

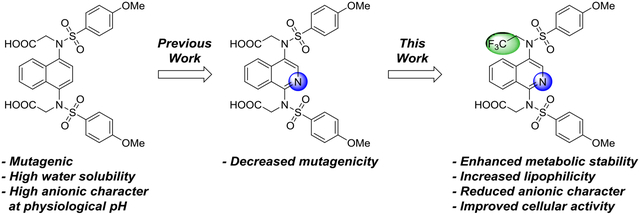
Abstract
Pharmacological activation of NRF2 (Nuclear factor erythroid 2-related factor 2) arises from blocking interaction of NRF2 with its negative regulator, KEAP1 (Kelch-like ECH-associated protein 1). We previously reported an isoquinoline-based NRF2 activator, but this compound showed negative logD7.4 and a −2 charge at physiological pH, which may have limited its membrane permeability. In this work, we report potent, metabolically stable analogs that result from replacing a carboxymethyl group at the 4-position with a fluoroalkyl group.
Keywords: KEAP1, NRF2, protein-protein interaction
Graphical Abstract
INTRODUCTION:
Regulation of the oxidative stress response is crucial for attenuating tissue and DNA damage from reactive molecules, such as reactive oxygen species (ROS), reactive nitrogen species (RNS), and electrophiles. The cellular antioxidant response is primarily governed by the KEAP1-NRF2 protein-protein interaction (PPI).1 KEAP1 (Kelch-like ECH-associated protein 1) negatively regulates NRF2 (Nuclear factor (erythroid-derived 2)-like 2) in the absence of oxidative or electrophilic stressors (Figure 1).2 In the presence of ROS, RNS, and electrophilic species, one or more of 27 cysteine residues on KEAP1 become covalently modified, inducing a conformation which no longer allows for NRF2 to be sequestered.2–3 This dissociation increases cellular levels of NRF2, which translocates to the nucleus and promotes the transcription of genes coding for antioxidant enzymes such as NADPH quinone oxidoreductase (NQO-1), heme oxygenase (HO-1), and glutathione-S-transferase (GST).1, 4–5
Figure 1.
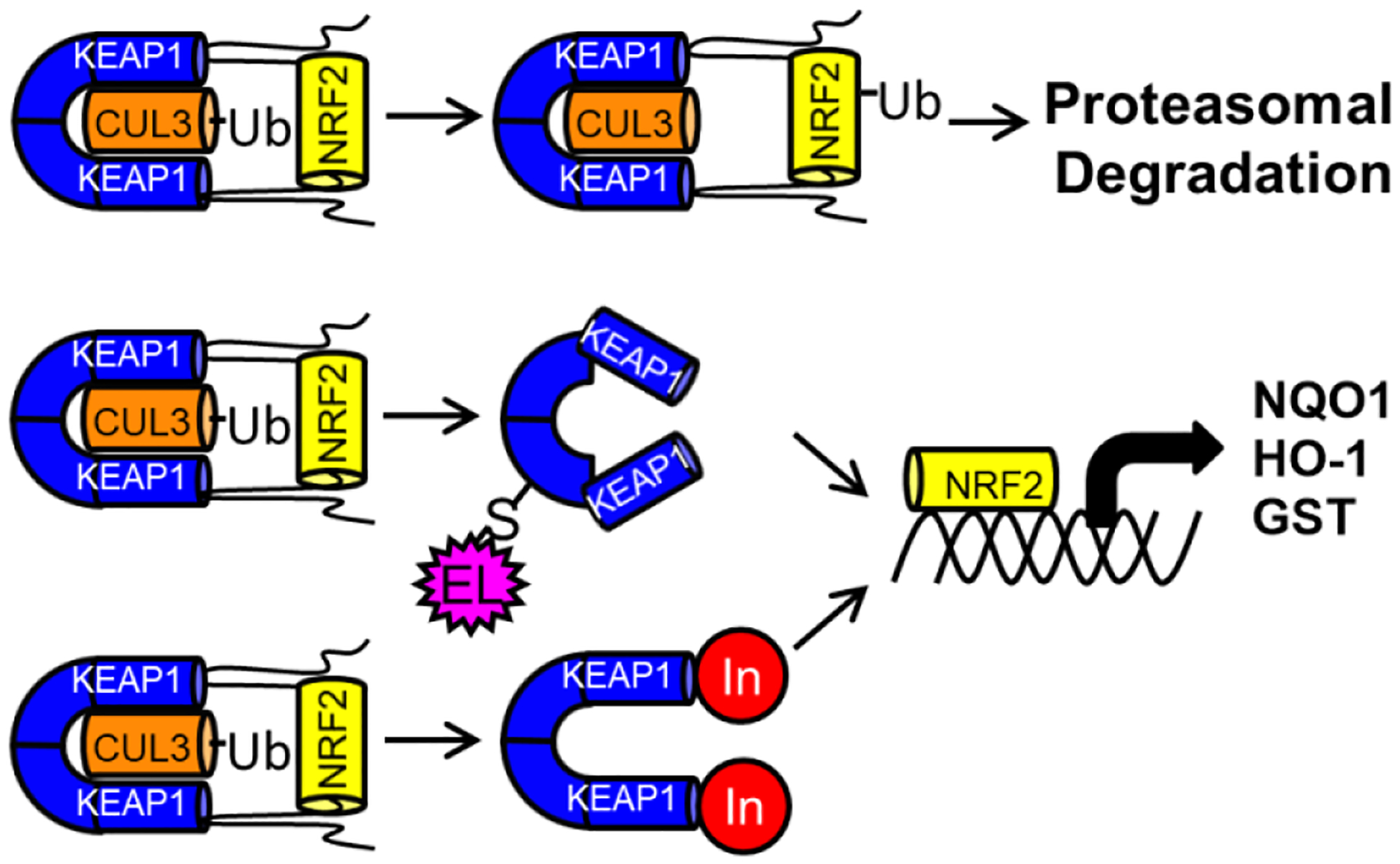
Under basal conditions, NRF2 is bound to KEAP1, ubiquitinated by CUL3, and degraded by the proteasome (top). In the presence of an electrophile (middle) or a non-covalent inhibitor (bottom), NRF2 translocates to the nucleus and transcribes cytoprotective genes, like NADPH quinone oxidoreductase 1 (NQO1), heme oxygenase-1 (HO-1), and glutathione S-transferase (GST).
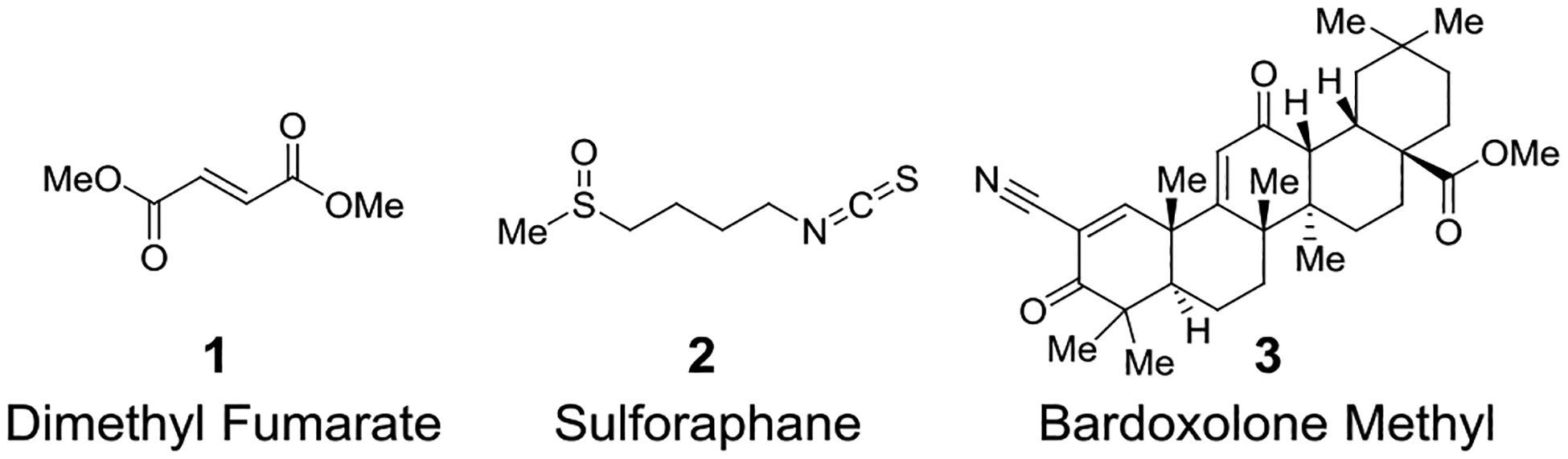
Misregulation of the KEAP1-NRF2 axis has been implicated in several disease states,1, 6 including rheumatoid arthritis7, chronic kidney disease8, multiple sclerosis9, and diabetic wound healing.10–11 Several therapies, such as dimethyl fumarate (1), sulforaphane (2), and bardoxolone methyl (3), that pharmacologically activate NRF2 in patients with chronic inflammatory diseases rely on electrophilic activation of NRF2 through covalent modification of KEAP1 (Chart 1).12 Electrophilic therapies may have non-trivial off-target effects due to their reactive nature.13 Furthermore, the lack of specificity in a pharmacologically active molecule may complicate the understanding of the compound’s mechanism of action.14 To more thoroughly understand the role NRF2 plays in chronic inflammatory diseases, selective and non-electrophilic inhibitors are being developed and investigated.
Chart 1.
There are several known non-electrophilic inhibitors which block the binding of NRF2 to KEAP1 (Chart 2), several of which have been validated recently.18 Our lab has been interested in the aryl bicyclic sulfonamide-based inhibitors (4a, 4b and 5a).19–20 Other non-electrophilic NRF2 activators have been reported by groups at Sanofi (6)21 and GlaxoSmithKline/Astex (7)22–23 and are shown in Chart 2. For further examples of non-electrophilic NRF2 activators, see recent reviews.6, 24–27
Chart 2.
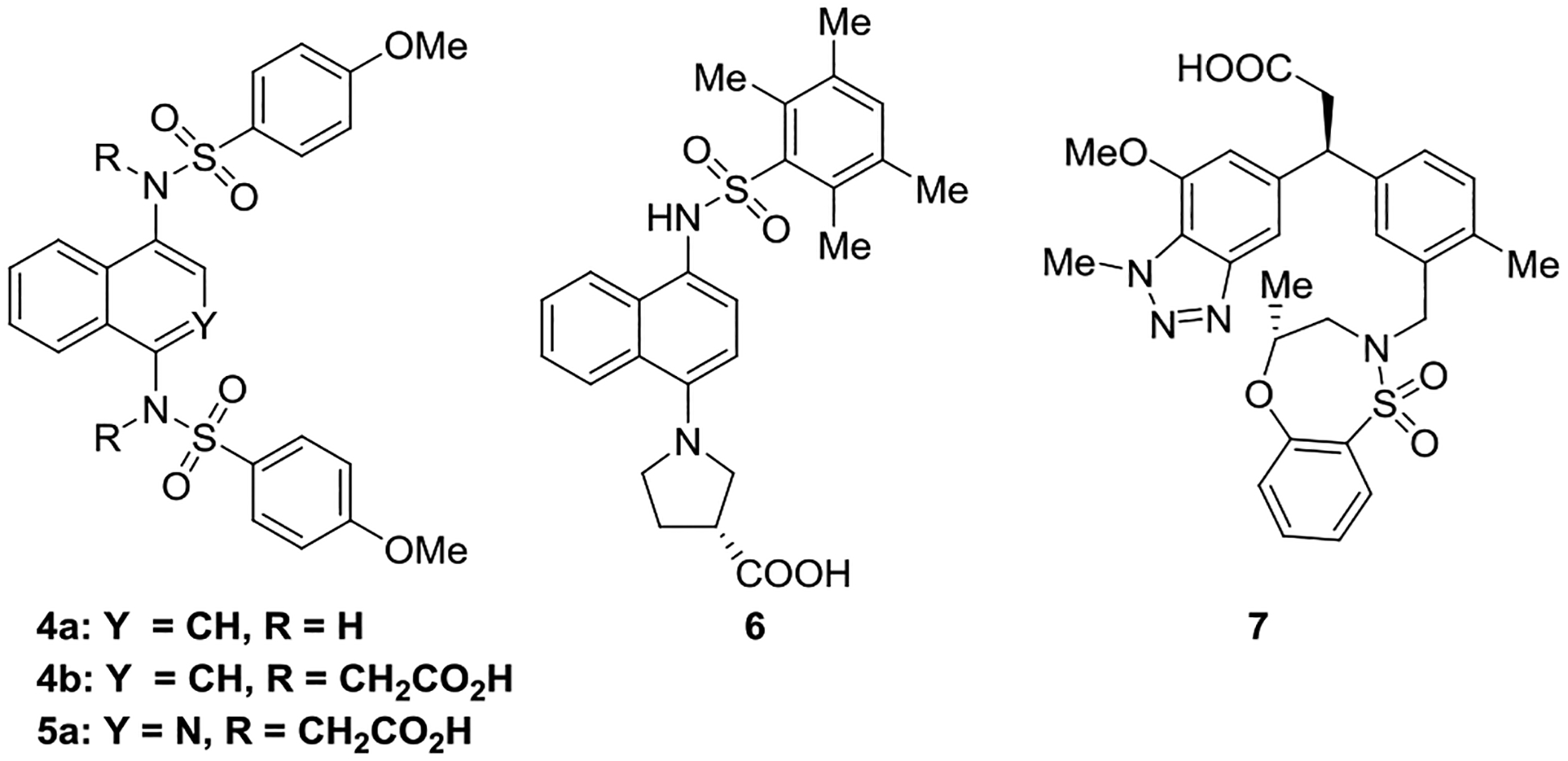
The premise of developing non-electrophilic NRF2 activators is based upon avoiding covalent reaction with cysteine nucleophiles from KEAP1, so it is important to also avoid activation to reactive metabolites. The 1,4-diaminonaphthalene scaffold, exemplified by unsubstituted 4a, may be oxidatively metabolized to quinone-type reactive metabolites. For this reason, we have invested substantial efforts in developing analogs of 1,4-bis-sulfonamide 4a and its bis-acid analog 4b that have electron-poor heterocyclic cores, but that afford a similar display of substituents. Recently, we disclosed an isoquinoline-based inhibitor 5a that possessed an improved safety profile over the previously known naphthalene-based inhibitors 4;28 however, isoquinoline 5a was rather hydrophilic, and, at physiological pH, possessed two negatively charged carboxylate groups, which may limit its membrane permeability. The work in this report describes modifications to isoquinoline 5a to impart more desirable physiochemical properties while maintaining potency and metabolic stability.
RESULTS:
Our investigation began with a brief survey of sulfonamides to determine whether 4-methoxybenzenesulfonamide was a suitable substitution for further modification. Symmetrically substituted isoquinoline bis-sulfonamides 5b and 5c (Table 1) were obtained through our previously reported procedure starting with 1-hydroxyisoquinoline.28 Synthesis of unsymmetrically substituted isoquinolines 5d and 5e called for the development of a new synthetic procedure in which 4-aminoisoquinoline (8) was sulfonylated, Boc-protected, and oxidized with m-chloroperoxybenzoic acid (mCPBA) to the isoquinoline-N-oxides 9a and 9b. The N-oxides underwent a hypervalent iodine-mediated formal [3 + 3] addition with the appropriate sulfonamide to provide unsymmetrically substituted isoquinoline bis-sulfonamides 10d and 10e (Scheme 1).29 From these intermediates, 5d and 5e were easily accessed, and these compounds were analyzed in a fluorescence anisotropy (FA) assay.30 Compound 5a maintained the highest binding affinity between the series of analogs (Table 1) and was selected for further optimization.
Table 1.
IC50, logD7.4, and Metabolic t1/2 of Isoquinoline NRF2 Activators
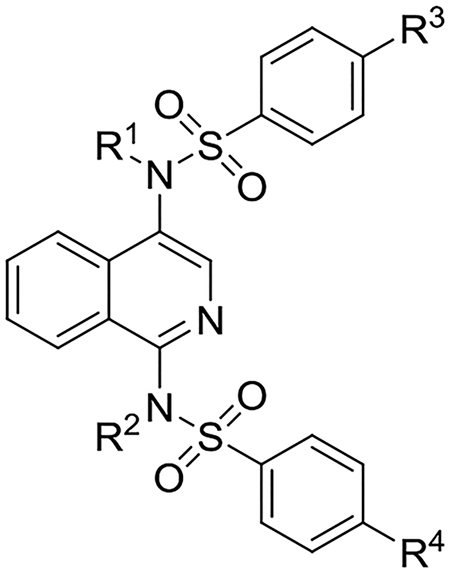 |
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Compound | R1 | R2 | R3 | R4 | IC50 (nM) (95% CI)a | logD7.4b | t½ (min)c |
| 1 | 5a | CH2COOH | CH2COOH | OMe | OMe | 63d (54–82) | −1.5 | 104 |
| 2 | 5b | CH2COOH | CH2COOH | F | F | 430 (233–566) | n.d. | n.d |
| 3 | 5c | CH2COOH | CH2COOH | OH | OH | 350 (291–380) | n.d. | 3 |
| 4 | 5d | CH2COOH | CH2COOH | OMe | F | 280 (251–352) | −1.9 | n.d |
| 5 | 5e | CH2COOH | CH2COOH | F | OMe | 240 (204–267) | −1.8 | n.d |
| 6 | 11a | CH2COOH | H | OMe | OMe | 1100 (807–1343) | −0.5 | n.d. |
| 7 | 11b | H | CH2COOH | OMe | OMe | 230 (210–280) | −1.6 | 37 |
| 8 | 11c | H | CH(CH3)COOH | OMe | OMe | 2300 (16002500) | −1.1 | n.d. |
| 9 | 12 | CH3 | CH2COOH | OMe | OMe | 1000 (770–1300) | 0.0 | 6 |
| 10 | 13a | CH2CF3 | CH2COOH | OMe | OMe | 73 (66–91) | 0.5 | 136 |
| 11 | 13b | CH2CF3 | CH2COOH | OMe | F | 130 (110–170) | 0.6 | >180 |
| 12 | 13c | CH2CF3 | CH2COOH | F | OMe | 120 (100–160) | 0.5 | >180 |
| 13 | 13d | CH2CF3 | CH2COOH | F | F | 420 (250–490) | 0.6 | >180 |
| 14 | 14 | CH2CHF2 | CH2COOH | OMe | OMe | 55 (50–70) | 0.2 | 97 |
| 15 | 15 | CH2CH2F | CH2COOH | OMe | OMe | 210 (190–240) | -0.1 | 58 |
IC50 values, shown in nM units with 95% confidence intervals, were determined by fluorescence anisotropy;
LogD values at pH 7.4 represent the log of the partition coefficient between n-octanol and pH 7.4 buffer and were determined by HPLC;
Half-lives (t1/2) in human liver microsomes in the presence of NADPH are given in minutes and were determined by LC-MS/MS.
IC50 value reported in Richardson et al.28 n.d.: not determined.
Scheme 1.
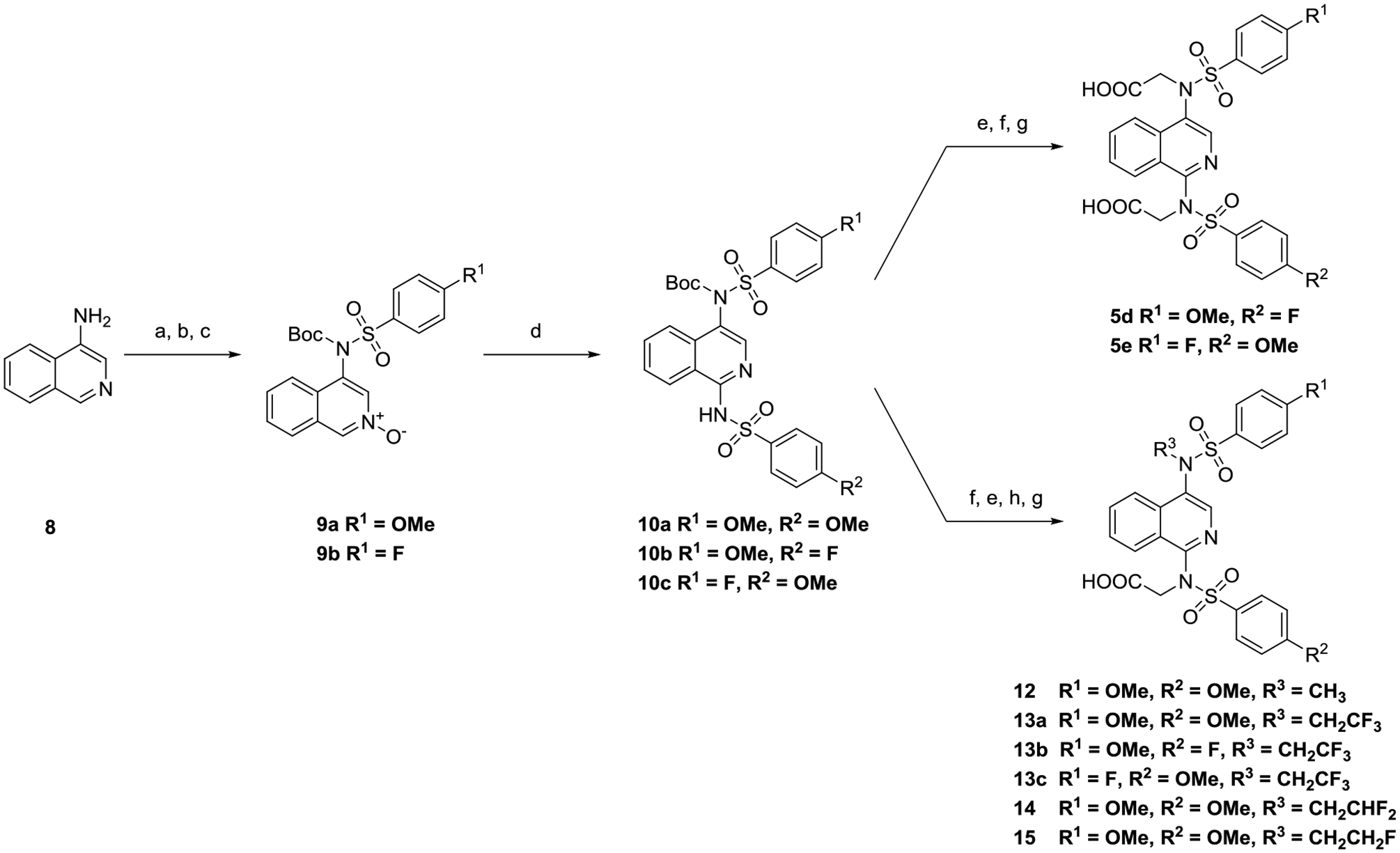
Synthesis of 1,4-Substituted Isoquinolinesa
aReagents: (a) 4-methoxybenzenesulfonyl chloride, pyridine, rt, 1 h; (b) Boc2O, N,N-dimethylaminopyridine, MeCN, rt, 2 h; (c) mCPBA, CHCl3, rt, 24 h; (d) 4-methoxybenzenesulfonamide or 4-fluorobenzenesulfonamide, PPh3, PhI(OAc)2, MeCN, 80°C, 1h; (e) CF3CO2H, CH2Cl2, 0 °C - rt, 2 h; (f) BrCH2CO2Et, K2CO3, Me2NCHO, rt, 48 h; (g) 15% NaOH, MeOH, rt, 4 h. (h) alkyl-X or alkyl-OTf, K2CO3, Me2NCHO or MeCN, rt, 18 – 48h
Next we set out to address the −2 charge of these bis-acid inhibitors by synthesizing mono-acidic analogs of bis-acid 5a. Small molecules possessing significant anionic character at physiological pH typically require active transport to cross cellular membranes.31 Previously, we described that mono-acid analogs of naphthalene 4 maintained good potency in vitro. Due to the reduction in symmetry in the isoquinoline core, both constitutional isomers—one where the acid is substituted off the 4-position sulfonamide (11a) and the other where the acid is substituted off the 1-position sulfonamide (11b)—were needed to determine whether there was a preference for the placement of the acidic moiety relative to the isoquinoline ring nitrogen. Modifying our syntheses in Scheme 1 allowed us to easily access 11a and 11b (see SI). Comparison of these two compounds in a FA assay indicated that 11b (IC50 = 230 nM) was a more potent inhibitor of KEAP1 than 11a (IC50 = 1.1 μM). While these compounds now only had one negatively charged group, they were still rather hydrophilic, possessing logD7.4 values of −1.6 and −0.5 respectively.
With mono-acid 11b suggesting the 1-position as the proper site for the carboxylate group, we turned our attention to increasing the lipophilicity of these compounds. Lipophilicity is a major factor in determining the membrane permeability of small molecules.32 Compounds with logP values below 1 may be resistant to crossing the amphipathic cell membrane through passive transport due to their significant water solubility.33–34 Due to the acidic carboxylate group that would be ionized at physiological pH, we found it fitting to measure the partition coefficient as logD7.4.35 We hypothesized that the addition of more aliphatic character (propionate 11c) or alkylation of the free sulfonamide (N-methyl sulfonamide 12) would likely increase the logD7.4. We saw a five-fold loss of inhibitory activity with each of these modifications (Table 1 entries 8 & 9), even though methylated sulfonamide 12 showed an increase in the logD7.4 to 0.0. Furthermore, it appeared that compounds of structure 12 were less metabolically stable than their bis-acid analogs.
To address the issues of potency, lipophilicity, and metabolic stability, we sought to replace the carboxylate in 5a with an electron-withdrawing yet lipophilic moiety. We installed small variably fluorinated alkyl chains, such as 2,2,2-trifluoroethyl 13a, 2,2-difluoroethyl 14, and 2-fluoroethyl 15. While all of these fluoroalkyl compounds showed nanomolar potency in the FA assay, 13a showed the greatest lipophilicity with a logD7.4 of 0.5 (Table 1, Entry 6). Gratifyingly, 13a also showed a significant improvement in metabolic stability compared to 11b. In an attempt to further increase the lipophilicity and metabolic stability of these inhibitors, we replaced one or both of the p-methoxybenzenesulfonamides with p-fluorobenzenesulfonamides, yielding compounds 13b, 13c, and 13d (entries 11–13, Table 1). These compounds were found to be much more metabolically stable than 13a, each possessing a t½ > 180 min; however, there were negligible gains in lipophilicity and a slight decrease in binding affinity associated with these modifications.
We obtained a crystal structure of 13a bound to the Kelch domain of KEAP1 (Figure 2). We previously reported20 a docking experiment with monoacidic 1,4-diamoninaphthalene-based inhibitors, in which we hypothesized that the carboxylate would bind near R483 of the Kelch domain pocket, rather than near R380. Interestingly, we observe in the crystal structure that the carboxylate of 13a interacts with R380, N414, and R415. The sulfonamide oxygens engage S508, S555, and S602 through hydrogen bonding. The distances from the pendant phenyl rings to Y334 and Y525 are 4.99 and 5.66 Å, respectively. These distances are outside the range typically seen for π-stacking interactions.
Figure 2.
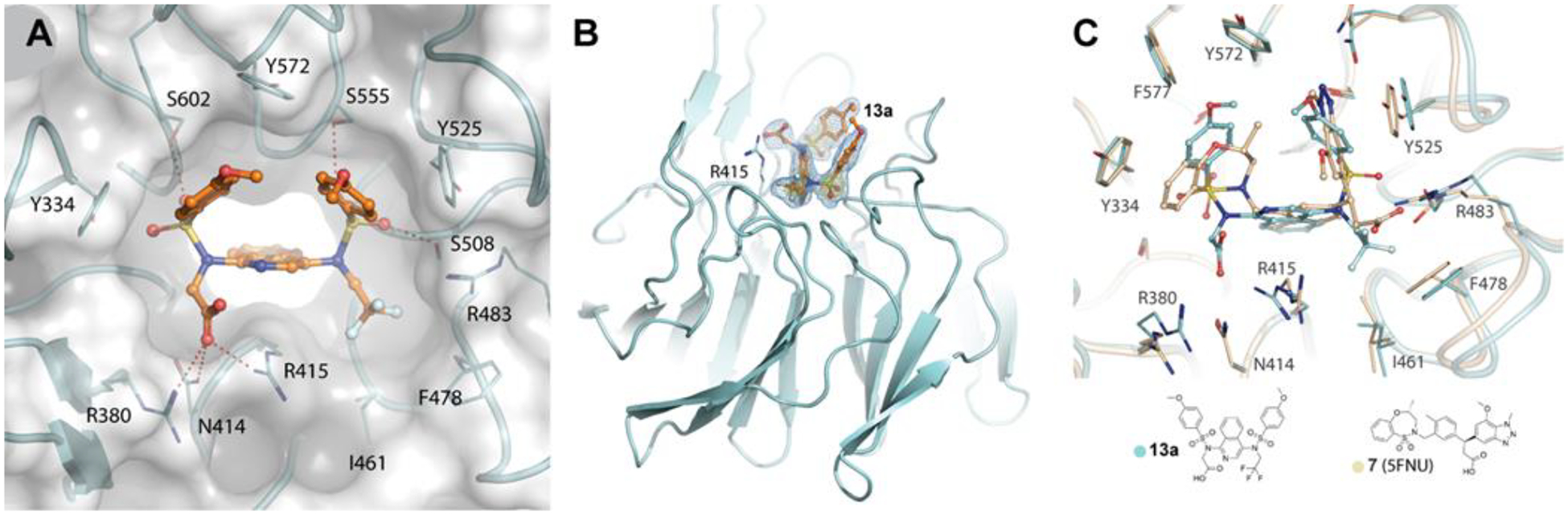
X-ray structure (PDB: 6UF0) of compound 13a bound to human Keap1 Kelch domain. (A, B) Interactions of 13a (orange) with the NRF-2 binding site of Keap1 (cyan ribbon). Polar interactions within 3 Å are denoted by dashed lines in (A). An Fo-Fc electron density omit map, with 13a removed from map calculations, is shown as blue mesh and contoured at 2.7s in (B). (C) Superposition of Keap1 structures bound to 13a (cyan) and compound 7 from pdb 5FNU (wheat).
Lastly, we determined the ability of these compounds to activate NRF2 in cultured keratinocytes. An inhibitor of the KEAP1/NRF2 interaction should lead to increased NRF2 protein levels, and this has been demonstrated by us and others.21–22, 28 To assess this, we measured the protein levels of NRF2 by Western blot in keratinocytes treated with compound or vehicle control (Figure 3). While 5a, 11b , and 13a have similar IC50 values in the fluorescence anisotropy assay, there was a notable increase in cellular levels of NRF2, which correlated with the decrease in negative charge from −2 to −1 and an increase in logD7.4 of −1.5 to 0.5 (Table 2). NQO1 expression was not substantially different among compounds 5a, 11b, and 13a; however, we observed a dose-related increase in NQO1 levels when HaCaT cells were treated with increasing concentrations of 13a (Figure 3B/Table 3). These data support the hypothesis that the modifications made to 5a, resulting in compound 13a, enhanced cell membrane permeability and cellular activity.
Figure 3.
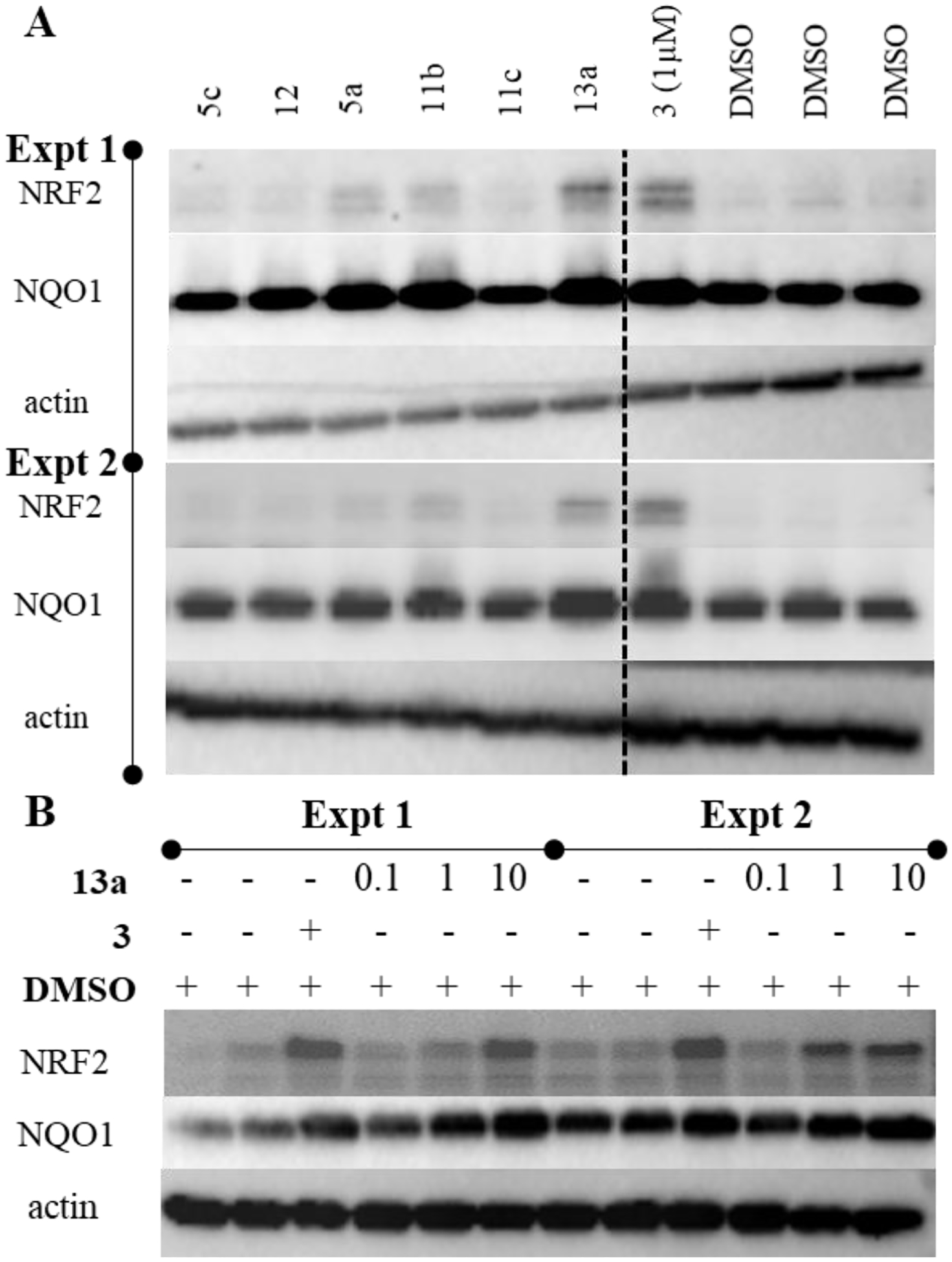
A) HaCaT cells were treated with vehicle control DMSO, 10 μM compound (5e, 12, 5a, 11b, 11c, 13a), or positive control 3 (1 μM) for 16 h. Cell lysates were prepared, equal amounts of protein was probed with antibodies specific for NRF2 or NQO1. Actin was used as a reference. Image was cropped at the dotted line to remove an unrelated compound. See the supplemental information for the uncropped images. NRF2 and NQO1 band intensities were quantified using NIH ImageJ software, and were normalized to that of respective actin band intensity. The normalized values of NRF2 and NQO1 of DMSO-treated samples was taken as one unit. The fold increase in NRF2 and NQO1 expression of two independent sets is presented relative to DMSO controls independently or combined. B) HaCaT cells were treated with DMSO, 0.1, 1, or 10 μM 13a, or 250 nM 3.
Table 2.
Quantification of Western Blot Data in Figure 3A.
| DMSO | 5c | 12 | 5a | 11b | 11c | 13a | 3 (1 μM) | |
|---|---|---|---|---|---|---|---|---|
| Fold Change | 1 | 1.60 | 1.46 | 2.88 | 3.77 | 1.5 | 7.30 | 8.93 |
| NRF2 | ||||||||
| Fold Change | 1 | 1.06 | 1.27 | 1.62 | 1.61 | 1.18 | 1.60 | 1.31 |
| NQO1 |
Data represent the average fold change, relative to DMSO control, of two biological replicates for immunoblots for NRF2 and NQO1 (see Figure 3A). Bardoxolone methyl (3) was used as positive control. β-actin was used as loading control.
Table 3.
Quantification of Western Blot Data in Figure 3B.
| DMSO | 3 (0.25 μM) | 13a (0.1 μM) | 13a (1.0 μM) | 13a (10 μM) | |
|---|---|---|---|---|---|
| Fold Change | 1 | 3.34 | 1.19 | 2.02 | 3.07 |
| NRF2 | |||||
| Fold Change | 1 | 1.43 | 1.15 | 1.36 | 1.59 |
| NQO1 |
Data represent the average fold change, relative to DMSO control, of two biological replicates for immunoblots for NRF2 and NQO1 (see Figure 3A). HaCaT cells were treated with 0.1, 1.0, or 10 μM 13a for 14 hours (see Figure 3B). Bardoxolone methyl (3) was used as positive control. β-actin was used as loading control.
DISCUSSION AND CONCLUSIONS:
In this report, we have described monoacidic inhibitors of the KEAP1/NRF2 interaction. The molecules are potent and have high metabolic stability, which is an important consideration because of the mechanistically undesirable NRF2 activation that may arise by reaction of reactive metabolites with sensor cysteines on KEAP1.
You and coworkers first described diacidic naphthalene 4b, which showed good in vitro activity.36 Diacid 4b was designed so that the carboxylic acids made contacts with two subpockets present where NRF2’s ETGE motif binds to KEAP1. We previously developed diacid isoquinoline 5a, but we wanted to reduce the number of negative charges, as a way to increase cellular potency of NRF2 activators. Our studies reveal that monoacidic NRF2 activators based on the isoquinoline scaffold can be developed. There is a slight preference for the negatively charged group to be placed at the 1-position of the isoquinoline, rather than the 4-position. Our studies also demonstrate that a fluoroalkyl group can be placed at the 4-position to increase lipophilicity, enhance cellular potency, and reduce metabolic liabilities.
The trends seen with the fluoroalkyl analogs in the metabolic studies follow a logical progression and can be readily explained. First, stability in human liver microsomes increases as more electronegative atoms are installed at the 4-position (i.e., CH2CF3 > CH2CHF2 ~ CH2CO2H > CH2CH2F > H > CH3). Additionally, the CH2CF3 group also shows increased lipophilicity, which may account for increased cell permeability. Second, converting either or both of the para-methoxy groups to para-fluoro groups shuts down metabolism altogether, implying O-dealkylation as a contributor to the metabolic profile. The p-methoxy groups may provide an attractive position for installing a metabolic soft spot if it is necessary to redirect metabolism away from a reactive metabolite. Third, converting the para-methoxy groups to para-hydroxy groups provides an opportunity for conjugative metabolism and causes rapid loss of the parent.
The crystal structure of isoquinoline 13a adds much to our understanding of the binding surface of the Kelch domain. It is instructive to first consider the binding of GSK compound 7 to the Kelch domain. In addition to interactions with S555, Q530, and Y525, GSK compound 7 engages R483 and S508 though the carboxylate moiety (Figure 2c). These same interactions are not apparent in the structure of 13a. Rather, the carboxylate binds on the opposite side of the binding pocket, relative to the carboxylate of compound 7. The carboxylate of isoquinoline 13a forms hydrogen bonds with the sidechains of R415, R380, and N414. To accommodate these new interactions, two amino acids move: R415 and R380 shift substantially between the structures of 7 and 13a. Another observation from the structure is that the trifluoroethyl group of 13a occupies a hydrophobic pocket made up of residues I461, F478, and G462. This hydrophobic pocket might dictate the orientation of isoquinoline 13a in the binding pocket. Overall, the structure of 13a suggests there is significant plasticity in the Kelch domain binding site which can give rise to different small molecule-binding orientations.
In conclusion, we have addressed molecular properties that support the ability of 2,2,2-trifluoroethyl monoacid isoquinoline 13a to cross cellular membranes while maintaining potency and metabolic stability. Specifically, we decreased the negative charge of bis-acid isoquinoline 5a at physiological pH and increased the overall lipophilicity through the incorporation of a 2,2,2-trifluoroethyl group as a replacement for a carboxymethyl group. This modification led to the synthesis of a collection of molecules which will be used in lead optimization for the treatment of diabetic wounds and other chronic inflammatory diseases.
EXPERIMENTAL SECTION:
General Considerations.
All starting materials and solvents were purchased from Sigma-Aldrich, Acros Organics, Fischer Scientific, ArkPharm, TCI America, or Matrix Scientific and used without further purification. Reactions were run without taking precautions to exclude air or moisture, unless otherwise noted. Compound 5a was synthesized as previously reported.28 4-methoxybenzenesulfonamide and 4-fluorobenzenesulfonamide were synthesized from the corresponding sulfonyl chlorides according to reported procedures.37–38 Compound identities were confirmed by 1H and 13C nuclear magnetic resonance (NMR) spectroscopy and high resolution mass spectrometry (HRMS). 1H and 13C NMR spectra were recorded on a Bruker 400 MHz or Bruker 900 MHz spectrometer using the corresponding residual solvent peak (CDCl3, 1H δ = 7.26 and 13C δ = 77.2; CD3COCD3, 1H δ = 2.05 and 13C δ = 29.2; CD3OD, 1H δ = 3.31 and 13C = 49.2; CD3CN, 1H δ = 1.96 and 13C δ = 118.3; DMSO-d6, 1H δ = 2.50 and 13C δ = 39.5)) as an internal standard. HRMS spectra were recorded on a Shimadzu LCMS-IT-TOF, and the molecular weight of the compounds was within 0.05% of calculated values. Purity of each of the final, tested compounds was determined by HPLC on a Shimadzu LC-20AB (Solvent system: gradient from 30% MeCN/70% H2O to 95% MeCN/5% H2O (+0.1% formic acid) over 13 min; Column: Shimadzu C18, 50 μm, 50 × 4.6 mm). Purity of each compound was ≥95% (UV, 254 nm) unless otherwise noted. Flash chromatography was performed using silica gel (230–400 mesh). Reactions were monitored by thin-layer chromatography (TLC) on silica gel GHLF plates (250 μm, Macherey-Nagel, Inc., Bethlehem, PA).
2,2’-(Isoquinoline-1,4-diylbis(((4-fluorophenyl)sulfonyl)azanediyl))diacetic acid (5b).
Diethyl 2,2’-(isoquinoline-1,4-diylbis(((4-fluorophenyl)sulfonyl)azanediyl))diacetate (16) (0.072 g, 0.11 mmol, 1 eq) was added to a flask and dissolved in THF (0.5 mL). 15% NaOH in H2O (wt/wt) (0.05 mL) was added, and the reaction mixture was stirred at room temperature for 18 h. Upon completion, the mixture was diluted with H2O (10 mL), and the pH was adjusted to 4 using HCl (2N). The aqueous mixture was extracted with EtOAc (3 × 10 mL), and the organic extract was washed with brine (10 mL), dried (Na2SO4), and evaporated. The crude product was purified using a C18 semi-preparative HPLC column (CH3CN:H2O with 0.1% formic acid; 0 – 17.5 min, 50% CH3CN; 17.5 – 19.5 min, 50 – 95% CH3CN; 19.5 – 26.5 min, 95% CH3CN; 26.5 – 29 min, 95 – 50% CH3CN; 29 – 36 min, 50% CH3CN). The combined fractions were concentrated to remove CH3CN and frozen. The product was then subjected to lyophilization to yield 0.016 g (25% yield) of 5b as a white solid. 1H NMR (400 MHz, CD3CN) δ 8.71 (d, J = 8.4 Hz, 1H), 8.18 (d, J = 8.4 Hz, 1H), 7.95 – 7.73 (m, 5H), 7.67 (dd, J = 8.8, 5.1 Hz, 2H), 7.30 (q, J = 8.6 Hz, 4H), 4.70–4.43 (m, 4H); 13C NMR (101 MHz, CD3CN) δ 169.2, 168.4, 165.6 (d, J = 253.8 Hz, 1 C), 165.6 (d, J = 253.5 Hz), 152.4, 142.0, 137.1, 134.4, 132.7, 132.4, 131.7, 131.4 (d, J = 9.7 Hz), 130.9 (d, J = 9.5 Hz), 128.6, 128.1, 127.8, 123.0, 116.4 (d, J = 22.7 Hz), 116.3 (d, J = 23.3 Hz), 52.6, 51.2 HRMS-ESI (+) (m/z): [M + H]+ calcd for C25H19F2N3O8S2, 592.060; found, 592.0659.
2,2’-(Isoquinoline-1,4-diylbis(((4-hydroxyphenyl)sulfonyl)azanediyl))diacetic acid (5c).
A solution of diethyl 2,2’-(isoquinoline-1,4-diylbis(((4-methoxyphenyl)sulfonyl)azanediyl))diacetate28 (0.100 g, 0.148 mmol, 1 eq) in CH2Cl2 (0.5 mL) was stirred at −78 °C for 0.5 h. 1 M BBr3 in CH2Cl2 (1.0 mL) was added dropwise to the flask. The reaction mixture was allowed to warm to room temperature and was stirred for 24 h. EtOH (5 mL) was added to quench the reaction, and the reaction mixture was diluted with EtOAc (10 mL) and washed with H2O (20 mL) and brine (20 mL). The crude mixture was purified by semi-preparative HPLC column: 0 – 35 min, 30% CH3CN; 35 – 36 min, 30 – 95% CH3CN; 36 – 43 min, 95% CH3CN; 43 – 45 min, 95 – 30% CH3CN; 45 – 50 min, 30% CH3CN). The combined fractions were concentrated to remove CH3CN and frozen. The product was then subjected to lyophilization to yield 0.030 g (24% yield) of (5c) as a white powder. 1H NMR (400 MHz, CD3OD) δ 8.78 (d, J = 8.7 Hz, 1 H), 8.11 (d, J = 8.7 Hz, 1 H), 8.00 – 7.92 (m, 1 H), 7.83 – 7.72 (m, 1 H), 7.73 – 7.67 (m, 1 H), 7.61 – 7.51 (m, 2 H), 7.45 – 7.34 (m, 2 H), 6.99 – 6.85 (m, 4 H), 4.63 – 4.50 (m, 1 H), 4.49 – 4.40 (m, 2 H), 4.38 – 4.28 (m, 1 H) 13C NMR (101 MHz, DMSO-d6 ) δ 170.2, 169.5, 162.5, 162.4, 152.6, 141.4, 137.3, 132.6, 131.2, 131.0, 130.5, 128.7, 128.2, 127.7, 126.0, 123.8, 116.0, 116.0, 114.3 HRMS-ESI (+) (m/z): [M + H]+ calcd for C25H21N3O10S2, 588.0747; found, 588.0742.
N-(1-((N-(Carboxymethyl)-4-fluorophenyl)sulfonamido)isoquinolin-4-yl)-N-((4-methoxyphenyl)sulfonyl)glycine (5d).
In a 20 mL screw-cap vial, ethyl N-(1-((N-(2-ethoxy-2-oxoethyl)-4-fluorophenyl)sulfonamido)isoquinolin-4-yl)-N-((4-methoxyphenyl)sulfonyl)glycinate (17) (0.025 mg, 0.037 mmol) was dissolved in tetrahydrofuran (1.0 mL), and 15% NaOH in H2O (wt/wt) (0.20 mL) was added. The reaction was stirred at room temperature for 2 hours. Upon completion, as judged by TLC, the reaction was diluted with H2O (10 mL). The solution was adjusted to pH 4 with 2N HCl, and a white precipitate formed. The suspension was extracted with EtOAc (3 × 10 mL). The combined organic extracts were washed with H2O (2 × 15 mL) and brine (2 × 15 mL), dried over Na2SO4, and concentrated to yield an off-white solid. The crude product was purified by semi-preparative HPLC (C18; CH3CN/H2O with 1% formic acid: 0 – 18.5 min, 50% CH3CN; 18.5 – 20.5 min, 50 – 95% CH3CN; 20.5 – 27.5 min, 95% CH3CN; 27.5 – 30 min, 95 – 50% CH3CN; 30 – 37 min, 50% CH3CN). The combined fractions were concentrated to remove CH3CN and frozen. The product was then subjected to lyophilization to yield 12 mg (54% yield) of 5d as an off-white solid. 1H NMR (400 MHz, CD3CN) δ 8.75 – 8.64 (m, 1H), 8.30 – 8.18 (m, 1H), 7.90 – 7.83 (m, 1H), 7.79 (s, 2H), 7.72 – 7.59 (m, 4H), 7.31 (s, 2H), 7.05 (d, J = 9.0 Hz, 2H), 4.48 (s, 4H), 3.90 (s, 3H); 13C NMR (101 MHz, CD3CN) δ 169.3, 168.5, 165.6 (d, J = 253.4 Hz), 163.7, 152.2, 141.8, 137.3, 132.8, 132.7 (d, J = 2.7 Hz), 131.5, 131.4 (d, J = 9.8 Hz), 130.1, 129.5, 128.5, 128.0, 127.7, 123.3, 116.2 (d, J = 23.0 Hz), 114.3, 55.6, 52.6, 51.3; HRMS-ESI (+) (m/z): [M + H]+ calcd for C26H22FN3O9S2, 604.0860; found, 604.0855.
N-(4-((N-(Carboxymethyl)-4-fluorophenyl)sulfonamido)isoquinolin-1-yl)-N-((4-methoxyphenyl)sulfonyl)glycine (5e).
In a 4 mL screw-cap vial, 4-fluoro-N-(1-((4-methoxyphenyl)sulfonamide)isoquinolin-4-yl)benzenesulfonamide (18) (45 mg, 0.092 mmol) and potassium carbonate (30 mg, 0.22 mmol) were placed, and dimethylformamide (0.75 mL) was added. Ethyl bromoacetate (30 μL, 0.27 mmol) was added to the stirred suspension, and the resulting mixture was allowed to stir for 72 hours. Upon complete consumption of starting material, as determined by LC-MS, the reaction was diluted with EtOAc (10 mL) and washed with H2O (20 mL), brine (20 mL) and dried over Na2SO4. Solvent was removed under reduced pressure to yield 44 mg (73% yield) of a yellow solid. The isolated intermediate, ethyl N-(4-((N-2(-ethoxy-2-oxoethyl)-4-fluorophenyl)sulfonamide)isoquinolin-1-yl)-N-((4-methoxyphenyl)sulfonyl)glycinate (44 mg, 0.066 mmol) was placed in a 20 mL screw-cap vial, dissolved in tetrahydrofuran (2 mL), and 15% NaOH in H2O (wt/wt) (0.35 mL) was added. The reaction was stirred at room temperature for 2 hours. The reaction was diluted with H2O (10 mL). The solution was adjusted to pH 4 with 2N HCl, and a white precipitate formed. The suspension was extracted with EtOAc (3 × 10 mL). The combined organic extracts were washed with H2O (2 × 15 mL) and brine (2 × 15 mL), dried over Na2SO4, and concentrated to yield an off-white solid. The crude product was purified by semi-preparative HPLC (C18; CH3CN/H2O with 1% formic acid: 0 – 18.5 min, 47% CH3CN; 18.5 – 20.5 min, 47 – 95% CH3CN; 20.5 – 27.5 min, 95% CH3CN; 27.5 – 30 min, 95 – 47% CH3CN; 30 – 37 min, 47% CH3CN). The combined fractions were concentrated to remove CH3CN and frozen. The product was then subjected to lyophilization to yield 12 mg (30% yield) of 5e as a white solid. 1H NMR (400 MHz, CDCl3) δ 8.74 – 8.64 (m, 1H), 8.23 – 8.10 (m, 1H), 7.86 (s, 2H), 7.78 (d, J = 3.9 Hz, 3H), 7.53 (d, J = 9.0 Hz, 2H), 7.29 (s, 2H), 7.06 (d, J = 8.9 Hz, 2H), 4.65 – 4.53 (m, 1H), 4.45 (d, J = 6.4 Hz, 3H), 3.92 (s, 3H) 13C NMR (101 MHz, CD3CN) δ 169.3, 168.7, 165.5 (d, J = 252.0 Hz), 163.8, 152.7, 141.9, 137.0, 134.4 (d, J = 3.0 Hz), 132.2, 131.6, 130.9 (d, J = 9.8 Hz), 130.6, 128.7, 128.0, 127.9, 127.8, 122.9, 116.3 (d, J = 22.0 Hz), 114.2, 55.6, 52.7, 51.1; HRMS-ESI (+) (m/z): [M + H]+ calcd for C26H22FN3O9S2, 604.0860; found, 604.0855.
General Procedure for the Synthesis of Intermediates 9a and 9b.
The appropriate compound, either 19a or 19b (1 equiv, 4.8 mmol) was dissolved in chloroform (15 mL), and a solution of mCPBA (4 equiv, 3.33 g, 19.3 mmol) in 45 mL of chloroform was added dropwise. After the addition was complete, the reaction was stirred at room temperature for 18 hours. 1 N NaOH (25 mL) was added to the reaction and stirred for 10 minutes. The layers were separated, and the organic layer was washed again with 1 N NaOH (25 mL), dried over MgSO4 and concentrated under reduced pressure to yield an off-white solid. The crude product was purified by column chromatography (silica gel; MeOH/EtOAc 0:100 to 10:90). Pure fractions were combined to yield isoquinoline N-oxide 9a or 9b as an off-white solid.
4-((N-(tert-Butoxycarbonyl)-4-methoxyphenyl)sulfonamido)isoquinoline 2-oxide (9a).
Off-white solid; yield 78% (1.63 g , 3.8 mmol); 1H NMR (400 MHz, CDCl3) δ 8.84 (s, 1H), 8.13 – 8.10 (m, 1H), 8.07 – 7.96 (m, 2H), 7.85 (dd, J = 6.1, 3.4 Hz, 1H), 7.81 – 7.75 (m, 1H), 7.71 – 7.63 (m, 2H), 7.08 – 7.00 (m, 2H), 3.91 (s, 3H), 1.29 (s, 9H).
4-((N-tert-Butoxylcarbonyl)-4-fluorophenyl)sulfonamide)isoquinoline 2-oxide (9b).
Off-white solid; yield 70% (0.363 g , 0.87 mmol); 1H NMR (400 MHz, CDCl3) δ 8.86 (s, 1H), 8.19 – 8.11 (m, 3H), 7.88 – 7.79 (m, 2H), 7.76 – 7.65 (m, 2H), 7.31 (dd, J = 8.9, 8.2 Hz, 2H), 1.32 (s, 9H).
General Procedure for the Synthesis of Intermediates 10a-c.
Compound 9a or 9b (1 equiv, 1.16 mmol), the appropriate sulfonamide (2 equiv, 2.3 mmol), diacetoxy iodobenzene (2 equiv, 2.3 mmol), and triphenylphosphine (2 equiv, 2.3 mmol) were placed in a flame-dried single-necked round-bottom flask. The flask was purged with argon, and acetonitrile (2.3 mL) was added. The resulting suspension was heated to 80 °C. After 18 hours the flask was removed from heat and allowed to cool to room temperature. H2O (20 mL) was added to the flask and the mixture was extracted with EtOAc (2 × 25 mL). The combined organic layers were washed with brine (25 mL) and dried over Na2SO4. Solvent was removed under reduced pressure to yield an orange oil. The crude material was purified by column chromatography (silica gel; EtOAc/Hexanes 0:100 to 50:50) which yielded a mixture of the product and residual sulfonamide. Pure product was obtained by a second column (silica gel; EtOAc/DCM 0:100 to 10:90) to yield 10a-c as off-white solids.
tert-Butyl (1-((4-methoxyphenyl)sulfonamido)isoquinolin-4-yl)((4-methoxyphenyl)sulfonyl)carbamate (10a).
Off-white solid; yield 37% (0.26 g, 0.43 mmol); 1H NMR (400 MHz, CDCl3) δ 12.06 (d, J = 6.1 Hz, 1H), 8.60 (d, J = 7.6 Hz, 1H), 8.04 – 7.99 (m, 2H), 7.99 – 7.93 (m, 2H), 7.75 (ddd, J = 8.2, 7.1, 1.3 Hz, 1H), 7.56 (ddd, J = 8.2, 7.2, 1.2 Hz, 1H), 7.51 (d, J = 8.1 Hz, 1H), 7.38 (d, J = 6.1 Hz, 1H), 7.05 – 7.00 (m, 2H), 7.00 – 6.95 (m, 2H), 3.92 (s, 3H), 3.85 (s, 3H), 1.33 (s, 9H).
tert-Butyl (1-((4-fluorophenyl)sulfonamido)isoquinolin-4-yl)((4-methoxyphenyl)sulfonyl)carbamate (10b).
Off-white solid; yield 54% (0.190 g, 0.32 mmol); 1H NMR (400 MHz, CDCl3) δ 12.04 – 11.91 (m, 1H), 8.59 (d, J = 8.3 Hz, 1H), 8.13 – 8.03 (m, 2H), 7.97 (d, J = 8.8 Hz, 2H), 7.81 – 7.72 (m, 1H), 7.60 (t, J = 7.7 Hz, 1H), 7.47 (d, J = 8.2 Hz, 1H), 7.30 (br. s., 1H), 7.21 (q, J = 8.5 Hz, 2H), 7.05 (d, J = 9.0 Hz, 2H), 3.95 (s, 3H), 1.36 (s, 9H).
tert-Butyl ((4-fluorophenyl)sulfonyl)(1-((4-methoxyphenyl)sulfonamide)isoquinolin-4-yl)carbamate (10c)
Off-white solid; yield 50% (0.135 g, 0.23 mmol); 1H NMR (400 MHz, CDCl3) δ 12.09 – 11.91 (m, 1H), 8.60 (d, J = 8.4 Hz, 1H), 8.11 – 8.05 (m, 2H), 8.04 – 7.97 (m, 2H), 7.77 – 7.69 (m, 1H), 7.59 (d, J = 7.3 Hz, 1H), 7.36 (d, J = 8.0 Hz, 1H), 7.28 – 7.22 (m, 3H), 7.05 – 6.96 (m, 2H), 3.88 (s, 3H), 1.35 (s, 9H).
General Procedure for the Synthesis of Compounds 11a-c, 12, 13a, 14 and 15.
In a 20 mL screw-cap vial, the appropriate ester 20a-20c, 21, 22a, 24, 25 (0.10 mmol) was dissolved in methanol (2 mL), and 15% aqueous NaOH (0.25 mL) was added. The reaction was stirred at room temperature for 4 hours. Upon completion the methanol was removed under reduced pressure, and the residual liquid was diluted with H2O (10 mL). The solution was adjusted to pH 4 with 2N HCl, and a white precipitate formed. The suspension was extracted with EtOAc (3 × 10 mL), washed with H2O (2 × 15 mL) and brine (2 × 15 mL), dried over Na2SO4, and concentrated to yield an off-white solid.
N-(1-((4-Methoxyphenyl)sulfonamido)isoquinolin-4-yl)-N-((4-methoxyphenyl)sulfonyl)glycine (11a).
The crude product was purified by HPLC (C18; CH3CN/H2O 60%:40%, isocratic) to yield 11a as an off-white solid. Yield 55% (31 mg, 0.056 mmol). 1H NMR (400 MHz, DMSO-d6) δ 12.88 (br. s., 1H), 11.89 (br. s., 1H), 8.35 (d, J = 7.8 Hz, 1H), 7.92 (d, J = 8.6 Hz, 2H), 7.82 – 7.73 (m, 1H), 7.69 (d, J = 8.1 Hz, 3H), 7.60 (t, J = 7.7 Hz, 1H), 7.46 (br. s., 1H), 7.08 (d, J = 8.8 Hz, 4H), 4.52 (d, J = 18.1 Hz, 1H), 4.21 (d, J = 17.6 Hz, 1H), 3.86 (s, 3H), 3.82 (s, 3H) 13C NMR (101 MHz, DMSO-d6) δ 170.6, 163.3, 162.4, 151.8, 136.1, 135.1, 134.0, 131.9, 130.4, 130.2, 128.7, 128.4, 127.5, 124.9, 123.3, 121.1, 114.9, 114.6, 56.2, 56.0, 52.6 HRMS-ESI (+) (m/z): [M + H]+ calcd for C25H23N3O8S2, 558.0999; found, 558.1006.
N-(4-((4-Methoxyphenyl)sulfonamido)isoquinolin-1-yl)-N-((4-methoxyphenyl)sulfonyl)glycine (11b).
The crude product was purified by HPLC (C18; CH3CN/H2O 60%:40%, isocratic) to yield 11b as an off-white solid. Yield 80% (38 mg, 0.068 mmol). 1H NMR (400 MHz, CDCl3) δ 8.61 – 8.53 (m, 1H), 8.04 (s, 1H), 7.94 – 7.83 (m, 1H), 7.77 – 7.64 (m, 4H), 7.60 – 7.48 (m, 3H), 7.00 – 6.93 (m, 2H), 6.92 – 6.85 (m, 2H), 4.47 (br. s., 2H), 3.89 (s, 3H), 3.82 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 170.0, 164.0, 163.6, 150.2, 134.2, 133.7, 132.4, 130.5, 129.9, 129.5, 129.1, 128.9, 127.9, 127.9, 120.9, 114.5, 114.5, 55.7, 52.3; HRMS-ESI (+) (m/z): [M + H]+ calcd for C25H23N3O8S2, 558.0960; found, 558.1011.
N-(4-((4-Methoxyphenyl)sulfonamido)isoquinolin-1-yl)-N-((4-methoxyphenyl)sulfonyl)alanine (11c).
Crude product was purified by HPLC (C18; CH3CN/H2O 60%:40%, isocratic) to yield 11c as a white solid. Yield 70% (2.4 mg, 0.0042 mmol). 1H NMR (900 MHz, CDCl3) δ 8.53 (d, J = 8.23 Hz, 1H), 8.15 (br. s., 1H), 8.04 (dd, J = 8.78, 2.20 Hz, 1H), 7.91 (t, J = 7.68 Hz, 1H), 7.84 (t, J = 7.41 Hz, 1H), 7.77 (d, J = 8.78 Hz, 2H), 7.57 (d, J = 8.23 Hz, 2H), 6.98 (d, J = 8.23 Hz, 2H), 6.94 (d, J = 8.78 Hz, 2H), 5.12 (q, J = 7.32 Hz, 1H), 3.90 (s, 3H), 3.85 (s, 3H), 1.16 (d, J = 7.68 Hz, 3H); 13C NMR (226 MHz, CDCl3) δ 172.3, 164.1, 163.8, 147.7, 133.7, 133.2, 132.5, 130.4, 130.0, 129.8, 129.7, 129.7, 129.6, 128.2, 128.0, 121.1, 114.6, 114.6, 59.3, 55.7, 55.7, 16.8; HRMS-ESI (+) (m/z): [M + H]+ calcd for C26H25N3O8S2, 572.1117; found, 572.1216; HPLC Purity: 93.3%.
N-(4-((4-Methoxy-N-methylphenyl)sulfonamido)isoquinolin-1-yl)-N-((4-methoxyphenyl)sulfonyl)glycine (12).
The crude product was purified by HPLC (C18; CH3CN/H2O 60%:40%, isocratic) to yield 12 as an off-white solid. Yield 52% (7 mg, 0.012 mmol). 1H NMR (400 MHz, CDCl3) δ 8.57 (d, J = 8.1 Hz, 1H), 8.24 (d, J = 8.3 Hz, 1H), 7.9 (ddd, J = 8.4, 6.9, 1.3 Hz, 1H), 7.85 – 7.78 (m, 1H), 7.74 – 7.66 (m, 3H), 7.65 – 7.58 (m, 2H), 7.13 – 6.91 (m, 4H), 4.40 – 4.63 (m, 2H), 3.91 (s, 3H), 3.89 (s, 3H), 3.33 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 169.5, 164.0, 163.6, 152.1, 138.0, 137.8, 135.9, 133.0, 130.5, 130.1, 129.4, 128.4, 128.4, 128.3, 127.6, 123.4, 114.5, 55.7, 55.7, 52.7, 39.7 HRMS-ESI (+) (m/z): [M + H]+ calcd for C26H25N3O8S2, 572.1117; found, 572.1142.
N-(4-((4-methoxy-N-(2,2,2-trifluoroethyl)phenyl)sulfonamido)isoquinolin-1-yl)-N-((4-methoxyphenyl)sulfonyl)glycine (13a).
The crude product was purified by HPLC (C18; MeCN/H2O 60:40 to 75:25) to yield 13a as an off-white solid. Yield 81% (21 mg, 0.033 mmol). 1H NMR (400 MHz, CDCl3) δ 8.70 (d, J = 8.3 Hz, 1H), 8.01 – 7.96 (m, 1H), 7.92 – 7.86 (m, 1H), 7.86 – 7.78 (m, 1H), 7.75 (s, 1H), 7.68 – 7.58 (m, 2H), 7.55 – 7.45 (m, 2H), 7.01 – 6.90 (m, 4H), 4.63 – 4.42 (m, 3H), 4.36 – 4.21 (m, 1H), 3.90 (s, 6H); 13C NMR (101 MHz, CDCl3) δ 170.3, 164.0, 163.9, 153.0, 140.8, 137.2, 133.1, 132.6, 130.5, 130.1, 129.2, 129.1, 128.6, 128.4, 127.8, 122.4 (q, J = 272 Hz), 123.7, 55.8, 55.7, 52.2 (q, J = 39.6 Hz), 52.4; HRMS-ESI (+) (m/z): [M + H]+ calcd for C27H24F3N3O8S2, 640.0990; found, 640.1037.
N-((4-Fluorophenyl)sulfonyl)-N-(4-((4-methoxy-N-(2,2,2-trifluoroethyl)phenyl)sulfonamido)isoquinolin-1-yl)glycine (13b).
In a 20 mL screw-cap vial, ethyl N-((4-fluorophenyl)sulfonyl)-N-(4-((4-methoxy-N-(2,2,2-trifluoroethyl)phenyl)sulfonamido)isoquinolin-1-yl)glycinate (22b) (44 mg, 0.067 mmol) was dissolved in tetrahydrofuran (1.5 mL), and 15% NaOH in H2O (wt/wt) (0.35 mL) was added. The reaction was stirred at room temperature for 6 hours. Upon completion, as judged by TLC, the reaction was diluted with H2O (10 mL). The solution was adjusted to pH 4 with 2N HCl, and a white precipitate formed. The suspension was extracted with EtOAc (3 × 10 mL). The combined organic extracts were washed with H2O (2 × 15 mL) and brine (2 × 15 mL), dried over Na2SO4, and concentrated to yield an off-white solid. The crude product was purified by semi-preparative HPLC (C18; CH3CN/H2O with 1% formic acid: 0 – 18.5 min, 60% CH3CN; 18.5 – 20.5 min, 60 – 95% CH3CN; 20.5 – 27.5 min, 95% CH3CN; 27.5 – 30 min, 95 – 60% CH3CN; 30 – 37 min, 60% CH3CN). The combined fractions were concentrated to remove CH3CN and frozen. The product was then subjected to lyophilization to yield to yield 10 mg (23% yield) of 13b as a white solid. 1H NMR (400 MHz, CD3CN) δ 8.74 (d, J = 8.4 Hz, 1H), 8.11 (d, J = 8.4 Hz, 1H), 7.91 (t, J = 7.6 Hz, 1H), 7.86 – 7.77 (m, 1H), 7.68 – 7.56 (m, 5H), 7.32 (t, J = 8.7 Hz, 2H), 7.07 (d, J = 8.9 Hz, 2H), 4.74 – 4.57 (m, 1H), 4.49 (d, J = 2.7 Hz, 2H), 4.40 – 4.23 (m, 1H), 3.91 (s, 3H); 13C NMR (101 MHz, CD3CN) δ 168.4, 165.6 (d, J=252.0 Hz), 164.1, 152.6, 141.4, 137.2, 132.5 (d, J = 2.9 Hz, 132.4, 132.0, 131.4 (d, J = 8.8 Hz), 130.3, 128.6, 128.35, 128.2, 128.0, 124.2 (d, J = 279.0 Hz), 122.7, 116.3 (d, J = 22.0 Hz), 114.6, 55.7, 52.6 (q, J=33.0 Hz), 51.2; HRMS-ESI (+) (m/z): [M + H]+ calcd for C26H21F4N3O7S2, 628.07835; found, 628.0833.
N-(4-((4-Fluoro-N-(2,2,2-trifluoroethyl)phenyl)sulfonamide)isoquinolin-1-yl)-N-((4-methoxyphenyl)sulfonyl)glycine (13c)
In a 4 mL screw-cap vial, ethyl N-(4-((4-fluorophenyl)sulfonamide)isoquinolin-1-yl)-N-((4-methoxyphenyl)sulfonyl)glycinate (23) (40 mg, 0.069 mmol) and potassium carbonate (12 mg, 0.083 mmol) were dissolved in DMF (1 mL), and 2,2,2-trifluoroethyl trifluoromethanesulfonate (13 μL, 0.082 mmol) was added. The reaction was stirred overnight. The reaction was diluted with H2O and extracted with EtOAc (3 × 7 mL). The combined organics were washed with H2O (2 × 20 mL) and brine (20 mL), dried over Na2SO4, and concentrated under reduced pressure to afford 35 mg (79% yield) of a white solid that did not require further purification. In a 20 mL screw-cap vial, ethyl N-(4-((4-fluoro-N-(2,2,2-trifluoroethyl)phenyl)sulfonamide)isoquinolin-1-yl)-N-((4-methoxyphenyl)sulfonyl)glycinate (22c) (35 mg, 0.053 mmol) was dissolved in tetrahydrofuran (2 mL), and 15% NaOH in H2O (wt/wt) (0.35 mL) was added. The reaction was stirred at room temperature for 5 hours. Upon completion, as judged by TLC, the reaction was diluted with H2O (10 mL). The solution was adjusted to pH 4 with 2N HCl acid, and a white precipitate formed. The suspension was extracted with EtOAc (3 × 10 mL). The combined organic extracts were washed with H2O (2 × 15 mL) and brine (2 × 15 mL), dried over Na2SO4, and concentrated to yield an off-white solid. The crude product was purified by semi-preparative HPLC (C18; CH3CN/H2O with 1% formic acid: 0 – 18.5 min, 60% CH3CN; 18.5 – 20.5 min, 60 – 95% CH3CN; 20.5 – 27.5 min, 95% CH3CN; 27.5 – 30 min, 95 – 60% CH3CN; 30–37 min, 60% CH3CN). The combined fractions were concentrated to remove CH3CN and frozen. The product was then subjected to lyophilization to yield 15 mg (35% yield) of 13c as a white solid. 1H NMR (400 MHz, CDCl3) δ 8.75 (d, J = 8.4 Hz, 1H), 8.05 (s, 1H), 7.94 – 7.87 (m, 1H), 7.87 – 7.80 (m, 1H), 7.76 (dd, J = 8.8, 5.0 Hz, 2H), 7.67 (s, 1H), 7.52 (d, J = 8.9 Hz, 2H), 7.32 (t, J = 8.8 Hz, 2H), 7.07 (d, J = 8.9 Hz, 2H), 4.74 – 4.59 13C NMR (101 MHz, CD3CN) δ 168.6, 165.8 (d, J = 252.0 Hz), 163.8, 153.3, 141.5, 136.8, 133.2 (d, J = 2.9 Hz), 132.0, 131.7, 131.1 (d, J = 10.3 Hz,), 130.6, 128.7, 128.3, 128.1, 127.7, 124.1 (d, J = 277.0 Hz), 122.4, 116.7 (d, J = 24.0 Hz), 114.2, 55.6, 52.6 (q, J = 33.3 Hz), 51.1; HRMS-ESI (+) (m/z): [M + H]+ calcd for C26H21F4N3O7S2, 628.0835; found, 628.0833.
N-(4-((4-Fluoro-N-(2,2,2-trifluoroethyl)phenyl)sulfonamido)isoquinolin-1-yl)-N-((4-fluorophenyl)sulfonyl)glycine (13d).
In a 4 mL screw-cap vial, ethyl N-(4-((4-fluoro-N-(2,2,2-trifluoroethyl)phenyl)sulfonamido)isoquinolin-1-yl)-N-((4-fluorophenyl)sulfonyl)glycinate (22d) (0.014 mg, 0.021 mmol) was dissolved in tetrahydrofuran (0.5 mL), and 15% NaOH in H2O (wt/wt) (0.20 mL) was added. The reaction was stirred at room temperature for 6 hours. Upon completion, as judged by TLC, the reaction was diluted with H2O (10 mL). The solution was adjusted to pH 4 with 2N HCl, and a white precipitate formed. The precipitate was filtered and dissolved in CH2Cl2 (5 mL). The organic layer was dried over Na2SO4 and concentrated to yield a yellow solid. The crude product was purified by semi-preparative HPLC: 0 – 18.5 min, 60% CH3CN; 18.5 – 20.5 min, 60 – 95% CH3CN; 20.5 – 27.5 min, 95% CH3CN; 27.5 – 30 min, 95 – 60% CH3CN; 30 – 37 min, 60% CH3CN). The combined fractions were concentrated to remove CH3CN and frozen. The product was then subjected to lyophilization to yield 6 mg (46% yield) of 13d as a white solid. 1H NMR (400 MHz, CD3CN) δ 8.83 – 8.69 (m, 1H), 8.10 – 7.99 (m, 1H), 7.95 – 7.86 (m, 1H), 7.86 – 7.78 (m, 1H), 7.75 (dd, J=8.9, 5.0 Hz, 2H), 7.69 – 7.60 (m, 3H), 7.31 (td, J=8.8, 5.6 Hz, 5H), 4.73 – 4.58 (m, 1H), 4.43 (d, J =1.2 Hz, 2H), 4.41 – 4.30 (m, 1H); 13C NMR (101 MHz, CD3CN) δ 169.1, 165.8 (d, J = 253.0 Hz), 165.6 (d, J = 252.0 Hz), 153.22, 141.5, 136.8, 133.2 (d, J = 3.0 Hz), 132.7 (d, J = 3.0 Hz), 131.9, 131.8, 131.4 (d, J = 9.8 Hz), 131.1 (d, J = 9.8 Hz), 128.8, 128.3, 128.1, 124.1 (d, J = 278.0 Hz), 122.3, 116.6 (d, J = 23.0 Hz), 116.2 (d, J = 23.0 Hz), 52.6 (q, J = 34.0 Hz), 51.7; HRMS-ESI (+) (m/z): [M + H]+ calcd for C25H18F5N3O6S2, 616.0635; found, 616.0630.
N-(4-((4-methoxy-N-(2,2-difluoroethyl)phenyl)sulfonamido)isoquinolin-1-yl)-N-((4-methoxyphenyl)sulfonyl)glycine (14).
The crude product was purified by HPLC (C18; MeCN/H2O + 0.1% formic acid 60:40 to 75:25) to yield 14 as an off-white solid. Yield 59% (10.2 mg, 0.016 mmol). 1H NMR (400 MHz, CDCl3) δ 8.66 (d, J = 8.1 Hz, 1H), 7.91 – 7.76 (m, 4H), 7.61 (d, J = 9.1 Hz, 2H), 7.54 (d, J = 9.1 Hz, 2H), 6.97 (d, J = 8.8 Hz, 4H), 6.09 (tt, J = 56, 4.0 Hz, 1H), 4.52 (br. s., 2H), 3.99 (dt, J = 13, 4.4 Hz, 2H), 3.89 (s, 6H); 13C NMR (101 MHz, CDCl3) δ 169.9, 164.0, 163.9, 152.7, 140.5, 137.3, 130.5, 130.0, 129.1, 128.9, 128.5, 128.2, 127.9, 122.5, 114.1, 114.4, 114.6, 55.8, 55.7, 54.0, 52.4; HRMS-ESI (+) (m/z): [M + H]+ calcd for C27H26F2N3O8S2, 622.1129; found, 622.1132.
N-(4-((N-(2-fluoroethyl)-4-methoxyphenyl)sulfonamido)isoquinolin-1-yl)-N-((4-methoxyphenyl)sulfonyl)glycine (15).
The crude product was purified by HPLC (C18; CH3CN/H2O + 0.1% formic acid 60:40 to 75:25) to yield 15 as an off-white solid. Yield 38% (3.8 mg, 0.0063 mmol). 1H NMR (400 MHz, CDCl3) δ 8.62 (d, J = 7.8 Hz, 1H), 8.11 (d, J = 8.3 Hz, 1H), 7.93 – 7.86 (m, 1H), 7.86 – 7.80 (m, 1H), 7.77 (s, 1H), 7.69 – 7.63 (m, 2H), 7.62 – 7.54 (m, 2H), 7.02 – 6.94 (m, 4H), 4.64 – 4.45 (m, 4H), 4.16 – 3.92 (m, 2H), 3.90 (s, 6H); 13C NMR (101 MHz, CDCl3) δ 169.1, 164.0, 163.6, 152.5, 139.7, 138.4, 133.8, 133.2, 130.5, 129.9, 129.6, 129.4, 128.4, 128.1, 127.9, 123.1, 114.5, 81.5 (d, J = 172 Hz), 55.7, 55.7, 52.7, 52.5 (d, J = 22 Hz) ; HRMS-ESI (+) (m/z): [M + H]+ calcd for C27H27FN3O8S2, 604.1223; found, 604.1224.
Diethyl 2,2’-(isoquinoline-1,4-diylbis(((4-fluorophenyl)sulfonyl)azanediyl))diacetate (16).
N,N’-(isoquinoline-1,4-diyl)bis(4-fluorobenzenesulfonamide) (26), K2CO3 (0.11 g, 0.85 mmol) and 18-crown-6 ether (0.019 g, 0.074 mmol) were added to a flask. DMF (1.5 mL) was added, and the mixture was stirred at room temperature for 0.5 hr. Ethyl bromoacetate (0.13 mL, 1.18 mmol) was added, and the mixture was stirred for 24 h at room temperature. The mixture was diluted with EtOAc (40 mL) and washed with H2O (30 mL) and brine (30 mL), dried (Na2SO4), and evaporated to yield a crude product, which was dissolved in CH2Cl2 (1 mL) and purified by column chromatography (silica gel; EtOAc/Hexanes, 10:90 to 100:0). The fractions were combined and concentrated in vacuo to yield 0.077 g (32 % yield) of 16 as a tan solid. 1H NMR (400 MHz, CDCl3) δ 8.90 – 8.77 (m, 1H), 8.07 (s, 1H), 8.00 – 7.86 (m, 1H), 7.86 – 7.73 (m, 4H), 7.73 – 7.58 (m, 2H), 7.26 – 7.09 (m, 4H), 4.80 (d, J = 18 Hz, 1H), 4.49 (d, J = 2.7 Hz, 2H), 4.29 – 3.94 (m, 5H), 1.24 (t, J = 7.2 Hz, 3H), 1.11 (t, J = 7.1 Hz, 3H).
Ethyl N-(1-((N-(2-ethoxy-2-oxoethyl)-4-fluorophenyl)sulfonamido)isoquinolin-4-yl)-N-((4-methoxyphenyl)sulfonyl)glycinate (17).
In a 4 mL screw-cap vial, 4-fluoro-N-(4-((4-methoxyphenyl)sulfonamido)isoquinolin-1-yl)benzenesulfonamide (27) (19 mg, 0.038 mmol), potassium carbonate (12 mg, 0.090 mmol) and 18-crown-6 ether (6 mg, 0.023 mmol) were placed, and dimethylformamide (0.50 mL) was added. Ethyl bromoacetate (12 μL, 0.10 mmol) was added to the stirred suspension, and the resulting mixture was allowed to stir for 48 hours. Upon complete consumption of starting material, as determined by LC-MS, the reaction was diluted with EtOAc (10 mL) and washed with H2O (20 mL), brine (20 mL) and dried over Na2SO4. Solvent was removed under reduced pressure to yield 25 mg (99% yield) of a yellow solid 17. The product did not require further purification. 1H NMR (400 MHz, CDCl3) δ 8.90 – 8.76 (m, 1H), 8.08 – 7.98 (m, 2H), 7.84 – 7.74 (m, 2H), 7.72 – 7.66 (m, 2H), 7.66 – 7.58 (m, 2H), 7.24 – 7.13 (m, 2H), 7.00 – 6.91 (m, 2H), 4.79 – 4.65 (m, 1H), 4.49 (d, J = 6.5 Hz, 2H), 4.29 (d, J = 16.3 Hz, 1H), 4.15 (dd, J = 7.1, 5.4 Hz, 2H), 4.06 (qd, J = 7.1, 1.7 Hz, 2H), 3.90 (s, 3H).
4-Fluoro-N-(1-((4-methoxyphenyl)sulfonamide)isoquinolin-4-yl)benzenesulfonamide (18).
In a 20 mL screw-cap vial, tert-Butyl ((4-fluorophenyl)sulfonyl)(1-((4-methoxyphenyl)sulfonamide)isoquinolin-4-yl)carbamate (10c) (65 mg, 0.11 mmol) was dissolved in CH2Cl2 (2 mL), and the resulting solution was cooled to 0 – 5 °C in an ice/water bath. Once cooled, trifluoroacetic acid (0.3 mL, 0.022 mmol) was added to the isoquinoline solution, and the reaction was allowed to slowly warm to room temperature. After 4 hours the reaction was quenched with sat. aq. NaHCO3 (4 mL), and the resulting mixture was allowed to stir for 30 minutes. The layers were separated and the aqueous layer was extracted with CH2Cl2 (6 mL). The combined organics were washed with brine (5 mL) and dried over Na2SO4 to yield 45 mg (85%) of 18 as a light yellow solid. The product did not require further purification. 1H NMR (400 MHz, CDCl3) δ 11.57 (br. s., 1H), 8.37 (d, J = 8.1 Hz, 1H), 7.97 – 7.91 (m, 3H), 7.81 (dd, J = 8.8, 5.0 Hz, 2H), 7.65 – 7.52 (m, 2H), 7.48 – 7.38 (m, 1H), 7.14 – 7.02 (m, 3H), 6.97 (d, J = 8.9 Hz, 2H).
General Procedure for the Synthesis of Intermediates 19a and 19b.
In a single-necked flask, 28a or 28b (1 equiv, 7.79 mmol) and DMAP (0.1 equiv, 0.78 mmol) were dissolved in CH3CN (105 mL). A solution of Boc2O (1.5 equiv, 11.9 mmol) in CH3CN (20 mL) was added in one portion, and the resulting mixture was stirred at room temperature for 2 hours. The reaction mixture was concentrated under reduced pressure to give an orange oil (19a) or a yellow oil (19b).
tert-Butyl isoquinolin-4-yl((4-methoxyphenyl)sulfonyl)carbamate (19a).
The crude product mixture was purified by column chromatography (silica gel; EtOAc/Hexanes 0:100 to 50:50). Pure fractions were combined to yield 19a as a yellow solid. Yield 66% (2.14 grams, 5.16 mmol). 1H NMR (400 MHz, CDCl3) δ 9.28 (s, 1H), 8.39 (s, 1H), 8.06 – 7.99 (m, 3H), 7.90 (d, J = 8.31 Hz, 1H), 7.78 (t, J = 7.70 Hz, 1H), 7.66 (t, J = 7.70 Hz, 1H), 7.03 (d, J = 9.05 Hz, 2H), 3.91 (s, 3H), 1.28 (s, 9H).
tert-Butyl ((4-fluorophenyl)sulfonyl)(isoquinolin-4-yl)carbamate (19b).
The crude product mixture was purified by column chromatography (silica gel; EtOAc/Hexanes 0:100 to 65:35). Pure fractions were combined to yield 19b as a yellow solid. Yield 90% (0.720 grams, 1.79 mmol). 1H NMR (400 MHz, CDCl3) δ 9.32 (s, 1H), 8.41 (s, 1H), 8.20 – 8.11 (m, 2H), 8.09 (d, J = 8.1 Hz, 1H), 7.94 – 7.85 (m, 1H), 7.82 (dd, J = 6.8, 1.2 Hz, 1H), 7.71 (dd, J = 8.1, 1.0 Hz, 1H), 7.29 (t, J = 8.6 Hz, 2H), 1.30 (s, 9H).
Ethyl N-(1-((4-methoxyphenyl)sulfonamido)isoquinolin-4-yl)-N-((4-methoxyphenyl)sulfonyl)glycinate (20a).
In a 4 mL screw-cap vial, N,N’-(isoquinoline-1,4-diyl)bis(4-methoxybenzenesulfonamide)28 (200 mg, 0.40 mmol), and K2CO3 (66 mg, 0.48 mmol), were dissolved in DMF and cooled to 0 °C, and ethyl bromoacetate (48 μL, 0.44 mmol) was added to the cold solution. The reaction was stirred for 6 hours, maintaining a temperature of 0 – 5 °C the entire time. After 6 hours, the reaction was diluted with H2O and extracted with ethyl acetate (3 × 10 mL). The combined organic fractions were washed with H2O (2 × 20 mL) and brine (2 × 20 mL), dried over Na2SO4, and concentrated under reduced pressure to yield an orange oil. The crude product was purified by column chromatography (silica gel; EtOAc/Hexanes, 0:100 to 40:60) to afford 59.6 mg (25% yield) of 20a as a yellow solid. 1H NMR (400 MHz, CDCl3) δ 11.73 (d, J = 6.1 Hz, 1H), 8.55 (d, J = 8.1 Hz, 1H), 7.99 – 7.93 (m, 2H), 7.78 – 7.72 (m, 4H), 7.55 (ddd, J = 8.3, 6.4, 1.7 Hz, 1H), 7.22 (d, J = 6.1 Hz, 1H), 7.01 – 6.94 (m, 4H), 4.86 (d, J = 18.1 Hz, 1H), 4.22 – 4.11 (m, 2H), 4.04 (d, J = 18.1 Hz, 1H), 3.92 (s, 3H), 3.87 (s, 3H), 1.25 (t, J = 7.1 Hz, 3H).
Ethyl N-(4-((4-methoxyphenyl)sulfonamido)isoquinolin-1-yl)-N-((4-methoxyphenyl)sulfonyl)glycinate (20b).
In a 20 mL screw-cap vial, ethyl N-(4-((N-(tertbutoxycarbonyl)-4-methoxyphenyl)sulfonamido)isoquinolin-1-yl)-N-((4-methoxyphenyl)sulfonyl)glycinate (29a) (75 mg, 0.11 mmol) was dissolved in dichloromethane (3 mL), and the resulting solution was cooled to 0 – 5 °C in an ice/water bath. Once cooled, trifluoroacetic acid (0.50 mL, 6.5 mmol) was added to the isoquinoline solution, and the reaction was allowed to slowly warm to room temperature. After 4 hours the reaction was quenched with sat. aq. NaHCO3 (3 mL), and the resulting mixture was allowed to stir for 30 minutes. The layers were separated, and the aqueous layer was extracted with CH2Cl2 (5 mL). The combined organics were washed with brine (5 mL) and dried over MgSO4 to yield 62 mg (97%) of 20b as a light yellow solid. The product did not require further purification. 1H NMR (400 MHz, CDCl3) δ 8.81 – 8.76 (m, 3H), 8.04 (s, 1H), 7.95 – 7.86 (m, 1H), 7.76 – 7.61 (m, 4H), 7.53 – 7.39 (m, 3H), 6.97 – 6.91 (m, 2H), 6.88 – 6.83 (m, 2H), 4.45 (s, 2H), 3.99 (q, J = 7.09 Hz, 2H), 3.88 (s, 3H), 3.85 – 3.70 (m, 3H), 1.03 (t, J = 7.09 Hz, 3H).
Ethyl N-(4-((4-methoxyphenyl)sulfonamido)isoquinolin-1-yl)-N-((4-methoxyphenyl)sulfonyl)alaninate (20c).
In a 20 mL screw-cap vial, ethyl N-(4-((N-(tertbutoxycarbonyl)-4-methoxyphenyl)sulfonamido)isoquinolin-1-yl)-N-((4-methoxyphenyl)sulfonyl)alaninate (29b) (13 mg, 0.019 mmol) was dissolved in CH2Cl2 (0.5 mL). The resulting solution was cooled to 0 – 5 °C in an ice/water bath. Once cooled, trifluoroacetic acid (0.25 mL, 3.3 mmol) was added to the isoquinoline solution, and the reaction was allowed to slowly warm to room temperature. After 2 hours the reaction was quenched with sat. aq. NaHCO3 (3 mL), and the resulting mixture was stirred for 30 minutes. The layers were separated, and the aqueous layer was extracted with CH2Cl2 (5 mL). The combined organics were washed with brine (5 mL) and dried over MgSO4 to yield a brown residue. The crude product was purified by column chromatography (silica gel; EtOAc/hexanes, 0/100 to 30/70) to give 3.6 mg (32% yield) of 20c as a light brown residue. 1H NMR (400 MHz, CDCl3) δ 8.74 (br. s., 1H), 8.11 (s, 1H), 7.91 – 7.84 (m, 1H), 7.77 – 7.61 (m, 6H), 6.96 (d, J = 9.05 Hz, 2H), 6.90 – 6.81 (m, 3H), 4.63 (d, J = 7.34 Hz, 1H), 4.10 (br. s., 2H), 3.90 (s, 3H), 3.81 (s, 3H), 1.27 – 0.98 (m, 6H).
Ethyl N-(4-((4-methoxy-N-methylphenyl)sulfonamido)isoquinolin-1-yl)-N-((4-methoxyphenyl)sulfonyl)glycinate (21).
In a screw-cap vial, ethyl N-(4-((4-methoxyphenyl)sulfonamido)isoquinolin-1-yl)-N-((4-methoxyphenyl)sulfonyl)glycinate (20b) (18.5 mg, 0.032 mmol) and potassium carbonate (6.5 mg, 0.05 mmol) were dissolved in CH3CN (0.2 mL), and iodomethane (2.94 μL, 0.05 mmol) was added. The reaction was stirred overnight. The reaction was diluted with ethyl acetate and filtered through a short pad of celite. The filtrate was concentrated under reduced pressure to yield a yellow oil. The crude product was purified by column chromatography (silica gel; EtOAc/Hexanes, 0:100 to 30:70) afford 14 mg (74%) of 21 an off-white solid. 1H NMR (400 MHz, CDCl3) δ 8.86 – 8.79 (m, 1H), 8.13 – 8.07 (m, 1H), 7.84 – 7.73 (m, 2H), 7.70 – 7.63 (m, 3H), 7.57 – 7.50 (m, 2H), 7.02 – 6.93 (m, 4H), 4.48 (br. s., 2H), 4.04 (q, J = 7.09 Hz, 2H), 3.90 (s, 3H), 3.89 (s, 3H), 3.32 (s, 3H), 1.07 (t, J = 7.09 Hz, 3H).
Ethyl N-(4-((4-methoxy-N-(2,2,2-trifluoroethyl)phenyl)sulfonamido)isoquinolin-1-yl)-N-((4-methoxyphenyl)sulfonyl)glycinate (22a).
In a 4 mL screw-cap vial, ethyl N-(4-((4-methoxyphenyl)sulfonamido)isoquinolin-1-yl)-N-((4-methoxyphenyl)sulfonyl)glycinate (20b) (30 mg, 0.051 mmol) and potassium carbonate (28.4 mg, 0.21 mmol) were dissolved in DMF, and 2,2,2-trifluoroethyl trifluoromethanesulfonate (27 μL, 0.18 mmol) was added. The reaction was stirred overnight. The reaction was diluted with water and extracted with ethyl acetate (3 × 10 mL). The combined organics were washed with water (2 × 20 mL) and brine (2 × 20 mL), dried over Na2SO4, and concentrated under reduced pressure to yield an orange oil. The crude product was purified by column chromatography (silica gel; EtOAc/Hexanes, 0:100 to 30:70) to afford 27.3 mg (80% yield) of 22a as an off-white solid. 1H NMR (400 MHz, CDCl3) δ 8.90 – 8.83 (m, 1H), 7.87 – 7.81 (m, 1H), 7.81 – 7.72 (m, 3H), 7.63 – 7.57 (m, 2H), 7.48 – 7.41 (m, 2H), 6.97 – 6.89 (m, 4H), 4.58 – 4.45 (m, 3H), 4.29 – 4.16 (m, 1H), 4.03 (q, J = 7.2 Hz, 2H), 3.88 (d, J = 5.1 Hz, 6H), 1.04 (t, J = 7.1 Hz, 3H).
Ethyl N-((4-fluorophenyl)sulfonyl)-N-(4-((4-methoxy-N-(2,2,2-trifluoroethyl)phenyl)sulfonamido)isoquinolin-1-yl)glycinate (22b).
In a 4 mL screw-cap vial, Ethyl N-((4-fluorophenyl)sulfonyl)-N-(4-((4-methoxyphenyl)sulfonamido)isoquinolin-1-yl)glycinate (30) (60 mg, 0.10 mmol) and potassium carbonate (17 mg, 0.12 mmol) were dissolved in DMF (0.5 mL), and 2,2,2-trifluoroethyl trifluoromethanesulfonate (23 μL, 0.15 mmol) was added. The reaction was stirred overnight. The reaction was diluted with H2O and extracted with EtOAc (3 × 10 mL). The combined organics were washed with H2O (2 × 20 mL) and brine (20 mL), dried over Na2SO4, and concentrated under reduced pressure to afford 47 mg (69% yield) of a white solid 22b that did not require further purification. 1H NMR (400 MHz, CDCl3) δ 8.86 (dd, J = 7.0, 2.2 Hz, 1H), 7.91 – 7.86 (m, 1H), 7.81 (tt, J = 7.3, 5.1 Hz, 2H), 7.75 (s, 1H), 7.63 (d, J = 8.9 Hz, 2H), 7.59 – 7.46 (m, 2H), 7.18 (t, J = 8.5 Hz, 2H), 6.95 (d, J = 8.9 Hz, 2H), 4.62 – 4.44 (m, 3H), 4.25 (dd, J = 15.7, 8.3 Hz, 2H), 4.06 (q, J = 7.1 Hz, 2H), 3.90 (s, 3H), 1.07 (t, J = 7.1 Hz, 3H).
Ethyl N-(4-((4-fluoro-N-(2,2,2-trifluoroethyl)phenyl)sulfonamido)isoquinolin-1-yl)-N-((4-fluorophenyl)sulfonyl)glycinate (22d).
In a 4 mL screw-cap vial, 4-fluoro-N-(1-((4-fluorophenyl)sulfonamido)isoquinolin-4-yl)-N-(2,2,2-trifluoroethyl)benzenesulfonamide (31) (37 mg, 0.063 mmol) and potassium carbonate (10 mg, 0.075 mmol) were placed, and dimethylformamide (0.75 mL) was added. Ethyl bromoacetate (17 μL, 0.11 mmol) was added to the stirred suspension, and the resulting mixture was allowed to stir for 48 hours. Upon complete consumption of starting material, as determined by LC-MS, the reaction was diluted with EtOAc (10 mL) and washed with H2O (20 mL) and brine (20 mL) and dried over Na2SO4. Solvent was removed under reduced pressure to yield a crude product which was dissolved in CH2Cl2 (0.8 mL) and purified by column chromatography (silica gel; EtOAc/Hexanes, 0:100 to 60:40). The fractions were combined and concentrated in vacuo to yield 0.014 g (35% yield) of 22d. 1H NMR (400 MHz, CDCl3) δ 8.92 – 8.83 (m, 1H), 7.87 – 7.76 (m, 4H), 7.75 – 7.68 (m, 2H), 7.62 – 7.49 (m, 2H), 7.18 (t, J = 8.5 Hz, 4H), 4.69 – 4.56 (m, 1H), 4.55 – 4.47 (m, 2H), 4.29 – 4.14 (m, 1H), 4.07 (q, J = 7.1 Hz, 2H), 1.09 (t, J = 7.1 Hz, 3H).
Ethyl N-(4-((4-fluorophenyl)sulfonamide)isoquinolin-1-yl)-N-((4-methoxyphenyl)sulfonyl)glycinate (23).
In a 20 mL screw-cap vial, ethyl N-(4-((N-tertobutoxycarbonyl)-4-fluorophenyl)sulfonamido)isoquinolin-1-yl)-N-((4-methoxyphenyl)sulfonyl)glycinate (32) (45 mg, 0.074 mmol) was dissolved in CH2Cl2 (2 mL), and the resulting solution was cooled to 0–5 °C in an ice/water bath. Once cooled, trifluoroacetic acid (0.10 mL, 1.4 mmol) was added to the isoquinoline solution, and the reaction was allowed to slowly warm to room temperature. After 4 hours the reaction was quenched with sat. aq. NaHCO3 (3 mL), and the resulting mixture was allowed to stir for 30 minutes. The layers were separated, and the aqueous layer was extracted with CH2Cl2 (5 mL). The combined organics were washed with brine (5 mL) and dried over Na2SO4 to yield 42 mg (99%) of 23 as a light yellow solid. The product did not require further purification. 1H NMR (400 MHz, CDCl3) δ 8.85 – 8.75 (m, 1H), 8.03 (s, 1H), 7.86 – 7.73 (m, 3H), 7.72 – 7.63 (m, 2H), 7.52 (d, J = 8.8 Hz, 2H), 7.30 (s, 1H), 7.09 (t, J = 8.5 Hz, 2H), 6.97 (d, J = 8.9 Hz, 2H), 4.47 (s, 2H), 4.02 (q, J = 7.1 Hz, 2H), 3.91 (s, 3H), 1.06 (t, J = 7.1 Hz, 3H).
Ethyl N-(4-((4-methoxy-N-(2,2-difluoroethyl)phenyl)sulfonamido)isoquinolin-1-yl)-N-((4-methoxyphenyl)sulfonyl)glycinate (24).
In a 4 mL screw-cap vial, ethyl N-(4-((4-methoxyphenyl)sulfonamido)isoquinolin-1-yl)-N-((4-methoxyphenyl)sulfonyl)glycinate (20b) (20 mg, 0.034 mmol) and potassium carbonate (18.9 mg, 0.14 mmol) were dissolved in DMF (0.2 mL), and 2,2-difluoroethyl trifluromethanesulfonate (16.2 μL, 0.12 mmol) was added. The reaction was stirred overnight. The reaction was diluted with H2O and extracted with ethyl acetate (3 × 10 mL). The combined organics were washed with H2O (2 × 20 mL) and brine (2 × 20 mL), dried over Na2SO4, and concentrated under reduced pressure to yield an orange oil. The crude product was purified by column chromatography (silica gel; EtOAc/Hexanes, 0:100 to 30:70) to afford 18 mg (81%) of 24 as an off-white solid. 1H NMR (400 MHz, CDCl3) δ 8.93 – 8.78 (m, 1H), 7.80 (s, 1H), 7.78 – 7.71 (m, 3H), 7.67 – 7.57 (m, 2H), 7.54 – 7.45 (m, 2H), 7.01 – 6.88 (m, 4H), 6.08 (tt, J = 56, 4.0 Hz, 1H), 4.51 (s, 2H), 4.08 – 3.92 (m, 4H), 3.89 (d, J = 4.7 Hz, 6H), 1.06 (t, J = 7.1 Hz, 3H).
Ethyl N-(4-((4-methoxy-N-(2-fluoroethyl)phenyl)sulfonamido)isoquinolin-1-yl)-N-((4-methoxyphenyl)sulfonyl)glycinate (25).
In a 4 mL screw-cap vial, ethyl N-(4-((4-methoxyphenyl)sulfonamido)isoquinolin-1-yl)-N-((4-methoxyphenyl)sulfonyl)glycinate (20b) (20 mg, 0.034 mmol) and potassium carbonate (18.9 mg, 0.14 mmol) were dissolved in DMF, and 1-iodo-2-fluoroethane (10 μL, 0.12 mmol) was added. The reaction was stirred overnight. The reaction was diluted with H2O and extracted with ethyl acetate (3 × 10 mL). The combined organics were washed with H2O (2 × 20 mL) and brine (2 × 20 mL), dried over Na2SO4, and concentrated under reduced pressure to yield a yellow oil. The crude product was purified by column chromatography (silica gel; EtOAc/Hexanes, 0:100 to 30:70) to afford 10.5 mg (49% yield) of 25 as an off-white solid. 1H NMR (400 MHz, CDCl3) δ 8.89 – 8.83 (m, 1H), 8.00 – 7.94 (m, 1H), 7.82 – 7.74 (m, 3H), 7.68 – 7.62 (m, 2H), 7.54 – 7.48 (m, 2H), 7.00 – 6.93 (m, 4H), 4.51 (d, J = 2.9 Hz, 2H), 4.63 – 4.40 (m, 2H), 4.10 – 3.97 (m, 4H), 3.91 (s, 3H), 3.90 (s, 3H), 1.08 (t, J = 7.1 Hz, 3H).
N,N’-(Isoquinoline-1,4-diyl)bis(4-fluorobenzenesulfonamide) (26).
Compound 26 was prepared from 1,4-dibromoisoquinoline analogously to our previously reported procedure.28 Pink solid; Yield 68% (0.570 g, 1.2 mmol); 1H NMR (400 MHz, CDCl3) δ 8.39 – 8.31 (m, 1H), 8.07 – 7.95 (m, 1H), 7.89 (dd, J = 8.7, 5.3 Hz, 3H), 7.81 – 7.66 (m, 3H), 7.64 – 7.55 (m, 1H), 7.49 – 7.29 (m, 5H), 7.22 – 7.13 (m, 1H).
4-Fluoro-N-(4-((4-methoxyphenyl)sulfonamido)isoquinolin-1-yl)benzenesulfonamide (27).
27 was isolated as a minor product during the formation of 10b. 1H NMR (400 MHz, CDCl3) δ 11.73 (br. s., 1H), 8.51 (d, J = 8.3 Hz, 1H), 7.84 – 8.18 (m, 2H), 7.79 – 7.69 (m, 3H), 7.70 – 7.62 (m, 1H), 7.59 – 7.49 (m, 1H), 7.23 – 7.12 (m, 2H), 7.07 (s, 1H), 6.99 – 6.86 (m, 2H), 6.40 (br. s., 1H), 3.87 (s, 3H).
N-(Isoquinolin-4-yl)-4-methoxybenzenesulfonamide (28a).
In a single-necked round-bottom flask, isoquinoline-4-amine (8) (1.50 g, 10.4 mmol) and 4-methoxybenzenesulfonyl chloride (2.47 g, 11.96 mmol) were dissolved in anhydrous pyridine (18 mL) and stirred at room temperature overnight. Upon completion of the reaction, determined by TLC, the reaction was diluted with H2O (50 mL) and extracted with EtOAc (25 mL). The layers were separated, and the aqueous layer was extracted with EtOAc (2 × 25 mL). The combined organics were washed with H2O (50 mL) and brine (2 × 50 mL), dried over Na2SO4, and concentrated under reduced pressure to give a brown oil. Toluene (50 mL) was added to the oil and removed under reduced pressure to yield an orange solid. The crude product was purified by recrystallization from toluene to give 2.50 grams (77% yield) of 28a as an orange solid. 1H NMR (400 MHz, CDCl3) δ 9.17 (s, 1H), 8.23 (s, 1H), 8.13 (d, J = 8.8 Hz, 1H), 8.03 (d, J = 8.1 Hz, 1H), 7.79 (ddd, J = 8.4, 7.0, 1.2 Hz, 1H), 7.74 – 7.66 (m, 3H), 6.92 – 6.86 (m, 2H), 3.83 (s, 3H).
4-Fluoro-N-(isoquinolin-4-yl)benzenesulfonamide (28b).
In a single-necked round-bottom flask, isoquinoline-4-amine (8) (0.560 g, 3.88 mmol) and 4-fluorobenzenesulfonyl chloride (0.90 g, 4.65 mmol) were dissolved in anhydrous pyridine (10 mL) and stirred at room temperature overnight. Upon completion of the reaction, determined by TLC, the reaction was diluted with H2O (35 mL) and extracted with EtOAc (25 mL). The layers were separated, and the aqueous layer was extracted with EtOAc (2 × 25 mL). The combined organics were washed with H2O (40 mL) and brine (2 × 50 mL), dried over Na2SO4, and concentrated under reduced pressure to give a brown oil. Toluene (50 mL) was added to the oil and removed under reduced pressure to yield an off-white solid. The crude product was purified by filtration and washing with CH2Cl2 (20 mL) to give 0.795 grams (68% yield) of 28b as an off-white solid. 1H NMR (400 MHz, DMSO-d6) δ 10.48 (br. s., 1H), 9.19 (s, 1H), 8.15 – 8.07 (m, 2H), 7.95 (d, J = 8.1 Hz, 1H), 7.76 – 7.60 (m, 4H), 7.40 – 7.28 (m, 2H).
Ethyl N-(4-((N-(tert-butoxycarbonyl)-4-methoxyphenyl)sulfonamido)isoquinolin-1-yl)-N-((4-methoxyphenyl)sulfonyl)glycinate (29a).
In a 4 mL screw-cap vial, tert-butyl (1-((4-methoxyphenyl)sulfonamido)isoquinolin-4-yl)((4-methoxyphenyl)sulfonyl)carbamate (10a) (100 mg, 0.17 mmol) and potassium carbonate (58 mg, 0.42 mmol) were placed, and acetonitrile (1.3 mL) was added. Ethyl bromoacetate (37 μL, 0.33 mmol) was added to the stirred suspension, and the resulting mixture was allowed to stir for 36 hours. Upon complete consumption of starting material as determined by LC-MS, the reaction was diluted with EtOAc (2 mL) and filtered through a small pad of celite, which was washed with EtOAc. The filtrate was concentrated under reduced pressure to yield an orange oil. The crude product was purified by column chromatography (silica gel; EtOAc/Hexanes 0:100 to 30:70). Pure fractions were combined and concentrated under reduced pressure to yield 75 mg (66% yield) of 29a as a light yellow solid. 1H NMR (400 MHz, CDCl3) δ 8.96 – 8.88 (m, 1H), 8.14 (s, 1H), 8.04 – 7.89 (m, 2H), 7.83 – 7.73 (m, 2H), 7.72 – 7.62 (m, 1H), 7.55 – 7.47 (m, 2H), 7.07 – 7.01 (m, 2H), 7.00 – 6.94 (m, 2H), 4.58 (s, 2H), 4.15 – 3.99 (m, 2H), 3.94 (s, 3H), 3.90 (s, 3H), 1.30 (s, 9H), 1.06 (t, J = 7.09 Hz, 3H).
Ethyl N-(4-((N-(tert-butoxycarbonyl)-4-methoxyphenyl)sulfonamido)isoquinolin-1-yl)-N-((4-methoxyphenyl)sulfonyl)alaninate (29b).
In a 4 mL screw-cap vial, tert-butyl (1-((4-methoxyphenyl)sulfonamido)isoquinolin-4-yl)((4-methoxyphenyl)sulfonyl)carbamate (10a) (190 mg, 0.32 mmol) and potassium carbonate (66 mg, 0.47 mmol) were placed, and CH3CN (1 mL) was added. Ethyl 2-bromopropionate (123 μL, 1.11 mmol) was added to the stirred suspension, and the resulting mixture was allowed to stir for 36 hours. Upon complete consumption of starting material as determined by LC-MS, the reaction was diluted with EtOAc (2 mL) and filtered through a small pad of Celite, washing with EtOAc. The filtrate was concentrated under reduced pressure to yield a yellow oil. The crude product was purified by column chromatography (silica gel; EtOAc/Hexanes 0:100 to 30:70). Pure fractions were combined and concentrated under reduced pressure to yield 12 mg (5% yield) of 29b as a light yellow residue. 1H NMR (400 MHz, CDCl3) δ 8.85 (br. s., 1H), 8.23 (d, J = 1.2 Hz, 1H), 8.01 (dd, J = 8.9, 3.1 Hz, 2H), 7.81 – 7.65 (m, 5H), 7.04 (dd, J = 9.1, 2.7Hz, 2H), 7.00 – 6.94 (m, 2H), 4.77 – 4.63 (m, 1H), 4.17 (q, J = 6.8 Hz, 2H), 3.95 (d, J = 1.2 Hz, 3H), 3.90 (s, 3H), 1.34 – 1.18 (m, 15H).
Ethyl N-((4-fluorophenyl)sulfonyl)-N-(4-((4-methoxyphenyl)sulfonamido)isoquinolin-1-yl)glycinate (30).
In a 20 mL screw-cap vial, ethyl N-(4-((N-(tert-butoxycarbonyl)-4-methoxyphenyl)sulfonamido)isoquinolin-1-yl)-N-((4-fluorophenyl)sulfonyl)glycinate (33) (0.10 mg, 0.15 mmol) was dissolved in CH2Cl2 (2 mL), and the resulting solution was cooled to 0–5 °C in an ice/water bath. Once cooled, trifluoroacetic acid (0.4 mL, 0.031 mmol) was added to the isoquinoline solution, and the reaction was allowed to slowly warm to room temperature. After 4 hours the reaction was quenched with sat. aq. NaHCO3 (4 mL), and the resulting mixture was allowed to stir for 30 minutes. The layers were separated, and the aqueous layer was extracted with CH2Cl2 (10 mL). The combined organics were washed with brine (5 mL) and dried over Na2SO4 to yield 84 mg (95% yield) of 30 as a light yellow solid. The product did not require further purification. 1H NMR (400 MHz, CDCl3) δ 8.87 – 8.75 (m, 1H), 8.04 (s, 1H), 7.93 – 7.82 (m, 1H), 7.82 – 7.66 (m, 4H), 7.65 – 7.53 (m, 2H), 7.24 – 7.13 (m, 2H), 6.96 – 6.86 (m, 2H), 6.82 (s, 1H), 4.47 (s, 2H), 4.03 (q, J = 7.2 Hz, 2H), 3.85 (s, 3H), 1.08 (t, J = 7.1 Hz, 3H).
4-Fluoro-N-(1-((4-fluorophenyl)sulfonamido)isoquinolin-4-yl)-N-(2,2,2-trifluoroethyl)benzenesulfonamide (31).
In a 4 mL screw-cap vial, N,N’-(isoquinoline-1,4-diyl)bis(4-fluorobenzenesulfonamide) (26) (100 mg, 0.21 mmol) and potassium carbonate (35 mg, 0.252 mmol) were dissolved in DMF (1.0 mL), and 2,2,2-trifluoroethyl trifluoromethanesulfonate (46 μL, 0.31 mmol) was added. The reaction was stirred overnight. The reaction was diluted with H2O and extracted with EtOAc (3 × 10 mL). The combined organics were washed with H2O (2 × 20 mL) and brine (20 mL), dried over Na2SO4, and concentrated under reduced pressure to afford a crude yellow oil, which was dissolved in CH2Cl2 (0.75 mL) and purified by column chromatography (silica gel; EtOAc/Hexanes, 0:100 to 50:50). The fractions were combined and concentrated in vacuo to yield 37 mg (31% yield) of 31 as an off-white solid. 1H NMR (400 MHz, CD3CN) δ 11.62 (br. s, 1H), 8.48 (d, J = 8.3 Hz, 1H), 8.09 – 7.99 (m, 2H), 7.91 – 7.77 (m, 3H), 7.74 – 7.69 (m, 1H), 7.68 – 7.61 (m, 1H), 7.36 – 7.23 (m, 4H), 7.09 – 6.95 (m, 1H), 4.51 – 4.26 (m, 2H).
Ethyl N-(4-((N-tertobutoxycarbonyl)-4-fluorophenyl)sulfonamido)isoquinolin-1-yl)-N-((4-methoxyphenyl)sulfonyl)glycinate (32).
In a 4 mL screw-cap vial, tert-butyl ((4-fluorophenyl)sulfonyl)(1-((4-methoxyphenyl)sulfonamide)isoquinolin-4-yl)carbamate (10c) (66 mg, 0.12 mmol) and potassium carbonate (24 mg, 0.18 mmol) were placed, and dimethylformamide (0.5 mL) was added. Ethyl bromoacetate (18.5 μL, 0.17 mmol) was added to the stirred suspension, and the resulting mixture was allowed to stir for 72 hours. Upon complete consumption of starting material, as determined by LC-MS, the reaction was diluted with EtOAc (10 mL) and washed with H2O (20 mL), brine (20 mL) and dried over Na2SO4. Solvent was removed under reduced pressure to yield a yellow mixture. The crude product was purified by column chromatography (silica gel; EtOAc/Hexanes 0:100 to 50:50). Pure fractions were combined and concentrated under reduced pressure to yield 46 mg (68% yield) of 32 as a light yellow solid. 1H NMR (400 MHz, CDCl3) δ 8.96 – 8.86 (m, 1H), 8.13 (s, 1H), 8.12 – 8.04 (m, 2H), 7.81 – 7.73 (m, 2H), 7.62 (dd, J = 6.5, 3.0 Hz, 1H), 7.53 – 7.43 (m, 2H), 7.33 – 7.25 (m, 2H), 7.00 – 6.90 (m, 2H), 4.58 (s, 2H), 4.04 (dd, J = 7.2, 4.0 Hz, 2H), 3.90 (s, 3H), 1.32 – 1.28 (m, 9H), 1.06 (t, J = 7.1 Hz, 3H).
Ethyl N-(4-((N-(tert-butoxycarbonyl)-4-methoxyphenyl)sulfonamido)isoquinolin-1-yl)-N-((4-fluorophenyl)sulfonyl)glycinate (33).
In a 4 mL screw-cap vial, tert-butyl (1-((4-fluorophenyl)sulfonamido)isoquinolin-4-yl)((4-methoxyphenyl)sulfonyl)carbamate (10b) (180 mg, 0.31 mmol) and potassium carbonate (55 mg, 0.40 mmol) were placed, and dimethylformamide (1 mL) was added. Ethyl bromoacetate (68 μL, 0.62 mmol) was added to the stirred suspension, and the resulting mixture was allowed to stir for 72 hours. Upon complete consumption of starting material, as determined by LC-MS, the reaction was diluted with EtOAc (15 mL) and washed with H2O (25 mL), brine (25 mL) and dried over Na2SO4. Solvent was removed under reduced pressure to yield a yellow solid. The crude product was purified by column chromatography (silica gel; EtOAc/Hexanes 0:100 to 50:50). Pure fractions were combined and concentrated under reduced pressure to yield 155 mg (83% yield) of 33 as a light yellow solid. 1H NMR (400 MHz, CDCl3) δ 8.95 – 8.82 (m, 1H), 8.13 (s, 1H), 8.05 – 7.94 (m, 2H), 7.85 – 7.75 (m, 2H), 7.74 – 7.66 (m, 1H), 7.62 – 7.53 (m, 2H), 7.19 (t, J = 8.5 Hz, 2H), 7.11 – 6.95 (m, 2H), 4.58 (s, 2H), 4.06 (dd, J = 7.1, 2.6 Hz, 2H), 3.95 (s, 3H), 1.38 – 1.29 (m, 9H), 1.08 (t, J = 7.2 Hz, 3H).
Metabolic Stability and Half-Life Studies.
The compound working solution was prepared by diluting a 10 mM DMSO stock of the compound to 0.05 mM in DMSO:Water (1:1, v/v). A potassium phosphate buffer was prepared at 100 mM (pH 7.4). A NADPH Regenerating system (Cofactor 5X) was prepared by making a solution containing NADP (1.7 mg/mL, 2.22 mM) and G6P (7.8 mg/mL, 27.6 mM) in 100 mM KPi. The resulting solution was aliquoted and stored at −20 °C. Before using, the solution was warmed to 37 °C and G6PDH (2.0 U/mL was added before use. The Stop solution consisted of MeCN containing 100 ng/mL of tolbutamide.
Human liver microsomes were thawed in a water bath (37 °C) and then kept on ice once thawed. 200 μL of stop solution was added to each well of a 96-deep well sample collection plate. The plate was covered with a sealing mat and kept on ice. Microsome mixtures were prepared with 0.625 mg/mL of the microsomes in potassium phosphate buffer. A base plate was set up with 1.0 mL glass vials and 7.5 μL of compound working solution was added per vial. 592.5 μL/vial of microsomal mixture was added and mixed by pipetting. 250 μL of the spiked microsomal mixtures were transferred into two parallel sets of 1 mL glass vials (+NADPH/− NADPH). 62.5 μL/vial of potassium phosphate buffer was added to the NADPH vials, mixed by pipetting and 50 μL transferred into the T= 0 wells of the sample collection plate. 62.5 μL/vial of cofactor solution was added to the +NADPH vials, mixed by pipetting, and placed into the incubator (37 °C, 5% CO2) with shaking at 200 rpm. For the +NADPH incubations, the solutions were mixed by pipetting and 50 μL was transferred at 5, 15, 30, and 45 minutes into the corresponding wells of the sample collection plate. For the -NADPH incubations, the solutions were mixed by pipetting and 50 μL was transferred at 45 minutes into the corresponding wells of the sample collection plate. The sample collection plate was vortexed at 1700 rpm for 3 minutes and centrifuged at 3500 rpm for 15 minutes. A new 96-deep well plate was used and 100 μL/well of ultrapure water was added. 100 μL/well of supernatant was taken from the sample collection plate and transferred into the corresponding wells of the final plate. The final plate was vortexed at 1700 rpm for 1 minute and the samples were analyzed by LC-MS/MS.
Cell culture and immunoblot analysis.
Human keratinocytes (HaCaT cell line) were cultured in Dubecco’s modified Eagle’s medium (DMEM) with 10% FBS and 1% antibiotics. Cells at 70–80% confluence were treated with various compounds as indicated for 16 h in the presence of the complete medium. Cells were lysed in a RIPA extraction buffer (Sigma, Cat # R0278) containing Halt Protease and Phosphatase Inhibitor cocktail (Cat # 1861280, Thermo Scientific) and 1 mM PMSF. Protein extract (~40 μg) was separated on a 10% SDS-PAGE, blotted onto membrane. The membrane was cut at ~50kD size and top part was probed with anti-NRF2 antibody (16396–1-AP, Proteintech, Rosemont, IL), and bottom part with anti-NQO1 antibody (11451–1-AP, Proteintech). Bottom blot was stripped and probed with anti-actin antibody (SC-58673, Santa Cruz Biotech, CA). The blots were developed using HyGLO™ Chemiluminescent Antibody Detection Reagent and ChemiDoc™ Gel Imaging System (Bio-Rad). NRF2 and NQO1 band intensities were quantified using NIH ImageJ software, and were normalized to that of respective actin band intensity. The normalized values of NRF2 and NQO1 of DMSO-treated samples was taken as one unit. The fold increase in NRF2 and NQO1 expression of two independent sets was presented relative to DMSO controls independently or combined.
Keap1 Kelch E540A/E542A purification and crystallization.
pET11a vector with human Keap1 Kelch domain (aa 321–609) mutant (E540A/E542A)39 in frame with N-terminal His6-tag and TEV protease cleavage site was obtained from Genscript. The construct was transformed into BL21(DE3) cells and maintained in media containing 50 μg/mL carbenicillin. Protein expression was induced using an auto-induction protocol modified from Studier.40 Briefly, 1 mL day cultures were used to inoculate a 2L flask of 500 mL of Super LB containing 50 ug/mL carbenicillin. Cells were grown for 24h at 20°C and then harvested by centrifugation.
All steps of Keap1 Kelch E540A/E542A purification were performed at 4 °C. Protein yield and purity at each step was monitored by Bradford assay and SDS-PAGE, respectively. Frozen cells pellets were lysed by sonication in Buffer A (20 mM Tris-HCl, pH 8, 0.5 M NaCl, 8 mM imidazole) containing Roche EDTA-free protease inhibitor tablets, 10 μg/mL DNaseI, and 10 μg/mL lysozyme. The lysate was clarified by centrifugation and loaded onto a 5-mL Talon cobalt column equilibrated with Buffer A. Following a 60-mL wash with Buffer A, bound His6-Keap E540A/E542A protein was eluted with Buffer B (20 mM Tris-HCl, pH 8.0, 0.5 M NaCl, 50 mM imidazole). Fractions containing His6-Keap E540A/E542A protein were pooled and incubated overnight with 1 mM DTT and 0.01 equivalents of TEV protease at 4 °C. The cleaved His6-tag and TEV protease were removed from Keap1 Kelch E540A/E542A by diluting the sample 10-fold with 20 mM Tris-HCl pH 8, 0.5 M NaCl and passing the sample over a 1-mL Talon cobalt column equilibrated with Buffer A. Column flow-through containing unbound Keap1 Kelch E540A/E542A was collected, concentrated, and further purified by size exclusion chromatography. Purified Keap1 E540A/E542A in 20 mM Tris-HCl, pH 7.5, 5 mM DTT was concentrated to 10 mg/mL and used immediately for co-crystallization experiments.
Crystals of Keap1 Kelch E540A/E542A complexed with Compound 16b from a previous publication20 were grown by hanging drop vapor diffusion at 12–16 °C. Prior to crystallization, 5–6 mg/mL Keap1 protein was incubated with 2-fold excess Compound 16b20 for 30 min on ice. Crystals of the complex were grown by mixing 2 μL of Keap1 E540A/E542A: Compound 16b20 with 0.4 μL of reservoir solution containing 3.5–3.8 M sodium formate, pH 7. Crystals grew overnight and were used to streak seed drops of Keap1 E540A/E542A: Compound 16b20 growing at lower saturation (1 μL Keap1 E540A/E542A: Compound 16b20 mixed with 0.3–0.5 μL of reservoir solution containing 3.1–3.6 M sodium formate, pH 7.0). To complex the solvent-accessible subunit (monomer A) of the Keap1 E540A/E542A: Compound 16b20 asymmetric unit, crystals were soaked in a solution of mother liquor containing 2 mM 13a for 3–4 days, leading to a crystal form containing two monomers of Keap1 Kelch E540A/E542A, with one monomer complexed to Compound 16b20 and one monomer complexed to 13a.
Data collection and structure refinement.
The high concentration of sodium formate in the crystallization solution was sufficient to cryo-protect crystals, which were flash-cooled in liquid nitrogen. Data were collected on a MAR300 detector at 0.979 Å at the Life Sciences Collaborative Access Team beamline 21-ID-F at the Advanced Photon Source, Argonne National Laboratory. Data indexing, integration, and scaling were performed using XDS,41 and phases were determined by molecular replacement using Molrep42 and a Keap1 Kelch structure (PDB entry: 1U6D) as search model. Rigid body refinement followed by iterative rounds of restrained refinement and model building were performed with CCP4i43 modules Refmac544 and Coot45. The coordinates and structure factors have been deposited with PDB accession code: 6UF0.
Supplementary Material
ACKNOWLEDGMENT
This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817). The authors acknowledge Alliance Pharma (Malvern, PA) for performing the liver microsomal stability studies. The authors are grateful to Dr. Benjamin Ramirez for his assistance with some of the NMR spectroscopy experiments.
Funding Sources
The authors would like to thank the following funders for their support of this work: National Institute of Arthritis, Musculoskele-tal and Skin Diseases to T.W.M. (1R01 AR069541-01A1) and National Heart, Lung, and Blood Institute to S.P.R. (1R01 HL136946-01).
ABBREVIATIONS
- DMF
N,N-dimethylformamide
- GST
glutathione-S-transferase
- HO-1
heme oxygenase
- KEAP1
Kelch-like ECH-associated protein 1
- NQO-1
NADPH quinone oxidoreductase
- mCPBA
m-chloroperoxybenzoic acid
- NRF2
Nuclear factor (erythroid-derived 2)-like 2
- PPI
protein-protein interaction
- RNS
reactive nitrogen species
- ROS
reactive oxygen species
Footnotes
The Supporting Information is available free of charge on the ACS Publications website.
The following files are available:
Fluorescence anisotropy assay details; LogD7.4 experimental details; metabolism data tables; Western blot and cellular assay details and quantification; crystal structure parameters; HPLC chromatogram traces for key target compounds (.PDF)
Molecular Formula strings (.CSV)
Any additional relevant notes should be placed here.
REFERENCES
- 1.Ahmed SMU; Luo L; Namani A; Wang XJ; Tang X, Nrf2 Signaling Pathway: Pivotal Roles in Inflammation. Biochimica et Biophysica Acta - Molecular Basis of Disease 2017, 1863, 585–597. [DOI] [PubMed] [Google Scholar]
- 2.Kobayashi A; Kang M-I; Okawa H; Ohtsuji M; Zenke Y; Chiba T; Igarashi K; Yamamoto M, Oxidative Stress Sensor Keap1 Functions as an Adaptor for Cul3-Based E3 Ligase To Regulate Proteasomal Degradation of Nrf2. Molecular and Cellular Biology 2004, 24, 7130–7139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cullinan SB; Gordan JD; Jin J; Harper JW; Diehl JA, The Keap1-BTB Protein is an Adaptor That Bridges Nrf2 to a Cul3-Based E3 Ligase: Oxidative Stress Sensing by a Cul3-Keap1 Ligase. Molecular and Cellular Biology 2004, 24, 8477–8486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Martin D; Rojo AI; Salinas M; Diaz R; Gallardo G; Alam J; Ruiz De Galarreta CM; Cuadrado A, Regulation of Heme Oxygenase-1 Expression through the Phosphatidylinositol 3-Kinase/Akt Pathway and the Nrf2 Transcription Factor in Response to the Antioxidant Phytochemical Carnosol. Journal of Biological Chemistry 2004, 279, 8919–8929. [DOI] [PubMed] [Google Scholar]
- 5.Itoh K; Chiba T; Takahashi S; Oyake T; Ishii T; Yamamoto M; Katoh Y; Hayashi N; Satoh K; Igarashi K; Hatayama I; Nabeshima Y. i., An Nrf2/Small Maf Heterodimer Mediates the Induction of Phase II Detoxifying Enzyme Genes through Antioxidant Response Elements. Biochemical and Biophysical Research Communications 2002, 236, 313–322. [DOI] [PubMed] [Google Scholar]
- 6.Cuadrado A; Rojo AI; Wells G; Hayes JD; Cousin SP; Rumsey WL; Attucks OC; Franklin S; Levonen AL; Kensler TW; Dinkova-Kostova AT, Therapeutic Targeting of the NRF2 and KEAP1 Partnership in Chronic Diseases. Nature Reviews Drug Discovery 2019, 18, 295–317. [DOI] [PubMed] [Google Scholar]
- 7.Ferrándiz ML; Nacher-Juan J; Alcaraz MJ, Nrf2 as a Therapeutic Target for Rheumatic Diseases. Biochemical Pharmacology 2018, 152, 338–346. [DOI] [PubMed] [Google Scholar]
- 8.Nezu M; Souma T; Yu L; Suzuki T; Saigusa D; Ito S; Suzuki N; Yamamoto M, Transcription Factor Nrf2 Hyperactivation In Early-Phase Renal Ischemia-Reperfusion Injury Prevents Tubular Damage Progression. Kidney International 2017, 91, 387–401. [DOI] [PubMed] [Google Scholar]
- 9.Gopal S; Mikulskis A; Gold R; Fox RJ; Dawson KT; Amaravadi L, Evidence of Activation of the Nrf2 Pathway in Multiple Sclerosis Patients Treated with Delayed-Release Dimethyl Fumarate in the Phase 3 DEFINE and CONFIRM Studies. Multiple Sclerosis Journal 2017, 23, 1875–1883. [DOI] [PubMed] [Google Scholar]
- 10.Rabbani PS; Ellison T; Waqas B; Sultan D; Abdou S; David JA; Cohen JM; Gomez-Viso A; Lam G; Kim C; Thomson J; Ceradini DJ, Targeted Nrf2 Activation Therapy with RTA 408 Enhances Regenerative Capacity of Diabetic Wounds. Diabetes Res Clin Pract 2018, 139, 11–23. [DOI] [PubMed] [Google Scholar]
- 11.Long M; Rojo de la Vega M; Wen Q; Bharara M; Jiang T; Zhang R; Zhou S; Wong PK; Wondrak GT; Zheng H; Zhang DD, An Essential Role of NRF2 in Diabetic Wound Healing. Diabetes 2016, 65 (3), 780–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Suzuki T; Yamamoto M, Molecular Basis of the Keap1-Nrf2 System. Free Radical Biology and Medicine 2015, 88, 93–100. [DOI] [PubMed] [Google Scholar]
- 13.Park BK; Boobis A; Clarke S; Goldring CEP; Jones D; Kenna JG; Lambert C; Laverty HG; Naisbitt DJ; Nelson S; Nicoll-Griffith DA; Obach RS; Routledge P; Smith DA; Tweedie DJ; Vermeulen N; Williams DP; Wilson ID; Baillie TA, Managing the Challenge of Chemically Reactive Metabolites in Drug Development. Nature Reviews Drug Discovery 2011, 10, 292–306. [DOI] [PubMed] [Google Scholar]
- 14.To C; Shilton BH; Di Guglielmo GM, Synthetic Triterpenoids Target the Arp2/3 Complex and Inhibit Branched Actin Polymerization. Journal of Biological Chemistry 2010, 285, 27944–27957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lan A; Li W; Liu Y; Zhang X; Zhou S; Palko O; Chen H; Kapita M; Prigge J; Schmidt E; Chen X; Sun Z; Chen XL, Chemoprevention of Oxidative Stress-Associated Oral Carcinogenesis by Sulforaphane Depends on NRF2 and the Isothiocyanate Moiety. Cancer Research 2016, 7, 53502–53514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McCullough PA; Ali S, Cardiac and Renal Function in Patients with Type 2 Diabetes Who have Chronic Kidney Disease: Potential Effects of Bardoxolone Methyl. Drug Design, Development and Therapy 2012, 6, 141–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de Zeeuw D; Akizawa T; Audhya P; Bakris GL; Chin M; Christ-Schmidt H; Goldsberry A; Houser M; Krauth M; Lambers Heerspink HJ; McMurray JJ; Meyer CJ; Parving H-H; Remuzzi G; Toto RD; Vaziri ND; Wanner C; Wittes J; Wrolstad D; Chertow GM, Bardoxolone Methyl in Type 2 Diabetes and Stage 4 Chronic Kidney Disease. New England Journal of Medicine 2013, 369, 2492–2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tran KT; Pallesen JS; Solbak SMO; Narayanan D; Baig A; Zang J; Aguayo-Orozco A; Carmona RMC; Garcia AD; Bach A, A Comparative Assessment Study of Known Small-Molecule Keap1-Nrf2 Protein-Protein Interaction Inhibitors: Chemical Synthesis, Binding Properties, and Cellular Activity. J Med Chem 2019, 62 (17), 8028–8052. [DOI] [PubMed] [Google Scholar]
- 19.Jiang ZY; Lu MC; Xu LL; Yang TT; Xi MY; Xu XL; Guo XK; Zhang XJ; You QD; Sun HP, Discovery of Potent Keap1-Nrf2 Protein-Protein Interaction Inhibitor Based on Molecular Binding Determinants Analysis. Journal of Medicinal Chemistry 2014, 57, 2736–2745. [DOI] [PubMed] [Google Scholar]
- 20.Jain AD; Potteti H; Richardson BG; Kingsley L; Luciano JP; Ryuzoji AF; Lee H; Krunic A; Mesecar AD; Reddy SP; Moore TW, Probing the Structural Requirements of Non-Electrophilic Naphthalene-Based Nrf2 Activators. European Journal of Medicinal Chemistry 2015, 103, 252–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Winkel AF; Engel CK; Margerie D; Kannt A; Szillat H; Glombik H; Kallus C; Ruf S; Güssregen S; Riedel J; Herling AW; Von Knethen A; Weigert A; Brüne B; Schmoll D, Characterization of RA839, a Noncovalent Small Molecule Binder to Keap1 and Selective Activator of Nrf2 Signaling. Journal of Biological Chemistry 2015, 290, 28446–28455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Davies TG; Wixted WE; Coyle JE; Griffiths-Jones C; Hearn K; McMenamin R; Norton D; Rich SJ; Richardson C; Saxty G; Willems HMG; Woolford AJA; Cottom JE; Kou JP; Yonchuk JG; Feldser HG; Sanchez Y; Foley JP; Bolognese BJ; Logan G; Podolin PL; Yan H; Callahan JF; Heightman TD; Kerns JK, Monoacidic Inhibitors of the Kelch-like ECH-Associated Protein 1: Nuclear Factor Erythroid 2-Related Factor 2 (KEAP1:NRF2) Protein-Protein Interaction with High Cell Potency Identified by Fragment-Based Discovery. Journal of Medicinal Chemistry 2016, 59, 3991–4006. [DOI] [PubMed] [Google Scholar]
- 23.Heightman TD; Callahan JF; Chiarparin E; Coyle JE; Griffiths-Jones C; Lakdawala AS; McMenamin R; Mortenson PN; Norton D; Peakman TM; Rich SJ; Richardson C; Rumsey WL; Sanchez Y; Saxty G; Willems HMG; Wolfe L; Woolford AJ-A; Wu Z; Yan H; Kerns JK; Davies TG, Structure–Activity and Structure-Conformation Relationships of Aryl Propionic Acid Inhibitors of the Kelch-like ECH-Associated Protein 1/Nuclear Factor Erythroid 2-Related Factor 2 (KEAP1/NRF2) Protein-Protein Interaction. Journal of Medicinal Chemistry 2019, 62, 4683–4702. [DOI] [PubMed] [Google Scholar]
- 24.Pallesen JS; Tran KT; Bach A, Non-covalent Small-Molecule Kelch-like ECH-Associated Protein 1-Nuclear Factor Erythroid 2-Related Factor 2 (Keap1-Nrf2) Inhibitors and Their Potential for Targeting Central Nervous System Diseases. Journal of Medicinal Chemistry 2018, 61, 8088–8103. [DOI] [PubMed] [Google Scholar]
- 25.Richardson BG; Jain AD; Speltz TE; Moore TW, Non-electrophilic Modulators of the Canonical Keap1/Nrf2 Pathway. Bioorg Med Chem Lett 2015, 25 (11), 2261–2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu MC; Ji JA; Jiang ZY; You QD, The Keap1-Nrf2-ARE Pathway As a Potential Preventive and Therapeutic Target: An Update. Med Res Rev 2016, 36 (5), 924–963. [DOI] [PubMed] [Google Scholar]
- 27.Wells G, Peptide and Small Molecule Inhibitors of the Keap1-Nrf2 Protein-Protein Interaction. Biochem Soc Trans 2015, 43 (4), 674–679. [DOI] [PubMed] [Google Scholar]
- 28.Richardson BG; Jain AD; Potteti HR; Lazzara PR; David BP; Tamatam CR; Choma E; Skowron K; Dye K; Siddiqui Z; Wang YT; Krunic A; Reddy SP; Moore TW, Replacement of a Naphthalene Scaffold in Kelch-like ECH-Associated Protein 1 (KEAP1)/Nuclear Factor (Erythroid-derived 2)-like 2 (NRF2) Inhibitors. Journal of Medicinal Chemistry 2018, 61, 8029–8047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yu X; Yang S; Zhang Y; Guo M; Yamamoto Y; Bao M, Intermolecular Amidation of Quinoline N-Oxides with Arylsulfonamides under Metal-Free Conditions. Organic Letters 2017, 19, 6088–6091. [DOI] [PubMed] [Google Scholar]
- 30.Inoyama D; Chen Y; Huang X; Beamer LJ; Kong AN; Hu L, Optimization of Fluorescently Labeled Nrf2 Peptide Probes and the Development of a Fluorescence Polarization Assay for the Discovery of Inhibitors of Keap1-Nrf2 Interaction. J Biomol Screen 2012, 17 (4), 435–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang NJ; Hinner MJ, Getting Across the Cell Membrane: An Overview for Small Molecules, Peptides, and Proteins. Methods Mol Biol 2015, 1266, 29–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wenlock MC; Austin RP; Barton P; Davis AM; Leeson PD, A Comparison of Physiochemical Property Profiles of Development and Marketed Oral Drugs. J Med Chem 2003, 46 (7), 1250–1256. [DOI] [PubMed] [Google Scholar]
- 33.Liu X; Testa B; Fahr A, Lipophilicity and its Relationship with Passive Drug Permeation. Pharm Res 2011, 28 (5), 962–977. [DOI] [PubMed] [Google Scholar]
- 34.DeGoey DA; Chen HJ; Cox PB; Wendt MD, Beyond the Rule of 5: Lessons Learned from AbbVie’s Drugs and Compound Collection. J Med Chem 2018, 61 (7), 2636–2651. [DOI] [PubMed] [Google Scholar]
- 35.Bhal SK; Kassam K; Peirson IG; Pearl GM, The Rule of Five Revisited: Applying log D in Place of log P in Drug-Likeness Filters. Mol Pharm 2007, 4 (4), 556–560. [DOI] [PubMed] [Google Scholar]
- 36.Jiang ZY; Lu MC; Xu LL; Yang TT; Xi MY; Xu XL; Guo XK; Zhang XJ; You QD; Sun HP, Discovery of Potent Keap1-Nrf2 Protein-Protein Interaction Inhibitor Based on Molecular Binding Determinants Analysis. Journal of Medicinal Chemistry 2014, 57 (6), 2736–2745. [DOI] [PubMed] [Google Scholar]
- 37.Kim J; Stahl SS, Cu-Catalyzed Aerobic Oxidative Three-Component Coupling Route to N-Sulfonyl Amidines via an Ynamine Intermediate. J Org Chem 2015, 80 (4), 2448–2454. [DOI] [PubMed] [Google Scholar]
- 38.Huang B; Wang X; Liu X; Chen Z; Li W; Sun S; Liu H; Daelemans D; De Clercq E; Pannecouque C; Zhan P; Liu X, Discovery of Novel DAPY-IAS Hybrid Derivatives as Potential HIV-1 Inhibitors Using Molecular Hybridization Based on Crystallographic Overlays. Bioorg Med Chem 2017, 25 (16), 4397–4406. [DOI] [PubMed] [Google Scholar]
- 39.Horer S; Reinert D; Ostmann K; Hoevels Y; Nar H, Crystal-Contact Engineering to Obtain a Crystal Form of the Kelch Domain of Human Keap1 Suitable for Ligand-Soaking Experiments. Acta Crystallogr Sect F Struct Biol Cryst Commun 2013, 69 (Pt 6), 592–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Studier FW, Protein Production by Auto-Induction in High-Density Shaking Cultures. Protein Expression and Purification 2005, 41 (1), 207–234. [DOI] [PubMed] [Google Scholar]
- 41.Kabsch W, XDS. Acta Crystallogr D Biol Crystallogr 2010, 66 (Pt 2), 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vagin A; Teplyakov A, Molecular Replacement with MOLREP. Acta Crystallogr D Biol Crystallogr 2010, 66 (Pt 1), 22–25. [DOI] [PubMed] [Google Scholar]
- 43.Potterton E; Briggs P; Turkenburg M; Dodson E, A Graphical User Interface to the CCP4 Program Suite. Acta Crystallogr D Biol Crystallogr 2003, 59 (Pt 7), 1131–1137. [DOI] [PubMed] [Google Scholar]
- 44.Murshudov GN; Vagin AA; Dodson EJ, Refinement of Macromolecular Structures by the Maximum-Likelihood Method. Acta Crystallogr D Biol Crystallogr 1997, 53 (Pt 3), 240–55. [DOI] [PubMed] [Google Scholar]
- 45.Emsley P; Lohkamp B; Scott WG; Cowtan K, Features and Development of Coot. Acta Crystallogr D Biol Crystallogr 2010, 66 (Pt 4), 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.