

Abstract
The green and environmentally friendly synthesis of highly valuable organic substances is one possibility for the utilization of laccases (EC 1.10.3.2). As reactants for the herein described syntheses, different o-substituted arylamines or arylthiols and 2,5-dihydroxybenzoic acid and its derivatives were used. In this way, the formation of phenothiazines, phenoxazines, and phenazines was achieved in aqueous solution mediated by the laccase of Pycnoporus cinnabarinus in the presence of oxygen. Two types of phenothiazines (3-hydroxy- and 3-oxo-phenothiazines) formed in one reaction assay were described for the first time. The cyclization reactions yielded C–N, C–S, or C–O bonds. The syntheses were investigated with regard to the substitution pattern of the reaction partners. Differences in C–S and C–N bond formations without cyclization are discussed.
Introduction
Laccases (EC 1.10.3.2, benzenediol/dioxygen oxidoreductase) are of general interest for biotechnological applications. The enzyme-mediated oxidations demand only atmospheric oxygen and no depletive cofactors such as NADPH. Laccases oxidize substrates by one-electron reactions via four copper atoms, which are situated in the catalytic center.1−4 The enzyme can oxidize a broad range of substrates such as phenols, thiols, or amines.5−11 The laccase-mediated oxidations result in the formation of radicals that can undergo three different reaction mechanisms. The first mechanism involves the cleavage, while the second and third mechanisms involve coupling/bond formation. The oxidized laccase substrate can react with molecules of the same kind (homomolecular reaction) or with other radicals or molecules, which can be either laccase substrates or nonlaccase substrates (heteromolecular reaction).9,12 The homomolecular reaction may result in the formation of C–C,13−15 C=C,16,17 C–O,18,19 N=N,12 or S–S bonds.10,11 The heteromolecular reaction allows the combination of compounds via C–C,20,21 C=C,17 C–O,22 C–N,23−35 C=N,17,23,25 or C–S.7,11,16,36 The bond formations result in the synthesis of di-, tri-, and polymers whereby novel ring-closure mechanisms forming cyclic products are also possible but less explored.7,37−39 These reactions are the basis for the derivatization of biologically active substances such as catechin,40 mithramycin,21 pyrimidines,41 epinephrine (adrenaline),39 azoles,33,42 penicillins, and cephalosporins.27,31
Other important targets for laccase-mediated reactions are phenothiazines, phenoxazines, and phenazines. Phenothiazines such as chlorpromazine are widely used in medicine as neuroleptics.43 Phenothiazines, phenoxazines, and phenazines have anticancer44−46 and antimicrobial47−49 activities. In addition, phenothiazines have been reported to possess multidrug resistance reverting activity.50,51 Phenothiazines are also part of dyes such as methylene blue or act as electron donors in photovoltaic cells.52,53 Phenoxazinones and phenazines can also be used in optoelectronics such as in organic light-emitting diodes.54,55 Chemical methods to synthesize phenothiazines comprise catalyses with transition metals such as copper iodide, iron salt, or palladium in organic solvents and in part at elevated temperatures above 90 °C.56−59 Alternatively, KI or Cs2CO3 in organic solvents can be used.60,61 The phenothiazines can be oxidized to the respective phenothiazinones, e.g., by K2Cr2O7 or Na2Cr2O7 in boiling acetic acid.62 Other biologically active heterocycles such as benzoxazepines can be synthesized in organic solvents at a temperature of 100 °C.63 The successful laccase-mediated synthesis of different substance classes (including in part also isomer formation) in comparison with traditional organic reactions catalyzed by oxidants such as silver oxide, sodium iodate, or manganese dioxide was described previously.24,64−68 This recommends the laccase for various industrial applications.69,70
The utilization of mild and environmentally friendly reaction conditions such as room temperature, atmospheric pressure, and the avoidance of organic solvents makes the laccase-mediated reaction a valuable tool in green chemistry for the synthesis of novel organic compounds as fine chemicals.
The literature of laccase-mediated syntheses is mainly focused on Michael addition reactions. Thus, the reaction of substituted hydroquinones and amines results in mono- or diaminated quinones24,25,32,67 or quinonimines.25,71 The formation of cyclic products may be a cascade reaction involving also Michael addition with subsequent 1,2-addition or addition–elimination reaction. After 1,2-addition, also spiro-cyclization or rearomatization is conceivable.37 Other reactions involve further laccase-mediated oxidations, additional Michael additions, or 1,3-additions.39,41
The introduction of newly synthesized substances that were formed during ring-closure mechanisms is a further possibility to broaden the application field for laccase-mediated reactions. Thus, we derivatized substituted para-hydroquinones using the laccase-mediated reaction with thiols and amines. The formed products were different homo- and heteromolecular products. The cyclization reactions resulted in the synthesis of phenothiazines, phenoxazines, or phenazines.
Phenoxazines and phenazines are, in contrast to phenothiazines, naturally occurring substances. Thus, fungi of the genus Pycnoporus produce orange-red pigments of the phenoxazinone type such as cinnabarin, tramesanguin, or cinnabarinic acid in the fruiting bodies.72,73 Phenoxazines can be synthesized by heating 2-aminophenol and 2-aminophenol hydrochloride in an equimolar mixture.74 Other methods comprise, e.g., potassium or cesium carbonate with copper in organic solvents.75,76 The phenazine pyocyanin is one of the pigments that is produced by most of the strains of the pathogenic bacterium Pseudomonas aeruginosa.77 The chemical production of phenazines may be accomplished, among others, by the Wohl–Aue reaction, which includes heating upon 200 °C, or the Beirut reaction. Transition metals may be used as catalysts similar to the syntheses of phenothiazines and phenoxazines (for reviews, see Laursen and Nielsen78 or Chaudhary and Khurana79).
We developed a one-pot reaction with a nonstoichiometric and environmentally friendly catalyst, which can be performed in an aqueous solution (with less than 5% methanol, which can be easily replaced by solvents such as ethanol80 due to similar characteristics) at room temperature. The reactions were performed in the presence of oxygen, at atmospheric pressure, and at a moderate pH value (pH 5). These characteristics of the introduced laccase-mediated reactions are valuable for the green synthesis of cyclic products as well as dimers and trimers. The use of reaction partners with amino, hydroxyl, and/or thiol groups presented the possibility to form new heterocycles bio-enzymatically.
Results
General Observations
The thiols and amines used in this study consist of a benzene ring with two functional groups in ortho-position to each other. The compounds contain a thiol group and/or amino group. Hydroxyl, amino, methyl, or nitro groups can be second substituents. The thiols and amines were incubated with 2,5-dihydroxybenzoic acid or with one of its derivatives and the laccase of Pycnoporus cinnabarinus. Oxidative C–N bond formation followed by cyclization was the predominant reaction, resulting in phenothiazines, phenoxazines, and phenazines.
All reactions were performed with 0.5 U laccase of P. cinnabarinus as the catalyst in sodium acetate buffer (pH 5) at room temperature with a reactant concentration of 1 mM. The reactions were analyzed in the course of an incubation time of 24 h.
Reactions of 2,5-Dihydroxybenzoic Acid and Its Derivatives
The laccase-mediated reactions of 2,5-dihydroxybenzoic acid (1a), 2,5-dihydroxy-N-(2-hydroxyethyl)benzamide (1b), and 2,5-dihydroxybenzoic acid methylester (1c) may result in the formation of the respective benzoquinone and/or a radical (2, Scheme 1).81 These intermediates can be attacked by water or solvents such as methanol, resulting in hydroxylated and methoxylated benzoquinonoid products.81,82 These side reactions may diminish the yield of products formed with additional coupling partners such as thiols and amines.
Scheme 1. Laccase-Catalyzed Reaction of 2,5-Dihydroxybenzoic Acid Derivatives 1a–1c for the Synthesis of the Respective Benzoquinone (2) Using 0.5 U Laccase of P. cinnabarinus as the Catalyst in Sodium Acetate Buffer (pH 5; Reactant Concentration, 1 mM).

Under the chosen reaction conditions, none of these products were formed in controls (without laccase).
Reactions of 2-Aminothiophenol with 2,5-Dihydroxybenzoic Acid
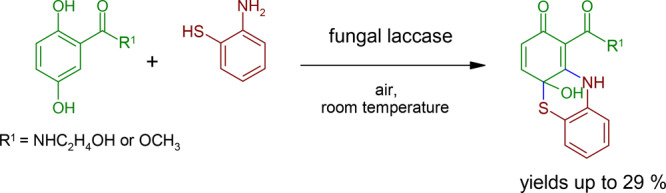
The reactions of 1a–1c with 2-aminothiophenol (3a) resulted in laccase-mediated formation of adducts (4a–4c) and phenothiazines (5a–5c, Scheme 2 and Table 1). The yields of the phenothiazines ranged from 5% (5c) to 29% (5b).
Scheme 2. Laccase-Catalyzed Reaction of 2,5-Dihydroxybenzoic Acid Derivatives 1a–1c with 2-Aminothiophenol 3a for the Synthesis of Adducts 4a–4c and the Cyclization Products 4a-Hydroxy-2-oxo-phenothiazines 5a–5c using 0.5 U Laccase of P. cinnabarinus as the Catalyst in Sodium Acetate Buffer (pH 5; Reactant Concentration, 1 mM).

Table 1. Reactions of 1a–1c with 3a and Formed Products Using 0.5 U Laccase of P. cinnabarinus as the Catalyst in Sodium Acetate Buffer (pH 5) within an Incubation Time of 24 h (Reactant Concentration, 1 mM).
| reactant 1 | R1 | reactant 3 | product | yielda |
|---|---|---|---|---|
| 1a | OH | 3a | 4a | - |
| 5a | - | |||
| 7a | - | |||
| 9a | - | |||
| 10a | - | |||
| 1b | NHC2H4OH | 3a | 4b | |
| 5b | 29% | |||
| 7b | - | |||
| 9b | 6% | |||
| 10b | ||||
| 1c | OCH3 | 3a | 4c | - |
| 5c | 5% | |||
| 7c, 8c | -, 5% | |||
| 9c | - | |||
| 10c |
-, not determined; no isolation of products was performed.
Under the chosen reaction conditions, none of these products were formed in controls (without laccase).
The products 4a–4c were not stable and cannot be isolated under the used conditions. Nevertheless, the recorded mass spectra of the reaction assays allowed the identification of the products (Figures S6, S8, S10, S12, Supporting Information).
The incubation of 3a resulted in the formation of a disulfide (6, Scheme 3).
Scheme 3. Laccase-Catalyzed or Autocatalytic Reaction of 2-Aminothiophenol (3a) for the Synthesis of the Disulfide 6 Using 0.5 U Laccase of P. cinnabarinus as the Catalyst in Sodium Acetate Buffer (pH 5; Reactant Concentration, 1 mM).

Retention time, UV–vis maxima, and LC/MS analyses in comparison with the commercially available reference 2-aminophenyl disulfide confirmed the proposed dimeric structure for 6 (Table S1, Supporting Information for product 6: Rf (HPLC) 11.54 min; UV−vis (MeOH) λmax 215, 334 nm; LC/MS m/z (rel. intensity) AP-ESI pos. mode [M + H]+ 249.0 (100); Table S2, Supporting Information for reference 2-aminophenyl disulfide: Rf (HPLC) 12.02 min, UV−vis (MeOH) λmax 218, 335 nm; LC/MS m/z (rel. intensity) AP-ESI pos. mode [M + H]+ 249.0 (100)). The two molecules of 3a are connected via a S–S bond.
The laccase-mediated reaction of 1a–1c and 6 resulted in the formation of 7a–7c (Scheme 4).
Scheme 4. Laccase-Catalyzed Reaction of 2,5-Dihydroxybenzoic Acid Derivatives 1a–1c with 2-Aminothiophenol 3a for the Synthesis of Adducts 7a–7c and 8c Using 0.5 U Laccase of P. cinnabarinus as the Catalyst in Sodium Acetate Buffer (pH 5; Reactant Concentration, 1 mM).

Products 7a–7c were not stable and cannot be isolated under the used conditions. Nevertheless, the recorded mass spectra of the reaction assays allowed the identification of the products (Figures S33, S35, S37, S39, S40 and Tables S11–S14, Supporting Information). In addition, the NMR data of 8c in the mixture with 5c support the structure of [2-[(2-aminophenyl)disulfanyl]anilino]-3,6-dihydroxy-benzoic acid.
The reactions of 1b and 3a or 2-aminophenyl disulfide (reference for product 6) were chosen to determine the laccase-mediated reaction in more detail: The reaction of 1b and 3a resulted in the formation of 6, 4b, 5b, and 7b. The amount of product 5b increased with concomitant decrease of products 4b and 7b (Figure 1). Products 4b and 7b peaked after 20 and 10 min, respectively, and decreased afterward, whereas the product 5b was detected after 20 min and increased rapidly up to 100 min. Afterward, 5b was stable within 24 h (data not shown).
Figure 1.

Reaction course of product formation for 4b (cross), 5b (filled triangle), and 7b (open circle) at equimolar concentrations (1 mM) of reactants 2,5-dihydroxy-N-(2-hydroxyethyl)benzamide (1b) and 2-aminothiophenol (3a) using 0.5 U laccase of P. cinnabarinus as the catalyst in sodium acetate buffer (pH 5; reactant concentration, 1 mM).
This supports the assumption that products 4b and/or 7b are possible reactants for the formation of 5b (Scheme 5).
Scheme 5. Laccase-Catalyzed Reactions of 2,5-Dihydroxybenzoic Acid Derivatives 1a–1c with 2-Aminothiophenol 3a, with Its Dimer 6 or with the Commercially Available Reference 2-Aminophenyl Disulfide (6(ref)) for the Synthesis of Adducts 4a–4c and 7a–7c and the Cyclization Products 4a-Hydroxy-2-oxo-phenothiazines 5a–5c Using 0.5 U Laccase of P. cinnabarinus as the Catalyst in Sodium Acetate Buffer (pH 5; Reactant Concentration, 1 mM).

The laccase-mediated reaction of 1b with 2-aminophenyl disulfide (reference for product 6) resulted also in the formation of products 4b, 5b, and 7b in similar product yields. The formation of products 4b and 5b require the cleavage of 2-aminophenyl disulfide or the 2-aminophenyl disulfide part in 7b (Scheme 5), but no 3a was detected in the homomolecular reaction of 2-aminophenyl disulfide. Nevertheless, in the heteromolecular reactions of 1b with 3a or 1b and 2-aminophenyl disulfide, a product with a retention time of 5.57 min was detected, which possessed a similar UV–vis spectrum as 3a. Additionally, the retention time of the product was only 1 min later compared with 3a (Rf (HPLC) 4.59 min). The concurrent formation of this product with the increase of product 5b may explain the synthesis of 4b and 5b in the reaction of 1b and 2-aminophenyl disulfide.
The disulfide 6 was detected in the reactions without addition of laccase but none of the further formed heteromolecular products.
A parallel pathway can be hypothesized for the reaction of 1a–1c with 3a because of the additional formation of products 9a–9c and 10a–10c (in parallel to products 4, 5, 7, and 8). For the formation of products 9 and 10, the amino group of 3a reacted with the C-2 atom of the aromatic ring of 1a–1c followed by a ring closure via the thiol group on the C-3 atom of the aromatic ring of 1a–1c. In contrast to that reaction, the formation of products 4, 5, 7, and 8 started with an attack of the amino group of 3a at the C-6 atom of the aromatic ring of 1a–1c. The attack at the C-6 atom of 1a–1c resulted in the formation of adducts 4a–4c, 7a–7c, and 8c and the cyclization products 4a-hydroxy-2-oxo-phenothiazines 5a–5c, whereas the attack at the C-2 atom of 1a–1c resulted in the formation of 3-hydroxy-phenothiazines 9a–9c. The product 9b was not detected in the reaction of 1b and 2-aminophenyl disulfide (6(ref)).
In the course of the LC/MS analyses, phenothiazines 9a–9c and 10a–10c were detected (Scheme 6).
Scheme 6. Laccase-Catalyzed Reaction of 2,5-Dihydroxybenzoic Acid Derivatives 1a–1c and 2-Aminothiophenol 3a for the Synthesis of 3-Hydroxy-10H-phenothiazines 9a–9c and 3-Oxo-phenothiazines 10a–10c Using 0.5 U Laccase of P. cinnabarinus as the Catalyst in Sodium Acetate Buffer (pH 5; Reactant Concentration, 1 mM).

The LC/MS spectra contain both masses for 3-oxo-phenothiazines and 3-hydroxy-10H-phenothiazines (such as for 9b: m/z (rel. intensity) AP-ESI pos. mode [M + H]+, 303.0 (5); for 10b: m/z (rel. intensity) AP-ESI pos. mode [M + H]+, 301.0 (100); Figure S44 and Table S16) but in very low intensity for 3-hydroxy-phenothiazines (9a–9c).
The NMR data of 9b clearly identify this product as 3-hydroxy-N-(2-hydroxyethyl)-10H-phenothiazine-1-carboxamide (Figures S44-S48 and Table S16), which may be easily transformed via oxidation to the respective 3-oxo-phenothiazine.
Reactions of 2-Aminophenol or ortho-Phenylenediamine with 2,5-Dihydroxybenzoic Acid Derivatives
In the laccase-catalyzed reaction of 1a–1c and 2-aminophenol (3b), the phenoxazines (11a–11c) were formed (Scheme 7 and Table 2). At first, amination in the ortho-position to the side chain—in the case of the product 11b in the ortho-position of the amide group—took place with subsequent bond formation between the hydroxyl group of 3b and C-5 of the aromatic ring.
Scheme 7. Laccase-Catalyzed Reaction of 2,5-Dihydroxybenzoic Acid Derivatives 1a–1c and 2-Aminophenol 3b for the Synthesis of Phenoxazines 11a–11c Using 0.5 U Laccase of P. cinnabarinus as the Catalyst in Sodium Acetate Buffer (pH 5; Reactant Concentration, 1 mM).

Table 2. Reactions of 1a–1c with 3b–3f and Formed Products Using 0.5 U Laccase of P. cinnabarinus as the Catalyst in Sodium Acetate Buffer (pH 5) within an Incubation Time of 24 h (Reactant Concentration, 1 mM).
| reactant 1 | R1 | reactant 3 | product | yielda |
|---|---|---|---|---|
| 1a | OH | 3b | 11a | - |
| 3c | 12a | - | ||
| 1b | NHC2H4OH | 3b | 11b | 8% |
| 3c | 12b | 71% | ||
| 3d | 13a | 15% | ||
| 14a | 15% | |||
| 15a | - | |||
| 3e | 13b | 16% | ||
| 14b | ||||
| 15b | - | |||
| 3f | 13c | - | ||
| 15c | - | |||
| 1c | OCH3 | 3b | 11c | - |
| 3c | 12c | 7% |
-, not determined; no isolation of products was performed.
The reactions of 1a–1c and ortho-phenylenediamine (3c) resulted in the formation of phenazines (12a–12c; Scheme 8) due to the two neighboring amino groups of 3c. The reaction of 3c and 1b resulted in almost one heteromolecular product, whereas for the reaction with 1c, at least two main products were detected. This may explain the small yield of 12c in comparison with 12b. (We were unable to isolate the second product of the reaction of 3c with 1c.)
Scheme 8. Laccase-Catalyzed Reaction of 2,5-Dihydroxybenzoic Acid Derivatives 1a–1c and ortho-Phenylenediamine 3c for the Synthesis of Phenazines 12a–12c Using 0.5 U Laccase of P. cinnabarinus as the Catalyst in Sodium Acetate Buffer (pH 5; Reactant Concentration, 1 mM).

In the course of the reactions of 3b or 3c with a second reactant and laccase, for 3b and 3c, also ring-closure mechanisms were detected. These reactions resulted in phenazines and phenoxazinones that were not further characterized due to only limited influence on the product formation with 1a–1c. Such phenazines and phenoxazinones formed from ortho-diamines and ortho-aminophenols with laccase were also described previously.83,84
Reactions of 2-Methylaniline, 2-Nitroaniline, or 2-Methylbenzenethiol with 2,5-Dihydroxybenzoic Acid Derivatives
Although all laccase-mediated reactions of 3a–3c resulted in the formation of cyclic products, we determined no cyclization between 2-methylaniline (3d), 2-nitroaniline (3e), or 2-methylbenzenethiol (3f) and 1b, respectively. In these reactions, different adducts were detected (Scheme 9). The adduct of the reaction of 1b and 3d was isolated as a mixture of the quinonoid (13a) and hydroquinonoid (14a) forms, which were also described previously for the reaction with 2-aminobenzoic acid.37 Similarly, 14b was described by NMR analyses, whereas 13b was only detected by LC/MS. For the formation of 13c–15c, a C–S bond was assumed.
Scheme 9. Laccase-Catalyzed Reaction of 2,5-Dihydroxy-N-(2-hydroxyethyl)benzamide 1b and 2-Methylaniline 3d, 2-Nitroaniline 3e, or 2-Methylbenzenethiol 3f for the Synthesis of Quinonoid 13a–13c or Hydroquinonoid 14a and 14b and Adducts 15a–15c Using 0.5 U Laccase of P. cinnabarinus as the Catalyst in Sodium Acetate Buffer (pH 5; Reactant Concentration, 1 mM).

Detailed Structural Characterization of Products
The various cyclic products possessed different UV–vis spectra. The phenothiazines (5a–5c) and phenoxazines (11a–11c) had an orange-brown color, whereas the phenazines (12a–12c) were red or yellow. The phenothiazine 9/10b was violet. Phenothiazines 5a–5c were characterized by three maxima under 300 nm and one intense maximum around 390 nm, whereas phenothiazines 9/10a–10c had an intense maximum around 240 nm, two to three maxima under 400 nm, and another one around 500 nm. The phenoxazines (11a–11c) had two maxima under 300 nm and one intense maximum around 380 nm. The phenazines (12a–12c) possessed one intense maximum around 250 nm and two maxima above 360 nm, which is in line with the literature.77
In the laccase-mediated reaction of 1b and 3a, the phenothiazine 5b was isolated for mass spectroscopy (LC/MS m/z AP-ESI pos. mode [M + H]+, 319.0 (100); neg. mode [M – H]−, 316.9 (100)). These mass data can be attributed to the amination of 3a on the quinonoid form of 1b and cyclization via the thiol group of 3a on the carbonyl group of the quinone (C-5 in the reactant and C-4a in the product), resulting in a six-membered nonaromatic ring. The NMR analyses confirmed the formation of two bonds between 1b and 3a (Figure 2).
Figure 2.

Product 5b: (A) HMBC spectrum of product 5b with HMBC correlation numbers: red, correlations that confirm the novel structure; blue, correlations that are similar to that of the parent compounds. (B) Numbering of C-atoms. (C) 1H (blue) and 13C (red) assignments (chemical shifts are expressed in d (ppm) calibrated on the resonances of the residual nondeuterated solvent DMSO) and multiplicity of the 1H signals and J values (black). J values are in Hz. (D) HMBC correlations (H → C): red, correlations that confirm the novel structure; blue, correlations that are similar to that of the parent compounds.
Multiplicity of H-4 and H-3 suggested that the amination step took place at C-10a. The HMBC correlations (Figure 2) of the proton H-10 unambiguously identified this amination at C-10a. A signal for the hydroxyl group at C-4a (7.53 ppm) was observed. This signal of 5b was a broad signal without any HMBC correlations, but this signal of 5c (7.50 ppm) showed HMBC correlations to C-4a (69.7 ppm) and C-10a (154.6 ppm), supporting the concept of cyclization of the thiol group at C-4a and the removal of the para-quinonoid character. The chemical shift to a higher field (69.6 ppm) of the C-4a of 5b resulted in the assumption of a nonaromatic, noncarbonyl carbon atom. Additionally, 13C NMR showed only one typical signal for quinones in the range of 180 ppm, indicating only one quinonoid carbonyl group for 5b. The chemical shifts within the transformed structure of 1b and 1c are similar to previously described structures.37 The analyses and the comparison led to the identification of 5b as 4a-hydroxy-N-(2-hydroxyethyl)-2-oxo-10H-phenothiazine-1-carboxamid.
Products 9b and 10b could only be isolated as a mixture with MS data AP-ESI in positive mode for the oxo structure and with NMR data for the hydroxyl structure. The molecular mass of 300 was attributed to the amination of 3a on the quinonoid form of 1b and cyclization via the thiol group of 3a, resulting in 10b. The postulated product structure of 9b was confirmed by NMR. 1H NMR spectral data of 9b showed characteristic signals for both reactants (Figure 3). Multiplicity of H-2 and H-4 suggested that the amination step took place at C-10a. The HMBC correlations (Figure 3) of the proton H-10 clearly confirmed this amination at C-10a. The HMBC spectrum showed also correlations between protons H-2 and H-4 and the aminated C-10a atom in the typical range of 130–150 ppm and between protons H-2 and H-4 and the hydroxylated C-3 atom (152.4 ppm), unambiguously showing 9b to be a nonquinonoid aminated and hydroxylated structure substituted at C-10a and C-4a. All MS and NMR results led to the identification of 10b as N-(2-hydroxyethyl)-3-oxo-phenothiazine-1-carboxamide and 9b as 3-hydroxy-N-(2-hydroxyethyl)-10H-phenothiazine-1-carboxamide.
Figure 3.

Product 9b: (A) HMBC spectrum of product 9b with HMBC correlation numbers: red, correlations that confirm the novel structure; blue, correlations that are similar to that of the parent compounds. (B) Numbering of C-atoms. (C) 1H (blue) and 13C (red) assignments (chemical shifts are expressed in d (ppm) calibrated on the resonances of the residual nondeuterated solvent DMSO) and multiplicity of the 1H signals and J values (black). J values are in Hz. (D) HMBC correlations (H → C): red, correlations that confirm the novel structure; blue, correlations that are similar to that of the parent compounds.
In the laccase-mediated reaction of 1b and 3b, the phenoxazine 11b was isolated. The mass spectroscopy data (LC/MS m/z AP-ESI pos. mode [M + H]+, 303.0 (100)) confirmed the formation of an adduct similar to that described above for 5b. The mass data can also be attributed to amination of 3b on the quinonoid form of 1b, but the cyclization took place via the hydroxyl group of 3b, yielding 11b. The carbonyl group (C-4a) of the quinone intermediate of 1b is the C-atom for cyclization for both 5b and 11b, resulting in six-membered nonaromatic rings with an ether bridge in the structure of 11b but with a thioether bridge for 5b. The NMR analyses of 11b also confirmed the formation of two bonds between 1b and 3b (Figure 4) as described for 5b. The HMBC correlations (Figure 4) of the proton H-10 also supported the amination at C-10a for 11b. The signal for the hydroxyl group at C-4a (8.20 ppm) showed HMBC correlations to C-4a (86.8 ppm), C-4 (137.5 ppm), and C-10a (156.1 ppm) and 1H-1H COSY correlations to H-3 (6.22 ppm), H-4 (6.92 ppm), and H-9 (7.43 ppm), supporting the concept of cyclization of the hydroxyl group of 3b at C-4a and the removal of the para-quinonoid character. The chemical shift to a higher field (86.8 ppm) of the C-4a of 11b resulted also in the assumption of a nonaromatic, noncarbonyl carbon atom. Additionally, as described for 5b, 13C NMR showed only one typical signal for quinones at 180 ppm, indicating only one quinonoid carbonyl group and the structure of 4a-hydroxy-N-(2-hydroxyethyl)-2-oxo-10H-phenoxazine-1-carboxamid for 11b.
Figure 4.

Product 11b: (A) HMBC spectrum of product 11b with HMBC correlation numbers: red, correlations that confirm the novel structure; blue, correlations that are similar to that of the parent compounds. (B) Numbering of C-atoms. (C) 1H (blue) and 13C (red) assignments (chemical shifts are expressed in d (ppm) calibrated on the resonances of the residual nondeuterated solvent DMSO) and multiplicity of the 1H signals and J values (black). J values are in Hz. (D) HMBC correlations (H → C): red, correlations that confirm the novel structure; blue, correlations that are similar to that of the parent compounds.
Discussion
The structural characterization of the products by MS and NMR analyses together with UV–vis data led to the description of cyclic products as well as different additional products (Scheme 10). The type of product was dependent on the coupling partners and the amount of reactive groups. Thus, the homomolecular reaction of 3a resulted in the formation of a disulfide (6). Heteromolecular products 4a–4c and disulfides 7a–7c and 8c were formed in the reactions of 3a with 1a–1c, whereby the disulfide 6 is part of adducts 7a–7c and 8c. The reactions of 1a–1c and coupling partners with one amino group and an adjacent thiol group (3a) or, instead, a hydroxyl group (3b) as well as substances with two amino groups (3c) resulted in cyclic products such as phenothiazines (5a–5c and 9/10a–10c), phenoxazines (11a–11c), or phenazines (12a–12c). Compounds with only one reactive group (3d–3f) for the laccase-mediated heteromolecular coupling resulted in quinonoid (13a−13c, and 15a−15c) and hydroquinonoid (14a, 14b) products. To the best of our knowledge, this is the first description of cyclic products 5a–5c and 9/10a–10c for laccase-mediated reactions.
Scheme 10. Possible Reaction Mechanisms.

All heteromolecular reactions started with the laccase-mediated oxidation of the respective 2,5-dihydroxybenzoic acid derivative, which resulted in the formation of quinonoid derivatives (2).
Two reaction pathways for the formation of different phenothiazines were described. The main pathway for products 5a–5c comprised the intermolecular Michael addition (1,4-addition) of 3a via the amino group on the quinonoid derivative 2 of 1a–1c. After the second oxidation, the heteromolecular products (4a–4c) were formed, which undergo intramolecular 1,2-addition. Similar reactions were described previously for 2,5-dihydroxybenzoic acid derivatives and five- or six-membered amines containing an amino group and carboxamide group.37 The key difference of the present research results is that C–S and C–N bonds were formed within one single product, whereas the bond formation of C–O and C–N bonds has been described until now. Another possibility is the dimerization of 3a, which is the prerequisite for the reaction with 1a–1c forming the disulfides 7a–7c, which may react to the phenothiazines (5a–5c) or additional products (4a–4c). In this case, a laccase-mediated bond cleavage may be conceivable, similar to the previously described cleavage processes for amino compounds.12 The second pathway for phenothiazine synthesis (9a–9c and 10a–10c) started with amination of 3a on the C-2 atom of the aromatic ring of 1a–1c with subsequent C–S bond formation via the thiol group and the aromatic ring of 1a–1c. But we were unable to rule out the possibility that the thiolation on the C-3 atom of the aromatic ring of 1a−1c took place first and afterward the amination. However, compared to all of our previous described reaction pattern, this is the first time that we detected bond formations on C-2 and C-3 atoms of the laccase substrate 2,5-dihydroxybenzoic acid and its derivatives (1a–1c). A plausible explanation is based on the different nature of the thiol group compared to the amino and hydroxyl groups.
However, it must be taken into account that the p-hydroquinone substrates are very different from 2,5-dihydroxybenzoic acid and its derivatives. Thus, a similar pathway was described for the laccase-mediated reactions of p-hydroquinone or its derivatives with 5-substituted-4-amino-3-mercapto-1,2,4-triazoles or 2-amino-thiophenol described by Bhalerao et al.7 and Cannatelli and Ragauskas,10 respectively. In the case of 9a–9c and 10a–10c, the reaction of the thiol group of the reaction partner took place on the carbonyl group of the quinonoid form of 1a–1c and the reaction of the amino group on the adjacent unsubstituted CH group, whereas Cannatelli and Ragauskas10 described the reaction of the amino group on the carbonyl group of the quinonoid form of the laccase substrates and the reaction of the thiol group on the adjacent unsubstituted CH group.
The authors proposed an addition–reaction of the amino group with the C-atom at the carbonyl group of the quinone, resulting in a quinonimine, with subsequent reaction of the thiol group with the adjacent C-atom of the benzene ring. Bhalerao et al.7 described the reaction of p-hydroquinone with 5-substituted-4-amino-3-mercapto-1,2,4-triazoles. The coupling partners were connected via C–S and C=N bonds in the formed benzothiadiazinones. Again, a quinonimine was formed due to the reaction of the amino group with the C-atom at the keto group of the quinone.
Bhalerao et al.7 as well as Cannatelli and Ragauskas10 used p-hydroquinone or its derivatives. This may explain the different reaction pathway for the herein used 2,5-dihydroxybenzoic acid derivatives.
The formation of products 7a–7c was supported by the NMR analyses of 5c. The disulfide (6) of 3a was described previously by Cannatelli and Ragauskas.10 For laccase-mediated reactions, disulfides 7a–7c are unusual. In most cases, the reaction of two amino compounds with dihydroxybenzoic acid derivatives proceeded via two C–N bonds in the para-position.32 The UV–vis spectra of products 7a–7c (7a: 210, 455 nm; 7b: 209, 492 nm; 7c: 210, 473 nm) resembled more monoaminated quinonamines with at least one absorption spectrum under 300 nm and one around 500 nm than diaminated quinonamines with at least two maxima under 400 nm.12,25,85 These absorption maxima were not only in line with those for the mono- and diaminated products (13a, 13b, and 15a, 15b) but also those for the monothiolated product 13c and dithiolated product 15c. These products were formed by nucleophilic addition of one or two molecules of 3f with 1b, forming C–S bonds. In this case, the thiol groups and not the amino groups (as for 13a, 13b, 15a, and 15b) were the donors in the Michael addition.86 The reaction of para-hydroquinones and thiols resulting in different adducts was described previously.11,36,87 Cyclization involving two SH groups, which resulted in 2,3-ethylenedithio-1,4-quinones with yields of 37–74%, was also described.38
In summary, the cyclic products described in our study were isolated with yields of 4% up to 68%. This confirms the complexity, in comparison to the so-called “easy” and “straightforward” laccase-catalyzed reactions of 2,5-dihydroxybenzoic acid derivatives and reaction partners with only one amino group,24,26−29 of such cyclization reactions accompanied by simultaneous formation of homomolecular products of the amines and by-products, which diminished the yields. Further studies will lead to strategies for a higher product yield through variations of reaction parameters such as the kind of laccase, pH value of the reaction assay, or concentration of reactants.
Conclusions
The introduced one-pot laccase-mediated reactions of cyclic products as well as additional products are consistent with the principles of green chemistry defined by Anastas and Warner.88 The utilization of laccases as nonstoichiometric catalysts in an aqueous solution with less than 5% solvent (which is only necessary for the solubilization of the reagents and can be replaced in most cases by water) and the room temperature make this process an attractive alternative to chemical syntheses.
Experimental Section
Enzymes
The used laccase was obtained from P. cinnabarinus SBUG-M 1044. The white rot fungus was isolated from an oak tree in northern Germany and is deposited at the strain collection of the Department of Biology of the University of Greifswald (SBUG).
Cultivation of P. cinnabarinus SBUG-M 1044 and crude preparation of laccase were carried out as we reported previously.19 This enzyme preparation contains only isoenzymes of laccase but no other enzymes and was used always in 20 mM sodium acetate buffer (SAB; pH 5) because of the pH optimum around pH 5.19,89
Measurement of Laccase Activity
The activity of laccase was determined spectrophotometrically at 420 nm with ABTS (2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) diammonium salt) as the substrate90 using the method described by Jonas et al.19 One unit (1 U) is defined as 1 μmol·mL–1·min–1.
Experimental Procedures
Analytical Procedure
For analytical experiments, amines, thiols (1 mM), and the respective dihydroxylated compound (1 mM) were incubated with laccase (activity, 0.5 U). Reaction mixtures were incubated with agitation at 200 rpm at room temperature in the dark. The reaction mixtures were analyzed by HPLC. The separation of the substances was achieved with a RP18 column at a flow rate of 1 mL/min. A solvent system consisting of methanol (eluent A) and 0.1% phosphoric acid (eluent B), starting from an initial ratio of 10% A and 90% B and reaching 100% methanol within 14 min, was used. Controls were performed without the addition of laccase.
Product Isolation
All reaction mixtures for product isolation were performed with laccase of P. cinnabarinus (final activity, 0.5 U) in SAB. Isolation steps were performed by solid-phase extraction with a RP18 silica gel column (60 mL, 10 g of adsorbent material, Phenomenex, Strata, Germany). The product 5b was isolated from the reaction mixture (320 mL) after an incubation period of 24 h (1b:3a, 1:1 mM assay). After charging the column with 40 mL of reaction mixture, 20 mL of methanol/distilled water (20:80, v/v) and 40 mL of methanol/distilled water (40:60, v/v) were used to remove undesired impurities. Elution of the orange fraction was performed with 30 mL of methanol. The product 5c (mixture with 8c) was isolated from the reaction mixture (400 mL) after an incubation period of 4.5 h (1c:3a, 1:1 mM assay). After charging the column with 40 mL of reaction mixture, 30 mL of methanol/distilled water (20:80, v/v) and 30 mL of methanol/distilled water (40:60, v/v) were used to remove undesired impurities. Elution of the orange fraction was performed with 30 mL of methanol.
The product 9/10b was isolated from the reaction mixture (320 mL) after an incubation period of 20 min (1b:3a, 1:1 mM assay). After charging the column with 40 mL of reaction mixture and washing steps with 20 mL of methanol/distilled water (20:80, v/v) and 40 mL of methanol/distilled water (50:50, v/v), the product was eluted with 20 mL of methanol/distilled water (50:50 v/v). The product 11b was isolated from the reaction mixture (280 mL) after an incubation period of 4 h (1b:3b, 1:1 mM assay). After charging the column with 40 mL of reaction mixture, 40 mL of methanol/distilled water (40:60, v/v), 15 mL of methanol/ distilled water (60:40, v/v), and 5 mL of methanol/distilled water (80:20, v/v) were used to remove undesired impurities. The product was eluted with additional 10 mL of methanol/distilled water (80:20, v/v). The product 12b was isolated from the reaction mixture (240 mL) after an incubation period of 20 min (1b:3c, 1:1 mM assay). After charging the column with 40 mL of reaction mixture, 20 mL of methanol/distilled water (20:80, v/v) and 50 mL of methanol/distilled water (60:40, v/v) were used to remove undesired impurities. The product was eluted with 25 mL of methanol. The product 12c was isolated from the reaction mixture (520 mL) after an incubation period of 3.5 h (1c:3c, 1:1 mM assay). After charging the column with 40 mL of reaction mixture, 40 mL of methanol/distilled water (40:60, v/v), 15 mL of methanol/distilled water (60:40, v/v), 5 mL of methanol/distilled water (80:20, v/v), and 10 mL of methanol/distilled water (80:20, v/v) were used to remove undesired impurities. The product was eluted with additional 20 mL of methanol/distilled water (80:20, v/v). The product 13/14a was isolated from the reaction mixture (120 mL) after an incubation period of 2 h (1b:3d, 1:1 mM assay). After charging the column with 40 mL of reaction mixture, 20 mL of distilled water, 20 mL of methanol/distilled water (10:90, v/v), 10 mL of methanol/distilled water (30:70, v/v), and 20 mL of methanol/distilled water (50:50, v/v) were used to remove undesired impurities. The product was eluted with additional with 40 mL of methanol (50:50, v/v). The product 13/14b was isolated from the reaction mixture (160 mL) after an incubation period of 2 h (1b:3e, 1:1 mM assay). After charging the column with 40 mL of reaction mixture, 40 mL of methanol/distilled water (20:80, v/v) was used to remove undesired impurities. The product was eluted with 50 mL of methanol (50:50, v/v).
For nuclear magnetic resonance (NMR) spectroscopy, the isolated products were dried by lyophilization. The lyophilized products and reaction mixtures were characterized using an LC/MS system. The atmospheric pressure ionization (API) mass spectrometry experiments were performed on an Agilent Series 1200 HPLC system with a diode array detector and Agilent 6120 quadrupole mass spectrometer (Waldbronn, Germany). The high-resolution mass spectra (HRMS) were recorded on a QExactive classic system (Thermo Scientific, Bremen, Germany).
4a-Hydroxy-N-(2-hydroxyethyl)-2-oxo-10H-phenothiazine-1-carboxamide (5b)
Synthesis and isolation as described above. Brown solid. Yield, 29.24% (29.77 mg). mp (decomposition) 160 °C. 1H NMR (DMSO-d6): δ 3.39 (m, J = 5.5, 2H, H-13), 3.53 (m, J = 5.2, J = 5.5, 2H, H-14), 4.77 (t, J = 5.2, 1H, OH, at C-14), 6.27 (d, J = 9.9, 1H, H-3), 6.82 (dd, J = 0.5, J = 9.9, 1H, H-4), 7.19 (m(t), J = 1.3, J = 7.6, 1H, H-7), 7.33 (m(t), J = 1.3, J = 8.0, 1H, H-8), 7.38 (m(d), J = 1.3, J = 8.0, 1H, H-9), 7.46 (m(d), J = 1.3, J = 7.6, 1H, H-6), 7.53 (s(broad), 1H, OH, at C-4a), 10.09 (t, J = 5.5, 1H, NH, at N-12), 14.56 (s, 1H, NH, at N-10). 13C NMR: δ 41.6 (C-13), 60.1 (C-14), 69.6 (C-4a), 94.8 (C-1), 116.3 (C-5a), 119.7 (C-9), 125.6 (C-7), 127.9 (C-8), 128.8 (C-6), 129.8 (C-3), 134.6 (C-9a), 137.8 (C-4), 157.4 (C-10a), 169.4 (C-11), 182.4 (C-2). HMBC correlations: H-3 (C-1, C-4, C-4a, C-11), H-4 (C-2, C-4a, C-5a, C-10a), H-6 (C-8, C-9a), H-7 (C-5a, C-6, C-9), H-8 (C-5a, C-6, C-9a), H-9 (C-5a, C-7), H-10 (C-1, C-4a, C-5a, C-9, C-9a, C-10a), H-12 (C-1, C-11, C-13), H-13 (C-11, C-14), H-14 (C-13), OH at C-14 (C-13, C-14). Rf (HPLC) 10.89 min; UV–vis (MeOH) λmax 252, 284, 300, 397 nm. MS m/z (rel. intensity) AP-ESI: pos. ion mode [M + H]+, 319.0 (100); AP-ESI: neg. ion mode [M – H]−, 316.9 (100). HRMS (ESI/Quadrupole-Orbitrap) m/z: [M + H]+ calcd for C15H15N2O4S, 319.0753; found, 319.0746.
4a-Hydroxy-N-(2-hydroxyethyl)-2-oxo-10H-phenothiazine-1-carboxylic Acid Methylester (5c)
Synthesis and isolation as described above. Orange solid. Mixture of 8c, 5c, and other inseparable substances. Yield, 15.56% (18.01 mg). The peak areas of the integrated 1H NMR signals are in a 1:1:1 ratio, so the yield distribution between 8c, 5c, and the other inseparable substances was given as 5.19, 5.19, and 5.19%. 1H NMR (DMSO-d6), 5c: δ 3.76 (s, 3H, H-12), 6.16 (d, J = 10.0, 1H, H-3), 6.77 (m(d), J = 10.0, J = 0.5, 1H, H-4), 7.11 (m(t), J = 7.5, J = 1.1, 1H, H-7), 7.27 (m(t), J = 7.9, J = 7.4, J = 1.3, 1H, H-8), 7.38 (m(d), J = 8.1, J = 1.1, 1H, H-6), 7.42 (m(d), J = 8.1, J = 1.0, 1H, H-9), 7.50 (s, 1H, OH, at C-4a), 11.42 (s, 1H, NH, H-10). 13C NMR, 5c: δ 57.3 (C-12), 69.7 (C-4a), 97.6 (C-1), 116.4 (C-5a), 119.3 (C-9), 125.0 (C-7), 127.7 (C-8), 128.8 (C-6), 129.9 (C-3), 135.2 (C-9a), 137.8 (C-4), 154.6 (C-10a), 168.9 (C-11), 179.6 (C-2). HMBC correlations, 5c: H-3 (C-1, C-4a), H-4 (C-2, C-4a, C-10a), H-6 (C-8, C-9a), H-7 (C-5a, C-8, C-9), H-8 (C-5a, C-6, C-9a), H-9 (C-5a, C-7), H-12 (C-11), OH at C-4a (C-4a, C-10a), H-10 (C-1, C-4a, C-5a, C-9, C-9a). Rf (HPLC) 11.13 min; UV–vis (MeOH) λmax 249, 280, 300, 382 nm. MS m/z (rel. intensity) AP-ESI: pos. ion mode [M + H]+, 290.1 (100); AP-ESI: neg. ion mode [M – H]−, 288.0 (100).
[2-[(2-Aminophenyl)disulfanyl]anilino]-3,6-dihydroxy-benzoic Acid (8c)
Mixture of 8c and 5c. 1H NMR (DMSO-d6), δ structure part of laccase substrate 1c within 8c: 3.60 (s, 3H, H-14), 6.62 (d, J = 8.8, 1H, H-5), 6.95 (s, 1H, NH, H-15), 6.89 (d, J = 8.8, 1H, H-4), 8.99 (s, 1H, OH at C-3), 9.34 (s, 1H, OH at C-6); structure part of the dimer 3 within 8c: 6.39 (m(d), J = 8.2, J = 1.0, 1H), 6.42 (m(t), J = 7.5, J = 7.3, J = 1.1, 1H), 6.60 (m(t), J = 8.2, J = 1.1, 1H), 6.72 (m(d), J = 8.1, J = 1.1, 1H), 6.73 (m(d), J = 8.2, J = 1.4, 1H), 7.00 (m(d), J = 7.7, J = 1.5, 1H), 7.07 (m(t), J = 7.6, J = 1.3), 7.09 (m(t), 1H). 13C NMR, δ structure part of laccase substrate 1c within 8c: 52.4 (C-14), 113.5 (C-5), 117.1 (C-1), 119.7 (C-4), 126.8 (C-2), 144.8 (C-3), 149.3 (C-6), 167.7 (C-13); structure part of the dimer 6 within 8c: 114.9 (1H 6.39), 115.3x (1H 6.72, 1H 6.73), 116.x (1H 6.42), 118.8 (1H 6.60), 131.6 (1H 7.07), 134.6 (1H 7.09), 135.9 (1H 7.00). HMBC correlations, structure part of laccase substrate 1c within 8c: H-4 (C-1, C-2, C-3, C-6), H-5 (C-1, C-3, C-6, C-13), H-14 (C-13), OH at C-3 (C-2, C-3, C-4), OH at C-6 (C-1, C-5, C-6), H-15 (C-3, C-8, C12); structure part of the dimer 6 within 8c: 6.42 (115.3x, 131.6, 135.9), 6.60 (120.2, 134.6), 6.72 (116.x), 6.73 (116.x), 7.00 (131.6, 150.2), 7.07 (115.3x, 117.1, 135.9, 150.2), 7.09 (131.6).
3-Hydroxy-N-(2-hydroxyethyl)-10H-phenothiazine-1-carboxamide (9b) and N-(2-Hydroxyethyl)-3-oxo-phenothiazine-1-carboxamide (10b)
Synthesis and isolation as described above. Violet solid. Yield, 6.39% (6.18 mg). mp 209 °C. 1H NMR (DMSO-d6), 9b: δ 3.30 (signal covered by the solvent signal, H-13), 3.54 (t, J = 6.0, 2H, H-14), 6.64 (d(s), J = 2.5, 1H, H-4), 6.66 (m(d), J = 8.0, J = 1.1, 1H, H-9), 6.80 (m(t), J = 7.5, J = 1.1, 1H, H-7), 6.97 (m(d), J = 7.6, J = 1.2, 1H, H-6), 7.00 (d(s), J = 2.6, 1H, H-2), 7.01 (m(t), J = 7.6, J = 1.2, 1H, H-8), 8.49 (t, J = 5.2, 1H, NH, H-12), 9.22 (s(broad), 1H, OH), 9.81 (s, 1H, NH, H-10). 13C NMR, 9b: δ 42.6 (C-13), 60.0 (C-14), 113.3 (C-2), 115.6 (C-9), 116.9 (C-5a), 117.1 (C-4), 120.1 (C-1), 122.5 (C-7), 126.7 (C-6), 128.1 (C-8), 135.6 (C-10a), 142.2 (C-9a), 152.4 (C-3), 168.5 (C-11). HMBC correlations, 9b: H-2 (C-3, C-4, C-10a, C-11), H-4 (C-2, C-3, C-10a), H-6 (C-8, C-9a), H-7 (C-5a, C-6, C-9), H-8 (C-6, C-9a), H-9 (C-5a, C-7), H-10 (C-1, C-5a, C-9), H-12 (C-11), H-13 (C-11, C-14), H-14 (C-13). 10b: Rf (HPLC) 8.86 min; Rf (LC/MS) 12.95 min; UV–vis (MeOH) λmax 234, 293, 385, 539 nm. MS m/z (rel. intensity) AP-ESI: pos. ion mode [M + H]+, 301.0 (100); [2M + Na]+, 622.9 (5). 9b: MS m/z (rel. intensity) AP-ESI: pos. ion mode [M + H]+, 303.0 (5).
4a-Hydroxy-N-(2-hydroxyethyl)-2-oxo-10H-phenoxazine-1-carboxamide (11b)
Synthesis and isolation as described above. Orange solid. Yield, 8.40% (7.06 mg). mp (decomposition) 165 °C. 1H NMR (DMSO-d6): δ 3.38 (m, J = 5.6 Hz, 2H, H-13), 3.53 (m, J = 5.8, 2H, H-14), 4.77 (t, J = 4.8, 1H, OH, at C-14), 6.22 (d, J = 10.1, 1H, H-3), 6.92 (d, J = 10.0, 1H, H-4), 7.11 (m, J = 7.6, J = 7.8, 1H, H-8), 7.14 (m, J = 7.5, 1H, H-6), 7.16 (m, J = 7.5, J = 7.6, 1H, H-7), 7.43 (d, J = 7.8, 1H, H-9), 8.20 (s, 1H, OH, at C-4a), 9.74 (t, J = 5.5, 1H, NH, H-12), 13.71 (s, 1H, NH, H-10). 13C NMR: δ 41.4 (C-13), 60.2 (C-14), 86.8 (C-4a), 95.8 (C-1), 118.1 (C-6), 118.3 (C-9), 123.5 (C-8), 125.8 (C-9a), 125.9 (C-7), 129.8 (C-3), 137.5 (C-4), 140.6 (C-5a), 156.1 (C-10a), 168.8 (C-11), 182.8 (C-2). HMBC correlations: H-3 (C-1, C-4a, C-11), H-4 (C-1, C-2, C-10a), H-6 (C-9a), H-7 (C-5a, C-9), H-8 (C-6, C-9a), H-9 (C-5a, C-7), H-10 (C-1, C-4a, C-5a, C-9, C-9a, C-10a), H-12 (C-1, C-11, C-13, C-14), H-13 (C-11, C-14), H-14 (C-13), OH at C-14 (C-13, C-14), OH at C-4a (C-4, C-4a, C-10a). 1H-1H COSY correlations: H-3 (H-4), H-4 (H-3), H-6 (H-7), H-7 (H-6), H-8 (H-9), H-9 (H-8), H-10 (H-4, H-6, H-9), H-12 (H-13, OH at C-14), H-13 (H-12, H-14), H-14 (H-13, OH at C-14), OH at C-14 (H-14), OH at C-4a (H-3, H-4, H-9). Rf (HPLC) 10.20 min; UV–vis (MeOH) λmax 208, 283, 387 nm. MS m/z (rel. intensity) AP-ESI: pos. ion mode [M + H]+, 303.0 (100); [M + Na]+, 324.9 (9); [2M + H]+, 626.9 (7). HRMS (ESI/Quadrupole-Orbitrap) m/z: [M + H]+ calcd for C15H15N2O5, 303.0981; found 303.0972; [M + Na]+ calcd for C15H14N2O5Na, 325.0800; found, 325.0792.
2-Hydroxy-N-(2-hydroxyethyl)-phenazine-1-carboxamide (12b)
Synthesis and isolation as described above. Yellow solid. Yield, 71.01% (48.28 mg). mp (decomposition) 170 °C. 1H NMR (DMSO-d6): δ 3.63 (m, J = 5.3, 2H, H-13), 3.76 (m, J = 5.3, 2H, H-14), 5.09 (s(broad), 1H, OH, at C-14), 7.56 (d, J = 8.9, 1H, H-3), 7.89 (m(t), J = 7.5, 1H, H-8), 7.98 (m(t), J = 1.0 Hz, J = 7.5, 1H, H-7), 8.18 (d, J = 8.7, 3H, H-4, H-6, H-9), 11.78 (s, 1H, NH, at N-12). 13C NMR (DMSO-d6): δ 42.0 (C-13), 59.9 (C-14), 102.6 (C-1), 128.4 (C-3, C-6), 129.6 (C-9), 130.1 (C-8), 132.4 (C-7), 135.7 (C-4), 139.9 (C-4a), 140.5 (C-5a, C-9a), 142.1 (C-2), 169.6 (C-10a), 170.8 (C-11). HMBC correlations: H-3 (C-1, C-4a), H-4 (C-2, C-10a), H-6 (C-8, C-9a), H-7 (C-5a, C-9), H-8 (C-6, C-9a), H-9 (C-5a, C-7). Rf (HPLC) 12.66 min; UV–vis (MeOH) λmax 213, 252, 360, 403 nm. MS m/z (rel. intensity) AP-ESI: pos. ion mode [M + H]+, 284.0 (100). HRMS (ESI/Quadrupole-Orbitrap) m/z: [M + H]+ calcd for C15H14N3O3, 284.1035; found, 284.1031.
2-Hydroxyphenazine-1-carboxylic Acid Methylester (12c)
Synthesis and isolation as described above. Red solid. Yield, 6.60% (10.31 mg). 1H NMR (DMSO-d6): δ 3.96 (s, 3H, H-12), 7.68 (d, J = 9.4, 1H, H-3), 7.85 (m, J = 8.5, J = 6.6, J = 1.3, 1H, H-8), 7.91 (m, J = 8.5, J = 6.6, J = 1.3, 1H, H-7), 8.10 (d, J = 8.5, J = 0.7, 1H, H-6), 8.18 (d, J = 9.4, 1H, H-4), 8.19 (d, J = 8.5, J = 0.7, 1H, H-9). 13C NMR (DMSO-d6): δ 52.5 (C-12), 113.9 (C-1), 126.9 (C-3), 129.0 (C-6), 129.7 (C-8), 129.9 (C-9), 131.6 (C-7), 132.3 (C-4), 138.9 (C-4a), 141.3 (C-9a), 142.5 (C-2), 143.0 (C-5a), 158.0 (C-10a), 167.4 (C-11). HMBC correlations: H-3 (C-1, C-4a), H-4 (C-2, C-10a), H-6 (C-8, C-9a), H-7 (C-5a, C-9), H-8 (C-6, C-9a), H-9 (C-5a, C-7). Rf (HPLC) 11.88 min; UV–vis (MeOH) λmax 217, 247, 360, 470 nm. MS m/z (rel. intensity) AP-ESI: pos. ion mode [M + H]+, 255.1 (100); AP-ESI: neg. ion mode [M – H]−, 253.0 (100).
N-(2-Hydroxyethyl)-2-(2-methylanilino)-3,6-dioxocyclohexa-1,4-diene-1-carboxamide (13a) and 3,6-Dihydroxy-N-(2-hydroxyethyl)-2-(2-methylanilino)benzamide (14a)
Synthesis and isolation as described above. Orange solid. Yield, 29.14% (10.50 mg). mp 156 °C. The peak area of the integrated 1H NMR signals are in a 1:1 ratio, so the yield distribution between 13a and 14a is given as 14.57 and 14.57%. 13a: N-(2-Hydroxyethyl)-2-(2-methylanilino)-3,6-dioxo-cyclohexa-1,4-diene-1-carboxamide; 1H NMR (DMSO-d6): δ 2.21 (s, 3H, H-8′), 3.23 (m, J = 5.6, 2H, H-9), 3.46 (m, J = 5.5, 2H, H-10), 4.76 (m, J = 5.1, 1H, OH at C-10), 6.72 (d, J = 10.1, 1H, H-4), 6.74 (d, J = 10.1, 1H, H-5), 7.12–7.17 (3 x m, 3H, H-3′, H-4′, H-5′), 7.25 (d, J = 7.4, 1H, H-6′), 9.40 (t(broad), 1H, H-8, NH). 13C NMR (DMSO-d6): δ 17.5 (C-8′), 41.0 (C-9), 59.3 (C-10), 102.4 (C-1), 125.3/126.1, (C-3′/C5′), 126.3 (C-4′), 130.2 (C-6′), 132.0 (C-1′), 133.2 (C-4), 138.1 (C-2′), 139.6 (C-5), 151.0 (C-2), 167.5 (C-7), 182.8 (C-3), 183.6 (C-6). HMBC correlations: H-4 (C-2, C-6), H-5 (C-1, C-3, C-7), H-8 (C-7, C-9), H-9 (C-7, C-10), H-10 (C-9), H-3′/H-4′/H-5′ (C-2′, C-3′, C-5′), H-6′ (C-2′, C-4′, C-8′), H-8′ (C-1′, C-2′, C-6′), OH at C-10 (C-9, C-10). Rf (HPLC) 8.89 min; UV–vis (MeOH) λmax 211, 260, 490 nm. MS m/z (rel. intensity) AP-ESI: pos. ion mode [M + H]+, 301.0 (100). HRMS (ESI/Quadrupole-Orbitrap) m/z: [M + H]+ calcd for C16H17N2O4, 301.1188; found, 301.1182; [M + Na]+ calcd for C16H16N2O4Na, 323.1008; found, 323.1001. 14a: 3,6-Dihydroxy-N-(2-hydroxyethyl)-2-(2-methylanilino)benzamide: 1H NMR (DMSO-d6): δ 2.29 (s, 3H, H-8′), 3.18 (m, J = 6.1, J = 5.5, 2H, H-9), 3.24 (m, J = 5.5, 2H, H-10), 4.64 (m, J = 5.2, 1H, OH at C-10), 6.23 (d, J = 7.9, 1H, H-3′), 6.62 (d, J = 8.9, 1H, H-5), 6.68 (m, J = 7.3, J = 0.6, 1H, H-5′), 6.91 (m, J = 7.5, J = 0.6, 1H, H-4′), 6.93 (d, J = 8.9, 1H, H-4), 7.03 (s, 1H, H-7′, NH), 7.07 (d, J = 7.2, 1H, H-6′), 8.88 (s, 1H, OH), 8.98 (t, J = 5.4, 1H, H-8, NH), 11.25 (s, 1H, OH). 13C NMR (DMSO-d6): δ 17.4 (C-8′), 41.2 (C-9), 59.1 (C-10), 112.9 (C-5), 113.3 (C-3′), 113.7 (C-1), 119.0 (C-5′), 119.7 (C-4), 124.6 (C-1′), 126.0 (C-4′), 128.4 (C-6), 129.7 (C-6′), 143.7 (C-2′), 144.2 (C-3), 151.6 (C-2), 168.0 (C-7). HMBC correlations: H-4 (C-1, C-2, C-3, C-6), H-5 (C-1, C-2, C-3, C-6, C-7), H-8 (C-7, C-9, C-10), H-9 (C-7, C-10), H-10 (C-9), H-3′ (C-1′, C-2′, C-5′), H-4′ (C-2′, C-6′), H-5′ (C-1′, C-3′), H-6′ (C-2′, C-4′, C-8′), H-7′ (C-3, C-3′), H-8′ (C-1′, C-2′, C-3′, C-6′), OH at C-10 (C-9, C-10), OH (C-3, C-4, C-6), OH (C-1, C-2).
N-(2-Hydroxyethyl)-2-(2-nitroanilino)-3,6-dioxocyclohexa-1,4-diene-1-carboxamide (13b) and 3,6-Dihydroxy-N-(2-hydroxyethyl)-2-(2-nitroanilino)benzamide (14b)
Synthesis and isolation as described above. Brown solid. Yield, 15.53% (8.23 mg). 14b: 3,6-Dihydroxy-N-(2-hydroxyethyl)-2-(2-nitroanilino)benzamide: 1H NMR (DMSO-d6): δ 3.16 (m, J = 5.9, 2H, H-9), 3.44 (m, 2H, H-10), 4.49 (s(broad), 1H, OH at C-10), 6.55 (m(d), J = 1.1, J = 8.7, 1H, H-3′), 6.72 (d, J = 8.3, 1H, H-5), 6.77 (m, J = 7.7, J = 1.2, 1H, H-5′), 6.85 (d, J = 8.3, 1H, H-4), 7.39 (m, J = 7.8, J = 1.4, 1H, H-4′), 8.05 (m, J = 1.4, J = 8.6, 1H, H-6′), 8.15 (t, J = 5.5, 1H, H-8, NH), 9.03 (s(broad), 1H, OH), 9.38 (s, 1H, H-7′, NH), 9.65 (s(broad), 1H, OH). 13C NMR (DMSO-d6): δ 41.9 (C-9), 59.9 (C-10), 115.5 (C-5), 117.7 (C-3′), 121.7 (C-1), 117.5 (C-5′), 118.6 (C-4), 123.7 (C-6), 133.2 (C-1′), 135.9 (C-4′), 125.8 (C-6′), 143.4 (C-2′), 145.5 (C-3), 148.7 (C-2), 166.3 (C-7). HMBC correlations: H-4 (C-2, C-3, C-5, C-6), H-5 (C-1, C-2, C-3, C-7), H-8 (C-7), H-9 (C-7, C-10), H-10 (C-9), H-3′ (C-1′, C-5′), H-4′ (C-2′, C-6′), H-5′ (C-1′, C-3′), H-6′ (C-1′, C-2′, C-4′), H-7′ (C-1, C-1′, C-3, C-3′). Rf (HPLC) 9.00 min; UV–vis (MeOH) λmax 209, 262, 483 nm. MS m/z (rel. intensity) AP-ESI: pos. ion mode [M + H]+, 331.9 (100); [M + Na]+, 353.9 (7).
Acknowledgments
Not applicable.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c00719.
1H NMR, 13C NMR, HSQC, HMBC spectra, 1H-NMR and 13C-NMR correlation, retention time, UV–vis, and MS-data (PDF)
The authors declare no competing financial interest.
Dedication
∥ This paper is dedicated to the memory of Prof. Dr. Frieder Schauer.
Supplementary Material
References
- Nakamura T. On the process of enzymatic oxidation of hydroquinone. Biochem. Biophys. Res. Commun. 1960, 2, 111–113. 10.1016/0006-291X(60)90198-4. [DOI] [Google Scholar]
- Solomon E. I.; Chen P.; Metz M.; Lee S.-K.; Palmer A. E. Oxygen binding, activation, and reduction to water by copper proteins. Angew. Chem., Int. Ed. 2001, 40, 4570–4590. . [DOI] [PubMed] [Google Scholar]
- Solomon E. I.; Augustine A. J.; Yoon J. O2 reduction to H2O by the multicopper oxidases. Dalton Trans. 2008, 30, 3921–3932. 10.1039/B800799C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munk L.; Andersen M. L.; Meyer A. S. Direct rate assessment of laccase catalysed radical formation in lignin by electron paramagnetic resonance spectroscopy. Enzyme Microb. Tech. 2017, 106, 88–96. 10.1016/j.enzmictec.2017.07.006. [DOI] [PubMed] [Google Scholar]
- Keilin D.; Mann T. Laccase, a Blue Copper-Protein Oxidase from the Latex of Rhus succedanea. Nature 1939, 143, 23–24. 10.1038/143023b0. [DOI] [Google Scholar]
- Bollag J.-M.; Shuttleworth K. L.; Anderson D. H. Laccase-mediated detoxification of phenolic compounds. Appl. Environ. Microbiol. 1988, 54, 3086–3091. 10.1128/AEM.54.12.3086-3091.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhalerao U. T.; Muralikrishna C.; Rani B. R. Laccase enzyme catalysed efficient synthesis of 3-Substituted-1,2,4-triazoto(4,3-b)(4,1,2)benzothiadiazine-8-ones. Tetrahedron 1994, 50, 4019–4024. 10.1016/S0040-4020(01)89677-0. [DOI] [Google Scholar]
- Thurston C. F. The structure and function of fungal laccases. Microbiology 1994, 140, 19–26. 10.1099/13500872-140-1-19. [DOI] [Google Scholar]
- Mikolasch A.; Schauer F. Fungal laccases as tools for the synthesis of new hybrid molecules and biomaterials. Appl. Microbiol. Biotechnol. 2009, 82, 605–624. 10.1007/s00253-009-1869-z. [DOI] [PubMed] [Google Scholar]
- Cannatelli M. D.; Ragauskas A. J. Ecofriendly syntheses of phenothiazones and related structures facilitated by laccase - a comparative study. Tetrahedron Lett. 2016, 57, 3749–3753. 10.1016/j.tetlet.2016.07.016. [DOI] [Google Scholar]
- Schlippert M.; Mikolasch A.; Hahn V.; Schauer F. Enzymatic thiol Michael addition using laccases: Multiple C-S bond formation between p-hydroquinones and aromatic thiols. J. Mol. Catal. B-Enzym. 2016, 126, 106–114. 10.1016/j.molcatb.2015.12.012. [DOI] [Google Scholar]
- Hahn V.; Mikolasch A.; Schauer F. Cleavage and synthesis function of high and low redox potential laccases towards 4-morpholinoaniline and aminated as well as chlorinated phenols. Appl. Microbiol. Biotechnol. 2014, 98, 1609–1620. 10.1007/s00253-013-4984-9. [DOI] [PubMed] [Google Scholar]
- Bollag J.-M.; Sjoblad R. D.; Minard R. D. Polymerization of phenolic intermediates of pesticides by a fungal enzyme. Experientia 1977, 33, 1564–1566. 10.1007/BF01933998. [DOI] [PubMed] [Google Scholar]
- Jonas U.; Hammer E.; Haupt E. T. K.; Schauer F. Characterisation of coupling products formed by biotransformation of biphenyl and diphenyl ether by the white rot fungus Pycnoporus cinnabarinus. Arch. Microbiol. 2000, 174, 393–398. 10.1007/s002030000220. [DOI] [PubMed] [Google Scholar]
- Ciecholewski S.; Hammer E.; Manda K.; Bose G.; Nguyen V. T. H.; Langer P.; Schauer F. Laccase-catalyzed carbon-carbon bond formation: oxidative dimerization of salicylic esters by air in aqueous solution. Tetrahedron 2005, 61, 4615–4619. 10.1016/j.tet.2005.03.007. [DOI] [Google Scholar]
- Benfield G.; Bocks S. M.; Bromley K.; Brown B. R. Studies of fungal and plant laccases. Phytochemistry 1964, 3, 79–88. 10.1016/S0031-9422(00)83998-9. [DOI] [Google Scholar]
- Simmons K. E.; Minard R. D.; Bollag J.-M. Oxidative co-oligomerization of guaiacol and 4-chloroaniline. Environ. Sci. Technol. 1989, 23, 115–121. 10.1021/es00178a016. [DOI] [PubMed] [Google Scholar]
- Leonowicz A.; Edgehill R. U.; Bollag J. M. The effect of pH on the transformation of syringic and vanillic acids by the laccases of Rhizoctonia praticola and Trametes versicolor. Arch. Microbiol. 1984, 137, 89–96. 10.1007/BF00414446. [DOI] [Google Scholar]
- Jonas U.; Hammer E.; Schauer F.; Bollag J.-M. Transformation of 2-hydroxydibenzofuran by laccases of the white rot fungi Trametes versicolor and Pycnoporus cinnabarinus and characterization of oligomerization products. Biodegradation 1997, 8, 321–327. 10.1023/A:1008220120431. [DOI] [PubMed] [Google Scholar]
- Bollag J.-M.; Liu S.-Y.; Minard R. D. Asymmetric diphenol formation by a fungal laccase. Appl. Environ. Microbiol. 1979, 38, 90–92. 10.1128/AEM.38.1.90-92.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anyanwutaku I. O.; Petroski R. J.; Rosazza J. P. N. Oxidative coupling of mithramycin and hydroquinone catalyzed by copper oxidases and benzoquinone. Implications for the mechanism of action of aureolic acid antibiotics. Bioorg. Med. Chem. 1994, 2, 543–551. 10.1016/0968-0896(94)80025-1. [DOI] [PubMed] [Google Scholar]
- Bollag J.-M.; Liu S.-Y. Copolymerization of halogenated phenols and syringic acid. Pestic. Biochem. Phys. 1985, 23, 261–272. 10.1016/0048-3575(85)90014-8. [DOI] [Google Scholar]
- Tatsumi K.; Wada S.; Ichikawa H.; Liu S. Y.; Bollag J.-M. Cross-coupling of a chloroaniline and phenolic acids catalyzed by a fungal enzyme. Wat. Sci. Tech. 1992, 26, 2157–2160. 10.2166/wst.1992.0685. [DOI] [Google Scholar]
- Manda K.; Hammer E.; Mikolasch A.; Niedermeyer T.; Dec J.; Jones A. D.; Benesi A. J.; Schauer F.; Bollag J.-M. Laccase-induced cross-coupling of 4-aminobenzoic acid with para-dihydroxylated compounds 2,5-dihydroxy-N-(2-hydroxyethyl)-benzamide and 2,5-dihydroxybenzoic acid methyl ester. J. Mol. Catal. B: Enzym. 2005, 35, 86–92. 10.1016/j.molcatb.2005.06.001. [DOI] [Google Scholar]
- Niedermeyer T. H. J.; Mikolasch A.; Lalk M. Nuclear amination catalyzed by fungal laccases: Reaction products of p-hydroquinones and primary aromatic amines. J. Org. Chem. 2005, 70, 2002–2008. 10.1021/jo048454s. [DOI] [PubMed] [Google Scholar]
- Manda K.; Hammer E.; Mikolasch A.; Gördes D.; Thurow K.; Schauer F. Laccase-induced derivatization of unprotected amino acid L-tryptophan by coupling with p-hydroquinone 2,5-dihydroxy-N-(2-hydroxyethyl)-benzamide. Amino Acids 2006, 31, 409–419. 10.1007/s00726-005-0276-8. [DOI] [PubMed] [Google Scholar]
- Mikolasch A.; Niedermeyer T. H. J.; Lalk M.; Witt S.; Seefeldt S.; Hammer E.; Schauer F.; Gesell M.; Hessel S.; Jülich W.-D.; Lindequist U. Novel penicillins synthesized by biotransformation using laccase from Trametes spec. Chem. Pharm. Bull. 2006, 54, 632–638. 10.1248/cpb.54.632. [DOI] [PubMed] [Google Scholar]
- Mikolasch A.; Niedermeyer T. H. J.; Lalk M.; Witt S.; Seefeldt S.; Hammer E.; Schauer F.; Salazar M. G.; Hessel S.; Jülich W.-D.; Lindequist U. Novel cephalosporins synthesized by amination of 2,5-dihydroxybenzoic acid derivatives using fungal laccases II. Chem. Pharm. Bull. 2007, 55, 412–416. 10.1248/cpb.55.412. [DOI] [PubMed] [Google Scholar]
- Mikolasch A.; Hessel S.; Salazar M. G.; Neumann H.; Manda K.; Go̅rdes D.; Schmidt E.; Thurow K.; Hammer E.; Lindequist U.; Beller M.; Schauer F. Synthesis of new N-analogous corollosporine derivatives with antibacterial activity by laccase-catalyzed amination. Chem. Pharm. Bull. 2008, 56, 781–786. 10.1248/cpb.56.781. [DOI] [PubMed] [Google Scholar]
- Mikolasch A.; Matthies A.; Lalk M.; Schauer F. Laccase-induced C–N coupling of substituted p-hydroquinones with p-aminobenzoic acid in comparison with known chemical routes. Appl. Microbiol. Biotechnol. 2008, 80, 389–397. 10.1007/s00253-008-1595-y. [DOI] [PubMed] [Google Scholar]
- Mikolasch A.; Wurster M.; Lalk M.; Witt S.; Seefeldt S.; Hammer E.; Schauer F.; Jülich W.-D.; Lindequist U. Novel beta-lactam antibiotics synthesized by amination of catechols using fungal laccase. Chem. Pharm. Bull. 2008, 56, 902–907. 10.1248/cpb.56.902. [DOI] [PubMed] [Google Scholar]
- Hahn V.; Mikolasch A.; Manda K.; Gördes D.; Thurow K.; Schauer F. Laccase-catalyzed carbon-nitrogen bond formation: coupling and derivatization of unprotected l-phenylalanine with different para-hydroquinones. Amino Acids 2009, 37, 315–321. 10.1007/s00726-008-0154-2. [DOI] [PubMed] [Google Scholar]
- Hahn V.; Mikolasch A.; Wende K.; Bartrow H.; Lindequist U.; Schauer F. Derivatization of the azole 1-aminobenzotriazole using laccase of Pycnoporus cinnabarinus and Myceliophthora thermophila: influence of methanol on the reaction and biological evaluation of the derivatives. Biotechnol. Appl. Biochem. 2010, 56, 43–48. 10.1042/BA20100078. [DOI] [PubMed] [Google Scholar]
- Mikolasch A.; Manda K.; Schlüter R.; Lalk M.; Witt S.; Seefeldt S.; Hammer E.; Schauer F.; Jülich W.-D.; Lindequist U. Comparative analyses of laccase-catalyzed amination reactions for production of novel β-lactam antibiotics. Biotechnol. Appl. Biochem. 2012, 59, 295–306. 10.1002/bab.1026. [DOI] [PubMed] [Google Scholar]
- Mikolasch A.; Hildebrandt O.; Schlüter R.; Hammer E.; Witt S.; Lindequist U. Targeted synthesis of novel β-lactam antibiotics by laccase-catalyzed reaction of aromatic substrates selected by pre-testing for their antimicrobial and cytotoxic activity. Appl. Microbiol. Biotechnol. 2016, 100, 4885–4899. 10.1007/s00253-016-7288-z. [DOI] [PubMed] [Google Scholar]
- Wellington K. W.; Bokako R.; Raseroka N.; Steenkamp P. A one-pot synthesis of 1,4-naphthoquinone-2,3-bis-sulfides catalysed by a commercial laccase. Green Chem. 2012, 14, 2567–2576. 10.1039/c2gc35926j. [DOI] [Google Scholar]
- Hahn V.; Davids T.; Lalk M.; Schauer F.; Mikolasch A. Enzymatic cyclizations using laccases: Multiple bond formation between dihydroxybenzoic acid derivatives and aromatic amines. Green Chem. 2010, 12, 879–887. 10.1039/b920081a. [DOI] [Google Scholar]
- Cannatelli M. D.; Ragauskas A. J. Laccase-catalyzed synthesis of 2,3-ethylenedithio-1,4-quinones. J. Mol. Catal. B-Enzym. 2015, 119, 85–89. 10.1016/j.molcatb.2015.05.016. [DOI] [Google Scholar]
- Hahn V.; Mikolasch A.; Kuhlisch C.; Schauer F. Laccase-mediated multi-step homo- and heteromolecular reactions of ortho-dihydroxylated aromatic compounds and mono- or diaminated substances resulting in C-C, C-O and C-N bonds. J. Mol. Catal. B-Enzym. 2015, 56–63. 10.1016/j.molcatb.2015.08.011. [DOI] [Google Scholar]
- Hosny M.; Rosazza J. P. N. Novel oxidations of (+)-catechin by horseradish peroxidase and laccase. J. Agric. Food Chem. 2002, 50, 5539–5545. 10.1021/jf020503j. [DOI] [PubMed] [Google Scholar]
- Abdel-Mohsen H. T.; Conrad J.; Harms K.; Nohr D.; Beifuss U. Laccase-catalyzed green synthesis and cytotoxic activity of novel pyrimidobenzothiazoles and catechol thioethers. RSC Adv. 2017, 7, 17427–17441. 10.1039/C6RA28102H. [DOI] [Google Scholar]
- Kudanga T.; Prasetyo E. N.; Sipilä J.; Nousiainen P.; Widsten P.; Kandelbauer A.; Nyanhongo G. S.; Guebitz G. Laccase-mediated wood surface functionalization. Eng. Life Sci. 2008, 8, 297–302. 10.1002/elsc.200800011. [DOI] [Google Scholar]
- Basta-Kaim A.; Budziszewska B.; Jaworska-Feil L.; Tetich M.; Kubera M.; Leśkiewicz M.; Otczyk M.; Lasoń W. Antipsychotic drugs inhibit the human corticotropin-releasing-hormone gene promoter activity in neuro-2A cells - an involvement of protein kinases. Neuropsychopharmacology 2006, 31, 853–865. 10.1038/sj.npp.1300911. [DOI] [PubMed] [Google Scholar]
- Takahashi K.; Takahashi I.; Morimoto M.; Tomita F. DC-86-M, a novel antitumor antibiotic. II. Structure determination and biological activities. J. Antibiot. 1986, 39, 624–628. 10.7164/antibiotics.39.624. [DOI] [PubMed] [Google Scholar]
- Zhao Y.-q.; Yin Y.-q.; Liu J.; Wang G.-h.; Huang J.; Zhu L.-j.; Wang J.-h. Characterization of HJ-PI01 as a novel Pim-2 inhibitor that induces apoptosis and autophagic cell death in triple-negative human breast cancer. Acta Pharmacol. Sin. 2016, 37, 1237–1250. 10.1038/aps.2016.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.; Ming C.; Zhang W.; Okechukwu P. N.; Morak-Mlodawska B.; Pluta K.; Jeleń M.; Akim A. M.; Ang K.-P.; Ooi K. K. 10H-3,6-Diazaphenothiazine induces G2/M phase cell cycle arrest and caspase-dependent apoptosis and inhibits cell invasion of A2780 ovarian carcinoma cells through the regulation of NF-kappa B and (BIRC6-XIAP) complexes. Drug Des. Dev. Ther. 2017, 11, 3045–3063. 10.2147/DDDT.S144415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waksman S. A.; Geiger W. B.; Reynolds D. M. Strain specificity and production of antibiotic substances. VII. Production of actinomycin by different actinomycetes. P. Natl. Acad. Sci. USA 1946, 32, 117–120. 10.1073/pnas.32.5.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger A.; Keller-schierlein W.; Brandl M.; Zähner H. Metabolites of microorganisms. 247. Phenazines from Streptomyces antibioticus, strain TÜ 2706. J Antibiot 1988, 41, 1542–1551. 10.7164/antibiotics.41.1542. [DOI] [PubMed] [Google Scholar]
- Bansode T. N.; Shelke J. V.; Dongre V. G. Synthesis and antimicrobial activity of some new N-acyl substituted phenothiazines. Eur. J. Med. Chem. 2009, 44, 5094–5098. 10.1016/j.ejmech.2009.07.006. [DOI] [PubMed] [Google Scholar]
- Kolaczkowski M.; Michalak K.; Motohashi N. Phenothiazines as potent modulators of yeast multidrug resistance. International Journal of Antimicrobial Agents 2003, 22, 279–283. 10.1016/S0924-8579(03)00214-0. [DOI] [PubMed] [Google Scholar]
- Bisi A.; Meli M.; Gobbi S.; Rampa A.; Tolomeo M.; Dusonchet L. Multidrug resistance reverting activity and antitumor profile of new phenothiazine derivatives. Bioorgan. Med. Chem. 2008, 16, 6474–6482. 10.1016/j.bmc.2008.05.040. [DOI] [PubMed] [Google Scholar]
- Fingerle M.; Hemgesberg M.; Lach S.; Thiel W. R.; Ziegler C. Symmetrically substituted phenothiazine as prospective candidate for UV responsive dye sensitized solar cells. Thin Solid Films 2015, 591, 8–12. 10.1016/j.tsf.2015.07.032. [DOI] [Google Scholar]
- Weng Y.-L.; Li Y.-C.; Chen C.-P.; Chang Y. J. Effect of intermolecular interaction with phenothiazine core on inverted organic photovoltaics by using different acceptor moiety. Dyes Pigm. 2017, 146, 374–385. 10.1016/j.dyepig.2017.07.027. [DOI] [Google Scholar]
- Xie Y.; Fujimoto T.; Dalgleish S.; Shuku Y.; Matsushita M. M.; Awaga K. Synthesis, optical properties and charge transport characteristics of a series of novel thiophene-fused phenazine derivatives. J. Mater. Chem. C 2013, 1, 3467–3481. 10.1039/c3tc30346b. [DOI] [Google Scholar]
- Pearson R. M.; Lim C.-H.; McCarthy B. G.; Musgrave C. B.; Miyake G. M. Organocatalyzed atom transfer radical polymerization using N-aryl phenoxazines as photoredox catalysts. J. Am. Chem. Soc. 2016, 138, 11399–11407. 10.1021/jacs.6b08068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma D.; Geng Q.; Zhang H.; Jiang Y. Angew. Chem., Int. Ed. 2010, 49, 1291–1294. 10.1002/anie.200905646. [DOI] [PubMed] [Google Scholar]
- Sun X.; Tu X.; Dai C.; Zhang X.; Zhang B.; Zeng Q. Palladium-catalyzed C-N cross coupling of sulfinamides and aryl halides. J. Org. Chem. 2012, 77, 4454–4459. 10.1021/jo3003584. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Wang H.; Yang B.; Fan R. Imino exchange reaction in a dearomatization strategy: synthesis of N-acyl diarylamines and phenothiazines from two anilines. Org. Chem. Front. 2014, 1, 1055–1057. 10.1039/C4QO00201F. [DOI] [Google Scholar]
- Hu W.; Zhang S. Method for the synthesis of phenothiazines via a domino iron-catalyzed C-S/C-N cross-coupling reaction. J. Org. Chem. 2015, 80, 6128–6132. 10.1021/acs.joc.5b00568. [DOI] [PubMed] [Google Scholar]
- Chen J.; Li G.; Xie Y.; Liao Y.; Xiao F.; Deng G.-J. Four-component approach to N-substituted phenothiazines under transition-metal-free conditions. Org. Lett. 2015, 17, 5870–5873. 10.1021/acs.orglett.5b03058. [DOI] [PubMed] [Google Scholar]
- Lin Y.-m.; Lu G.-p.; Wang R.-K.; Yi W.-b. Radical route to 1,4-benzothiazine derivatives from 2-aminobenzenethiols and ketones under transition-metal-free conditions. Org. Lett. 2016, 18, 6424–6427. 10.1021/acs.orglett.6b03324. [DOI] [PubMed] [Google Scholar]
- Bodea C.; Raileanu M. Über Phenthiazone. I. Chlor-phenthiazone und deren Leukoverbindungen. Justus Liebigs Ann. Chem. 1958, 614, 171–176. 10.1002/jlac.19586140120. [DOI] [Google Scholar]
- Zheng X.; Liu B.; Yang F.; Hu Q.; Yao L.; Hu Y. Access to benzoxazepines and fully substituted indoles via HDDA coupling. Org. Lett. 2020, 22, 956–959. 10.1021/acs.orglett.9b04499. [DOI] [PubMed] [Google Scholar]
- Intra A.; Nicotra S.; Riva S.; Danieli B. Significant and unexpected solvent influence on the selectivity of laccase-catalyzed coupling of tetrahydro-2-naphthol derivatives. Adv. Synth. Catal. 2005, 347, 973–977. 10.1002/adsc.200505043. [DOI] [Google Scholar]
- Ponzoni C.; Beneventi E.; Cramarossa M. R.; Raimondi S.; Trevisi G.; Pagnoni U. M.; Riva S.; Forti L. Laccase-catalyzed dimerization of hydroxystilbenes. Adv. Synth. Catal. 2007, 349, 1497–1506. 10.1002/adsc.200700043. [DOI] [Google Scholar]
- Bruyneel F.; Payen O.; Rescigno A.; Tinant B.; Marchand-Brynaert J. Laccase-mediated synthesis of novel substituted phenoxazine chromophores featuring tuneable water solubility. Chem. – Eur. J. 2009, 15, 8283–8295. 10.1002/chem.200900681. [DOI] [PubMed] [Google Scholar]
- Hahn V.; Mikolasch A.; Manda K.; Gördes D.; Thurow K.; Schauer F. Derivatization of amino acids by fungal laccases: Comparison of enzymatic and chemical methods. J. Mol. Catal. B: Enzym. 2009, 60, 76–81. 10.1016/j.molcatb.2009.04.002. [DOI] [Google Scholar]
- Pickel B.; Constantin M.-A.; Pfannstiel J.; Conrad J.; Beifuss U.; Schaller A. An Enantiocomplementary Dirigent Protein for the Enantioselective Laccase-Catalyzed Oxidative Coupling of Phenols. Angew. Chem., Int. Ed. 2010, 49, 202–204. 10.1002/anie.200904622. [DOI] [PubMed] [Google Scholar]
- Kunamneni A.; Camarero S.; García-Burgos C.; Plou F. J.; Ballesteros A.; Alcalde M. Engineering and applications of fungal laccases for organic synthesis. Microb. Cell. Fact. 2008, 32. 10.1186/1475-2859-7-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunamneni A.; Plou F. J.; Ballesteros A.; Alcalde M. Laccases and their applications: A patent review. Recent Pat. Biotechnol. 2008, 2, 10–24. 10.2174/187220808783330965. [DOI] [PubMed] [Google Scholar]
- Tatsumi K.; Freyer A.; Minard R. D.; Bollag J.-M. Enzymatic coupling of chloroanilines with syringic acid, vanillic acid and protocatechuic acid. Soil Biol. Biochem. 1994, 26, 735–742. 10.1016/0038-0717(94)90266-6. [DOI] [Google Scholar]
- Gripenberg J. Fungus pigments. I. Cinnabarin, a colouring matter from Trametes cinnabarina Jacq. Acta Chem. Scand. 1951, 5, 590–595. 10.3891/acta.chem.scand.05-0590. [DOI] [Google Scholar]
- Sullivan G.; Henry E. D. Occurrence and distribution of phenoxazinone pigments in the genus Pycnoporus. J. Pharm. Sci. 1971, 60, 1097–1098. 10.1002/jps.2600600725. [DOI] [PubMed] [Google Scholar]
- Gilman H.; Moore L. O. The preparation of some 10-substituted phenoxazines. J. Am. Chem. Soc. 1957, 79, 3485–3487. 10.1021/ja01570a048. [DOI] [Google Scholar]
- Bonvicino G.; Yogodzinski L. H.; Hardy R. A. Synthesis of 2-Chloro-10-(3-dimethylaminopropyl)phenoxazine: a The o-phenoxyaniline route; b a modification of turpin reaction. J. Org. Chem. 1961, 26, 2797–2803. [Google Scholar]
- Liu N.; Wang B.; Chen W.; Liu C.; Wang X.; Hu Y. A general route for synthesis of N-aryl phenoxazines via copper(I)-catalyzed N-, N-, and O-arylations of 2-aminophenols. RSC Adv. 2014, 4, 51133–51139. 10.1039/C4RA09593F. [DOI] [Google Scholar]
- Turner J. M.; Messenger A. J. Occurrence, biochemistry and physiology of phenazine pigment production. Adv. Microb. Physiol. 1986, 27, 211–275. 10.1016/S0065-2911(08)60306-9. [DOI] [PubMed] [Google Scholar]
- Laursen J. B.; Nielsen J. Phenazine natural products: Biosynthesis, synthetic analogues, and biological activity. Chem. Rev. 2004, 104, 1663–1686. 10.1021/cr020473j. [DOI] [PubMed] [Google Scholar]
- Chaudhary A.; Khurana J. M. Synthetic routes for phenazines: an overview. Res. Chem. Intermediat. 2018, 44, 1045–1083. 10.1007/s11164-017-3152-8. [DOI] [Google Scholar]
- Prat D.; Wells A.; Hayler J.; Sneddon H.; McElroy C. R.; Abou-Shehada S.; Dunn P. J. CHEM21 selection guide of classical- and less classical-solvents. Green Chem. 2015, 17, 288–296. [Google Scholar]
- Manda K.; Gördes D.; Mikolasch A.; Hammer E.; Schmidt E.; Thurow K.; Schauer F. Carbon-oxygen bond formation by fungal laccases: cross-coupling of 2,5-dihydroxy-N-(2-hydroxyethyl)-benzamide with the solvents water, methanol, and other alcohols. Appl. Microbiol. Biotechnol. 2007, 76, 407–416. 10.1007/s00253-007-1024-7. [DOI] [PubMed] [Google Scholar]
- Hahn V.; Mikolasch A.; Wende K.; Bartrow H.; Lindequist U.; Schauer F. Synthesis of model morpholine derivatives with biological activities by laccase-catalysed reactions. Biotechnol. Appl. Biochem. 2009, 54, 187–195. 10.1042/BA20090219. [DOI] [PubMed] [Google Scholar]
- Eggert C.; Temp U.; Dean J. F. D.; Eriksson K. E. L. Laccase-mediated formation of the phenoxazinone derivative, cinnabarinic acid. FEBS Lett. 1995, 376, 202–206. 10.1016/0014-5793(95)01274-9. [DOI] [PubMed] [Google Scholar]
- Sousa A. C.; Oliveira M. C.; Martins L. O.; Robalo M. P. Towards the rational biosynthesis of substituted phenazines and phenoxazinones by laccases. Green Chem. 2014, 16, 4127–4136. 10.1039/C4GC00901K. [DOI] [Google Scholar]
- Chakraborty M.; McConville D. B.; Niu Y.; Tessier C. A.; Youngs W. J. Reactions of primary and secondary amines with substituted hydroquinones: Nuclear amination, side-chain amination, and indolequinone formation. J. Org. Chem. 1998, 63, 7563–7567. 10.1021/jo980541v. [DOI] [PubMed] [Google Scholar]
- Mather B. D.; Viswanathan K.; Miller K. M.; Long T. E. Michael addition reactions in macromolecular design for emerging technologies. Prog. Polym. Sci. 2006, 31, 487–531. 10.1016/j.progpolymsci.2006.03.001. [DOI] [Google Scholar]
- Wellington K. W.; Gordon G. E. R.; Ndlovu L. A.; Steenkamp P. Laccase-catalyzed C-S and C-C coupling for a one-pot synthesis of 1,4-naphthoquinone sulfides and 1,4-naphthoquinone sulfide dimers. Chem. Cat. Chem. 2013, 5, 1570–1577. [Google Scholar]
- Anastas P. T.; Warner J. C.. Green Chemistry: Theory and Practice; Oxford University Press: New York, 1998. [Google Scholar]
- Eggert C.; Temp U.; Eriksson K. E. L. The ligninolytic system of the white rot fungus Pycnoporus cinnabarinus: Purification and characterization of the laccase. Appl. Environ. Microbiol. 1996, 62, 1151–1158. 10.1128/AEM.62.4.1151-1158.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourbonnais R.; Paice M. G. Oxidation of nonphenolic substrates - an expanded role for laccase in lignin biodegradation. FEBS Lett. 1990, 267, 99–102. 10.1016/0014-5793(90)80298-W. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.