

Abstract
Sphingosine kinase 1 (SphK1) is an oncogenic lipid kinase that catalyzes the formation of sphingosine-1-phosphate via phosphorylation of sphingosine and known to play a crucial role in angiogenesis, lymphocyte trafficking, signal transduction pathways, and response to apoptotic stimuli. SphK1 has received attention because of its involvement in varying types of cancer and inflammatory diseases such as rheumatoid arthritis, diabetes, renal fibrosis, pulmonary fibrosis, asthma, and neurodegenerative disorders. In the malignancies of breast, lung, uterus, ovary, kidney, and leukemia, overexpression of SphK1 has been reported and thus considered as a potential drug target. In this study, we have performed virtual high-throughput screening of ∼90,000 natural products from the ZINC database to find potential SphK1-inhibitors. Initially, the hits were selected by applying absorption, distribution, metabolism, excretion, and toxicity properties, Lipinski’s rule, and PAINS filters. Further, docking analysis was performed to estimate the binding affinities and specificity to find safe and effective preclinical leads against SphK1. Two compounds, ZINC05434006 and ZINC04260971, bearing appreciable binding affinity and SphK1 selectivity were selected for 100 ns molecular dynamics (MD) simulations under explicit water conditions. The all-atom MD simulation results suggested that the ZINC05434006 and ZINC04260971 binding induces a slight structural change and stabilizes the SphK1 structure. In conclusion, we propose natural compounds, ZINC05434006 and ZINC04260971, as potential inhibitors of SphK1, which may be further exploited as potential leads to develop effective therapeutics against SphK1-associated diseases including cancer after in vitro and in vivo validations.
1. Introduction
Sphingosine-1-phosphate (S1P) is a pleiotropic sphingolipid mediator that acts as a key player in the regulation of many central biological processes, including cell proliferation, apoptosis, angiogenesis, lymphocyte trafficking, and inflammation.1 S1P is synthesized from the sphingosine molecule by an ATP-dependent phosphorylation reaction catalyzed by sphingosine kinase (SphK).2 SphK is an evolutionary-conserved, diverse class of lipid kinase and does not share sequence homology with other lipid kinases. Two isoforms of SphK, SphK1 and SphK2, have been recognized in humans, both possessing five conserved domains (C1–C5). Domains C1–C3 and C5 share sequence homology with diacylglycerol kinase (DGK) and ceramide kinase, whereas domain C4 is unique to SphK.3 Both isoforms catalyze the same biological reaction, conversion of sphingosine to S1P, despite originating from the different genes.4 However, they show distinct subcellular location, tissue distribution, and substrate specificity.5 SphK1 is found in the cytoplasm and translocated to the plasma membrane when activated. However, SphK2 is explicitly present in the nucleus.4
SphK1 is synthesized as a 384-amino acid residue long polypeptide that lacks a transmembrane domain but contains various phosphorylation sites for kinase and three calcium/calmodulin-binding sequences. It is ubiquitously expressed in the heart, brain, kidney, liver, spleen, and lungs. SphK1 is normally found in the cytosol but gets translocated to the plasma membrane upon activation. A diverse range of external stimuli act as activators of SphK1 such as tumor necrosis factor α, a proinflammatory cytokine, and various several growth factors, including nerve growth and epidermal growth factors (EGFs).6 Upon stimulation by these agonist molecules, SphK1 is phosphorylated at Ser225 by an extracellular signal-regulated kinase that leads to its activation and translocation to the plasma membrane.7 Activated SphK1 further catalyzes the phosphorylation of sphingosine, resulting in a transient increase in the intracellular levels of S1P, which acts as a bioactive lipid molecule having both extracellular function and intracellular targets.8 S1P binds to a family of five G protein-coupled receptors (S1PR1–5) and activates several downstream signaling pathways regulating various cellular processes.9 S1PRs are differentially expressed in various cell types that explain the diverse signaling and hence the biological functions of S1P. In addition to interacting with S1PRs placed on the plasma membrane, S1P also functions as an intracellular messenger and acts on specific intracellular targets.
Structurally, SphK1 is a monomeric protein with two domains: N-terminal domain (NTD) and C-terminal domain (CTD). The NTD adopts an α/β fold formed by a fundamental core of a twisted parallel β-sheet with flanking α-helices on either side. On the other hand, CTD possesses a sandwiched antiparallel β-sheet assembly formed by 11 β-strands with four α-helices present on one side of the sheet. The ATP-binding site of SphK1 resides deeper inside the cavity between NTD and CTD, whereas the lipid-binding pocket is buried in the hydrophobic core of CTD.3 The crystal structure of SphK110 reveals the presence of an ATP-binding pocket that involves five conserved domains and a structural motif S79GDG82X17–21K103. Arg185 forms the ATP-binding site along with other amino acid residues that play a crucial role in catalysis.
An upregulation of SphK1 is reported in different malignancies, including lung, brain, breast, colorectal, and prostate cancers.11−14 A close link between the overexpression of SphK1 to malignant tissues such as angiogenesis and tumorigenesis is reported.15 S1P is known to be involved in many biological processes and plays a crucial role in breast cancer development.16 The SphK1 action is mediated by some developmental factors such as estradiol (E2), EGF, vitamins, and cytokines IL1 and IL6.17 S1P/SphK1 signaling has been associated with the pathophysiology of various metabolic and inflammatory diseases, including pulmonary fibrosis, diabetes, obesity, rheumatoid arthritis, sepsis, and Alzheimer’s disease.18−22 All these findings revealed that SphK1 is an attractive drug target for the development of effective therapeutic molecules to address cancer and other SphK1-associated diseases.23,24 Numerous synthetic molecules such as SK1-I, PF-543, and FTY-720 have been developed that target the SphK1/S1P signaling axis and showed inhibitory action in μM to nM ranges. SK1-I, an inhibitor of SphK1, has demonstrated to inhibit the growth of glioblastoma multiforme cell lines and causes a reduction in the tumor growth and vascularization in a mice model.25 FTY-720 is an FDA-approved sphingosine analog generally used for the treatment of multiple sclerosis. It has been used to increase the sensitivity of prostate cancer cells and murine xenografts toward radiotherapy.26
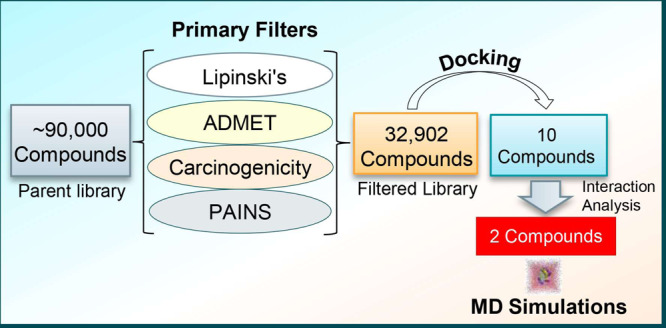
The known kinase inhibitors are often plagued with specificity and selectivity issues where the off-targeting leads to deleterious effects.27−29 Most of the inhibitors are checked during the initial phases of clinical trials because of the toxicity concerns.30 Thus, it is imperative to design inhibitors that are target-specific and safer. Structure-based drug discovery is a smart and relevant approach to identify lead molecules that exhibit high affinity and target selectivity.31−34 We adopted a systematic structure-based drug design approach as illustrated in Figure 1.
Figure 1.
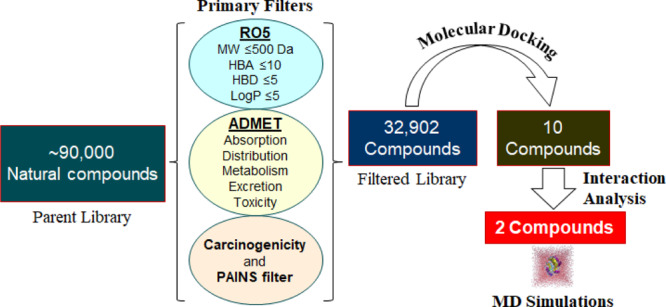
Pipeline used in this study illustrates the process of filtering compounds using a virtual high-throughput screening approach (left to right). RO5: Lipinski’s rule of five; MW: molecular weight; HBA: hydrogen bond acceptor; and HBD: hydrogen bond donor.
In this work, we virtually screened a pool of 90,000 natural compounds by applying Lipinski’s rule of five, absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties, carcinogenicity, and PAINS filter. Subsequently, we have used a structure-based virtual high-throughput screening (vHTS) approach to dock filtered compounds with SphK1 by utilizing the molecular docking approach.35,36 The binding affinity and interaction analysis was carried out to explore the binding pattern of the selected compounds with SphK1. We further evaluated the structural dynamics and stability of SphK1 and its docked complexes with the selected compounds by utilizing all-atom molecular dynamics (MD) simulations and principal component analysis (PCA). We performed MD simulations on three systems, one apo and two ligand-bound states of SphK1 for 100 ns, to describe their interaction and conformational dynamics of SphK1 under an explicit solvent environment.
2. Results and Discussion
2.1. Filtration of Natural Compounds
First, the physicochemical parameters for all compounds present in the ZINC library of natural products were calculated through the SwissADME webtool and Discovery Studio. From the library of ∼90,000 compounds, a total of 32,902 compounds were selected after all the applied filters. The filtered compounds follow Lipinski’s rule of five (molecular weight ≤ 500 Da, log P ≤ 5, number of hydrogen bond donor ≤ 5, and hydrogen bond acceptor ≤ 10) and the bioavailability score of a minimum of 0.55. All these compounds do not possess any carcinogenic and PAINS patterns. The physicochemical parameters and their scores for the finally selected two compounds along with known SphK1 inhibitors are presented in Table 1. The identified compounds show similar properties when compared to the known SphK1 inhibitors PF-543 and SKI-II. All the four, two identified compounds, ZINC05434006 and ZINC04260971, and two known SphK1 inhibitors, PF-543 and SKI-II, follow Lipinski’s rule.
Table 1. Physicochemical Properties of the Selective Compounds and Known Inhibitors against SphK1 Following Lipinski’s Rule.
| s. no. | compound ID | molecular formula | molecular weight | rotatable bond | H-bond acceptor | H-bond donor | log P |
|---|---|---|---|---|---|---|---|
| 1 | ZINC05434006 | C27H25N5O4 | 460.53 | 5 | 7 | 2 | 4.48 |
| 2 | ZINC04260971 | C26H28N4O4 | 483.52 | 6 | 7 | 2 | 4.26 |
| 3 | PF-543 | C27H31NO4S | 465.60 | 9 | 5 | 1 | 4.50 |
| 4 | SKI-II | C15H11ClN2OS | 302.80 | 3 | 4 | 2 | 4.80 |
The filtered compounds on the basis of their physicochemical, ADMET, and other druglike properties were selected. The ADMET properties of compounds were predicted through the pkCSM web server.37 These properties show all the parameters of the selected compounds, ZINC05434006 and ZINC04260971, and the known SphK1 inhibitor, PF-543, within the range of drug ability (Table 2). The identified compounds ZINC05434006 and ZINC04260971 show better ADMET properties than the known SphK1 inhibitors, PF-543 and SKI-II, as they have higher GI absorption compared with PF-543 and SKI-II (Table 2).
Table 2. ADMET Properties of the Selected Compounds and Known SphK1 Inhibitors, PF-543 and SKI-II.
| absorption |
distribution | metabolism | excretion | toxicity | ||
|---|---|---|---|---|---|---|
| compound ID | GI absorption (%) | water solubility | BBB/CNS permeation | CYP2D6 substrate | OCT2 substrate | AMES/skin sens. |
| ZINC05434006 | 100.00 | soluble | no | no | no | no |
| ZINC04260971 | 100.00 | soluble | no | no | no | no |
| PF-543 | 93.21 | soluble | no | no | no | no |
| SKI-II | 88.05 | soluble | no | yes | no | no |
2.2. Structure-Based Virtual Screening
To find and select the high-affinity inhibitors of SphK1, the structure-based virtual screening was carried out on the filtered compounds employing the molecular docking approach. Here, we noticed that several compounds possess an appreciable binding affinity to the SphK1. We selected the top 10 hits out of 32,902 compounds showing considerably higher binding affinities to SphK1. Selected hits show affinity in the range of −11.8 to −12.2 kcal/mol to SphK1 (Table 3). The binding affinity for the finally selected two compounds ZINC05434006 and ZINC04260971 were estimated as −12.0 and −11.9 kcal/mol, respectively. Both compounds showed to have a higher affinity toward SphK1 as compared with known SphK1 inhibitors, PF-543 and SKI-II, as they show an affinity of −9.1 and −8.8 kcal/mol, respectively (Table 3). The free energy of binding was also estimated through AutoDock 4, which uses the AMBER-based scoring function in docking calculations. The free energy of binding for ZINC05434006 and ZINC04260971 for SphK1 was calculated as −11.16 and −11.03 kcal/mol, respectively, while the free energy of binding for PF-543 and SKI-II toward SphK1 was calculated as −9.32 and −8.94 kcal/mol, respectively. This comparison of predicted binding affinities from two different tools suggests that the selected compounds show a significant affinity to the SphK1.
Table 3. List of the Top 10 Hits and Known SphK1 Inhibitors, PF-543 and SKI-II, with Their Binding Affinity toward SphK1.
| s. no. | compound ID | binding affinity (kcal/mol) |
|---|---|---|
| 1 | ZINC12898623 | –12.2 |
| 2 | ZINC02149103 | –12.1 |
| 3 | ZINC05434006 | –12.0 |
| 4 | ZINC04260971 | –11.9 |
| 5 | ZINC05433944 | –11.9 |
| 6 | ZINC03839231 | –11.8 |
| 7 | ZINC06623685 | –11.8 |
| 8 | ZINC08296863 | –11.8 |
| 9 | ZINC08788772 | –11.8 |
| 10 | ZINC12898106 | –11.8 |
| 11 | PF-543 | –9.1 |
| 12 | SKI-II | –8.8 |
2.3. Molecular Docking
To find the specific compounds bind to the substrate-binding site of SphK1, a detailed interaction analysis was carried while using Discovery Studio. The possible binding conformers of the top 10 hits were split and their interaction toward the SGK1 binding pocket was analyzed in detail. We identified ZINC05434006 and ZINC04260971, which interact with a set of functionally critical residues of SphK1 (Figure 2). Many residues of the SphK1 kinase domain participate in the protein–ligand interaction. A detailed binding pattern of these compounds with SphK1 is illustrated in Figure 2.
Figure 2.

Interaction of the selected compounds ZINC05434006 and ZINC04260971 along with known SphK1 inhibitors PF-543 and SKI-II toward SphK1. (A) Overall structural representation of SphK1 complexed with the selected compounds and known inhibitors. (B) Residues of substrate-binding pocket participating in hydrogen bonding to the selected compounds. (C) Surface potential representation of SphK1 binding pocket occupied by the selected compounds and known inhibitors. Asp178 substrate—binding site; Asp81—active site; and Arg191—ATP binding site.
We analyzed the interactions of selected inhibitors with the functionally important residues of the binding pocket of SphK1. Both the compounds interact with the substrate-binding (Asp178), active site (Asp81), and ATP-binding site (Arg191) residues of SphK1 (Figure 3). Both the compounds share hydrogen bonds with the substrate-binding site (Asp178) and offer many common interactions as a pattern shared by the known inhibitors PF-543 and SKI-II. The hydrogen bonding is strong enough to keep the compounds inside the SphK1 pocket. We observed many specific interactions between the critically important residues of SphK1 and the identified compounds, which suggest their strong and stable binding with SphK1. The SphK1 complexes with the selected compounds were stabilized by many noncovalent interactions offered by the lipid substrate binding site (Asp 178), active site (Asp81), and ATP-binding site (Arg191) of SphK1. Interestingly, both the compounds make hydrogen-bonded interaction to Asp178. We observed that ZINC05434006 and ZINC04260971 interact with the substrate-binding site of SphK1 and mimic the pose of cocrystallized known SphK1 inhibitors PF-543 and SKI-II. Analysis of interaction pattern suggests that the compounds ZINC05434006 and ZINC04260971 may serve as ATP/substrate-competitive inhibitors of SphK1.
Figure 3.

: Two-dimensional (2D) plot of SphK1 interactions with (A) ZINC05434006, (B) ZINC04260971, (C) PF-543, and (D) SKI-II.
2.4. MD Simulations
Three systems, apo-SphK1, SphK1-ZINC05434006-, and SphK1-ZINC04260971-bound complexes, were subjected to all-atom MD simulations for 100 ns. The potential energy of SphK1 apo, SphK1-ZINC05434006, and SphK1-ZINC04260971 was calculated as −890,000, −879,465, and −880,921 kJ/mol, respectively. Other dynamic parameters such as systems volume, density, kinetic energy, enthalpy, and total energy were further calculated to ascertain the equilibration and stability of the systems where we found no major changes when comparing the free SphK1 and its complexes (Table 4).
Table 4. Systematic and Energetic Parameters for apo-SphK1, SphK1-ZINC05434006, and SphK1-ZINC04260971 Systems.
| system | rmsd (nm) | RMSF (nm) | Rg (nm) | SASA (nm2) | enthalpy (kJ/mol) | density (g/L) |
|---|---|---|---|---|---|---|
| SphK1 | 0.30 | 0.13 | 1.96 | 145.61 | –744,675 | 1028.44 |
| SphK1-ZINC05434006 | 0.26 | 0.12 | 1.99 | 147.20 | –735,691 | 1029.05 |
| SphK1-ZINC04260971 | 0.28 | 0.11 | 2.00 | 147.05 | –736,909 | 1029.11 |
2.5. Structural Deviations and Compactness
Binding of any compound to their receptor (protein) can induce large conformational changes. Root-mean-square deviation (rmsd) is used to estimate the structural dynamics of protein.38−40 To assess the structural dynamics of SphK1 in free and complex states, we calculated the rmsd of all the systems. The average rmsd of SphK1 in apostate, SphK1-ZINC05434006, and SphK1-ZINC04260971 complex was found at 0.30, 0.26, and 0.28 nm, respectively (Table 4). The rmsd of all the three systems shows that SphK1 gets stabilized after ZINC05434006 and ZINC04260971 binding. The ZINC05434006 and ZINC04260971 binding leads to a fewer conformational change in the SphK1 structure from its native one (Figure 4A). The rmsd of SphK1 complexed with ZINC05434006 and ZINC04260971 shows a slight decrease that is equilibrated throughout the simulation trajectory, suggesting durable stability of these protein–ligand complexes (Figure 4A, upper panel). The probability distribution function (PDF) plots for rmsd show a minor decrease in the rmsd value of ligand-bound systems of SphK1 compared with SphK1 alone (Figure 4A, lower panel).
Figure 4.
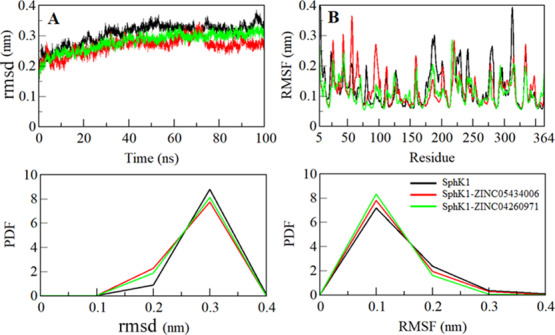
Dynamics of the SphK1 structure as a function of time. (A) RMSD plot of SphK1 before and after compound binding. (B) Residual fluctuation plot of SphK1 and upon ZINC05434006 and ZINC04260971 binding.
To explore the residual vibrations in free SphK1 and its ligand-bound states, the fluctuations of each residue were plotted as root-mean-square fluctuation (RMSF). Random residual fluctuations present in SphK1 can be observed in different regions from N- to C-termini (Figure 4B). The fluctuations of the SphK1 backbone were compared with each residue after the ZINC05434006 and ZINC04260971 binding. The fluctuations were found to be minimum at several residues in the case of SphK1-ZINC05434006 and SphK1-ZINC04260971 complexes (Table 5). Initially, the RMSF of the SphK1-ZINC05434006 system was increased up to that of Gly160, but thereafter, it is minimized up to Val325, including the substrate-binding site in SphK1. The RMSF suggested that the fluctuations of the residues are reduced in the region where ZINC05434006 and ZINC04260971 are binding (Figure 4B, upper panel). A slight increase in fluctuations of SphK1 after ligand binding was noticed, which might be due to ligand adjustment in the SphK1 binding pocket.
Table 5. Finally Selected Compounds against SphK1 and Their Chemical Properties.

The PDF analysis suggested a slight increase in the PDF of SphK1 RMSF while in the ligand-bound state (Figure 4B, lower panel). The higher fluctuations observed in the region of residue 25–125 flap particularly in the SphK1-ZINC05434006 complex can be interrelated with the docking results, where fewer close interactions are formed between the protein–ligand at this region (Figure 3A). These residual fluctuations suggest increased dynamics of internal vibration in SphK1 while in complexes with ZINC05434006. The decreased fluctuations in both the complexes, particularly in the region of residue 175–325 flap, suggest that this region of SphK1 directly binds with the ligands and forms many close interactions with ZINC05434006 and ZINC04260971, thereby showing a reduced RMSF in comparison to the other regions of SphK1.
The radius of gyration (Rg) is a structural parameter associated with the overall conformation and three-dimensional (3D) structure of a protein, which has been utilized to get insights into their compactness and folding behavior.41−43 We estimated the stability of apo-SphK1, SphK1-ZINC05434006, and SphK1-ZINC04260971 systems by calculating their Rg values. The average values of Rg for apo SphK1, SphK1-ZINC05434006, and SphK1-ZINC04260971 complexes were calculated as 1.96, 1.99, and 2.00 nm, respectively. The Rg plot depicts a minor increase in Rg values while in the case of the bound states. This increase in Rg is possibly due to the SphK1 packing while its binding pocket is occupied by ZINC05434006 and ZINC04260971. Here, no structural swift was observed in SphK1 in after compound binding, where Rg attains a stable equilibrium and thus suggests stability of the complexes throughout the trajectory (Figure 5A).
Figure 5.
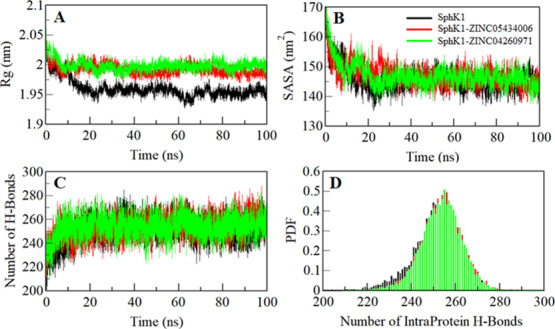
Structural compactness of SphK1. (A) Time evolution of the radius of gyration. (B) SASA plot of SphK1 as a function of time. (C) Time evolution of stability of intramolecular hydrogen bonds formed within SphK1, where (D) shows the PDF of intramolecular hydrogen bonds.
The solvent-accessible surface area (SASA) is the surface area of a protein that is accessible to its surrounding solvent.44 SASA is one of the fundamental properties of a protein that is utilized to evaluate its structural folding–unfolding dynamic under the solvent environment.45,46 We have calculated and investigated the SASA of SphK1, SphK1-ZINC05434006, and SphK1-ZINC04260971 to explore their folding behavior during simulation. The average values of SASA for SphK1, SphK1-ZINC05434006, and SphK1-ZINC04260971 were calculated as 145.61, 147.20, and 147.05 nm2, respectively. We observed a slight increase in the SASA of SphK1 in the presence of ZINC05434006 and ZINC04260971, which is projected due to the exposure of some inner residues to the surface (Figure 5B). Overall, the SASA immediately achieved an equilibrium after 20 ns without any shift during the entire simulation, which suggests folding stability of SphK1 before and after ligand binding.
Intramolecular hydrogen bonds (H-bonds) within a protein play an essential role to stabilize its 3D structure and overall conformation.47−49 To validate the stability of apo-SphK1, SphK1-ZINC05434006, and SphK1-ZINC04260971 complexes, we computed the dynamics of intramolecular H-bonds paired within 0.35 nm. The computed average number of intramolecular H-bonds formed within SphK1 apo, SphK1-ZINC05434006, and SphK1-ZINC04260971 were estimated as 251, 254, and 253, respectively (Figure 5C). A few numbers of H-bonds increased in SphK1 after compound binding, which might be due to the higher compactness of some intramolecular space within the protein. The PDF analysis of hydrogen bond dynamics indicates that the complexes of SphK1-ZINC05434006 and SphK1-ZINC04260971 are stable with a minimal change (Figure 5D).
2.6. Hydrogen Bonding Analysis
The H-bonds formed between a protein and ligand can be explored to get insights into the stability of a protein–ligand complex to understand the molecular recognition and specificity of interactions.47 To evaluate the complex stability, we have studied the dynamics of intermolecular H-bonds of ZINC05434006 and ZINC04260971 with SphK1 paired within 0.35 nm. Our analysis suggests an average of two H-bonds were shared by ZINC05434006 and ZINC04260971 to SphK1, which were stable throughout the trajectory (Figure 6). Both the compounds bind in the SphK1 binding pocket with two to three H-bonds with mutability and up to two H-bonds with higher stability, supporting the molecular docking result. The PDF of H-bonding suggests that ZINC05434006 and ZINC04260971 bind to SphK1 with one and two H-bonds, respectively, with higher stability and distribution throughout the simulation trajectory (Figure 6, lower panel).
Figure 6.
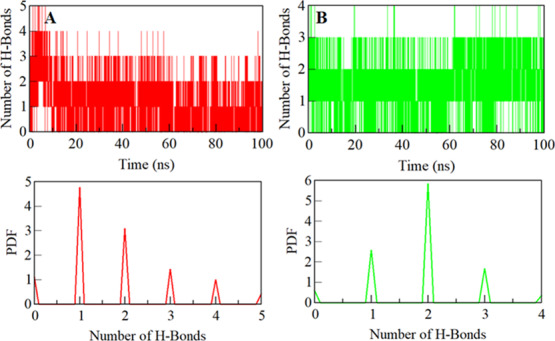
Stability of hydrogen bonds formed. (A) Intermolecular hydrogen bonds between compounds ZINC05434006 and (B) ZINC04260971 with SphK1 (the lower panel shows the probability of distribution of hydrogen bonding as a PDF).
2.7. Principal Component and Free Energy Landscapes
The dynamic movement of a protein structure can be explored through its phase space behavior. We performed PCA to investigate the conformational dynamics of apo-SphK1, SphK1-ZINC05434006, and SphK1-ZINC04260971 via investigating their collective motions while utilizing essential dynamics approach.50 The PCA helps to understand the dynamic motion and overall flexibility of a protein and its docked complexes with the small molecules in a conformational subspace. The conformational sampling along the eigenvector (EV) 1 and EV2 projected by the Cα atom of SphK1 before and after ZINC05434006 and ZINC04260971 binding in the essential phase space is illustrated in Figure 7A. Here, we observed that the SphK1-ZINC05434006 and SphK1-ZINC04260971 complex occupied the same conformational subspace. However, the overall flexibility of the SphK1-ZINC05434006 complex was slightly increased at both EVs with overlapping of stable clusters with the subspace of SphK1 in the free state (Figure 7B). The overall PCA including the atomic fluctuations suggests that SphK1 is quite stable during the simulation after compound binding.
Figure 7.
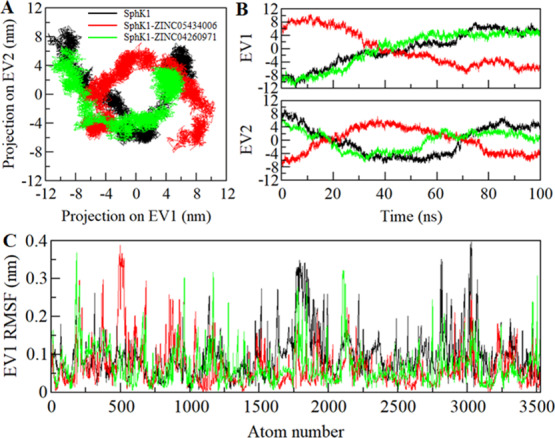
Essential dynamics showing SphK1 conveying PCA. (A) 2D projections of trajectories on two EVs illustrating different projections of SphK1. (B) Projections of trajectories on EVs concerning time. (C) Atomic fluctuations of SphK1 on EV 1.
To further investigate the conformational behavior of SphK1, Gibbs free energy landscapes (FELs) were generated using the first two EVs. Figure 8 shows the FELs of apo SphK1, SphK1-ZINC05434006, and SphK1-ZINC04260971 complex systems. The deeper blue color in the plots illustrates the structural conformations with lower free energy. SphK1 shows a single global minimum restricted to a single local basin. However, SphK1 while in complex with ZINC05434006 acquired multiple minima and with varying conformational states, while the SphK1-ZINC04260971 system did not progress to multiple global minima, showing the overall single global minimum limited to a single basin. This PCA suggests that the presence of ZINC05434006 and ZINC04260971 does not affect much the conformation and the position of the sampled subspace of SphK1 (Figure 8B–C).
Figure 8.

Gibbs energy landscapes of (A) free SphK1, (B) SphK1-ZINC05434006, and (C) SphK1-ZINC04260971.
Altogether, the physicochemical and ADMET properties, interaction analysis in comparison of the known SphK1 inhibitors (PF-543 and SKI-II), and MD simulation studies suggest that the identified two natural compounds, ZINC05434006 and ZINC04260971, can be further utilized as a potent scaffold in development of high-affinity inhibitors of SphK1 (Table 5).
3. Conclusions
SphK1 is an important enzyme that regulates the sphingolipid rheostat accountable for determining cell fate. The development of targeted therapeutics to address the clinical management of cancer without being cytotoxic has received growing interest. Natural compounds have been acknowledged to be effective and curable in many complex diseases including cancer and neurodegenerative diseases. Here, we performed structure-based virtual screening of natural compounds against SphK1 to identify its potent inhibitors, which can further be used in drug development against cancer. The identified two natural compounds (ZINC05434006 and ZINC04260971) were selected by evaluating their druglike properties, binding affinities, and a specific interaction toward the SphK1 binding pocket. MD simulation studies further revealed that ZINC05434006 and ZINC04260971 strongly bind to SphK1 and forms a stable complex with minimal structural dynamics. Both compounds bearing anticancer activity and kinase inhibitor potency as predicted through the PASS analysis. This study predicted a considerable inhibitory potential of identified natural compounds that could be a starting point for the development of new SphK1 inhibitors. Experimental studies are further required before the clinical implications of these compounds.
4. Materials and Methods
4.1. Computational Environment and Web Resources
Computational tools, including MGL Tools,51 AutoDock Vina,52 Discovery Studio visualizer,53 and the GROMACS package,54 were employed for screening and simulation studies. Various web resources such as NCBI,55 Protein Data Bank (PDB),56 ZINC database,57 SwissADME,58 and standalone QtGrace59 were used in retrieving and analyzing the data. Structural coordinates of human SphK1 were taken from the PDB (PDB ID: 3VZB, resolution: 2.0 Å)10 and refined further through the MGL tools. A library of ∼90,000 compounds containing natural products and their derivatives was downloaded from the ZINC database.
4.2. Filtration of Natural Compounds
Initially, the ZINC library containing ∼90,000 was filtered based on their physicochemical properties following Lipinski’s rule of five. The PAINS screening was done to filter those compounds having PAINS patterns with a high tendency of binding with multiple targets. The filtered compounds were further screened for their carcinogenicity and ADMET properties. Compounds that were noncarcinogenic and possess acceptable ADMET properties were selected further to process with the structure-based virtual screening.
4.3. Structure-Based Virtual Screening
The atomic coordinates of the 3D structure of human SphK1 were downloaded from the PDB (PDB ID: 3VZB).10 The cocrystallized d-sphingosine and water were removed, and the structure was subsequently refined in MGL tools. The docking was performed using AutoDock Vina, with a structurally blind search space having a grid size of 50, 58, and 56 Å, centralized at 53.15, 51.79, and −1.54 for X, Y, and Z coordinates, respectively. The filtered library which contains 32,902 compounds was subjected to screen with structure-based molecular docking to select compounds with higher binding affinities for SphK1. The compounds with higher docking scores were subjected to splitting to generate all possible docked conformers and further analyzed through Discovery Studio for detailed interaction to SphK1. In interaction analysis, the only compounds having a specific interaction toward the substrate binding site of SphK1 were selected.
4.4. MD Simulations
All-atom MD simulations were performed on SphK1 before and after the binding of the identified compounds, ZINC05434006 and ZINC04260971, for 100 ns at the molecular mechanics level at 300 K utilizing GROMOS 54A7 force field in the GROMACS 5.1.2 package. The topology parameters for compounds ZINC05434006 and ZINC04260971 were produced in the PRODRG and complexed into the protein topology to make SphK1-ZINC05434006 and SphK1-ZINC04260971 complex systems. All apo-SphK1, SphK1-ZINC05434006, and SphK1-ZINC04260971 were solvated using the simple point charge (spc216) in a cubic box. All the three systems were minimized to remove steric clashes by utilizing 1500 steps of the steepest descent approach. The temperature of all the three systems was then raised from 0 to 300 K during the equilibrium phase of 100 ps at a constant volume with a stable pressure of 1 bar. The final MD run was set to 100,000 ps for all the three systems, and the resulting trajectory files were studied through the GROMACS utilities and plotted using the QtGrace tool.
Acknowledgments
T.M. is thankful to the University Grants Commission, India, for the award of Maulana Azad National Senior Research Fellowship (F1-17.1/2017-18/MANF-2017-18-UTT-87495). The authors sincerely thank the Department of Science and Technology, Government of India, for the FIST support (FIST program no. SR/FST/LSI-541/2012). M.F.A., A.H., and M.T.R. acknowledge the generous support from the Deanship of Scientific Research at King Saud University, Riyadh, Kingdom of Saudi Arabia, for funding their research group (grant no. RGP-1441-150).
Author Contributions
D.S.J. and T.M. have equally contributed. All authors have read and agreed to publish the final version of the manuscript. T.M., K.A, D.S.J., and M.I.H. conceptualized the study; T.M. and D.S.J. developed the methodology; M.T.R. and A.H. developed the software; D.S.J., A.H., and M.I.H. validated the study; M.F.A. and G.M.H. performed the formal analysis; T.M., K.A, and P.G. investigated the study; M.F.A. and D.S.J. brought the resources; T.M., P.G., and G.M.H. performed data curation; T.M., K.A., D. S.J., and M.I. H. wrote and prepared the original draft; D.S.J., P.G., and M.I.H. wrote, reviewed, and edited the manuscript; M.F.A., G.M.H., and A.H. visualized the study; M.F.A. and M.I.H. supervised the study; M.F.A. and M.I.H. involved in project administration; and M.F.A. and A.H. involved in funding acquisition.
M.I.H. acknowledges the Indian Council of Medical Research for financial assistance (project no. BIC/12(01)/2015).
The authors declare no competing financial interest.
References
- Pyne N. J.; Pyne S. Sphingosine 1-phosphate and cancer. Nat. Rev. Cancer 2010, 10, 489–503. 10.1038/nrc2875. [DOI] [PubMed] [Google Scholar]
- Maceyka M.; Harikumar K. B.; Milstien S.; Spiegel S. Sphingosine-1-phosphate signaling and its role in disease. Trends Cell Biol. 2012, 22, 50–60. 10.1016/j.tcb.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.; Min X.; Xiao S.-H.; Johnstone S.; Romanow W.; Meininger D.; Xu H.; Liu J.; Dai J.; An S.; Thibault S.; Walker N. Molecular Basis of Sphingosine Kinase 1 Substrate Recognition and Catalysis. Structure 2013, 21, 798–809. 10.1016/j.str.2013.02.025. [DOI] [PubMed] [Google Scholar]
- Maceyka M.; Sankala H.; Hait N. C.; Le Stunff H.; Liu H.; Toman R.; Collier C.; Zhang M.; Satin L. S.; Merrill A. H.; Milstien S.; Spiegel S. SphK1 and SphK2, Sphingosine Kinase Isoenzymes with Opposing Functions in Sphingolipid Metabolism. J. Biol. Chem. 2005, 280, 37118–37129. 10.1074/jbc.m502207200. [DOI] [PubMed] [Google Scholar]
- Melendez A. J.; Carlos-Dias E.; Gosink M.; Allen J. M.; Takacs L. Human sphingosine kinase: molecular cloning, functional characterization and tissue distribution. Gene 2000, 251, 19–26. 10.1016/s0378-1119(00)00205-5. [DOI] [PubMed] [Google Scholar]
- Hannun Y. A.; Obeid L. M. Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 175. 10.1038/nrm.2017.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitson S. M.; Moretti P. A. B.; Zebol J. R.; Lynn H. E.; Xia P.; Vadas M. A.; Wattenberg B. W. Activation of sphingosine kinase 1 by ERK1/2 mediated phosphorylation. EMBO J. 2003, 22, 5491–5500. 10.1093/emboj/cdg540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne S. G.; Milstien S.; Spiegel S. Sphingosine-1-phosphate: dual messenger functions. FEBS Lett. 2002, 531, 54–57. 10.1016/s0014-5793(02)03480-4. [DOI] [PubMed] [Google Scholar]
- Okamoto Y.; Wang F.; Yoshioka K.; Takuwa N.; Takuwa Y. Sphingosine-1-Phosphate-Specific G Protein-Coupled Receptors as Novel Therapeutic Targets for Atherosclerosis. Pharmaceuticals 2011, 4, 117–137. 10.3390/ph4010117. [DOI] [Google Scholar]
- Wang Z.; Min X.; Xiao S.-H.; Johnstone S.; Romanow W.; Meininger D.; Xu H.; Liu J.; Dai J.; An S.; Thibault S.; Walker N. Molecular basis of sphingosine kinase 1 substrate recognition and catalysis. Structure 2013, 21, 798–809. 10.1016/j.str.2013.02.025. [DOI] [PubMed] [Google Scholar]
- Hait N. C.; Maiti A. The Role of Sphingosine-1-Phosphate and Ceramide-1-Phosphate in Inflammation and Cancer. Mediators Inflammation 2017, 2017, 4806541. 10.1155/2017/4806541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W.; Yu C.-P.; Xia J.-t.; Zhang L.; Weng G.-X.; Zheng H.-q.; Kong Q.-l.; Hu L.-j.; Zeng M.-S.; Zeng Y.-x.; Li M.; Li J.; Song L.-B. Sphingosine Kinase 1 Is Associated with Gastric Cancer Progression and Poor Survival of Patients. Clin. Cancer Res. 2009, 15, 1393–1399. 10.1158/1078-0432.ccr-08-1158. [DOI] [PubMed] [Google Scholar]
- Ogretmen B. Sphingolipid metabolism in cancer signalling and therapy. Nat. Rev. Cancer 2018, 18, 33–50. 10.1038/nrc.2017.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L.; Wang Z.; Lin Y.; Chen Z.; Liu H.; Chen Y.; Wang N.; Song X. Sphingosine kinase 1 enhances the invasion and migration of non-small cell lung cancer cells via the AKT pathway. Oncol. Rep. 2015, 33, 1257–1263. 10.3892/or.2014.3683. [DOI] [PubMed] [Google Scholar]
- Shida D.; Takabe K.; Kapitonov D.; Milstien S.; Spiegel S. Targeting SphK1 as a new strategy against cancer. Curr. Drug Targets 2008, 9, 662–673. 10.2174/138945008785132402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu Y.; Furuya H.; Tamashiro P. M.; Iino K.; Chan O. T. M.; Goodison S.; Pagano I.; Hokutan K.; Peres R.; Loo L. W. M.; Hernandez B.; Naing A.; Chong C. D. K.; Rosser C. J.; Kawamori T. Genetic deletion of sphingosine kinase 1 suppresses mouse breast tumor development in an HER2 transgenic model. Carcinogenesis 2018, 39, 47–55. 10.1093/carcin/bgx097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maczis M. A.; Maceyka M.; Waters M. R.; Newton J.; Singh M.; Rigsby M. F.; Turner T. H.; Alzubi M. A.; Harrell J. C.; Milstien S.; Spiegel S. Sphingosine kinase 1 activation by estrogen receptor α36 contributes to tamoxifen resistance in breast cancer. J. Lipid Res. 2018, 59, 2297–2307. 10.1194/jlr.m085191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J.; Li J.; Chen Z.; Li J.; Chen Q.; Gong W.; Liu P.; Huang H. Sphingosine kinase 1 mediates diabetic renal fibrosis via NF-κB signaling pathway: involvement of CaCl2. Oncotarget 2017, 8, 88988–89004. 10.18632/oncotarget.21640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milara J.; Navarro R.; Juan G.; Peiró T.; Serrano A.; Ramón M.; Morcillo E.; Cortijo J. Sphingosine-1-phosphate is increased in patients with idiopathic pulmonary fibrosis and mediates epithelial to mesenchymal transition. Thorax 2012, 67, 147–156. 10.1136/thoraxjnl-2011-200026. [DOI] [PubMed] [Google Scholar]
- Nagahashi M.; Yamada A.; Katsuta E.; Aoyagi T.; Huang W.-C.; Terracina K. P.; Hait N. C.; Allegood J. C.; Tsuchida J.; Yuza K.; Nakajima M.; Abe M.; Sakimura K.; Milstien S.; Wakai T.; Spiegel S.; Takabe K. Targeting the SphK1/S1P/S1PR1 axis that links obesity, chronic inflammation and breast cancer metastasis. Cancer Res. 2018, 78, 1713. 10.1158/0008-5472.can-17-1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czubowicz K.; Jęśko H.; Wencel P.; Lukiw W. J.; Strosznajder R. P. The Role of Ceramide and Sphingosine-1-Phosphate in Alzheimer’s Disease and Other Neurodegenerative Disorders. Mol. Neurobiol. 2019, 56, 5436–5455. 10.1007/s12035-018-1448-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng M. L.; Wadham C.; Sukocheva O. A. The role of sphingolipid signalling in diabetes-associated pathologies (Review). Int. J. Mol. Med. 2017, 39, 243–252. 10.3892/ijmm.2017.2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta P.; Khan F. I.; Roy S.; Anwar S.; Dahiya R.; Alajmi M. F.; Hussain A.; Rehman M. T.; Lai D.; Hassan M. I. Functional implications of pH-induced conformational changes in the Sphingosine kinase 1. Spectrochim. Acta, Part A 2020, 225, 117453. 10.1016/j.saa.2019.117453. [DOI] [PubMed] [Google Scholar]
- Gupta P.; Mohammad T.; Dahiya R.; Roy S.; Noman O. M. A.; Alajmi M. F.; Hussain A.; Hassan M. I. Evaluation of binding and inhibition mechanism of dietary phytochemicals with sphingosine kinase 1: Towards targeted anticancer therapy. Sci. Rep. 2019, 9, 18727. 10.1038/s41598-019-55199-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L.; Weng W.; Sun Z.-X.; Fu X.-J.; Ma J.; Zhuang W.-F. SphK1 inhibitor II (SKI-II) inhibits acute myelogenous leukemia cell growth in vitro and in vivo. Biochem. Biophys. Res. Commun. 2015, 460, 903–908. 10.1016/j.bbrc.2015.03.114. [DOI] [PubMed] [Google Scholar]
- Lim K. G.; Tonelli F.; Li Z.; Lu X.; Bittman R.; Pyne S.; Pyne N. J. FTY720 Analogues as Sphingosine Kinase 1 Inhibitors. J. Biol. Chem. 2011, 286, 18633–18640. 10.1074/jbc.m111.220756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naz F.; Shahbaaz M.; Bisetty K.; Islam A.; Ahmad F.; Hassan M. I. Designing New Kinase Inhibitor Derivatives as Therapeutics Against Common Complex Diseases: Structural Basis of Microtubule Affinity-Regulating Kinase 4 (MARK4) Inhibition. OMICS: J. Integr. Biol. 2015, 19, 700–711. 10.1089/omi.2015.0111. [DOI] [PubMed] [Google Scholar]
- Naz H.; Jameel E.; Hoda N.; Shandilya A.; Khan P.; Islam A.; Ahmad F.; Jayaram B.; Hassan M. I. Structure guided design of potential inhibitors of human calcium-calmodulin dependent protein kinase IV containing pyrimidine scaffold. Bioorg. Med. Chem. Lett. 2016, 26, 782–788. 10.1016/j.bmcl.2015.12.098. [DOI] [PubMed] [Google Scholar]
- Voura M.; Khan P.; Thysiadis S.; Katsamakas S.; Queen A.; Hasan G. M.; Ali S.; Sarli V.; Hassan M. I. Probing the Inhibition of Microtubule Affinity Regulating Kinase 4 by N-Substituted Acridones. Sci. Rep. 2019, 9, 1676. 10.1038/s41598-018-38217-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross S.; Rahal R.; Stransky N.; Lengauer C.; Hoeflich K. P. Targeting cancer with kinase inhibitors. J. Clin. Invest. 2015, 125, 1780–1789. 10.1172/jci76094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan M. I.; Kumar V.; Singh T. P.; Yadav S. Structural model of human PSA: a target for prostate cancer therapy. Chem. Biol. Drug Des. 2007, 70, 261–267. 10.1111/j.1747-0285.2007.00553.x. [DOI] [PubMed] [Google Scholar]
- Hoda N.; Naz H.; Jameel E.; Shandilya A.; Dey S.; Hassan M. I.; Ahmad F.; Jayaram B. Curcumin specifically binds to the human calcium-calmodulin-dependent protein kinase IV: fluorescence and molecular dynamics simulation studies. J. Biomol. Struct. Dyn. 2016, 34, 572–584. 10.1080/07391102.2015.1046934. [DOI] [PubMed] [Google Scholar]
- Khan N. S.; Khan P.; Ansari M. F.; Srivastava S.; Hasan G. M.; Husain M.; Hassan M. I. Thienopyrimidine-Chalcone Hybrid Molecules Inhibit Fas-Activated Serine/Threonine Kinase: An Approach To Ameliorate Antiproliferation in Human Breast Cancer Cells. Mol. Pharm. 2018, 15, 4173–4189. 10.1021/acs.molpharmaceut.8b00566. [DOI] [PubMed] [Google Scholar]
- Khan P.; Queen A.; Mohammad T.; Smita; Khan N. S.; Hafeez Z. B.; Hassan M. I.; Ali S. Identification of alpha-Mangostin as a Potential Inhibitor of Microtubule Affinity Regulating Kinase 4. J. Nat. Prod. 2019, 82, 2252–2261. 10.1021/acs.jnatprod.9b00372. [DOI] [PubMed] [Google Scholar]
- Mohammad T.; Batra S.; Dahiya R.; Baig M. H.; Rather I. A.; Dong J. J.; Hassan I. Identification of High-Affinity Inhibitors of Cyclin-Dependent Kinase 2 Towards Anticancer Therapy. Molecules 2019, 24, 4589. 10.3390/molecules24244589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammad T.; Siddiqui S.; Shamsi A.; Alajmi M. F.; Hussain A.; Islam A.; Ahmad F.; Hassan M. I. Virtual Screening Approach to Identify High-Affinity Inhibitors of Serum and Glucocorticoid-Regulated Kinase 1 among Bioactive Natural Products: Combined Molecular Docking and Simulation Studies. Molecules 2020, 25, 823. 10.3390/molecules25040823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pires D. E. V.; Blundell T. L.; Ascher D. B. pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. 10.1021/acs.jmedchem.5b00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beg A.; Khan F. I.; Lobb K. A.; Islam A.; Ahmad F.; Hassan I. High throughput screening, docking and molecular dynamics studies to identify potential inhibitors of human calcium/calmodulin-dependent protein kinase IV. J. Biomol. Struct. Dyn. 2018, 37, 2179–2192. 10.1080/07391102.2018.1479310. [DOI] [PubMed] [Google Scholar]
- Gulzar M.; Syed S. B.; Khan F. I.; Khan P.; Ali S.; Hasan G. M.; Taneja P.; Hassan M. I. Elucidation of interaction mechanism of ellagic acid to the integrin linked kinase. Int. J. Biol. Macromol. 2019, 122, 1297–1304. 10.1016/j.ijbiomac.2018.09.089. [DOI] [PubMed] [Google Scholar]
- Gupta P.; Mohammad T.; Khan P.; Alajmi M. F.; Hussain A.; Rehman M. T.; Hassan M. I. Evaluation of ellagic acid as an inhibitor of sphingosine kinase 1: A targeted approach towards anticancer therapy. Biomed. Pharmacother. 2019, 118, 109245. 10.1016/j.biopha.2019.109245. [DOI] [PubMed] [Google Scholar]
- Khan F. I.; Aamir M.; Wei D.-Q.; Ahmad F.; Hassan M. I. Molecular mechanism of Ras-related protein Rab-5A and effect of mutations in the catalytically active phosphate-binding loop. J. Biomol. Struct. Dyn. 2017, 35, 105–118. 10.1080/07391102.2015.1134346. [DOI] [PubMed] [Google Scholar]
- Khan F. I.; Shahbaaz M.; Bisetty K.; Waheed A.; Sly W. S.; Ahmad F.; Hassan M. I. Large scale analysis of the mutational landscape in beta-glucuronidase: A major player of mucopolysaccharidosis type VII. Gene 2016, 576, 36–44. 10.1016/j.gene.2015.09.062. [DOI] [PubMed] [Google Scholar]
- Khan P.; Parkash A.; Islam A.; Ahmad F.; Hassan M. I. Molecular basis of the structural stability of hemochromatosis factor E: A combined molecular dynamic simulation and GdmCl-induced denaturation study. Biopolymers 2016, 105, 133–142. 10.1002/bip.22760. [DOI] [PubMed] [Google Scholar]
- Ito A.; Mukaiyama A.; Itoh Y.; Nagase H.; Thøgersen I. B.; Enghild J. J.; Sasaguri Y.; Mori Y. Degradation of interleukin 1β by matrix metalloproteinases. J. Biol. Chem. 1996, 271, 14657–14660. 10.1074/jbc.271.25.14657. [DOI] [PubMed] [Google Scholar]
- Rodier F.; Bahadur R. P.; Chakrabarti P.; Janin J. Hydration of protein–protein interfaces. Proteins: Struct., Funct., Bioinf. 2005, 60, 36–45. 10.1002/prot.20478. [DOI] [PubMed] [Google Scholar]
- Ali S.; Hassan M.; Islam A.; Ahmad F. A review of methods available to estimate solvent-accessible surface areas of soluble proteins in the folded and unfolded states. Curr. Protein Pept. Sci. 2014, 15, 456–476. 10.2174/1389203715666140327114232. [DOI] [PubMed] [Google Scholar]
- Hubbard R. E.; Kamran Haider M.. Hydrogen Bonds in Proteins: Role and Strength. eLS; John Wiley & Sons, Ltd., 2001. [Google Scholar]
- Naz F.; Khan F. I.; Mohammad T.; Khan P.; Manzoor S.; Hasan G. M.; Lobb K. A.; Luqman S.; Islam A.; Ahmad F.; Hassan M. I. Investigation of molecular mechanism of recognition between citral and MARK4: A newer therapeutic approach to attenuate cancer cell progression. Int. J. Biol. Macromol. 2018, 107, 2580–2589. 10.1016/j.ijbiomac.2017.10.143. [DOI] [PubMed] [Google Scholar]
- Jameel E.; Naz H.; Khan P.; Tarique M.; Kumar J.; Mumtazuddin S.; Ahamad S.; Islam A.; Ahmad F.; Hoda N.; Hassan M. I. Design, synthesis, and biological evaluation of pyrimidine derivatives as potential inhibitors of human calcium/calmodulin-dependent protein kinase IV. Chem. Biol. Drug Des. 2017, 89, 741–754. 10.1111/cbdd.12898. [DOI] [PubMed] [Google Scholar]
- Amadei A.; Linssen A. B. M.; Berendsen H. J. C. Essential dynamics of proteins. Proteins: Struct., Funct., Bioinf. 1993, 17, 412–425. 10.1002/prot.340170408. [DOI] [PubMed] [Google Scholar]
- Jacob R. B.; Andersen T.; McDougal O. M. Accessible high-throughput virtual screening molecular docking software for students and educators. PLoS Comput. Biol. 2012, 8, e1002499 10.1371/journal.pcbi.1002499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott O.; Olson A. J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biovia D. S.Discovery Studio Modeling Environment; Dassault Systèmes: San Diego, 2015. [Google Scholar]
- Abraham M. J.; Murtola T.; Schulz R.; Páll S.; Smith J. C.; Hess B.; Lindahl E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- Wheeler D. L.; Barrett T.; Benson D. A.; Bryant S. H.; Canese K.; Chetvernin V.; Church D. M.; DiCuccio M.; Edgar R.; Federhen S. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2006, 35, D5–D12. 10.1093/nar/gkl1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman H. M.; Bourne P. E.; Westbrook J.; Zardecki C.. The protein data bank. Protein Structure; CRC Press, 2003; pp 394–410. [Google Scholar]
- Sterling T.; Irwin J. J. ZINC 15–ligand discovery for everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. 10.1021/acs.jcim.5b00559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daina A.; Michielin O.; Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner P.Grace-5.1. 22/qtGrace v 0.2. 4. 2018.