. PLoS genetics 14, e1007486, doi: 10.1371/journal.pgen.1007486 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Ait Saada A, Lambert SAE & Carr AM Preserving replication fork integrity and competence via the homologous recombination pathway. DNA Repair (Amst) 71, 135–147, doi: 10.1016/j.dnarep.2018.08.017 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Raschle M et al. Mechanism of replication-coupled DNA interstrand crosslink repair. Cell 134, 969–980 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Amunugama R et al. Replication Fork Reversal during DNA Interstrand Crosslink Repair Requires CMG Unloading. Cell reports 23, 3419–3428, doi: 10.1016/j.celrep.2018.05.061 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Klein Douwel D et al. XPF-ERCC1 acts in Unhooking DNA interstrand crosslinks in cooperation with FANCD2 and FANCP/SLX4. Mol Cell 54, 460–471, doi: 10.1016/j.molcel.2014.03.015 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Knipscheer P et al. The Fanconi anemia pathway promotes replication-dependent DNA interstrand cross-link repair. Science 326, 1698–1701, doi: 10.1126/science.1182372 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Sogo JM, Lopes M & Foiani M Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science 297, 599–602 (2002). [DOI] [PubMed] [Google Scholar]
- 244.Ray Chaudhuri A et al. Topoisomerase I poisoning results in PARP-mediated replication fork reversal. Nature structural & molecular biology 19, 417–423, doi: 10.1038/nsmb.2258 (2012). [DOI] [PubMed] [Google Scholar]
- 245.Zellweger R et al. Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. The Journal of cell biology 208, 563–579, doi: 10.1083/jcb.201406099 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Kile AC et al. HLTF’s Ancient HIRAN Domain Binds 3’ DNA Ends to Drive Replication Fork Reversal. Mol Cell 58, 1090–1100, doi: 10.1016/j.molcel.2015.05.013 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Moldovan GL et al. Inhibition of homologous recombination by the PCNA-interacting protein PARI. Mol Cell 45, 75–86, doi: 10.1016/j.molcel.2011.11.010 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Dungrawala H et al. RADX Promotes Genome Stability and Modulates Chemosensitivity by Regulating RAD51 at Replication Forks. Mol Cell 67, 374–386 e375, doi: 10.1016/j.molcel.2017.06.023 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Schlacher K et al. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145, 529–542, doi:S0092–8674(11)00375–8 [pii] 10.1016/j.cell.2011.03.041 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Schlacher K, Wu H & Jasin M A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 22, 106–116, doi: 10.1016/j.ccr.2012.05.015 S1535–6108(12)00214–0 [pii] (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Kolinjivadi AM et al. Smarcal1-Mediated Fork Reversal Triggers Mre11-Dependent Degradation of Nascent DNA in the Absence of Brca2 and Stable Rad51 Nucleofilaments. Mol Cell 67, 867–881 e867, doi: 10.1016/j.molcel.2017.07.001 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Taglialatela A et al. Restoration of Replication Fork Stability in BRCA1- and BRCA2-Deficient Cells by Inactivation of SNF2-Family Fork Remodelers. Mol Cell 68, 414–430 e418, doi: 10.1016/j.molcel.2017.09.036 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Vujanovic M et al. Replication Fork Slowing and Reversal upon DNA Damage Require PCNA Polyubiquitination and ZRANB3 DNA Translocase Activity. Mol Cell 67, 882–890 e885, doi: 10.1016/j.molcel.2017.08.010 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Duxin JP & Walter JC What is the DNA repair defect underlying Fanconi anemia? Current opinion in cell biology 37, 49–60, doi: 10.1016/j.ceb.2015.09.002 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Huang J et al. The DNA translocase FANCM/MHF promotes replication traverse of DNA interstrand crosslinks. Mol Cell 52, 434–446, doi: 10.1016/j.molcel.2013.09.021 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Semlow DR, Zhang J, Budzowska M, Drohat AC & Walter JC Replication-Dependent Unhooking of DNA Interstrand Cross-Links by the NEIL3 Glycosylase. Cell 167, 498–511 e414, doi: 10.1016/j.cell.2016.09.008 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Zhang J et al. DNA interstrand cross-link repair requires replication-fork convergence. Nature structural & molecular biology 22, 242–247, doi: 10.1038/nsmb.2956 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Larsen NB, Sass E, Suski C, Mankouri HW & Hickson ID The Escherichia coli Tus-Ter replication fork barrier causes site-specific DNA replication perturbation in yeast. Nat Commun 5, 3574, doi: 10.1038/ncomms4574 (2014). [DOI] [PubMed] [Google Scholar]
- 259.Larsen NB et al. Stalled replication forks generate a distinct mutational signature in yeast. Proc Natl Acad Sci U S A 114, 9665–9670, doi: 10.1073/pnas.1706640114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Ceccaldi R, Sarangi P & D’Andrea AD The Fanconi anaemia pathway: new players and new functions. Nat Rev Mol Cell Biol 17, 337–349, doi: 10.1038/nrm.2016.48 (2016). [DOI] [PubMed] [Google Scholar]
- 261.Nakanishi K et al. Homology-directed Fanconi anemia pathway cross-link repair is dependent on DNA replication. Nature structural & molecular biology 18, 500–503, doi: 10.1038/nsmb.2029 nsmb.2029 [pii] (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Foster SS, Balestrini A & Petrini JH Functional interplay of the Mre11 nuclease and Ku in the response to replication-associated DNA damage. Mol Cell Biol 31, 4379–4389, doi: 10.1128/MCB.05854-11 MCB.05854–11 [pii] (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Balestrini A et al. The Ku heterodimer and the metabolism of single-ended DNA double-strand breaks. Cell reports 3, 2033–2045, doi: 10.1016/j.celrep.2013.05.026 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Chanut P, Britton S, Coates J, Jackson SP & Calsou P Coordinated nuclease activities counteract Ku at single-ended DNA double-strand breaks. Nat Commun 7, 12889, doi: 10.1038/ncomms12889 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Balmus G et al. ATM orchestrates the DNA-damage response to counter toxic non-homologous end-joining at broken replication forks. Nat Commun 10, 87, doi: 10.1038/s41467-018-07729-2 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Vriend LE et al. Distinct genetic control of homologous recombination repair of Cas9-induced double-strand breaks, nicks and paired nicks. Nucleic Acids Res, doi: 10.1093/nar/gkw179 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Bhat KP & Cortez D RPA and RAD51: fork reversal, fork protection, and genome stability. Nature structural & molecular biology 25, 446–453, doi: 10.1038/s41594-018-0075-z (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Frock RL et al. Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nature biotechnology 33, 179–186, doi: 10.1038/nbt.3101 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Tubbs A et al. Dual Roles of Poly(dA:dT) Tracts in Replication Initiation and Fork Collapse. Cell 174, 1127–1142 e1119, doi: 10.1016/j.cell.2018.07.011 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Shastri N et al. Genome-wide Identification of Structure-Forming Repeats as Principal Sites of Fork Collapse upon ATR Inhibition. Mol Cell 72, 222–238 e211, doi: 10.1016/j.molcel.2018.08.047 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Petermann E, Orta ML, Issaeva N, Schultz N & Helleday T Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol Cell 37, 492–502, doi:S1097–2765(10)00072–9 [pii] 10.1016/j.molcel.2010.01.021 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Heller RC & Marians KJ Replisome assembly and the direct restart of stalled replication forks. Nat Rev Mol Cell Biol 7, 932–943 (2006). [DOI] [PubMed] [Google Scholar]
- 273.Carr AM & Lambert S Replication Stress-Induced Genome Instability: The Dark Side of Replication Maintenance by Homologous Recombination. J Mol Biol, doi:S0022–2836(13)00271–4 [pii] 10.1016/j.jmb.2013.04.023 (2013). [DOI] [PubMed] [Google Scholar]
- 274.Lambert S et al. Homologous recombination restarts blocked replication forks at the expense of genome rearrangements by template exchange. Mol Cell 39, 346–359, doi: 10.1016/j.molcel.2010.07.015 (2010). [DOI] [PubMed] [Google Scholar]
- 275.Lambert S, Watson A, Sheedy DM, Martin B & Carr AM Gross chromosomal rearrangements and elevated recombination at an inducible site-specific replication fork barrier. Cell 121, 689–702 (2005). [DOI] [PubMed] [Google Scholar]
- 276.Julan M, Oehler J, Morrow CA, Osman F & Whitby MC Factors affecting template switch recombination associated with restarted DNA replication. eLife 8:e41697, doi: 10.7554/eLife.41697 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Sotiriou SK et al. Mammalian RAD52 Functions in Break-Induced Replication Repair of Collapsed DNA Replication Forks. Mol Cell 64, 1127–1134, doi: 10.1016/j.molcel.2016.10.038 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Nik-Zainal S et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 534, 47–54, doi: 10.1038/nature17676 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Menghi F et al. The tandem duplicator phenotype as a distinct genomic configuration in cancer. Proc Natl Acad Sci U S A 113, E2373–2382, doi: 10.1073/pnas.1520010113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]