

Abstract
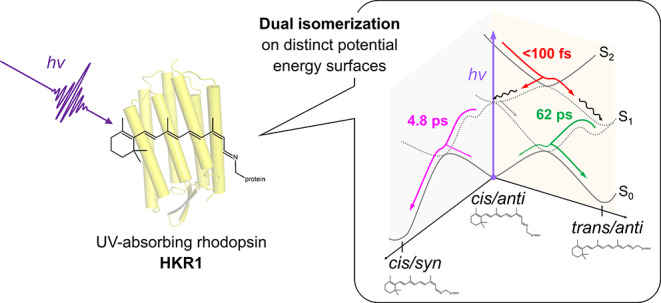
UV-absorbing rhodopsins are essential for UV vision and sensing in all kingdoms of life. Unlike the well-known visible-absorbing rhodopsins, which bind a protonated retinal Schiff base for light absorption, UV-absorbing rhodopsins bind an unprotonated retinal Schiff base. Thus far, the photoreaction dynamics and mechanisms of UV-absorbing rhodopsins have remained essentially unknown. Here, we report the complete excited- and ground-state dynamics of the UV form of histidine kinase rhodopsin 1 (HKR1) from eukaryotic algae, using femtosecond stimulated Raman spectroscopy (FSRS) and transient absorption spectroscopy, covering time scales from femtoseconds to milliseconds. We found that energy-level ordering is inverted with respect to visible-absorbing rhodopsins, with an optically forbidden low-lying S1 excited state that has Ag– symmetry and a higher-lying UV-absorbing S2 state of Bu+ symmetry. UV-photoexcitation to the S2 state elicits a unique dual-isomerization reaction: first, C13=C14 cis–trans isomerization occurs during S2–S1 evolution in <100 fs. This very fast reaction features the remarkable property that the newly formed isomer appears in the excited state rather than in the ground state. Second, C15=N16 anti–syn isomerization occurs on the S1–S0 evolution to the ground state in 4.8 ps. We detected two ground-state unprotonated retinal photoproducts, 13-trans/15-anti (all-trans) and 13-cis/15-syn, after relaxation to the ground state. These isomers become protonated in 58 μs and 3.2 ms, respectively, resulting in formation of the blue-absorbing form of HKR1. Our results constitute a benchmark of UV-induced photochemistry of animal and microbial rhodopsins.
Introduction
Rhodopsins are light-sensing photoreceptor proteins widely distributed in Eukaryota, Bacteria, and Archaea,1−3 capable of absorbing a wide range of wavelengths. Most animal and microbial rhodopsins bind a retinal chromophore with a protonated Schiff base, which enables tuning their absorbance maxima throughout the visible part of the electromagnetic spectrum, from the blue to the red.1,3 Rhodopsins rely on light-driven isomerization of the retinal chromophore for their photoactivated functions. The light-driven excited-state dynamics of protonated retinal Schiff base (RSB) in animal and microbial rhodopsins have been reasonably well established after decades of extensive research, where photoexcitation from the ground state to the lowest-lying excited state is followed by ultrafast photoisomerization via a conical intersection to a ground-state product state,1,4−12 from which particular inter- and intraprotein reactions proceed further.
The absorption spectral range of protonated RSBs at the short-wavelength side has a limit around 410–430 nm.13 To enable UV-light sensing below this range, some animals including insects, amphibians, fish, birds, and mammals have evolved rhodopsins with a stable unprotonated RSB.14−17 The electronic structure of unprotonated RSBs differs fundamentally from their protonated counterparts with respect to electron distribution, which raises the question through which mechanism their photoactivation occurs. Animal UV-absorbing rhodopsins have long resisted in-depth characterization because of prohibiting difficulties in expression and protein purification. The discovery of a UV-absorbing rhodopsin in the eukaryotic alga Chlamydomonas reinhardtii has alleviated this situation.18 Histidine kinase rhodopsin 1 (HKR1) is a unique microbial rhodopsin that has photochromic bistable dark states: a blue-absorbing state (Rh-Bl, λmax ≈ 485 nm) and a UV-absorbing state (Rh-UV, λmax ≈ 380 nm), binding retinal via a protonated and an unprotonated RSB, respectively18 (Figure 1). The Rh-Bl and Rh-UV states can be reversibly photoconverted by blue and UV light, respectively. In this work, we identify the pathway and mechanism by which the Rh-UV state is transformed to the Rh-Bl state. We present the comprehensive photodynamics of the Rh-UV state through transient absorption (TA) and femtosecond stimulated Raman spectroscopy (FSRS)19 from the femtosecond to the submillisecond time scales, thereby identifying unusual isomerization reaction dynamics of the UV-absorbing rhodopsin.
Figure 1.

Retinal conformers in HKR1. (a) UV-absorbing (Rh-UV) and blue-absorbing (Rh-Bl) states of HKR1. (b) Steady-state absorption spectra of Rh-UV (black solid line) and Rh-Bl (blue dashed line) states.
Results and Discussion
Excited-State Ordering, Interconversions, and Relaxation
It is known from previous work that the Rh-UV state may occur in two different conformations, Rh-UV1 with C13=C14 cis/C15=N16 anti (hereby described as 13-cis/15-anti) and Rh-UV2 with an equal mixture of 13-trans/15-syn and 13-cis/15-anti conformation20 of which Rh-UV1 is by far the most dominant under our experimental conditions and designated as Rh-UV in the following (see Methods for more details), although a small contribution of Rh-UV2 cannot be totally excluded. The Rh-Bl state occurs in two protonated retinal conformers (13-trans/15-anti and 13-cis/15-syn) with almost equal contribution18 (Figure 1a). To investigate how the UV-light-driven photodynamics control conversion from Rh-UV to Rh-Bl, femto- to submillisecond TA spectroscopy and FSRS were applied on the Rh-UV state. Globally fitted TA spectra—evolution-associated difference spectra (EADS) and decay-associated difference spectra (DADS)—are shown in Figures 2a and S1, respectively, while selected kinetic traces are shown in Figure S2. For the global analysis, six components were required: 40 fs, 370 fs, 4.8 ps, 62 ps, 58 μs, and infinite. The time constants and spectral features of the 370 fs, 4.8 ps, and 62 ps components correspond to a previously reported TA study.18 With the extended time window of our improved setup, an additional 58 μs component could be identified. Because of sparse time sampling around time zero in the current data (which resulted from an experimental trade-off between capturing ultrafast dynamics and that at sub-ms time scales in a single experiment while maintaining reasonable data-collection times), as well as an instrument response function of 50 fs, there is an uncertainty in the fitted time constant that assumes a value of 40 fs here. An attempt to fit the data with one less component resulted in a similar time constant of 65 fs but an absence of the 370 fs component (Figure S3) and showed significant misfits in the first picosecond. This demonstrates that the ultrafast component is genuine and that both the 40 and 370 fs components are strictly required in the analysis, even if the time sampling is sparse. Given the sparse time sampling around zero, we cautiously assign a <100 fs time constant to the fastest component. It does not correspond to a cross-phase modulation or coherent artifact around time zero, as with the use of electronically synchronized pump and probe lasers no well-defined phase relation exists between pump and probe pulses, which washes out any such effects. We also reanalyzed the ultrafast data of ref (18), thereby including a distinct ultrafast time constant (Figure S4): in ref (18), a so-called pulse follower giving an instantaneous response was included ad hoc in the analysis to account for the signals around time zero. In the reanalyzed data (Figure S4) we identified a 110 fs lifetime, with an EADS similar to that shown in Figure 2a, supporting the notion that the ultrafast component is genuine. However, with an instrument response function of 80 fs (full width at half maximum) vs 50 fs for the current data set, as well as an elevated noise level with respect to the current data, the ultrafast component is less well resolved in the data of ref (18); we continue to define this time constant as <100 fs.
Figure 2.

Transient absorption (TA) spectroscopy and FSRS of the Rh-UV state of HKR1 upon 400 nm excitation. (a) Evolution-associated difference spectra (EADS) of the TA signals. The amplitude of the first EADS (black line) is scaled down by 2. The EADS of the 4.8 ps component scaled on that of the 62 ps component is shown as a dashed magenta line. (b) First four EADS (top) and decay-associated difference spectra (DADS, bottom) of the FSRS signals. (c) Time traces at 1690 cm–1 (top) and 1720 cm–1 (bottom). Open dots and solid lines show the raw data and fitting, respectively.
The <100 fs EADS (black line, Figure 2a) represents the state at time zero prepared by the excitation pulse and shows a stimulated emission band in the blue region (less than ∼470 nm) and a pronounced excited-state absorption at >470 nm. Notably, the stimulated emission shows a vibronic structure with an energy spacing that matches that of the absorption spectrum (Figure 1b), which supports our notion that this constitutes a genuine molecular state rather than a cross-phase modulation artifact. It evolves to the second EADS in <100 fs (blue line, Figure 2a) that has two broad positive bands at ∼470 and ∼590 nm. In 370 fs, these bands sharpen, while their peak positions do not significantly change (blue to magenta line evolution). In 4.8 ps, the positive signals decayed to a lower amplitude while maintaining spectral shape (magenta to green line evolution), after which the signal disappeared almost completely in 62 ps (green to red line evolution). Finally in 58 μs, a positive band at ∼480 nm appeared at low amplitude (red to cyan line evolution). A negative band at ∼400 nm was observed in all components at varying amplitudes and is due to the ground-state bleach of the Rh-UV state.
FSRS spectra were obtained with a preresonant 800 nm Raman pump upon actinic excitation of Rh-UV at 400 nm. Selected raw FSRS spectra are shown in Figure S5a. The FSRS spectra were globally fitted by applying the same time constants as for the TA experiments (Figures 2b and S5b). From the EADS and DADS (Figure 2b), it is obvious that a strong band near 1712 cm–1 band appeared in 370 fs. After the ∼1712 cm–1 band rise, it decayed with two components peaking at 1696 and 1716 cm–1, as observed in the DADS, with time constants of 4.8 and 62 ps, respectively (Figure 2b). In Figure 2c, the kinetic traces at 1690 (top) and 1720 cm–1 (bottom) are shown with fitted curves. Note that the 4.8 and 62 ps EADS show a slight broad negative baseline in the region between 1600 and 1800 cm–1. This is a consequence of the watermarking approach, which causes broad negative features to appear on either side of strong positive bands (the “Mexican hat” effect), and should be interpreted as an incomplete baseline removal rather than genuine negative signals.21
The excited-state signals derived from TA spectroscopy and FSRS of the Rh-UV state differ significantly from those observed in visible-absorbing microbial rhodopsins, both in spectral signature and lifetimes. The pronounced positive band near 1712 cm–1 (Figure 2b) observed in FSRS has not been observed in visible-absorbing rhodopsins,8,9,22−24 and its unusually high frequency points to a specific origin. Similar highly upshifted bands are generally observed and well-established in the optically forbidden S1 (2Ag–) excited state of linear polyenes of similar conjugation length, where they were assigned to an ethylenic C=C stretch vibration that was unusually highly upshifted, which resulted from vibronic coupling between the S1 (2Ag–) excited and S0 (1Ag–) ground states.25,26 Notably, such a band was also observed for an unprotonated RSB analogue in solution with picosecond Raman spectroscopy peaking at 1738 cm–1, where the ethylenic C=C stretch vibration was upshifted by ∼200 cm–1 and assigned to vibronic coupling in analogy to the electronic structure in linear polyenes.27 We accordingly assign the positive signal near 1712 cm–1 to a large upshift of the C=C ethylenic stretch that results from population of the optically forbidden S1 (2Ag–) state in HKR1.
In linear polyenes, the excited-state energy level ordering involves an optically forbidden low-lying state, according to S2 (1Bu+) – S1 (2Ag–) – S0 (1Ag–), where S2 is optically allowed and S1 is optically forbidden. Here, the 1Bu+ state has an “ionic” charge-transfer character, while the 2Ag– state (which has the same symmetry as the ground-state S0) exhibits “covalent” character with charges distributed evenly along the conjugated polyene chain.28,29 For linear polyenes, this energy-level ordering has been demonstrated for a wide range of conjugation lengths of N = 3 to N > 15.30−32 Early experimental and theoretical studies indicated that in RSB protonation leads to Bu+/Ag– energy-level inversion with respect to linear polyenes,33 implying an optically allowed Bu+-like state as the lowest-lying excited state, later confirmed by numerous computational studies.4,34−38 According to this model, unprotonated opsin-bound RSB would have an energy-level ordering similar to that of linear polyenes. In unprotonated RSB in solution, the radiative rate of the S1 state was determined to be very small at (260 ns)−1 – (450 ns)−1, which confirms the optically forbidden nature of this transition and is consistent with an Ag– character.39,40 Here we experimentally determine that this simple picture likely applies to HKR1, i.e., the optically allowed 1Bu+-like state is located higher than the optically forbidden 2Ag–-like state, resulting in S0–S2 photoexcitation in Rh-UV HKR1.
We note that, even if the molecular symmetry of RSB does not entirely conform with those of linear polyenes, the Ag–/Bu+ character of their excited-state manifold remains largely valid, as has been confirmed with structure-based calculations for protonated RSB, where the Bu+ state lies below 2Ag–.4,36 Such structure-based calculations on UV-absorbing rhodopsins with unprotonated RSB will have to await high-resolution X-ray structures. Likewise, geometric distortion that results from binding to the protein does not appreciably affect the overall ground- and excited-state characters of RSB, which retain their Ag– character in the ground state and the Ag– and Bu+ designations of their excited states.4,36
Recent theoretical work on the UV-absorbing Siberian hamster cone pigment found a 1Bu+ state ∼0.3 eV lower than the 2Ag– state, essentially reproducing the situation for animal rhodopsin binding protonated RSB.41 Our data cannot be explained with the energy-level ordering or spacings reported there: if the 1Bu+ state absorbs a photon and the 2Ag– state would lie 0.3 eV above it, the latter will not get populated, which is contrary to our experimental observations. We note that the calculations were performed on a homology model of bovine rhodopsin with the RSB in a 11-cis isomeric configuration, which differs significantly from HKR1 microbial rhodopsin with a 13-cis RSB. Even so, given the inexact structural modeling, it remains to be seen if that work represents an accurate description of UV rhodopsins in the animal kingdom.
A band near 1700 cm–1 was observed in the excited state of unprotonated RSB in solution using pump-degenerate four-wave mixing (DFWM) experiments,42 where it was assigned to a C=N stretch vibration. However, we consider it highly unlikely that the positive band near 1712 cm–1 in Figure 2b represents a C=N vibration, given that C=N bands in unprotonated RSB are located at much lower frequencies around 1610–1630 cm–1.43,44 In HKR1, the C=N mode was identified at 1614 cm–1 in the molecular ground state using resonant Raman spectroscopy,18 and we are not aware of any viable physical mechanism that could cause this C=N mode to upshift by 100 cm–1 in the S1 excited state. In addition, the C=N band has a very low Raman activity (6% of the C=C ethylenic stretch),18 and hence, it is very unlikely to be the cause of the much larger signals at ∼1710 cm–1 in Figure 2b. In contrast, vibronic coupling between S0 and S1 states is a well-established and often experimentally observed mechanism to dramatically upshift C=C stretch vibrations in the S1 state of linear polyenes and related molecules,25−27,45−49 so our interpretation of the signals ∼1710 cm–1 constitutes the most likely rationalization.
The <100 fs decay of the S2 signal implies that the S2 (1Bu+) → S1 (2Ag–) transition proceeds in <100 fs. The spectral sharpening of TA spectra (Figure 2a) in 370 fs is likely due to vibrational cooling of the S1 (2Ag–) state. From the absorption and fluorescence spectra of RSB in organic solvent, the S2 – S1 energy gap may be estimated at ∼4000 cm–1.40 Hence, a large amount of heat is dissipated in the molecule upon S2–S1 conversion, which will cause vibrational cooling to occur.
We assigned the unusually upshifted FSRS band at ∼1712 cm–1 that appears within 370 fs (Figure 2b) as a C=C stretching band of the optically forbidden S1 (2Ag–) state. The S1 C=C stretching modes appeared in 370 fs, not in <100 fs (Figure 2b), even though the S1 state is formed with the latter time constant as demonstrated by our TA results (Figure 2a). Most likely, intramolecular vibrational energy redistribution is required prior to the occurrence of S1–S0 vibronic coupling.
Identification of an Isomerization Reaction in the S2–S1 Evolution
Having established the nature of the excited states that are formed in HKR1 upon UV excitation, we are in a position to discuss the isomerization pathways that ultimately result in the blue-light-absorbing HKR1 state shown in Figure 1. We first note that the S1 state (2Ag–) shows a biphasic decay with time constants of 4.8 and 62 ps, indicating two separate S1 fractions with distinct lifetimes. The TA signatures of these states are nearly identical, as shown in Figure 2a, where the 4.8 ps EADS (magenta dotted line) was scaled on the 62 ps EADS (green line). In principle, an unprotonated RSB also has access to a n−π* state where one electron from the lone-pair nitrogen of the RSB is promoted to the chromophore π-electron system. Population of n−π* states has been detected in retinal in solution.50 However, the electronic absorption spectra (ESA) of the two distinct S1 states are nearly identical; hence, we consider it very likely that they represent similar π–π* transitions.
In FSRS, the DADS of the 4.8 and 62 ps components show positive bands at 1696 and 1716 cm–1, respectively (Figure 2b), confirming the existence of two distinct S1 species. The significant difference in the S1 C=C stretch frequency implies that two structurally distinct S1 states are formed following excitation to the S2 state. We recall that the retinal conformer of the Rh-UV state is homogeneous 13-cis/15-anti under our background illumination conditions, having only one C=C stretch band at ∼1566 cm–118 (at 1567 cm–1 in Figure S6). From these observations, we hypothesize that retinal isomerization may have taken place during the S2–S1 conversion and ensuing vibrational-cooling process. Hereby, one fraction undergoes isomerization and the other fraction maintains its isomeric state, resulting in two distinct S1 C=C frequencies corresponding to two minima on the S1 excited-state potential energy surface, as schematically indicated in Figure 3. In an unprotonated RSB analogue in solution, cis isomers were downshifted in their S1 C=C frequency with respect to the all-trans isomer by up to 11 cm–1,27 supporting this interpretation.
Figure 3.
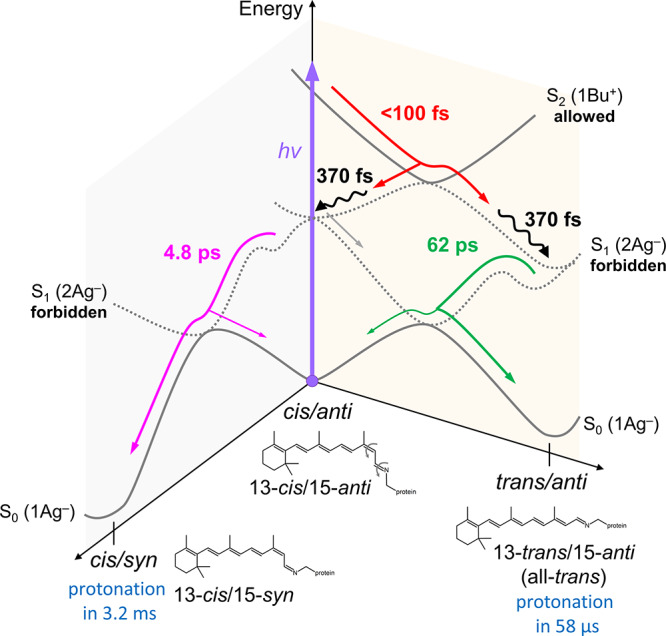
Excited-state reaction model of Rh-UV HKR1. The red arrow indicates the isomerization pathway on the S2–S1 surfaces, including a reactive fraction to 13-trans/15-anti (all-trans) and a nonreactive fraction back to 13-cis/15-anti. The magenta arrow indicates the isomerization pathways on the S1–S0 surfaces from 13-cis/15-anti to 13-cis/15-syn. The wiggly black lines indicate vibrational cooling on the S1 potential energy surface. The green arrow indicates internal conversion from the S1 to the S0 state while maintaining the 13-trans/15-anti isomeric state.
Because the 4.8 ps DADS in FSRS is essentially all-positive (magenta line, bottom panel, Figure 2b), the 4.8 ps component represents pure decay of the 1696 cm–1 band. Thus, the 4.8 ps spectral evolution does not represent a band shift of the S1 C=C stretch but rather a separate decay component of the S1 state, as we observed for the 4.8 ps component in the TA data (Figures 2a and S1). This observation rules out the possibility that the 4.8 ps component is due to vibrational cooling: if this were the case, the 1696 cm–1 band would represent population of the ν = 1 vibrational level in the S1 state, which is downshifted with respect to the ν = 0 vibrational level due to anharmonicity in the potential energy well. During vibrational cooling, the band would shift to the ν = 0 vibrational level at 1716 cm–1. The 1716 cm–1 band would then rise upon decay of the 1696 cm–1 band, which would manifest itself as a pronounced negative amplitude component ∼1716 cm–1 in the 4.8 ps DADS, which is not observed experimentally. Also, no such rise at 1716 cm–1 is observed in the raw FSRS spectra (Figure S5a). To further strengthen this argument, in the TA results the 4.8 ps EADS has an identical spectral shape to that of the 62 ps EADS (Figure 2a, see scaled 4.8 ps EADS), which is not consistent with a vibrational-cooling process. Instead, the TA experiments clearly indicated vibrational cooling in 370 fs, as evidenced from the sharpening of the S1 excited-state absorption with that time constant (Figure 2a). Therefore, we conclude that vibrational cooling is completed in 370 fs and eliminate the possibility that it occurs in 4.8 ps.
It is important to note here that the ultrafast UV–vis spectral dynamics upon isomerization and vibrational cooling in HKR1 cannot be directly compared with those of canonical animal or microbial rhodopsins that bind a protonated RSB. For instance, photorhodopsin/bathorhodopsin and the J/K intermediates in rhodopsins with a protonated RSB are accompanied by characteristic bathochromic shifts after isomerization.1,5,51 In HKR1, the isomerized photoproduct is formed on an entirely different electronic state, namely, the S1 state (2Ag–) of unprotonated RSB, as opposed to canonical rhodopsins where the S0 state (1Ag–) of protonated RSB is populated, which results in entirely different spectroscopic signatures. A similar issue pertains to the FSRS spectra of HKR1 on the picosecond time scale. We note that most of the Raman bands show a significant shift on the picosecond time scale (Figure S5). However, one cannot directly compare these results on HKR1 with those of canonical rhodopsins: in the latter, the isomerization results in a ground-state species, and the Raman frequency shifts with respect to the reactant state become apparent immediately, allowing for in-depth interpretation of the retinal structure.8,9 In HKR1, the system resides in the S1 excited state after isomerization, which by itself will generally shift most frequencies because the overall bond order changes due to the π–π* character of the optical transition. This will overwhelm any effect of isomerization on the Raman bands, precluding a meaningful comparison with the reactant spectrum. For that reason, only the upshifted S1 C=C band near 1700 cm–1 shown in Figure 2b gives useful information because this specific vibrational band is shifted completely out of its frequency region of the reactant state. The fact that we observe two distinct Raman frequencies here proves that there are two retinal conformers in the S1 state.
Additional Isomerization Reaction in the S1–S0 Evolution
The S1 state shows a biphasic decay to the S0 ground state with time constants of 4.8 and 62 ps, and after this event the ground-state photoproducts will become apparent. The 58 μs EADS in TA (red line, Figures 2a, 4a, and S7), which appears after the S1 state decay in 62 ps, has a very low amplitude. This observation indicates that the main fraction of photoproducts corresponds to UV-absorbing unprotonated RSB species that significantly overlap with that of Rh-UV, causing their positive and negative signals to cancel out in the difference spectrum.18 Still, the 58 μs EADS shows a very small absorption at 450–600 nm, as depicted in Figure S7, which indicates that a small fraction of unprotonated retinal becomes protonated and acquires an absorption spectrum peaking at 520 nm immediately after decay of the S1 excited state. We estimate this fraction as 4% of the totally formed photoproducts based on the amplitude of the EADS in the TA (Figures 4a and S7) and flash photolysis (Figure S8a) data as explained in the figure caption of Figure S8.
Figure 4.

Transient absorption (TA) and stimulated Raman spectra of the 58 μs and infinite components. (a) EADS of the TA signals. The last two components from Figure 2a are shown. (b) A ns−μs time trace at 500 nm of the TA experiments. The open dots and the solid line show raw data and a fitting curve, respectively. (c) FSRS spectra taken at 12.5 ns and 100 μs time delays with extensive data averaging. The thin and thick lines indicate raw and smoothed spectra, respectively.
To characterize the photoproduct(s) formed after S1 state decay, we performed FSRS experiments at 12.5 ns and 100 μs time delays with extensive data averaging (Figure 4c). In the 12.5 ns spectrum (red line), a positive band at 1528 cm–1 is observed along with a negative Rh-UV ground-state bleach signal at 1559 cm–1. In addition, a positive band at 1574 cm–1 with a shoulder at 1584 cm–1 is clearly resolved. The observation of three distinct positive C=C bands (1528, 1574, and 1584 cm–1) indicates that at least three retinal conformers were formed after the excited-state evolution. Interestingly, the 1574 cm–1 band is at a nearly identical spectral position as the 1576 cm–1 band observed for all-trans unprotonated RSB in frozen solution44 and may thus be assigned as unprotonated all-trans. Following the same work, the 1584 cm–1 band likely corresponds to the unprotonated 13-cis conformer. This observation indicates that two distinct unprotonated retinal conformers exist in the photoproduct ground states, which appear upon S1–S0 relaxation proceeding in 4.8 and 62 ps. Therefore, in addition to isomerization on evolving from the S2 to the S1 state, another isomerization may occur on evolving from the S1 to the S0 state. Otherwise, only one unprotonated ground-state product would be observed because the nonisomerized 13-cis conformer in the S1 state (see Figure 3) would return to its original parent ground state that has its C=C frequency at 1567 cm–1 and would not contribute to the difference signals.
The 1528 cm–1 band has a higher amplitude than those at 1574/1584 cm–1, but its frequency is obviously too low for a unprotonated RSB species (see ref (43) for the relation between absorption wavelength and retinal C=C stretch frequency). Instead, we interpret this 1528 cm–1 band as arising from the minor fraction of directly formed protonated RSB (red lines, Figures 4a and S7). Despite its low transient concentration and minor contribution to the total product yield of 4%, it gives a larger FSRS signal due to the more favorable resonance conditions with the 800 nm Raman pump through its absorption band at 520 nm. Its isomeric state remains unknown at this point.
Dual Isomerization on Distinct Potential Energy Surfaces: Lifetimes and Pathways
With FSRS, we concluded that two isomerization reactions may occur in the HKR1 UV-absorbing state upon absorption of UV light: upon evolution from S2 to S1 and with the transition from S1 to S0. An open question here is which isomerization, C13=C14 cis–trans or C15=N16 anti–syn, occurs during the subsequent S2 – S1 – S0 electronic energy-state transition cascade. In general, photoisomerization proceeds via conical intersections,4,52 and there are three possibilities for dual photoisomerization:
-
(i)
C13=C14 cis–trans isomerization with the S2–S1 transition; C15=N16 anti–syn isomerization with the S1–S0 transition (Figure 3).
-
(ii)
C15=N16 anti–syn isomerization with the S2–S1 transition; C13=C14 cis–trans isomerization with the S1–S0 transition (Figure S9a).
-
(iii)
Parallel isomerizations of C13=C14 cis–trans and C15=N16 anti–syn with the S2–S1 transition (Figure S9b).
If both cis−trans and anti−syn isomerization reactions would take place sequentially along a single pathway, a single 13-trans/15-syn product would be formed. This contradicts with the observed two unprotonated photoproducts in HKR1 (Figure 4c) and the known isomeric composition of the Rh-Bl state.18,20 Therefore, the possibility of sequential cis−trans and anti−syn isomerization along a single pathway is excluded. We consider option (iii) unlikely because it would result in three distinct S1 conformers with distinct decay rates (Figure S9b), while we observe only two such conformers (Figure 2a and b).
In protonated RSB in microbial rhodopsins, the 1Bu+ state drives C13=C14 trans–cis isomerization on femtosecond time scales but not C15=N16 anti–syn. Because the 1Bu+ is populated upon S2 excitation in HKR1, we accordingly suggest that the first photoisomerization involves C13=C14 cis to trans isomerization similarly to protonated RSB. A nonreactive fraction relaxes to the 13-cis form in the S1 state. This is consistent with the presence of two distinct S1 C=C bands, where the all-trans conformer has a higher frequency than the cis conformer, as observed in a RSB analogue.27 We hence assign the 1716 cm–1 band to the 13-trans and the 1696 cm–1 band to the 13-cis unprotonated RSB in the S1 state. This reaction features the remarkable property that the newly formed isomer appears in the excited state rather than in the ground state. Isomerization in <100 fs is unusually fast for a microbial rhodopsin,1 but similarly fast isomerizations have been observed in channelrhodopsin 1 from Chlamydomonas augustae (CaChR1)24 and animal rhodopsin.5,6
As discussed in the previous section, both 13-trans/15-anti (all-trans) and 13-cis/15-syn isomers are likely formed as ground-state photoproducts, indicating that C15=N16 anti/syn isomerization may occur upon S1–S0 relaxation in 4.8 or 62 ps, presumably via another conical intersection. A conical intersection between 2Ag– and 1Ag– electronic states is more complex than that between 1Bu+ and 1Ag– and features two adjacent 90° twisted bonds, also referred to as “CH3-kink” or “Hula-twist” conical intersections, as opposed to the simple one-bond flip intersections that are found for Bu+/Ag– potential energy surfaces.4 As indicated in Figure 3, two distinct positions at the S1 potential energy surface are populated upon decay of the S2 state, i.e., 13-trans/15-anti and 13-cis/15-anti. It is likely that these two conformers relax to the S0 ground state via two distinct conical intersections (CIs), giving rise to the two S1 decay time constants. The evolution on the potential energy surface to reach the conical intersections is unlikely to be barrierless, given the 4.8 and 62 ps lifetimes. Hence, the first CI is reached in 4.8 ps after passing a barrier or a flat region, and the second CI is reached in 62 ps upon passing a higher barrier. At this point, it is difficult to unambiguously identify specific reaction paths to the ground state, as the FSRS signal of the deprotonated ground-state photoproducts has much lower amplitude than the S1 state, and they only become apparent after full S1 decay; from the kinetics alone one cannot assign either of the 4.8 and 62 ps components to an isomerization reaction. However, under the assumption that the isomeric composition of Rh-Bl follows from the photoreactions of HKR1-UV, we hypothesize that the 4.8 ps decay corresponds to the 13-cis/15-anti fraction in the S1 state that undergoes C15=N16 anti–syn isomerization, producing the 13-cis/15-syn ground-state conformer (Figure 3). We then assign the 62 ps component to the 13-trans/15-anti S1 conformer that passes a different conical intersection and relaxes back to 13-trans/15-anti without undergoing isomerization, as schematically depicted in Figure 3.
Biphasic Protonation in 58 μs and 3.2 ms
After formation of the two unprotonated ground-state species discussed earlier, the system further evolves through proton transfer to RSB, finally resulting in the two Rh-Bl states. The significant rise of the TA signals at ∼450–600 nm with 58 μs (Figure 4a and b) implies a protonation reaction proceeding in 58 μs. A reanalysis of the previously published flash photolysis data18 reveals two rise components of 81 μs and 3.2 ms at ∼450–600 nm (Figure S8). We hence conclude that protonation of the ground-state photoproducts occurs in two distinct phases of ∼60–80 μs and 3.2 ms. We note that the observed protonation dynamics are much slower than that reported for the photoactivated M state of bacteriorhodopsin53 and proteorhodopsin,54 where they proceed on the ns to hundreds of ns time scales. Strikingly, the 1574 cm–1 band in the 12.5 ns FSRS spectrum (red line, Figure 4c) has disappeared in the 100 μs FSRS spectrum (cyan line), while the 1584 cm–1 band has remained. A prominent band at 1540 cm–1 has come up in the 100 μs FSRS spectrum (cyan line, Figure 4c), consistent with partial protonation of the RSB. Thus, only the 1574 cm–1 band disappears in the 58 μs evolution, and it is replaced by a band at 1540 cm–1. In the steady-state stimulated Raman spectrum of the Rh-Bl state (Figure S6), two conformers in the protonated Rh-Bl state at 1540 and 1550 cm–1 are observed, similar to the previous resonance Raman spectroscopy work.18Figure S10 reproduces the FSRS spectrum at 100 μs (cyan line), compared with the steady-state Rh-Bl minus Rh-UV spectrum (black line), clearly showing that the 1540 cm–1 band has formed in the former but the 1550 cm–1 band has not. On the basis of an extensive resonant Raman study of channelrhodopsin-2, the 1540 cm–1 band may be assigned to the 13-trans species, whereas the 1550 cm–1 band is associated with the 13-cis/15-syn conformer.55 The 1540 cm–1 band observed in the 100 μs FSRS spectrum (Figure 4c) is thus assigned to 13-trans conformer, implying that protonation of 13-trans/15-anti RSB proceeds in 58 μs. Likely the other 1550 cm–1 band appeared in 3.2 ms, which is out of our FSRS temporal range, implying that protonation of 13-cis/15-syn conformer proceeds in 3.2 ms. With the 3.2 ms process, the Rh-UV to Rh-Bl transformation is complete. The two distinct protonation channels as correlated with the distinct retinal isomers are indicated in the reaction scheme of Figure 3. A light-driven C15=N16 syn–anti isomerization of an unprotonated retinal conformer followed by protonation reactions was observed in a rhodopsin mimic protein by steady-state spectroscopy and X-ray crystallography,56 but it is not clear through which excited-state pathways these reactions proceed.
Concluding Remarks
We investigated the UV-light-induced photoreaction of a UV-absorbing form (Rh-UV) of histidine kinase rhodopsin 1 (HKR1), utilizing femtosecond-to-millisecond transient absorption spectroscopy and FSRS. Immediately after UV-light absorption, the unprotonated RSB in HKR1 is excited to the optically allowed S2 (Bu+) state, indicating that Bu+/Ag– energy level inversion occurs with respect to the protonated RSB in visible-absorbing rhodopsins. We propose a dual photoisomerization that involves cis/trans isomerization in <100 fs with the S2–S1 transition and an anti/syn isomerization with the S1–S0 conversion in 4.8 ps. After the dual isomerization on distinct potential energy surfaces, which forms two unprotonated ground-state photoproducts, protonation reactions were detected in 58 μs and 3.2 ms for the 13-trans/15-anti isomer and, presumably, the 13-cis/15-syn isomer, respectively, completing the formation of the blue-light absorbing (Rh-Bl) states of HKR1. The model shown in Figure 3 has been constructed on the basis of the data presented in this work and the known isomeric compositions and their Raman signatures of the HKR1 Rh-UV and Rh-Bl states.18 Furthermore, we have assumed that the dual isomers of the Rh-Bl state are formed directly from the UV-induced reaction of the single-isomer Rh-UV state, i.e., we assume that they do not follow from slower dark adaption processes such as is the case, for instance, in bacteriorhodopsin. This assumption is substantiated by the observation of dual RSB isomers and protonation phases throughout the photoreaction, from the very first events on the femtosecond time scale that indicate two isomeric conformers in the optically forbidden S1 state (Figure 2), to the observation of two distinct unprotonated ground-state photoproducts (Figure 4) and the biphasic protonation on the micro- to millisecond time scales that involve selective RSB isomers (Figures 4 and S8). Even though the order in which the C13=C14 cis–trans and C15=N16 anti–syn isomerizations occur has not been rigorously demonstrated, the scheme of Figure 3, which proposes initial C13=C14 cis–trans isomerization on a <100 fs time scale, is consistent with known properties of trans and cis isomers of an unprotonated RSB analogue in the S1 state,27 rendering this reaction model the most likely one.
In conclusion, this work reports on the heretofore unknown excited-state dynamics and isomerization pathways and mechanisms of a UV-absorbing rhodopsin. Our findings constitute a benchmark of the photoreaction dynamics of an unprotonated retinal conformer in the broad classes of UV-absorbing rhodopsins that are widely dispersed in all kingdoms of life.
Methods
Sample Preparation
The protein samples were expressed and purified as reported previously.18 The purified samples were concentrated in 20 mM Tris buffer at pH 8.0 containing 150 mM NaCl and 0.03% β-d-maltoside (DDM) to OD (/cm) ∼10 and ∼30 at 380 nm for transient absorption and FSRS experiments, respectively. The samples were filled in a homemade sample holder that has two 2 mm thick CaF2 plates, and the sample thickness was set as 400 μm with an appropriate sample spacer. The sample holder was set on a Lissajous scanner that ensures sample refreshment after each laser shot with a time interval of 60 s between successive exposures to the laser pulses.57,58 HKR1 can assume two distinct UV forms called Rh-UV1 and Rh-UV2, depending on illumination conditions.20,59 Rh-UV1 is formed on blue light background illumination, whereas Rh-UV2 substantially coaccumulates on green or orange background illumination and is red-shifted with respect to Rh-UV1. In this work, the Rh-UV1 state was prepared with blue light (470 nm) illumination and indicated as Rh-UV for brevity.
Transient Absorption Spectroscopy
Transient absorption measurements were performed with a femtosecond-to-submillisecond pump–probe setup as reported previously.37,60 A CaF2 plate was used for supercontinuum white light generation, and a selected wavelength region, 375−750 nm, was detected by a photodiode array. The time delay was varied up to 500 μs at 167 data points with a minimal temporal step of 50 fs, generated by an optical delay and an electronic delay as reported previously.23,37,60 The diameters of the pump and the probe beams at the sample position were ∼200 and ∼70 μm, respectively. The wavelength of the pump beam was centered at 400 nm, and the power was attenuated to ∼300 nJ. The instrumental response function was ∼50 fs, estimated from global analysis. The sample was illuminated with a ∼100 mW LED (λmax ≈ 470 nm) during the experiments.
Watermarked Transient Stimulated Raman Spectroscopy
Femtosecond to submillisecond time-resolved stimulated Raman experiments were performed with the watermarked, nearly baseline-free stimulated Raman setup reported previously.21,23 The Raman pump (∼800 nm, ∼12 μJ) and Raman probe (∼840–960 nm) were spatiotemporally overlapped at the sample position with a diameter of ∼100 μm. Actinic pump (∼400 nm, ∼400 nJ) was focused on the protein sample to a diameter of ∼150 μm with a time delay from −500 ps to 400 μs at 72 data points with a minimum temporal step of 50 fs, generated by an optical delay and an electronic delay as reported previously.27,37,60 The instrument response function was ∼70 fs, estimated from global analysis. The sample was illuminated with a ∼100 mW LED (λmax ≈ 470 nm) during the experiments. Raman pumps pass through a specially designed chopper blade for the watermarking approach,21 which produces 14 Raman pump sequences whose wavelengths are slightly shifted from each other.21 As a result, 14 different stimulated Raman experiments are effectively performed simultaneously, which makes the baseline-free watermarking approach possible. The sample exposure time to the beams was ∼1.5 h in total for the time-resolved stimulated Raman experiment. For Figures 4c, S6, and S10, baseline correction was applied using a Gaussian curve to remove the “Mexican-hat” shape artifacts.21 We note here that potential pump–repump/dump–probe effects by the 800 nm Raman pump are filtered out by the watermarking technique.21 The 800 nm Raman pump is not resonant with any of the HKR1 transient states (see Figure 2a), except possibly for the very short-lived S2 state, and we do not expect significant dump/repump effects to occur nonetheless.
Global Analysis Methodology
Global analysis was performed for the transient absorption spectra and the transient stimulated Raman spectra using the Glotaran program.60,61 With global analysis, all wavelengths/wavenumbers were analyzed simultaneously with a set of common time constants.62 A kinetic model was applied consisting of sequentially interconverting, evolution-associated difference spectra (EADS), i.e., 1 → 2 → 3 → ···, in which the arrows indicate successive monoexponential decays of a time constant, which can be regarded as the lifetime of each EADS.62 The first EADS corresponds to the difference spectrum at time zero. The first EADS evolves into the second EADS with time constant τ1, which in turn evolves into the third EADS with time constant τ2, etc. The procedure clearly visualizes the evolution of the intermediate states of the protein.63 Decay-associated difference spectra (DADS) indicate the spectral changes with parallel decay channels and independent decay time constants. It is important to note that the parallel and the sequential analyses are mathematically equivalent and yield identical time constants.64 The standard errors in the time constants were <5% for the transient absorption and <10% for the transient stimulated Raman results.61,63
Acknowledgments
Y.H., J.W., and J.T.M.K. were supported by the Chemical Sciences Council of The Netherlands Organization for Scientific Research (NWO) through a VICI Grant and a Middelgroot Investment Grant to J.T.M.K. M.B., M.L., and P.H. were supported by the German Research Foundation (DFG: Leibniz, SFB1078 & UniCat); P.H. is a Hertie Professor and supported by the Hertie Foundation.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c03229.
Decay-associated difference spectra, selected transient absorption time traces with fitting curves, transient absorption data, comparison of global fitting of the transient absorption spectra, FSRS spectra, ground-state stimulated Raman spectra, transient absorption EADS, flash photolysis experiments, and alternative excited-state reaction models (PDF)
Author Present Address
∥ Y.H.: School of Applied and Engineering Physics, Cornell University, Ithaca, NY 14853, United States
The authors declare no competing financial interest.
Supplementary Material
References
- Ernst O. P.; Lodowski D. T.; Elstner M.; Hegemann P.; Brown L. S.; Kandori H. Microbial and animal rhodopsins: structures, functions, and molecular mechanisms. Chem. Rev. 2014, 114 (1), 126–63. 10.1021/cr4003769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shichida Y.; Matsuyama T. Evolution of opsins and phototransduction. Philos. Trans. R. Soc., B 2009, 364 (1531), 2881–2895. 10.1098/rstb.2009.0051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumauchi M.; Ebry T.. Visual Pigments as Photoreceptors. In Handbook of Photosensory Receptors; Briggs W. R., Spudich J. L., Eds.; Wiley: Weinheim, Germany, 2006; pp 43–76. [Google Scholar]
- Gozem S.; Luk H. L.; Schapiro I.; Olivucci M. Theory and Simulation of the Ultrafast Double-Bond lsomerization of Biological Chromophores. Chem. Rev. 2017, 117 (22), 13502–13565. 10.1021/acs.chemrev.7b00177. [DOI] [PubMed] [Google Scholar]
- Polli D.; Altoe P.; Weingart O.; Spillane K. M.; Manzoni C.; Brida D.; Tomasello G.; Orlandi G.; Kukura P.; Mathies R. A.; Garavelli M.; Cerullo G. Conical intersection dynamics of the primary photoisomerization event in vision. Nature 2010, 467 (7314), 440–3. 10.1038/nature09346. [DOI] [PubMed] [Google Scholar]
- Johnson P. J. M.; Halpin A.; Morizumi T.; Prokhorenko V. I.; Ernst O. P.; Miller R. J. D. Local vibrational coherences drive the primary photochemistry of vision. Nat. Chem. 2015, 7 (12), 980–986. 10.1038/nchem.2398. [DOI] [PubMed] [Google Scholar]
- Schapiro I.; Ryazantsev M. N.; Frutos L. M.; Ferre N.; Lindh R.; Olivucci M. The Ultrafast Photoisomerizations of Rhodopsin and Bathorhodopsin Are Modulated by Bond Length Alternation and HOOP Driven Electronic Effects. J. Am. Chem. Soc. 2011, 133 (10), 3354–3364. 10.1021/ja1056196. [DOI] [PubMed] [Google Scholar]
- Shim S.; Dasgupta J.; Mathies R. A. Femtosecond time-resolved stimulated Raman reveals the birth of bacteriorhodopsin’s J and K intermediates. J. Am. Chem. Soc. 2009, 131 (22), 7592–7. 10.1021/ja809137x. [DOI] [PubMed] [Google Scholar]
- Kukura P.; McCamant D. W.; Yoon S.; Wandschneider D. B.; Mathies R. A. Structural observation of the primary isomerization in vision with femtosecond-stimulated Raman. Science 2005, 310 (5750), 1006–9. 10.1126/science.1118379. [DOI] [PubMed] [Google Scholar]
- Nogly P.; Weinert T.; James D.; Carbajo S.; Ozerov D.; Furrer A.; Gashi D.; Borin V.; Skopintsev P.; Jaeger K.; Nass K.; Bath P.; Bosman R.; Koglin J.; Seaberg M.; Lane T.; Kekilli D.; Brunle S.; Tanaka T.; Wu W. T.; Milne C.; White T.; Barty A.; Weierstall U.; Panneels V.; Nango E.; Iwata S.; Hunter M.; Schapiro I.; Schertler G.; Neutze R.; Standfuss J. Retinal isomerization in bacteriorhodopsin captured by a femtosecond x-ray laser. Science 2018, 361 (6398), eaat0094. 10.1126/science.aat0094. [DOI] [PubMed] [Google Scholar]
- Nass Kovacs G.; Colletier J. P.; Grunbein M. L.; Yang Y.; Stensitzki T.; Batyuk A.; Carbajo S.; Doak R. B.; Ehrenberg D.; Foucar L.; Gasper R.; Gorel A.; Hilpert M.; Kloos M.; Koglin J. E.; Reinstein J.; Roome C. M.; Schlesinger R.; Seaberg M.; Shoeman O. L.; Stricker M.; Boutet S.; Haacke S.; Heberle J.; Heyne K.; Domratcheva T.; Barends T. R. M.; Schlichting I. Three-dimensional view of ultrafast dynamics in photoexcited bacteriorhodopsin. Nat. Commun. 2019, 10, 3177. 10.1038/s41467-019-10758-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frutos L. M.; Andruniow T.; Santoro F.; Ferre N.; Olivucci M. Tracking the excited-state time evolution of the visual pigment with multiconfigurational quantum chemistry. Proc. Natl. Acad. Sci. U. S. A. 2007, 104 (19), 7764–7769. 10.1073/pnas.0701732104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt D. M.; Wilkie S. E.; Bowmaker J. K.; Poopalasundaram S. Vision in the ultraviolet. Cell. Mol. Life Sci. 2001, 58 (11), 1583–1598. 10.1007/PL00000798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salcedo E.; Zheng L.; Phistry M.; Bagg E. E.; Britt S. G. Molecular basis for ultraviolet vision in invertebrates. J. Neurosci. 2003, 23 (34), 10873–8. 10.1523/JNEUROSCI.23-34-10873.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y.; Radlwimmer F. B.; Yokoyama S. Molecular genetics and the evolution of ultraviolet vision in vertebrates. Proc. Natl. Acad. Sci. U. S. A. 2001, 98 (20), 11731–6. 10.1073/pnas.201257398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyanagi M.; Kawano E.; Kinugawa Y.; Oishi T.; Shichida Y.; Tamotsu S.; Terakita A. Bistable UV pigment in the lamprey pineal. Proc. Natl. Acad. Sci. U. S. A. 2004, 101 (17), 6687–91. 10.1073/pnas.0400819101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita T.; Ohuchi H.; Tomonari S.; Ikeda K.; Sakai K.; Shichida Y. Opn5 is a UV-sensitive bistable pigment that couples with Gi subtype of G protein. Proc. Natl. Acad. Sci. U. S. A. 2010, 107 (51), 22084–9. 10.1073/pnas.1012498107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luck M.; Mathes T.; Bruun S.; Fudim R.; Hagedorn R.; Tran Nguyen T. M.; Kateriya S.; Kennis J. T.; Hildebrandt P.; Hegemann P. A photochromic histidine kinase rhodopsin (HKR1) that is bimodally switched by ultraviolet and blue light. J. Biol. Chem. 2012, 287 (47), 40083–90. 10.1074/jbc.M112.401604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukura P.; McCamant D. W.; Mathies R. A. Femtosecond stimulated Raman spectroscopy. Annu. Rev. Phys. Chem. 2007, 58, 461–488. 10.1146/annurev.physchem.58.032806.104456. [DOI] [PubMed] [Google Scholar]
- Luck M.; Bruun S.; Keidel A.; Hegemann P.; Hildebrandt P. Photochemical chromophore isomerization in histidine kinase rhodopsin HKR1. FEBS Lett. 2015, 589 (10), 1067–1071. 10.1016/j.febslet.2015.03.024. [DOI] [PubMed] [Google Scholar]
- Kloz M.; WeiSsenborn J.; Polivka T.; Frank H. A.; Kennis J. T. M. Spectral watermarking in femtosecond stimulated Raman spectroscopy: resolving the nature of the carotenoid S* state. Phys. Chem. Chem. Phys. 2016, 18 (21), 14619–28. 10.1039/C6CP01464J. [DOI] [PubMed] [Google Scholar]
- McCamant D. W.; Kukura P.; Mathies R. A. Femtosecond stimulated Raman study of excited-state evolution in bacteriorhodopsin. J. Phys. Chem. B 2005, 109 (20), 10449–10457. 10.1021/jp050095x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hontani Y.; Inoue K.; Kloz M.; Kato Y.; Kandori H.; Kennis J. T. The photochemistry of sodium ion pump rhodopsin observed by watermarked femto- to submillisecond stimulated Raman spectroscopy. Phys. Chem. Chem. Phys. 2016, 18 (35), 24729–36. 10.1039/C6CP05240A. [DOI] [PubMed] [Google Scholar]
- Schnedermann C.; Muders V.; Ehrenberg D.; Schlesinger R.; Kukura P.; Heberle J. Vibronic Dynamics of the Ultrafast all-trans to 13-cis Photoisomerization of Retinal in Channelrhodopsin-1. J. Am. Chem. Soc. 2016, 138 (14), 4757–62. 10.1021/jacs.5b12251. [DOI] [PubMed] [Google Scholar]
- Simpson J. H.; McLaughlin L.; Smith D. S.; Christensen R. L. Vibronic Coupling in Polyenes - High-Resolution Optical Spectroscopy of All-trans-2,4,6,8,10,12,14-hexadecaheptaene. J. Chem. Phys. 1987, 87 (6), 3360–3365. 10.1063/1.452978. [DOI] [Google Scholar]
- Christensen R. L.; Kohler B. E. Vibronic Coupling in Polyenes - High-Resolution Optical Spectroscopy of 2,10-Dimethylundecapentaene. J. Chem. Phys. 1975, 63 (5), 1837–1846. 10.1063/1.431560. [DOI] [Google Scholar]
- Mukai Y.; Hashimoto H.; Koyama Y.; Kuroda S.; Hirata Y.; Mataga N. S1-State and T1-State Properties of Normal-Butylamine Schiff-Bases of Isomeric Retinylideneacetaldehyde As Revealed by Transient Absorption and Transient Raman Spectroscopies and by HPLC Analysis of Triplet-Sensitized Isomerization. J. Phys. Chem. 1991, 95 (26), 10586–10592. 10.1021/j100179a019. [DOI] [Google Scholar]
- Tavan P.; Schulten K. the Low-Lying Electronic Excitations in Long Polyenes - A PPP-MRD-CL Study. J. Chem. Phys. 1986, 85 (11), 6602–6609. 10.1063/1.451442. [DOI] [Google Scholar]
- Tavan P.; Schulten K. Electronic Excitations in Finite and Infinite Polyenes. Phys. Rev. B: Condens. Matter Mater. Phys. 1987, 36 (8), 4337–4358. 10.1103/PhysRevB.36.4337. [DOI] [PubMed] [Google Scholar]
- Christensen R. L.; Enriquez M. M.; Wagner N. L.; Peacock-Villada A. Y.; Scriban C.; Schrock R. R.; Polivka T.; Frank H. A.; Birge R. R. Energetics and Dynamics of the Low-Lying Electronic States of Constrained Polyenes: Implications for Infinite Polyenes. J. Phys. Chem. A 2013, 117 (7), 1449–1465. 10.1021/jp310592s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen R. L.; Galinato M. G. I.; Chu E. F.; Howard J. N.; Broene R. D.; Frank H. A. Energies of Low-Lying Excited States of Linear Polyenes. J. Phys. Chem. A 2008, 112 (49), 12629–12636. 10.1021/jp8060202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garavelli M.; Celani P.; Bernardi F.; Robb M. A.; Olivucci M. Force fields for ’’ultrafast’’ photochemistry: The S-2 (1B(u))->S-1(2A(g))->S-0 (1A(g)) reaction path for all-trans-hexa-1,3,5-triene. J. Am. Chem. Soc. 1997, 119 (47), 11487–11494. 10.1021/ja971280u. [DOI] [Google Scholar]
- Birge R. R. 2-Photon Spectroscopy of Protein-Bound Chromophores. Acc. Chem. Res. 1986, 19 (5), 138–146. 10.1021/ar00125a003. [DOI] [Google Scholar]
- Cembran A.; Bernardi F.; Olivucci M.; Garavelli M. Counterion controlled photoisomerization of retinal chromophore models: a computational investigation. J. Am. Chem. Soc. 2004, 126 (49), 16018–16037. 10.1021/ja048782+. [DOI] [PubMed] [Google Scholar]
- Cembran A.; Bernardi F.; Olivucci M.; Garavelli M. Excited-state singlet manifold and oscillatory features of a nonatetraeniminium retinal chromophore model. J. Am. Chem. Soc. 2003, 125 (41), 12509–12519. 10.1021/ja030215j. [DOI] [PubMed] [Google Scholar]
- Marin M. D.; Agathangelou D.; Orozco-Gonzalez Y.; Valentini A.; Kato Y.; Abe-Yoshizumi R.; Kandori H.; Choi A.; Jung K. H.; Haacke S.; Olivucci M. Fluorescence Enhancement of a Microbial Rhodopsin via Electronic Reprogramming. J. Am. Chem. Soc. 2019, 141 (1), 262–271. 10.1021/jacs.8b09311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hontani Y.; Marazzi M.; Stehfest K.; Mathes T.; van Stokkum I. H. M.; Elstner M.; Hegemann P.; Kennis J. T. M. Reaction dynamics of the chimeric channelrhodopsin C1C2. Sci. Rep. 2017, 7 (1), 7217. 10.1038/s41598-017-07363-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dokukina I.; Weingart O. Spectral properties and isomerisation path of retinal in C1C2 channelrhodopsin. Phys. Chem. Chem. Phys. 2015, 17 (38), 25142–25150. 10.1039/C5CP02650D. [DOI] [PubMed] [Google Scholar]
- Bachilo S. M.; Bondarev S. L.; Gillbro T. Fluorescence properties of protonated and unprotonated Schiff bases of retinal at room temperature. J. Photochem. Photobiol., B 1996, 34 (1), 39–46. 10.1016/1011-1344(95)07271-3. [DOI] [Google Scholar]
- Bachilo S. M.; Gillbro T. Fluorescence of retinal Schiff base in alcohols. J. Phys. Chem. A 1999, 103 (15), 2481–2488. 10.1021/jp983646a. [DOI] [Google Scholar]
- Bonvicini A.; Demoulin B.; Altavilla S. F.; Nenov A.; El-Tahawy M. M. T.; Segarra-Marti J.; Giussani A.; Batista V. S.; Garavelli M.; Rivalta I. Ultraviolet vision: photophysical properties of the unprotonated retinyl Schiff base in the Siberian hamster cone pigment. Theor. Chem. Acc. 2016, 135 (4), 110. 10.1007/s00214-016-1869-x. [DOI] [Google Scholar]
- Kraack J. P.; Buckup T.; Motzkus M. Coherent High-Frequency Vibrational Dynamics in the Excited Electronic State of All-Trans Retinal Derivatives. J. Phys. Chem. Lett. 2013, 4 (3), 383–387. 10.1021/jz302001m. [DOI] [PubMed] [Google Scholar]
- Doukas A. G.; Aton B.; Callender R. H.; Ebrey T. G. Resonance Raman Studies of Bovine Metarhodopsin-I and Metarhodopsin-II. Biochemistry 1978, 17 (12), 2430–2435. 10.1021/bi00605a028. [DOI] [PubMed] [Google Scholar]
- Aton B.; Doukas A. G.; Callender R. H.; Becher B.; Ebrey T. G. Resonance Raman Studies of Purple Membrane. Biochemistry 1977, 16 (13), 2995–2999. 10.1021/bi00632a029. [DOI] [PubMed] [Google Scholar]
- Nagae H.; Kuki M.; Zhang J. P.; Sashima T.; Mukai Y.; Koyama Y. Vibronic coupling through the in-phase, C=C stretching mode plays a major role in the 2A(g)(−) to 1A(g)(−) internal conversion of all-trans-beta-carotene. J. Phys. Chem. A 2000, 104 (18), 4155–4166. 10.1021/jp9924833. [DOI] [Google Scholar]
- Kuici M.; Nagae H.; Cogdell R. J.; Shimada K.; Koyama Y. Solvent Effect on Spheroidene in Nonpolar and Polar Solutions and the Environment of Spheroidene in the Light-Harvesting Complexes of Rhodobacter Sphaeroides 2.4.1 As Revealed by the Energy of the (1)A(G)(−)- B-1(U)+ Absorption and the Frequencies of the Vibronically Coupled C=C Stretching Raman Lines in the (1)A(G)(−) and 2(1)A(G)(−) States. Photochem. Photobiol. 1994, 59 (1), 116–124. 10.1111/j.1751-1097.1994.tb05009.x. [DOI] [Google Scholar]
- Hashimoto H.; Koyama Y. the C=C Stretching Raman Lines of Beta-Carotene Isomers in the S1 State As Detected by Pump-Probe Resonance Raman-Spectroscopy. Chem. Phys. Lett. 1989, 154 (4), 321–325. 10.1016/0009-2614(89)85363-1. [DOI] [Google Scholar]
- Hashimoto H.; Koyama Y.; Hirata Y.; Mataga N. S1 and T1 Species of Beta-Carotene Generated by Direct Photoexcitation from the All-Trans, 9-Cis, 13-Cis, and 15-Cis Isomers as Revealed by Picosecond Transient Absorption and Transient Raman Spectroscopies. J. Phys. Chem. 1991, 95 (8), 3072–3076. 10.1021/j100161a022. [DOI] [Google Scholar]
- Granville M. F.; Holtom G. R.; Kohler B. E. High-Resolution One and Two Photon Excitation-Spectra of trans,trans-1,3,5,7-Octatetraene. J. Chem. Phys. 1980, 72 (9), 4671–4675. 10.1063/1.439802. [DOI] [Google Scholar]
- Ishii K.; Takeuchi S.; Tahara T. A 35-fs time-resolved absorption study of all-trans retinal in a nonpolar solvent: Ultrafast photophysics revisited. Chem. Phys. Lett. 2006, 418 (4–6), 307–310. 10.1016/j.cplett.2005.11.002. [DOI] [Google Scholar]
- Peteanu L. A.; Schoenlein R. W.; Wang Q.; Mathies R. A.; Shank C. V. the 1st Step in Vision Occurs in Femtoseconds - Complete Blue and Red Spectral Studies. Proc. Natl. Acad. Sci. U. S. A. 1993, 90 (24), 11762–11766. 10.1073/pnas.90.24.11762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B. G.; Martinez T. J. Isomerization through conical intersections. Annu. Rev. Phys. Chem. 2007, 58, 613–634. 10.1146/annurev.physchem.57.032905.104612. [DOI] [PubMed] [Google Scholar]
- Kalisky O.; Lachish U.; Ottolenghi M. Time Resolution of a Back Photoreaction in Bacteriorhodopsin. Photochem. Photobiol. 1978, 28 (2), 261–263. 10.1111/j.1751-1097.1978.tb07705.x. [DOI] [Google Scholar]
- Eckert C. E.; Kaur J.; Glaubitz C.; Wachtveitl J. Ultrafast Photoinduced Deactivation Dynamics of Proteorhodopsin. J. Phys. Chem. Lett. 2017, 8 (2), 512–517. 10.1021/acs.jpclett.6b02975. [DOI] [PubMed] [Google Scholar]
- Bruun S.; Stoeppler D.; Keidel A.; Kuhlmann U.; Luck M.; Diehl A.; Geiger M. A.; Woodmansee D.; Trauner D.; Hegemann P.; Oschkinat H.; Hildebrandt P.; Stehfest K. Light-Dark Adaptation of Channelrhodopsin Involves Photoconversion between the all-trans and 13-cis Retinal Isomers. Biochemistry 2015, 54 (35), 5389–400. 10.1021/acs.biochem.5b00597. [DOI] [PubMed] [Google Scholar]
- Nosrati M.; Berbasova T.; Vasileiou C.; Borhan B.; Geiger J. H. A Photoisomerizing Rhodopsin Mimic Observed at Atomic Resolution. J. Am. Chem. Soc. 2016, 138 (28), 8802–8808. 10.1021/jacs.6b03681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandre M. T.; Domratcheva T.; Bonetti C.; van Wilderen L. J.; van Grondelle R.; Groot M. L.; Hellingwerf K. J.; Kennis J. T. Primary reactions of the LOV2 domain of phototropin studied with ultrafast mid-infrared spectroscopy and quantum chemistry. Biophys. J. 2009, 97 (1), 227–37. 10.1016/j.bpj.2009.01.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groot M. L.; van Wilderen L.; Di Donato M. Time-resolved methods in biophysics. 5. Femtosecond time-resolved and dispersed infrared spectroscopy on proteins. Photochemical & Photobiological Sciences 2007, 6 (5), 501–507. 10.1039/b613023b. [DOI] [PubMed] [Google Scholar]
- Luck M.; Hegemann P. The two parallel photocycles of the Chlamydomonas sensory photoreceptor histidine kinase rhodopsin 1. J. Plant Physiol. 2017, 217, 77–84. 10.1016/j.jplph.2017.07.008. [DOI] [PubMed] [Google Scholar]
- Ravensbergen J.; Abdi F. F.; van Santen J. H.; Frese R. N.; Dam B.; van de Krol R.; Kennis J. T. M. Unraveling the Carrier Dynamics of BiVO4: A Femtosecond to Microsecond Transient Absorption Study. J. Phys. Chem. C 2014, 118 (48), 27793–27800. 10.1021/jp509930s. [DOI] [Google Scholar]
- Snellenburg J. J.; Laptenok S. P.; Seger R.; Mullen K. M.; van Stokkum I. H. M. Glotaran: A Java-Based Graphical User Interface for the R Package TIMP. J. Stat Softw 2012, 49 (3), 1–22. 10.18637/jss.v049.i03. [DOI] [Google Scholar]
- van Stokkum I. H. M.; Larsen D. S.; van Grondelle R. Global and target analysis of time-resolved spectra. Biochim. Biophys. Acta, Bioenerg. 2004, 1657 (2–3), 82–104. 10.1016/j.bbabio.2004.04.011. [DOI] [PubMed] [Google Scholar]
- Kennis J. T. M.; Groot M. L. Ultrafast spectroscopy of biological photoreceptors. Curr. Opin. Struct. Biol. 2007, 17 (5), 623–630. 10.1016/j.sbi.2007.09.006. [DOI] [PubMed] [Google Scholar]
- Toh K. C.; Stojkovic E. A.; van Stokkum I. H.; Moffat K.; Kennis J. T. Fluorescence quantum yield and photochemistry of bacteriophytochrome constructs. Phys. Chem. Chem. Phys. 2011, 13 (25), 11985–97. 10.1039/c1cp00050k. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.