

Abstract
Triggered by Offset, Multiplexed, Accurate mass, High resolution, and Absolute Quantitation (TOMAHAQ) is a recently introduced targeted proteomics method that combines peptide and sample multiplexing. TOMAHAQ assays enable sensitive and accurate multiplexed quantification by implementing an intricate data collection scheme that comprises multiple MSn scans, mass inclusion lists, and data-driven filters. Consequently, manual creation of TOMAHAQ methods can be time-consuming and error prone, while the resulting TOMAHAQ data may not be compatible with common mass spectrometry analysis pipelines. To address these concerns we introduce TomahaqCompanion, an open-source desktop application that enables rapid creation of TOMAHAQ methods and analysis of TOMAHAQ data. Starting from a list of peptide sequences, a user can perform each step of TOMAHAQ assay development including (1) generation of priming run target list, (2) analysis of priming run data, (3) generation of TOMAHAQ method file, and (4) analysis and export of quantitative TOMAHAQ data. We demonstrate the flexibility of TomahaqCompanion by creating a variety of methods testing TOMAHAQ parameters (e.g., number of SPS notches, run length, etc.). Lastly, we analyze an interference sample comprising heavy yeast peptides, a standard human peptide mixture, TMT11-plex, and super heavy TMT (shTMT) isobaric labels to demonstrate ~10–200 attomol limit of quantification within a complex background using TOMAHAQ.
Keywords: TOMAHAQ, isobaric labeling, targeted proteomics, TMT, SPS-MS3, quantitative proteomics, computational analysis, open source, Orbitrap, mass spectrometry
Graphical Abstract

INTRODUCTION
Due to their sensitivity and quantitative accuracy, targeted proteomics methods such as single, multiple, or parallel reaction monitoring (SRM, MRM, and PRM, respectively) have traditionally been employed to validate observations found within discovery proteomics experiments.1–5 Targeted methods are advantageous for this purpose, as a small number of analytes can be monitored across a large number of cell types, treatments, and/or time points.6 Targeted assays can generally be separated into three tiers based on their intended purpose.7 In practice many targeted measurements are categorized as either clinically validated (Tier 1) or well-characterized research assays (Tier 2), which both require labeled internal standards and demonstrate a high level of precision.7 Advances in speed and sensitivity of mass spectrometers has allowed “Tier 2” assays to monitor hundreds of targets within a single experiment—this is commonly referred to as peptide multiplexing.8,9 These assays offer superb quantitation, but have limited throughput as only one experimental sample is analyzed within each MS analysis.
Sample multiplexing utilizing isobaric labels offers an avenue to analyze multiple samples in one analysis and has become the technology of choice for discovery proteomics experiments in which entire proteomes or PTM-omes of batched samples are analyzed simultaneously.10–12 Combining sample and peptide multiplexing within targeted proteomics experiments would enable analysis of hundreds of samples in a rapid fashion and facilitate the analysis of more cell types, treatments, time points, etc. An assay of this structure would be considered a “Tier 3” assay because it enables discovery in a targeted mode.7 In principle, hundreds of analytes could be monitored across hundreds of samples within a short time period to reveal protein signatures or elucidate mechanism of action.
Due to the higher levels of precursor coisolation that occur when targeting low level analytes within a complex background, initial experiments to combine sample and peptide multiplexing analyzed targets were performed with enriched subproteomes.13–15 Recently, Triggered by Offset, Multiplexed, Accurate mass, High resolution, and Absolute Quantitation (TOMAHAQ) was introduced to enable accurate quantification of low-level precursors in unfractionated, complex mixtures (Figure 1A).16 Robust targeting is accomplished by monitoring “trigger peptides”, standard peptides labeled with a structurally identical, but isotopically distinct version of the isobaric reagent used for sample multiplexing. To ensure target peptides are analyzed only when they are eluting, trigger peptides are placed on an inclusion list and identified in real time by matching ≥5 fragment ions at high mass accuracy (inSeq).17 To maintain quantitative accuracy in the presence of interfering peptides, MS2 spectra are analyzed to determine which b- or y-ion fragments ions should be isolated for SPS-MS3 analysis.18,19
Figure 1.
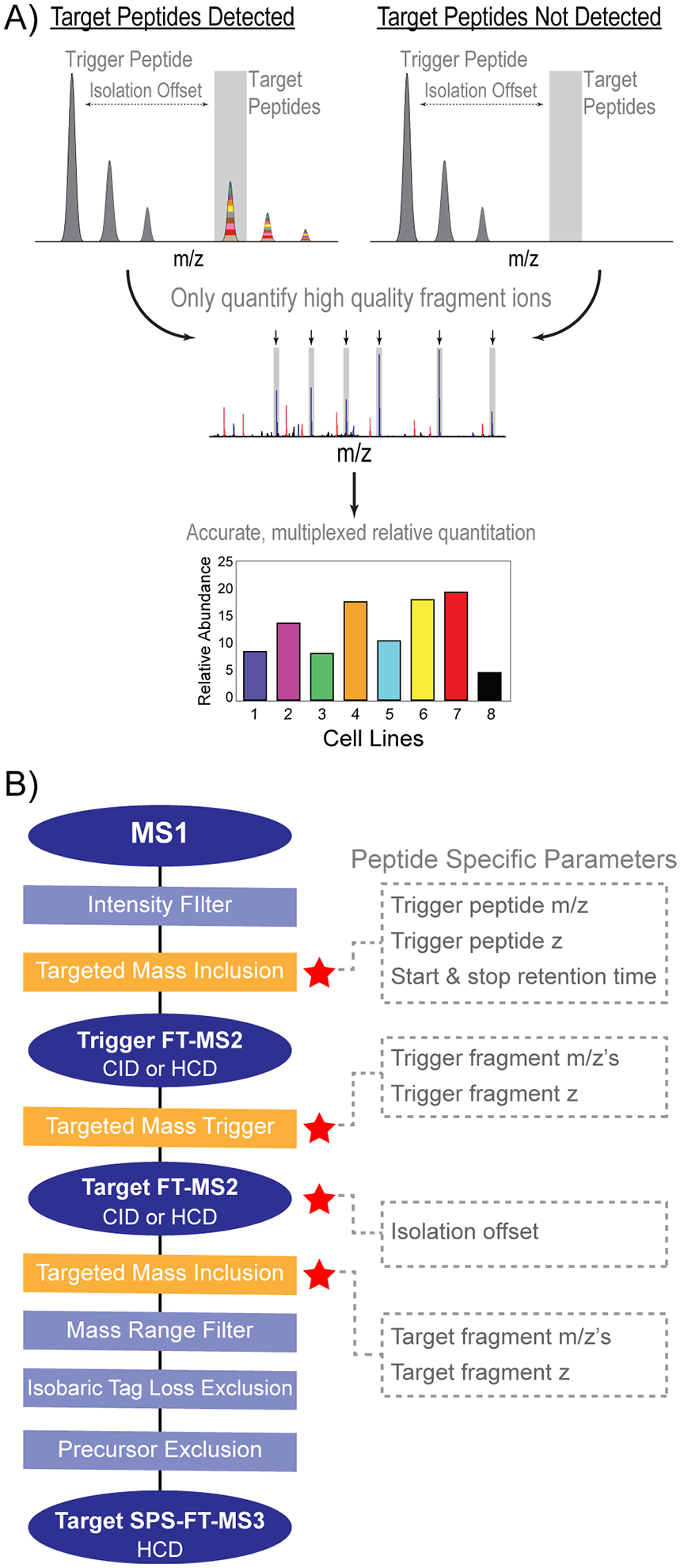
TOMAHAQ scheme and instrument method structure. (A) TOMAHAQ experiments monitor synthetic trigger peptides that are spiked into a mixture of multiplexed samples. Monitoring trigger peptides enables quantification of target peptides even if they are not visible in an MS1 spectrum. Target MS2 spectra are analyzed in order to select b- or y-fragment ions for accurate MS3 analysis. (B) TOMAHAQ instrument method structure. MSn scans (blue ovals) are collected for either the synthetic trigger peptide or multiplexed target peptides. Instrument filters (rectangles) have either defined values for all peptides (light blue) or peptide specific values (orange). For filters or scans where peptide specific features are required, the particular values that need to be changed are indicated.
The original version of TOMAHAQ utilized custom instrument code to perform the complex scan structure of TOMAHAQ and implemented filters that are not currently available in the method editor (e.g., SPS ion purity).16 In principle, TOMAHAQ methods can be created using the standard method editor; however, manual construction of these methods is time-consuming and error prone. This is due to the intricate structure of the method, which includes an MS1 survey scan, an MS2 trigger scan, an MS2 target scan, an MS3 target scan, two targeted inclusion lists, one targeted mass trigger list, and a number of filters (Figure 1B). Furthermore, analyzing TOMAHAQ data with typical mass spectrometry data analysis pipelines is challenging due to the unique structure of the scans within the raw data.
To address these challenges we have developed TomahaqCompanion, an open source Windows desktop application written in C# that facilitates all aspects of a TOMAHAQ experiment, including: (1) creation of priming run inclusion list, (2) analysis of priming run data, (3) automatic creation of TOMAHAQ method file, as well as (4) extraction, manual curation, and export of quantitative data. We demonstrate the flexibility of TomahaqCompanion by creating a number of TOMAHAQ methods varying parameters believed to be important in quantitative accuracy (e.g., number of SPS notches). Lastly, we use TomahaqCompanion to analyze a standard interference model to estimate a ~10–200 amol limit of quantitation for peptides analyzed with TOMAHAQ.
METHODS
TomahaqCompanion Program
TomahaqCompanion (TC) is a Windows desktop application written in the C# programming language that utilizes the C# Mass Spectrometry Library (CSMSL) and vendor specific library (MSFileReader, ThermoFisher Scientific) to create mass lists and TOMAHAQ instrument methods, as well as analyze TOMAHAQ priming run and experimental data (Figure 2A). TC is an open source application and the source code is available within a GIT repository, TomahaqCompanion, which can be accessed through the TOMAHAQ web portal (https://gygi.med.harvard.edu/publications/tomahaq). The web portal also has a link to the TomahaqCompanionProgram repository, which contains the current release of TC as well as the target peptide list, TOMAHAQ templates, TOMAHAQ method files, and TC output files associated with the work presented here. The TomahaqCompanionProgram is the recommended repository for typical use of TC. Currently, TC is licensed under GNU public license V3. TC facilitates the three elements of a TOMAHAQ experiment: (1) priming run data collection of a trigger peptide mixture, (2) TOMAHAQ method creation and TOMAHAQ data collection, and (3) TOMAHAQ data curation and analysis (Figure 2B). The specifics of each step are detailed in the sections below.
Figure 2.
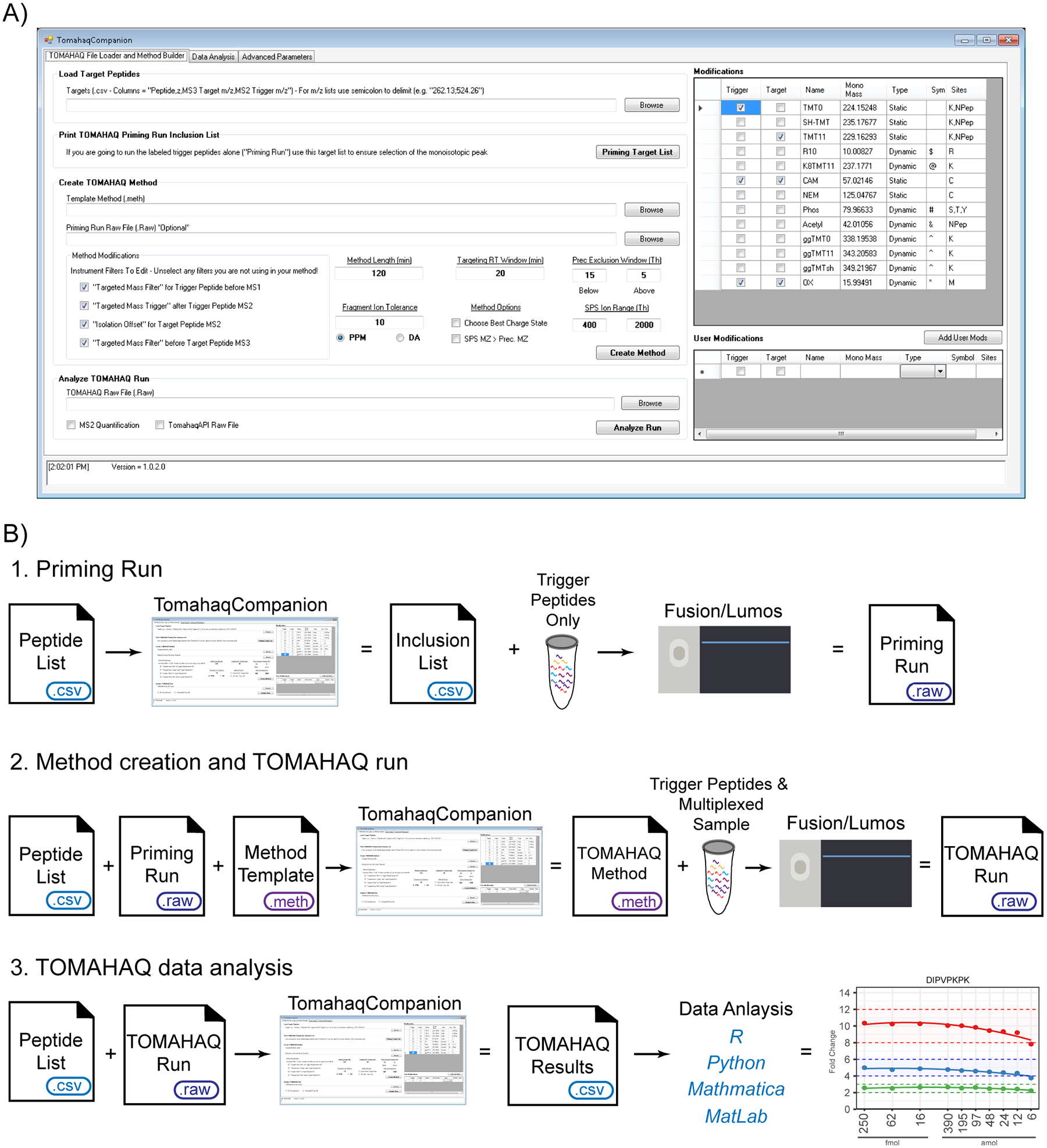
TomahaqCompanion program and workflow. (A) TomahaqCompanion graphical user interface. (B) TomahaqCompanion workflow: (1) Priming run analysis of trigger peptides alone, (2) Creation of a TOMAHAQ method file and analysis of experimental samples within a TOMAHAQ run, and (3) Analysis of TOMAHAQ data through TomahaqCompanion and export to external data analysis software (e.g., R).
Importing Peptides and Setting Modifications
TomahaqCompanion (TC) was written to allow for maximal flexibility when designing TOMAHAQ assays. The simplest input for TC is a comma separated value (.csv) file that contains a single column labeled “Peptide”, which contains one peptide sequence per trigger/target peptide pair (Figure S1). However, users can specify additional parameters including charge state (“z”), MS2 trigger ions (“MS2 Trigger m/z”), SPS-MS3 target ions (“MS3 Target m/z”), the corresponding protein (“Protein”), as well as start and stop retention times (“StartTime” and “StopTime”) (Figure S1). TOMAHAQ experiments may analyze peptides comprised of various combinations of stable isotope enriched amino acids, mass tags, or a variety of post-translational modifications (PTMs). TC allows users specify if a modification is present on the trigger, target, or both peptides using the check box list within the graphical user interface (GUI) (Figure 3). TC displays a number of predetermined modifications, but user specified modifications may be added through the GUI. As with a database search, TC utilizes two classes of modifications, “Static” or “Dynamic”. Static modifications are automatically added to peptides at the indicated amino acid or terminus. Dynamic modifications are only added to peptides at amino acid residues followed by the symbol corresponding to the modification. For example, if “#” is set to represent a dynamic phosphorylation modification, then “PEPS#TIDEEEK” represents a phosphopeptide with a modified serine (Figure 3A).
Figure 3.

Peptide modifications within TomahaqCompanion graphical user interface. (A) TOMAHAQ modifications for an experiment utilizing TMT0 and TMT11 with phosphopeptides. (B) TOMAHAQ modifications for an experiment utilizing heavy amino acids (lysine or arginine) to impart the offset mass. (C) TOMAHAQ modifications for an experiment analyzing variable lysine modifications (e.g., ubiquitin remnant and acetylation).
It is important to note that modifications will only be added to a trigger or target peptide if the corresponding check box is selected within the GUI. This allows users to configure TOMAHAQ assays that utilize various isotopic labels to create the mass offset (Figure 3A,B). For example, a typical TOMAHAQ experiment comprises trigger peptides labeled with TMT0 and target peptides labeled with TMT11 to encode a 5.0104 Da mass shift per TMT label (Figure 3A). Here, the TMT0 and TMT11 modifications are set to static and the check boxes are selected for the trigger and target peptides, respectively. Mass offsets can also be induced as heavy amino acids (e.g., lysine or arginine) allowing the same isobaric label to be used for both trigger and target peptides (Figure 3B). In these configurations TMT11 is selected for both the trigger and target peptides as a static modification, whereas heavy arginine (R10, 10.0083 Da) and heavy lysine plus TMT11 (K8TMT11, 237.1771 Da) are set as dynamic modifications with check boxes only selected for the trigger peptides.
TC only allows one modification per residue and modifications are added in the order they appear in the table (top to bottom). This results in modifications lower in the table replacing those higher in the table if they modify the same residue within the selected trigger or target peptide. This is particularly important for modifications of lysine residues such as isotopically heavy lysine, ubiquitin remnant, and acetylation (Figure 3B,C). For these, users must consider if the chemical tag will or will not label the modified lysine residue. In cases where the chemical tag is capable of labeling the modified lysine residue (e.g., heavy lysine, ubiquitin remnant) the mass of the tag must be added to the mass of the modification and provided as a single value. If the tag differs between the trigger and target peptides, a separate modification must be provided for each (e.g., ggTMT0 338.1954 Da vs ggTMT11 343.2058 Da) (Figure 3C). This is not the case for acetylation, where the modification blocks the addition of a chemical label. In these cases, the modification retains its typical mass (42.0106 Da). Because it is lower positioned in the modification table, it will replace any static modification on a terminus or amino acid marked with the “&” symbol (Figure 3C). While combinations of modifications can become complex, TC allows users to save the user modification configurations such that they can be easily reused.
Priming Run Analysis and TOMAHAQ Method Creation
Once peptides are selected and synthesized, the first step in a TOMAHAQ experiment is analysis of the trigger peptides without any background in what is called the “priming run” (Figure 2B.1). Although a priming run is not required, priming run data are important, as they allow TC to determine the retention time of target peptides as well as the b- and y-fragment ions to use for InSeq identification and targeted SPS-MS3 quantification.17 Once the peptide file has been chosen and user modifications configured, the user simply presses the “Priming Target List” button. This click exports .csv file that can be directly loaded into the “Targeted Mass List” filter of an FT-MS2 method (Figure 2B.1). If retention time scheduling will be used, the method should be the same length and employ the same gradient as the TOMAHAQ analyses the user will perform.
Once the trigger peptides are analyzed, priming run data (.raw) can be loaded into TC along with a template TOMAHAQ method provided in proprietary vendor .meth format (Figure 2B.2). The template method contains a single node that depicts the scans and filters required for a TOMAHAQ analysis (Figure 1B). Example template methods and experimental templates are stored within the TC GIT repository. In the template method, mass lists and isolation offsets are set to default values that will be rewritten upon method creation. Before creating a method, the user should adjust the fragment ion tolerance within the TC GUI to match the priming run data. For MS2 data collected in the ion trap, users can choose Dalton (Da) mass tolerances to extract ions (Figure 2A). At this time, the length and retention time window used within the TOMAHAQ method can be changed. TC allows users to prefilter targeted SPS-ions to reduce method file size. These filters include “precursor exclusion filter”, “SPS-ion range filter”, and “SPS-ion m/z > precursor m/z”. The precursor exclusion filter allows users to exclude a region around the precursor m/z value that frequently contains interfering neutral loss peaks from contaminating peptides. The SPS-ion range filter ensures that y1 ions are not selected for MS3 analysis. An additional SPS-ion filter enables users to only select ions that are above the precursor m/z, as these ions tend to be more specific and help limit interference. Users can configure TC to “choose best charge state”, which places the most intense charge state for each peptide onto the target list. This is ideal for target lists with a large number of peptides. For smaller numbers of targets, inspecting the MS/MS fragmentation of each charge state can ensure that the optimal charge state with the best fragmentation is selected.
Given the configurable nature of TC, automated method creation extends beyond TOMAHAQ to other types of targeted MS experiments that involve defining filters or mass offsets within a method file. For TOMAHAQ, four parameters are altered from the template method: (1) targeted inclusion list, (2) fragment ion trigger list, (3) target peptide isolation offset, and (4) target peptide SPS-ion target list (Figure S2A). All of these filters must be present in the template method for proper method creation; however, a user can omit a filter by deleting it from the template method and unchecking the corresponding box in the TC GUI (Figure S2B) to create a variety of targeted-offset mass methods. For example, to create a parallel reaction monitoring (PRM) assay that utilizes a synthetic peptide as a trigger, a user would set the appropriate modifications within the GUI and use a template method file that does not contain an MS3 scan (Figure S2B). The output method would monitor the synthetic peptides, perform inSeq confirmation of a sequence, and trigger offset FT-MS2 for PRM analysis. In this way, any combination of the four filters can be employed so long as the template method has a matching configuration.
Automatic creation of native method (.meth) files takes seconds with TomahaqCompanion. To accomplish this, TC loads peptide sequences, applies modifications, and calculates trigger and target peptide m/z values before analyzing the priming run to find the most intense peak matching a target m/z value (i.e., within 10 ppm). TC then extracts MS/MS data, matches fragment ions, creates a consensus target peptide spectrum with fragment m/z values adjusted for offset mass shifts, filters potential SPS-ions based on user input, and exports all calculated values to .xml file that can be used to edit a template method (Figure S3A). If TC is installed on an instrument computer, TC will automatically open the .xml file and edit the template method to create a TOMAHAQ method file in the native vendor format (Figure S3A). Alternatively, users can create an .xml file on a noninstrument computer and later use that .xml file along with a template method file to create a TOMAHAQ method using another instance of TC (Figure S3B). The method created by TomahaqCompanion can be directly loaded onto the instrument for analysis of TOMAHAQ samples.
Automatic method creation averages spectral data from five scans surrounding the peak apex; however, users can manually inspect priming run data and decide which scans and/or peptides should be used to create the TOMAHAQ method. This is accomplished within the TC GUI via the “Analysis” tab (Figure S4). For each trigger peptide, the extracted ion chromatogram (XIC) is displayed, as well as the currently selected MS/MS scan, and a consensus spectrum comprising the calculated target peptide ions (Figure S4). Users can select the scans on the MS1 XIC or via checkboxes within the scan list. Additionally, users can select peptides they would like included in the TOMAHAQ method—a useful feature if a peptide does not behave as expected and must be removed from the analysis. Once peptides and scans are selected, users can select the “Update Method w/Selected Scans” button within the TC GUI (Figure S4). This will use the current settings to create a new .xml file and new method that reflect the changes made within the GUI.
TOMAHAQ Run Analysis and Data Export
Once TOMAHAQ data are collected, a raw data file can be loaded directly into TC to extract trigger and target peptide XICs. These data are plotted along with points representing trigger peptide MS2, target peptide MS2, and target peptide MS3 scans (Figure S5). The GUI allows users to interact with TOMAHAQ data by selecting points to display the target MS2 spectrum, along with the reporter ion signal-to-noise value from the corresponding target MS3 spectrum (Figure S5). Visualizing both together helps with assessment of SPS-ion purity within target MS2 scans. Users can visualize matching b- or y- fragment ions, which are marked with red stars (matched ion used for SPS-MS3) or blue stars (matched ion not used for SPS-MS3) (Figure S5). All data within the run are exported to a .csv upon completion of TC analysis, but users can select specific scans for export by highlighting dots in the XIC plot or by checking the boxes in the grid at the bottom of the program (Figure S5). This allows users to select the data that is nearest to the peak apex, or the MS2 that contains the least amount of interference. Similarly, specific peptides can be selected for export by checking/unchecking the boxes next to the peptides within the TC GUI. Data can be exported to a .csv file for facile import into a statistical analysis program, such as R.
EXPERIMENTS
TOMAHAQ Standard Mixture
All experiments utilized Pierce Retention Time Calibration (PRTC) peptide mixture (Thermo Scientific Pierce Protein Biology, Rockford, IL) for both the trigger and target peptides for TOMAHAQ analysis. PRTC peptides were first dried, desalted, and dried again before being resuspended and split into nine aliquots. Each aliquot was labeled with either TMT11-plex20 or a recently described version of TMT termed super heavy-TMT (shTMT)21 (200 mM HEPES pH 8.5, 28% ACN, 5:1 label:peptide ratio, 1 h reaction time). For TMT11-plex labeling, samples 128C and 129N were omitted as blank channels. After quenching with hydroxylamine, a pool of target peptides was created by mixing 10:5:2.5:1:0:0:1:2.5:5:10 before samples were dried, desalted, and dried again. Five aliquots of HeLa Protein Digest Standard (Thermo Scientific Pierce Protein Biology, Rockford, IL) were labeled with TMT11-plex reagents (126–128C; 200 mM HEPES pH 8.5, 28% ACN, 4:1 label:peptide ratio, 1 h reaction time) and mixed 1:1:1:1:1:0:0:0:0:0, dried, desalted, and dried again.
The shTMT labeled trigger peptides were mixed with the 5-plex HeLa mixture such that the shTMT peptides were 250 fmol/μL and the HeLa digests were 200 ng/μL within each channel to serve as the background for target peptide dilutions. Target peptides dilutions were tested in a range from 250 fmol/μL to 61 amol/μL in the most abundant TMT11-plex channel.
Mass Spectrometry and LC Methods
For all experiments, peptides were separated using a Dionex UltiMate 3000 RSLCnano Proflow system (Thermo Fisher Scientific, Sunnyvale, CA) with a gradient of 2% buffer A (98% H20, 2% ACN with 0.1% formic acid) to 35% buffer B (98% ACN, 2% H20, 0.1% formic acid) with a flow rate of 450 nL/min. Samples were separated over a 25 cm capillary column (100 μm I.D.) packed with Waters nanoAcquity M-Class BEH (1.7 μm) material (New Objective, Woburn, MA). All samples were analyzed using an Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher Scientific, San Jose, CA).
For priming run analysis, MS1 survey scans were performed in the Orbitrap (resolution = 60 000) using quadrupole isolation to select a mass range of 350–1350 Th (max IT = 50 ms, AGC target = 4 × 105). Selected precursors were excluded for 2 s after selection (±5 ppm) and isotopes excluded from analysis. To ensure that monoisotopic peaks were selected, TomahaqCompanion was used to make an inclusion list of peptide neutral mass (M) as well as charge range (z = 2–5). Selected peptides were isolated using the quadrupole (0.4 Th isolation window) before fragmentation by collisionally induced dissociation (CID) at a normalized collision energy (NCE) of 30. The resulting fragment ions were analyzed in the Orbitrap (resolution 15 000; AGC target = 2 × 105; max IT = 35 ms).
For TOMAHAQ analysis, MS1 survey scans were collected in the Orbitrap (resolution = 60 000) using the quadrupole to isolate a range from 400–1050 Th (max IT = 50 ms, AGC target = 1 × 106). Trigger peptides were placed on an inclusion list with their corresponding m/z and z. Matching peaks were chosen for MS2 if observed within 15 ppm of the correct m/z. Trigger peptides were selected using the quadrupole (isolation window = 0.4 Th), fragmented by CID (NCE = 30), and the resulting fragments analyzed in the Orbitrap (resolution = 15 000; AGC target = 2 × 105; max IT = 35 ms). A “Product Ion Trigger” filter was used to match trigger peptide fragment ions (15 ppm) and prompt an MS2 on the target peptide cluster if ≥5 fragment ions were matched.17 Target peptides were selected using the quadrupole (isolation window = 0.4 Th) at a predetermined mass offset, fragmented by CID (NCE = 30), and the resulting fragments were analyzed in the Orbitrap (resolution = 60 000; AGC target = 2 × 105; max IT = 900 ms). Target peptide MS2 spectra were further filtered using “Precursor Ion Exclusion” (Low = 70, High = 5), “Isobaric Tag Exclusion” (Reagent Tag Type = TMT), “Precursor Selection Range” (400–2000), and a “Targeted Mass Inclusion List” (target peptide SPS m/z and z values). Up to six targeted SPS ions were then isolated by synchronous precursor selection (SPS, SPS isolation window = 2 m/z) before HCD fragmentation (NCE = 55) and analysis of fragments in the Orbitrap (resolution = 60 000; AGC target = 1 000 000; max IT = 2500 ms). For TOMAHAQ parameter evaluation all parameters were kept constant except the one being evaluated.
All mass spectrometry data files are publicly available within PeptideAtlas (PASS01295).
Data Analysis
All data were manually analyzed and curated within TomahaqCompanion and then exported into .csv files, which were further analyzed using custom scripts written in R.22
RESULTS
A Standard Mixture for the Evaluation of TOMAHAQ
Currently, there is no standard sample or experimental design to evaluate the TOMAHAQ method. An ideal TOMAHAQ standard would comprise reagents that are readily available, standardized, and easy to prepare. One hurdle has been the use of synthetic trigger peptides, which can be costly to generate and are typically specific to a particular assay. Recently a number of synthetic, heavy peptide mixtures have been introduced as internal standards used for retention time calibration.23,24 We surmised that peptides such as these could be leveraged to create a standard mixture for optimizing TOMAHAQ experiments. The standard mixture comprised the Pierce Retention Time Calibration (PRTC) mixture, which contains 15 yeast peptides labeled with heavy lysine (+8) or arginine (+10) residues, as well as a commercially available HeLa cell digest. Isotopically labeled PRTC peptides were used as both trigger and target peptides, which required the use of a chemical label to induce a mass offset. Typically, TMT0 is used for this purpose; however, we have noted for peptides that receive only one isobaric label, isotopes from nearby trigger peptides can be coisolated during analysis. This can skew quantitative results by contaminating SPS-MS3 scans with reporter ion signal from the trigger peptides. To avoid this, a recently described version of TMT termed super heavy-TMT (shTMT) was used. shTMT has the same structure as TMT11-plex reagents, but is 6.0138 Da heavier (Figure S6A).21 Labeling with shTMT results in trigger peptides that are heavier in mass than target peptides, which is advantageous for peptides bearing only one isobaric label (e.g., peptides that end in arginine) since isotopes from trigger peptides do not contaminate target peptide signals (Figure S6B). Using these components an interference model was constructed where the PRTC peptides were labeled with TMT11 and mixed 10:5:2:1::0:0:1:2:5:10 for target peptides, HeLa digest peptides labeled with TMT11 and mixed 1:1:1:1:1:0:0:0:0:0 to provide interference, and a separate aliquot of PRTC peptides labeled with shTMT to act as trigger peptides (Figure 4A).
Figure 4.
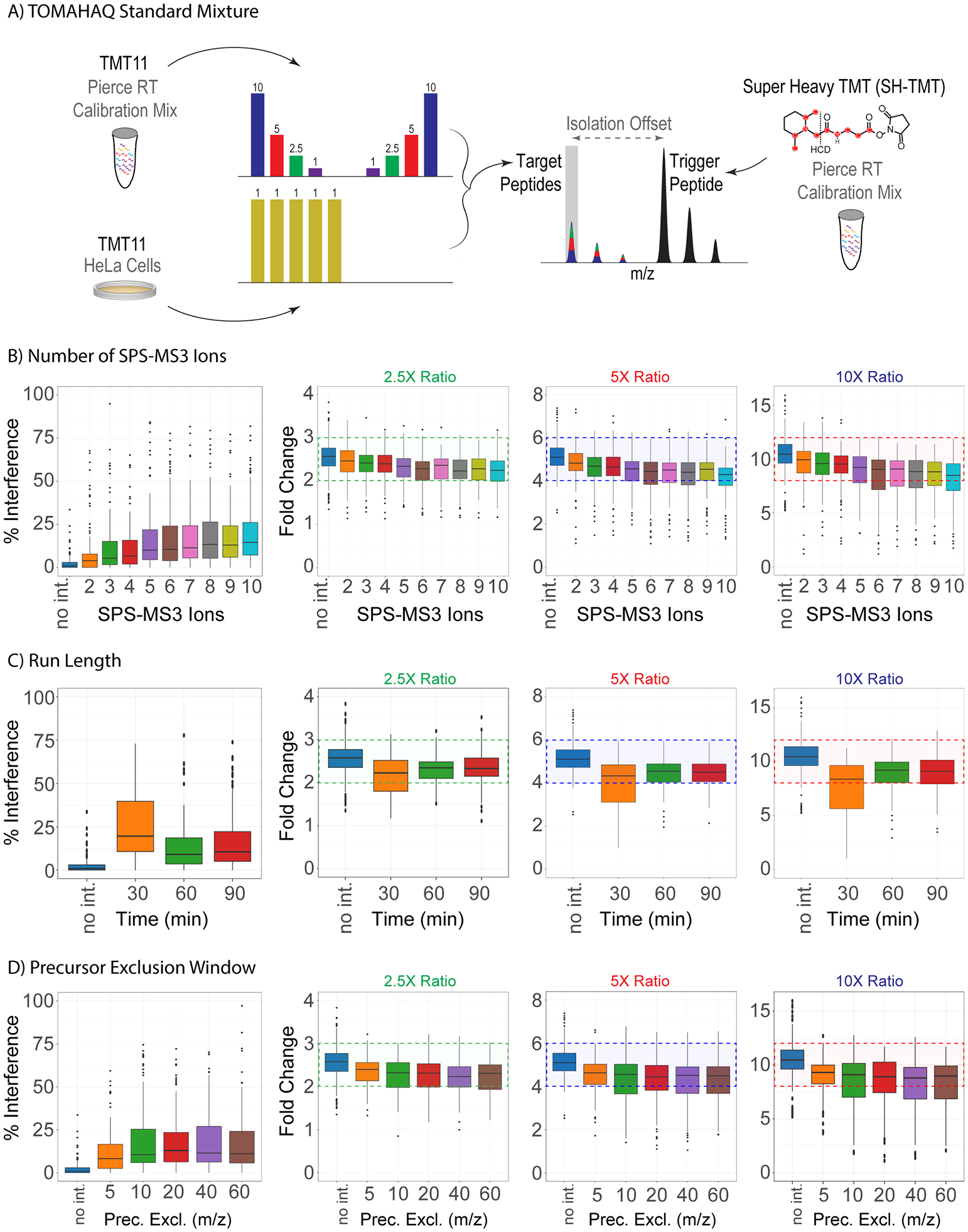
Optimization of TOMAHAQ method parameters. (A) TOMAHAQ standard mixture consisting of synthetic retention time calibration peptides labeled with super heavy-TMT (shTMT) to act as trigger peptides or TMT11 to act as target peptides (126 to 128N and 129C to 131N) and HeLa standard peptide digest labeled with TMT11 (126 to 128C) to act as interference. (B–D) Optimization of TOMAHAQ method parameters including synchronous precursor selection (SPS), run length, and precursor exclusion window. Percent interference is calculated by dividing the interference only channel (128C) by the lowest quantitative channel (128N). Except for no interference (no int.) data, all ratios are derived from measurements that contained interference. Box plots summarize all quantified spectra with signal-to-noise above 400. Each dot represents a single spectrum for a target peptide. For each box, the median (line), inner quartile range (box), outer quartile range (whiskers), and outliers (points) are displayed.
TomahaqCompanion Enables Evaluation of TOMAHAQ Method Parameters
We used TC to create a number of methods investigating parameters believed to be important in quantitative accuracy of TOMAHAQ including number of SPS notches, run length, and precursor exclusion window (Figure 4B–D). Two metrics were used to evaluate each parameter: (1) interference percentage, defined as the ratio of the interference only channel (TMT128C) to the lowest quantitative channel containing interference (TMT128N), and (2) the ratios measured in each quantitative channel that contains interference.
The coisolation of peptide precursors is a well-studied challenge within isobaric label based multiplexed experiments.25 While extensive fractionation,26 minimized isolation widths,27,28 or ion mobility29,30 can minimize the amount of precursor interference, the most common way to improve quantitative accuracy is gas-phase purification.18,31 The most common gas-phase purification method is synchronous precursor selection MS3 (SPS-MS3), a method involving precursor peptide fragmentation followed by simultaneous isolation of multiple fragments ions.19 Within this scheme the number of fragments selected (SPS ions) impacts both the purity and signal intensity of the resulting quantification. For global shotgun experiments, this value is typically set between 8 and 10 SPS ions. However, for targeted applications we surmised that increasing the number of SPS ions could dramatically impact quantitation under the premise that increasing the number of SPS ions increases the chance of isolating interfering ions. TC was used to construct methods allowing isolation of 2–10 SPS ions for MS3 quantification. Analysis was performed on a sample with ~400 amol of peptide in the lowest quantitative channel (Figure 4B). Increasing number of SPS ions quickly increased the median interference percentage from 2 to 6 SPS ions (3.9–10.5%) and then more slowly from 6 to 10 SPS ions (10.5–14.5%). As the amount of interference increased, median measured ratios decreased for each of the comparisons: 2.5-fold (2.47–2.25), 5-fold (4.83–4.29), and 10-fold ratios (9.94–8.50). Interestingly, the amount of reporter ion signal measured as measured by ion flux (signal-to-noise per ms injection time) in quantitative MS3 scans appeared to plateau around 6 SPS-ions before decreasing at 9 SPS-ions (Figure S7A). However, these changes were not significant. We also tested the raw signal-to-noise for each quantitative result and found that 6 to 9 SPS-MS3 ions returned significantly (t test, two sided, Bonferroni correction) more signal than using 2 SPS-MS3 ions (Figure S7C). Given these results, our recommendation for most methods is to 6 SPS-ions as a middle point between decreasing accuracy and increasing signal for TOMAHAQ experiments. However, for experiments where accuracy is paramount, utilizing 2 SPS-MS3 ions will incur the least amount of precursor interference and return the most accurate result.
Targeted assays are typically employed across many samples, making the total time dedicated to an analysis an important consideration. TOMAHAQ was originally proposed as a 2 h mass spectrometry analysis, but the original version did not determine the impact of run length on quantitative accuracy. Run lengths of 30, 60, and 90 min were tested where the time included loading, analysis, washing and equilibration of the column (Figure 4C). A marked decrease in the percentage of interference was observed upon extending runs from 30 to 60 min (19.6–9.1%), with little additional change from 60 to 90 min (9.1–10.5%). The high level of interference led to decreased accuracy in the 30 min runs as compared to the 60 or 90 min analyses: 2.5 fold (2.23 vs 2.34 or 2.33), 5-fold (4.32 vs 4.54 or 4.49), and 10-fold ratios (8.37 vs 9.20 or 9.09). This result is expected, as ineffective chromatographic separation leads to increased coelution globally and a higher occurrence of precursor coisolation. While shorter gradients increase maximum peak height, the net effect of peptide coelution outweighs benefits from shorter gradient analyses. From these data we conclude that 60 and 90 min runs return similar quantitative results. The 60 min run length was chosen for method development, but longer runs (90–120 min) are recommended for analysis of experimental samples.
SPS-MS3 was developed to maximize the amount of quantitative signal for reporter ions by isolating multiple fragment ions (and regions) after peptide fragmentation. In addition to the precursor peptide, neutral loss ions from the precursor are often found in the region around the precursor. As neutral loss ions from coisolated precursors could also theoretically be in this region, it is typically excluded from SPS-ion selection with the presumption that a larger exclusion window will limit precursor coisolation. However, data has not been demonstrated to determine the optimal size of this exclusion window. To test the exclusion window size around the precursor ion for SPS-ion selection, TC was used to construct methods excluding 5, 10, 20, 40, and 60 m/z below the precursor ion (Figure 4D). Interestingly, increasing the exclusion window from 5 to 60 m/z caused an increase in the interference percentage (8.2–11.1%) and a slight decrease in accuracy: 2.5 fold (2.40–2.31), 5-fold (4.63–4.49), and 10-fold (9.31–8.97) ratios. While the median ratio did not change drastically, the smallest exclusion window exhibits the lowest variation, invalidating our original hypothesis that larger exclusion windows within the neutral loss region would return more accurate quantitative data.
Determination of TOMAHAQ Limit of Quantification
In the initial description of TOMAHAQ, a simple interference mixture was used to demonstrate that peptides present at 400 amol or 100 amol on column were accurately quantified with TOMAHAQ as compared to MS2 and data-dependent MS3 analysis.16 Despite these results, it was still unclear what limit of detection or quantification could be expected from a TOMAHAQ assay. To address this question, TMT11 peptides from the TOMAHAQ standard were diluted into a mixture of TMT11 labeled HeLa peptides and shTMT labeled trigger peptides (Figure 5A). The trigger peptides were held at a constant 250 fmol on column, the HeLa interference held constant at 200 ng/channel, and the lowest channel within the target peptides ranged from 25 fmol to 6 amol. For dilution experiments, 14 of the 15 PRTC peptides were analyzed. One peptide (LTILEELR) was not chosen for MS/MS during the priming run causing TC to exclude it from TOMAHAQ method generation. Each sample mixture was analyzed with an identical 60 min TOMAHAQ analysis (total run time). As target peptides are diluted into the background interference, median interference percentage increased from 0% to 36% (Figure 5B).
Figure 5.

Dilution series of TOMAHAQ standard. (A) Amount on column throughout the dilution series for the quantitative channels in the TOMAHAQ standard. (B) Interference percentage for samples within the dilution series. (C) Fold change for each peptide measured within the dilution series. Data are a mean of the all measurements with a signal-to-noise above 400 and MS2 isolation specificity above 0.5. If no measurements met these criteria, a point is not shown.
By examining the quantitative ratio for each peptide, the level of interference as well as the quantitative accuracy was found to be peptide dependent across the dilution series (Figure 5C). Nearly all peptides (12 of 14) were quantifiable at 195 amol on column and 8 of the 14 peptides were quantifiable at the 97 amol level. One peptide (DIPVPKPK) demonstrated excellent accuracy for all ratios at all dilutions and was quantifiable down to 6 amol on column. Five of the remaining peptides were detected at the 6 amol level and exhibited a change greater than 4-fold. Interestingly, one peptide (SSAAPPPPPR) consistently demonstrated a depressed ratio for the 10-fold and 2.5 fold comparisons, but remained accurate at 5-fold throughout the dilution series—this effect was detected in each experiment, although the reason for this was not determined. One possibility is that this peptide was “overlabeled” by the isobaric tag in particular aliquots. Off target labeling is dependent on the sequence of the peptide, which could explain why this phenomenon was only detected for one peptide within the mixture.32 Taken together, these results demonstrate the benefit of TomahaqCompanion and the ability of TOMAHAQ to perform quantitative analysis of peptides in the 6 to 197 amol range in a complex background mixture.
DISCUSSION
Obtaining accurate quantification within targeted isobaric labeling experiments is challenging. Inherently, abundant precursor interferences impact low level peptides such that obtaining accurate quantification of these peptides within an unfractionated sample is difficult without advanced methods such as TOMAHAQ. Despite the advantages of TOMAHAQ, the difficulty of creating methods and a lack of data analysis tools has hindered the community from broadly adopting TOMAHAQ assays. Here, we introduce TomahaqCompanion (TC) as an open source tool that enables facile development, optimization, and utilization of TOMAHAQ assays. Foremost among its functionality is the ability to generate instrument methods in the native vendor format. Making TOMAHAQ methods by hand for just a small number of peptides is time-consuming and error prone, while TC has the ability to make TOMAHAQ methods for hundreds of peptides in just seconds. Combining the ability to make methods with rich data analysis functionalities enables users to quickly evaluate the performance of TOMAHAQ assays and assess the quality of TOMAHAQ data.
TC is the first step toward making TOMAHAQ experiments more accessible. We are currently developing an instrument API implementation of TOMAHAQ that will work in conjunction with TC. Implementing TOMAHAQ through the API will allow most peptide-specific data to be stored on a desktop computer, rather than the embedded instrument computer. This implementation will remove restrictions on the number of peptides that can be analyzed by TOMAHAQ, although we have not yet determined how many peptides can practically be targeted. Like all targeted methods the maximum number of peptides will be determined by the retention time window, number of points across the peak, and maximum dwell time (i.e., injection time). TOMAHAQ utilizes long dwell times for endogenous peptides, yet unlike other targeted methods can yield quantitative results in as little as one scan. With optimized retention time windows it is likely that TOMAHAQ could match or exceed other advanced targeted proteomics methods such as IS-PRM.
In addition to introducing a software platform that facilitates TOMAHAQ experiments, this paper introduces a TOMAHAQ standard mixture that combines commercially available standard heavy peptides and HeLa whole cell digest into a sample useful for characterizing TOMAHAQ methods. For our mixture, the use of super heavy-TMT (shTMT) enabled more accurate quantification of peptides bearing only a single isobaric label (e.g., arginine ending peptides). However, a comparable mixture could also be created using TMT0 and TMT6–11 as the pair of isobaric labels for the TOMAHAQ assay. TC was used to test parameters that impact quantitative accuracy, demonstrating that 5–6 SPS ions, longer runs (60 to 90 min), and smaller precursor exclusion windows for SPS ion selection yield more accurate data. Lastly we used TC to analyze a dilution series of the TOMAHAQ standard mixture and found that nearly all peptides were quantified at ~200 amol on column, many were quantified at ~100 amol on column, and the very best peptide was accurately quantified at 6 amol on column. This peptide dependence of TOMAHAQ for quantification is expected given the known effects of peptide sequence, RT, m/z, and z. Interestingly, similar conclusions were made in an experiment comparing MS2 and MS3 PRM reporting that the limit of quantification for both methods appears to be peptide dependent.33
Supplementary Material
ACKNOWLEDGMENTS
We thank Derek J. Bailey for assistance with the C# Mass Spectrometry Library (CSMSL) and Robert A. Everley for suggestions during the development of TomahaqCompanion. This work was supported in part by the NIH Grant GM67945 (S.P.G).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jproteome.8b00767.
Figures S1–S7 (PDF)
The authors declare the following competing financial interest(s): J.C., J.R., R.V., and R.B. are employees of Thermo Fisher Scientific, developer and distributor of the Orbitrap Fusion/Lumos mass spectrometers. C.M.R. and D.S.K. are employees of Genentech.
REFERENCES
- (1).Keshishian H; Addona T; Burgess M; Kuhn E; Carr SA Quantitative, Multiplexed Assays for Low Abundance Proteins in Plasma by Targeted Mass Spectrometry and Stable Isotope Dilution. Mol. Cell. Proteomics 2007, 6 (12), 2212–2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Picotti P; Bodenmiller B; Mueller LN; Domon B; Aebersold R Full Dynamic Range Proteome Analysis of S. Cerevisiae by Targeted Proteomics. Cell 2009, 138 (4), 795–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Peterson AC; Russell JD; Bailey DJ; Westphall MS; Coon JJ Parallel Reaction Monitoring for High Resolution and High Mass Accuracy Quantitative, Targeted Proteomics. Mol. Cell. Proteomics 2012, 11 (11), 1475–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Schoenherr RM; Saul RG; Whiteaker JR; Yan P; Whiteley GR; Paulovich AG Anti-Peptide Monoclonal Antibodies Generated for Immuno-Multiple Reaction Monitoring-Mass Spectrometry Assays Have a High Probability of Supporting Western Blot and ELISA. Mol. Cell. Proteomics 2015, 14 (2), 382–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Carnielli CM; Macedo CCS; De Rossi T; Granato DC; Rivera C; Domingues RR; Pauletti BA; Yokoo S; Heberle H; Busso-Lopes AF; et al. Combining Discovery and Targeted Proteomics Reveals a Prognostic Signature in Oral Cancer. Nat. Commun 2018, 9 (1), 3598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Keenan AB; Jenkins SL; Jagodnik KM; Koplev S; He E; Torre D; Wang Z; Dohlman AB; Silverstein MC; Lachmann A; et al. The Library of Integrated Network-Based Cellular Signatures NIH Program: System-Level Cataloging of Human Cells Response to Perturbations. Cell Syst. 2018, 6 (1), 13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Carr SA; Abbatiello SE; Ackermann BL; Borchers C; Domon B; Deutsch EW; Grant RP; Hoofnagle AN; Hüttenhain R; Koomen JM; et al. Targeted Peptide Measurements in Biology and Medicine: Best Practices for Mass Spectrometry-Based Assay Development Using a Fit-for-Purpose Approach. Mol. Cell. Mol. Cell. Proteomics 2014, 13 (3), 907–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Yan W; Luo J; Robinson M; Eng J; Aebersold R; Ranish J Index-Ion Triggered MS2 Ion Quantification: A Novel Proteomics Approach for Reproducible Detection and Quantification of Targeted Proteins in Complex Mixtures. Mol. Cell. Proteomics 2011, 10 (3), M110.005611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Gallien S; Kim SY; Domon B Large-Scale Targeted Proteomics Using Internal Standard Triggered-Parallel Reaction Monitoring (IS-PRM). Mol. Cell. Proteomics 2015, 14 (6), 1630–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Rose CM; Venkateshwaran M; Volkening JD; Grimsrud PA; Maeda J; Bailey DJ; Park K; Howes-Podoll M; den Os D; Yeun LH; et al. Rapid Phosphoproteomic and Transcriptomic Changes in the Rhizobia-Legume Symbiosis. Mol. Cell. Proteomics 2012, 11 (9), 724–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Rose CM; Isasa M; Ordureau A; Prado MA; Beausoleil SA; Jedrychowski MP; Finley DJ; Harper JW; Gygi SP Highly Multiplexed Quantitative Mass Spectrometry Analysis of Ubiquitylomes. Cell Syst. 2016, 3 (4), 395–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Samie M; Lim J; Verschueren E; Baughman JM; Peng I; Wong A; Kwon Y; Senbabaoglu Y; Hackney JA; Keir M; et al. Selective Autophagy of the Adaptor TRIF Regulates Innate Inflammatory Signaling. Nat. Nat. Immunol 2018, 19 (3), 246–254. [DOI] [PubMed] [Google Scholar]
- (13).Savitski MM; Fischer F; Mathieson T; Sweetman G; Lang M; Bantscheff M Targeted Data Acquisition for Improved Reproducibility and Robustness of Proteomic Mass Spectrometry Assays. J. Am. Soc. Mass Spectrom 2010, 21 (10), 1668–1679. [DOI] [PubMed] [Google Scholar]
- (14).Everley RA; Kunz RC; McAllister FE; Gygi SP Increasing Throughput in Targeted Proteomics Assays: 54-Plex Quantitation in a Single Mass Spectrometry Run. Anal. Chem 2013, 85 (11), 5340–5346. [DOI] [PubMed] [Google Scholar]
- (15).Curran TG; Zhang Y; Ma DJ; Sarkaria JN; White FM MARQUIS: A Multiplex Method for Absolute Quantification of Peptides and Posttranslational Modifications. Nat. Commun 2015, 6 (1), 5924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Erickson BK; Rose CM; Braun CR; Erickson AR; Knott J; McAlister GC; Wühr M; Paulo JA; Everley RA; Gygi SP A Strategy to Combine Sample Multiplexing with Targeted Proteomics Assays for High-Throughput Protein Signature Characterization. Mol. Cell 2017, 65 (2), 361–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Bailey DJ; Rose CM; McAlister GC; Brumbaugh J; Yu P; Wenger CD; Westphall MS; Thomson JA; Coon JJ Instant Spectral Assignment for Advanced Decision Tree-Driven Mass Spectrometry. Proc. Natl. Acad. Sci. U. S. A 2012, 109 (22), 8411–8416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Ting L; Rad R; Gygi SP; Haas W MS3 Eliminates Ratio Distortion in Isobaric Multiplexed Quantitative Proteomics. Nat. Methods 2011, 8 (11), 937–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).McAlister GC; Nusinow DP; Jedrychowski MP; Wühr M; Huttlin EL; Erickson BK; Rad R; Haas W; Gygi SP MultiNotch MS3 Enables Accurate, Sensitive, and Multiplexed Detection of Differential Expression across Cancer Cell Line Proteomes. Anal. Chem 2014, 86 (14), 7150–7158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).McAlister GC; Huttlin EL; Haas W; Ting L; Jedrychowski MP; Rogers JC; Kuhn K; Pike I; Grothe RA; Blethrow JD; et al. Increasing the Multiplexing Capacity of TMTs Using Reporter Ion Isotopologues with Isobaric Masses. Anal. Chem 2012, 84 (17), 7469–7478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Paulo JA; Navarrete-Perea J; Erickson AR; Knott J; Gygi SP An Internal Standard for Assessing Phosphopeptide Recovery from Metal Ion/Oxide Enrichment Strategies. J. Am. Soc. Mass Spectrom 2018, 29 (7), 1505–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018; p 1. [Google Scholar]
- (23).Escher C; Reiter L; MacLean B; Ossola R; Herzog F; Chilton J; MacCoss MJ; Rinner O Using IRT, a Normalized Retention Time for More Targeted Measurement of Peptides. Proteomics 2012, 12 (8), 1111–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Holman SW; McLean L; Eyers CE RePLiCal: A QconCAT Protein for Retention Time Standardization in Proteomics Studies. J. Proteome Res 2016, 15 (3), 1090–1102. [DOI] [PubMed] [Google Scholar]
- (25).Ow SY; Salim M; Noirel J; Evans C; Rehman I; Wright PC ITRAQ Underestimation in Simple and Complex Mixtures: “The Good, the Bad and the Ugly. J. Proteome Res 2009, 8 (11), 5347–5355. [DOI] [PubMed] [Google Scholar]
- (26).Niu M; Cho J-H; Kodali K; Pagala V; High AA; Wang H; Wu Z; Li Y; Bi W; Zhang H; et al. Extensive Peptide Fractionation and y 1 Ion-Based Interference Detection Method for Enabling Accurate Quantification by Isobaric Labeling and Mass Spectrometry. Anal. Chem 2017, 89 (5), 2956–2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Savitski MM; Sweetman G; Askenazi M; Marto JA; Lang M; Zinn N; Bantscheff M Delayed Fragmentation and Optimized Isolation Width Settings for Improvement of Protein Identification and Accuracy of Isobaric Mass Tag Quantification on Orbitrap-Type Mass Spectrometers. Anal. Chem 2011, 83 (23), 8959–8967. [DOI] [PubMed] [Google Scholar]
- (28).Roumeliotis TI; Weisser H; Choudhary JS Evaluation of a Dual Isolation Width Acquisition (DIWA) Method for Isobaric Labelling Ratio Decompression. bioRxiv 2018, 387878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Sturm RM; Lietz CB; Li L Improved Isobaric Tandem Mass Tag Quantification by Ion Mobility Mass Spectrometry. Rapid Commun. Mass Spectrom 2014, 28 (9), 1051–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Pfammatter S; Bonneil E; McManus FP; Prasad S; Bailey DJ; Belford M; Dunyach J-J; Thibault P A Novel Differential Ion Mobility Device Expands the Depth of Proteome Coverage and the Sensitivity of Multiplex Proteomic Measurements. Mol. Cell. Proteomics 2018, 17, 2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Wenger CD; Lee MV; Hebert AS; McAlister GC; Phanstiel DH; Westphall MS; Coon JJ Gas-Phase Purification Enables Accurate, Multiplexed Proteome Quantification with Isobaric Tagging. Nat. Methods 2011, 8 (11), 933–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Böhm G; Prefot P; Jung S; Selzer S; Mitra V; Britton D; Kuhn K; Pike I; Thompson AH Low-PH Solid-Phase Amino Labeling of Complex Peptide Digests with TMTs Improves Peptide Identification Rates for Multiplexed Global Phosphopeptide Analysis. J. Proteome Res 2015, 14 (6), 2500–2510. [DOI] [PubMed] [Google Scholar]
- (33).Remes PM; Huguet R Comparison of Peptide Parallel Reaction Monitoring via MS2 and MS3Methods In 66th ASMS Conference on Mass Spectrometry and Allied Topics; 218AD. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.