

Abstract
Acetaminophen/paracetamol (APAP) overdose is the leading cause of drug-induced acute liver failure (ALF) in the United States and Europe. The progression of the disease is attributed to sterile inflammation induced by the release of high mobility group box 1 (HMGB1) and the interaction with receptor for advanced glycation end products (RAGE). A specific, effective, and safe approach to neutralize the proinflammatory activity of HMGB1 is highly desirable. Here, we found that a heparan sulfate (HS) octadecasaccharide (18-mer-HP or hepatoprotective 18-mer) displays potent hepatoprotection by targeting the HMGB1/RAGE axis. Endogenous HS proteoglycan, syndecan-1, is shed in response to APAP overdose in mice and humans. Furthermore, purified syndecan-1, but not syndecan-1 core protein, binds to HMGB1, suggesting that HMGB1 binds to HS polysaccharide side chains of syndecan-1. Last, we compared the protection effect between 18-mer-HP and N-acetyl cysteine, which is the standard of care to treat APAP overdose. We demonstrated that 18-mer-HP administered 3 hours after a lethal dose of APAP is fully protective; however, the treatment of N-acetyl cysteine loses protection. Therefore, 18-mer-HP may offer a potential therapeutic advantage over N-acetyl cysteine for late-presenting patients. Synthetic HS provides a potential approach for the treatment of APAP-induced ALF.
INTRODUCTION
Sterile inflammation is a natural process that initiates tissue repair in response to cellular damage. However, exaggerated inflammation after an initial insult often damages surrounding healthy tissues and is a key contributor to many disease processes. High mobility group box 1 (HMGB1) is a DNA binding protein that regulates transcription and is released from the nucleus during necrosis (1, 2). Extracellularly, HMGB1 is a damage-associated molecular pattern (DAMP) protein that acts as a proinflammatory molecule, orchestrating migration and activation of inflammatory cells to the injury site (3). HMGB1-dependent inflammation has been associated with ischemia-reperfusion and drug-induced liver injury (4).
Acetaminophen (APAP), also known as paracetamol, is a widely used analgesic and is the active pharmaceutical ingredient of Tylenol. Ingestion of a supratherapeutic dose causes acute liver failure (ALF) (5). The misuse of Vicodin or Percocet, coformulations of opioids and APAP, can also cause ALF. In the United States, nearly 50% of drug-induced liver injury has been attributed to APAP toxicity (6), which accounts for ~80,000 emergency room visits annually (7). The mechanism for APAP toxicity begins with its metabolic conversion to the reactive chemical species, N-acetyl-p-benzoquinone imine (NAPQI), which causes hepatocyte necrosis (8). Necrotic hepatocytes release HMGB1, which drives chemotaxis of neutrophils through the receptor for advanced glycation end products (RAGE), activating sterile inflammation and amplifying liver injury (4).
Heparan sulfate (HS) is a sulfated polysaccharide abundant on the cell surface and in the extracellular matrix. It primarily exists in the form of HS proteoglycan that contains a core protein and HS polysaccharides. Syndecans, including syndecan-1, syndecan-2, syndecan-3, and syndecan-4, are common HS proteoglycans on the cell surface (9). HS proteoglycans participate in various aspects of inflammation including chemokine presentation and neutrophil transendothelial migration (10–13). The functions of HS proteoglycans are dominated by the HS polysaccharide chains, which are composed of disaccharide repeating units of glucuronic acid (GlcA) or iduronic acid (IdoA) linked to glucosamine (GlcN) residues that carry sulfo groups. The chain length and sulfation pattern of HS determine its biological functions (14). Many DAMPs, including HMGB1, are HS-binding proteins (15). In the current study, we present the synthesis of a HS octadecasaccharide (18-mer-HP or hepatoprotective 18-mer) to exploit the anti-inflammatory effect. We demonstrate that the 18-mer-HP protects against APAP-induced ALF by neutralizing the proinflammatory activity of HMGB1/RAGE axis in a murine model. Our findings uncover the use of carbohydrate-based compounds to curb ALF caused by APAP overdose.
RESULTS
Homogeneous 18-mer-HP is synthesized using the chemoenzymatic approach
HS isolated from natural sources is a highly complex mixture with different polysaccharide chain lengths and sulfation patterns. The lack of structurally homogeneous HS oligosaccharides, especially long HS oligosaccharides that display similar functions to full-length endogenous HS, hampers the efforts to exploit the applications of HS as a therapeutic agent (16). We have recently developed a highly efficient chemoenzymatic method to synthesize HS oligosaccharides (17–19). Here, we synthesized HS oligosaccharides, including 323 mg of 18-mer-HP (Fig. 1A and fig. S1). This represents one of the longest HS oligosaccharides synthesized to date. The structure of 18-mer-HP, GlcNS-GlcA-GlcNS-(IdoA2S-GlcNS)7-GlcA-pNP, was confirmed by high-resolution mass spectrometry (MS) and nuclear magnetic resonance (NMR). The purity was determined to be >98% by high-resolution anion exchange high-performance liquid chromatography (HPLC; figs. S2 to S4 and table S1).
Fig. 1. 18-mer-HP protects from liver injury after APAP overdose.
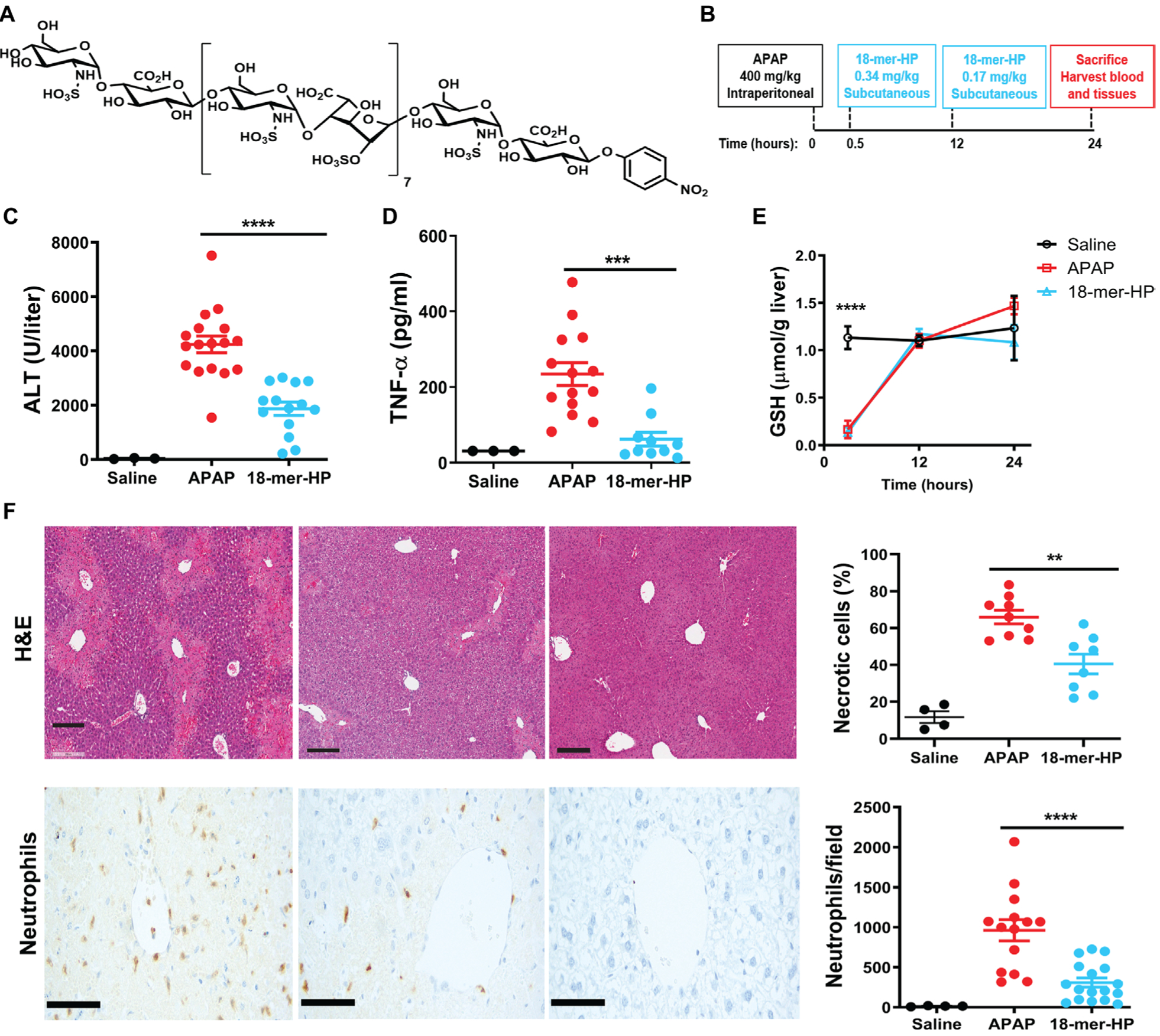
(A) Chemical structure of 18-mer-HP. (B) Murine model experimental design schematic including time, dose, and administration route of APAP and 18-mer-HP. (C) Plasma ALT concentrations from mice treated with saline, APAP alone, and APAP + 18-mer-HP. (D) Plas- ma TNF-α concentrations are decreased in the 18-mer-HP—treated group compared to APAP alone. (E) 18-mer-HP— and APAP-treated mice both have decreased GSH concentrations immediately after APAP overdose compared to the saline control mice. 18-mer-HP does not affect APAP’s metabolism to NAPQI. Saline n= 3, APAP n = 6, 18-mer-HP n = 6. (F) Representative images of hematoxylin and eosin (H&E) staining of formalin-fixed paraffin-embedded liver tissues and neutrophil immunohistochemistry (IHC). Quantitation of H&E-and IHC neutrophil—stained liver tissues from 100× fields are shown on the right. Scale bars, 200 μm. Data represent means ± SEM (C, D, E, and F). **P < 0.01, ***P < 0.001, ****P < 0.0001 by one-way ANOVA followed by Tukey’s post hoc test.
18-mer-HP protects from liver injury induced by APAP
The hepatoprotective effects of 18-mer-HP were examined in a murine model of APAP-induced ALF (Fig. 1B). Mice treated with 18-mer-HP after APAP overdose had healthier livers than APAP control mice as indicated by a lower plasma concentration of alanine aminotransferase (ALT), a biomarker of liver damage (Fig. 1C). 18-mer-HP treatment reduced plasma concentrations of tissue necrosis factor—α (TNF-α) (Fig. 1D), suggesting attenuation of local and systemic inflammation. Hepatic glutathione (GSH) concentrations in APAP control and 18-mer-HP—treated mice were essentially the same during the course of the studies (Fig. 1E). GSH concentrations decrease as NAPQI forms (8, 20); therefore, the data suggest that 18-mer-HP does not affect the metabolic conversion of APAP to the cytotoxic intermediate, NAPQI. These results demonstrate that 18-mer-HP ameliorates liver injury by reducing the inflammatory responses rather than by affecting APAP metabolism. In addition, 18-mer-HP treatment reduced the percentage of necrotic liver area and hepatic neutrophil infiltration compared to APAP control mice (Fig. 1F).
18-mer-HP targets HMGB1/RAGE axis to attenuate inflammation
HMGB1 has recently been implicated as an important mediator in animal models of APAP toxicity; therefore, we sought to determine whether 18-mer-HP’s hepatoprotection is due to targeting HMGB1 (4). Three lines of evidence suggest that 18-mer-HP targets the HMGB1/RAGE axis to reduce inflammation. First, we discovered that 18-mer-HP diminishes the HMGB1-mediated neutrophil infiltration in two in vivo models. In an air pouch model, injection of recombinant HMGB1 induced extensive neutrophil infiltration (Fig. 2A), an effect that was reduced by coadministration of 18-mer-HP. 18-mer-HP also reduced neutrophil infiltration in a peritonitis model induced by intraperitoneal injection of liver lysate (fig. S5), a process known to be mediated by HMGB1 (4).
Fig. 2. 18-mer-HP targets HMGB1/RAGE axis to decrease inflammation and liver injury.
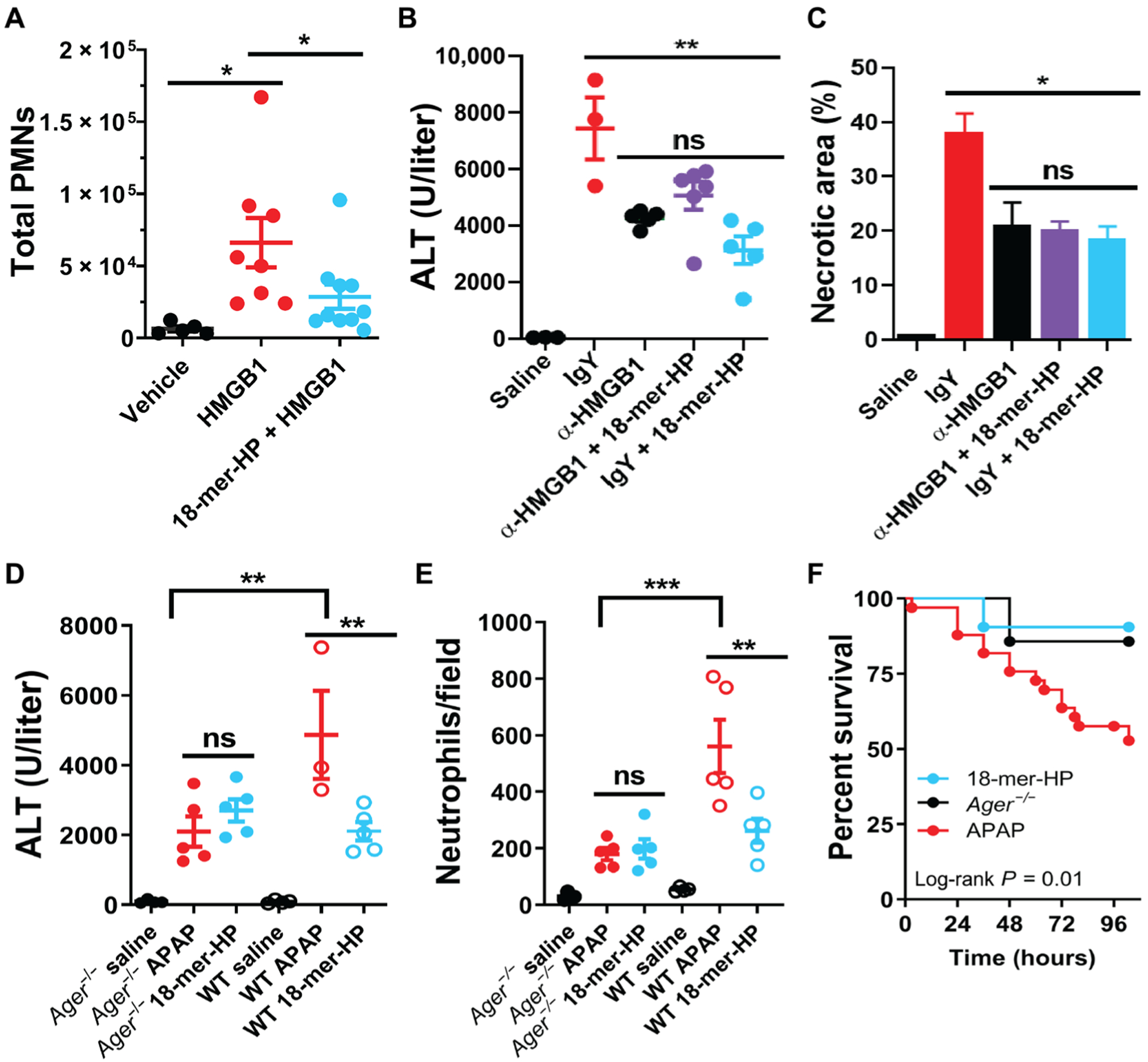
(A) 18-mer-HP prevents neutrophil infiltration induced by HMGB1 in the air pouch model. Number of polymorphonuclear neutrophils (PMNs) migrating to the air pouch was determined by flow cytometry. (B) HMGB1-neutralizing antibody (α-HMGB1; 4 mg/kg) decreases ALT at 24 hours after APAP. 18-mer-HP (0.34 mg/kg) in combination with α-HMGB1 (4 mg/kg) shows no further protection compared to α-HMGB1 or 18-mer-HP alone. (C) α-HMGB1 decreases liver necrotic area at 24 hours after APAP as determined by H&E staining. 18-mer-HP in the presence of α-HMGB1 shows no further protection compared to α-HMGB1 or 18-mer-HP alone. (D and E) 18-mer-HP lacks anti-inflammatory effects in Ager−/− mice as demonstrated by ALT (D) and neutrophil infiltration (E). (F) 18-mer-HP improves survival after a lethal overdose of APAP (600 mg/kg) comparable to the protection seen in Ager−/− mice (APAP, n = 33; 18-mer, n = 21; Ager−/− mice, n = 7). *P < 0.05, **P < 0.01, and ***P < 0.001 by one-way ANOVA followed by Dunnett’s post hoc test (A) and by one-way ANOVA followed by Tukey’s post hoc test (B to E). ns, not significant.
Second, it has been reported that HMGB1-neutralizing antibody (α-HMGB1) protects against liver injury from APAP overdose (21). We demonstrate that 18-mer-HP and α-HMGB1 display a similar hepatoprotective effect in the mouse model (Fig. 2, B and C). A combination treatment of α-HMGB1 and 18-mer-HP did not decrease ALT or necrotic liver area compared to either treatment alone (Fig. 2, B and C), suggesting that both 18-mer-HP and α-HMGB1 achieve their hepatoprotection by targeting the same biological process.
Third, RAGE-deficient mice (Ager−/−) were used to demonstrate that 18-mer-HP targets the HMGB1/RAGE axis. Because the interaction of HMGB1 and RAGE is essential for the proinflammatory response in APAP overdose (4), the hepatoprotective effect of 18-mer-HP is expected to be dependent on the presence of RAGE. 18-mer-HP treatment in Ager−/− mice was incapable of lowering the ALT concentration (Fig. 2D). As reported previously (4), after APAP overdose, wild-type (WT) mice had robust hepatic neutrophil infiltration, whereas Ager−/− mice had lower neutrophil infiltration (Fig. 2E). Whereas 18-mer-HP reduced neutrophil infiltration in WT mice, it had no effect on Ager−/− mice. The data suggest that 18-mer-HP lost its protective effect in the absence of RAGE.
Mortality studies using a lethal dose of APAP (600 mg/kg) were conducted in both WT and Ager−/− mice. For WT mice, 18-mer-HP decreased mortality after APAP overdose, resulting in 90% survival at 96 hours compared to 53% survival in the APAP WT group (Fig. 2F). For Ager −/− mice, the survival was already 86% in the absence of the treatment with 18-mer-HP, suggesting that Ager −/− mice are insensitive to APAP-induced liver injury. The observation is consistent with the liver injury after APAP overdose being amplified through the HMGB1/RAGE axis (4).
HMGB1 may exist in different redox states, and each redox form has distinct activity. A fully reduced HMGB1 (all thiol) form acts as a chemoattractant, and an oxidized HMGB1 (disulfide) form has cytokine-inducing activity (22–24). Our recombinant HMGB1 preparation contained both reduced and oxidized forms of HMGB1 (figs. S6 to S8). The recombinant HMGB1 protein was expected to be able to perform both chemokine and cytokine functions, namely, inducing neutrophil migration and stimulating macrophages. Contrary to its inhibitory effect on the chemokine functions of HMGB1, inducing neutrophil migration (Fig. 2A), 18-mer-HP appears to have no direct effect on the cytokine function of HMGB1, which is mainly mediated by the effects of TLR4 on macrophages (fig. S9) (25).
Binding to HMGB1 and hepatoprotection depend on the length of HS oligosaccharide
To investigate the structure-activity relationship between HS oligosaccharide and HMGB1, we used oligosaccharides that varied in chain length but maintained the same disaccharide repeating unit of 2-O-sulfo-iduronic acid (IdoA2S) and N-sulfo-glucosamine (GlcNS). For this purpose, we synthesized 6-and 12-mer to complement the 18-mer-HP (Fig. 3A and fig. S10). The biotinylated versions of the compounds were used to pull down endogenous HMGB1 from mouse liver lysate and visualized by immunoblotting for HMGB1 (Fig. 3B and fig. S11A). Only 18-mer-HP was successful in pulling down HMGB1 from liver lysate. To demonstrate that 18-mer-HP engages HMGB1 in the APAP overdose model, we pulled down circulating HMGB1 from APAP overdose mouse plasma using 18-mer-HP, whereas pulldown with the 6-mer served as a negative control (Fig. 3C and fig. S11, B and D). The HMGB1/18-mer-HP binding was also examined by using 18-mer-HP to compete with radiolabeled [35S]HS binding to HMGB1 (Fig. 3D). The 18-mer-HP’s half maximum inhibitory concentration (IC50) was 658 nM, whereas the 6-mer did not show any inhibition even at a concentration of 12,500 nM, again suggesting that 6-mer is incapable of binding to HMGB1. Last, we showed that 18-mer-HP binds to HMGB1 at the site of injury in situ (Fig. 3E). Using liver tissue sections from APAP overdose mice, we incubated oligosaccharides or phosphate-buffered saline (PBS) with the tissue before anti-HMGB1 staining. HMGB1 staining was apparent in the nucleus (dark brown) and outside the cell (diffuse brown staining). Incubation with 18-mer-HP at 10 and 100 μM decreased HMGB1 staining. Consistent with the binding results, 6-mer (100 μM) incubation did not affect HMGB1 staining.
Fig. 3. Size of HS oligosaccharide is important for HS/HMGB1 interaction and hepatoprotection.
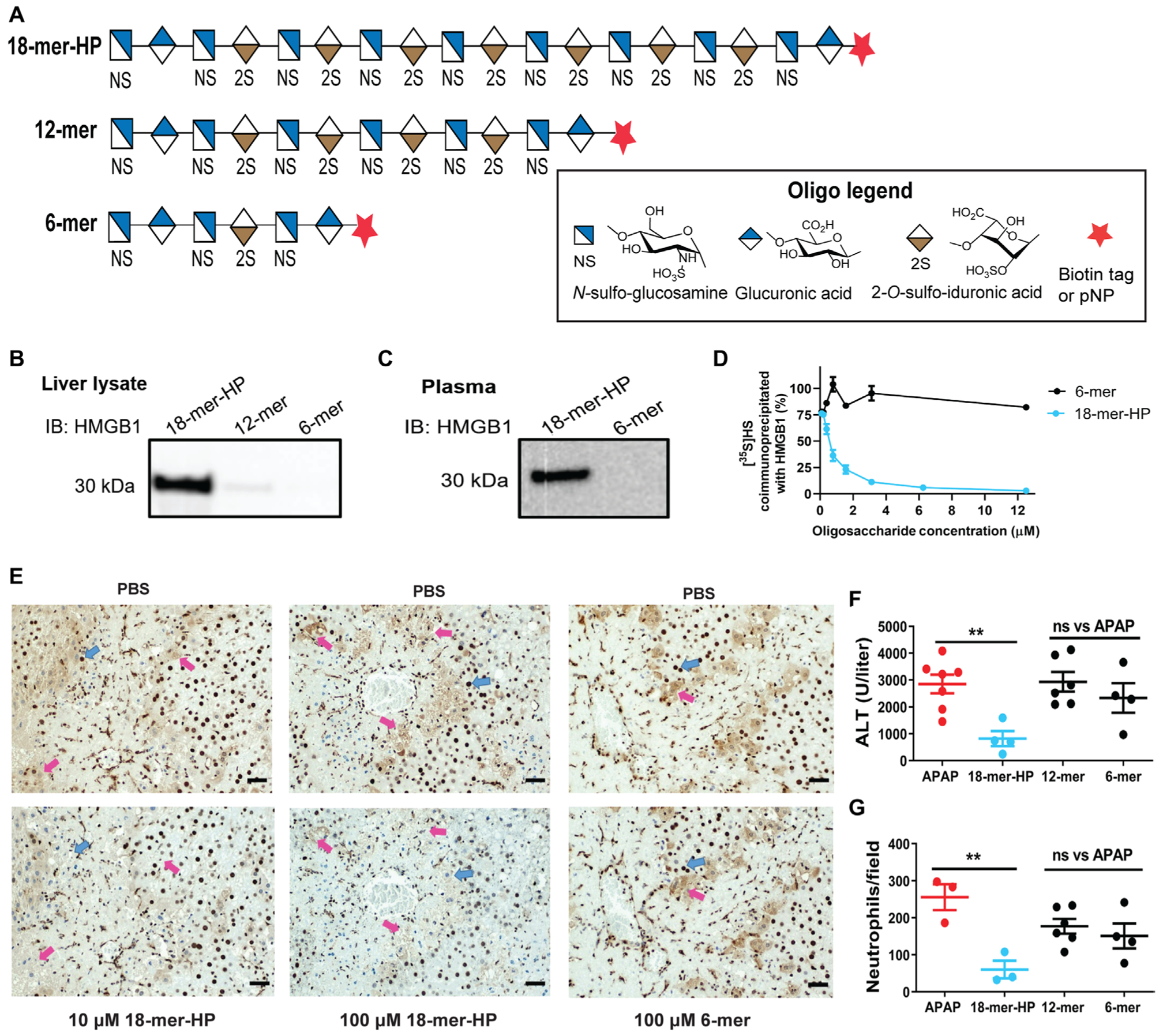
(A) Symbolic structures of HS oligosaccharides. (B) Western analysis of HMGB1 pulldown from mouse liver lysate by 18-mer-HP, 12-mer, and 6-mer biotinylated oligosaccharides using avidin-Sepharose. Full image is presented in fig. S11A. IB, immunoblotting. (C) Western analysis of plasma circulating HMGB1 pulldown from APAP overdose mouse plasma by 18-mer-HP and 6-mer biotinylated oligosaccharides. A higher—molecular weight protein band was also detected, possibly because of the cross-link with other plasma proteins from the action of transglutaminase-2 (fig. S11B) (41). We detected the presence of trasglutaminase-2, which is known to bind to heparin (42), in the plasma sample (fig. S11D). (D) HMGB1 binding competition assay using 35S-labeled HS with oligosaccharides as competitor ligands. (E) Liver sections from mice treated with APAP (400 mg/kg) and euthanized at 24 hours, stained with anti-HMGB1 antibody (top). Blue arrows indicate HMGB1 staining in the nucleus, and pink arrows indicate diffuse HMGB1 staining outside the cell. Incubation with 18-mer-HP before anti-HMGB1 results in decreased staining (bottom) compared to incubation with PBS (top). 6-mer has no effect on anti-HMGB1 staining. (F and G) 18-mer-HP decreases ALT (F) and neutrophil infiltration (G) after APAP overdose, whereas 12-and 6-mer do not. Data represent means ± SEM. **P < 0.01 by one-way ANOVA followed by Dunnett’s posttest.
We also tested the hepatoprotective effect of these three oligosaccharides in the APAP overdose mouse model. Only 18-mer-HP decreased plasma ALT concentrations and neutrophil infiltration in the livers of mice challenged with APAP (Fig. 3, F and G). The lack of hepatoprotection observed in APAP mice treated with 6-and 12-mer was consistent with the inability of these two compounds to bind to HMGB1. Together, these data provide evidence for a length requirement greater than 12 saccharides to bind to HMGB1 and display hepatoprotection.
Anticoagulant 18-mer-AXa does not display hepatoprotection
Heparin is an analog of HS and is an anticoagulant. To examine the effect of anticoagulant activity on hepatoprotection, another octadecasaccharide, 18-mer-AXa, was synthesized with a sequence of GlcNS6S-GlcA-GlcNS3S6S-(IdoA2S-GlcNS6S)7-GlcA-pNP/biotin (figs. S12 to S14 and table S2). The structure of 18-mer-AXa carries additional 3-O-and 6-O-sulfo groups compared with 18-mer-HP (Fig. 4A). Administration of 18-mer-AXa, but not 18-mer-HP, to APAP overdose mice reduced the activity of factor Xa (FXa) in plasma, confirming that 18-mer-AXa displays anticoagulant activity (Fig. 4B). Biotinylated 18-mer-AXa, like 18-mer-HP, was successful in pulling down HMGB1 from liver lysate (Fig. 4C and fig. S15). The binding affinity (KD) of recombinant HMGB1 to 18-mer-HP and 18-mer-AXa was determined to be 187 and 65 nM, respectively, by surface plasmon resonance (SPR) using recombinant human HMGB1 (table S3). Further binding affinity analysis using SPR showed that both reduced and oxidized HMGB1 display very similar binding affinity to 18-mer-HP and 18-mer-AXa (table S3).
Fig. 4. 18-mer-AXa binds to HMGB1 and displays anti-FXa activity.
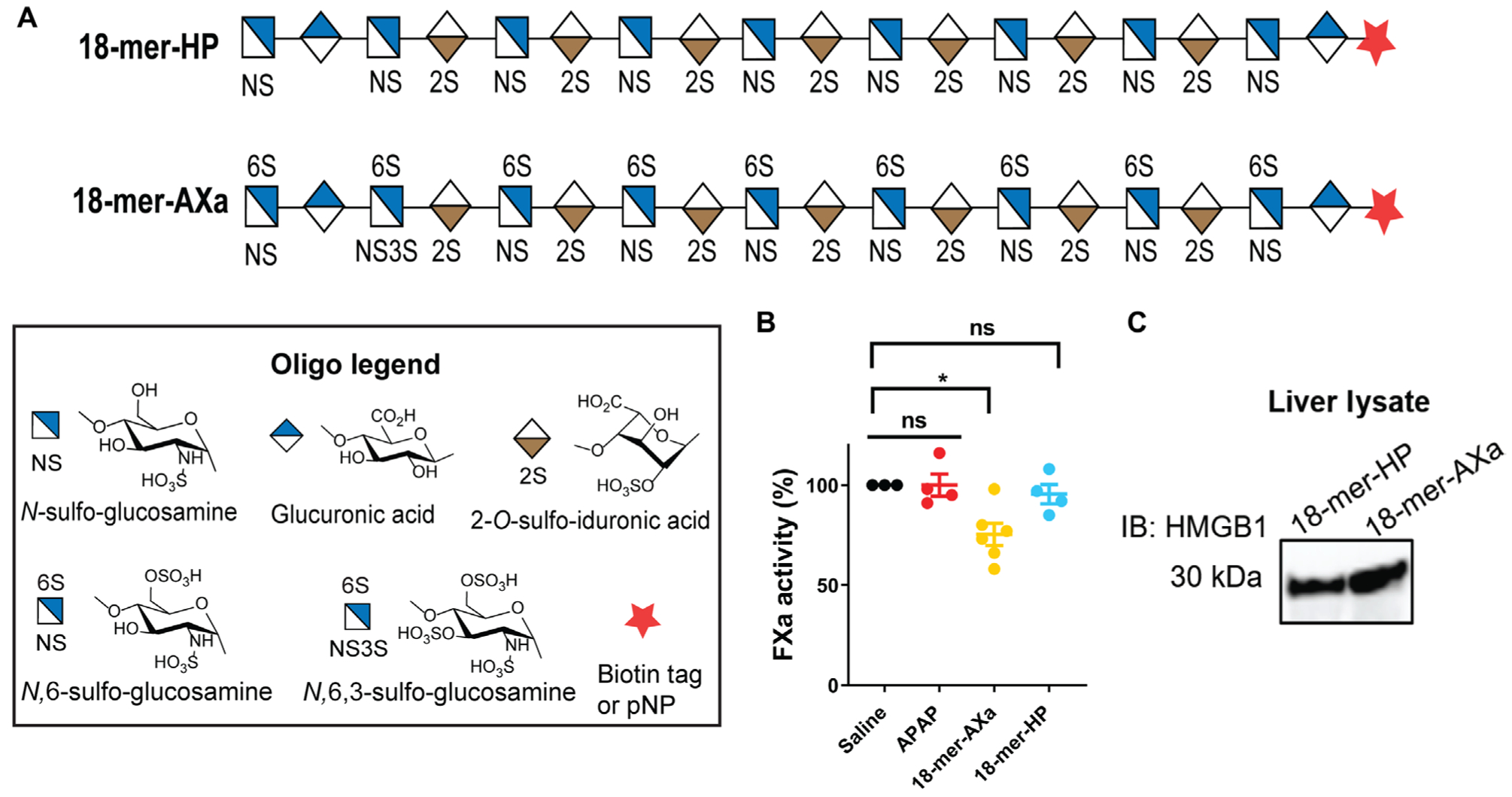
(A) Symbolic structure of 18-mer-HP and 18-mer-AXa. (B) Factor Xa (FXa) activity measured in plasma from saline-treated mice and APAP overdose mice treated with saline (APAP control group), 18-mer-AXa, or 18-mer-HP. Plasma was collected at the time of sacrifice, which was 12 hours after the second oligosaccharide injections and 24 hours after APAP overdose. (C) Western analysis of HMGB1 pulldown from liver lysate by 18-mer-HP and 18-mer-AXa biotinylated oligosaccharides using avidin-Sepharose. Full image is presented in fig. S15. Data represent means ± SEM. *P < 0.05 by one-way ANOVA followed my Dunnett’s posttest (B).
Although 18-mer-AXa binds to HMGB1 with high affinity, it does not display the hepatoprotective effect in APAP-challenged mice, as measured by ALT and neutrophil infiltration (Fig. 5, A and B and fig. S16). Further examination suggested that 18-mer-AXa treatment impaired liver regeneration after injury. The liver regeneration was assessed by measuring Ki-67—positive cells (24, 26). We observed an increased number of Ki-67—positive cells, which peaked at 72 hours and returned to baseline at 120 hours, in the APAP overdose group (Fig. 5C). The pattern of Ki-67—positive cells in the 18-mer-HP—treated group followed a very similar time course to the APAP overdose group, but the number of Ki-67 cells in the 18-mer-HP—treated group was less than that of the APAP control group because of reduced damage (Fig. 5C). In contrast, the 18-mer-AXa—treated group did not show an increase in Ki-67—positive cells at 72 hours despite suffering more severe liver damage compared to 18-mer-HP—treated group (Fig. 5C and fig. S17), suggesting that the liver regeneration process was likely impaired after the treatment with 18-mer-AXa.
Fig. 5. 18-mer-AXa impairs liver regeneration after APAP overdose.
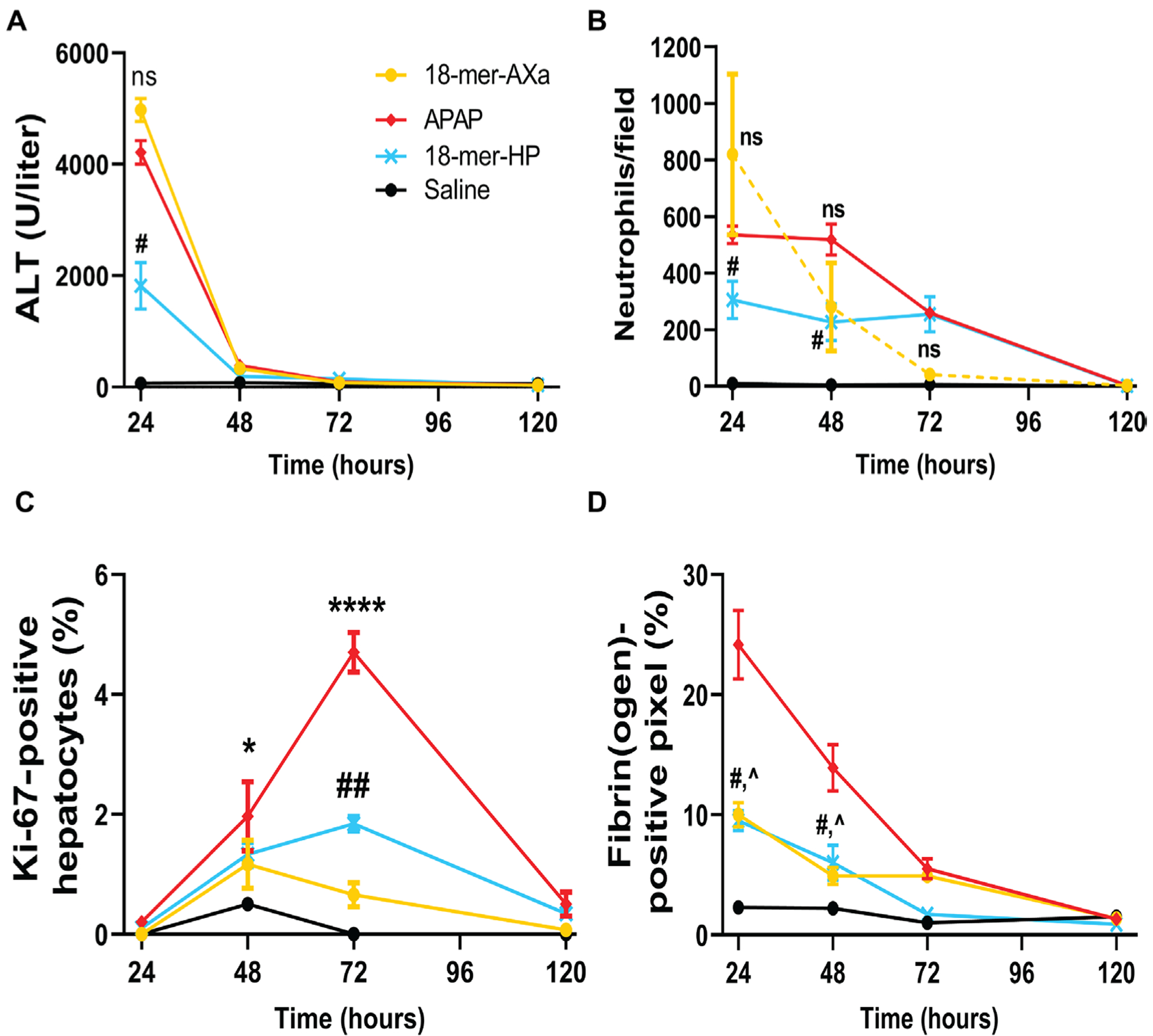
18-mer-AXa or 18-mer-HP was administered every 12 hours for 120 hours after APAP overdose. (A) ALT is increased after APAP overdose. At 24 hours, 18-mer-HP decreased ALT compared to APAP (#P = 0.0121), whereas there was no difference between 18-mer-AXa and APAP. (B) Neutrophils accumulate in the liver of APAP and 18-mer-AXa—treated mice up to 48 hours after APAP overdose. At 24 and 48 hours, 18-mer-HP decreased neutrophil infiltration (#P = 0.0355 and #P = 0.0301 at 24 and 48 hours, respectively). (C) APAP overdose mice began to increase proliferating hepatocytes at 48 hours (*P = 0.0280) and peaked at 72 hours (****P < 0.0001) before returning to baseline. 18-mer-HP treatment resulted in mild liver regeneration with an increase at 72 hours compared to saline (##P = 0.0045). In contrast, 18-mer-AXa treatment did not result in significant changes in Ki-672014positive hepatocytes at any time point after APAP overdose. (D) At 24 and 48 hours, APAP overdose resulted in elevated fibrin(ogen) deposits in the liver. 18-mer-HP and 18-mer-AXa have decreased the concentration of firbrin(ogen) at 24 hours (#P = 0.01 and ^P = 0.02, respectively) and 48 hours (#P = 0.05 and ^P = 0.02). n = 3 for all groups at all time points. Data represent means ± SEM. One-way ANOVA was followed by Dunnett’s posttest.
We tested whether the lack of liver regeneration in 18-mer-AXa—treated group is attributed to reduction in fibrin accumulation at the injury site. Kopec and colleagues (27) reported that fibrin (ogen) is required to initiate liver regeneration after APAP overdose in mice. A lower amount of fibrin(ogen) deposition was observed in the livers from 18-mer-AXa—treated mice compared to the APAP group (Fig. 5D), despite both groups suffering from severe liver damage (Fig. 5A). The amount of fibrin(ogen) in the 18-mer-AXa– treated group was 2.3-fold lower at 24 hours and 2.8-fold lower at 48 hours compared to the APAP group (Fig. 5D and fig. S18; ^P = 0.0209 and ^P = 0.0212 at 24 and 48 hours, respectively). Like 18-mer-AXa, anticoagulant unfractionated heparin also lacked hepatoprotection after APAP overdose (fig. S19).
Endogenous syndecan-1 is shed after APAP overdose
We next investigated the potential role of syndecan-1, a common HS proteoglycan found on hepatocytes, in response to APAP overdose. Syndecan-1 consists of a core protein attached with HS chains and is shed from the cell surface by matrix metalloproteases under pathological conditions (9). A recent report demonstrated that Syndecan-1−/− mice were highly sensitive to liver damage caused by APAP toxicity (20). The authors also demonstrated that purified syndecan-1 and HS polysaccharide, but not the core protein of syndecan-1, rendered protection in Syndecan-1−/− mice after APAP overdose. Furthermore, they reported that syndecan-1 also displayed protection in APAP overdose WT mice (20). Although the authors did not show that HMGB1 binds to syndecan-1, their data offered strong evidence for the functional role of syndecan-1 in protecting against APAP-induced liver injury.
After APAP overdose in mice, we observed a rapid increase of plasma syndecan-1 ectodomain as early as 3 hours, which coincided with the increased plasma concentration of HMGB1 (Fig. 6A). Loss of cell surface syndecan-1 was confirmed by immunostaining liver sections (fig. S20). Treatment with 18-mer-HP reduced shed syndecan-1 and plasma HMGB1 concentrations (Fig. 6A). We also observed an increase in plasma syndecan-1 concentrations in patients with APAP-induced ALF (Fig. 6B). Shed syndecan-1 concentrations in patients with APAP-ALF were ~200-fold higher than in APAP overdose mice as measured by core protein analysis. HS chain analysis confirmed the elevated amounts of shed syndecan-1 in patients with ALF (fig. S21A). HMGB1 and ALT were also higher in patients with APAP-ALF than in the healthy control group (fig. S21, B and C), consistent with a previous report (28).
Fig. 6. Endogenous syndecan-1 is shed in response to liver injury after APAP overdose and binds to HMGB1.
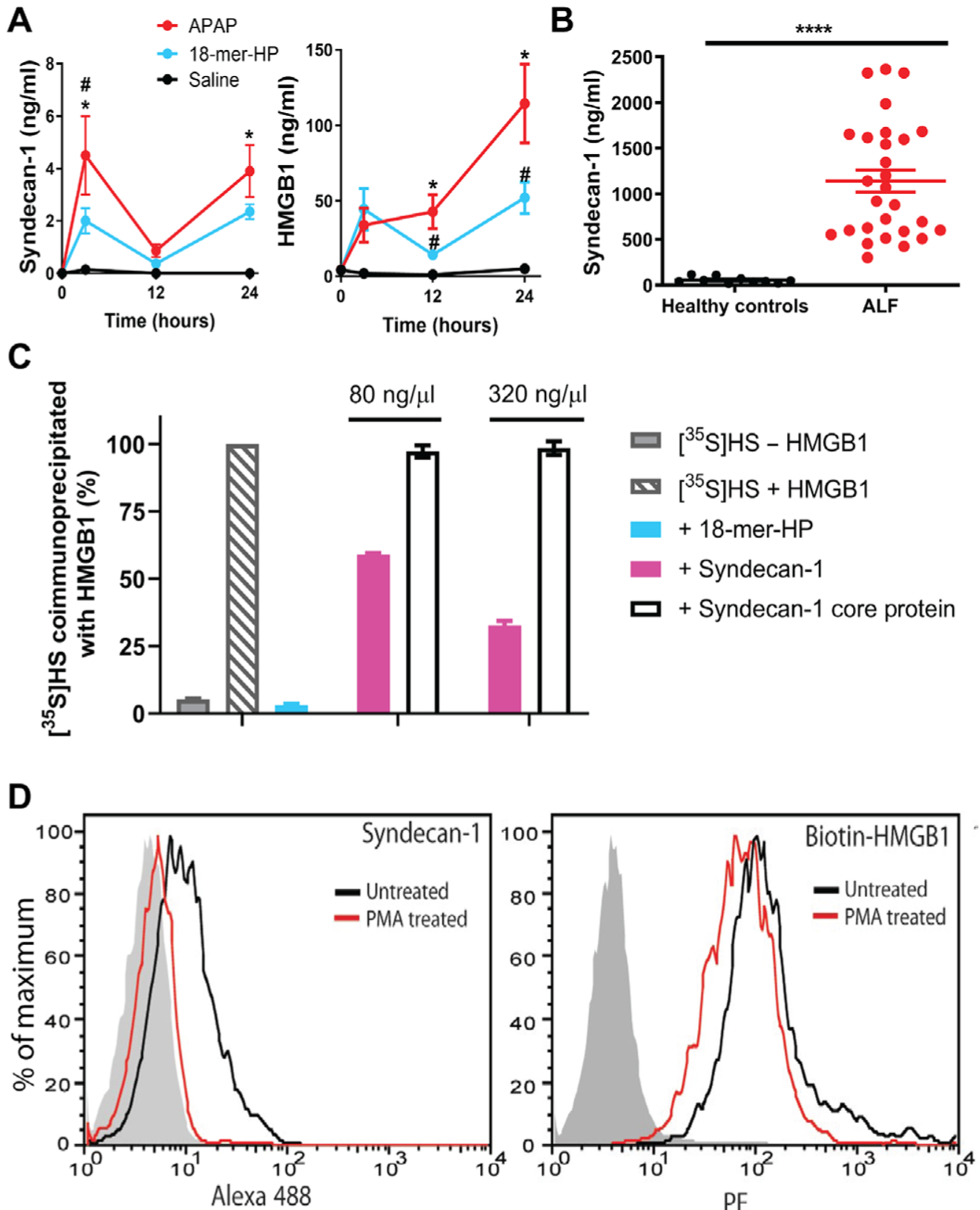
(A) Mouse plasma syndecan-1 and HMGB1 concentrations after APAP overdose with or without 18-mer-HP treatment or only saline as a control. At 3 hours after APAP overdose, plasma syndecan-1 concentrations were elevated in APAP mice compared to saline (*P = 0.0152) and 18-mer-—treated mice (#P = 0.0444). Plasma syndecan-1 concentrations decreased at 12 hours and then rose again at 24 hours in the APAP group compared to saline (*P = 0.0215). Plasma HMGB1 concentrations were elevated in the APAP group at 12 (*P = 0.0354 versus saline, #P = 0.0354 versus 18-mer-HP) and 24 hours (*P = 0.0169 versus saline, #P = 0.0306 versus 18-mer); n = 2 to 3 for saline, n = 5 to 6 for APAP, and n = 5 to 7 for 18-mer-HP at all time points. (B) Plasma syndecan-1 in healthy individuals and in patients with APAP-ALF. (C) HMGB1 binding competition assay using [35S]HS with competitor ligands 18-mer-HP, syndecan-1, and syndecan-1 core protein. (D) Syndecan-1 shedding reduces the binding of HMGB1 to Hep3B cells. Left: PMA treatment induces syndecan-1 shedding. Right: Binding of HMGB1 to Hep3B cells before and after syndecan-1 shedding. The shaded histogram is from cells stained with isotype control rat IgG and secondary antibody (left) or with streptavidin—R-phycoerythrin (PE) only (right). Data represent means ± SEM. One-way ANOVA was followed by Sidak’s post hoc test (A) and by unpaired Student’s t test (B).
We sought to determine whether syndecan-1 directly interacts with HMGB1 and whether the interaction involves HS chains. First, we demonstrated that purified syndecan-1 competes with the binding of 35S-labeled HS to HMGB1 in an immunoprecipitation assay in a dose-dependent manner (Fig. 6C). We also demonstrated that syndecan-1 core protein, which lacks HS polysaccharide side chains, is unable to compete for the binding of 35S-labeled HS to recombinant HMGB1. These results suggest that shed syndecan-1 binds to HMGB1 through its HS chains. In addition, the IC50 values determined from the coimmunoprecipitation assay were ~500 to 550 nM for syndecan-1 and 658 nM for 18-mer-HP, suggesting that syndecan-1 and 18-mer-HP have similar binding affinities for HMGB1. We demonstrated that purified syndecan-1 has no anti-FXa activity (fig. S22) and thereby no anticoagulant activity.
We next demonstrated that hepatocyte cell surface syndecan-1 binds to HMGB1, and the shedding process reduces binding of HMGB1 to the human hepatocyte cell line Hep3B. When the Hep3B cells are treated with phorbol 12-myristate 13-acetate (PMA), it induced syndecan-1 shedding as previously reported (29). About 85% of syndecan-1 was shed from hepatocyte surface after the PMA treatment (Fig. 6D, left). In addition, binding of biotinylated HMGB1 was reduced by ~50% compared to untreated cells after syndecan-1 shed (Fig. 6D, right; relative fluorescence unit = 67 versus 132).
Delayed treatment with 18-mer-HP is protective
N-acetyl cysteine (NAC) is a NAPQI-neutralizing antioxidant that is the standard of care for the treatment of APAP overdose. However, NAC treatment is only effective if given within 8 hours after APAP ingestion in patients (30). Because sterile inflammation occurs after NAPQI-induced cell damage, we examined the possibility of delaying 18-mer-HP treatment after APAP toxicity. In the mouse model, we examined the efficacy of delayed treatment with 18-mer-HP at 3-, 6-, and 9-hour time points after a sublethal dose of APAP (400 mg/kg) and compared the protection with NAC (Fig. 7A). NAC lost its protective effect when administered at 3 and 6 hours after APAP in mice, whereas the 18-mer-HP was still able to decrease ALT. However, 18-mer-HP lost its protection at the 9-hour point (Fig. 7B). The number of infiltrating neutrophils was increased 13-fold from 3 to 12 hours after APAP overdose, suggesting that there is a rapid increase in neutrophil infiltration during this time window (fig. S23A). Extensive damage had already occurred at 12 hours after APAP intoxication, as measured by ALT concentrations (fig. S23B).
Fig. 7. Delayed treatment with 18-mer-HP after APAP overdose remains effective.
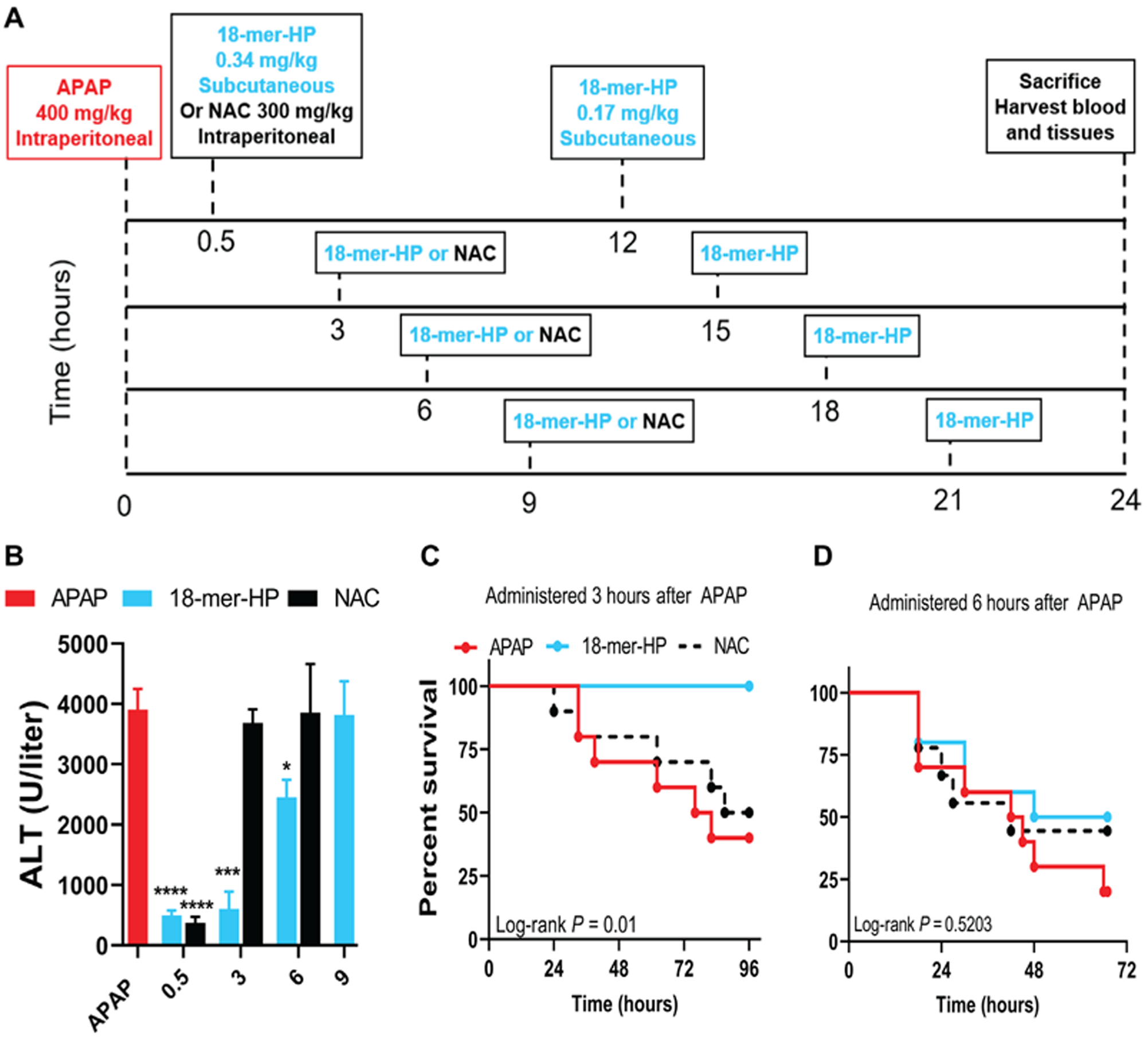
(A) Murine model experimental design schematic including time, dose, and route of administration of APAP, NAC, and 18-mer-HP. All groups received APAP at time 0 and were euthanized at 24 hours. The dosing remained the same in all 18-mer-HP—treated groups in that the first injection was 18-mer-HP (0.34 mg/kg), and the second injection was 18-mer-HP (0.17 mg/kg). NAC (300 mg/kg) was injected once in the indicated groups because of the known mechanism of action and was not used in the 9-hour delay group because no protection was seen at 3 and 6 hours. (B)18-mer-HP decreased ALT when given at 0.5, 3, and 6 hours after APAP (APAP, n = 24; 18-mer, n = 5 to 7; NAC, n = 6 to 9). Data represent means ± SEM. *P < 0.05, ***P < 0.001, and ****P < 0.0001 by two-way ANOVA followed by Tukey’s post hoc test. (C) 18-mer-HP administered 3 hours after APAP (600 mg/kg) increased the survival rate compared to both APAP and NAC given at 3 hours after APAP. (n = 10 for all groups). (D) 18-mer-HP administered 6 hours after APAP (600 mg/kg) had no protective effect (18-mer-HP, n = 10; NAC, n = 9; APAP, n = 10).
We conducted survival studies for 3-and 6-hour delayed treatment with 18-mer-HP or NAC after a lethal dose of APAP (600 mg/kg) (Fig. 7, C and D). We found that delayed treatment with 18-mer-HP administered at 3 hours after APAP injection was fully protective, whereas NAC was unable to provide any protection compared to untreated mice. However, at the 6-hour time point, 18-mer-HP did not improve survival. Overall, our data suggest that 18-mer-HP treatment has a potential advantage for late-presenting patients with APAP overdose by providing a wider therapeutic window than NAC.
DISCUSSION
Here, we describe the use of synthetic HS to treat APAP-induced ALF by targeting HMGB1-mediated sterile inflammation. Previous studies have shown that heterogeneous heparin derivatives protect mice from ALF, but the underlying mechanism has been unclear (20, 31). The availability of homogeneous oligosaccharides has made it possible to identify candidate targets underlying this hepatoprotective effect. In addition to neutralizing HMGB1, HS can also activate liver repair (20) and modulate the activity of chemokines (32). Although 18-mer-HP could also contribute to these functions, our data suggest that 18-mer-HP’s neutralization of HMGB1/RAGE-mediated damage is a critical factor for hepatoprotection in the APAP model.
Heparin is a widely used anticoagulant. Heparin derivatives display a wide range of therapeutic potential and are well tolerated (33). Treatments for cancer and sickle cell disease using chemically modified heparin are currently under investigation in clinical trials (34). The use of homogeneous oligosaccharides allowed us to increase the selectivity of binding to HMGB1 while eliminating off-target effects. Although a humanized HMGB1-neutralizing antibody was successfully used to treat APAP-induced liver injury in mice (35), it has not been approved to treat APAP-induced liver injury in humans. Small-molecule HMGB1 inhibitors are also available (36). For example, glycyrrhizin is protective, but only as a prophylactic agent (4), casting doubt on its therapeutic utility for the treatment of APAP overdose. Finding a safe and effective HMGB1 inhibitor to treat APAP-induced ALF is an ongoing clinical priority.
APAP-induced liver injury activates coagulation, resulting in fibrin formation, which is required to begin the liver regeneration process (27). The lack of hepatoprotection from 18-mer-AXa is due to its anticoagulant activity that disrupts the fibrin formation at the injury site. Mice treated with 18-mer-HP also displayed a decrease in fibrin(ogen) formation compared to the APAP group. However, this fibrin(ogen) reduction was expected because there was less liver damage in 18-mer-HP—treated group compared to APAP group.
The relationship between syndecan-1 shedding and HMGB1 release in humans and mice after APAP overdose underscores an endogenous protective pathway. When syndecan-1 is shed from hepatocytes, soluble syndecan-1 may attenuate sterile inflammation by displacing extracellular HMGB1 from the injury sites and/or interacting with circulating HMGB1. We discovered that 18-mer-HP displaces extracellular HMGB1 from necrotic liver tissue in APAP overdose mice and binds to plasma HMGB1. During extensive liver damage, it is likely that shed syndecan-1 is inadequate to neutralize all HMGB1, and the addition of 18-mer-HP, a mimetic of HS on syndecan-1, provides further protection. Direct evidence from an in vivo model to prove that shed syndecan-1 displays protection through HMGB1/RAGE is still lacking at present. However, one plausible hypothesis is that the 18-mer-HP potentiates the host anti-inflammatory effect mediated by syndecan-1.
Our study provides a potential approach to treat late-presenting patients with APAP overdose; however, the outcome of delayed treatment with 18-mer-HP depends on the extent of damage. In mice, extensive liver damage was detected at the 12-hour time point, explaining why only partial protection was observed 6 hours after APAP overdose. Therefore,18-mer-HP should be administered when damage to the liver is below a certain threshold to display its full protective effect. The time frame of effective delayed treatment with 18-mer-HP remains to be determined in patients.
Chemoenzymatic synthesis has greatly reduced production costs for homogeneous HS oligosaccharides. A similar 12-mer compound is now synthesized in gram scale using the chemoenzymatic method at reasonable costs (19), and the synthetic scale is about 100,000-fold larger than the original synthesis first reported 8 years ago (16). Additional efforts are underway to further improve the chemoenzymatic synthesis. Because HMGB1 has been implicated in diverse disease states including cancers, stroke, arthritis (37), and sepsis (38), HS-based therapeutics deserve further studies as inhibitors of HMGB1.
MATERIALS AND METHODS
Study design
This study was designed to synthesize a HS octadecasaccharide (18-mer-HP) and evaluate its anti-inflammatory effect in an APAP-induced liver failure murine model. We demonstrate the chemoenzymatic synthesis of two HS 18-mers, including 18-mer-HP (hepatoprotective compound) and 18-mer-AXa (anticoagulant compound). The structures of two 18-mers were confirmed using both NMR and high resolution MS. Demonstration of the binding between HMGB1 and biotinylated 18-mer-HP and 18-mer-AXa was achieved using an avidin-agarose column, followed by Western analysis. The binding affinity between HMGB1 and 18-mer-HP or 18-mer-AXa was determined by SPR. The hepatoprotective effect of the synthesized 18-mers was evaluated in an APAP-induced liver failure murine model. The liver damage was assessed by two methods, including plasma ALT concentration and examination of hematoxylin and eosin—stained liver sections. The inflammation responses after APAP overdose were assessed by determining hepatic neutrophil infiltration. FXa activity was used as a surrogate to assess the anticoagulant activity of 18-mer-HP, 18-mer-AXa, 12-mer, and 6-mer in vitro and in vivo. The murine studies were not blinded. Animals were randomly assigned to the control and treated groups. The number of animals in each experiment was determined on the basis of previous experiences. Anonymous patient ALF plasma samples were obtained from the Acute Liver Failure Study Group (ALFSG) biorepository to determine the plasma concentration of shed syndecan-1. The analyses were stopped after analyzing 31 patient samples because a clear statistical difference between healthy control group (n = 11) and patients with ALF was observed.
Mouse model of APAP liver injury
All animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of University of North Carolina at Chapel Hill (Chapel Hill, NC) and the IACUC of the University at Buffalo (Buffalo, NY). Ager−/− mice were originally gifted by A. Bierhaus (University of Heidelberg, Heidelberg, Germany) (39). C57BL/6 J or Ager−/− mice were fasted overnight (12 to 15 hours) to deplete GSH stores before APAP (Sigma) administration. Fresh APAP was dissolved in warm (~50°C) sterile 0.9% sodium chloride solution (sterile saline), cooled to 37°C, and injected intraperitoneally at 400 or 600 mg/kg. The plasma samples were collected 24 hours after APAP administrations for measuring ALT concentration. In general, the concentration of ALT reached its highest point at 24 hours after APAP overdoes. There was a decrease in the plasma concentration of ALT after 24 hours, which is characteristic of the model. In some experiments, mice were injected subcutaneously with 9.5 μM HS oligosaccharide in ~200 μl of sterile saline at 30 min after APAP and followed by injections every 12 hours with 4.75 μM HS oligosaccharide in ~200 μl or equivalent volumes of sterile saline until the end of the experiment (either 24 or 120 hours).
To compare the 18-mer-HP’s effectiveness to NAC, NAC (300 mg/kg) at pH 7.5 in sterile saline was injected intraperitoneally 30 min after APAP. In a separate experiment, NAC (300 mg/kg) was administered at 3 or 6 hours after APAP. In the 18-mer-HP—treated groups, 18-mer-HP (0.34 mg/kg; 9.5 μM in 200 μl) was administered 3, 6, or 9 hours after APAP, with a second dose of 18-mer-HP (0.17 mg/kg; 4.75 μM in 200 μl) 12 hours later. At 24 hours after APAP, mice were euthanized, and blood and liver tissue were collected.
In the survival study [APAP (600 mg/kg)], C57Bl/6 J mice were injected 30 min after APAP with 18-mer-HP (0.4 mg/kg), followed by repeat injections every 12 hours or equivalent volumes of sterile saline for 96 hours. Ager−/− mice were subjected to the same APAP (600 mg/kg) treatment. In some experiments, 18-mer-HP (0.4 mg/kg) or NAC (300 mg/kg) was administered at 3 or 6 hours after APAP (600 mg/kg). 18-mer-HP treatment or equivalent volumes of sterile saline were repeated every 12 hours for 96 hours or until 20% survival was reached in any group.
Evaluation of APAP-induced liver injury
Plasma ALT was measured using the ALT Infinity reagent (Thermo Fisher Scientific) following the manufacturer’s instructions. Plasma TNF-α was measured using Mouse TNF-α DuoSet Kit (R&D Systems) according to the manufacturer’s instructions. Plasma HMGB1 concentrations were determined using the HMGB1 ELISA Kit (Tecan US) according to the manufacturer’s instructions. Plasma syndecan-1 concentrations were determined using Mouse Syndecan-1 ELISA (Cell Sciences) according to the manufacturer’s instructions. Hepatic GSH concentrations were determined using the Glutathione Assay Kit (Cayman Chemicals) according to the manufacturer’s instructions.
Mouse models of inflammation: Peritonitis and air pouch
We used two models to study in vivo neutrophil migration: peritonitis and an air pouch inflammation model. For the peritonitis model, 30 mg of liver lysate (see below for preparation of lysate) was injected into the peritoneal cavity of the mouse in the absence or presence of 20 μg of 18-mer-HP. After 20 hours, the mice were euthanized via inhalation of isoflurane, and the peritoneal cavity was washed with 10 ml of ice-cold PBS. The peritoneal lavage was used to determine neutrophil migration using flow cytometry.
In the air pouch technique, 3 ml of sterile air was injected under the skin on the back of the mouse. Three days later, the pouch was refilled with sterile air. On the sixth day, 5 μg of recombinant HMGB1 in the absence or presence of 22.3 μg of 18-mer-HP was injected into the air pouch. Control mice were injected with bovine serum albumin (2 mg/ml) in PBS. After 4 hours, mice were euthanized by isoflurane inhalation, and the air pouch was washed with PBS only. The lavage was used to determine neutrophil migration using flow cytometry.
HMGB1-neutralizing antibody in APAP mouse model
Anti-HMGB1 chicken immunoglobulin Y (IgY)—neutralizing polyclonal antibody (100 μg) (Tecan) per mouse was intraperitoneally administered 2 hours after APAP (400 mg/kg). As a control, 100 μg of anti-IgY antibody (Tecan) per mouse was intraperitoneally administered 2 hours after APAP (400 mg/kg). In some groups, 18-mer-HP (0.34 mg/kg) was subcutaneously injected 30 min after APAP, and then, anti—HMGB1-neutralizing antibody or anti-IgY antibody was administered 2 hours after APAP. 18-mer-HP—treated mice received a second dose of 18-mer-HP (0.17 mg/kg) at 12 hours after APAP. All mice were euthanized at 24 hours, and blood was drawn for analysis.
APAP-ALF patient plasma analysis
APAP-ALF patient plasma, containing EDTA (1.8 mg/ml), was obtained from the ALFSG biorepository. Details on the study design and collection methods were described previously (40). Briefly, starting in 1998, adult patients who met the inclusion and exclusion criteria were enrolled in the ALFSG registry. Plasma samples were obtained on admission to the registry. In our study, we only analyzed plasma from patients who overdosed on APAP. Clinical data, including the estimated quantity of APAP ingested, estimated time from ingestion to hospitalization, intentionality of overdose, and patient demographics (age, gender, race, comorbidities, etc.), were not revealed for this study. Plasma syndecan-1 concentrations were measured using an enzyme-linked immunosorbent assay (ELISA) kit (Human Syndecan-1 ELISA, Cell Sciences) according to the manufacturer’s protocol. Plasma HMGB1 concentrations were measured using an HMGB1 ELISA according to the manufacturer’s protocol. Plasma ALT concentrations were measured using ALT Infinity Reagent.
Statistical analysis
All data are expressed as means ± SEM. Statistical significance of differences between experimental and control groups was analyzed by two-tailed unpaired Student’s t test, between multiple groups by one-way analysis of variance (ANOVA) followed by Dunnett’s or Tukey’s multiple comparisons test, and Kaplan-Meier survival curves by log-rank test using GraphPad Prism software (version 7.03; GraphPad Software Inc.). Original data are provided in data file S1.
Supplementary Material
Data file S1. Original data.
References (43–46)
Fig. S1. Scheme for the chemoenzymatic synthesis of 18-mer-HP.
Fig. S2. DEAE-HPLC chromatogram and high-resolution MS spectrum of 18-mer-HP.
Fig. S3. Structure characterization of 18-mer-HP by NMR.
Fig. S4. Structure characterization of 18-mer-HP by 2D NMR.
Fig. S5. Neutrophil infiltration with or without 18-mer-HP treatment in a peritonitis model.
Fig. S6. MS characterization of unmodified recombinant HMGB1.
Fig. S7. MS characterization of recombinant HMGB1 after iodoacetamide modification.
Fig. S8. Recombinant HMGB1 characterization by SDS-polyacrylamide gel electrophoresis.
Fig. S9. 18-mer-HP does not inhibit macrophage activation by HMGB1.
Fig. S10. Chemical structures of biotinylated and nonbiotinylated oligosaccharides: 6-mer, 12-mer, 18-mer-HP, and 18-mer-AXa.
Fig. S11. Full lanes of Western blot analysis from endogenous HMGB1 pulldown and preparation of plasma HMGB1.
Fig. S12. DEAE-HPLC chromatogram and high-resolution MS spectrum of 18-mer-AXa.
Fig. S13. Structure characterization of 18-mer-AXa by NMR.
Fig. S14. Structure characterization of 18-mer-AXa by 2D NMR.
Fig. S15. Full lanes of Western blot analysis of HMGB1 affinity-purified from liver lysate using biotinylated 18-mer-HP and 18-mer-AXa.
Fig. S16. Neutrophil infiltration into liver tissue after APAP overdose determined at 24, 48, 72, and 120 hours after APAP overdose in mice.
Fig. S17. Hepatocyte proliferation determined by Ki-67-positive hepatocyte staining at 24, 48, 72, and 120 hours after APAP overdose in mice.
Fig. S18. Fibrin(ogen) deposition in liver tissue determined at 24, 48, 72, and 120 hours after APAP overdose in mice.
Fig. S19. Unfractionated heparin does not display hepatoprotection.
Fig. S20. APAP overdose results in shed syndecan-1 from the hepatocyte surface.
Fig. S21. Analysis of syndecan-1 and the plasma concentration of ALT and HMGB1 in patients with ALF.
Fig. S22. Determination of the anticoagulant activity of syndecan-1.
Fig. S23. Hepatic neutrophil numbers in the liver and ALT after APAP overdose increase over time.
Table S1. 1H NMR assignment for 18-mer-HP.
Table S2. 1H NMR assignment for 18-mer-AXa in parts per million.
Table S3. Summary of kinetic data from SPR analysis with recombinant HMGB1 and reduced and disulfide HMGB1 with 18-mer-AXa and 18-mer-HP.
Acknowledgments:
We thank R. Sanderson (UAB) for providing anti-mouse syndecan-1 antibody, A. Bierhaus (University of Heidelberg, Heidelberg, Germany) for providing Ager−/− mice, and D. Noubouossie for critically reviewing the manuscript.
Funding: This work is supported by grants from NIH (GM102137 to J.L., HL094463 to J.L., GM128484 to Y.X., and AR070179 to D.X.) and Eshelman Innovation Institute. K.A. is a recipient of predoctoral fellowships from Pharmaceutical Research and Manufacturers of America Foundation and from American Foundation for Pharmaceutical Education. R.J.L.’s lab received funding from three NIH grants: DK111958 [to K. J. McCarthy, principal investigator (PI)], HL125371 (to E. P. Schmidt, PI), and CA207717 (to Y. Yamaguchi, PI). Human samples were supplied by ALFSG, which receives funding from the National Institute of Diabetes and Digestive and Kidney Diseases grant U-01 58369 (to W. M. Lee, PI). Additional NIH grants were available to our team during the study: R01 HL144970 (to J.L.), R41 GM123792 (to J.L.), R44 GM134738 (to V.P.), and R44 HL139187 (to V.P.).
Footnotes
Competing interests: J.L. and Y.X. are founders of Glycan Therapeutics, LLC. V.P. is an employee of Glycan Therapeutics, LLC and has the equity. K.A., Y.X., J.L., D.X., and R.P. are inventors for a U.S. patent (US 62/581,443). All other authors declare that they have no competing interests.
Data and materials availability: All data associated with this study are present in the paper or the Supplementary Materials.
REFERENCES AND NOTES
- 1.Zitvogel L, Kepp O, Kroemer G, Decoding cell death signals in inflammation and immunity. Cell 140, 798–804 (2010). [DOI] [PubMed] [Google Scholar]
- 2.Chen GY, Nuñez G, Sterile inflammation: Sensing and reacting to damage. Nat. Immunol 10, 826–837 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bianchi ME, Crippa MP, Manfredi AA, Mezzapelle R, Querini PR, Venereau E, High-mobility group box 1 protein orchestrates responses to tissue damage via inflammation, innate and adaptive immunity, and tissue repair. Immunol. Rev 280, 74–82 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Huebener P, Pradere J-P, Hernandez C, Gwak G-Y, Caviglia JM, Mu X, Loike JD, Schwabe RF, The HMGB1/RAGE axis triggers neutrophil-mediated injury amplification following necrosis. J. Clin. Invest 125, 539–550 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heard KJ, Acetylcystein for acetaminophen poisoning. N. Engl. J. Med 359, 285–292 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee WM, Acetaminophen toxicity: Changing perceptions on a social/medical issue. Hepatology 46, 966–970 (2007). [DOI] [PubMed] [Google Scholar]
- 7.Blieden M, Paramore LC, Shah D, Ben-Joseph R, A perspective on the epidemiology of acetaminophen exposre and toxicity in the United States. Expert. Rev. Clin. Pharmacol 7, 341–348 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Mossanen JC, Tacke F, Acetaminophen-induced acute liver injury in mice. Lab. Anim 49, 30–36 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Park PW, Reizes O, Bernfield M, Cell surface heparan sulfate proteoglycans: Selective regulators of ligand-receptor encounters. J. Biol. Chem 275, 29923–29926 (2000). [DOI] [PubMed] [Google Scholar]
- 10.Proudfoot AE, Handel TM, Johnson Z, Lau EK, LiWang P, Clark-Lewis I, Borlat F, Wells TN, Kosco-Vilbois MH, Glycosaminoglycan binding and oligomerization are essential for the in vivo activity of certain chemokines. Proc. Natl. Acad. Sci. U.S.A 100, 1885–1890 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang L, Fuster M, Sriramarao P, Esko JD, Edothelial heparan sulfate deficiency impairs L-selectin-and chemokine-mediated neutrophil trafficking during inlammatoray responses. Nat. Immunol 6, 902–910 (2005). [DOI] [PubMed] [Google Scholar]
- 12.Axelsson J, Xu D, Kang BN, Nussbacher JK, Handel TM, Ley K, Sriramarao P, Esko JD, Inactivation of heparan sulfate 2-O-sulfotransferase accentuates neutrolphil infiltration during acute inflammation in mice. Blood 120, 1742–1751 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sarris M, Masson J-B, Maurin D, Van der Aa LM, Boudinot P, Lortat-Jacob H, Herbomel P, Inflammatory chemokines direct and restrict leukocyte migration within live tissues as glycan-bound gradients. Curr. Biol 22, 2375–2382 (2012). [DOI] [PubMed] [Google Scholar]
- 14.Gama CI, Tully SE, Sotogaku N, Clark PM, Rawat M, Vaidehi N, Goddard III WA, Nishi A, Hsieh-Wilson LC, Sulfation patterns of glycosaminoglycans encode molecular recognition and activity. Nat. Chem. Biol 2, 467–473 (2006). [DOI] [PubMed] [Google Scholar]
- 15.Xu D, Young J, Song D, Esko JD, Heparan sulfate is essential for high mobility group protein 1 (HMGB1) signaling by the receptor for advanced glycation end products (RAGE). J. Biol. Chem 286, 41736–41744 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu J, Linhardt RJ, Chemoenzymatic synthesis of heparan sulfate and heparin. Nat. Prod. Rep 31, 1676–1685 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu Y, Masuko S, Takieddin M, Xu H, Liu R, Jing J, Mousa SA, Linhardt RJ, Liu J, Chemoenzymatic synthesis of homogeneous ultra-low molecular weight heparin. Science 334, 498–501 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu Y, Cai C, Chandarajoti K, Hsieh P-H, Li L, Pham TQ, Sparkenbaugh EM, Sheng J, Key NS, Pawlinski R, Harris EN, Linhardt RJ, Liu J, Homogeneous and reversible low-molecular weight heparins with reversible anticoagulant activity. Nat. Chem. Biol 10, 248–250 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu Y, Chandarajoti K, Zhang X, Pagadala V, Dou W, Hoppensteadt DM, Sparkenbaugh EM, Cooley B, Daily S, Key NS, Severynse-Stevens D, Fareed J, Linhardt RJ, Pawlinski R, Liu J, Synthetic oligosaccharides can replace animal-sourced low-molecular weight heparins. Sci. Transl. Med 9, eaan5954 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nam EJ, Hayashida K, Aquino RS, Couchman JR, Kozar RA, Liu J, Park PW, Syndecan-1 limits the progression of liver injury and promotes liver repairin acetaminophen-induced liver injury. Hepatology 66, 1601–1615 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Antoine DJ, Williams DP, Kipar A, Laverty H, Park BK, Diet restriction inhibits apoptosis and HMGB1 oxidation and promotes inflammatory cell recruitment during acetaminophen hepatotoxicity. Mol. Med 16, 479–490 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 22.Yang H, Lundbäck P, Ottosson L, Erlandsson-Harris H, Venereau E, Bianchi ME, Al-Abed Y, Andersson U, Tracey KJ, Antoine DJ, Redox modification of cysteine residues regulates the cytokine activity of high mobility group box-1 (HMGB1). Mol. Med 18, 250–259 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 23.Venereau E, Casalgrandi M, Schiraldi M, Antoine DJ, Cattaneo A, De Marchis F, Liu J, Antonelli A, Preti A, Raeli L, Shams SS, Yang H, Varani L, Andersson U, Tracey KJ, Bachi A, Uguccioni M, Bianchi ME, Mutually exclusive redox forms of HMGB1 promote cell recruitement of proinflammatory cytokin release. J. Exp. Med 209, 1519–1528 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tirone M, Tran NL, Ceriotti C, Gorzanelli A, Canepari M, Bottinelli R, Raucci A, Di Maggio S, Santiago C, Mellado M, Saclier M, François S, Careccia G, He M, De Marchis F, Conti V, Ben Larbi S, Cuvellier S, Casalgrandi M, Preti A, Chazaud B, Al-Abed Y, Messina G, Sitia G, Brunelli S, Bianchi ME, Vénéreau E, High mobility group box 1 orchestrates tissue regeneration via CXCR4. J. Exp. Med 215, 303–318 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang H, Hreggvidsdottir HS, Palmblad K, Wang H, Ochani M, Li J, Lu B, Chavan S, Rosas-Ballina M, Al-Abed Y, Akira S, Bierhaus A, Erlandsson-Harris H, Andersson U, Tracey KJ, A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. Proc. Natl. Acad. Sci. U.S.A 107, 11942–11947 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee G, Espirito Santo AI, Zwingenberger S, Cai L, Vogl T, Feldmann M, Horwood NJ, Chan JK, Nanchahal J, Fully reduced HMGB1 accelerates the regeneration of multiple tissues by transitioning stem cells to Galert. Proc. Nati. Acad. Sci. U.S.A 115, E4463–E4472 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kopec AK, Joshi N, Cline-Fedewa H, Wojcicki AV, Ray JL, Sullivan BP, Froehlich JE, Johnson BF, Flick MJ, Luyendyk JP, Fibrin(ogen) drives repair after acetaminophen-induced liver injury via leukocyte aMb2 integrin-dependent upregulation of Mmp12. J. Hepatol 66, 787–797 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Antoine DJ, Dear JW, Lewis PS, Platt V, Coyle J, Masson M, Thanacoody RH, Gray AJ, Webb DJ, Moggs JG, Bateman DN, Goldring CE, Park BK, Mechanistic biomarkers provide early and sensitive detection of acetaminophen-induced acute liver injury at first presentation to hospital. Hepatology 58, 777–787 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Deng Y, Foley EM, Gonzales JC, Gordts PL, Li Y, Esko JD, Shedding of syndecan-1 from human hepatocytes alters very low density lipoprotein clearance. Hepatology 55, 277–286 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bailey GP, Najafi J, Elamin ME, Waring WS, Thomas SH, Archer JR, Wood DM, Dargan PI, Delays during the administration of acetylcysteine for the treatment of paraacetamol overdose. Br. J. Clin. Pharmacol 62, 1358–1363 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wildhagen KCAA, García de Frutos P, Reutelingsperger CP, Schrijver R, Aresté C, Ortega-Gómez A, Deckers NM, Hemker HC, Soehnlein O, Nicolaes GAF, Nonanticoagulant heparin prevents histone-mediated cytotoxicity in vitro and improves survival in sepsis. Blood 123, 1098–1101 (2014). [DOI] [PubMed] [Google Scholar]
- 32.Monneau Y, Arenzana-Seisdedos F, Lortat-Jacob H, The sweet spot: How GAGs help chemokines guide migrating cells. J. Leukoc. Biol 99, 935–953 (2016). [DOI] [PubMed] [Google Scholar]
- 33.Shriver Z, Raguram S, Sasisekharan R, Glycomics: A pathway to a class of new and improved therapeutics. Nat. Rev. Drug Discov 3, 863–873 (2004). [DOI] [PubMed] [Google Scholar]
- 34.Paderi J, Prestwich GD, Panitch A, Boone T, Stuart K, Glycan therapeutics: Resurrecting an almost pharma-forgotten drug class. Adv. Ther 1, 1800082 (2018). [Google Scholar]
- 35.Lundbäck P, Lea JD, Sowinska A, Ottosson L, Fürst CM, Steen J, Aulin C, Clarke JI, Kipar A, Klevenvall L, Yang H, Palmblad K, Park BK, Tracey KJ, Blom AM, Andersson U, Antoine DJ, Harris HE, A novel high mobility group box 1 neutralizing chimeric antibody attenuates drug-induced liver injury and postinjury inflammationin mice. Hepatology 64, 1699–1710 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yamamoto T, Tajima Y, HMGB1 is a promising therapeutic target for acute liver failure. Expert Rev. Gatroenterol. Hepatol 11, 673–682 (2017). [DOI] [PubMed] [Google Scholar]
- 37.Venereau E, De Leo F, Mezzapelle R, Careccia G, Musco G, Bianchi ME, HMGB1 as biomarker and drug target. Pharmacol. Res 111, 534–544 (2016). [DOI] [PubMed] [Google Scholar]
- 38.Yang H, Liu H, Zeng Q, Imperato GH, Addorisio ME, Li J, He M, Cheng KF, Al-Abed Y, Harris HE, Chavan SS, Andersson U, Tracey KJ, Inhibition of HMGB1/RAGE-mediated endocytosis by HMGB1 antagonist box A, anti-HMGB1 antibodies, and cholinergic agonists suppresses inflammation. Mol. Med 25, 13 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liliensiek B, Weigand MA, Bierhaus A, Nicklas W, Kasper M, Hofer S, Plachky J, Gröne HJ, Kurschus FC, Schmidt AM, Yan SD, Martin E, Schleicher E, Stern DM, Gü Hämmerling G, Nawroth PP, Arnold B, Receptor for advanced glycation end products (RAGE) regulates sepsis but not the adaptive immune response. J. Clin. Invest 113, 1641–1650 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reuben A, Tillman H, Fontana RJ, Davern T, McGuire B, Stravitz RT, Durkalski V, Larson AM, Liou I, Fix O, Schilsky M, McCashland T, Hay JE, Murray N, Shaikh OS, Ganger D, Zaman A, Han SB, Chung RT, Smith A, Brown R, Crippin J, Harrison ME, Koch D, Munoz S, Reddy KR, Rossaro L, Satyanarayana R, Hassanein T, Hanje AJ, Olson J, Subramanian R, Karvellas C, Hameed B, Sherker AH, Robuck P, Lee WM, Outcomes in adults with acute liver failure between 1998 and 2013: An observational cohort study. Ann. Intern. Med 164, 724–732 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Willis WL, Wang L, Wada TT, Gardner M, Abdouni O, Hampton J, Valiente G, Young N, Ardoin S, Agarwal S, Freitas MA, Wu L-C, Jarjour WN, The proinflammatory protein HMGB1 is a substrate of transglutaminase-2 and forms high-molecular weight complexes with autoantigens. J. Biol. Chem 293, 8394–8409 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang Z, Collighan RJ, Pytel K, Rathbone DL, Li X, Griffin M, Characterizationof Heparin-binding Site of Tissue Transglutaminase ITS IMPORTANCE IN CELL SURFACE TARGETING, MATRIX DEPOSITION, AND CELL SIGNALING. J. Biol. Chem 287, 13063–13083 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu R, Collighan RJ, Pytel K, Rathbone DL, Li X, Griffin M, Chemoenzymatic design of heparan sulfate oligosaccharides. J. Biol. Chem 285, 34240–34249 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu D, Moon A, Song D, Pedersen LC, Liu J, Engineering sulfotransferases to modify heparan sulfate. Nat. Chem. Biol 4, 200–202 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hsieh P-H, Xu Y, Keire DA, Liu J, Chemoenzymatic synthesis and structural characterization of 2-O-sulfated glucuronic acid containing heparan sulfate hexasaccharides. Glycobiology 24, 681–692 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Park PW, Isolation and functional analysis of syndecans. Methods Cell Biol. 143, 317–333 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data file S1. Original data.
References (43–46)
Fig. S1. Scheme for the chemoenzymatic synthesis of 18-mer-HP.
Fig. S2. DEAE-HPLC chromatogram and high-resolution MS spectrum of 18-mer-HP.
Fig. S3. Structure characterization of 18-mer-HP by NMR.
Fig. S4. Structure characterization of 18-mer-HP by 2D NMR.
Fig. S5. Neutrophil infiltration with or without 18-mer-HP treatment in a peritonitis model.
Fig. S6. MS characterization of unmodified recombinant HMGB1.
Fig. S7. MS characterization of recombinant HMGB1 after iodoacetamide modification.
Fig. S8. Recombinant HMGB1 characterization by SDS-polyacrylamide gel electrophoresis.
Fig. S9. 18-mer-HP does not inhibit macrophage activation by HMGB1.
Fig. S10. Chemical structures of biotinylated and nonbiotinylated oligosaccharides: 6-mer, 12-mer, 18-mer-HP, and 18-mer-AXa.
Fig. S11. Full lanes of Western blot analysis from endogenous HMGB1 pulldown and preparation of plasma HMGB1.
Fig. S12. DEAE-HPLC chromatogram and high-resolution MS spectrum of 18-mer-AXa.
Fig. S13. Structure characterization of 18-mer-AXa by NMR.
Fig. S14. Structure characterization of 18-mer-AXa by 2D NMR.
Fig. S15. Full lanes of Western blot analysis of HMGB1 affinity-purified from liver lysate using biotinylated 18-mer-HP and 18-mer-AXa.
Fig. S16. Neutrophil infiltration into liver tissue after APAP overdose determined at 24, 48, 72, and 120 hours after APAP overdose in mice.
Fig. S17. Hepatocyte proliferation determined by Ki-67-positive hepatocyte staining at 24, 48, 72, and 120 hours after APAP overdose in mice.
Fig. S18. Fibrin(ogen) deposition in liver tissue determined at 24, 48, 72, and 120 hours after APAP overdose in mice.
Fig. S19. Unfractionated heparin does not display hepatoprotection.
Fig. S20. APAP overdose results in shed syndecan-1 from the hepatocyte surface.
Fig. S21. Analysis of syndecan-1 and the plasma concentration of ALT and HMGB1 in patients with ALF.
Fig. S22. Determination of the anticoagulant activity of syndecan-1.
Fig. S23. Hepatic neutrophil numbers in the liver and ALT after APAP overdose increase over time.
Table S1. 1H NMR assignment for 18-mer-HP.
Table S2. 1H NMR assignment for 18-mer-AXa in parts per million.
Table S3. Summary of kinetic data from SPR analysis with recombinant HMGB1 and reduced and disulfide HMGB1 with 18-mer-AXa and 18-mer-HP.