

Abstract
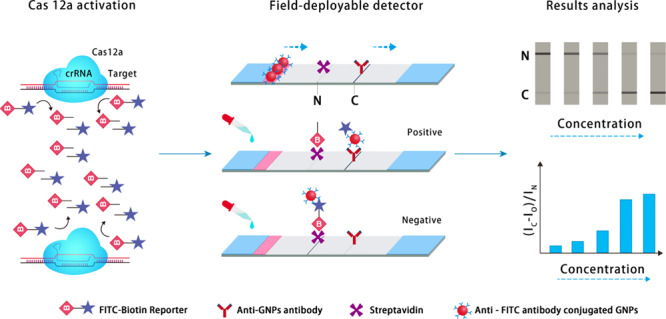
Field-deployable detectors of disease biomarkers provide a simple and fast analysis of clinical specimens. However, most of the existing field-deployable diagnostics have poor sensitivity and are not suitable for the detection of biomarkers with low abundance. Herein, we report a highly sensitive and rapid colorimetric readout paper-based assay for pathogen detection by integrating the unique collateral activity of a Cas12a-activated universal field-deployable detector (CUFD). The collateral effect of Cas12a results in a nonspecific destruction of a fluorophore biotin-labeled ssDNA reporter for the CUFD. This technique can quantify seven different kinds of pathogens in blood samples without any purification procedure, with sensitivity as low as 10 aM for the Shigella dysenteriae DNA. This CUFD technique has significant potential for the detection of pathogenic DNA as well as other types of DNA or RNA targets at the point-of-care application.
Introduction
Field-deployable detectors, such as lateral flow detection (LFD), are broadly employed in various areas because of their simplicity (one-step procedure), rapidity (5–10 min), and low cost (1–10 $).1,2 However, early works of LFD usually suffered from a high background signal, poor sensitivity, and a narrowly single target, not synchronous multiple targets.3,4 The performance of current methods for LFD-based biomarker detection has been rapidly upgraded by incorporating numerous kinds of enzymes as a reporter system.5−7
Cas12a is a member of CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats)-associated enzymes.8−10 It has revealed unique collateral DNase activity (destruction of nearby nontargeted DNAs) making it a fascinating tool for pathogenic diagnostics. By equipping Cas12a with a fluorophore quencher (FQ)-labeled reporter system, Doudna and her colleagues first created a DNA Endonuclease-Targeted CRISPR Trans Reporter (DETECTR) for ultrasensitive and specific analysis of human papillomavirus (HPV) in a clinical environment.11 Although FQ-labeled reporter systems have been employed by us and other groups,12−15 they usually need specialized and costly equipment restraining them from point-of-care diagnostics or in resource-restrained areas. Recently, a Cas12a-based electrochemical biosensor was designed for viral nucleic acid detection, which is more cost-saving than fluorescence-based platforms.16 Many research groups endeavor to develop field-deployable sensing platforms with good portability. Inspired by DETECTR, DNA probe-modified gold nanoparticles (GNPs) have been used to generate colorimetric readouts that can be judged by the naked eye for the detection of the red blotch viral DNA in grapevines.17,18 Another naked-eye readout of analytes was achieved by Cas12a-mediated visual assays to show that single nucleotide polymorphism (SNP) targets could be detected via Pt nanoparticle (PtNP)-catalyzed oxygen generation on a volumetric bar-chart chip.19
Recently, it was found that Cas12a can degrade DNA in a smart gel, thereby translating the recognition event into material property changes. For example, upon activation, Cas12a modulated the permeability of a smart gel-based microfluidic chip with both direct visual and electronic readouts for nucleic acid detection.20 However, it is hard to integrate with downstream hardware devices for accurate result analysis and processing. Here, we developed a rapid visual platform, named Cas12a-activated universal field-deployable detector (CUFD), by a combination of paper strips in a one-pot test. By coupling the CUFD assay with the molecular amplification technique, we illustrated the naked-eye observation platform to detect pathogenic infections in diverse clinical blood specimens collected from hospitals (Figure 1). The CUFD read-out depends on the degradation of the FITC–biotin reporter, allowing for diagnoses using commercial paper strips. In detail, an abundant amount of FITC–biotin reporters first bind to specific antiFITC antibody–GNP conjugates at the conjugate pad of the strip, when they diffuse over the area with the biotin ligand molecule at the noncleavaged line (N line) and produce a black line over several minutes. When the FITC–biotin reporter is destructed, the not-captured GNP conjugates pass through the N line and will be captured in the cleavaged line (C line) by the antiGNP antibody. The ratio (IC – Io)/IN indicates the amount of unleashed FITC molecules as well as the amount of nucleic acid targets in the system. The goal of the CUFD technique was fabricating a direct visual read-out that enables naked-eye observation and minimal clinical training to operate, which provides the translation of gene-editing enzymes for portable applications and precision medicine.
Figure 1.

Cas12a-activated field-deployable detector for nucleic acids and bacterial diagnostics. When the FITC (fluorescein isothiocyanate)–biotin reporter is destructed, the not-captured GNP conjugates pass through the noncleavaged line (N line) and will be captured in the cleavaged line (C line) by the antiGNP antibody. The ratio (IC – Io)/IN indicates the amount of unleashed FITC molecules as well as the amount of nucleic acid targets in the system. Here, IC, IN, and Io are the intensity of the cleavaged line, non-cleavaged line, and negative control, respectively.
Results and Discussion
The CUFD employs its collateral cleavage activity as the signal generation mechanism. This CUFD technique is both robust and highly sensitive, not only because Cas12a cannot tolerate the mismatches between the target and the nonseed site of gRNA in the first 18 nucleotides21 but also because Cas12 employs a single RuvC domain for RNA-guided cis-/trans-ssDNA or dsDNA cleavage in the presence of a specific DNA target in vitro.22−24 The Cas12a-based CUFD readout depends on the degradation of the FITC–biotin reporter, allowing diagnoses using commercial paper strips. This principle can be applied to develop a rapid and portable paper assay for nucleic acid detection in pathogenic diagnostics. The goal of the CUFD technique was fabricating a direct visual readout with minimal training to operate.
To verify the key collateral activity of the CUFD, Cas12a was biosynthesized and its activity was optimized (Figures 2 and S1). Both gel analysis (Figure 2) and FQ-labeled reporter assay (Figure S1) were used to optimize Cas12a activity in different buffers (NEB buffers, CutSmart buffer, and HEPES buffer), pH values (4.0–11.0), metal ions (Mg2+, Ca2+, Mn2+, Co2+, Ni2+, Zn2+, and Cu2+), incubation times (5–60 min), nucleic acid lengths (299–900 bp), and serum concentration contents (0–30%) (Figures 2 and S1). A band just above 180 kD was induced by IPTG, consistent with the size of the MBP–Cas12a fusion (192.1 kD). Upon the addition of TEV protease, a lower molecular weight band appeared, consistent with the size of free LbCas12a of 143.7 kD (Figure 2a). Clear DNA cleavage was observed at 5 min; with the prolongation of the reaction time, the substrates gradually decreased and the products gradually increased (Figure 2b). Compared with AsCas12a and FnCas12a, LbCas12a has better cleavage activity (Figure 2c). Cas12a had a better cleavage activity in Buffers QG1–5 and a worse cleavage activity in Buffer QG6 (Figure 2d). DNA cleavage was observed in the presence of Mg2+, Ca2+, and Mn2+; the addition of Co2+, Ni2+, Zn2+, and Cu2+ to the reaction did not result in DNA cleavage (Figure 2e). As displayed in Figures 2 and S1, under the optimized conditions of 10 mM Mg2+ in NEB buffer 2.1 (pH7.0), the real time fluorescence response of a Cas12a-based system has the highest response intensity. Also, the length of the target nucleic acid ranging from 299 to 900 bp seems to not influence the result, as indicated by Figures S1c and 2f.
Figure 2.

Cas12a characterization and optimization. (a) Stepwise purification of Coomassie blue-stained acrylamide gel of Cas12a. (b) Cas12a cleavage activity with crRNA (1 and 2) incubation ranging from 0 to 60 min. (c) Cas12a cleavage activity from Francisella novicida (Fn), Acidaminococcus sp. (As), and Lachnospiraceae bacterium ND2006 (Lb), as confirmed with different crRNAs (1 and 2). (d) Cas12a cleavage activity with both crRNA (1 and 2) in different buffers (Buffer QG1/NEBuffer 1.1; Buffer QG2/NEBuffer 2.1; Buffer QG3/NEBuffer 3.1; Buffer QG4/CutSmart Buffer; Buffer QG5/NEBuffer 3; Buffer QG6/1 × cleavage buffer/20 mM HEPES (pH 7.5), 150 mM KCl, 10 mM MgCl2, 1% glycerol, and 0.5 mM DTT). (e) Cas12a cleavage activity with different metal ions with 1 or 10 mM concentration. (f) Cas12a cleavage activity with crRNA (1, 2, and S as control) for different target lengths from 299 to 900 bp. crRNA 1: TAATTTCTACTCTTGTAGATAGGAGTGTTCAGTCTCCGTGAAC; crRNA 2: TAATTTCTACTCTTGTAGATCTGATGGTCCATGTCTGTTACTC.
We next studied the feasibility of Cas12a-collateral cleavage assay as a generic way to detect the presence of a series of pathogens (Figures 3 and S3). The highly conservative species-specific gene regions (iap gene for Listeria monocytogenes,25khe gene for Klebsiella pneumoniae,26tuf gene for Streptococcus pyogenes,27nuc gene for Staphylococcus aureus,28chuA for Escherichia coli,29 and invA for S. typhi(30)) of seven pathogens were used for pathogenic detection and phenotyping. Different lengths of pathogenic genera ranging from 178 to 418 bp were selected as target regions, and specific primers were screened using MegAlign software. The primer specificity of seven different pathogens was then confirmed by gel electrophoresis (Figures 3a–c and S2). Figures 3, S2 and S3 display the genotype of target genes from seven bacteria via Cas12a-collateral cleavage combined with different specific primers or crRNA species. The Cas12a-based fluorescence method can selectively detect each pathogen with ultralow cross-reactivity and minimal off-target efficiency. The discrimination factors of the method based on primers or crRNAs using the Wilcoxon test were between 0.65 and 1.27 or 0.95 and 1.34, respectively.31,32 The coefficient of variation of intraassay and interassay obtained using this method for L. monocytogenes with the same concentration is 11.1 and 8.2%, respectively (Figure S4). This result indicates a high reproducibility.
Figure 3.

Cas12a-based differential detection for nucleic acid detection from pathogenic bacteria. (a–c) Gel imaging of typical bacterial amplicons, including L. monocytogenes (a), K. pneumoniae (b), and S. pyogenes (c). (d–k) Fluorescence response for the differential detection of various bacterial types, including L. monocytogenes (d), K. pneumoniae (e), S. pyogenes (f), S. aureus (g), E. coli (h), S. dysenteriae (i),S. typhi (j), and specificity differential detection for seven pathogenic bacteria (k) dependent on different specific crRNAs (n = 3 technical replicates; bars represent mean ± s.d.).
We then examined the compatibility of the field-deployable detection with the CRISPR-Cas12a system (Figure 4). To study the quantification capability of the system, the synthetic S. dysenteriae DNA was diluted using a buffer solution. For the target of S. dysenteriae, the limit of detection (LOD) of the system could reach 102 aM using CUFD or 10 aM using the fluorescent readout (Figure 4a,b,e). Moreover, S. dysenteriae can be specifically detected using the CUFD (Figure 4f). We also found that Cas12a can tolerate up to two mismatches in the crRNA/target complex (Figure 4c,d). Remarkably, the Cas12a-based fluorescence method (10 aM of LOD) was much more sensitive than qPCR, as the nucleic acid targets with attomole concentration could hardly be measured by qPCR (Figure S5). The LOD of the fluorescent readout for other organisms ranges from 102 to 104 aM (Figure S6). The high sensitivity of CUFD was attributed to the catalytic capability of Cas12a to amplify the recognition event with the target DNA sequence. We found that CUFD enables the specific detection of S. dysenteriae and other bacteria in samples (Figures 4 and S7). These results demonstrated that the CUFD could be used as an ultrasensitive and accurate analysis tool for pathogenic diagnostics and profiling and accommodate it to be a point-of-care or on-site platform.
Figure 4.

Adapting Cas12a for LFD. (a–d) Kinetics (a) and correlation (b) of Cas12a-based detection for the S. dysenteriae DNA synthetic target and SNP analysis (c,d) for nucleotide mismatch using the Cas12a-based fluorescence method; (e,f) CUFD results from Figure S9 achieve the LOD (e) and specific detection (f) of S. dysenteriae (n = 3 technical replicates, bars represent mean ± s.d.). Negative control is dH2O. L. monocytogenes (L.M.), K. pneumoniae (K.P.), S. pyogenes (S.P.), S. aureus (S.A.), E. coli (E.C.), and S. dysenteriae (S.D.).
Having verified that LFD is fully compatible with Cas12a, we further applied our CUFD assay for diagnosing pathogenic infections without an extraction step. To measure the nucleic acid target directly from bacterial samples via LFD, we designed a rapid CUFD, a technique to lyse bacterial cells and neutralize the reaction system with the utilization of 1 M NaOH and Tris–HCl. The rapid CUFD permitted the sensitive detection of pathogenic cells from a sample mimicking infection at 104 CFU/mL for S. dysenteriae (Figure S8). The total time of assay was ca. 1 h with the fluorescent readout and colorimetric methods using the CUFD. The assay clearly leads to less variability with minimal requirement for complicated sample preparation and nucleic acid extraction.
We finally challenged our CUFD assay by analyzing the clinical blood samples collected from the hospital (Figure 5). Six samples from patients with suspected infections and one negative sample were obtained and detected by the traditional culture technique (assay time, 1–3 days) as well as by the CUFD (assay time, 1–2 h). Before bacterial DNA extraction, the samples were first placed in tubes containing the ACK lysing buffer (Gibco) to remove the red blood cells. The procedure for DNA extraction using the Bacterial DNA Kit (OMEGA) was then continued as for a single bacterial culture. These bacterial DNA were detected in parallel with the CUFD and the Cas12a-based fluorescent tool. Figure 5 displays the results from both the assay. In these cases, four samples were infected with K. pneumoniae and two samples with E. coli, as confirmed by both methods. The Cas12a-based fluorescence technique (Figure 5a) and colorimetric methods (Figure 5b,c) demonstrated good specificity and low-background response for pathogenic diagnostics in the serum samples. The results were in good agreement with that measured by the technique, indicating the potential of the CUFD for infectious disease diagnosis. Although the results of the CUFD are promising, further investigations on larger prospective clinical applications are required.
Figure 5.

Diagnosis of pathogens using the CUFD in blood samples. Detection of bacteria by the Cas12a-based fluorescence method (a) and the CUFD method (b,c) using seven specific crRNA probes for each bacteria type. Clinical specimens (1 mL for each sample) were processed. One of the seven clinical specimens was negative control (NC), which conformed with the standard culture method. (n = 3 technical replicates, bars represent mean ± s.d.).
Rapid and highly sensitive bacterial diagnosis is crucial for enhancing clinical patient care with suitable drug utilization, avoiding the mushrooming of infectious diseases, and confirming the source of pathogenic infection. To date, numerous diagnostic techniques for pathogenic nucleic acids have been developed, each of which varies in specificity, simplicity, and sensitivity.33−37 Strategies depending on time-of-flight mass spectrometry (TOF-MS) have displayed useful promise as a rapid tool for pathogenic identification.38−40 However, TOF-MS-based instruments are too high-priced for resource-poor conditions. Recently, a magneto-DNA nanoparticle platform for the rapid detection of clinical pathogens allows specific detection of 13 bacterial species, with sensitivity down to a single bacterium in 2 h41 Several technical issues, such as the susceptibility for false positive results, have been huge barriers for its utilization. Here, our CUFD allowing a direct and simple readout of targets with excellent advantages, such as low cost and excellent sensitivity, would be exceptionally beneficial for medical diagnostics.
Conclusions
Together, we created a CUFD platform that generates a direct visual strip readout for pathogenic diagnostics with minimal instrumentation and sample manipulation. This method fills the gap in the current nucleic acid diagnostic for point-of-care techniques for the portable diagnosis of pathogenic diseases. The CUFD system as a general platform can also be conveniently transferable to a lot of other important biomarkers. This platform has great potential to distinctly decrease the analysis time and the test cost for clinical precision medicine in developing areas. Further investigation integrated with an automated device for the direct and accurate readout can extend its utilization to other fields such as quantitative biology.
Experimental Section
Cas12a Expression and Purification
The cloning vector (6His-MBP-TEV-huLbCas12a) was a gift from Feng Zhang (Addgene plasmid # 90096). The transformed E. coli strain transetta DE3 (TransGen Biotech) obtained using an expression vector with His6 tag was incubated at 37 °C in Terrific Broth medium with shaking at 180 rpm until the formation of an early stationary phase (OD600 = 0.6–1.0). To obtain the protein target of interest, the culture with isopropyl-β-d-thiogalactopyranoside (IPTG, 1 mM) was further shaken for 12 h at 21 °C. Bacterial cells were obtained by centrifugation at 10,000 rpm for 5 min at 4 °C and then destructed using a sonicator. The debris was dismantled by centrifugation at 10,000 rpm for 15 min at 4 °C, and the supernatant was added to Bio-Scale Mini Nuvia IMAC Ni-Charged Cartridges (Bio-Rad Laboratories, Inc.). Fractions containing protein were concentrated and then further purified on ENrich SEC 650 high-resolution size exclusion columns (Bio-Rad Laboratories, Inc.) and HiTrap Heparin HP columns (GE Healthcare), respectively.
Bacteria Culture and Nucleic Acid Preparation
All bacteria were provided by the China Center of Industrial Culture Collection (CICC). Bacterial cells are grown to log-phase and harvested by centrifugation at 6000 rpm. for 10 min. After DNA extraction using the Bacterial DNA Kit (E.Z.N.A. Bacterial DNA Kit, OMEGA), the final DNA yield was detected using a NanoDrop microvolume spectrophotometer (Thermo Scientific) and agarose gel electrophoresis (DYY-6D, Beijing Liuyi Biotechnology Co., Ltd).
Rapid Preparation without Nucleic Acid Extraction
S. dysenteriae (50 μL) were mixed with 0.5 M NaOH (50 μL) for 10 min at 60 °C. The supernatant fluid (50 μL) was mixed with 50 μL of 0.5 M Tris–HCl (pH 8.0). The solution was directly used in PCR.
crRNA Preparation
For crRNA synthesis (Table S1), DNA oligonucleotides containing the T7 promoter, repeat, and spacer sequences from Sangon Biotech were annealed with a short T7 primer to form the in vitro transcription templates. Then, the crRNAs were synthesized by incubating at 37 °C for 16 h with the T7 quick high yield RNA synthesis kit (NTP buffer mix, 10 μL; template, 1 μg; T7 RNA polymerase mix, 2 μL; nuclease-free water up to 30 μL). Synthesized crRNAs were purified using the Monarch RNA Cleanup Kit (New England Biolabs). All crRNA sequences used in this assay are shown in Table S1.
Primer Design
Specific virulence gene sequences of each bacterium from the NCBI database were screened to gain a target region of interest (100–500 nucleotides in length) for hybridization using MegAlign software (DNASTAR). All oligonucleotides used for the primers were provided by Sangon Biotech. Primer specificity was performed by agarose gel electrophoresis.
Collateral Fluorescence Assays
Cas12a-mediated collateral fluorescence assays were carried out in cleavage buffer (NEBuffer 2.1) consisting of 250 nM Cas12a, 25 nM crRNA, 125 nM FQ-reporter, 15.0 μL of Target. The reaction (90 μL, 96-well microplate) was incubated using a fluorescence plate reader (BioTek H1 microplate reader) for up to 120 min at 37 °C with fluorescence measurements taken every 2 min (custom ssDNA FQ reporter = λex: 494 nm; λem: 520 nm).
CUFD Assay for Nucleic Acids and Bacteria
The CUFD assay was carried out in cleavage buffer (NEBuffer 2.1) consisting of 60 nM Cas12a, 60 nM crRNA, 1 μM lateral flow-reporter, 3.0 μL (20 μL reactions) of target for 15–20 min at 37 °C. LFD was achieved using commercially available detection strips (Milenia HybriDetect 1, TwistDx, Cambridge, UK). Cas12a detection reactions were diluted in HybriDetect assay buffer (100 μL), and then the strips were inserted and incubated for 2–5 min at room temperature. The strips were finally photographed via a camera and analyzed using ImageJ software (National Institutes of Health).
Acknowledgments
We gratefully acknowledge the support from the National Key Research and Development Program of China (no. 2018YFD0900704), the Hainan Provincial Natural Science Foundation of China (no. 418QN206), the National Natural Science Foundation of China (nos. 41866002 and 21621003), the Open Fund of Shandong Key Laboratory of Corrosion Science (KLCS201910), and the Research Foundation of Hainan University (no. KYQD(ZR)1711).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c01911.
Materials and methods, optimization of Cas12a activities, bacteria genomic DNA amplification using specific primers, collateral fluorescence assays, reproducibility of collateral fluorescence assays, bacterial detection using qPCR, sensitivity analysis using collateral fluorescence assays, and rapid CUFD assay for bacteria (PDF)
Author Contributions
Y.S. and H.L. contributed equally to this work. Y.S., Y.W. and J.L. conceived this study. Y.S., H.L., and Y.S. performed the experiments and analyzed the data. C.L., Y.W., and H.L. designed the experiments. X.H. and F.S. conducted Cas12a purification. X.G., Y.L., and A.W. assisted with crRNA synthesis and analysis. Y.W., H.L., K.Z., and J.L. wrote the manuscript, which was read and approved by all authors.
The authors declare no competing financial interest.
Supplementary Material
References
- Berlina A. N.; Zherdev A. V.; Dzantiev B. B. ELISA and Lateral Flow Immunoassay for the Detection of Food Colorants: State of the Art. Crit. Rev. Anal. Chem. 2019, 49, 209–223. 10.1080/10408347.2018.1503942. [DOI] [PubMed] [Google Scholar]
- Raeisossadati M. J.; Danesh N. M.; Borna F.; Gholamzad M.; Ramezani M.; Abnous K.; Taghdisi S. M. Lateral flow based immunobiosensors for detection of food contaminants. Biosens. Bioelectron. 2016, 86, 235–246. 10.1016/j.bios.2016.06.061. [DOI] [PubMed] [Google Scholar]
- Hu J.; Wang S.; Wang L.; Li F.; Pingguan-Murphy B.; Lu T. J.; Xu F. Advances in paper-based point-of-care diagnostics. Biosens. Bioelectron. 2014, 54, 585–597. 10.1016/j.bios.2013.10.075. [DOI] [PubMed] [Google Scholar]
- Ahmed S.; Bui M.-P. N.; Abbas A. Paper-based chemical and biological sensors: Engineering aspects. Biosens. Bioelectron. 2016, 77, 249–263. 10.1016/j.bios.2015.09.038. [DOI] [PubMed] [Google Scholar]
- Han G.-R.; Ki H.; Kim M.-G. Automated, Universal, and Mass-Producible Paper-Based Lateral Flow Biosensing Platform for High-Performance Point-of-Care Testing. ACS Appl. Mater. Interfaces 2020, 12, 1885. 10.1021/acsami.9b17888. [DOI] [PubMed] [Google Scholar]
- Dai T.; Hu T.; Yang X.; Shen D.; Jiao B.; Tian W.; Xu Y. A recombinase polymerase amplification-lateral flow dipstick assay for rapid detection of the quarantine citrus pathogen in China, Phytophthora hibernalis. Peerj 2019, 7, e8083 10.7717/peerj.8083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myhrvold C.; Freije C. A.; Gootenberg J. S.; Abudayyeh O. O.; Metsky H. C.; Durbin A. F.; Kellner M. J.; Tan A. L.; Paul L. M.; Parham L. A.; Garcia K. F.; Barnes K. G.; Chak B.; Mondini A.; Nogueira M. L.; Isern S.; Michael S. F.; Lorenzana I.; Yozwiak N. L.; MacInnis B. L.; Bosch I.; Gehrke L.; Zhang F.; Sabeti P. C. Field-deployable viral diagnostics using CRISPR-Cas13. Science 2018, 360, 444–448. 10.1126/science.aas8836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- East-Seletsky A.; O’Connell M. R.; Knight S. C.; Burstein D.; Cate J. H. D.; Tjian R.; Doudna J. A. Two distinct RNase activities of CRISPR-C2c2 enable guide-RNA processing and RNA detection. Nature 2016, 538, 270–273. 10.1038/nature19802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S. Y.; Cheng Q. X.; Wang J. M.; Li X. Y.; Zhang Z. L.; Gao S.; Cao R. B.; Zhao G. P.; Wang J. CRISPR-Cas12a-assisted nucleic acid detection. Cell Discovery 2018, 4, 20. 10.1038/s41421-018-0028-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernabé-Orts J. M.; Casas-Rodrigo I.; Minguet E. G.; Landolfi V.; Garcia-Carpintero V.; Gianoglio S.; Vázquez-Vilar M.; Granell A.; Orzaez D. Assessment of Cas12a-mediated gene editing efficiency in plants. Plant Biotechnol. J. 2019, 17, 1971–1984. 10.1111/pbi.13113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J. S.; Ma E.; Harrington L. B.; Da Costa M.; Tian X.; Palefsky J. M.; Doudna J. A. CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science 2018, 360, 436–439. 10.1126/science.aar6245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gootenberg J. S.; Abudayyeh O. O.; Lee J. W.; Essletzbichler P.; Dy A. J.; Joung J.; Verdine V.; Donghia N.; Daringer N. M.; Freije C. A.; Myhrvold C.; Bhattacharyya R. P.; Livny J.; Regev A.; Koonin E. V.; Hung D. T.; Sabeti P. C.; Collins J. J.; Zhang F. Nucleic acid detection with CRISPR-Cas13a/C2c2. Science 2017, 356, 438–442. 10.1126/science.aam9321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K.; Deng R.; Li Y.; Zhang L.; Li J. Cas9 cleavage assay for pre-screening of sgRNAs using nicking triggered isothermal amplification. Chem. Sci. 2016, 7, 4951–4957. 10.1039/c6sc01355d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Teng X.; Zhang K.; Deng R.; Li J. RNA Strand Displacement Responsive CRISPR/Cas9 System for mRNA Sensing. Anal. Chem. 2019, 91, 3989–3996. 10.1021/acs.analchem.8b05238. [DOI] [PubMed] [Google Scholar]
- Zhang K.; Deng R.; Teng X.; Li Y.; Sun Y.; Ren X.; Li J. Direct Visualization of Single-Nucleotide Variation in mtDNA Using a CRISPR/Cas9-Mediated Proximity Ligation Assay. J. Am. Chem. Soc. 2018, 140, 11293–11301. 10.1021/jacs.8b05309. [DOI] [PubMed] [Google Scholar]
- Dai Y.; Somoza R. A.; Wang L.; Welter J. F.; Li Y.; Caplan A. I.; Liu C. C. Exploring the Trans-Cleavage Activity of CRISPR-Cas12a (cpf1) for the Development of a Universal Electrochemical Biosensor. Angew. Chem., Int. Ed. 2019, 58, 17399. 10.1002/anie.201910772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B.; Wang R.; Wang D.; Wu J.; Li J.; Wang J.; Liu H.; Wang Y. Cas12aVDet: A CRISPR/Cas12a-Based Platform for Rapid and Visual Nucleic Acid Detection. Anal. Chem. 2019, 91, 12156–12161. 10.1021/acs.analchem.9b01526. [DOI] [PubMed] [Google Scholar]
- Li Y.; Mansour H.; Wang T.; Poojari S.; Li F. Naked-Eye Detection of Grapevine Red-Blotch Viral Infection Using a Plasmonic CRISPR Cas12a Assay. Anal. Chem. 2019, 91, 11510–11513. 10.1021/acs.analchem.9b03545. [DOI] [PubMed] [Google Scholar]
- Shao N.; Han X.; Song Y.; Zhang P.; Qin L. CRISPR-Cas12a Coupled with Platinum Nanoreporter for Visual Quantification of SNVs on a Volumetric Bar-Chart Chip. Anal. Chem. 2019, 91, 12384–12391. 10.1021/acs.analchem.9b02925. [DOI] [PubMed] [Google Scholar]
- English M. A.; Soenksen L. R.; Gayet R. V.; de Puig H.; Angenent-Mari N. M.; Mao A. S.; Nguyen P. Q.; Collins J. J. Programmable CRISPR-responsive smart materials. Science 2019, 365, 780–785. 10.1126/science.aaw5122. [DOI] [PubMed] [Google Scholar]
- Kleinstiver B. P.; Sousa A. A.; Walton R. T.; Tak Y. E.; Hsu J. Y.; Clement K.; Welch M. M.; Horng J. E.; Malagon-Lopez J.; Scarfò I.; Maus M. V.; Pinello L.; Aryee M. J.; Joung J. K. Engineered CRISPR–Cas12a variants with increased activities and improved targeting ranges for gene, epigenetic and base editing. Nat. Biotechnol. 2019, 37, 276. 10.1038/s41587-018-0011-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gootenberg J. S.; Abudayyeh O. O.; Kellner M. J.; Joung J.; Collins J. J.; Zhang F. Multiplexed and portable nucleic acid detection platform with Cas13, Cas12a, and Csm6. Science 2018, 360, 439–444. 10.1126/science.aaq0179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S.-Y.; Cheng Q.-X.; Liu J.-K.; Nie X.-Q.; Zhao G.-P.; Wang J. CRISPR-Cas12a has both cis- and trans-cleavage activities on single-stranded DNA. Cell Res. 2018, 28, 491–493. 10.1038/s41422-018-0022-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swarts D. C.; van der Oost J.; Jinek M. Structural Basis for Guide RNA Processing and Seed-Dependent DNA Targeting by CRISPR-Cas12a. Mol. Cell 2017, 66, 221–233. 10.1016/j.molcel.2017.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuenscher M. D.; Köhler S.; Bubert A.; Gerike U.; Goebel W. The iap gene of Listeria monocytogenes is essential for cell viability, and its gene product, p60, has bacteriolytic activity. J. Bacteriol. 1993, 175, 3491–3501. 10.1128/jb.175.11.3491-3501.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox B. L.; Schiffer H.; Dagget G.; Beierschmitt A.; Sithole F.; Lee E.; Revan F.; Halliday-Simmonds I.; Beeler-Marfisi J.; Palmour R.; Soto E. Resistance of Klebsiella pneumoniae to the innate immune system of African green monkeys. Vet. Microbiol. 2015, 176, 134–142. 10.1016/j.vetmic.2015.01.001. [DOI] [PubMed] [Google Scholar]
- Picard F. J.; Ke D.; Boudreau D. K.; Boissinot M.; Huletsky A.; Richard D.; Ouellette M.; Roy P. H.; Bergeron M. G. Use of tuf Sequences for Genus-Specific PCR Detection and Phylogenetic Analysis of 28 Streptococcal Species. J. Clin. Microbiol. 2004, 42, 3686–3695. 10.1128/jcm.42.8.3686-3695.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brakstad O. G.; Aasbakk K.; Maeland J. A. Detection of Staphylococcus aureus by polymerase chain reaction amplification of the nuc gene. J. Clin. Microbiol. 1992, 30, 1654–1660. 10.1128/jcm.30.7.1654-1660.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spurbeck R. R.; Dinh P. C. Jr.; Walk S. T.; Stapleton A. E.; Hooton T. M.; Nolan L. K.; Kim K. S.; Johnson J. R.; Mobley H. L. T. Escherichia coli Isolates That Carryvat,fyuA,chuA, andyfcVEfficiently Colonize the Urinary Tract. Infect. Immun. 2012, 80, 4115–4122. 10.1128/iai.00752-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galán J. E.; Curtiss R. 3rd Distribution of the invA, -B, -C, and -D genes of Salmonella typhimurium among other Salmonella serovars: invA mutants of Salmonella typhi are deficient for entry into mammalian cells. Infect. Immun. 1991, 59, 2901–2908. 10.1128/iai.59.9.2901-2908.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limbut W.; Hedström M.; Thavarungkul P.; Kanatharana P.; Mattiasson B. Capacitive biosensor for detection of endotoxin. Anal. Bioanal. Chem. 2007, 389, 517–525. 10.1007/s00216-007-1443-4. [DOI] [PubMed] [Google Scholar]
- Limbut W.; Thavarungkul P.; Kanatharana P.; Asawatreratanakul P.; Limsakul C.; Wongkittisuksa B. Comparative study of controlled pore glass, silica gel and poraver for the immobilization of urease to determine urea in a flow injection conductimetric biosensor system. Biosens. Bioelectron. 2004, 19, 813–821. 10.1016/j.bios.2003.08.007. [DOI] [PubMed] [Google Scholar]
- Shah K.; Bentley E.; Tyler A.; Richards K. S. R.; Wright E.; Easterbrook L.; Lee D.; Cleaver C.; Usher L.; Burton J. E.; Pitman J. K.; Bruce C. B.; Edge D.; Lee M.; Nazareth N.; Norwood D. A.; Moschos S. A. Field-deployable, quantitative, rapid identification of active Ebola virus infection in unprocessed blood. Chem. Sci. 2017, 8, 7780–7797. 10.1039/c7sc03281a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereiro I.; Bendali A.; Tabnaoui S.; Alexandre L.; Srbova J.; Bilkova Z.; Deegan S.; Joshi L.; Viovy J.-L.; Malaquin L.; Dupuy B.; Descroix S. A new microfluidic approach for the one-step capture, amplification and label-free quantification of bacteria from raw samples. Chem. Sci. 2017, 8, 1329–1336. 10.1039/c6sc03880h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y.; Qiao L.; Prudent M.; Bondarenko A.; Gasilova N.; Möller S. B.; Lion N.; Pick H.; Gong T.; Chen Z.; Yang P.; Lovey L. T.; Girault H. H. Sensitive and fast identification of bacteria in blood samples by immunoaffinity mass spectrometry for quick BSI diagnosis. Chem. Sci. 2016, 7, 2987–2995. 10.1039/c5sc04919a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng R.; Tang L.; Tian Q.; Wang Y.; Lin L.; Li J. Toehold-initiated Rolling Circle Amplification for Visualizing Individual MicroRNAs In Situ in Single Cells. Angew. Chem., Int. Ed. 2014, 126, 2421–2425. 10.1002/ange.201309388. [DOI] [PubMed] [Google Scholar]
- Deng R.; Zhang K.; Li J. Isothermal Amplification for MicroRNA Detection: From the Test Tube to the Cell. Acc. Chem. Res. 2017, 50, 1059–1068. 10.1021/acs.accounts.7b00040. [DOI] [PubMed] [Google Scholar]
- Wunschel S. C.; Jarman K. H.; Petersen C. E.; Valentine N. B.; Wahl K. L.; Schauki D.; Jackman J.; Nelson C. P.; White E. Bacterial analysis by MALDI-TOF mass spectrometry: An inter-laboratory comparison. J. Am. Soc. Mass Spectrom. 2005, 16, 456–462. 10.1016/j.jasms.2004.12.004. [DOI] [PubMed] [Google Scholar]
- Biswas S.; Rolain J.-M. Use of MALDI-TOF mass spectrometry for identification of bacteria that are difficult to culture. J. Microbiol. Methods 2013, 92, 14–24. 10.1016/j.mimet.2012.10.014. [DOI] [PubMed] [Google Scholar]
- Guo L.; Ye L.; Zhao Q.; Ma Y.; Yang J.; Luo Y. Comparative study of MALDI-TOF MS and VITEK 2 in bacteria identification. J. Thorac. Dis. 2014, 6, 534–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung H. J.; Castro C. M.; Im H.; Lee H.; Weissleder R. A magneto-DNA nanoparticle system for rapid detection and phenotyping of bacteria. Nat. Nanotechnol. 2013, 8, 369–375. 10.1038/nnano.2013.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.