

Abstract

Leishmaniasis is a chronic disease caused by protozoa of the distinct Leishmania genus transmitted by sandflies of the genus Phlebotomus (old world) and Lutzomyia (new world). Among the molecular factors that contribute to the virulence and pathogenesis of Leishmania are metalloproteases, e.g., glycoprotein 63 (gp63), also known as leishmanolysin or major surface protease (MSP). This protease is a zinc-dependent metalloprotease that is found on the surface of the parasite, abundant in Leishmania promastigote and amastigote. This study describes the prediction of three-dimensional (3D) structures of leishmanolysin (UniProt ID A0A088RJX7) of Leishmania panamensis employing a homology modeling approach. The 3D structure prediction was performed using the SWISS-MODEL web server. The tools PROCHECK, Molprobyty, and Verify3D were used to check the quality of the model, indicating that they are reliable. Best docking configurations were identified applying AutoDock Vina in PyRx 0.8 to obtain a potential antileishmanial activity. Biflavonoids such as lanaroflavone, podocarpusflavone A, amentoflavone, and podocarpusflavone B showed good scores among these molecules. Lanaroflavone appears to be the most suitable compound from binding affinity calculations.
1. Introduction
Leishmaniases, a neglected spectrum of diseases considered to have negative implications and many consequences, are caused by protozoa parasites of the genus Leishmania,1 transmitted by female sandflies of Phlebotomus (old world) and Lutzomyia (new world) as its vector. The Leishmania species are generally zoonotic and carried by rodents and canines, which are their main reservoir hosts.2 The Leishmania parasites infect 10–12 million people worldwide, causing a spectrum of diseases known as leishmaniases. It has been estimated that 350 million people are at the risk of contracting leishmaniasis, and 2 million cases are reported each year.3 Some scientific advances have been achieved in the treatment, diagnosis, and prevention of leishmaniasis in the last 10 years, and the costs of several key medicines have been reduced.4 Conventional chemotherapeutics for leishmaniasis are based on antimonial sodium stibogluconate (Pentostam), miltefosine (Miltex), and meglumine antimoniate (Glucantime). There are adverse side effects associated with the compounds, and drug resistance is emerging. However, these medications are not very effective or available, with some reports describing the toxic effects of drugs in patients. Researchers have observed that there is an increased incidence of multidrug resistance (MDR) in leishmaniasis. The absence of vaccines has led to attention being focused on the identification of novel targets and the development of an alternative drug. Therefore, the development of novel agents against these parasites is extremely significant.5,6
Many important enzymes are present in the Leishmania parasite, which are considered potential targets for the development of new therapeutic agents. Among the enzymes of Leishmania are metalloproteases, especially those belonging to metzincins, which contain zinc atoms in their structure, like the so-called leishmanolysin.7 Glycoprotein 63 (gp63) or leishmanolysin is a 63 kDa protein and a metalloprotease from the M8 family (subclan MA(M), metzincins), which contributes to the virulence and pathogenesis of the Leishmaniaparasite and is found on its surface.8 It was isolated for the first time in 1980 and described genetically and biochemically as a surface antigen expressed in promastigotes of the Leishmania species, having a range of substrates, including casein, gelatin, albumin, hemoglobin, and fibrinogen.7 The components of this protease include a sequence motif HExxHxxGxxH and an N-terminal propeptide, which renders the proenzyme inactive in the course of the translation and is eliminated during the maturation and activation. gp63 is the most abundant surface protein of promastigotes and contains 1% of the total parasite proteome but is upregulated in amastigotes.9,10 gp63 has been reported to interact with the fibronectin receptor and therefore could further aid the adherence of the parasite to macrophages.11 From these diverse findings, it is clear that by cleavage and/or degradation of various proteins, gp63 can profoundly affect the macrophage functions, favoring the survival of Leishmania.12,13
Even though glycoproteins have been one of the most important targets of the Leishmania parasite, few relevant studies with glycoprotein have been performed. In the search for new drugs for the treatment of leishmaniasis, Shaukat et al.14 conducted a study of benzimidazole derivatives, whose results both in vitro and in silico through molecular docking showed that these compounds could serve as the basis for the future treatment of leishmaniasis.14
Computational methods have been successfully applied to predict protein structures and ligand–protein interactions. Molecular docking is a method that is used to predict the preferred orientation of predominantly small organic molecules (ligands) within biological macromolecules (proteins). In this sense, natural products can be a valuable alternative to provide huge diversity of chemical structures for biological screening tests of Leishmania species; flavonoids are promising drug candidates for the treatment of all forms of leishmaniasis.15 This study reports a three-dimensional (3D) structure of the leishmanolysin protein, gp63, from Leishmania panamensis that was built through homologous modeling and optimized using molecular dynamics (MD) simulation. The leishmanolysin protein from L. major (ID PDB: 1LML) was used as a template. The structure was validated to check the quality of the models, indicating that they are reliable. Based on the 3D structure, the ligand-binding modes of flavonoid compounds were elucidated using molecular docking in proteins both crystallized and built by homology modeling. The docking analysis revealed that gp63 from L. major and L. panamensis can structurally accommodate various flavonoid-like ligands and helped us in the molecular recognition of the design of the possible inhibitors and provided new knowledge that can be used to treat the disease caused by the Leishmania species.
2. Results and Discussion
2.1. Homology Model
The structural models of leishmanolysin from L. panamensis were built by comparative modeling using the crystal structure of gp63 of L. major (PDB ID: 1LML) as the template (Figure 1).
Figure 1.

Homology model of leishmanolysin. The template structure of L. major (PDB ID: 1LML) is color coded in cyan (A) and the homology model of L. panamensis (lpgp63) is shown in red (B). (C) Superimposition of the homology model with the template. The active site and the zinc atom (gray) are highlighted in the squares on the right-hand side.
The alignment (Figure S1) between leishmanolysin proteins of L. major and L. panamensis indicated the presence of conserved regions throughout the protein under study. The structures were selected based on their similarity, identity, and number of gaps. The alignment is consistent with the experimental results. The comparison with the L. major leishmanolysin sequence showed the identity displayed 74.8% with Lpgp63.
These results are within the range of 59–71% reported previously by Sutter et al. for a variety of L. major leishmanolysin proteins coded by chromosome 10.16 The HEXXHXXGXXH motif of the active site, typical of the zinc-dependent metalloproteinases, is highly conserved. There is an insertion of 62 residues between glycine and the third histidine in each of the proteins modeled for the Leishmania species, as reported by Yao et al.8 The position of the active site is highlighted with two of the histidines being part of the α-helix (H8) and the other in a loop in the central domain. The zinc atom of the protein lies between the N-terminal and central domains. The alignment is consistent with experimental outcomes reporting that nine disulfide bridges in the template structure are completely aligned with the leishmanolysin sequence.17 The high conservation of the sequence is also reflected in the similarity of some properties.
2.2. Molecular Dynamics Simulation
Molecular dynamics simulations were employed to optimize and establish the stability of the protein constructed by homology modeling. The MD trajectory was analyzed by computing the root-mean-square deviation (RMSD) of Cα (Figure 2A). The molecular system reached stable states during 200 ns simulations.
Figure 2.

Optimization of the homology model by MD simulations. (A) Cα RMSD values of proteins 1LML (cyan) and lpgp63 (red) of L. major and L. panamensis, respectively, were plotted. (B) Root-mean-square fluctuation (RMSF) values of particular residues. Comparison along a 200 ns MD simulation.
The leishmanolysin of L. major, 1LML protein, converged in the time frame after 200 ns of MD simulation. The average RMSD value was 1.482 Å ± 0.103, and these outcomes were similar to those reported by Sutter et al. For the protein that was built by homology, Lpgp63, the final equilibrium was reached in the MD simulation with an average of 1.909 Å ± 0.207 nm.
The root-mean-square fluctuation (RMSF) values of the modeled structures generated during the MD simulation were calculated to characterize the mobility of particular residues (Figure 2B). As expected, the RMSF plot shows a variation in amino acid residues between the different leishmanolysin proteins of L. major and L. panamensis. In the model, the greatest fluctuations were observed in the N- and C-terminal domains, while the central domain where the active site is located remains rigid, as reported by Bianchini et al.18 On the other hand, are evidenced mainly in the change of the length of the amino acids in the active site concerning the zinc atom. It is observed in this study that the bond distance of zinc to the Nε2 atoms of HIS residues undergoes variations.17−19 It is additionally mentioned that the zinc atom shortens its distance to the OE2 of the GLU residue (Figure S2).
A Ramachandran plot was built, and it showed that 92.1 and 91.1% of residues were in the most favored regions and 7.1 and 8% were in the additional allowed regions. After the MD refinement, a checking process was also undertaken. The Ramachandran plot of the 1LML protein and model built by homology showed that 96.0 and 96.3% of residues were found in the most favored allowed regions and 4 and 3.7% were in the additional allowed region (Figure S3). Rodriguez et al. report that the homology model of L. braziliensis shows percentage of residues in favorable regions very similar to those reported in this article for the study of leishmanolysin.20,21
The analysis of the models with ProSA-web service shows a Z-score between −8.27 and −9.81 (acceptable values are below 0.5). The global score was between −0.98 and −1.49. Otherwise, the quality of the models was confirmed by the Verify3D analysis in an average of 91% of the residues with a score >2 in the 3D/one-dimensional (1D) profile.
2.3. Molecular Docking
Molecular docking has been implemented to reveal the interaction between flavonoid compounds and leishmanolysin proteins of L. major and L. panamensis. Table 1 shows 24 flavonoid molecules to have the lowest binding energy docked at the active site for the two leishmanolysin proteins of L. major and L panamensis, respectively. The chemical structures are indicated in the Supporting Information, Table S1. Inspection of the docked structures shows the most favorable interactions within the active site, particularly between hydrogen bonds and hydrophobic interactions.
Table 1. Binding Affinity (kcal/mol) of the Most Interesting Compounds Obtained by Docking with Different Leishmanolysin of the Leishmania Species.
| molecule | 1LML | Lpgp63 |
|---|---|---|
| amphotericin B | –11.5 | –10.7 |
| lanaroflavone | –10.5 | –9.9 |
| podocarpusflavone A | –10.1 | –9.7 |
| amentoflavone | –9.9 | –9.7 |
| podocarpusflavone B | –9.6 | –9.1 |
| pseudotsuganol | –9.5 | –9.2 |
| tetrahydrorobustaflavone | –9.4 | –9.7 |
| 2,3-dehydrosilibinin | –8.7 | –8.3 |
| rhuschalcone VI | –8.6 | –8.4 |
| epigallocatechin | –8.6 | –7.3 |
| agathisflavone | –8.4 | –8.6 |
| SN00000577 | –8.4 | –7.2 |
| methyltetrahydroamentoflavone | –8,4 | –8.5 |
| SN00000355 | –8.4 | –6.4 |
| SN00000558 | –8.3 | –7.2 |
| abyssinone IV | –8.2 | –7.6 |
| quercitrin | –8.2 | –7.5 |
| SN00000365 | –8.2 | –7.2 |
| 4-hidroxylonchocarpine | –8.1 | –7,2 |
| (α-naphthoflavone) SN00000328 | –8.1 | –7.3 |
| bipinnatone A | –8.0 | –6.5 |
| medicagenina | –8.0 | –7.5 |
| SN00000367 | –8.0 | –7.3 |
| SN00000357 | –8.0 | –6.8 |
| SN00157618 | –8.0 | –8.1 |
Docking of selected ligand molecules and reference compounds onto the binding site (pocket) of the leishmanolysin (gp63) protein and the homologous protein constructed was performed by AutoDock Vina in the PyRx 0.8 module. After that, for each molecule, the best pose based on its conformation and docking binding energy was selected. Since leishmanolysin structures do not contain a co-crystallized ligand, the study was conducted employing the active site reported by Schlagenhouf et al.10 The binding site comprised amino acids HIS264, GLU265, HIS268, HIS334, and MET345 and a Zn atom, and the grid box was centered on these residues. As reported by Shaukat et al.,14 amphotericin B was also used as a reference molecule. The antiparasitic activity of amphotericin B is related to the ability of this antibiotic to form permeable channels in the cellular membrane of the pathogen. These channels cause a leakage of monovalent ions and small organic molecules from the cellular membrane.14,22 In both amastigotes and promastigotes, leishmanolysin is present on the surface, and it is possible that amphotericin will interact with this protein. The docking performed with the antibiotic using the gp63 protein showed that it interacts with the amino acids GLU220 and GLU265 and the Zn atom for the case of gp63 of L. major. The proteins constructed by the homology of L. panamensis follow the same scheme described above (GLU218, GLU263, and Zn) (Figure S4).
The docking pose of lanaroflavone exhibited the lowest binding affinity, followed by podocarpusflavone A, amentoflavone, and podocarpusflavone B when docking was performed with 1LML and Lpgp63 (Figure 3). The most significant interactions between the ligands and proteins were hydrophobic, H-bonding, metal-contact, and π-stacking (π–π T-shaped) interactions. Table 2 summarizes the principal interactions between the first four ligands with the lowest binding affinity and each of the proteins studied. Lanaroflavone, podocarpusflavone, and amentoflavone formed an H-bond with residues LEU224, ALA225, SER418, and GLN341 within the active site, while podocarpusflavone B interacted with GLY222, SER418, and SER465 through H-bonds. All ligands presented hydrophobic links, especially π-stacking, with VAL261, HIS264, LEU257, PRO460, and ALA350 with the rings of biflavonoid compounds. Podocarpusflavone B interacted with amino acids GLY222, SER418, and SER465 through H-bonds.
Figure 3.

Docking poses of the binding interaction of molecules in the active site of leishmanolysin from L. major: (A) lanoraflavone, (B) amentoflavone, (C) podocarpusflavone A, and (D) podocarpusflavone B.
Table 2. H-Bond Interaction Information from Docking Calculations between Ligands and Modeled Proteins.
| 1LML |
Lpgp63 |
|||
|---|---|---|---|---|
| molecule | H-bond | distance (Å) | H-bond | distance (Å) |
| LEU224 NH···O=C-4 | 1.98 | LEU222 NH···C=O-4 | 3.00 | |
| ALA225 C=O···OH-5 | 2.80 | ALA223 C=O···OH-5 | 2.24 | |
| amentoflavone | ALA349 NH···O-1 | 2.19 | ASP416 O···OH-5″ | 2.80 |
| SER448 OH···OH-4‴ | 2.31 | |||
| THR459 OH···C=O-4″ | 2.77 | |||
| THR459 OH···OH-5″ | 2.80 | |||
| LEU224 NH···O=C-4 | 1.97 | LEU222 NH···C=O-4 | 2.27 | |
| ALA225 C=0···OH-5 | 2.49 | ALA223 NH···C=O-4 | 2.68 | |
| ALA225 NH···O=C-4 | 2.74 | ALA223 C=O···OH-5 | 2.65 | |
| lanaroflavone | HIS264 NH···OH-7 | 2.81 | LYS339 NH···C=O-4″ | 2.98 |
| ALA346 C=O···OH-7 | 2.92 | |||
| GLN341 C=O···OH-4″ | 2.98 | |||
| ALA349 NH···O-1 | 2.61 | |||
| LEU224 NH···O=C-4 | 1.96 | LEU222 NH···C=O-4 | 2.02 | |
| ALA225 C=0···OH-5 | 2.64 | ALA223 C=O···OH-5 | 2.16 | |
| podocarpusflavone A | ALA349 NH···O-1 | 2.40 | ||
| SER418 OH···OH-4″ | 2.09 | |||
| GLY222 C=O···OH-5 | 2.91 | LEU222 NH···C=O-4 | 2.01 | |
| podocarpusflavone B | SER418 OH···OH-4″ | 2.44 | ||
| SER465 OH···OH-4″ | 2.77 | |||
An interaction with the zinc atom was observed in lanoraflavone where the oxygen of the hydroxyl group in position 7 favorably interacts with this metal. In podocarpusflavone A and amentoflavone, the O–metal bond of the same hydroxyl group in position 7 was favorable, perhaps because of the distance between the two atoms, while in podocarpusflavone B, there was no interaction with the zinc as was observed.
In the case of leishmanolysin, lpgp63, of L. panamensis constructed by homology, the four molecules mentioned above presented the same pattern of interactions as in gp63 of L. major (Table 2). Lanaroflavone, amentoflavone, and podocarpusflavone A and B interact through H-bonds with residues LEU222, ALA223, and ALA334. Regarding hydrophobic interactions, the amino acids involved are ILE255, HIS262, and VAL347. The zinc atom has a metallic interaction with the hydroxyl group of the flavonoids (Figure 4).
Figure 4.

Docking poses of the molecules in the active site of leishmanolysin from L. panamensis built by the homology model: (A) lanoraflavone, (B) amentoflavone, (C) podocarpusflavone A, and (D) podocarpusflavone B.
2.4. Solvent-Accessible Surface Area (ASA)
Figure 5A,B shows the solvent-accessible surface area for the two proteins studied, 1LML and lpgp63, respectively. The graph shows that the docked molecules produce an alteration in the access to solvent, being more noticeable in amentoflavone. The HIS264 and GLU265 residues decrease their ASA values with respect to the native protein. The HIS334 residue does not show significant changes for lanaroflavone, podocarpusflavone A, and podocarpusflavone B, while in amentoflavone, the value of the ASA is drastically decreased. On the other hand, the zinc atom is also significantly affected when the docked compounds are compared to the native protein. Table S2 shows the ASA values for all protein residues that interact with docked molecules.
Figure 5.
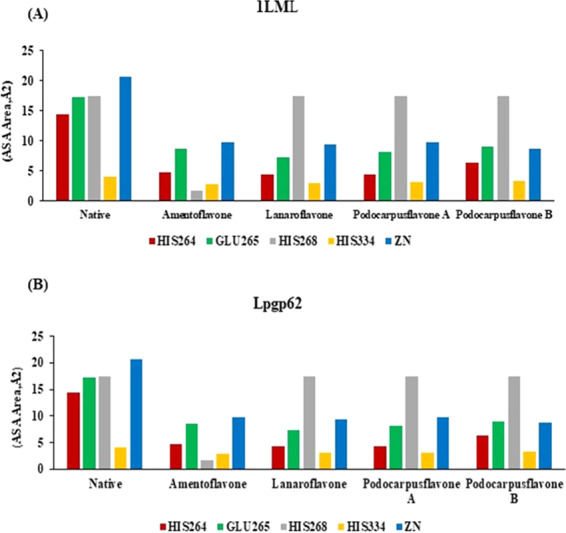
Solvent-accessible surface area (ASA) for residues from the active site and the zinc atom: (A) 1LML and (B) lpgp63.
2.5. Validation
The molecular docking performed with each of the models and the crystallographic structure was validated by the receiver operating characteristic (ROC) curve. The area under the curve (AUC) for 1LML and LpGp63 was 0.926 ± 0.026 and 0.928 ± 0.017, respectively (Figure S5). The ROC curve discriminates among active ligands and decoy structures, indicating the good overall predictive performance (AUC > 0.5) of docking for the models of lpgp63 and 1LML.
Since the calculation of the enrichment factor (EF1%) represents another method to evaluate the performance of virtual screening in discriminating active from inactive compounds, we performed a docking study with a decoy set including 86 active and 90 inactive molecules. Particularly, analyzing the top 1% of the ranking obtained by sorting the scores in ascending order, it was possible to find good EF values. For 1LML and LpGp63, the EF values were 45 and 44, respectively, which show that the model demonstrated sufficient discriminating capability for exploring the top 1% of docked compounds.
The correlation analysis performed for 32 flavonoids presented a linear relationship between theoretical free binding energy (kcal/mol) vs pIC50 for 1LML and the model developed by homology (Figure S5). Both correlations indicate a value of R > 0.5. Therefore, we can conclude that the outcome achieved in the virtual screening with AutoDock Vina for the two proteins tested was favorable (Table S3).
Flavonoids represent a broad family of polyphenolic compounds found in vegetables and fruits. It is generally accepted that these compounds are safe and nontoxic. The most common flavonoids are flavones and isoflavones. Naturally occurring flavones have been reported to have leishmanicidal activity.23 Various flavonoids have demonstrated activity against amastigotes in macrophage infection, such as quercetin that reduces intracellular load by 70% at 45 μM.15,23
Lanaroflavone has an ether bridge that links two fragments to form the biflavonoid (bipigenina, C″–O–C−). This allows its free rotation and the compound can better fit the binding pocket of the protein. By contrast, a C3′–C8′′ bond present in the other biflavonoids provides certain rigidity to these structures and the interaction with the binding pocket is less favored. Some studies have indicated that the hydroxyl and methoxyl groups at the rings of flavonoids are involved in their bioactivities.24−27 Lanaroflavone has a C–4‴–O–C–8 junction that permits better rotation (the substitution of C-4‴ seems to have higher relevance regarding the antiplasmodial and leishmanicidal activities).25 However, the C3″–C8′′ interflavonyl linkage of amentoflavone and podocarpusflavone A and B is more restricted,27 allowing lanaroflavone to be oriented in the active site of leishmanolysin.
Weniger et al.24 have reported that lanaroflavone exhibited optimum antiplasmodial activity in vitro. When compared with the strains of Plasmodium falciparum K1 chloroquine-resistant presents a moderate antileishmanial activity of L. donovani in amastigotes with an IC50 of 0.2 and 3.9 μg mL–1, respectively.24 In amentoflavone, the substitution of C-4‴ seems to have high relevance for the antiplasmodial and leishmanicidal activities.25 Little antileishmanial activity compared to promastigotes in L. donovani was reported for podocarpusflavone A and B.27 For biflavonoids having a C–C interflavonyl linkage, there is also a tendency to enhance the antimalarial activity with increasing number of methoxyl groups in the molecule, as observed in amentoflavone and in biflavonoids, such as methyltetrahydroamentoflavone and podocarpusflavone A, whose methoxy group in position 4″ interacts with the 1LML protein of L. major, as was described above.28−31 Recently, biflavonoids have come to the forefront of the polyphenol family for their antituberculosis activities.32
In recent pharmacological studies, they are also reported to possess antinociceptive, anti-inflammatory, and cytotoxic properties.33,34 Only a few studies indicate their antiprotozoal activity, but recently, antiplasmodial activity was established for two compounds of this kind.
Furthermore, it was found that amentoflavone was not active on axenic amastigotes of L. donovani in the study by Weniger et al.,25 and it showed poor activity on promastigotes of the same species according to del Rayo Camacho et al.27L. amazonensis displayed excellent antileishmanial activity for intracellular amastigotes 48 h after the aforementioned treatment. Therefore, the antileishmanial action of this compound may be mediated by the host cell. This action highlights the properties of the compound as a prodrug, which needs to be metabolized by the cell to exert its effect.35
The molecules pseudotsuganol and rhuschalcone VI have a binding affinity of −9.6 and −8.7 kcal/mol, respectively. These two compounds presented activity against L. major. Further, it was shown that these two molecules exhibit notable attraction for the nucleoside hydrolase enzyme of Leishmania. Methyltetrahydroamentoflavone (−8.4 kcal/mol) has a positive interaction energy with L. major methionyl-tRNA synthetase, according to the same authors.36
The analyzed compounds, in general, demonstrate affinity for the proteins considered. Although they are molecules of natural origin, Lipinski’s rules are not so relevant in predicting their drug-likeness, as reported for other biologically interesting natural products. We believe that this is because nature has learned to maintain low hydrophobicity and intermolecular H-bond donating potential when it needs to make biologically active compounds with significant molecular weight and considerable numbers of rotatable bonds. In most cases, natural products do not necessarily abide by Lipinski’s rule because they are thought to enter the human body not by passive diffusion but by more complex mechanisms like active transportation and hence are not expected to comply with the rules for bioavailability.37 Besides, natural products are more likely to resemble biosynthetic intermediates or endogenous metabolites than purely synthetic compounds and hence take advantage of active transport mechanisms.
3. Conclusions
In this study, the 3D structures of different Leishmania spp. gp63 proteins were constructed by homology modeling using the crystal structure of leishmanolysin (gp63) of L. major as a template. The reliability of three models was assessed by Ramachandran plots, ProSA-web, PROCHECK, Verify3D, and molprobity. As expected, the resulting structure was similar to that of the template. The data reported here are in agreement with what has been suggested by some authors, with the N-terminal and C-terminal regions being the regions of most appreciable variation. The findings indicate a more rigid central domain, although some regions (especially the loop region) were extremely flexible, despite certain points where protein mobility was observed.
Docking studies made it possible to predict affinity, activity, binding, and the orientation of the binding of flavonoids with leishmanolysin proteins. The analysis was based on the evaluation of their binding energy. Compounds like flavonoids, chalcones, and bioflavonoids showed great affinity to the leishmanolysin protein from L. major. Lanaroflavone, the molecule with the lowest binding energy, also showed good antileishmanial activity, despite violating the rule of five (Lipinski’s rules). Flavonoid derivatives are natural products that exhibit promising antileishmanial activity and deserve further research.
4. Experimental Section and Computational Methods
4.1. Homology Model
The protein sequence of gp63 from L. panamensis was retrieved from the UniProt database. The Uniprot accession number is A0A088RJX7 with 594 amino acid residues. The crystal structure of gp63 from L. major, employed as a template, was retrieved from the protein database PDB (PDB ID: 1LML).10 The model was generated using the SWISS-MODEL server.38 The best model was selected based on QMEAN and Z-score of ProSA-web. The quality of 3D prediction was assessed using Molprobity,39 ProSA-web,40 and Verify3D.41 The final representation was visualized using Discovery Studio Visualizer.42 The gp63 protein of L. major was obtained from the PDB database with PDB ID 1LML; all water molecules were removed and hydrogen atoms were added. On the other hand, the protonation states of the surrounding histidine residues for both proteins were assigned using PROPKA;43 they were selected as neutral compared to those reported by Sutter et al. (Figure 5).
4.2. Molecular Dynamics Simulation
Molecular dynamics (MD) simulations of 200 ns were performed for the modeled protein and the template from L. panamensis and L. major, respectively, using the GROMACS 2016.5 package;44 all simulations were carried out using Amber99sb as a force field.45 Both macromolecules were solvated by a cubic periodic box in which each protein was solvated with TIP3P water under periodic boundary conditions (PBCs).46 The system was neutralized, and the ionic strength (0.1 mol L–1) of the medium was adjusted by adding Na+ and Cl– ions, keeping the number of particles constant. After these steps, energy minimization of the systems was performed until convergence, which was followed by equilibration with pressure and temperature (NVT and NPT ensembles) kept constant at 300 K and 1.0 bar, respectively; equilibration periods were 1.0 ns, production runs were of 10 ns duration, and the V-rescale thermostat and Parrinello–Rahman thermostat were used. The LINCS47 and SETTLE48 algorithms were employed to determine bond lengths of hydrogen atom distance constraints, respectively, whereas long-range interactions were calculated employing the particle-mesh Ewald (PME) method,49,50 used to constrain the geometry of the water molecules. The equilibrated system was subjected to the final MD production run of 10 ns, applying the periodic boundary conditions (PBCs) and integrating the equation of motion every 2 fs. GROMACS and VMD51 software packages were used to analyze the MD trajectories. Root-mean-square deviation (RMSD) of Cα residues and root-mean-square fluctuation (RMSF) were also calculated. In total, the production time was 200 ns for each system.
4.3. Ligand Database and Preparation
In total, 5470 drug-like compounds (chalcones, flavonoids, and biflavonoids) were retrieved from the Super Natural II database52 as a single file in spatial data file (SDF) format. This file was imported in Open Babel53 and all ligand structures were converted into PDB format. Then, the compounds were optimized with density functional theory (DTF) at the B3LYP/6-31G level using the Gaussian 09 package.54 Amphotericin B was obtained from the PubChem database55 and employed as reference molecules in molecular docking studies according to the suggestions by Saukat et al.
4.4. Molecular Docking
Molecular docking was performed using AutoDock Vina56 in PyRx 30.8 (virtual screening tools).57 Then, the resulting collection of potential ligands was docked into the leishmanolysin proteins. Hydrogen atoms were added to all proteins, and partial atomic charges were calculated. Initially, flexible-ligand docking was done. The grid box size was set to 35 × 35 × 35 points with a spacing of 0.375 Å. For the calculation, 150 runs of the Lamarckian genetic algorithm (LGA)58 with 25 000 000 evaluations and 270 000 generations were performed.
4.5. Validation
To validate docking, scoring functions were assessed using different parameters like the area under the curve (AUC), receiver operating characteristic (ROC) curve, and enrichment factor (EF1%) according to Meekyum et al.59 A dataset of 176 molecules with strong, moderate, and weak antileishmanial activity was employed to validate and to assess the selectivity and sensibility of our methodology. A linear correlation analysis was performed between the biological activity (pIC50) and the affinity of flavonoids reported and the calculated affinity values were, in addition, used for validation of the virtual screening done by AutoDock Vina (Figure S6). The statistical significance of the AUC value of different models was evaluated with a p test with a 95% confidence limit.
Acknowledgments
This study was supported by the University of Cartagena, Cartagena; the National Administrative Department of Science, Technology, and Innovation of Colombia, Bogotá; and COLCIENCIAS, Grant 512-2012, the University of Cartagena (Cartagena).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c01584.
Homology modeling, molecular dynamics simulation, and docking were performed on leishmanolysin of Leishmania panamensis using gp63 of L. major as a template. A series of flavonoids were docked into the active site of gp63 from L. major and L. panamensis, and their scores were in the order lanaroflavone > amentoflavone > podocarpusflavone A > podocarpusflavone B (PDF)
Author Contributions
J.M.-C., H.G.-E., A.P., and L.C.-C. conceived and designed the experiments; J.M.-C., A.P.H.G.-E., and L.C.-C. performed the experiments; J.M.-C., H.G.-E., R.V.-R., and M.L.S.G. analyzed the data; R.V.-R. was also responsible for the correspondence of the manuscript. All authors discussed, edited, and approved the final version of the manuscript.
This work was supported by the Vice-Rectory for Research of the University of Cartagena (Grant RC 03381 DE 2016); the National Program for Doctoral Formation, Colciencias, 727- 2015; and the Doctoral program in biomedical sciences of the University of Cartagena.
The authors declare no competing financial interest.
Supplementary Material
References
- Alves C. R.; Santos de Souza R.; Charret K.d.S.; Monteiro de Castro Cortes L.; Pereira de Sá Silva M.; Barral-Veloso L.; Gonçalves Oliveira L. F.; Souza da Silva F. Understanding serine proteases implications on Leishmania spp lifecycle. Exp. Parasitol. 2018, 184, 67–81. 10.1016/j.exppara.2017.11.008. [DOI] [PubMed] [Google Scholar]
- Loría-Cervera E. N.; Andrade-Narváez F. The role of monocytes/macrophages in Leishmania infection: a glance at the human response. Acta Trop. 2020, 207, 105456 10.1016/j.actatropica.2020.105456. [DOI] [PubMed] [Google Scholar]
- WHO . Leishmaniasis: Situation and Trends; World Health Organization, 2015. [Google Scholar]
- Nagle A. S.; Khare S.; Kumar A. B.; Supek F.; Buchynskyy A.; Mathison C. J. N.; Molteni V.; et al. Recent Developments in Drug Discovery for Leishmaniasis and Human African Trypanosomiasis. Chem. Rev. 2014, 114, 11305–11347. 10.1021/cr500365f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Griensven J.; Balasegaram M.; Meheus F.; Alvar J.; Lynen L.; Boelaert M. Combination therapy for visceral leishmaniasis. Lancet Infect. Dis. 2010, 10, 184–194. 10.1016/S1473-3099(10)70011-6. [DOI] [PubMed] [Google Scholar]
- Mueller Y.; Nguimfack A.; Cavailler P.; Couffignal S.; Rwakimari J. B.; Loutan L.; Chappuis F. Safety and effectiveness of amphotericin B deoxycholate for the treatment of visceral leishmaniasis in Uganda. Ann. Trop. Med. Parasitol. 2008, 102, 11–19. 10.1179/136485908X252142. [DOI] [PubMed] [Google Scholar]
- Passalacqua T. G.; Torres F.; Nogueira C. T.; Almeida L.; Cistia M.; Dos Santos M. B.; Regasini L. O.; Graminha M. A. S.; Marchetto R.; Zottis A. The 2′,4′-dehydroxychalcone could be explored to develop new inhibitors against the glicerol-3-phosphate dehydrogenase from Leishmania species. Bioorg. Med. Chem. Lett. 2015, 4–3568. 10.1016/j.bmcl.2015.06.085. [DOI] [PubMed] [Google Scholar]
- Yao C.; Donelson J. E.; Wilson M. E. The major surface protease (MSP or GP63) of Leishmania sp. Biosynthesis, regulation of expression, and function. Mol. Biochem. Parasitol. 2003, 132, 1–16. 10.1016/S0166-6851(03)00211-1. [DOI] [PubMed] [Google Scholar]
- Barret A. J. Classification of peptidase. Method Enzymol. 1994, 44, 1–15. [DOI] [PubMed] [Google Scholar]
- Schlagenhauf E.; Etges R.; Metcalf P. The Crystal structure of the Leishmania major surface proteinase leishmanolysin (gp63). Structure 1998, 6, 1035–1045. 10.1016/S0969-2126(98)00104-X. [DOI] [PubMed] [Google Scholar]
- Brittingham A.; Morrison C. J.; McMaster W. R.; McGwire B. S.; Chang K.-P.; Mosser D. M. Role of the Leishmania surface protease GP63 in complement fixation, cell adhesion, and resistance to complement mediated lysis. Parasitol. Today 1995, 11, 445–446. 10.1016/0169-4758(95)80054-9. [DOI] [PubMed] [Google Scholar]
- Mosser D. M.; Edelson P. J. The mouse macrophage receptor for C3bi (CR3) is a major mechanism in the phagocytosis of Leishmania promastigotes. J. Immunol. 1985, 135, 2785–2789. [PubMed] [Google Scholar]
- Olivier M.; Atayde V. D.; Isnard A.; Hassani K.; Shio M. T. Leishmania virulence factors: focus on the metalloprotease GP63. Microbes Infect. 2012, 14, 1377–1389. 10.1016/j.micinf.2012.05.014. [DOI] [PubMed] [Google Scholar]
- Shaukat A.; Mirza H. M.; Ansari A. H.; Yasinzai M.; Zaidi S. Z.; Dilshad S.; Ansari F. L. Benzimidazole derivatives: synthesis, leishmanicidal effectiveness, and molecular docking studies. Med. Chem. Res. 2013, 22, 3606–3620. 10.1007/s00044-012-0375-5. [DOI] [Google Scholar]
- Cheuka P.; Mayoka G.; Mutai P.; Chibale K. The Role of Natural Products in Drug Discovery and Development against Neglected Tropical Diseases. Molecules 2016, 22, 58. 10.3390/molecules22010058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutter A.; Antunes D.; Silva-Almeida M.; Garcia de Souza Costa M.; Caffarena E. R. Structural insights into leishmanolysins encoded on chromosome 10 of Leishmania (Viannia) braziliensis. Mem. Inst. Oswaldo Cruz 2017, 112, 617–625. 10.1590/0074-02760160522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M. O.; Nichols S. E.; Wang Y.; McCammon J. A. Effects of histidine protonation and rotameric states on virtual screening of M. tuberculosis RmlC. J. Comput.-Aided Mol. Des. 2013, 27, 235–246. 10.1007/s10822-013-9643-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchini G.; Bocedi A.; Ascenzi P.; Gavuzzo E.; Mazza F.; Aschi M. Molecular dynamics simulation of Leishmania major surface metalloprotease GP63 (leishmanolysin). Proteins 2006, 64, 385–390. 10.1002/prot.21009. [DOI] [PubMed] [Google Scholar]
- Auld D. S. Zinc coordination sphere in biochemical zinc sites. BioMetals 2001, 14, 271–313. 10.1023/A:1012976615056. [DOI] [PubMed] [Google Scholar]
- Rodríguez L. A.; Peña G. S. Caracterización in silico de la proteína Leishmanolisina (gp63) de Leishmania braziliensis y búsqueda de epitopes de utilidad para el desarrollo de vacunas. Ágora Rev. Cent. 2015, 2, 227–232. 10.21679/arc.v2i2.43. [DOI] [Google Scholar]
- Waghmare S.; Buxi A.; Nandurkar Y.; Shelke A.; Chavan R. In silico sequence analysis, homology modeling and function annotation of leishmanolysin from Leishmania donovani. J. Parasit. Dis. 2016, 40, 1266–1269. 10.1007/s12639-015-0665-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baginski M.; Czub J. Amphotericin B and Its New Derivatives – Mode of Action. Curr. Drug Metab. 2009, 10, 459–469. 10.2174/138920009788898019. [DOI] [PubMed] [Google Scholar]
- Tasdemir D.; Kaiser M.; Brun R.; Yardley V.; Schmidt T. J.; Tosun F.; Rüed P. Antitrypanosomal and Antileishmanial Activities of Flavonoids and Their Analogues: In Vitro, In Vivo, Structure-Activity Relationship, and Quantitative Structure-Activity Relationship Studies. Antimicrob. Agents Chemother. 2006, 50, 1352–1364. 10.1128/AAC.50.4.1352-1364.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weniger B.; Vonthron-Senécheau C.; Arango G. J.; Kaiser M.; Brun R.; Anton R. A bioactive biflavonoid from Campnosperma panamense. Fitoterapia 2004, 75, 764–767. 10.1016/j.fitote.2004.09.015. [DOI] [PubMed] [Google Scholar]
- Weniger B.; Vonthron-Sénécheau C.; Kaiser M.; Brun R.; Anton R. Comparative antiplasmodial, leishmanicidal and antitrypanosomal activities of several biflavonoids. Phytomedicine 2006, 13, 176–180. 10.1016/j.phymed.2004.10.008. [DOI] [PubMed] [Google Scholar]
- Kumar S.; Pandey A. K. Chemistry and Biological Activities of Flavonoids: An Overview. Sci. World J. 2013, 1–16. 10.1155/2013/162750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Rayo Camacho M.; Mata R.; Castaneda P.; Kirby G. C.; Warhurst D. C.; Croft S. L.; Phillipson J. D. Bioactive compounds from Celaenodendron mexicanum. Planta Med. 2000, 66, 463–468. 10.1055/s-2000-8598. [DOI] [PubMed] [Google Scholar]
- Mbwambo Z. H.; Kapingu M. C.; Moshi M. J.; Machumi F.; Apers S.; Cos P.; Ferreira D.; Marais J. P. J.; Berghe D. V.; Maes L.; Vlietinck A.; Pieters L. Antiparasitic Activity of Some Xanthones and Biflavonoids from the Root Bark of Garciniali vingstonei. J. Nat. Prod. 2006, 69, 369–372. 10.1021/np050406v. [DOI] [PubMed] [Google Scholar]
- Jacob V.; Hagai T.; Soliman K. Structure-activity relationships of flavonoids. Curr. Org. Chem. 2011, 15, 2641–2657. 10.2174/138527211796367309. [DOI] [Google Scholar]
- Xiao J.; Kai G.; Yamamoto K.; Chen X. Advance in dietary polyphenols as α-glucosidases inhibitors: a review on structure-activity relationship aspect. Crit. Rev. Food Sci. Nutr. 2013, 53, 818–836. 10.1080/10408398.2011.561379. [DOI] [PubMed] [Google Scholar]
- Yu S.; Yan H.; Zhang L.; Shan M.; Chen P.; Ding A.; Li S. A Review on the Phytochemistry, Pharmacology, and Pharmacokinetics of Amentoflavone, a Naturally-Occurring Biflavonoid. Molecules 2017, 22, 299 10.3390/molecules22020299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y.-M.; Flavin M. T.; Cassidy C. S.; Mar A.; Chen F.-C. Biflavonoids as novel antituberculosis agents. Bioorg. Med. Chem. Lett. 2001, 11, 2101–2104. 10.1016/S0960-894X(01)00382-1. [DOI] [PubMed] [Google Scholar]
- Wong I. L. K.; Chan K.-F.; Chan T. H.; Chow L. M. C. Flavonoid Dimers as novel, potent antileishmanial agents. J. Med. Chem. 2012, 55, 8891–8902. 10.1021/jm301172v. [DOI] [PubMed] [Google Scholar]
- da Silva E. R.; do Carmo Maquiaveli C.; Magalhaes P. P. The leihmanicidal flavonols quercetin and quercetrin target Leishmania (Leishmania) amazonensis arginase. Exp. Parasitol. 2012, 130, 183–188. 10.1016/j.exppara.2012.01.015. [DOI] [PubMed] [Google Scholar]
- Kim H. P.; Son K. H.; Chang H. W.; Kang S. S. Effects of naturally occurring flavonoids on inflammatory responses and their action mechanisms. Nat. Prod. Sci. 2000, 6, 170–178. [Google Scholar]
- Suárez A. I.; Díaz B. M.; Delle Monache F.; Compagnone R. S. Biflavonoids from Podocalyxloranthoides. Fitoterapia 2003, 74, 473–475. 10.1016/S0367-326X(03)00065-0. [DOI] [PubMed] [Google Scholar]
- Khanna V.; Ranganathan S. Structural diversity of biologically interesting datasets: a scaffold analysis approach. J. Cheminf. 2011, 3, 30. 10.1186/1758-2946-3-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biasini M.; et al. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014, 42, 252–258. 10.1093/nar/gku340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen V. B.; Arendall W. B. III; Headd J. J.; Keedy D. A.; Immormino R. M.; Kapral G. J.; Murray L. W.; Richardson J. S.; Richardson D. C. Mol Probity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, D66, 12–21. 10.1107/s0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiederstein M.; Sippl M. J. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. 10.1093/nar/gkm290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowie J.; Lüthy R.; Eisenberg D. A method to identify protein sequences that fold into a known three-dimensional structure. Science 1991, 253, 164–70. 10.1126/science.1853201. [DOI] [PubMed] [Google Scholar]
- Dassault Systèmes BIOVIA. Discovery Studio Modeling Environment, Release 2017; Dassault Systèmes: San Diego, 2016.
- Dolinsky T. J.; Nielsen J. E.; McCammon J. A.; Baker N. A. PDB PQR: an automated pipeline for the setup, execution, and analysis of Poison-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004, 32, W665–W667. 10.1093/nar/gkh381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abraham M. J.; Murtola T.; Schulz R.; Pall S.; Smith J. C.; Hess B.; Lindahl E. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- Hornak V.; Abel R.; Okur A.; Strockbine B.; Roitberg A.; Simmerling C. Comparison of multiple AMBER force fields and development of improved protein backbone parameters. Proteins 2006, 65, 712–725. 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark P.; Nilsson L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J. Phys. Chem. A 2001, 105, 9954–e9960. 10.1021/jp003020w. [DOI] [Google Scholar]
- Hess B.; Bekker H.; Berendsen H. J. C.; Fraaije J. G. E. M. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. 10.1002/(sici)1096-987x(199709)18:123.0.co;2-h. [DOI] [Google Scholar]
- Miyamoto S.; Kollman P. A. Settle an analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. 10.1002/jcc.540130805. [DOI] [Google Scholar]
- Darden T.; York D.; Pedersen L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089. 10.1063/1.464397. [DOI] [Google Scholar]
- York D. M.; Wlodawer A.; Pedersen L. G.; Darden T. A. Atomic-level accuracy in simulations of large protein crystals. Proc. Natl. Acad. Sci. U.S.A. 1994, 91, 8715–8718. 10.1073/pnas.91.18.8715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD: visual molecular dynamics. J. Mol. Graphics 1996, 14, 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Banerjee P.; Erehman J.; Gohlke B.-O.; Wilhelm T.; Preissner R.; Dunkel M. Super Natural II: A database of natural products. Nucleic Acids Res. 2015, 43, D935–D939. 10.1093/nar/gku886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Boyle N. M.; Banck M.; James C. A.; Morley C.; Vandermeersch T.; Hutchison G. R. Open Babel: An open chemical toolbox. J. Cheminf. 2011, 3, 33. 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becke A. D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. 1988, 38, 3098. 10.1103/PhysRevA.38.3098. [DOI] [PubMed] [Google Scholar]
- Kim S.; Chen J.; Cheng T.; Gindulyte A.; He J.; He S.; Li Q.; Shoemaker B. A.; Thiessen P. A.; Yu B.; Zaslavsky L.; Zhang J.; Bolton E. E. PubChem 2019 update: improved access to chemical data. Nucleic Acids Res. 2019, 47, D1102–1109. 10.1093/nar/gky1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott O.; Olson A. J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallakyan S.; Olson A. J. Small-molecule library screening by docking with PyRx. Methods Mol. Biol. 2015, 1263, 243–250. 10.1007/978-1-4939-2269-7_19. [DOI] [PubMed] [Google Scholar]
- Morris G. M.; Goodsell D. S.; Halliday R. S.; Huey R.; Hart W. E.; Belew R. K.; Olson A. J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. 10.1002/(sici)1096-987x(19981115)19:143.0.co;2-b. [DOI] [Google Scholar]
- Kim M. O.; Nichols S. E.; Wang Y.; McCammon J. A. Effects of histidine protonation and rotameric states on virtual screening of M. tuberculosis RmlC. J. Comput.-Aided Mol. Des. 2013, 27, 235–246. 10.1007/s10822-013-9643-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.