

Abstract
Supramolecular gels have potential in many areas. In many cases, a major drawback is that the gels are formed at a high rate. As a result, nonoptimal, kinetically trapped self-assembled structures are often formed, leading to gels that can be hard to reproduce and control. One method to get around kinetic trapping is annealing. Thermal annealing is one possibility, but it is not always desirable to heat the gels. Here, we describe a method to anneal pH-triggered gels after they are formed. We employ a reaction relay in a peptide-based hydrogel system to anneal the structures by a controlled and uniform pH change. Our method allows us to prepare gels with more controlled properties. We show that this can be used to enable homogeneous “molding and casting” of the hydrogels. This method of annealing is more effective in improving gel robustness than a conventional heat–cool cycle.
Introduction
Supramolecular, self-assembled materials are formed by discrete molecules under the influence of various noncovalent forces like hydrogen bonding, π–π stacking, and van der Waals interactions. Such noncovalent interactions are individually weak; however, when they work in tandem, robust self-assembly can occur. Supramolecular gels are formed when these self-assembled structures interact to form a network that traps the solvent.1−8 As a consequence of this network, the materials behave as viscoelastic solids. Disruption of these noncovalent forces in gel architectures most often results in a return to the solution state.
Gels are attracting attention because of their high-end applications in many areas including environmental remediation, sensors, structuring, optoelectronics, catalysis, and tissue engineering.6,7,9−14 Clearly, in all of these cases, the gels need different specific properties.15−19 In order to form gels, usually a trigger such as pH, temperature, light, cosolvent (solvent switch), or addition of salt is applied to the solution, suspension, or dispersion of the gelator molecules. As a consequence, the solubility of the molecules in solution significantly decreases, and so aggregation occurs. In many cases, the self-assembly process occurs at a high rate,20 meaning that the self-assembled structures do not have time to reach their targeted/global thermodynamic minimum and often exist in a kinetically trapped state.17,18,21 Because of this, the final properties of the gels can depend significantly upon the method of preparation.17 Therefore, during self-assembly, if the environmental conditions are not well controlled, inhomogeneous gels with irreproducible properties can be formed. For gels to be used effectively for high value applications like biomaterials and optoelectronics, we need to overcome this kinetic trapping.6,11,15,16,22−27
One potential way to get around this kinetic trapping is annealing. Annealing is widely applied for many systems to drive the kinetically trapped structures toward their local thermodynamic minimum.18,28−34 This is usually accomplished by performing a heat–cool cycle on the kinetically trapped state.32−36 In the case of supramolecular gels, thermal annealing, which results in subsequent sol formation and regelling, can be used to adapt the properties for both single and multicomponent gel systems.18,30−32,34−40 While thermal annealing is suitable for some systems, there are limitations in terms of volume of the gels that can be annealed as well as the rate of temperature change. The final properties of the gels can differ with subtle variation of cooling rates during annealing,30,35,36,39−41 and hence, maintaining a homogeneous temperature change inside the system throughout the process is often difficult due to temperature gradients within the gel as well as at the liquid-container junction. Consequently, homogeneous “molding and casting” (melting followed by flowing and then solidification) of gels is not possible using thermal annealing. There can also be issues where solvent evaporation leads to a change in the solvent composition that may drive the system toward another kinetically trapped state. Finally, thermal sensitivity of various functional groups present on the gelator backbone is also sometimes an important factor.42−44
An alternative approach is to anneal the gel locally under mild conditions. To exemplify this, instead of a heat–cool cycle, here we employ a reaction relay to anneal a peptide-based hydrogel by a controlled and uniform pH change of the medium (Figure 1). There is significant interest at the moment in preparing transient gels, which is relevant here.45−49 To construct the consecutive reaction relay, we simultaneously incorporate two competing triggers. The first trigger drives the system to form an initially out-of-equilibrium gel, followed by increasing the pH of the medium to form a transient hydrogel; the second trigger leads to reassembly by a subsequent decrease in the pH. This method enables us to prepare homogeneous and reproducible gels by establishing an optimal balance of the physical interactions between the molecules that lead to the formation of a final gel, which is both stiffer and contains a different network structure. The method further allows autonomous programming of homogeneous “molding and casting” of the corresponding hydrogel assemblies in time and control over diffusion of encapsulated materials.50
Figure 1.
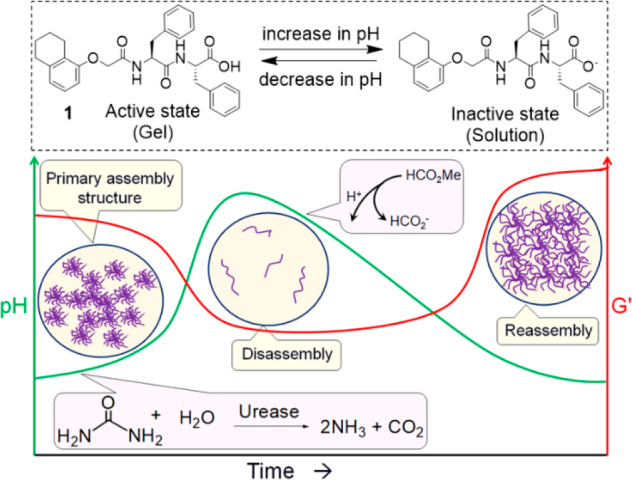
(Top) The functionalized dipeptide gelator (1) used here which forms a gel at low pH and a viscous solution at high pH in 20/80 DMSO/water (v/v). (Bottom) Cartoon representing annealing of peptide hydrogel of 1 using a consecutive reaction relay. The enzymatic reaction is responsible for increasing the pH (green line), while the hydrolysis of methyl formate lowers the pH. The solidlike properties are represented by the storage modulus (G′). The reaction proceeds in 20/80 DMSO/water (v/v).
Results and Discussion
Dipeptide 1 is a previously known hydrogelator,51 which forms transparent gels in DMSO/H2O (20/80, v/v) at a concentration of 2 mg/mL (Figure S1). The mechanical properties of the gel were evaluated by oscillatory rheological experiments (Figure S1). The gel is stable until a strain of ∼8% (the critical strain) after which the storage modulus (G′) starts to decrease; the gel completely converts to a sol after ∼105% strain (the yield point). Frequency sweep measurements show that G′ is considerably higher than the corresponding loss modulus (G″), and both are frequency independent. The viscoelastic nature of the gels arises from a network of spherulitic domains of fibers as can be seen using confocal microscopy imaging (Figure S2).
As a first step toward annealing of the DMSO/water gel of 1, we need to drive the self-assembly to a nongelled state in a transient manner. To do this, we used a method that we have recently reported,52 exploiting the urease-catalyzed hydrolysis of urea to produce NH3 as base to increase the pH.53−60 The pH of the initial gel is around pH 4.3. At a pH above the apparent pKa of 1 (ca. 6.4, Figure S3), deprotonation of the carboxylic acid leads to a micellar dispersion of 1, and hence the slow increase in pH leads to the gradual transformation of the gel to a solution state (Figure 2a, 2b). As per our previous work, the lifetime of the transient gel could be controlled by decreasing the concentration of urea and thereby decreasing the rate of pH increase (Figures 2a, S4).52
Figure 2.
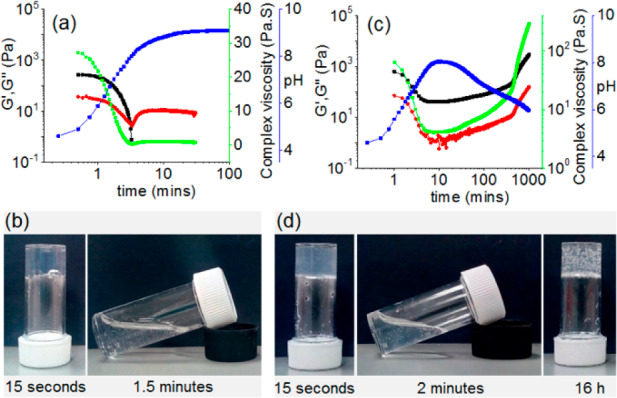
Variation of pH (blue), G′ (black), G″ (red), and complex viscosity (green) with time for 1 from the enzymatic reaction in the absence (a) and presence (c) of methyl formate. (b) and (d) represent the phase change of 1 with time associated with the enzymatic reactions performed in the absence (a) and presence (c) of methyl formate, respectively. The white structures in the gels are air bubbles, not precipitation. For (a)–(d), the initial reaction conditions are as follows: [urease] = 0.2 mg/mL, [urea] = 0.02 M. Solvent is 20/80 DMSO/water (v/v), and [1] = 2 mg/mL. For (c) and (d), the volume of methyl formate used is 100 μL.
To anneal the gel, we need to drive the disassembly kinetics toward reassembly by reducing the pH of the medium (Figure 1). To do this, we utilize the base-promoted saponification of methyl formate to formic acid.60 This leads to a decrease in the pH sufficiently below the pKa of 1 to lead to regelation. Using the same conditions as for the experiments shown in Figure 2a, but with the inclusion of 100 μL of methyl formate, results in a gel-to-sol-to-gel transition (Figure 2c, 2d). At the early time, the production of NH3 is faster than the hydrolysis of methyl formate. As a result, the pH of the medium increases steadily and reaches a pH of 8.0 after 10 min. At high pH, the hydrolysis of methyl formate becomes dominant, and the production of formic acid results in a reduction in pH of the sample.
Following the changes in state using rheology, initially G′ was considerably higher than G″ corroborating the hydrogel formation at the early stages (Figure 2c, 2d). With time, as the pH of the medium increases, the gel started to collapse shown by the decrease in both rheological moduli. In the high pH regime, both G′ and G″ reached a minimum and became almost constant for a significant period of time. As the hydrolysis of methyl formate starts to dominate and the pH of the medium begins to decrease, both G′ and G″ start to increase slowly. At this point, a visual transformation of a free-flowing solution to a gel was observed. Viscosity data recorded with time also depends on the pH changes and demonstrates the sol formation and regelling (Figure 2c). Interestingly, the final values of both rheological moduli and the viscosity were considerably higher than for the initially formed gel. Hence, our annealing approach here leads to an increase in the robustness of the material.
Importantly, it is possible to control the rate of annealing by adjusting the rate of pH change (Figure S5). A decrease in urea concentration (keeping all other parameters fixed) significantly reduces the rate of pH increase. There was also a slight reduction in the maximum pH (pH 7.7). However, no significant change in the rate of pH decrease was noticed. Similarly, an increase in methyl formate concentration (keeping all other parameters fixed) causes a substantial reduction in the rate of pH increase as well as the maximum pH. In this case, during the reaction, the maximum pH of the medium reduced from pH 8 (at 100 μL of methyl formate) to pH 7.4 (using 150 μL). Time sweep rheology as well as viscosity data correlates with the changes in pH (Figures S5, S6). The decrease in the rate of pH increase directly translates into delay in the decrease of both G′ and G″ and thereby emphasizes an increase in the lifetime of the primary assembled structures (Figures S5, S6). However, at high pH, no significant change in the viscosity of the solutions was noticed (Figure S5). There were also no considerable differences in the final pH values (pH 5.9–6.0). In both cases, the viscosity and rheological moduli of the final gels increase compared to the gel obtained from a high rate of initial pH increase.
For such systems, drying often leads to artifactual changes in structure.61 Hence, to probe the structures underpinning the different states, we used confocal microscopy and small angle neutron scattering (SANS) that can be used directly in the gel state. Confocal microscopy imaging was conducted at different time intervals for probing the development of the microstructure of the gels. For this purpose, we choose a system where the rate of pH change was sufficiently low. Confocal studies show the existence of high density of spherulitic domains of fibers in the initial gel at early stages (Figure 3).
Figure 3.

Time dependent confocal microscopy images of 1 involving the urea–urease reaction in the presence of methyl formate (scale bars represent 20 μm): after (i) 5 min, (ii) 10 min, (iii) 15 min, (iv) 17 min, (v) 20 min, (vi) 25 min, (vii) 30 min, (viii) 32 min, (ix) 35 min, (x) 40 min, (xi) 7 h, and (xii) 16 h. The initial reaction conditions are as follows: [1] = 2 mg/mL, [urease] = 0.2 mg/mL, [urea] = 0.02 M, volume of methyl formate = 150 μL. Solvent is 20/80 DMSO/water (v/v).
As the pH of the medium increases, the density of these domains started to decrease indicating the destruction of the intermolecular associations. The fibrous structures completely disappeared around the same time as when the pH reaches its maximum value (and rheological moduli reach their minimum values), and as expected the gel is converted to a free-flowing solution. At longer times as the pH decreases once again, a fibrous network reforms leading to regelling. The rate of annealing is likely to affect the microstructures of the final gels (Figures 3 and 4). Gels annealed at a slow rate of pH change were found to contain a higher density of long fibers and relatively fewer spherulitic domains than the gel annealed at a higher rate.
Figure 4.
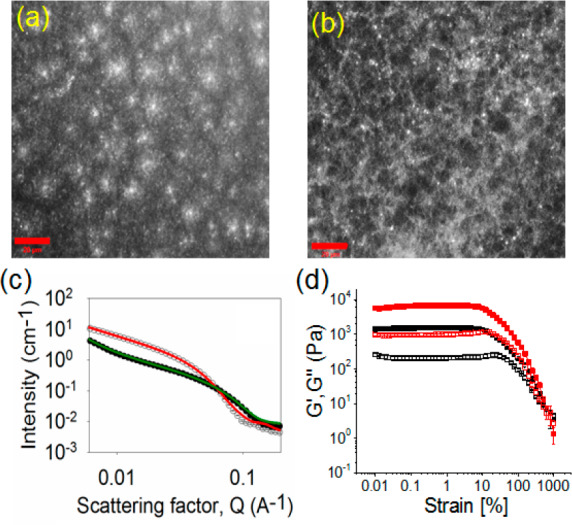
(a) and (b) Confocal fluorescence microscopy images (scale bars represent 20 μm) of the hydrogels of 1 obtained after annealing involving the following initial conditions: (a) [urease] = 0.2 mg/mL, [urea] = 0.02 M, volume of methyl formate = 100 μL; (b) [urease] = 0.2 mg/mL, [urea] = 0.01 M, volume of methyl formate = 100 μL. (c) SANS scattering data for the hydrogels of 1 (in the presence of urease) before (open symbol) and after annealing (close symbol). For annealing, initial conditions are [urease] = 0.2 mg/mL, [urea] = 0.02 M, volume of methyl formate = 100 μL. The lines represent the fit to the data before (red) and after (green) annealing. (d) Strain sweep experiments of the hydrogels of 1 obtained after annealing involving the urease–urea reaction under different conditions: (black data) [urease] = 0.2 mg/mL, [urea] = 0.02 M, volume of methyl formate = 100 μL; (red data) [urease] = 0.2 mg/mL, [urea] = 0.01 M, volume of methyl formate = 100 μL. The closed symbols represent G′, and the open symbols represent G″. In all cases, solvent is 20/80 DMSO/water (v/v) and [1] = 2 mg/mL.
The differences in microstructure of the gels obtained under different conditions were verified using SANS (Figures 4c, S7). This technique allows proving the primary structures underlying the network by fitting the scattering data to a model. SANS data for gel 1 were best fitted to an elliptical cylinder model for the gels obtained before and after annealing involving reaction relay. The elliptical cylinder model was combined with a power law component in order to fit the low Q region, which is influenced by the fractal scattering from the network.62 First, to prove if the presence of the enzyme had an effect on the fiber formation during gelation, SANS data for gel 1 before annealing were compared in the absence and presence of enzyme. Both data sets were fitted to an elliptical cylinder with radii of 3.22 ± 0.02 nm and 3.29 ± 0.03 nm for the gels with and without enzyme, respectively, axis ratios of 1.82 ± 0.02 and 1.85 ± 0.03, respectively, and lengths of 677 ± 6.40 and 732 ± 26.50 nm, respectively. The power law exponents are 2.25 ± 0.04 and 2.45 ± 0.08, respectively. These data show that the enzyme does not perturb the fiber formation. For the gels obtained after annealing using reaction relay at different rates, the scattering data could be also fitted to an elliptical cylinder with radii in the range of 1.74 ± 0.04 to 2.07 ± 0.03 nm, axes ratios between 1.75 ± 0.04 and 2.04 ± 0.07, and lengths in the range of 560 ± 12.50 to 1001 ± 13.10 nm. These results show that annealing results in an ∼40–50% reduction in the radius of the elliptical cylinders. Further description of the models and the fitting parameters are provided in the SI (Table S1 and Figure S7).
The annealing approach results in a change in the gel structure from spherulitic domains to a more uniform distribution of fibers, which ultimately leads to the formation of homogeneous hydrogels with improved mechanical properties. A decrease in rate of annealing resulted in a stiffer gel with a G′ that was ∼2–4 times higher depending on the rate of annealing used (Figures 4d, S8, S9). In comparison to the initially formed gel, irrespective of the rate of pH change, all the gels obtained after annealing could withstand considerably higher strains (>2–3 times increase in gel strength) as well as high crossover points (yield points are >400% strain where G″ > G′) (Figure S10, Table S2). Annealing also causes a substantial increase in robustness of the gels (∼2–7 times stiffer).
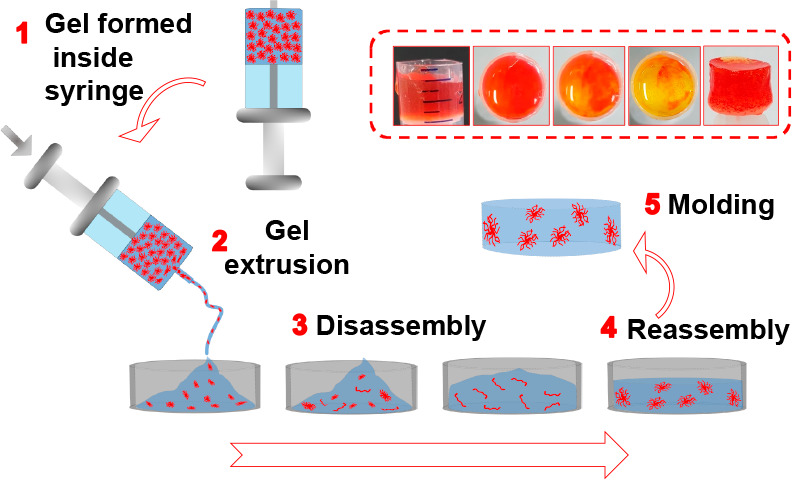
Next, we show the applicability of these gels in two areas. We have previously shown that the underlying microstructure determines whether or not such gels can be 3D printed.51 Gels with an underlying spherulitic microstructure such as the initial gels of 1 can be printed effectively, while those with a more uniform fiber network cannot.51 Hence, lines of gel were 3D printed for the different scenarios. Thin and continuous lines were printed in both the presence and absence of the enzyme from the gel formed from 1 before annealing (Figure S11). However, as expected from the changes in the underlying microstructure,51 we found that printing the gels obtained after annealing resulted in less uniform printed lines and, therefore, are not suitable for 3D printing (Figures S12, S13). Since we get a solution as an intermediate during annealing, it is possible to use our annealing method for a molding and casting process. Initially, we tested the free-flowing nature of the intermediate material before the regelation causes immobilization of the system again (Figure S14). We then used an automated 3D printer as an injector to drive the gel extrusion onto a mold. The ability to mold dipeptide 1 was evaluated under different conditions. The hydrogel of 1 (in the absence and presence of urease) before annealing was extruded 16 h after the gelation occurs (Figure S15, S16) as well as immediately after the gel was formed and then left to stand for 16 h (Figures S17, S18).
In all cases, the extruded gels do not adapt to the shape of the container and, therefore, are not suitable for molding. On the other hand, molding was performed employing the annealing method (Figures 5, S19–21). Here, two different annealing rates were used for demonstration by simply varying the concentration of methyl formate. In both situations, the initially formed gels were extruded immediately after formation (Figures 5, S19). After the extrusion, the gel changes from the initial distorted shape, producing a homogeneous solution inside the mold. With further time, regelling occurs, and homogeneous gels were formed that conform to the shape of the mold. We envisage that this method could be used for 3D print gels that slowly adapt to a second network that itself could not be effectively printed.
Figure 5.

Experiment showing autonomous programming of homogeneous “molding and casting” of the hydrogel of 1 (2 mg/mL) involving the following initial conditions: [urease] = 0.2 mg/mL, [urea] = 0.02 M, volume of methyl formate = 100 μL. An initially formed gel inside a syringe (a) was immediately extruded after 30 s of preparation (b). With time, the gel progressively converted to a homogeneous solution and acquired the shape of the container (c–e). Photographs were taken after 1 min (c), 2 min (d), and 5 min (e) of extrusion. Then the system was left undisturbed for 16 h (f). (g)–(i) represent photographs of the molded gel obtained after the experiment. The white structures in the gel are air bubbles, not precipitation. Solvent is 20/80 DMSO/water (v/v).
It is also possible to use this annealing approach to drive mixing. If a dye is encapsulated in gels of 1 in the absence of enzyme, slow diffusion occurs as would be expected from Fickian diffusion; this can be shown by forming a monolith where one side only contains the dye (Figure 6). If the annealing triggers are included, the rate of mixing is increased, as would be expected by the dissolution of the network restricting diffusion during the annealing. Using a pH-sensitive dye also allows the change in the pH during annealing to be visualized, and this can be used to show the rate of pH change is different where each side contains different amounts of the annealing trigger. In all cases, with further time, regelation causes formation of a single gel material. This method could be used, for example, to allow controlled mixing of encapsulated molecules or objects within different gel environments.
Figure 6.

Controlling diffusion in gels. (a) Two hemispheres prepared from gels formed from 1 with no enzyme present do not evolve with time. After 1 h, limited diffusion of the dye (methyl red) trapped in one gel to the other is observed (b). Where the enzymatic trigger (using 100 μL of methyl formate) is incorporated in the right-hand gel (c) with no trigger in the left-hand gel, diffusion is enhanced by the annealing of this gel (d). Note the color after 1 h is yellow as the pH is still above the pKa of the dye. Where the trigger is incorporated in both sides of the gel (e), mixing is further enhanced after 1 h (f). When different amounts of trigger are incorporated in both hemispheres (g), incorporation of the dye allows this to be visualized. (g, h) show the gels immediately after formation, (i) shows the gels after 1 min, and (j) shows the gels after 3 min. The red to orange color is more apparent in the left-hand side which has the higher amount of trigger. After annealing, the gels can be removed from the mold; (k) shows the gel in (h) after 16 h. For these images, gels at a concentration of 1 of 2 mg/mL were used. Methyl red (0.05 mg/mL) is used to dye the gels. For (e) and (g), volumes of methyl formate used are 100 and 150 μL for left and right hemispheres, respectively. For (c), (e), and (g), the initial concentration of urease is 0.2 mg/mL, and urea is 0.02 M.
Finally, as a comparison to other annealing methods, we performed a heat–cool operation on our hydrogel system in the absence of enzyme (Figure S22) and hence at a constant pH. Confocal studies revealed that a dramatic change in the microstructures from spherulitic fibers to relatively discrete spherical aggregates occurs on thermal annealing (Figure S23). SANS data also confirm that thermal annealing led to the underlying structures becoming significantly larger. The scattering data is dominated by a flexible elliptical cylinder (Figure S24), with an overall radius of 3.80 ± 0.02 nm, a Kuhn length of 20.81 ± 0.48 nm, and a length greater than that which can be effectively probed by this technique (Table S1 and Figure S24). Gels formed by thermal annealing exhibited inferior mechanical properties in terms of both gel stiffness and gel strength (Figure S25) as compared to our pH annealing method. These results again emphasize the effectiveness of our method in improving the gel properties.
Conclusions
We have successfully developed a new method to anneal peptide hydrogels with a high degree of control over the final properties of the gels by a reaction relay. To construct this, we simultaneously incorporate two competing triggers of which one is responsible for disassembly by increasing the pH and the second trigger drives the system to the original state by reducing the pH. Kinetic control over annealing is achieved in a number of ways that allows us to prepare homogeneous and reproducible gels with improved mechanical properties. We also establish that our annealing approach is more effective in improving gel robustness than thermal annealing for the present system. Unlike a conventional heat–cool cycle, where temperature is an issue, our method further allows autonomous programming of homogeneous “molding and casting” of the corresponding hydrogel in time.
Experimental Section
Materials
Compound 1 was synthesized as described previously.51 Urease (J61455 Urease, Jack Beans, minimum 45.0 units/mg solid) and urea (ultrapure 99%) were obtained from Alfa Aesar. Methyl formate was purchased from Sigma-Aldrich. Deionized water was used throughout all experiments.
Preparation of Solutions
Stock solution of 1 was prepared in DMSO at a concentration of 10 mg/mL by stirring. The enzyme and urea were highly soluble in H2O and therefore did not require stirring. The stock solution of the enzyme was prepared at a concentration of 0.254 mg/mL in H2O. The enzyme concentration in the stock solution was determined from the mass (in mg) dissolved in a known volume of H2O. Stock solution of the urea was prepared in H2O in the concentration of 2 M. Solutions of gelator, urease, and urea were prepared freshly before each experiment. The enzyme-catalyzed reactions involving gelator 1 were performed either in the presence or absence of methyl formate. For the enzymatic reactions in the absence of methyl formate, 1.580 mL of the urease solution was transferred to the vial containing 0.40 mL of the gelator solution and urea (either 20 μL of urea or a mixture of 10 μL of urea and 10 μL of H2O) and left undisturbed. Therefore, the final ratio of DMSO and H2O was 20:80, the concentration of 1 was 2 mg/mL, the concentration of urease was 0.2 mg/mL, and the initial concentration of urea was 0.02 and 0.01 M as required. A similar method was used to perform the enzymatic reactions in the presence of methyl formate. In this case, urease solution was added to the mixture of 1, urea, and methyl formate (either 100 or 150 μL), and the same solutions of 1, urease, and urea in respected volumes were used as above.
pH Measurements
A FC200 pH probe from HANNA instruments with a 6 mm × 10 mm conical tip was used for pH measurements. The stated accuracy of the pH measurements is ±0.1. For the urea–urease reaction involving the gelator, the reaction mixtures were prepared as described above in a 2 mL volume in a 7 mL Sterilin vial, and the pH change was monitored with time. The temperature was maintained at 25 °C during the measurement by using a circulating water bath.
pKa determination was carried out by recording the pH values after each addition of HCl (0.1 M) to the solution of 1 (concentration is 2 mg/mL) containing 1 mol equiv of NaOH (0.1 M) in 20% DMSO in H2O. During the titration, to prevent any gel formation, the solution was stirred continuously. The experimental temperature was 25 °C.
Hydrogel Formation and Annealing
Hydrogels of 1 were prepared under different conditions. For the solvent switch method, 1.6 mL of water was added to 0.4 mL of a DMSO solution of 1 to form the hydrogel. The samples were left overnight before measurements were carried out. Thermal annealing was performed by heating and cooling the gels typically at 1 °C/min. The final gelator concentration was 2 mg/mL.
Hydrogels were also prepared in the presence of the enzyme following the same procedure as mentioned earlier. To anneal the gels involving enzymatic reaction, the gelation experiments were carried out by adding urease solutions to the mixture of 1, urea, and methyl formate as mentioned above. The samples were then left to stand overnight to allow gelation.
Rheological Measurements
All rheological measurements were undertaken on an Anton Paar Physica MCR 301 rheometer at 25 °C. Strain, frequency, temperature, and time sweeps were performed using a vane and cup geometry. Strain sweeps were performed at 10 rad/s from 0.01% to 1000% strain. Frequency sweeps were carried out from 1 rad/s to 100 rad/s at 0.5% strain. All gels were left ∼16 h before being measured. Time sweeps were performed at an angular frequency of 10 rad/s and with a strain of 0.5%. For all experiments, gels were prepared as mentioned earlier in a 2 mL volume in 7 mL Sterilin vials.
For the temperature sweeps, gels were prepared as mentioned earlier in metal rheology cups and left overnight before measurements were carried out. G′ and G″ were recorded at a strain of 0.5% and a frequency of 10 rad/s within the temperature range of 25–80 °C. The heating and cooling rate was 1 °C/min.
Confocal Microscopy
A Zeiss LSM710 confocal microscope (Zeiss, Gottingen, Germany) with an LD EC Epiplan NEUFLUAR 50X, 0.55 DIC (Carl Zeiss, White Plains, NY, USA) objective was used for imaging. Samples were prepared as mentioned earlier containing Nile blue (2 μL/mL of a 0.1 wt % solution) in CELLview Culture dishes (35 mm diameter) and were excited at 633 nm using a He-Ne laser. Images were captured using the software Carl Zeiss ZEN 2011 v7.0.3.286.
UV–Vis Measurements
Data were collected on an Agilent Cary 60 UV–vis spectrophotometer. All UV–vis experiments were carried out in the presence of 20 μL of 0.005 M pyrene (in DMSO). Samples were prepared in a PMMA cuvette with a path length of 1 cm by following the same procedure as mentioned before.
Small Angle Neutron Scattering
SANS measurements were carried out on a SANS2D instrument, at ISIS Neutron Source in UK. We have used a 4 m sample-to-detector distance, a beam size of 8 mm, and a typical Q-range from 0.004 A–1 to 0.8 A–1 set by time-of-flight mode with wavelengths from 1.75 to 16.5 A. The samples were prepared inside a quartz cuvette of 2 mm path length by following similar methods described earlier and placed on a multiple slot sample changer with controlled temperature. We have subtracted the background of D2O scattering considering the appropriate volume fraction for each sample, and the scattering data was normalized to the absolute scale.
3D Printing and Molding Experiments
The 3D printing experiments were carried out with the same 3D printer previously reported by our research group.51 For the experiments, gels were prepared in a 2 mL volume in a 12 mL syringe following the same procedure as mentioned above. Before printing, some parameters need to be optimized in order to achieve high-quality 3D printed lines, among which we highlight the volume of the gel extruded, the speed of extrusion, the printer movement speed, and the printing height. For the different printing scenarios, each parameter was optimized. Different gel volumes (325–500 μL) and shear rates in the range of 250 s–1 to 7K s–1 were used in order to find the more suitable values at which the gel was the thinnest and continuous. All the lines were printed at 50 mm in length from a height of 3 mm. The diameter of the nozzle used for extrusion was 2.2 mm.
For molding experiments, initially the gels were prepared in a 2 mL volume in a 12 mL syringe following the same procedure as mentioned above. Then the gels were injected into a 24 mL syringe (diameter is 1.65 cm) operating as the mold using an automated 3D printer. We used a 3D printer for the molding experiments so that we could control the rate of extrusion, to make sure the same shear was applied for all the injection processes. The speed of extrusion was 1 mL· s–1 in all cases. After extrusion, gels (or sol as applied respectively) were left in the mold to settle before being transferred into a glass slide to prove the capability of the molding process.
Acknowledgments
S.P. thanks the Royal Society and SERB of India for a Newton International Fellowship. A.M.F.C. and E.R.C. thank the University of Glasgow for funding. D.A. thanks the EPSRC for a Fellowship (EP/L021978/1). This work benefitted from SasView software, originally developed by the DANSE project under NSF award DMR-0520547.63 SasView also contains code developed with funding from the EU Horizon 2020 programme, under the SINE2020 project Grant No. 654000. Experiments at ISIS Neutron and Muon Source were supported by a beamtime allocation RB1820004 from the Science and Technology Facilities Council. The primary data are available.64
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.chemmater.0c01483.
Rheology, confocal microscopy, photographs of 3D printed gels, SANS data, and fitting parameters (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Terech P.; Weiss R. G. Low Molecular Mass Gelators of Organic Liquids and the Properties of Their Gels. Chem. Rev. 1997, 97 (8), 3133–3160. 10.1021/cr9700282. [DOI] [PubMed] [Google Scholar]
- Amabilino D. B.; Smith D. K.; Steed J. W. Supramolecular materials. Chem. Soc. Rev. 2017, 46 (9), 2404–2420. 10.1039/C7CS00163K. [DOI] [PubMed] [Google Scholar]
- Estroff L. A.; Hamilton A. D. Water Gelation by Small Organic Molecules. Chem. Rev. 2004, 104 (3), 1201–1218. 10.1021/cr0302049. [DOI] [PubMed] [Google Scholar]
- Frisch H.; Besenius P. pH-Switchable Self-Assembled Materials. Macromol. Rapid Commun. 2015, 36 (4), 346–363. 10.1002/marc.201400623. [DOI] [PubMed] [Google Scholar]
- Draper E. R.; Adams D. J. Low-Molecular-Weight Gels: The State of the Art. Chem. 2017, 3 (3), 390–410. 10.1016/j.chempr.2017.07.012. [DOI] [Google Scholar]
- Li J.; Wong W.-Y.; Tao X.-m. Recent advances in soft functional materials: preparation, functions and applications. Nanoscale 2020, 12 (3), 1281–1306. 10.1039/C9NR07035D. [DOI] [PubMed] [Google Scholar]
- Dastidar P. Designing Supramolecular Gelators: Challenges, Frustrations, and Hopes. Gels 2019, 5 (1), 15. 10.3390/gels5010015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creton C. 50th Anniversary Perspective: Networks and Gels: Soft but Dynamic and Tough. Macromolecules 2017, 50 (21), 8297–8316. 10.1021/acs.macromol.7b01698. [DOI] [Google Scholar]
- Danks A. E.; Hall S. R.; Schnepp Z. The evolution of ‘sol–gel’ chemistry as a technique for materials synthesis. Mater. Horiz. 2016, 3 (2), 91–112. 10.1039/C5MH00260E. [DOI] [Google Scholar]
- Okesola B. O.; Smith D. K. Applying low-molecular weight supramolecular gelators in an environmental setting – self-assembled gels as smart materials for pollutant removal. Chem. Soc. Rev. 2016, 45 (15), 4226–4251. 10.1039/C6CS00124F. [DOI] [PubMed] [Google Scholar]
- Mehwish N.; Dou X.; Zhao Y.; Feng C.-L. Supramolecular fluorescent hydrogelators as bio-imaging probes. Mater. Horiz. 2019, 6 (1), 14–44. 10.1039/C8MH01130C. [DOI] [Google Scholar]
- Christoff-Tempesta T.; Lew A. J.; Ortony J. H. Beyond Covalent Crosslinks: Applications of Supramolecular Gels. Gels 2018, 4 (2), 40. 10.3390/gels4020040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echeverria C.; Fernandes S. N.; Godinho M. H.; Borges J. P.; Soares P. I. P. Functional Stimuli-Responsive Gels: Hydrogels and Microgels. Gels 2018, 4 (2), 54. 10.3390/gels4020054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Geng L.; Wang G.; Chu H.; Wei H. Self-Healable Gels for Use in Wearable Devices. Chem. Mater. 2017, 29 (21), 8932–8952. 10.1021/acs.chemmater.7b02895. [DOI] [Google Scholar]
- Draper E. R.; Adams D. J. Controlling the Assembly and Properties of Low-Molecular-Weight Hydrogelators. Langmuir 2019, 35 (20), 6506–6521. 10.1021/acs.langmuir.9b00716. [DOI] [PubMed] [Google Scholar]
- Ardoña H. A. M.; Draper E. R.; Citossi F.; Wallace M.; Serpell L. C.; Adams D. J.; Tovar J. D. Kinetically Controlled Coassembly of Multichromophoric Peptide Hydrogelators and the Impacts on Energy Transport. J. Am. Chem. Soc. 2017, 139 (25), 8685–8692. 10.1021/jacs.7b04006. [DOI] [PubMed] [Google Scholar]
- Raeburn J.; Zamith Cardoso A.; Adams D. J. The importance of the self-assembly process to control mechanical properties of low molecular weight hydrogels. Chem. Soc. Rev. 2013, 42 (12), 5143–5156. 10.1039/c3cs60030k. [DOI] [PubMed] [Google Scholar]
- Tantakitti F.; Boekhoven J.; Wang X.; Kazantsev R. V.; Yu T.; Li J.; Zhuang E.; Zandi R.; Ortony J. H.; Newcomb C. J.; Palmer L. C.; Shekhawat G. S.; de la Cruz M. O.; Schatz G. C.; Stupp S. I. Energy landscapes and functions of supramolecular systems. Nat. Mater. 2016, 15, 469. 10.1038/nmat4538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhiman S.; George S. J. Temporally Controlled Supramolecular Polymerization. Bull. Chem. Soc. Jpn. 2018, 91 (4), 687–699. 10.1246/bcsj.20170433. [DOI] [Google Scholar]
- Adams D. J.; Butler M. F.; Frith W. J.; Kirkland M.; Mullen L.; Sanderson P. A new method for maintaining homogeneity during liquid–hydrogel transitions using low molecular weight hydrogelators. Soft Matter 2009, 5 (9), 1856–1862. 10.1039/b901556f. [DOI] [Google Scholar]
- Liu K.; Levy A.; Liu C.; Olivier J.-H. Tuning Structure–Function Properties of π-Conjugated Superstructures by Redox-Assisted Self-Assembly. Chem. Mater. 2018, 30 (6), 2143–2150. 10.1021/acs.chemmater.8b00518. [DOI] [Google Scholar]
- Panettieri S.; Ulijn R. V. Energy landscaping in supramolecular materials. Curr. Opin. Struct. Biol. 2018, 51, 9–18. 10.1016/j.sbi.2018.02.001. [DOI] [PubMed] [Google Scholar]
- Shao T.; Falcone N.; Kraatz H.-B. Supramolecular Peptide Gels: Influencing Properties by Metal Ion Coordination and Their Wide-Ranging Applications. ACS Omega 2020, 5 (3), 1312–1317. 10.1021/acsomega.9b03939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deen G. R.; Loh X. J. Stimuli-Responsive Cationic Hydrogels in Drug Delivery Applications. Gels 2018, 4 (1), 13. 10.3390/gels4010013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walther A. Viewpoint: From Responsive to Adaptive and Interactive Materials and Materials Systems: A Roadmap. Adv. Mater. 2020, 32, 1905111. 10.1002/adma.201905111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho K. G.; Lee J. I.; Lee S.; Hong K.; Kang M. S.; Lee K. H. Light-Emitting Devices Based on Electrochemiluminescence Gels. Adv. Funct. Mater. 2020, 1907936. 10.1002/adfm.201907936. [DOI] [Google Scholar]
- Das S.; Presselt M. Progress and development in structural and optoelectronic tunability of supramolecular nonbonded fullerene assemblies. J. Mater. Chem. C 2019, 7 (21), 6194–6216. 10.1039/C9TC00889F. [DOI] [Google Scholar]
- Jiang J.; Jacobs A. G.; Wenning B.; Liedel C.; Thompson M. O.; Ober C. K. Ultrafast Self-Assembly of Sub-10 nm Block Copolymer Nanostructures by Solvent-Free High-Temperature Laser Annealing. ACS Appl. Mater. Interfaces 2017, 9 (37), 31317–31324. 10.1021/acsami.7b00774. [DOI] [PubMed] [Google Scholar]
- Yan Y.; Huang J.; Tang B. Z. Kinetic trapping – a strategy for directing the self-assembly of unique functional nanostructures. Chem. Commun. 2016, 52 (80), 11870–11884. 10.1039/C6CC03620A. [DOI] [PubMed] [Google Scholar]
- Barker E. C.; Martin A. D.; Garvey C. J.; Goh C. Y.; Jones F.; Mocerino M.; Skelton B. W.; Ogden M. I.; Becker T. Thermal annealing behaviour and gel to crystal transition of a low molecular weight hydrogelator. Soft Matter 2017, 13 (5), 1006–1011. 10.1039/C6SM02431A. [DOI] [PubMed] [Google Scholar]
- Yip H.-L.; Ma H.; Tian Y.; Acton O.; Tucker N. M.; Jen A. K. Y. Solvent-vapor annealing-induced growth, alignment, and patterning of π-conjugated supramolecular nanowires. J. Mater. Res. 2011, 26 (2), 311–321. 10.1557/jmr.2010.7. [DOI] [Google Scholar]
- Fuentes-Caparrós A. M.; de Paula Gómez-Franco F.; Dietrich B.; Wilson C.; Brasnett C.; Seddon A.; Adams D. J. Annealing multicomponent supramolecular gels. Nanoscale 2019, 11 (7), 3275–3280. 10.1039/C8NR09423C. [DOI] [PubMed] [Google Scholar]
- Yoshida T.; Takahashi M.; Hatakeyama T.; Hatakeyama H. Annealing induced gelation of xanthan/water systems. Polymer 1998, 39 (5), 1119–1122. 10.1016/S0032-3861(97)00266-8. [DOI] [Google Scholar]
- Boekhoven J.; Brizard A. M.; Stuart M. C. A.; Florusse L.; Raffy G.; Del Guerzo A.; van Esch J. H. Bio-inspired supramolecular materials by orthogonal self-assembly of hydrogelators and phospholipids. Chem. Sci. 2016, 7 (9), 6021–6031. 10.1039/C6SC01021K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llusar M.; Escuder B.; López-Castro J. D. D.; Trasobares S.; Monrós G. Transcription of Nanofibrous Cerium Phosphate Using a pH-Sensitive Lipodipeptide Hydrogel Template. Gels 2017, 3 (2), 23. 10.3390/gels3020023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari H.; Armitage S. E.; Kline S. R.; Damodaran K. K.; Kennedy S. R.; Atwood J. L.; Steed J. W. Fluorous ‘ponytails’ lead to strong gelators showing thermally induced structure evolution. Soft Matter 2015, 11 (43), 8471–8478. 10.1039/C5SM01865J. [DOI] [PubMed] [Google Scholar]
- Wang L.; Shi X.; Wu Y.; Zhang J.; Zhu Y.; Wang J. A multifunctional supramolecular hydrogel: preparation, properties and molecular assembly. Soft Matter 2018, 14 (4), 566–573. 10.1039/C7SM02358H. [DOI] [PubMed] [Google Scholar]
- Rubert Pérez C. M.; Rank L. A.; Chmielewski J. Tuning the thermosensitive properties of hybrid collagen peptide–polymer hydrogels. Chem. Commun. 2014, 50 (60), 8174–8176. 10.1039/C4CC03171G. [DOI] [PubMed] [Google Scholar]
- Fujiwara J.; Takahashi M.; Hatakeyama T.; Hatakeyama H. Gelation of hyaluronic acid through annealing. Polym. Int. 2000, 49 (12), 1604–1608. . [DOI] [Google Scholar]
- Puigmartí-Luis J.; Pérez del Pino Á.; Laukhina E.; Esquena J.; Laukhin V.; Rovira C.; Vidal-Gancedo J.; Kanaras A. G.; Nichols R. J.; Brust M.; Amabilino D. B. Shaping Supramolecular Nanofibers with Nanoparticles Forming Complementary Hydrogen Bonds. Angew. Chem., Int. Ed. 2008, 47 (10), 1861–1865. 10.1002/anie.200704864. [DOI] [PubMed] [Google Scholar]
- Trujillo-Ramírez D.; Lobato-Calleros C.; Jaime Vernon-Carter E.; Alvarez-Ramirez J. Cooling rate, sorbitan and glyceryl monostearate gelators elicit different microstructural, viscoelastic and textural properties in chia seed oleogels. Food Res. Int. 2019, 119, 829–838. 10.1016/j.foodres.2018.10.066. [DOI] [PubMed] [Google Scholar]
- Höck S.; Marti R.; Riedl R.; Simeunovic M. Thermal Cleavage of the Fmoc Protection Group. Chimia 2010, 64 (3), 200–202. 10.2533/chimia.2010.200. [DOI] [PubMed] [Google Scholar]
- Genaev A. M.; Shchegoleva L. N.; Salnikov G. E.; Shernyukov A. V.; Shundrin L. A.; Shundrina I. K.; Zhu Z.; Koltunov K. Y. Acid-Catalyzed Versus Thermally Induced C1–C1′ Bond Cleavage in 1,1′-Bi-2-naphthol: An Experimental and Theoretical Study. J. Org. Chem. 2019, 84 (11), 7238–7243. 10.1021/acs.joc.9b00915. [DOI] [PubMed] [Google Scholar]
- Yang S.; Chen J.-S.; Körner H.; Breiner T.; Ober C. K.; Poliks M. D. Reworkable Epoxies: Thermosets with Thermally Cleavable Groups for Controlled Network Breakdown. Chem. Mater. 1998, 10 (6), 1475–1482. 10.1021/cm970667t. [DOI] [Google Scholar]
- Rieß B.; Grötsch R. K.; Boekhoven J. The Design of Dissipative Molecular Assemblies Driven by Chemical Reaction Cycles. Chem. 2020, 6 (3), 552–578. 10.1016/j.chempr.2019.11.008. [DOI] [Google Scholar]
- Wang G.; Liu S. Strategies to Construct a Chemical-Fuel-Driven Self-Assembly. ChemSystemsChem. 2020, 10.1002/syst.201900046. [DOI] [Google Scholar]
- Singh N.; Formon G. J. M.; De Piccoli S.; Hermans T. M. Devising Synthetic Reaction Cycles for Dissipative Nonequilibrium Self-Assembly. Adv. Mater. 2020, 32, 1906834. 10.1002/adma.201906834. [DOI] [PubMed] [Google Scholar]
- Singh N.; Lainer B.; Formon G. J. M.; De Piccoli S.; Hermans T. M. Re-programming Hydrogel Properties Using a Fuel-Driven Reaction Cycle. J. Am. Chem. Soc. 2020, 142 (9), 4083–4087. 10.1021/jacs.9b11503. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Piskorz T. K.; Lovrak M.; Mendes E.; Guo X.; Eelkema R.; van Esch J. H. Transient Supramolecular Hydrogels Formed by Aging-Induced Seeded Self-Assembly of Molecular Hydrogelators. Adv. Sci. 2020, 7, 1902487. 10.1002/advs.201902487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randall C. L.; Gultepe E.; Gracias D. H. Self-folding devices and materials for biomedical applications. Trends Biotechnol. 2012, 30 (3), 138–146. 10.1016/j.tibtech.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolan M. C.; Fuentes Caparrós A. M.; Dietrich B.; Barrow M.; Cross E. R.; Bleuel M.; King S. M.; Adams D. J. Optimising low molecular weight hydrogels for automated 3D printing. Soft Matter 2017, 13 (45), 8426–8432. 10.1039/C7SM01694H. [DOI] [PubMed] [Google Scholar]
- Panja S.; Adams D. J. Gel to gel transitions by dynamic self-assembly. Chem. Commun. 2019, 55 (68), 10154–10157. 10.1039/C9CC05154F. [DOI] [PubMed] [Google Scholar]
- Hu G.; Pojman J. A.; Scott S. K.; Wrobel M. M.; Taylor A. F. Base-Catalyzed Feedback in the Urea–Urease Reaction. J. Phys. Chem. B 2010, 114 (44), 14059–14063. 10.1021/jp106532d. [DOI] [PubMed] [Google Scholar]
- Bubanja I. N.; Bánsági T.; Taylor A. F. Kinetics of the urea–urease clock reaction with urease immobilized in hydrogel beads. React. Kinet., Mech. Catal. 2018, 123 (1), 177–185. 10.1007/s11144-017-1296-6. [DOI] [Google Scholar]
- Heuser T.; Weyandt E.; Walther A. Biocatalytic Feedback-Driven Temporal Programming of Self-Regulating Peptide Hydrogels. Angew. Chem., Int. Ed. 2015, 54 (45), 13258–13262. 10.1002/anie.201505013. [DOI] [PubMed] [Google Scholar]
- Heuser T.; Steppert A.-K.; Molano Lopez C.; Zhu B.; Walther A. Generic Concept to Program the Time Domain of Self-Assemblies with a Self-Regulation Mechanism. Nano Lett. 2015, 15 (4), 2213–2219. 10.1021/nl5039506. [DOI] [PubMed] [Google Scholar]
- Heinen L.; Walther A. Temporal control of i-motif switch lifetimes for autonomous operation of transient DNA nanostructures. Chem. Sci. 2017, 8 (5), 4100–4107. 10.1039/C7SC00646B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinen L.; Heuser T.; Steinschulte A.; Walther A. Antagonistic Enzymes in a Biocatalytic pH Feedback System Program Autonomous DNA Hydrogel Life Cycles. Nano Lett. 2017, 17 (8), 4989–4995. 10.1021/acs.nanolett.7b02165. [DOI] [PubMed] [Google Scholar]
- Cingil H. E.; Meertens N. C. H.; Voets I. K. Temporally Programmed Disassembly and Reassembly of C3Ms. Small 2018, 14 (46), 1802089. 10.1002/smll.201802089. [DOI] [PubMed] [Google Scholar]
- Panja S.; Patterson C.; Adams D. J. Temporally-Programmed Transient Supramolecular Gels. Macromol. Rapid Commun. 2019, 40 (15), 1900251. 10.1002/marc.201900251. [DOI] [PubMed] [Google Scholar]
- Mears L. L. E.; Draper E. R.; Castilla A. M.; Su H.; Zhuola; Dietrich B.; Nolan M. C.; Smith G. N.; Doutch J.; Rogers S.; Akhtar R.; Cui H.; Adams D. J. Drying Affects the Fiber Network in Low Molecular Weight Hydrogels. Biomacromolecules 2017, 18 (11), 3531–3540. 10.1021/acs.biomac.7b00823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colquhoun C.; Draper E. R.; Schweins R.; Marcello M.; Vadukul D.; Serpell L. C.; Adams D. J. Controlling the network type in self-assembled dipeptide hydrogels. Soft Matter 2017, 13 (9), 1914–1919. 10.1039/C6SM02666D. [DOI] [PubMed] [Google Scholar]
- http://www.sasview.org/ (accessed 2019-12-01).
- Adams D. J.et al. Electrochemically Growing Hydrogels to Control Stiffness. STFC ISIS Neutron and Muon Source, 2019; 10.5286/ISIS.E.RB1920493. [DOI]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.