

Abstract
DM1 is an autosomal dominant multisystemic disease caused by an unstable CTG repeat expansion in the 3’-untranslated region (UTR) of the DMPK gene. The complex variant DMPK expanded the alleles containing CAG, CCG, CTC and/or GGC interruptions repetition sequences have been reported in 3-8% of DM1 patients. To date, very few information is available about the frequency and clinical consequences of pre-mutated DMPK variant allele. In this study, we describe a three-generation Italian family showing the segregation of an interrupted DMPK allele within the premutation range. TP-PCR with primers complementary to CCG repetitions and direct sequencing allow us to identify a hetero-triplet (CTG)6(CCGCTG)15(CTG)5 repeat structure. The haplotype analysis demonstrated that this variant allele is associated with the European founder DM1 haplotype. The pyrosequencing analysis of the CpG islands contained in the flanking regions of the CTG array, did not show the presence of a cis effect of the CCG interruptions on the methylation profile of the DM1 locus. The analysis of both meiotic transmissions, one maternal and one paternal, revealed the intrafamilial stability of the DM1 premutation among relatives. Our findings further support the hypothesis of a stabilizing effect of CCG interruptions on the mutational dynamics of the DM1 locus, also in intermediate DMPK alleles.
Key words: DMPK variant alleles, premutation, TP-PCR analysis, methylation
Introduction
Myotonic dystrophy type 1 (DM1, OMIM #160900) is the most common form of adult muscular dystrophy, with a prevalence of 12.5/100,000 and an autosomal dominant mode of inheritance 1,2.
Patients with DM1 show a progressive multisystemic disease affecting mainly skeletal muscle, heart and the central nervous system 3. DM1 is caused by the expansion of an unstable CTG trinucleotide repeat located in the 3′ untranslated region of the DMPK gene, on chromosome 19q13.3 4,5.The number of CTGs is polymorphic in the general population, with a range of 5 to about 37 repeats, a premutation range from 38 to 49 triplets and increases to 50 and up to many thousand in patients 6. Germline instability is the major factor determining the pronounced anticipation seen in DM1 and depend on the sex of the transmitting parent. Intermediate alleles can be stably inherited for several generations, especially if transmitted by the mother, passages through male germline almost invariably lead to a large increase into the full disease range 7,8. On the contrary, alleles longer than 80 CTGs tend to expand when transmitted through affected mothers and, depending on the mutation size, may lead to the congenital form of the disease. In the last years, variant (CAG)n, (CCG)n, (CTC)n and (CGG)n repeats interspersed within the CTG array have been reported, with an overall frequency of about 3-8% in DM1 patients 9-13. These variant alleles greatly alter the mutational dynamics and the phenotypic manifestation of the disease, leading to important practical consequences on DM1 genetic testing and counseling. Interruptions of the repeated tract have been observed in normal and intermediate alleles of other trinucleotide repeats (TNRs) diseases, such as spinocerebellar ataxia 1 (SCA1) and fragile X syndrome, where one or more interruptions must be lost before expansion can occur. In DM1, the search for variant repeats in a large set of DMPK normal alleles did not reveal any interruptions, which instead have been detected only within the intermediate alleles of four individuals with discordant clinical phenotypes 14-17. The analysis of a larger set of individuals is therefore warranted to assess the frequency and the possible causal or modifying effect on DM1 phenotype of variant DMPK intermediate alleles.
In this work we describe a three-generation Italian family in which a single 41 repeats interrupted allele showing a (CTG)6(CCGCTG)15(CTG)5 configuration segregates. Interestingly, the length and interruption pattern of this allele remained stable through either paternal and maternal transmissions, with no apparent consequences on the phenotype. The haplotype and methylation analysis of the DM1 locus demonstrated its association with the European founder DM1 haplotype and the absence of in cis epigenetic effects on the genomic region surrounding the CTG array.
Materials and methods
Patients collection and DNA extraction
Samples were obtained from an Italian family referred to the Genetic Unit of the Children’s University Hospital “A. Meyer”, Florence for DM1 and PWS genetic testing. Major clinical data available from patients and their pedigree are summarized in Figure 1. Genomic DNA was extracted from peripheral blood leukocytes using EZ1 DNA Blood Kit (Qiagen, Germany) according to standard procedures. Informed consents were obtained from all individuals participating to this study (Ethical Approval register number: CS_02/2019).
Figure 1.
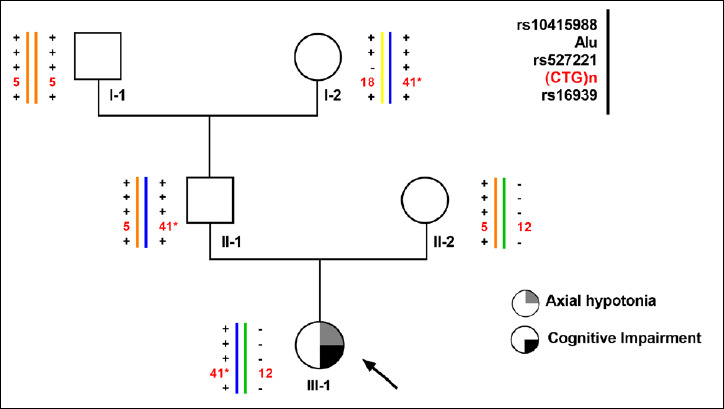
Pedigree and haplotype analysis of the DM1 locus of our three-generation Italian family. The proband is marked with an arrow.* This value corresponds to the apparent number of CTG repetitions.
Short range PCR (SR-PCR), triplet repeated-primed PCR (TP-PCR) and direct sequencing of DMPK alleles
DNA of each patient was analyzed and characterized using SR-PCR and TP-PCR with synthetic fluorescently labeled primers flanking and within the CTG repetitions, as described 16. The interruptions of the CTG array were detected with P4 internal primers, specific for the variant motif (CCGCTG)n, according to published protocols 14. The characterization of the interruption motifs was obtained using Sanger direct sequencing of gel-purified (Gel Extraction kit, Qiagen, Germany) SR-PCR products corresponding to the premutated DMPK alleles. Direct sequencing was performed using the BigDye Terminator Cycle Sequencing Ready Reaction kit and the sequences were analyzed with ABI 3130xl Automated Sequencer (ThermoFisher Scientific, Massachusetts, USA).
Haplotype analysis
The haplotype analysis of the DM1 locus was performed using four biallelic polymorphic markers as previously described 18. The presence of a 1-kb Alu insertion/deletion polymorphism was typed by PCR using a three-primers protocol 19. The following additional 3 single base pair polymorphic markers rs10415988 [Taq1] (T/C) in 15kbCEN, rs527221 [Bpm1] (G/C) in DMPK exon 10 and rs16939 [HinfI] (T/G) in intron 9 were typed by PCR and Sanger direct sequencing.
DMPK methylation analysis
Bisulfite conversion DNA (1 μg in 20 μl) has been obtained using EZ DNA Methylation-Gold Kit (Zymo Research, Irvine, CA, USA) according to the manufacturer’s instructions. The genomic DNA was quantified by the Qubit® 2.0 Fluorometer (ThermoFisher Scientific, Massachusetts, USA).
The methylation study was carried out on DMPK genomic region as previously described 20. A 189 bp fragment (10 CpG sites) in 5’ end region of CTG array was amplified by PCR from bisulfite-treated DNA using CTCF-1F (5’- GGAAGATTGAGTGTTCGGGGTA -3’) and CTCF-1R (5’- Biotinylated -GGGTTTTTGTAGTCGGGAATG -3’) primers. For 3’ end region, a 173 bp fragment (6 CpG sites) was amplified using CTCF-2F (5’- TAAATTGTAGGTTTGGGAAG -3’) and CTCF-2R (5’-biotyinylated- GGGAAATTTGTTTTTGTTAAA -3’) primers. PCR conditions: 95°C for 5 min, followed by 50 cycles of 95°C for 30 sec, 55°C for 30 sec and 72°C for 30 sec with final extension of 5 min at 72°C. The pyrosequencing analysis was performed on a PyroMark Q24 (Qiagen, Germany) with following sequencing primers: CT1-S (5’- GGGTTTTCGTTTAGTTTTAGTTTTG -3’) for 5’ end and CTCF-2F for 3’ end regions. The methylation percentage at each CpG sites was quantified by the PyroMark Q24 software, version 2.0.7 (Qiagen, Germany).
Results
Phenotypes
The proband (Subject III-1) is the 16 year-old-second-born female of a couple of healthy and unrelated parents. The perinatal period was characterized by a marked axial hypotonia, minor hypertonia in the lower limbs, respiratory insufficiency, poor sucking, and frequent apneas. She had facial hypotonus and micro-retro-gnathia. A child-neuropsychiatrist evaluation diagnosed a medium/severe cognitive impairment, with relational and learning problems at 6 years of age. No myotonic phenomenon was elicited in the proband nor in her parents on physical examination. Her father (II-1) is a 54 years old ex-sportsman (still practicing amateur marathon) and has a normal electromyography (EMG). The paternal grandmother (I-2) is 84 years old and reported only a senile cataract with no other signs of DM1. According to the proband’s clinical phenotype, the genetic testing for Prader-Willi syndrome (PWS) – and DM1 as differential diagnosis – was requested for III-1. The PWS/AS-region analysis showed a maternal uniparental heterodisomy (UPD) of chromosome 15 (data not shown), confirming the diagnosis of PWS in the proband.
Detection of DMPK premutated alleles and characterization of variant non CTG interruptions
As SR-PCR analysis of the proband’s DNA showed a 12 and an apparent 41 CTG alleles at the DM1 locus, the DMPK molecular analysis was extended to all the available family members. The results (see Figure 1), indicated that only the proband’s father and the grandmother (II-1 and I-2 respectively) were carriers of the 41 triplets DMPK allele. In order to exclude the presence of a DM1 expansion not detectable with SR-PCR analysis, a bi-directional TP-PCR, with primers P4-CTG (3’- end of CTG array) and P4-CAG (5’- end of the CTG array) was then performed. TP-PCR analysis did not reveal the presence of a pathological CTG expansion of the DMPK gene in subjects I-2, II-1 and III-1. However, the electrophoresis profiles were atypical, characterized by gaps in the continuous 3-base-pairs ladder signal, strongly suggesting an atypical interruption within the CTG repeated array (Figure 2A). A second round of TP-PCR, using P4-CCGCTG primer, confirmed the presence of atypical CCG interruption which remains stable through the meiotic transmissions (Figure 2B). The Sanger sequencing of the SR-PCR products confirmed a (CTG)6(CCGCTG)15(CTG)5 (HGVS nomenclature: NM_001081563.2: c.*224_*283CTG[6]CCGCTG[15]CTG[5]) interruption motif in the proband (III-1), in her father (II-1) and in the grandmother (I-2) (Figure 2C).
Figure 2.
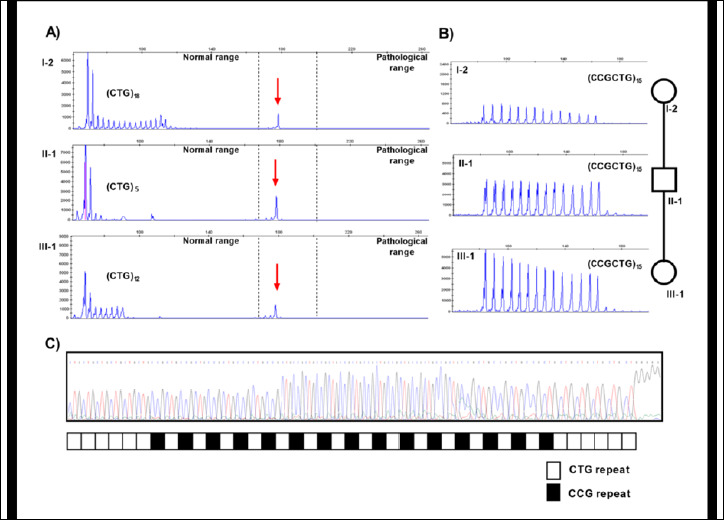
Molecular characterization of the DM1 locus: TP-PCR profiles of DMPK alleles of I-2, II-1 and III-1 with primers complementary to the CTG (A) and CCG (B) repetitions. Red arrows indicate the amplification of the apparent (CTG)41 DMPK allele. C) Sequencing analysis and structure of DMPK premutated interrupted alleles. Each square represents a single CCG repeat.
Haplotypes analysis
To determine the haplotype in linkage disequilibrium with the DM1 interrupted premutated alleles, all family members were analyzed with 4 genetic markers closely flanking the (CTG)n repeat: rs16936 in intron9 [HinfI], rs527221 in exon10 [BpmI], rs10415988 [TaqI] and 1-Kb Alu insertion/deletion. Their polymorphisms were used to construct compound haplotypes as follow: TaqI site present (+)/absent (-); 1-kb Alu insertion (+)/deletion (-); BpmI site present (+)/absent (-) and HinfI site present (+)/absent (-).As a result, three possible haplotype combinations were identified in this family: haplotype (+ + + +) linked to the (CTG)5 and (CTG)6(CCGCTG)15(CTG)5 DMPK alleles, haplotype (- - - -) linked to (CTG)12 allele, and haplotype (+ + - +) linked to the (CTG)18 allele (Fig. 1).
CpG methylation profile
In order to test a possible in cis effects of the CCG interruptions on the DMPK locus already reported in DM1 patients 20, we performed a methylation analysis on two CpG islands flanking the unstable CTG tract. The first CpG island, that contains 10 CpG sites, was localized in the upstream region of the (CTG)n expansion, while the second CpG island was in the downstream region of CTG array including 6 CpG sites (Fig. 3).
Figure 3.
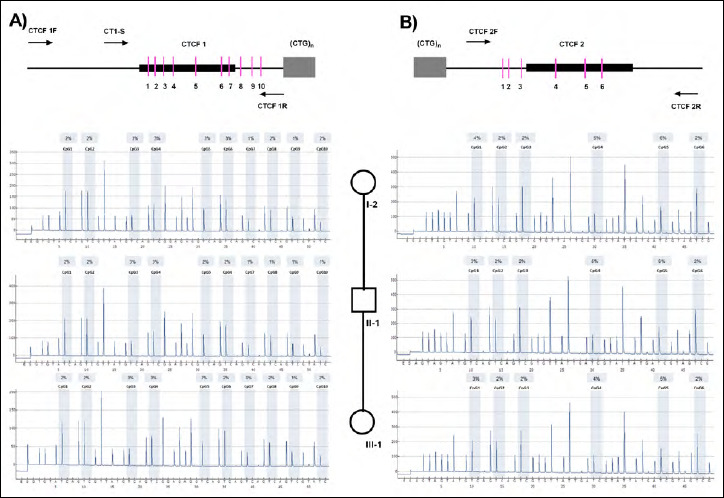
Methylation analysis of regions 5’ and 3’ to the CTG array. Up-panel: genomic structure upstream (A) and downstream (B) of the CTG repeat in the DMPK gene. The CpG islands are represented as bars, CTCF1 binding site as black box, CTG repeat region as gray box and the PCR primers used in this study are indicated as arrows. Down-panel: Pyrosequencing profiles of I-2, II-1 and III-1 samples, respectively 5’ (A) and 3’ (B) regions to the CTG array.
By pyrosequencing analysis, a homogeneous 2-4% hypomethylation level with no significant differences among I-2, II-1 and III-1 samples was found in the upstream region (Fig. 3A). Equally, in the downstream region of the (CTG)n expansion an average methylation level of 3% with no significant differences in I-2, II-1 and III-1 individuals was found (Fig. 3B). These results indicate that the presence of CCG interruptions in the DM1 locus do not influence the methylation levels of the genomic regions flanking the (CTG)n array.
Discussion
The presence of atypical interruptions in the DM1 locus can influence the phenotype in several simple repeat expansion disorders 11. To date, very few patients have been described not carrying CTG interruption in DM1 alleles containing more than 35 repetitions and the effects on the mutational dynamics and phenotypic outcome are still subject of debate. The first DMPK interrupted allele was described by Leeflang et al. 17, in a sperm donor carrying apparent 37 CTG repetitions and a (CTG)4(CCGCTG)16(CTG) hetero-triplet structure. Musova et al. 14, reported three individuals with a similar repeated structure in intermediated DMPK alleles. However the contribution to the phenotype remains unclear because of the simultaneous occurrence of other neuromuscular conditions 14. Our proband has been also referred for DM1 genetic testing because of a neuromuscular phenotype, which was associated to a maternal uniparental heterodisomy of PWS/AS critical region on chromosome 15q, confirming the diagnosis of PWS. The molecular characterization of the DM1 locus revealed the presence of a premutated DMPK allele with apparent 41 CTG repetitions in a heterozygous state. The combined use of SR-PCR and bidirectional TP-PCR allows us to detect CCG interruptions of the CTG tract with a (CTG)6(CCGCTG)15(CTG)5 structure. The extension of the molecular analysis in all the available family members established that the premutated interrupted DMPK allele was paternally inherited and derived from the proband’s grandmother. The interruption we have defined may explain the anomalous meiotic stability of this allele through both maternal and paternal transmission, as demonstrated by direct sequencing of SR-PCR products in II-1 and I-2 DNA samples. The linkage analysis also showed that the (CTG)6(CCGCTG)15(CTG)5 DMPK allele (HGVS nomenclature: NM_001081563.2: c.*224_*283CTG[6]CCGCTG[15]CTG[5]) is associated with the same chromosomal haplotype as pathogenic alleles present in affected DM1 patients (defined A-haplotype) 21. We can speculate that the hexamer at the DM1 locus originated from at least one mutation of the CTG to CCG, combined with subsequent slippage of the hexamer. The novelty of our work report is that no clinical signs of DM1 have been detected in the proband’s father and grandmother, in an apparent normal clinical status despite their advanced age. This allow us to conclude that the DMPK (CTG)6(CCGCTG)15(CTG)5 premutated allele does not have phenotypic consequences in the analyzed individuals. Our family enlarges the set of individuals so far described who carry the variant DMPK premutations and may help to assess the frequency and the possible clinical effects of these very rare alleles.
Figures and tables
References
- 1.Harper P. Myotonic dystrophy, 3rd ed. London: W.B. Saunders; 2001. [Google Scholar]
- 2.Vanacore N, Rastelli E, Antonini G, et al. An age-standardized prevalence estimates and a sex and age distribution of myotonic dystrophy types 1 and 2 in the Rome province, Italy. Neuroepidemiology 2016;46:191-7. https://doi.org/10.1159/000444018 10.1159/000444018 [DOI] [PubMed] [Google Scholar]
- 3.Harper PS. Myotonic dystrophy. Oxford, NY: Oxford University Press; 2009. [Google Scholar]
- 4.Brook JD, McCurrach ME, Harley HG, et al. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeats at the 3’ end of a transcript encoding a protein kinase family member. Cell 1992;69:799-808. https://doi.org/10.1016/0092-8674(92)90418-c 10.1016/0092-8674(92)90418-c [DOI] [PubMed] [Google Scholar]
- 5.Mahadevan M, Tsilfidis C, Sabourin L, et al. Myotonic dystrophy mutation: an unstable CTG repeat in the 3’untraslated region on the gene. Science 1992;225:1253-5. https://doi.org/10.1126/science.1546325 10.1126/science.1546325 [DOI] [PubMed] [Google Scholar]
- 6.Kamsteeg EJ, Kress W, Catalli C, et al. Best practice guidelines and recommendations on the molecular diagnosis of myotonic dystrophy types 1 and 2. Eur J Hum Genet 2012;20:1203-8. https://doi.org/10.1038/ejhg.2012.108 10.1038/ejhg.2012.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barcelo JM, Mahadevan MS, Tsilfidis C, et al. Intergenerational stability of the myotonic dystrophy protomutation. Hum Mol Genet 1993;2:705-9. https://doi.org/10.1093/hmg/2.6.705 10.1093/hmg/2.6.705 [DOI] [PubMed] [Google Scholar]
- 8.Simmons Z, Thornton CA, Seltzer WK, et al. Relative stability of a minimal CTG repeat expansion in large kindred with myotonic dystrophy. Neurology 1998;50:1501-4. https://doi.org/10.1212/wnl.50.5.1501 10.1212/wnl.50.5.1501 [DOI] [PubMed] [Google Scholar]
- 9.Santoro M, Masciullo M, Silvestri G, et al. Myotonic dystrophy type 1: role of CCG, CTC and CGG interruptions within DMPK alleles in the pathogenesis and molecular diagnosis. Clin Genet 2017;92:355-64. https://doi.org/10.1111/cge.1294 10.1111/cge.1294 [DOI] [PubMed] [Google Scholar]
- 10.Pesovic J, Peric S, Brkusanin M, et al. Repeat interruptions modify age at onset in myotonic dystrophy type 1 by stabilizing DMPK expansions in somatic cells. Front Genetics 2018;9:1-14. https://doi.org/10.3389/fgene.2018.00601 10.3389/fgene.2018.00601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cumming SA, Hamilton MJ, Robb Y, et al. De novo repeat interruptions are associated with reduced somatic instability and mild or absent clinical features in myotonic dystrophy type 1. Eur J Hum Genet 2018;26:1635-47. https://doi.org/10.1038/s41431-018-0156-9 10.1038/s41431-018-0156-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tome S, Dandelot E, Dogan C, et al. Unusual association of a unique CAG interruption in 5’ if DM1 CTG repeats with intergenerational contractions and low somatic mosaicism. Hum Mutat 2018;39:970-82. https://doi.org/10.1002/humu.23531 10.1002/humu.23531 [DOI] [PubMed] [Google Scholar]
- 13.Ballester-Lopez A, Koehorst E, Almendrote M, et al. A DM1 family with interruptions associated with atypical symptoms and late onset but not with a milder phenotype. Hum Mutat 2019;14 https://doi.org/10.1002/humu.23932 10.1002/humu.23932 [DOI] [PubMed] [Google Scholar]
- 14.Musova Z, Mazanec R, Krepelova A, et al. Highly unstable sequence interruptions on the CTG repeat in the myotonic dystrophy gene. Am J Med Genet 2009;149A:1365-74. https://doi.org/10.1002/ajmg.a.32987 10.1002/ajmg.a.32987 [DOI] [PubMed] [Google Scholar]
- 15.Santoro M, Masciullo M, Pietrobono R, et al. Molecular, clinical, and muscle studies in myotonic dystrophy type 1 (DM1) associated with novel variant CCG expansion. J Neurol 2013;260:1245-57. https://doi.org/10.1007/s00415-012-6779-9 10.1007/s00415-012-6779-9 [DOI] [PubMed] [Google Scholar]
- 16.Botta A, Rossi G, Marcaurelio M, et al. Identification and characterization of 5’ CCG interruptions in complex DMPK expanded alleles. Eur J Hum Genet 2017;25:257-61. https://doi.org/10.1038/ejhg.2016.148 10.1038/ejhg.2016.148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leeflang E, Arnheim N. A novel repeat structure at the myotonic dystrophy locus in a 37-repeat allele with unexpectedly high stability. Hum Mol Genet 1995;4:135-6. https://doi.org/10.1093/hmg/4.1.135 10.1093/hmg/4.1.135 [DOI] [PubMed] [Google Scholar]
- 18.Kumar A, Agarwal S, Pradhan S. Haplotype analysis and LD detection at DM1 locus. Gene 2015;567:45-50. https://doi.org/10.1016/j.gene.2015.04.069 10.1016/j.gene.2015.04.069 [DOI] [PubMed] [Google Scholar]
- 19.Mahadevan MS, Foitzik MA, Surh LC, et al. Characterization and polymerase chain reaction (PCR) detection of an Alu deletion polymorphism in total linkage disequilibrium with myotonic dystrophy. Genomics 1193;15:446-8. https://doi.org/10.1006/geno.1993.1087 10.1006/geno.1993.1087 [DOI] [PubMed] [Google Scholar]
- 20.Santoro M, Fontana L, Masciullo M, et al. Expansion size and presence of CCG/CTC/CGG sequence interruptions in the expanded CTG array are independently associated to hypermethylation at the DMPK locus in myotonic dystrophy type 1 (DM1). Biochim Biophys 2015;1852:2645-52. https://doi.org/10.1016/j.bbadis.2015.09.007 10.1016/j.bbadis.2015.09.007 [DOI] [PubMed] [Google Scholar]
- 21.Martorell L, Monckton DG, Sanchez A, et al. Frequency and stability of the myotonic dystrophy type 1 premutation. Neurology 2001;56:3 https://doi.org/10.1212/wnl.56.3.328 10.1212/wnl.56.3.328 [DOI] [PubMed] [Google Scholar]