

Abstract

Proton-coupled electron transfer (PCET) from tyrosine produces a neutral tyrosyl radical (Y•) that is vital to many catalytic redox reactions. To better understand how the protein environment influences the PCET properties of tyrosine, we have studied the radical formation behavior of Y32 in the α3Y model protein. The previously solved α3Y solution NMR structure shows that Y32 is sequestered ∼7.7 ± 0.3 Å below the protein surface without any primary proton acceptors nearby. Here we present transient absorption kinetic data and molecular dynamics (MD) simulations to resolve the PCET mechanism associated with Y32 oxidation. Y32• was generated in a bimolecular reaction with [Ru(bpy)3]3+ formed by flash photolysis. At pH > 8, the rate constant of Y32• formation (kPCET) increases by one order of magnitude per pH unit, corresponding to a proton-first mechanism via tyrosinate (PTET). At lower pH < 7.5, the pH dependence is weak and shows a previously measured KIE ≈ 2.5, which best fits a concerted mechanism. kPCET is independent of phosphate buffer concentration at pH 6.5. This provides clear evidence that phosphate buffer is not the primary proton acceptor. MD simulations show that one to two water molecules can enter the hydrophobic cavity of α3Y and hydrogen bond to Y32, as well as the possibility of hydrogen-bonding interactions between Y32 and E13, through structural fluctuations that reorient surrounding side chains. Our results illustrate how protein conformational motions can influence the redox reactivity of a tyrosine residue and how PCET mechanisms can be tuned by changing the pH even when the PCET occurs within the interior of a protein.
Introduction
Protein redox chemistry is at the heart of many biologically important processes such as photosynthesis, respiration, and nitrogen fixation. The large oxidoreductase class of enzymes uses a range of organic molecules and metallocofactors for catalytic and long-range electron transfer (ET) reactions.1−7 Some oxidoreductases use tyrosine (Y), tryptophan (W), cysteine, glycine, and possibly methionine as high potential one-electron redox mediators.7−18 Y and W are of particular interest because these residues can form spatially organized chains in which high potential, one-electron oxidizing equivalents are moved over large distances.14−18 Gray and Winkler have even suggested that Y/W-based radical transfer (“hole hopping”) pathways may be quite common and potentially serve as an important protective mechanism against oxidative damage.17,18 Under physiological conditions, ET from Y is typically coupled to proton transfer (PT) in a proton-coupled electron transfer (PCET) reaction, resulting in the formation of a neutral radical species.8,10,19−23 The thermodynamics and kinetics involved in radical formation and decay in these amino acids have direct implications for biocatalytic multistep ET/PCET processes. Thus, the study of such processes is important for (i) understanding how proteins effectively and functionally move highly oxidizing holes over large distances and (ii) directing the design of more effective biomimetic catalysts for applications such as the production of solar fuels.24−27
Due to the complexity and size of many enzymes, it is extremely challenging to experimentally resolve the thermodynamic and kinetic behavior of a single amino-acid residue. Simplified biomimetic molecular systems that contain Y or W, but lack a protein scaffold, have proven useful in shedding light on PCET kinetics and mechanisms of radical formation in aqueous buffer.28−43 However, the thermodynamics, kinetics, and mechanisms of Y radical formation may not necessarily reflect the behavior that would be observed for Y in a protein environment. The α3X family of model proteins bridges the gap between small-molecule model systems and enzymes by providing a well-defined protein environment wherein the formation of a single amino-acid radical can be experimentally resolved.44 Thus, the α3X proteins provide a unique opportunity to study PCET quantitatively in a protein environment.
The α3X model protein
system is based on a 65-residue,
single-stranded three-helix bundle (α3) with a buried,
redox-active residue (X32) at position 32 (Figure 1, α3Y). Tyrosine
(Y32),45,46 tryptophan (W32),47,48 and a number of unnatural amino acids such as mercaptophenols,49,50 fluorotyrosines (FnY32, where n = 2 or 3),51,52 and aminotyrosine53 have been placed in position 32. Structural
studies using circular dichroism spectroscopy and solution nuclear
magnetic resonance (NMR) spectroscopy have shown that the α3X proteins remain thermodynamically stable and well-folded
in the pH range of ∼5–10.44,45,48,49 A key advantage of
the α3X system is that residue 32 can be reversibly
oxidized and reduced.54 This property has
allowed the determination of true thermodynamic reduction potentials
( ) for
Y32, W32, and
a range of Y analogues incorporated at site 32.48,50−54 Pourbaix diagrams that map
) for
Y32, W32, and
a range of Y analogues incorporated at site 32.48,50−54 Pourbaix diagrams that map 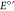 of Y32 and W32 as
a function of pH were obtained from protein film square-wave voltammograms
collected between pH 5.5 and 10.48,52 The slopes
of the Y32 and W32
of Y32 and W32 as
a function of pH were obtained from protein film square-wave voltammograms
collected between pH 5.5 and 10.48,52 The slopes
of the Y32 and W32 vs pH plots were consistent with a 1e–/1H+ redox process where oxidation is coupled
to the release of a proton to give Y32• and W32•, respectively. The α3Y and α3W proteins were also interrogated
by transient absorption (TA) spectroscopy upon oxidation by flash-quench
generated [Ru(bpy3)]3+ (bpy = 2,2′-bipyridine).46,48 Transient spectra confirmed the formation of the neutral radical
species (Y32• and W32•) when α3Y and α3W were oxidized. From TA kinetic studies, the PCET rate constants
of Y32• and W32• formation at pH 5.5 and 8.5 were determined. In α3Y, a significant kinetic isotope effect was observed at both pH 5.5
and 8.5, suggesting that proton transfer is participating in the rate
limiting step. PCET was tentatively proposed to proceed by either
a concerted or stepwise proton-first (PTET) mechanism with water as
the proton acceptor.46
vs pH plots were consistent with a 1e–/1H+ redox process where oxidation is coupled
to the release of a proton to give Y32• and W32•, respectively. The α3Y and α3W proteins were also interrogated
by transient absorption (TA) spectroscopy upon oxidation by flash-quench
generated [Ru(bpy3)]3+ (bpy = 2,2′-bipyridine).46,48 Transient spectra confirmed the formation of the neutral radical
species (Y32• and W32•) when α3Y and α3W were oxidized. From TA kinetic studies, the PCET rate constants
of Y32• and W32• formation at pH 5.5 and 8.5 were determined. In α3Y, a significant kinetic isotope effect was observed at both pH 5.5
and 8.5, suggesting that proton transfer is participating in the rate
limiting step. PCET was tentatively proposed to proceed by either
a concerted or stepwise proton-first (PTET) mechanism with water as
the proton acceptor.46
Figure 1.

The α3Y protein is composed of a single chain
of 65 amino acids: GSR(1)- -LGGGG-
-LGGGG- Y(32)
Y(32) -LGGGGE-
-LGGGGE- , where
helices 1, 2, and 3 are shown in
, where
helices 1, 2, and 3 are shown in  ,
,  , and
, and  , respectively. The protein ensemble
structure of α3Y (PDB ID 2MI7)46 shows very
little structural deviation in the 32 lowest energy states and is
consistent with a globally stable and well-defined protein.
, respectively. The protein ensemble
structure of α3Y (PDB ID 2MI7)46 shows very
little structural deviation in the 32 lowest energy states and is
consistent with a globally stable and well-defined protein.
The present work significantly extends the previous studies of tyrosine radical formation in α3Y by refining several mechanistic details concerning the formation of the Y32•. Although it was evident from the previous study46 that PT is involved in the rate limiting step of radical formation, it was unclear if Y32 oxidation proceeded by a concerted PCET or stepwise PTET mechanism, what the primary proton acceptor was, and how the mechanism was affected by pH. With the additional pH dependent radical formation kinetics data presented herein, we are able to resolve distinct mechanistic regimes of PCET in Y32. We also report kinetic data for Y32• formation as a function of buffer concentration. The obtained results clearly eliminate buffer, and point to H2O, as the primary proton acceptor for Y32. Finally, the solution NMR structure of α3Y shows that Y32 is situated in the hydrophobic core of the protein (Figure 1). The buried Y32 residue exhibits an effective solvent accessible surface area of zero (0.2 ± 0.2%). A residue depth analysis showed that the atoms associated with Y32 and with its aromatic side chains have an average depth of 7.7 ± 0.3 and 8.1 ± 0.4 Å, respectively.46 Based on the static α3Y structure, it was thus unclear how water gained access in order to accept the phenolic proton released from Y32 upon oxidation. To resolve this conundrum, we present molecular dynamics (MD) simulations that illustrate how structural fluctuations in the protein ensemble facilitate the proton transfer step in PCET. These combined results provide a detailed mechanistic framework that will contribute to the overall understanding of Y-based redox chemistry in enzymes.
Materials and Methods
Sample Preparation
α3Y was expressed and purified as described previously.45 Lyophilized protein was dissolved in phosphate buffer, KPi (KH2PO4 from Sigma Life Science ≥99.0% purity, K2HPO4 from ACROS Organics 99+% purity), containing 40 mM KCl (Alfa Aesar 99.0–100.5% purity). In experiments where kinetics was measured as a function of buffer concentration, the solutions were prepared with the following concentrations: [KPi] = 20–460 mM, [α3Y] = 391–486 μM, [[Ru(bpy)3]Cl2] (bpy = 2,2′-bipyridine) = 26–65 μM, and [[Co(NH3)5Cl]Cl2] = 4 mM. In experiments where kinetics was measured as a function of pH, solutions were prepared to the following concentrations: [KPi] = 20–40 mM, [α3Y] = 391–907 μM. 40 mM KPi was used to minimize pH fluctuations upon decomposition of [Co(NH3)Cl]Cl2, which generates NH3 upon quenching of *[Ru(bpy)3]Cl2 (* denotes excited species). Control experiments were carried out in the absence of α3Y, where [[Ru(bpy)3]Cl2] = 26–65 μM and [[Co(NH3)5Cl]Cl2] = 3–4 mM. Concentrations of α3Y, [Ru(bpy)3]Cl2, and [Co(NH3)5Cl]Cl2 were determined spectrophotometrically using a UV/vis spectrometer (Agilent 8453 diode array or Cary 50) using the following extinction coefficients: α3Y (ε277 = 1490 M–1 cm–1),44 [Ru(bpy)3]Cl2 (ε452 = 14600 M–1 cm–1),55 and [Co(NH3)5Cl]Cl2 (ε532 = 52 M–1 cm–1).48 In all experiments, photosensitizer and quencher solutions were prepared separately and mixed under dark conditions to prevent formation of [Ru(bpy)3]3+ and subsequent initiation of PCET prior to flash-quench experiments. The solution pH was adjusted with 0.1–1 M NaOH and 0.1–1 M HCl and measured using a Metrohm 654 pH meter and a calibrated Metrohm LL Biotrode pH-electrode.
Transient Absorption Methods
The transient absorption (TA) setup has been described previously.46 In short, the sample was excited using a Nd:YAG laser (Quantel, BrilliantB) with the laser light passed through an OPO that was tuned to 460 nm. The probe light was directed at a right angle to the excitation light and was provided by a 150 W unpulsed Xe lamp that was passed through a monochromator (Applied Photophysics, pbp Spectra Kinetic Monochromator 05-109) prior to reaching the sample. The monochromator was selected for either 410 or 450 nm light, with 2–3 mm slit openings and 4.65 nm/mm bandpass giving fwhm = 9.3 and 13.95 nm, respectively. A second monochromator was placed after the sample and directed the probe light to the detector (PMT, Hamamatsu R928). The signal was digitized using an Agilent Technologies Infiniium digital oscilloscope (600 MHz). TA traces were produced within the Applied Photophysics LKS software package.
Samples were contained in a 4 × 10 mm cuvette where the probe light was led through the 10 mm path length. Oxygen was excluded from the sample during measurement by first gently purging the solution with high purity Ar for 10 min prior to measuring and then by maintaining a positive pressure of Ar during flash photolysis. TA experiments were carried out at 22–23 (±1) °C.
A change of ca. 0.1–0.8 pH units was observed for individual samples used during flash photolysis with larger changes occurring in samples having lower buffer concentrations. The pH of analyzed solutions was measured before and after the flash photolysis experiments. The range of pH values during a given flash photolysis experiment and the average pH were calculated from a linear interpolation of the change in pH as a function of the number of laser flashes supplied to the sample. The first 10 shots were not used for fitting; these traces were not reproducible due to the presence of an impurity that is rapidly consumed under oxidizing conditions (Figure S1).46 Each sample received ca. 30 laser shots, and 10–26 laser shots were averaged to obtain kinetic traces from which rate constants were extracted. The power of each laser shot was 9–12 mJ. Approximately 1–6 μM [Ru(bpy)3]3+ was formed per shot. Data were analyzed using Matlab version 2018b.
Computational Methods
The solution NMR structure for α3Y served as the starting point for all simulations and was obtained from the Protein Data Bank (PDB code: 2MI7).46 As the NMR ensemble includes 32 structures, both the last conformer and the medoid (i.e., the most representative of the ensemble average, conformer 19) were selected. Independent simulations starting with each of these structures are referred to as “Traj. 1” and “Traj. 2”, respectively. Each initial protein structure was solvated with explicit TIP3P56 water in a periodic rectilinear box. The net positive charge of the protein at pH 7.0 was neutralized with Cl– ions, followed by adding Na+ and Cl– ions to achieve a salt concentration of ∼150 mM. After the careful equilibration procedure described in the Supporting Information, a 1 μs MD trajectory in the canonical (NVT) ensemble was propagated for each initial structure. This simulation procedure was conducted with two different force fields, CHARMM3657 and AMBER ff14SB.58 The results with these two force fields exhibit the same qualitative trends, but the CHARMM force field is known to slightly overstabilize helical structures.59−61 Therefore, the AMBER ff14SB simulations are featured in the main text, while the analogous CHARMM simulations are discussed in the Supporting Information. The root-mean-square deviations (RMSDs) for the backbone Cα atoms relative to the initial structure were computed to confirm the structural stability over the 1 μs trajectories (Figure S6). The root-mean-square fluctuations (RMSFs) of the Cα positions per residue were also computed to identify the most flexible regions of the protein (Figure S6). A hydrogen-bonding analysis for Y32 was also conducted over all of the trajectories.
Results and Discussion
Radical Formation Kinetics of Y32
The kinetics
of Y32 oxidation was investigated between pH 5.7 and pH
9.0. This pH range was chosen on the basis that there is no significant
change in the α-helical content, global stability, and tertiary
structure of α3Y.45,46Figure 2A summarizes the processes
that occur during laser flash photolysis under the conditions used
here. [Ru(bpy)3]2+ is excited and then oxidatively
quenched by the sacrificial quencher [Co(NH3)5Cl]Cl2 (kq = 9 × 108 M–1 s–1)46 to form [Ru(bpy)3]3+ (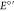 ([Ru(bpy)3]3+/2+)
= 1260 mV vs NHE). [Ru(bpy)3]3+ subsequently
oxidizes Y32; this oxidation is coupled to proton loss,
which yields Y32•. The Y32 Pourbaix diagram provides an
([Ru(bpy)3]3+/2+)
= 1260 mV vs NHE). [Ru(bpy)3]3+ subsequently
oxidizes Y32; this oxidation is coupled to proton loss,
which yields Y32•. The Y32 Pourbaix diagram provides an 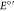 (pH 7.0)
value of 986 ± 3 mV for the
neutral tyrosine Y32(O•/OH) redox pair
and a pH independent E° value of 749 mV for
the tyrosinate Y32(O•/O–) redox pair (see ref (52), Table S2, for a Nernst analysis of the α3Y Pourbaix
diagram).48,52
(pH 7.0)
value of 986 ± 3 mV for the
neutral tyrosine Y32(O•/OH) redox pair
and a pH independent E° value of 749 mV for
the tyrosinate Y32(O•/O–) redox pair (see ref (52), Table S2, for a Nernst analysis of the α3Y Pourbaix
diagram).48,52
Figure 2.

(A) The mechanism of Y32–O• generation by laser flash-quench photolysis. (B) Single TA traces collected at pH 6.5 in 20 mM KPi buffer. Purple circles represent data recorded at 410 nm, shown with a black line fit. The dark gray solid line shows the Y32• component of the fit, and the light gray line shows the [Ru(bpy)3]2+ bleach component of the fit. Green circles represent data collected at 450 nm with a black line fit to a single exponential decay. Blue dots represent data collected at 410 nm in the absence of α3Y.
Figure 2B shows TA kinetic traces obtained from an α3Y-containing sample at 450 nm (green) and 410 nm (purple). The bleach at 450 nm is due to the depletion of [Ru(bpy)3]2+ as a result of oxidative quenching with [Co(NH3)5Cl]Cl2 (step 2, Figure 2A) that produces [Ru(bpy)3]3+. Y32 oxidation by [Ru(bpy)3]3+ (corresponding to step 3 in Figure 2A) replenishes [Ru(bpy)3]2+, as indicated by the recovery to the prepulse baseline of the TA signal at 450 nm. Both [Ru(bpy)3]2+ and Y32• absorb at 410 nm, resulting in a bleach after the laser flash that grows to a positive signal as [Ru(bpy)3]2+ recovers and Y32• is formed. On a slower time scale, Y32• dimerizes following second-order kinetics, as shown through fluorescence measurements.46 Kinetic traces collected at 410 nm were therefore fit to a model of pseudo-first-order formation followed by second-order decay. The fitting routine used to extract the pseudo-first-order rate constants (kobs) associated with radical formation is described in detail in the Supporting Information, page S5.62 Second-order PCET rate constants were calculated from kPCET = kobs/[α3Y]. The yield of Y32• formation ranged from 0.52 to 0.75, which is consistent with previous observations.46
The irreversible quenching produces Co2+(aq), NH4+(aq), and Cl–(aq).63 The Co2+ ions formed in the quenching event (step 2, Figure 2A) react to generate Co-oxides. These complexes scatter light and exhibit broad absorption spectra in the UV and visible regions (Supporting Information of ref (46)). The blue trace in Figure 2B was collected in the absence of α3Y. The slow increase observed after the laser flash is due to reduction of [Ru(bpy)3]3+ by water, Co2+ (aq), and Co-oxides formed under oxidizing conditions.46 We note that it is critical to minimize exposure to probe light when measuring on the ms to s time scale. A monochromator was placed before the sample to filter probe light centered at 450 and 410 nm. A control experiment in the absence of protein confirmed that the reduction of [Ru(bpy)3]3+ by side reactions is significantly slower than the observed rate constants in the presence of α3Y. Control experiments using α3X with redox inactive phenylalanine at position 32 have previously shown similar slow kinetics.46 As PCET rates associated with Y32• formation are significantly faster than Co-oxide formation, kinetic traces obtained in the presence of the protein can be attributed solely to Y32• formation (Figure S2). Additional control experiments without α3Y also confirmed that Co-oxide formation does not increase with buffer concentration (Figure S3).
PCET Rate Constants as a Function of pH and Buffer Concentration
Bimolecular rate constants for oxidation of Y32 by [Ru(bpy)3]3+ were determined at pH values from 5.7 to 9.0 using kinetics from ns laser flash photolysis, Figure 3. The data shows a steep pH dependence at high pH where a 10-fold increase in rate constant per pH unit is observed. The pH dependence is weaker below ca. pH 7.5. The phenomena giving rise to the observed trend in PCET rate constants (kPCET) are discussed below.
Figure 3.

Rate constants of PCET versus pH for α3Y (black circles). Solid black squares represent data from ref (46). The purple solid line is a fit to eq 2, where kYOH = 2.6 × 104 M–1 s–1 and kYO– = 1.4 × 108 M–1 s–1 (R2 = 0.87). The dashed lines show the pH independent and pH dependent terms of the fit. Vertical error bars correspond to ± one standard deviation. Horizontal error bars correspond to the change in pH over the course of the flash photolysis experiment.
The fractions of tyrosine and tyrosinate (fYOH and fYO–, respectively) change with pH, and kPCET can be expressed as a sum of these fractions multiplied by their respective oxidation rate constants (kYOH and kYO–):
| 1 |
fYOH is close to unity (>0.99) throughout the pH 5.7–9.0 range studied and can therefore be treated as independent of pH. fYO– can be approximated as 10pH–pKa from the Henderson–Hasselbalch equation, thus obtaining the following relation:
| 2 |
kPCET as a function of pH was fit to eq 2, using an apparent pKa of 11.3 for Y32,44 shown in purple (solid line, Figure 3). The fit resulted in kYOH = 2.6 × 104 M–1 s–1 and kYO– = 1.4 × 108 M–1 s–1.
The equilibrium concentration of deprotonated Y32 at pH 8–9 is similar to, or even lower than, that of [Ru(bpy)3]3+ created in one flash (roughly 0.25 μM compared to 1–6 μM, respectively, for each species at pH 8). Still TA kinetics from pH 8–9 showed single exponential behavior (pseudo-first-order conditions), suggesting that fYO– is constant during the experiment. This means that the equilibrium between protonated and deprotonated Y32 is fast on the time scale of Y32 oxidation in these experiments (ca. 0.5 s), and the PCET mechanism dominating at pH > 8 is a rapid pre-equilibrium PT with subsequent ET. Rate-limiting PT, or concerted PCET, to OH– or buffer species can be excluded (see the Supporting Information, page S8). The observed increase in kPCET at pH > 8 is due to the increasing equilibrium fraction of tyrosinate (fYO–). From the rate constants determined for protonated and deprotonated Y32, we note that the difference in reactivity between the tyrosinate/tyrosine forms of α3Y and freely solvated phenolate/phenol in aqueous solution is small, within experimental accuracy. With [Ru(bpy)3]3+ as oxidant in both cases, kYO–/kYOH ≈ 5000 (from eqs 1 and 2) for Y32, and the corresponding ratio for phenolate/phenol is kPhO–/kPhOH ≈ 9000.64 Even if both rate constants are about 10 times smaller for the protein, the ratios are similar and of the same order of magnitude. ET from Y32 is expected to be slower than ET from phenol due to a greater electron donor–acceptor distance and a lower diffusion rate constant. The near identical kYO–/kYOH and kPhO–/kPhOH ratios suggest that there is no additional kinetic obstacle for proton transfer from Y32, despite its average location of 7.7 ± 0.3 Å inside the protein.
At pH < 7.5, the PCET oxidation of protonated Y32 (kYOH) is the dominant contribution to kPCET. A significant kinetic isotope effect (KIE = 2.5 ± 0.5) was observed for PCET rate constants at pH 5.5, from which an ETPT mechanism can be excluded.46 With the extended pH dependent data (Figure 3), PTET can also be excluded. The observed pH dependence of kYOH is much weaker than the factor of 10 per pH unit predicted for a PTET mechanism with OH–, HPO42–, or PO43– as proton acceptor. Thus, we can now establish that the oxidation of the neutral Y32 in α3Y by external [Ru(bpy)3]3+ proceeds as a concerted PCET mechanism. Even a concerted PCET with OH–, HPO42–, or PO43– as primary proton acceptor would have given a 10-fold increase in rate constant per pH unit, following the first-order dependence on the proton acceptor concentration. This prediction seems to exclude those species.
To confirm that no buffer species were directly involved in the reaction, kPCET for Y32 was measured in pH 6.5 phosphate buffer with concentration ranging from 20 to 460 mM (Figure 4). If phosphate acts as the primary proton acceptor at pH < 7.5, kPCET is expected to increase with the phosphate concentration in the buffer. At pH 6.5, approximately 17% of the buffer exists in the HPO42– form (H2PO4– pKa = 7.2), which means that [HPO42–] was varied from 3.3 to 76 mM (20-fold increase). pH 6.5 was chosen for two reasons. First, the Co-oxide formation is slow enough that concentrations of α3Y could be kept to <500 μM without kinetic interference from the Co-oxide reactions. Second, at pH 6.5, only 0.002% of Y32 is in its deprotonated form, indicating that kYOH ≫ kYO– × 10pH–pKa, thus ensuring that PCET from protonated Y32 dominates. The PCET rate constants for Y32 as a function of buffer concentration are compared to PCET rate constants previously reported for a covalently linked ruthenium photosensitizer–tyrosine model complex,32 Ru–Y (chemical structure in Figure S5). PCET rate constants for Ru–Y are given in Figure 4 (black squares); PCET proceeds with an intramolecular ET, and therefore, the rate constants are of first order. Ru–Y exhibits a buffer independent region at low phosphate concentrations (<1 mM) and a region that is first order in phosphate concentrations above 10 mM, indicating that phosphate (HPO42–) acts as the primary proton acceptor only at high enough concentrations of buffer and that water is the primary proton acceptor at low buffer concentrations.32 In contrast, kPCET in Y32 is clearly independent of [KPi] even at higher phosphate concentrations (Figure 4, purple circles), showing that phosphate is not the primary proton acceptor in the oxidation of Y32. Thus, the absence of viable proton acceptors nearby Y32 in α3Y and the exclusion of OH– and buffer species strongly suggest that water (H2O) is the primary proton acceptor in the concerted PCET reaction of α3Y (kYOH).
Figure 4.

PCET rate constants measured in phosphate buffer. Error bars are shown when the error is larger than the marker. The left y-axis represents first-order rate constants for Ru–Y, and the right y-axis, second-order rate constants for α3Y. Black squares show data collected for Ru–Y in pH 7 phosphate buffer with a fit to the data using kobs = kb + fb[buffer]kb32 (dashed black line). Purple circles show data for α3Y collected in pH 6.5 phosphate buffer with a linear, constant value fit to the data (solid purple line).
From a thermodynamic perspective, HPO42– is a much better proton acceptor than water (H3O+, pKa = 0 per definition). Despite the thermodynamic advantage of PT to phosphate, the strongly distance dependent PT step requires that the proton acceptor penetrates the protein to get near the Y32 OH group. The finding that buffer does not act as a proton acceptor suggests that the HPO42– or PO43– do not come in close enough contact with Y32. This could be due to their larger size compared to water, as well as their negative charge.
The kinetic data are, however, not completely independent of pH below 7.5 where it deviates from the fit. Having excluded buffer dependence, another possible reason for the pH dependence of kYOH is that the global surface charge of the protein changes with pH. α3Y and α3W have an isoelectric point of ∼8, as shown by isoelectric gel electrophoresis.48 As the pH increases, amino acid residues become negatively charged, until the isoelectric point is reached and the protein has an overall neutral charge. For comparison, the oxidation of α3W was assigned to an ET-limited ETPT mechanism at pH 5.5 and 8.548 where PCET rate constants should be pH independent. PCET rate constants for α3W increased by a factor of 1.3 between pH 5.5 and 8.5, corresponding to a factor of 1.1 increase per pH unit.48 α3Y and α3W have the same protein sequence with the exception of site 32 and similar three-helix bundle solution structures.46,48 The protein surface charge interactions with [Ru(bpy)3]3+ should therefore be nearly identical for α3Y and α3W. Inclusion of a 1.1 factor increase per pH unit to the otherwise pH independent term kYOH only marginally improves the fit of pH dependent PCET rate constants for α3Y (Figure S4). Thus, the change in electrostatic interaction between α3Y and [Ru(bpy)3]3+ does not explain the weak pH dependence observed at pH < 7.5. We note that a similarly weak dependence of the concerted PCET reaction for aqueous Ru–Y has been reported,41 but a theoretical explanation is still lacking.
The weak pH dependence for kPCET at low pH, where kYOH dominates, could be due to protein conformational motions. Increasing the pH may favor conformational motions that permit water access to Y32 to a greater extent. Although studying the dependence of protein conformational motions on pH is challenging, MD simulations can provide more general insights into equilibrium conformational motions of the protein and surrounding water. Herein, MD simulations were performed to investigate the possible influence of protein motions on the PCET reactivity of Y32, particularly focusing on water accessibility.
Protein Molecular Dynamics Simulations
The solvent accessibility of Y32 in α3Y was explored through 1 μs MD simulations. Although the α3 protein scaffold was designed to sequester Y32,44 these simulations show that H2O can reach the Y32 site through structural fluctuations in nearby side chains. This phenomenon was observed for two different starting structures and two different force fields, suggesting that Y32 is able to briefly hydrogen bond to water. In the solution NMR structure, surrounding nonpolar side chains occlude water access to Y32 (Figure 5A). Using the AMBER ff14SB force field, MD simulations revealed protein conformations in which the side chain of L12 had rotated, forming a small void that transiently allowed water access and hydrogen-bonding interactions with Y32 (Figure 5B). Similar conformations were observed with the CHARMM36 force field and are shown in Figure S7. An additional conformation sampled with the AMBER ff14SB force field, in which Y32 has rotated to face outward, and the hydrophobic side chains have repacked the core, is shown in Figure S8. The prevalence of these transient side chain fluctuations and the associated hydrogen bond between Y32 and water is force field dependent, and thus, the probability of sampling these conformations cannot be established definitively from these simulations.
Figure 5.

Illustration of side chain motions observed in an MD trajectory propagated with the AMBER ff14SB force field. (A) The starting structure from the α3Y solution NMR structure.46 (B) A configuration from the MD trajectory in which two water molecules transiently reside within hydrogen-bonding distance to Y32. The backbone is depicted as tubes, and the residues occluding water access in panel A or bordering the water channel in panel B are depicted as spheres.
The water occupancy around Y32 was analyzed by computing the hydration number around Y32. Here the hydration number is defined as the number of water molecules with the oxygen atom within 3.0 Å of the Y32 hydroxyl oxygen. The average hydration number for Y32 was computed to be 0.49 and 0.35 for Traj. 1 and Traj. 2, respectively. These fractional values of the average hydration number indicate that, in a majority of the sampled protein configurations, Y32 is hydrogen bonded to either no water molecules or one water molecule (Figure 6). Two water molecules were within 3.0 Å of the Y32 hydroxyl group for a small number of configurations (Figure 6). The hydrogen-bonding interaction of the Y32 hydroxyl group with water was analyzed by defining a hydrogen bond according to the criteria of a heavy atom distance less than or equal to 3.0 Å and a donor–hydrogen–acceptor angle greater than or equal to 135°. The percentage of a given trajectory with Y32 forming at least one hydrogen bond to a water molecule was computed to be 38.2 and 27.7% for Traj. 1 and Traj. 2, respectively. The differences between these percentages and the average hydration numbers arise from the configurations with Y32 simultaneously hydrogen bonded to two water molecules. Y32 is also observed to hydrogen bond to the backbone carbonyl oxygen atoms of L12 and L58, the carbonyl of V9, or the carboxylate of the nearby E13 (Table 1). For a majority of the MD trajectories, these hydrogen bonds within the protein form when Y32 is not hydrogen bonded to water, although simultaneous hydrogen-bonding interactions are possible (Figure S8). The analogous breakdown of hydrogen bonds for the trajectories propagated with the CHARMM36 force field is given in Table S3.
Figure 6.

Histograms of hydration number for MD trajectories propagated with the AMBER ff14SB force field.
Table 1. Hydrogen-Bonding Interactions Involving Y32 for MD Trajectories of α3Y Propagated with the AMBER ff14SB Force Field.
| V9:O | E13:Oε1,ε2 | L58:O | L12:O | WAT:Oa | |
|---|---|---|---|---|---|
| Traj. 1 | 54.1% | 24.0% | 5.12% | 38.2% | |
| Traj. 2 | 39.38% | 21.56% | 3.98% | 3.32% | 27.7% |
The numbers reported reflect the sum of the hydrogen-bonding interactions with Y32 acting as a H-bond acceptor or donor and can include contributions from multiple water molecules.
For the hydrogen bonds between Y32 and water or E13, the donor–acceptor distance fluctuates and samples shorter distances that would enable proton transfer. Specifically, the MD trajectories propagated with the AMBER ff14SB force field sampled hydrogen bond donor–acceptor distances in the range 2.5–2.75 Å for around 10% of the trajectory, indicating that proton transfer could occur in either case (Figure 7). However, the distributions of distances sampled during the MD trajectories differ between water and E13, with water sampling shorter distances more frequently, as reflected by the percentages in Table 1 and the distributions in Figures 7 and S9.
Figure 7.

Histograms of O–O distances between the hydroxyl oxygen of Y32 and the closest water molecule (A) and the hydroxyl oxygen of Y32 and the closest carboxylate oxygen of E13 (B) for the first MD trajectory (Traj. 1) propagated with the AMBER ff14SB force field.
The hydrogen bonding between Y32 and water is significantly less prevalent for the trajectories propagated with the CHARMM36 force field (Table S3 and Figure S10), most likely due to overstabilization of the compact α-helix. Y32 was observed to be significantly more flexible for the trajectories propagated with the AMBER force field compared to those propagated with the CHARMM force field, as indicated by the Y32 RMSFs given in Table S4. Because of this diminished flexibility, when Y32 is not hydrogen bonded to water, it hydrogen bonds only to the backbone carbonyl oxygen atoms of L12 and L58 for the trajectories propagated with the CHARMM36 force field (Table S3). Despite the quantitative differences observed for the two different force fields, all of the trajectories exhibited the same qualitative trends. In particular, all of the trajectories illustrate that Y32 can become accessible to water through rotations and fluctuations of the surrounding nonpolar side chains.
The interaction between the −OH group of Y32 and the E13 carboxylate group observed with the AMBER ff14SB force field could result in proton transfer to E13, which is deprotonated in the MD simulations. α3Y may not be structurally well-defined at pH values below the pKa of E13 (∼4.3) which hinders us from determining rate constants in a pH range where E13 is protonated to rule out this residue as a potential proton acceptor. The E13 interaction has not been sampled with the CHARMM36 force field, which may be due to its overstabilization of helical structures. At present, it is not possible to determine which force field represents the experimental conditions more accurately. Despite these differences between the two force fields, the formation of the water cavity is observed with both of them.
General Discussion
Tyrosine becomes strongly acidic upon oxidation, resulting in a deprotonated neutral radical.19 The deprotonation mechanism can vary depending on the surroundings. To activate buried Y residues for redox chemistry, enzymes have evolved the placement of an internal proton acceptor within hydrogen-bonding distance. Such an acceptor can act to shuttle the proton back and forth upon redox cycling of the tyrosine (e.g., the YZ–histidine and YD–histidine pairs in PSII).11,65 In other cases, proton channels with several acid/base groups connect the PCET reaction with proton transport to or from the bulk solvent. For Y residues close to the protein surface, Brønsted bases in solution, such as water itself, may act as the primary proton acceptor. Water assisted PCET has recently been suggested to facilitate radical transfer between the α and β subunits in E. coli ribonucleotide reductase (RNR).66 In RNR, radical transfer occurs reversibly over >32 Å67 between a network of Ys where each radical transfer step is likely a concerted PCET mechanism. The radical transfer chain crosses over the α and β subunits of the protein where the distance between the donating and accepting tyrosines is >5 Å. Water is found between the subunits and is believed to aid the radical transfer. There is still much to learn about single water molecules or small water clusters acting as proton acceptors in PCET reactions, and RNR is not the only case reported thus far.68
The α3Y kinetic data and MD simulations presented in this report have shed light on the radical formation process. Y32 is occluded from solvent by nonpolar side chains and residues on average 7.7 ± 0.3 Å below the protein surface. The Y32 pocket is composed of hydrophobic residues that cannot act as proton acceptors. Our MD simulations show that protein fluctuations can transiently form a cavity in the protein that allows water to approach within hydrogen-bonding distance of the Y32 side chain. Within the time scale of our simulations, Y32 interacts with one water molecule at a time, although for a small number of configurations, two water molecules were within 3 Å of the Y32 oxygen. It is likely that such water molecules serve as the primary proton acceptor. In a later step, the proton would then be transferred further to bulk water. The small size of the cavity is likely the reason that phosphate buffer (HPO42–/PO43–) cannot compete as the primary proton acceptor. It should be noted that one of the force fields used in the MD simulations also suggests the possibility of E13 as a potential proton acceptor, which will be further examined in future studies.
Conclusions
The rate constants of Y32• formation have been determined as a function of pH and buffer concentration. The PCET mechanism is most likely a combination of PTET via Y32– (dominating at high pH) and concerted PCET (dominating at low pH) with water as the primary proton acceptor across the entire pH range studied. Our results show how the rate constants and PCET mechanism of a buried Y residue can be influenced by the protein environment in combination with the solution pH. The primary proton acceptor is either water or a glutamate (E13) located ≥6.7 Å from Y32 in the NMR structure. Molecular dynamics simulations show that water access to Y32 is facilitated by structural fluctuations of nearby side chains, forming a transient cavity. It should be noted that, while the cavity allowing for water access was seen in all trajectories using two different force fields, the E13 interaction was only seen with one force field (AMBER ff14SB). Local protein fluctuations allow for approach of the primary proton acceptor to form a transient hydrogen bond with Y32. This enables rapid oxidation of Y32 in spite of its location 7.7 ± 0.3 Å from the protein surface.
The α3X family of proteins strikes a balance between small model systems and enzymes, making it ideal for mechanistic and quantitative PCET studies. The α3X model proteins are structurally well-defined and exhibit the characteristic cooperative behavior of natural proteins. At the same time, the α3X proteins provide unambiguous kinetic and thermodynamic details of PCET that are highly challenging and often not possible to obtain from the natural enzyme systems. Our results clearly illustrate the importance of protein conformational motions in mediating PCET. Future PCET studies on the α3X model system will address how radical formation is affected by modulating the microenvironment of the radical site including solvent exposure and hydrogen-bonding interactions. Such studies can provide key insights into PCET amino acid radical behavior in natural systems.
Acknowledgments
C.R.R. is supported by the National Science Foundation Graduate Research Fellowship Program under Grant No. DGE1752134 and by the National Institutes of Health Grant 5T32GM06754 3-12. This work was also supported by National Institutes of Health Grants GM056207 (S.H.-S.) and GM79190 (C.T). Computational resources of the Yale Center for Research Computing (YCRC) were used for processing trajectories. Funding was also provided by the Swedish Research Council Grants 2016-04271 (L.H.) and 2017-04992 (S.D.G).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c04655.
Additional TA kinetics, data fitting routines, tabulated rate constants, kinetic derivations, computational details, and additional analysis of MD trajectories (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Selles Vidal L.; Kelly C. L.; Mordaka P.ł M.; Heap J. T. Review of NAD(P)H-dependent oxidoreductases: Properties, engineering and application. Biochim. Biophys. Acta, Proteins Proteomics 2018, 1866, 327–347. 10.1016/j.bbapap.2017.11.005. [DOI] [PubMed] [Google Scholar]
- Ferrer-Sueta G.; Campolo N.; Trujillo M.; Bartesaghi S.; Carballal S.; Romero N.; Alvarez B.; Radi R. Biochemistry of peroxynitrite and protein tyrosine nitration. Chem. Rev. 2018, 118, 1338–1408. 10.1021/acs.chemrev.7b00568. [DOI] [PubMed] [Google Scholar]
- Page C. C.; Moser C. C.; Chen X.; Dutton P. L. Natural engineering principles of electron tunnelling in biological oxidation–reduction. Nature 1999, 402, 47–52. 10.1038/46972. [DOI] [PubMed] [Google Scholar]
- Winkler J. R.; Gray H. B. Electron flow through metalloproteins. Chem. Rev. 2014, 114, 3369–3380. 10.1021/cr4004715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman B. M.; Lukoyanov D.; Yang Z.-Y.; Dean D. R.; Seefeldt L. C. Mechanism of nitrogen fixation by nitrogenase: the next stage. Chem. Rev. 2014, 114, 4041–4062. 10.1021/cr400641x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Chakraborty S.; Hosseinzadeh P.; Yu Y.; Tian S.; Petrik I.; Bhagi A.; Lu Y. Metalloproteins containing cytochrome, iron–sulfur, or copper redox centers. Chem. Rev. 2014, 114, 4366–4469. 10.1021/cr400479b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broderick J. B.; Duffus B. R.; Duschene K. S.; Shepard E. M. Radical S-adenosylmethionine enzymes. Chem. Rev. 2014, 114, 4229–4317. 10.1021/cr4004709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migliore A.; Polizzi N. F.; Therien M. J.; Beratan D. N. Biochemistry and theory of proton-coupled electron transfer. Chem. Rev. 2014, 114, 3381–3465. 10.1021/cr4006654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordlund P.; Sjöberg B.-M.; Eklund H. Three-dimensional structure of the free radical protein of ribonucleotide reductase. Nature 1990, 345, 593. 10.1038/345593a0. [DOI] [PubMed] [Google Scholar]
- Tommos C.; Babcock G. T. Proton and hydrogen currents in photosynthetic water oxidation. Biochim. Biophys. Acta, Bioenerg. 2000, 1458, 199–219. 10.1016/S0005-2728(00)00069-4. [DOI] [PubMed] [Google Scholar]
- Hammarström L.; Styring S. Proton-coupled electron transfer of tyrosines in Photosystem II and model systems for artificial photosynthesis: the role of a redox-active link between catalyst and photosensitizer. Energy Environ. Sci. 2011, 4, 2379–2388. 10.1039/c1ee01348c. [DOI] [Google Scholar]
- Blomberg M. R. Mechanism of oxygen reduction in cytochrome c oxidase and the role of the active site tyrosine. Biochemistry 2016, 55, 489–500. 10.1021/acs.biochem.5b01205. [DOI] [PubMed] [Google Scholar]
- Wikström M.; Krab K.; Sharma V. Oxygen activation and energy conservation by cytochrome c oxidase. Chem. Rev. 2018, 118, 2469–2490. 10.1021/acs.chemrev.7b00664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarboush N. A.; Jensen L. M.; Yukl E. T.; Geng J.; Liu A.; Wilmot C. M.; Davidson V. L. Mutagenesis of tryptophan199 suggests that hopping is required for MauG-dependent tryptophan tryptophylquinone biosynthesis. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 16956–16961. 10.1073/pnas.1109423108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z.; Tan C.; Guo X.; Li J.; Wang L.; Sancar A.; Zhong D. Determining complete electron flow in the cofactor photoreduction of oxidized photolyase. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 12966–12971. 10.1073/pnas.1311073110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minnihan E. C.; Nocera D. G.; Stubbe J. Reversible, long-range radical transfer in E. coli class Ia ribonucleotide reductase. Acc. Chem. Res. 2013, 46, 2524–2535. 10.1021/ar4000407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray H. B.; Winkler J. R. Hole hopping through tyrosine/tryptophan chains protects proteins from oxidative damage. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 10920–10925. 10.1073/pnas.1512704112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray H. B.; Winkler J. R. Living with oxygen. Acc. Chem. Res. 2018, 51, 1850–1857. 10.1021/acs.accounts.8b00245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon W. T.; Murphy D. Determination of the acidity constants of some phenol radical cations by means of electron spin resonance. J. Chem. Soc., Faraday Trans. 2 1976, 72, 1221–1230. 10.1039/f29767201221. [DOI] [Google Scholar]
- Mayer J. M. Proton-coupled electron transfer: a reaction chemist’s view. Annu. Rev. Phys. Chem. 2004, 55, 363–390. 10.1146/annurev.physchem.55.091602.094446. [DOI] [PubMed] [Google Scholar]
- Reece S. Y.; Nocera D. G. Proton-coupled electron transfer in biology: results from synergistic studies in natural and model systems. Annu. Rev. Biochem. 2009, 78, 673–699. 10.1146/annurev.biochem.78.080207.092132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammes-Schiffer S. Introduction: Proton-Coupled Electron Transfer. Chem. Rev. 2010, 110, 6937–6938. 10.1021/cr100367q. [DOI] [PubMed] [Google Scholar]
- Weinberg D. R.; Gagliardi C. J.; Hull J. F.; Murphy C. F.; Kent C. A.; Westlake B. C.; Paul A.; Ess D. H.; McCafferty D. G.; Meyer T. J. Proton-coupled electron transfer. Chem. Rev. 2012, 112, 4016–4093. 10.1021/cr200177j. [DOI] [PubMed] [Google Scholar]
- Rakowski DuBois M.; DuBois D. L. The roles of the first and second coordination spheres in the design of molecular catalysts for H 2 production and oxidation. Chem. Soc. Rev. 2009, 38, 62–72. 10.1039/B801197B. [DOI] [PubMed] [Google Scholar]
- Magnuson A.; Anderlund M.; Johansson O.; Lindblad P.; Lomoth R.; Polivka T.; Ott S.; Stensjö K.; Styring S.; Sundström V.; Hammarström L. Biomimetic and microbial approaches to solar fuel generation. Acc. Chem. Res. 2009, 42, 1899–1909. 10.1021/ar900127h. [DOI] [PubMed] [Google Scholar]
- Appel A. M.; et al. Frontiers, opportunities, and challenges in biochemical and chemical catalysis of CO2 fixation. Chem. Rev. 2013, 113, 6621–6658. 10.1021/cr300463y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauchfuss T. B. Diiron azadithiolates as models for the [FeFe]-hydrogenase active site and paradigm for the role of the second coordination sphere. Acc. Chem. Res. 2015, 48, 2107–2116. 10.1021/acs.accounts.5b00177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnuson A.; Berglund H.; Korall P.; Hammarström L.; Åkermark B.; Styring S.; Sun L. Mimicking electron transfer reactions in photosystem II: Synthesis and photochemical characterization of a ruthenium (II) tris (bipyridyl) complex with a covalently linked tyrosine. J. Am. Chem. Soc. 1997, 119, 10720–10725. 10.1021/ja972161h. [DOI] [Google Scholar]
- Sjödin M.; Styring S.; Åkermark B.; Sun L.; Hammarström L. Proton-Coupled Electron Transfer from Tyrosine in a Tyrosine- Ruthenium- tris-Bipyridine Complex: Comparison with TyrosineZ Oxidation in Photosystem II. J. Am. Chem. Soc. 2000, 122, 3932–3936. 10.1021/ja993044k. [DOI] [Google Scholar]
- Sjödin M.; Styring S.; Wolpher H.; Xu Y.; Sun L.; Hammarström L. Switching the redox mechanism: models for proton-coupled electron transfer from tyrosine and tryptophan. J. Am. Chem. Soc. 2005, 127, 3855–3863. 10.1021/ja044395o. [DOI] [PubMed] [Google Scholar]
- Reece S. Y.; Nocera D. G. Direct tyrosine oxidation using the MLCT excited states of rhenium polypyridyl complexes. J. Am. Chem. Soc. 2005, 127, 9448–9458. 10.1021/ja0510360. [DOI] [PubMed] [Google Scholar]
- Irebo T.; Reece S. Y.; Sjödin M.; Nocera D. G.; Hammarström L. Proton-coupled electron transfer of tyrosine oxidation: Buffer dependence and parallel mechanisms. J. Am. Chem. Soc. 2007, 129, 15462–15464. 10.1021/ja073012u. [DOI] [PubMed] [Google Scholar]
- Ishikita H.; Soudackov A. V.; Hammes-Schiffer S. Buffer-Assisted Proton-Coupled Electron Transfer in a Model Rhenium- Tyrosine Complex. J. Am. Chem. Soc. 2007, 129, 11146–11152. 10.1021/ja072708k. [DOI] [PubMed] [Google Scholar]
- Fecenko C. J.; Thorp H. H.; Meyer T. J. The Role of Free Energy Change in Coupled Electron- Proton Transfer. J. Am. Chem. Soc. 2007, 129, 15098–15099. 10.1021/ja072558d. [DOI] [PubMed] [Google Scholar]
- Irebo T.; Johansson O.; Hammarström L. The rate ladder of proton-coupled tyrosine oxidation in water: A systematic dependence on hydrogen bonds and protonation state. J. Am. Chem. Soc. 2008, 130, 9194–9195. 10.1021/ja802076v. [DOI] [PubMed] [Google Scholar]
- Song N.; Stanbury D. M. Proton-coupled electron-transfer oxidation of phenols by hexachloroiridate (IV). Inorg. Chem. 2008, 47, 11458–11460. 10.1021/ic8015595. [DOI] [PubMed] [Google Scholar]
- Costentin C.; Louault C.; Robert M.; Savéant J.-M. The electrochemical approach to concerted proton—electron transfers in the oxidation of phenols in water. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 18143–18148. 10.1073/pnas.0910065106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonin J.; Costentin C.; Louault C.; Robert M.; Routier M.; Savéant J.-M. Intrinsic reactivity and driving force dependence in concerted proton–electron transfers to water illustrated by phenol oxidation. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 3367–3372. 10.1073/pnas.0914693107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M.-T.; Hammarström L. Proton-coupled electron transfer from tryptophan: A concerted mechanism with water as proton acceptor. J. Am. Chem. Soc. 2011, 133, 8806–8809. 10.1021/ja201536b. [DOI] [PubMed] [Google Scholar]
- Bonin J.; Costentin C.; Louault C.; Robert M.; Savéant J.-M. Water (in water) as an intrinsically efficient proton acceptor in concerted proton electron transfers. J. Am. Chem. Soc. 2011, 133, 6668–6674. 10.1021/ja110935c. [DOI] [PubMed] [Google Scholar]
- Irebo T.; Zhang M.-T.; Markle T. F.; Scott A. M.; Hammarström L. Spanning Four Mechanistic Regions of Intramolecular Proton-Coupled Electron Transfer in a Ru(bpy)32+–Tyrosine Complex. J. Am. Chem. Soc. 2012, 134, 16247–16254. 10.1021/ja3053859. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Swierk J. R.; Megiatto J. D.; Sherman B.; Youngblood W. J.; Qin D.; Lentz D. M.; Moore A. L.; Moore T. A.; Gust D.; Mallouk T. E. Improving the efficiency of water splitting in dye-sensitized solar cells by using a biomimetic electron transfer mediator. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 15612–15616. 10.1073/pnas.1118339109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dongare P.; Maji S.; Hammarström L. Direct evidence of a tryptophan analogue radical formed in a concerted electron- proton transfer reaction in water. J. Am. Chem. Soc. 2016, 138, 2194–2199. 10.1021/jacs.5b08294. [DOI] [PubMed] [Google Scholar]
- Tommos C.; Skalicky J. J.; Pilloud D. L.; Wand A. J.; Dutton P. L. De novo proteins as models of radical enzymes. Biochemistry 1999, 38, 9495–9507. 10.1021/bi990609g. [DOI] [PubMed] [Google Scholar]
- Martínez-Rivera M. C.; Berry B. W.; Valentine K. G.; Westerlund K.; Hay S.; Tommos C. Electrochemical and structural properties of a protein system designed to generate tyrosine Pourbaix diagrams. J. Am. Chem. Soc. 2011, 133, 17786–17795. 10.1021/ja206876h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover S. D.; Jorge C.; Liang L.; Valentine K. G.; Hammarström L.; Tommos C. Photochemical tyrosine oxidation in the structurally well-defined α3Y protein: proton-coupled electron transfer and a long-lived tyrosine radical. J. Am. Chem. Soc. 2014, 136, 14039–14051. 10.1021/ja503348d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Q.-H.; Tommos C.; Fuentes E. J.; Blomberg M. R.; Dutton P. L.; Wand A. J. Structure of a de novo designed protein model of radical enzymes. J. Am. Chem. Soc. 2002, 124, 10952–10953. 10.1021/ja0264201. [DOI] [PubMed] [Google Scholar]
- Glover S. D.; Tyburski R.; Liang L.; Tommos C.; Hammarström L. Pourbaix Diagram, Proton-Coupled Electron Transfer, and Decay Kinetics of a Protein Tryptophan Radical: Comparing the Redox Properties of W32• and Y32• Generated Inside the Structurally Characterized α3W and α3Y Proteins. J. Am. Chem. Soc. 2018, 140, 185–192. 10.1021/jacs.7b08032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay S.; Westerlund K.; Tommos C. Moving a Phenol Hydroxyl Group from the Surface to the Interior of a Protein: Effects on the Phenol Potential and pKa. Biochemistry 2005, 44, 11891–11902. 10.1021/bi050901q. [DOI] [PubMed] [Google Scholar]
- Tommos C.; Valentine K. G.; Martínez-Rivera M. C.; Liang L.; Moorman V. R. Reversible phenol oxidation and reduction in the structurally well-defined 2-mercaptophenol-α3C protein. Biochemistry 2013, 52, 1409–1418. 10.1021/bi301613p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravichandran K. R.; Liang L.; Stubbe J.; Tommos C. Formal reduction potential of 3, 5-difluorotyrosine in a structured protein: insight into multistep radical transfer. Biochemistry 2013, 52, 8907–8915. 10.1021/bi401494f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravichandran K. R.; Zong A. B.; Taguchi A. T.; Nocera D. G.; Stubbe J.; Tommos C. Formal reduction potentials of difluorotyrosine and trifluorotyrosine protein residues: Defining the thermodynamics of multistep radical transfer. J. Am. Chem. Soc. 2017, 139, 2994–3004. 10.1021/jacs.6b11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W.; Kasanmascheff M.; Huynh M.; Quartararo A.; Costentin C.; Bejenke I.; Nocera D. G.; Bennati M.; Tommos C.; Stubbe J. Properties of site-specifically incorporated 3-aminotyrosine in proteins to study redox-active tyrosines: Escherichia coli ribonucleotide reductase as a paradigm. Biochemistry 2018, 57, 3402–3415. 10.1021/acs.biochem.8b00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry B. W.; Martínez-Rivera M. C.; Tommos C. Reversible voltammograms and a Pourbaix diagram for a protein tyrosine radical. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 9739–9743. 10.1073/pnas.1112057109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juris A.; Balzani V.; Barigelletti F.; Campagna S.; Belser P. l.; von Zelewsky A. v. Ru (II) polypyridine complexes: photophysics, photochemistry, eletrochemistry, and chemiluminescence. Coord. Chem. Rev. 1988, 84, 85–277. 10.1016/0010-8545(88)80032-8. [DOI] [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. 10.1063/1.445869. [DOI] [Google Scholar]
- Huang J.; MacKerell A. D. Jr CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. 10.1002/jcc.23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier J. A.; Martinez C.; Kasavajhala K.; Wickstrom L.; Hauser K. E.; Simmerling C. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löw C.; Weininger U.; Lee H.; Schweimer K.; Neundorf I.; Beck-Sickinger A. G.; Pastor R. W.; Balbach J. Structure and dynamics of helix-0 of the N-BAR domain in lipid micelles and bilayers. Biophys. J. 2008, 95, 4315–4323. 10.1529/biophysj.108.134155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patapati K. K.; Glykos N. M. Three force fields’ views of the 310 helix. Biophys. J. 2011, 101, 1766–1771. 10.1016/j.bpj.2011.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best R. B.; Mittal J.; Feig M.; MacKerell A. D. Jr Inclusion of many-body effects in the additive CHARMM protein CMAP potential results in enhanced cooperativity of α-helix and β-hairpin formation. Biophys. J. 2012, 103, 1045–1051. 10.1016/j.bpj.2012.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien J.-Y. Kinetic analysis of irreversible consecutive reactions. J. Am. Chem. Soc. 1948, 70, 2256–2261. 10.1021/ja01186a078. [DOI] [Google Scholar]
- Endicott J. F.; Caspari G.; Hughes R. G.; Hoffman M. Z. Flash photolytic, isotope tracer, and chemical scavenging studies of the ultraviolet photochemistry of pentaamminecobalt (III) chloride. Evidence for excited-state intermediates. J. Am. Chem. Soc. 1970, 92, 6801–6810. 10.1021/ja00726a015. [DOI] [Google Scholar]
- Sjödin M.; Irebo T.; Utas J. E.; Lind J.; Merényi G.; Åkermark B.; Hammarström L. Kinetic effects of hydrogen bonds on proton-coupled electron transfer from phenols. J. Am. Chem. Soc. 2006, 128, 13076–13083. 10.1021/ja063264f. [DOI] [PubMed] [Google Scholar]
- Lubitz W.; Reijerse E. J.; Messinger J. Solar water-splitting into H2 and O2: design principles of photosystem II and hydrogenases. Energy Environ. Sci. 2008, 1, 15–31. 10.1039/b808792j. [DOI] [Google Scholar]
- Nick T. U.; Ravichandran K. R.; Stubbe J.; Kasanmascheff M.; Bennati M. Spectroscopic evidence for a H bond network at Y356 located at the subunit interface of active E. coli ribonucleotide reductase. Biochemistry 2017, 56, 3647–3656. 10.1021/acs.biochem.7b00462. [DOI] [PubMed] [Google Scholar]
- Kang G.; Taguchi A. T.; Stubbe J.; Drennan C. L. Structure of a trapped radical transfer pathway within a ribonucleotide reductase holocomplex. Science 2020, 368, 424–427. 10.1126/science.aba6794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne R. L.; Zhu H.; Iavarone A. T.; Blackburn N. J.; Klinman J. P. Interdomain long-range electron transfer becomes rate-limiting in the Y216A variant of tyramine β-monooxygenase. Biochemistry 2013, 52, 1179–1191. 10.1021/bi3013609. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.