

Summary
The discovery of TMEM173/STING-dependent innate immunity has recently provided guidance for the prevention and management of inflammatory disorders. Here, we show that myeloid TMEM173 occupies an essential role in regulating coagulation in bacterial infections through a mechanism independent of type I interferon response. Mechanistically, TMEM173 binding to ITPR1 controls calcium release from the endoplasmic reticulum in macrophages and monocytes. The TMEM173-dependent increase in cytosolic calcium drives Gasdermin D (GSDMD) cleavage and activation, which triggers the release of F3, the key initiator of blood coagulation. Genetic or pharmacological inhibition of the TMEM173-GSDMD-F3 pathway blocks systemic coagulation and improves animal survival in three models of sepsis (cecal ligation and puncture or bacteremia with Escherichia coli or Streptococcus pneumoniae infection). The upregulation of the TMEM173 pathway correlates with the severity of disseminated intravascular coagulation and mortality in patients with sepsis. Thus, TMEM173 is a key regulator of blood clotting during lethal bacterial infections.
Introduction
Sepsis, which was recently recognized by the World Health Organization as a global health priority (Reinhart et al., 2017), is a challenging clinical syndrome caused by infection (Singer et al., 2016) and accounts for approximately 15% of in-hospital deaths (Rhee et al., 2017). One of the pathologic hallmarks of sepsis is the excessive activation of the immune and coagulation systems by pathogens, particularly bacteria (≥80%) (Gando et al., 2016, Hotchkiss et al., 2016). However, the mechanisms underlying the interplay between coagulation and innate immune pathways, namely immunocoagulation, are poorly understood (Esmon, 2005, Samuels et al., 2018). Myeloid cells, such as monocytes and macrophages, are central innate immune cells not only responsible for eradicating invading pathogens but also instrumental for initiating both inflammatory and coagulant responses (Cavaillon and Adib-Conquy, 2005, Levi and van der Poll, 2010, Pawlinski and Mackman, 2010, Rothmeier et al., 2015, van Dam-Mieras et al., 1985, Wu et al., 2019a). Finally, the excessive activation of host innate immune and coagulation responses can lead to multi-organ failure and death (Gando et al., 2016, Hotchkiss et al., 2016). Thus, an understanding of the mechanisms underlying the excessive immunocoagulation response that accompany death due to sepsis could reveal therapeutic targets to treat sepsis.
Pathogen-associated molecular pattern molecules (PAMPs) from microbes and damage-associated molecular pattern molecules (DAMPs) from hosts serve as so-called “signal 0s” or “danger signals” to initiate the innate immune response (Bianchi, 2007, Gallucci and Matzinger, 2001, Matzinger, 1994, Tang et al., 2012). Thus, we reasoned that specialized innate immune sensors couldcontribute to excessive coagulation in sepsis. Transmembrane protein 173 (TMEM173, also known as STING) is an endoplasmic reticulum (ER)-associated immune adaptor protein that initiates or amplifies inflammation responses to PAMPs (e.g., bacterial cyclic dinucleotide [CDN]) or DAMPs (e.g., host DNA) during infection and tissue injury (Ishikawa and Barber, 2008, Sun et al., 2009, Zhong et al., 2008). The excessive activation of TMEM173 is implicated in various inflammatory diseases or conditions, including sepsis and septic shock (Ge et al., 2019, Heipertz et al., 2017, Hu et al., 2019, Li et al., 2019, Zeng et al., 2017). However, it is unknown whether the TMEM173 pathway also triggers excessive coagulation during infections.
In the present study, we demonstrate that myeloid TMEM173 occupies an essential role in the dysregulated coagulation leading to lethal sepsis. Mechanically, these effects are independent of a classical TMEM173-induced type I IFN response in monocytes and macrophages. Instead, TMEM173 drives coagulation through ER stress-initiated activation of gasdermin D (GSDMD), an effector of pyroptosis (Aglietti et al., 2016, Chen et al., 2016, Ding et al., 2016, He et al., 2015, Kayagaki et al., 2015, Shi et al., 2015), and the subsequent release of coagulation factor III (F3, also known as tissue factor), an initiator of the coagulation cascade (Grover and Mackman, 2018, Levi et al., 2006, Zelaya et al., 2018). TMEM173-dependent systemic coagulation is implicated in septic death in mice and humans. These findings establish a paradigm for TMEM173-triggered coagulation during lethal infection.
Results
TMEM173 Mediates Coagulation Activation in Lethal Infection
Disseminated intravascular coagulation (DIC) is a common complication of sepsis, responsible for multiple-organ failure and poor outcomes (Gando et al., 2016, Hotchkiss et al., 2016). Cecal ligation and puncture (CLP)-induced polymicrobial sepsis is the most-used experimental model for mimicking the progression and characteristics of human sepsis, including the occurrence of DIC (Lewis et al., 2016, Song et al., 2013). To determine the effects of TMEM173 on lethal coagulation, we used a severe mouse model of sepsis, which was induced by CLP with resuscitation fluid alone but not combined with antibiotic treatment. Consistent with previous observations that TMEM173 is a significant driving force for septic death (Heipertz et al., 2017, Zeng et al., 2017), the global deletion of Tmem173 (Tmem173−/−) protected against CLP-induced septic death (Figure S1A). To determine whether TMEM173 is required for systemic coagulation, we assayed blood markers for DIC diagnosis—including prothrombin time (PT), activated partial thromboplastin time (APTT), plasma fibrinogen (FIB), platelet count, and D-dimer—in wild-type (WT) and Tmem173−/− mice. The platelet count (Figure S1B) and fibrinogen concentration (Figure S1C) were reduced, whereas PT (Figure S1D), APTT (Figure S1E), and D-dimer (Figure S1F) were elevated during CLP-induced sepsis in WT mice in comparison with Tmem173−/− mice. Consequently, the level of fibrin deposition (another clinical feature consistent with DIC) in various tissues—such as lung, liver, and spleen—after coagulation activation were reduced in CLP-induced septic Tmem173−/− mice (Figure S1G). Furthermore, the plasma level of F3 (the primary initiator of blood coagulation) was blocked in CLP-induced septic Tmem173−/− mice (Figure S1H), supporting the hypothesis that TMEM173 is a positive regulator of DIC.
Cyclic GMP-AMP (cGAMP) synthase (cGAS) is a cytosolic DNA sensor that activates innate immune responses through amplifying the production of the second messenger cGAMP, which can activate the adaptor TMEM173 in response to virus infection and DNA damage (Ablasser and Chen, 2019, Sun et al., 2013). Unlike Tmem173−/−, the deletion of cGAS (cGAS−/−) failed to protect against CLP-induced septic death (Figure S1A) and systemic coagulation activation (Figures S1B–S1H). These findings suggest that a cGAS-independent TMEM173 pathway (Costa Franco et al., 2018, Holm et al., 2016, Movert et al., 2018, Suschak et al., 2016) could promote systemic coagulation activation in polymicrobial sepsis.
The conditional ablation of TMEM173 in mice found by using Cre/loxP technology revealed that TMEM173 expressed by myeloid cells (Tmem173Mye−/−), but not by T cells (Tmem173T−/−), played a major role in mediating CLP-induced septic death and coagulation activation (Figure 1). The deletion of Tmem173 in myeloid cells, but not in T cells, prolonged animal survival (Figure 1A) associated with increased platelet count (Figure 1B) and fibrinogen concentration (Figure 1C); decreased blood PT (Figure 1D), APTT (Figure 1E), and D-dimer (Figure 1F); reduced fibrin expression in liver, lung, and spleen (Figure 1G); as well as decreased plasma F3 (Figure 1H). As expected, serum markers of tissue damage, such as blood urea nitrogen (BUN) and glutamic pyruvic transaminase (GPT, also known as ALT), were lower in CLP-induced septic Tmem173Mye−/− mice than in WT controls (Figures 1I and 1J). The deletion of myeloid TMEM173 resulted in similar protective effects when mice were infected intraperitoneally with Escherichia coli (E. coli) or Streptococcus pneumoniae (S. pneumoniae), the leading causes of human septic death by Gram-negative or Gram-positive bacterial infection, respectively (Figure S2). Together, these findings demonstrate that myeloid TMEM173 is responsible for DIC and multi-organ failure in sepsis.
Figure 1. Myeloid TMEM173 Is Required for CLP-Induced Systemic Coagulation and Septic Death.
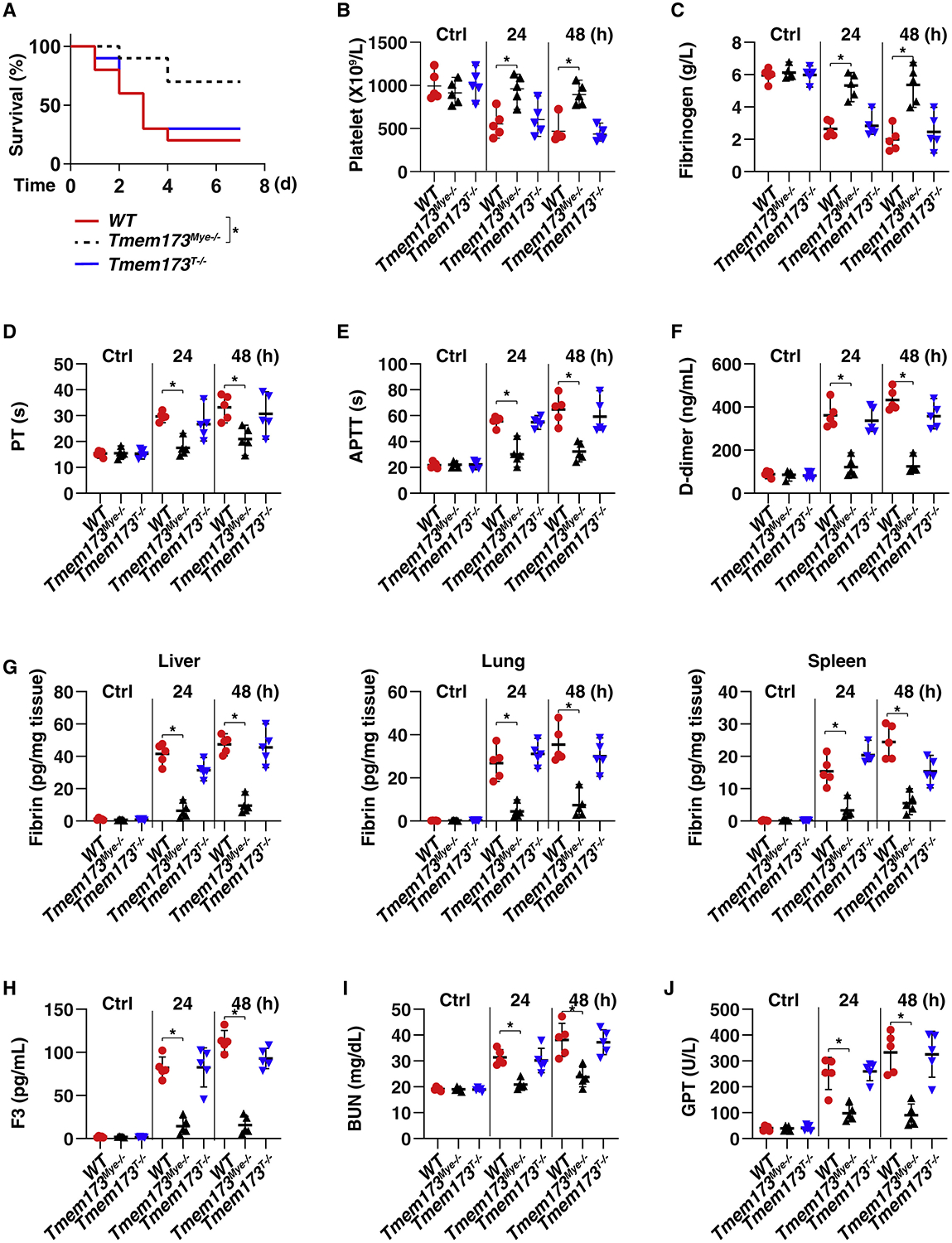
(A) Survival of the indicated mice in CLP-induced sepsis (n = 10 mice per group; *p < 0.05, Kaplan-Meier survival analysis).
(B–J) In parallel, the levels of blood markers of DIC ([B], platelet; [C], fibrinogen; [D], PT; [E], APTT; [F], D-dimer), tissue fibrin (G), plasma F3 (H), and markers of tissue injury ([I], BUN; [J], GPT) were assayed (n = 5 mice per group; *p < 0.05, t test). See also Figures S1 and S2.
TMEM173-Mediated Coagulation Activation Is Independent of Type I IFN Response
One of the major functions of TMEM173 is to induce type I IFN (IFNα and IFNβ) production in response to infection and tissue injury (Barber, 2015, Motwani et al., 2019). The deletion of the common type I IFN receptor, IFNAR1 (Ifnar1−/−), has been shown to increase animal survival from 20% to 50% after CLP-induced sepsis (Dejager et al., 2014). Therefore, we examined the role of type I IFN response in coagulation activation in sepsis. Interestingly, although the deletion of Ifnar1 in mice mildly increased septic animal survival from 20% to 40% (Figure 2A), it failed to affect CLP-induced changes in DIC markers: platelets (Figure 2B), fibrinogen (Figure 2C), PT (Figure 2D), APTT (Figure 2E), D-dimer (Figure 2F), fibrin (Figure 2G), and F3 (Figure 2H), indicating that IFNAR1 is not required for systemic coagulation activation in sepsis. In line with these findings, IFNα and IFNβ did not induce F3 release in primary and immortalized monocytes or macrophages, including THP1 (a human monocyte cell line), primary mouse bone-marrow-derived macrophages (BMDMs), or primary human peripheral blood mononuclear cell-derived macrophages (HPBMs) (Figure 2I). In contrast, E. coli and S. pneumoniae infection induced F3 release in both monocytes and macrophages (Figures 2I and S3A).
Figure 2. TMEM173-Mediated Coagulation Activation Is Independent of Type I IFN Response.
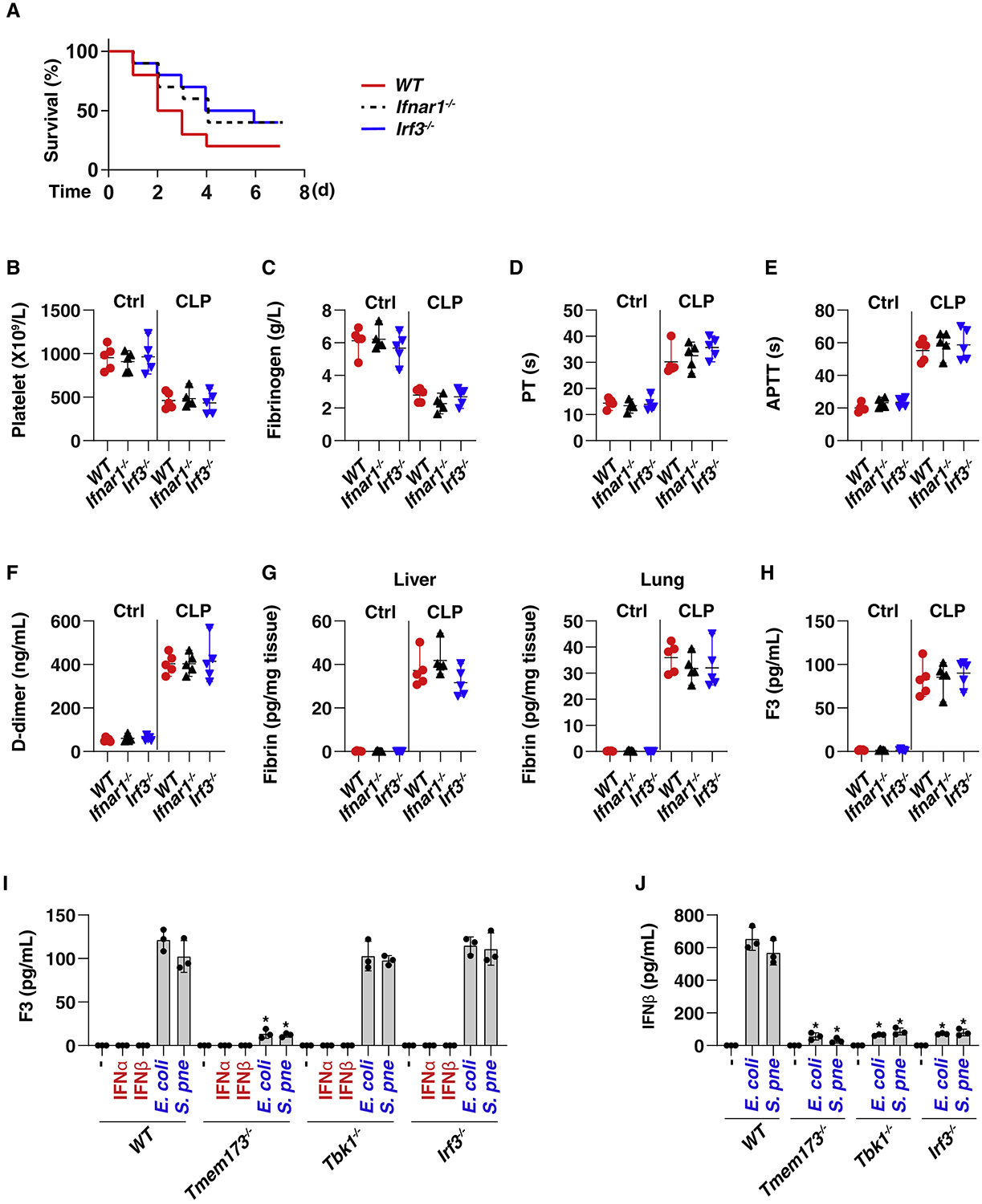
(A) Survival of the indicated mice in CLP-induced sepsis (n = 10 mice per group).
(B–H) The levels of blood markers of DIC ([B]–[F], as in Figures 1B–1F), tissue fibrin (G), and plasma F3 (H) were assayed at 48 h in indicated CLP-induced septic mice (n = 5 mice per group; *p < 0.05, t test).
(I and J) Analysis of F3 (I) and IFNβ (J) release in THP1, BMDM (Figure S3), and HPBM (Figure S3) cells after treatment with IFNα (5 ng/mL), IFNβ (5 ng/mL), E. coli (25 MOI), or S. pneumoniae (25 MOI) infection for 24 h. Data are presented as mean ± SD. See also Figure S3.
TMEM173-induced type I IFN signaling requires the activation of TANK binding kinase 1 (TBK1) and interferon regulatory factor 3 (IRF3) (Liu et al., 2015, Shang et al., 2019, Tanaka and Chen, 2012). Consistently, the E. coli and S. pneumoniae infection-induced IFNβ release was blocked after knockout or knockdown of Tmem173, Tbk1, and Irf3 in THP1 (Figure 2J), BMDM (Figure S3B), and HPBM (Figure S3B) cells. In contrast, only the loss of Tmem173, but not of Tbk1 and Irf3, blocked E. coli and S. pneumoniae infection-induced F3 release in primary or immortalized monocytes and macrophages (Figures 2I and S3A). Like Ifnar1−/−, IRF3 deletion (Irf3−/−) also mildly reduced animal death, but it failed to affect coagulation activation in CLP-induced septic mice (Figures 2A–2H). Together, these findings indicate that TMEM173-mediated F3 release and systemic coagulation activation are independent of the type I IFN response during polymicrobial sepsis, although IFNα and IFNβ could contribute to sepsis through their complex immunomodulating effects (Bolívar et al., 2018, Tilg and Peschel, 1996).
TMEM173-Mediated Coagulation Activation Relies on Calcium Release from the ER
Pathological ER stress can lead to cell pathology and subsequent tissue dysfunction in sepsis (Khan et al., 2015). Many factors, such as nutrient depletion, hypoxia, and oxidative stress, contribute to infection-induced ER stress in host cells, including various immune cells, in response to infection (Zhang, 2010). Given that TMEM173 is an ER-associated membrane protein (Ishikawa and Barber, 2008), we determined whether ER stress is involved in TMEM173-mediated F3 release. Infection by E. coli or S. pneumoniae (but not IFNα or IFNβ stimulation) increased the mRNA expression of ER stress markers—such as DNA damage-inducible transcript 3 (Ddit3, also known as Chop), heat-shock protein family A member 5 (Hspa5, also known as Bip), X-box binding protein 1 (Xbp1), eukaryotic translation initiation factor 2 alpha kinase 3 (Eif2ak3, also known as Perk), ER-to-nucleus signaling 1 (Ern1, also known as Ire1), activating transcription factor 4 (Atf4), ER degradation-enhancing alpha-mannosidase-like protein 1 (Edem1), and derlin-1 (Derl1)—in THP1 (Figure 3A) and BMDM cells (Figure S4A). The elevation of these ER stress markers was blocked by the deletion of Tmem173 but not by the deletion of Tbk1 and Irf3, indicating that TMEM173-mediated ER stress is independent of TBK1 and IRF3 (Figures 3A and S4A). The deletion of Tmem173, Tbk1, or Irf3 did not affect E. coli- or S. pneumoniae-induced F3 mRNA upregulation (Figure 3A), indicating that TMEM173-mediated F3 release is independent of the transcription modulation of F3 expression. Additionally, circulating monocytes from CLP-induced Tmem173−/− mice displayed decreased mRNA expression of ER stress markers (Ddit3 and Hspa5) (Figure 3B) and F3 release in the supernatants (Figure 3C), but not mRNA expression of F3 (Figure 3B). E. coli or S. pneumoniae infection-induced F3 release was rescued by ER stress inhibitors, such as tauroursodeoxycholic acid (TUDCA) or 4-phenyl butyric acid (4PBA) (Soustek et al., 2018) (Figures 3D and S4B). These findings suggest that TMEM173-mediated ER stress is involved in F3 release.
Figure 3. TMEM173-Mediated Coagulation Activation Relies on ER Calcium Release.
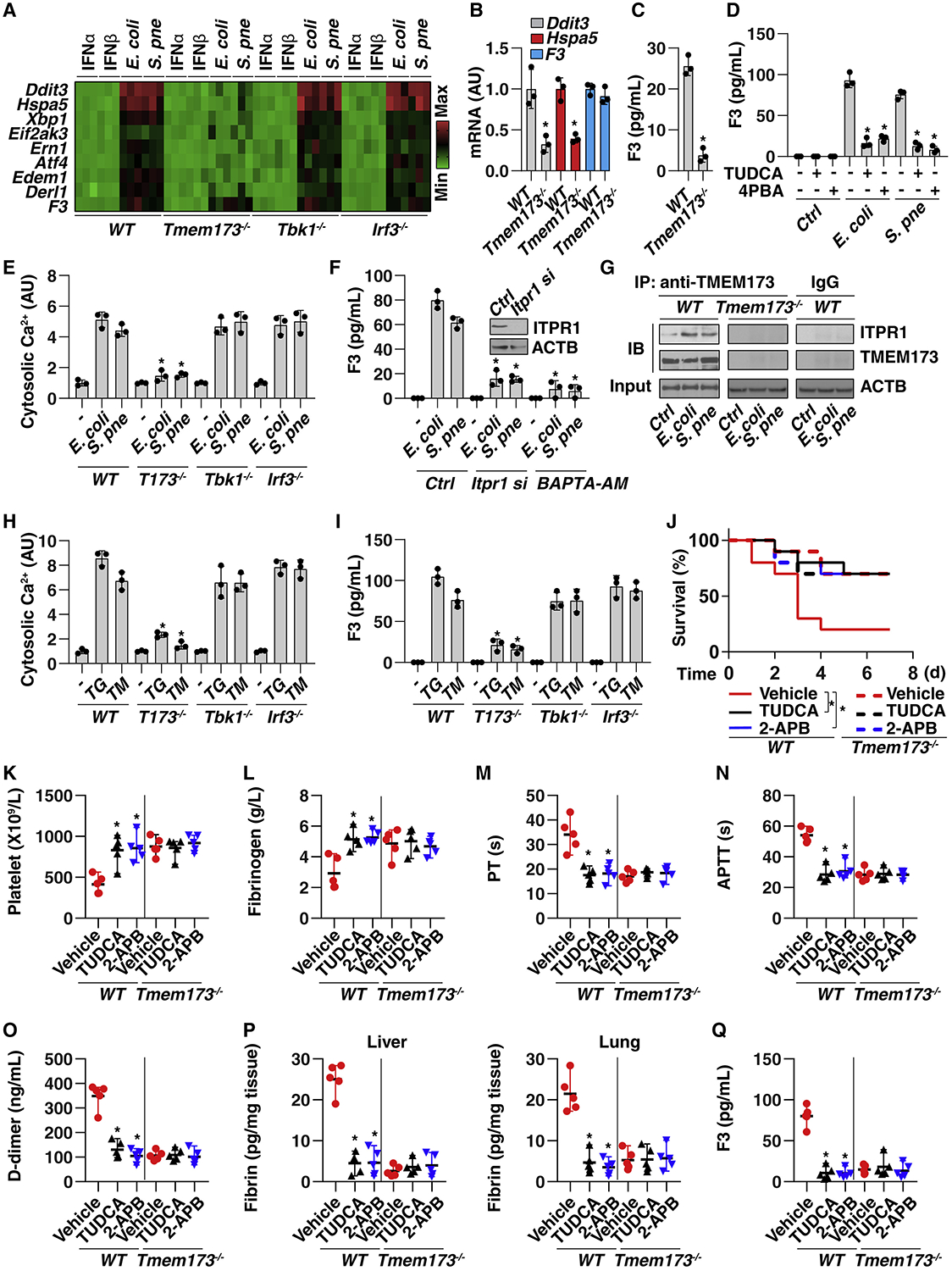
(A) Heatmap of gene mRNA changes in THP1 cells after treatment with IFNα (5 ng/mL), IFNβ (5 ng/mL), E. coli (25 MOI), or S. pneumoniae (25 MOI) infection for 24 h.
(B) Analysis of mRNA expression of the indicated gene in monocytes from CLP-induced (for 48 h) WT or Tmem173−/− mice (n = 3; *p < 0.05 versus WT group, t test).
(C) Analysis of F3 levels in the supernatants of monocytes from CLP-induced (for 48 h) WT or Tmem173−/− mice (n = 3 mice per group; *p < 0.05 versus WT group, t test).
(D) Analysis of F3 release in THP1 cells after E. coli (25 MOI) or S. pneumoniae (25 MOI) infection in the absence or presence of TUDCA (50 μM) or 4PBA (1 mM) for 24 h (n = 3 wells per group; *p < 0.05 versus E. coli or S. pneumoniae group, t test).
(E) Analysis of intracellular Ca2+ in indicated THP1 cells after E. coli (25 MOI) or S. pneumoniae (25 MOI) infection for 24 h (n = 3 wells per group; *p < 0.05 versus WT group, t test).
(F) Analysis of F3 release in indicated WT or Itpr1-knockdown THP1 cells after E. coli (25 MOI) or S. pneumoniae (25 MOI) infection in the absence or presence of BAPTA-AM (10 μM) for 24 h (n = 3 wells per group; *p < 0.05 versus control group, t test).
(G) Analysis of TMEM173-ITPR1 interaction by IP in indicated THP1 cells after E. coli (25 MOI) or S. pneumoniae (25 MOI) infection for 24 h.
(H and I) Analysis of intracellular Ca2+ (H) and F3 (I) release in indicated THP1 cells after thapsigargin (“TG,” 1 μM) and tunicamycin (“TM,” 1.0 μg/mL) treatment for 24 h (n = 3 wells per group; *p < 0.05 versus WT group, t test).
(J) Administration of TUDCA (200 mg/kg) or 2-APB (20 mg/kg) prevented CLP-induced animal death in WT mice, but there was no further protective effect for TUDCA and 2-APG in Tmem173−/− mice (n = 10 mice per group; *p < 0.05, Kaplan-Meier survival analysis).
(K–Q) In parallel, the levels of blood markers of DIC ([K]–[O], as in Figures 1B–1F and 2B–2F), tissue fibrin (P), and plasma F3 (Q) were assayed (n = 5 mice per group; *p < 0.05 versus vehicle group, t test).
Data are presented as mean ± SD ([B], [C], [D], [E], [F], [H], and [I]). See also Figure S4.
Increased cytosolic calcium influx from the ER is an endogenous signal that drives cell death and immune responses (Bettigole and Glimcher, 2015, Todd et al., 2008). E. coli- or S. pneumoniae-induced cytosolic calcium upregulation was observed in WT, Tbk1−/−, and Irf3−/− cells, but not in Tmem173−/− THP1 (Figure 3E) or BMDM cells (Figure S4C), indicating that TMEM173 activation regulates cytosolic calcium influx during ER stress. The administration of a calcium chelator (BAPTA-AM) or inhibition of inositol 1,4,5-trisphosphate receptor type 1 (ITPR1, also known as IP3R, the primary calcium release channel of the ER (Prole and Taylor, 2019) by siRNA or by a pharmacological inhibitor (2-APB) blocked E. coli- or S. pneumoniae-induced F3 release in THP1 (Figure 3F) and BMDM cells (Figure S4D). Moreover, E. coli or S. pneumoniae infection increased the interaction between ITPR1 and TMEM173 in THP1 (Figure 3G) and BMDM cells (Figure S4E), indicating the ITPR1-TMEM173 protein complex contributes to calcium homeostasis and F3 release during infection. In addition to infection, the chemical activation of ER stress and calcium release by thapsigargin and tunicamycin (Figures 3H and S4F) induced F3 release (Figures 3I and S4G), an effect that was blocked in Tmem173−/− but not in Tbk1−/− or Irf3−/− cells. In vivo, the inhibition of ER stress or calcium release by TUDCA or 2-APB protected against CLP-induced septic death (Figure 3J) and coagulation activation (Figures 3K–3Q) in wild-type mice, but there was no further protective effect for TUDCA and 2-APG on the Tmem173−/− mice. Collectively, these findings suggest that TMEM173 interacts with ITPR1 to promote the ER calcium release, leading to subsequent F3 release and coagulation activation in sepsis.
ATP2A2-Dependent ER Calcium Uptake Limits TMEM173-Mediated F3 Release
One step of TMEM173-mediated TBK1 activation and type I IFN production consists in its accumulation in the ER-Golgi intermediate compartment (ERGIC) (Dobbs et al., 2015, Gui et al., 2019, Mukai et al., 2016). This process is controlled through a number of ER-associated proteins, including positive (e.g., ZDHHC1) (Zhou et al., 2014) and negative regulators (e.g., STIM1) (Srikanth et al., 2019). As expected, the knockdown of Zdhhc1 decreased, whereas the knockdown of Stim1 by siRNA increased IFNβ release in THP1 (Figures 4A and 4B) and BMDM (Figures S5A and S5B) cells during E. coli or S. pneumoniae infection, supporting the notion that the translocation of TMEM173 into ERGIC is critical for TMEM173-mediated type I IFN production (Dobbs et al., 2015). However, the knockdown of Zdhhc1 or Stim1 failed to affect E. coli- or S. pneumoniae-induced F3 release in THP1 (Figure 4C) and BMDM (Figure S5C) cells, indicating that TMEM173-mediated F3 release is independent of TMEM173 trafficking to the ERGIC.
Figure 4. ATP2A2-Dependent ER Calcium Uptake Limits TMEM173-Mediated F3 Release.
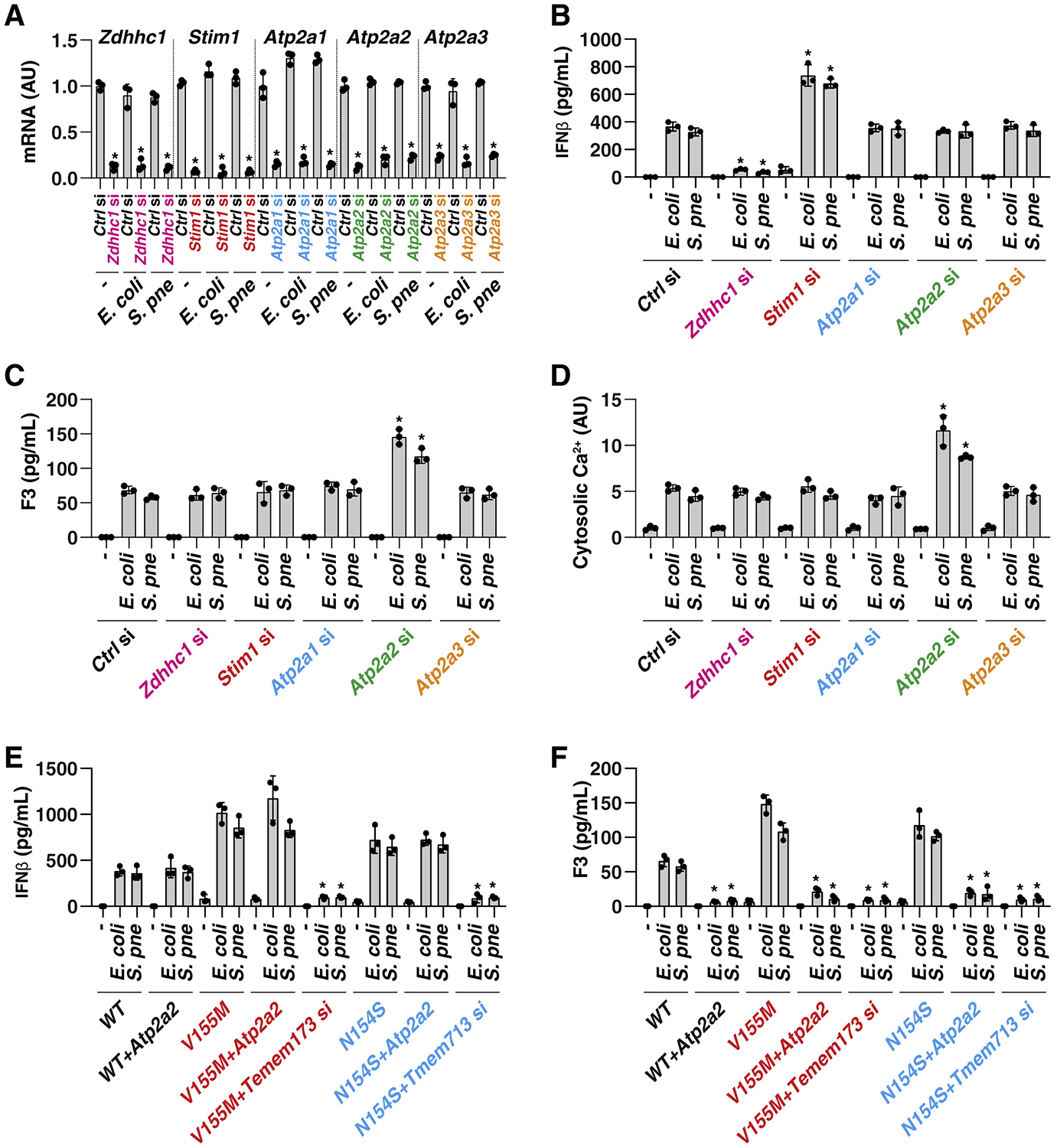
(A) Analysis of gene mRNA expression in indicated gene knockdown THP1 cells after E. coli (25 MOI) or S. pneumoniae (25 MOI) infection for 24 h (n = 3 wells per group; *p < 0.05 versus control siRNA group, t test).
(B–D) Analysis of IFNβ release (B), F3 release (C), and cytosolic calcium (D) in indicated THP1 cells after E. coli (25 MOI) or S. pneumoniae (25 MOI) infection for 24 h (n = 3 wells per group; *p < 0.05 versus control siRNA group, t test).
(E and F) Analysis of IFNβ (E) and F3 (F) release in indicated WT and TMEM173 mutation (V155M and N154S) THP1 cells after E. coli (25 MOI) or S. pneumoniae (25 MOI) infection for 24 h in the absence or presence of Atp2a2 overexpression or Tmem173 knockdown (n = 3 wells per group; *p < 0.05 versus control group, t test). Data are presented as mean ± SD. See also Figure S5.
Unlike ZDHCC1, STIM1 is a calcium-sensitive protein that has the ability to mediate calcium entry in many circumstances (Zhang et al., 2005). Interestingly, the knockdown of Stim1 and Zdhcc1 had no influence on the regulation of E. coli- or S. pneumoniae-induced calcium release from the ER to cytosol in THP1 (Figure 4D) and BMDM (Figure S5D) cells, indicating that STIM1- and ZDHCC1-independent pathways might contribute to ER calcium mobilization during bacterial infection.
The ATPase sarcoplasmic/ER Ca2+ transporting (ATP2A, also known as SERCA) proteins, including ATP2A1–3, are additional pumps that transport calcium ions from the cytoplasm into the ER (Periasamy and Kalyanasundaram, 2007). We therefore determined the function of the ATP2A family in the regulation of F3 release. Indeed, the knockdown of Atp2a2 (but not that of Atp2a1 or Atp2a3) enhanced the level of cytosolic calcium and F3 release (but not IFNβ release) in THP1 (Figures 4A–4D) and BMDM (Figures S5A–S5D) cells after infection by E. coli or S. pneumoniae.
Several variants of TMEM173 have been described in the human population. In particular, the V155M and N154S variant results in a gain-of-function mutation with the constitutive activation of TMEM173 in patients with STING-associated vasculopathy with onset in infancy (SAVI), a disorder involving abnormal inflammation with vasculitic rash and vasoocclusive processes (Liu et al., 2014). In comparison with WT cells, THP1 cells with TMEM173-V155M and TMEM173-N154S mutations exhibited increased IFNβ and F3 production during E. coli or S. pneumoniae infection (Figures 4E and 4F), suggesting these mutations provide a gain-of-function phenotype for both type I IFN and F3 release. Moreover, the overexpression of Atp2a2 limited E. coli- or S. pneumoniae-induced release of F3, but not IFNβ, in cells with TMEM173-V155M and TMEM173-N154S mutations (Figures 4E and 4F). As expected, the knockdown of Tmem173 inhibited E. coli- or S. pneumoniae-induced release of both F3 and IFNβ in cells with TMEM173-V155M and TMEM173-N154S mutations (Figures 4E and 4F). Together, these findings suggest that ATP2A2-mediated, but not STIM1-mediated, ER calcium uptake limits F3 release during TMEM173 activation.
TMEM173-Mediated GSDMD Cleavage Determines F3 Release and Lethal coagulation
Pyroptosis is a highly inflammatory form of regulated cell death that occurs depending on the cleavage of GSDMD by both inflammasome-dependent and inflammasome-independent mechanisms (Deng et al., 2018a, He et al., 2015, Kambara et al., 2018, Kayagaki et al., 2015, Orning et al., 2018, Sarhan et al., 2018, Shi et al., 2015, Taabazuing et al., 2017). The production of N-terminal fragments of GSDMD (GSDMD-N) plays a direct role in driving F3 release in macrophages and monocytes (Wu et al., 2019a, Yang et al., 2019). However, the functional relationship between TMEM173 and GSDMD remains unknown, although TMEM173 can promote pytoptosis and other forms of regulated cell death (Brault et al., 2018, Gaidt et al., 2017, Gulen et al., 2017, Larkin et al., 2017, Zierhut et al., 2019). The generation of GSDMD-N after E. coli or S. pneumoniae infection was absent in Tmem173−/−, but not in Tbk1−/− and Irf3−/− THP1 cells (Figure 5A). The knockdown of Itpr1 blocked GSDMD-N formation, whereas the knockdown of Atp2a2 increased GSDMD-N production in response to E. coli or S. pneumoniae infection (Figure 5B). Moreover, the overexpression of Atp2a2 limited E. coli- and S. pneumoniae-induced GSDMD-N formation in TMEM173-V155M and TMEM173-N154S cells (Figure 5C), indicating that an intracellular calcium signal was involved in GSDMD cleavage downstream of TMEM173 activation.
Figure 5. TMEM173-Mediated GSDMD Cleavage Promotes F3 Release.
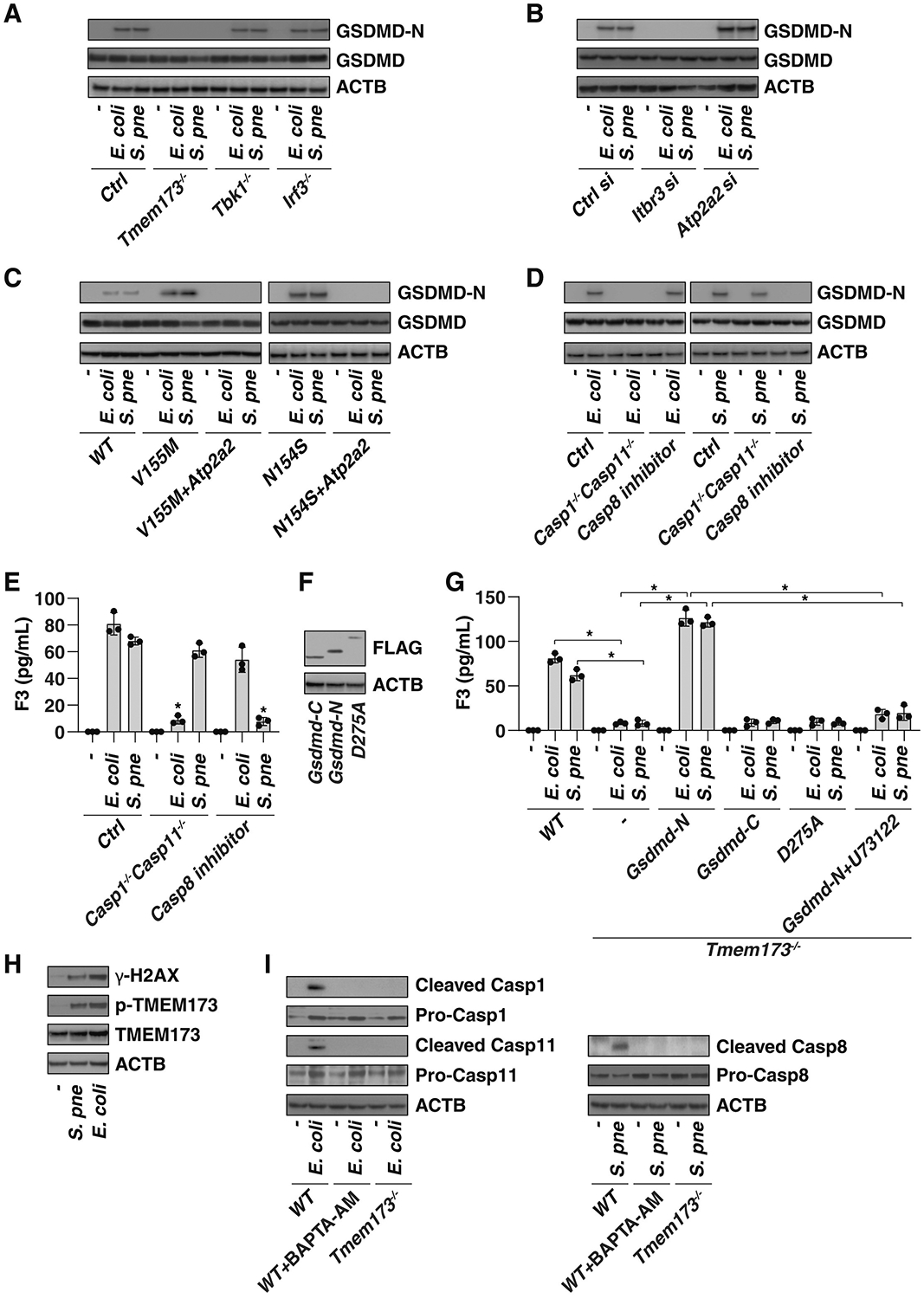
(A–C) Western blot analysis of GSDMD-N production in indicated knockout (A), knockdown (B), or mutated (C) THP1 cells after E. coli (25 MOI) or S. pneumoniae (25 MOI) infection for 24 h.
(D and E) Analysis of GSDMD-N production (D) and F3 release (E) in indicated BMDM cells after E. coli (25 MOI) or S. pneumoniae (25 MOI) infection for 24 h in the absence or presence of the CASP8 inhibitor Z-IETD-FMK (20 μM) (n = 3 wells per group; *p < 0.05 versus control group, t test).
(F) Cell lysates were analyzed by anti-FLAG antibody in Tmem173−/− THP1 cells after transfection indicated GSDMD mutants.
(G) Analysis of F3 release in indicated THP1 cells after E. coli (25 MOI) or S. pneumoniae (25 MOI) infection for 24 h in the absence or presence of the PLC inhibitor U73122 (10 μM) (n = 3 wells per group; *p < 0.05, t test).
(H) Western blot analysis of protein expression in THP1 cells after E. coli (25 MOI) or S. pneumoniae (25 MOI) infection for 24 h.
(I) Western blot analysis of protein expression in indicated THP1 cells after E. coli (25 MOI) or S. pneumoniae (25 MOI) infection in the absence or presence of BAPTA-AM (10 μM) for 24 h.
Data are presented as mean ± SD ([E] and [G]).
Although inflammatory caspases, including CASP1 and CASP11 (CASP4 and CASP5 in humans), are most often responsible for GSDMD cleavage (He et al., 2015, Kayagaki et al., 2015, Shi et al., 2015), there has been persistent evidence that some noninflammatory caspases, such as CASP8, can also influence GSDMD-N production in macrophages (Orning et al., 2018, Sarhan et al., 2018). The double depletion of Casp1 and Casp11 (Casp1−/−Casp11−/−), but not Z-IETD-FMK (a CASP8 inhibitor), prevented E. coli infection-induced GSDMD-N formation (Figure 5D) and F3 release (Figure 5E) in BMDMs. In contrast, S. pneumoniae-induced GSDMD-N formation (Figure 5D) and F3 release (Figure 5E) were inhibited by the CASP8 inhibitor, but were not prevented in Casp1−/−Casp11−/− cells exposed to S. pneumoniae. These findings suggest that the different caspase required for GSDMD-N production depends on the type of bacterial infection.
To establish the direct relationship between GSDMD-N and F3 release, we used plasmids expressing GSDMD mutants to continue this line of investigation. Transfection with Gsdmd-N (1–276), but not Gsdmd-C (277–487) or GSDMD cleavage mutant (D275A), restored F3 release in activated Tmem173−/− and Gsdmd−/− BMDMs (Figures 5F and 5G). Notably, the overexpression of GSDMD-N was not sufficient to cause F3 release because the basic expression of F3 is relatively low under basic conditions (Osterud, 1998, Yang et al., 2019). Our previous study indicates that phospholipase C gamma 1 (PLCG1) activation in the plasma membrane is required for GSDMD-N–mediated pyroptosis via Ca2+ elevation (Kang et al., 2018). The pharmacological inhibition of Plcg1 by U73122 inhibited Gsdmd-N-induced F3 release (Figure 5G). These findings further suggest that GSDMD-N is an intracellular regulator of IF release.
Given that DNA damage is an important trigger of TMEM173 activation (Li and Chen, 2018), we assayed the level of γ-H2AX, a marker of DNA damage, during E. coli and S. pneumoniae infection in THP1 cells. Consistent with previous findings that DNA damage is increased in host cells during bacterial infection (Žgur-Bertok, 2013), the level of γ-H2AX was upregulated by E. coli and S. pneumoniae (Figure 5H). As expected, the phosphorylation of TMEM173 on Ser366, an event of TMEM173 activation (Tanaka and Chen, 2012), was also upregulated by E. coli and S. pneumoniae (Figure 5H). These findings indicate that bacterial infection-induced DNA damage could cause TMEM173 activation.
We next examined the impact of TMEM173 on caspase activation during bacterial infection. Like the administration of the calcium chelator BAPTA-AM, the knockout of Tmem173 in THP1 cells inhibited the production of cleaved CASP1/CASP11 or cleaved CASP8 in response to E. coli or S. pneumoniae, respectively (Figure 5I). These finding indicate that TMEM173-mediated calcium signals are required for individual CASP activation in a bacterial type-dependent manner.
Although the global deletion of Gsdmd protects against LPS-induced coagulation activation in mice (Wu et al., 2019a, Yang et al., 2019), the function of GSDMD-N in driving coagulation activation in bacterial sepsis still remains unknown. Accordingly, we used GsdmdI105N/I105N mice (which bear a GSDMD with a cleavage-site mutation that renders the protein resistant to proteolytic activation by caspase) (Kayagaki et al., 2015) and the PLC inhibitor U73122 to examine the role of GSDMD-N in coagulation. GsdmdI105N/I105N mice and WT mice treated with the PLC inhibitor U73122 exhibited protection against death (Figure 6A), systemic coagulation activation (Figures 6B–6H), and tissue injury (Figures 6I and 6J) similar to that seen with anti-mouse F3 antibody after CLP. Similar protective effects on animal survival of GsdmdI105N/I105N mice and treatment with anti-mouse F3 antibody or U73122 in WT mice were obtained after infection with E. coli (Figure S6A) or S. pneumoniae (Figure S6B) or in a low-grade sepsis model, in which mice received antibiotics plus fluid resuscitation after CLP (Figure S6C). Collectively, these findings suggest that the formation of GSDMD-N promotes the activation of systemic coagulation responsible for lethal sepsis.
Figure 6. Inhibition of GSDMD and F3 Protects Mice against CLP-Induced Polymicrobial Sepsis.
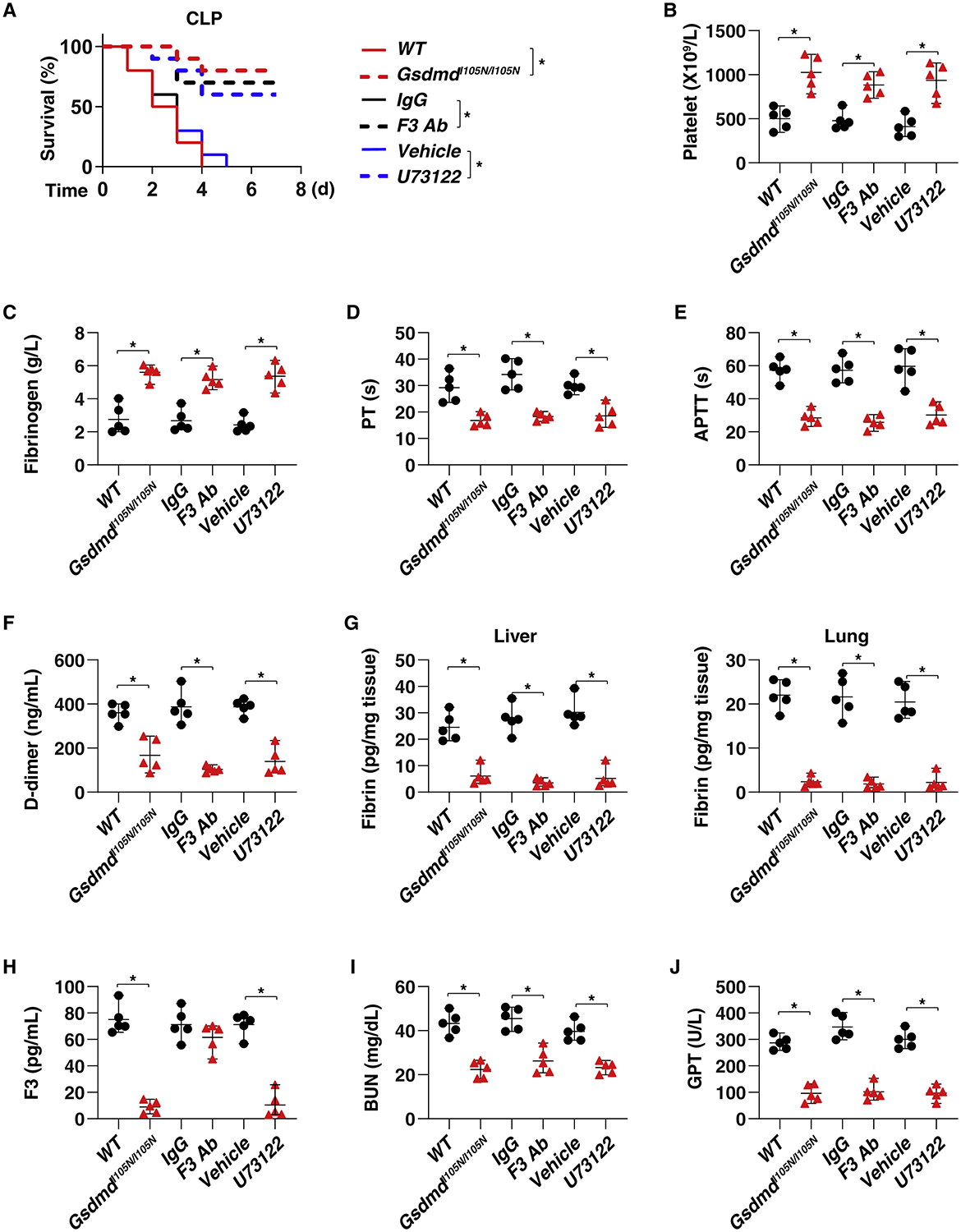
(A) Survival of the indicated mice in CLP-induced sepsis with or without treatment of IgG, anti-F3 antibody (10 mg/kg), or U73122 (30 mg/kg) (n = 10 mice per group; *p < 0.05, Kaplan-Meier survival analysis).
(B–J) In parallel, the levels of blood markers of DIC ([B]–[F], as in as in Figures 1B–1F, 2B–2F, and 3K–3O), tissue fibrin (G), plasma F3 (H), and markers of tissue injury ([I], BUN; [J], GPT) were assayed at 48 h (n = 5 mice per group; *p < 0.05, t test). See also Figure S6.
The TMEM173-GSDMD Pathway Correlates with the Severity of DIC in Patients with Sepsis
We next investigated the clinical relevance of dysregulated TMEM173-GSDMD pathway in sepsis. Our study focused on a cohort of 40 patients with bacterial sepsis, who were diagnosed according to the Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3) (Singer et al., 2016). Patient demographic and clinical characteristics are shown in Table S1. The International Society of Thrombosis and Hemostasis scoring algorithm was further used to stratify patients with sepsis into two groups, those with (n = 24, 60%) and without (n = 16, 40%) DIC, on the basis of laboratory analyses of platelet counts, prothrombin time-international normalized ratio (PT-INR), D-dimer, and fibrinogen (Patel et al., 2019). Sepsis patients with DIC had a higher 28-day mortality rate (33.33%) and plasma levels of proinflammatory mediators (tumor necrosis factor [TNF], interleukin 6 [IL6], interleukin 1 alpha [IL1A], interleukin 1 beta [IL1B], and high-mobility group box 1 [HMGB1]) than did the non-DIC group (Table S1). There was also a strong correlation between the sequential organ-failure assessment (SOFA) and DIC scores in patients with sepsis (Figure 7A). PBMC TMEM173 and GSDMD mRNA levels also significantly correlated with SOFA (Figure 7B) and DIC (Figure 7C) scores. The expression of PBMC TMEM173 and GSDMD mRNA levels in the DIC and non-survivor groups was higher than in the non-DIC (Figure 7D) and survivor groups (Figure 7E). There was a significant positive correlation between TMEM173 and GSDMD mRNA expression and plasma proinflammatory mediator levels (TNF, IL6, IL1A, IL1B, and HMGB1) (Figure S7). Moreover, the protein levels of p-TMEM173, TMEM173, GSDMD-N, and GSDMD in the DIC group were higher than in the non-DIC groups (Figure 7F). These findings further support a potential pathogenic role for the TMEM173-GSDMD pathway in promoting lethal DIC during sepsis.
Figure 7. Association of the TMEM173-GSDMD Axis with the Severity of DIC in Patients with Sepsis.
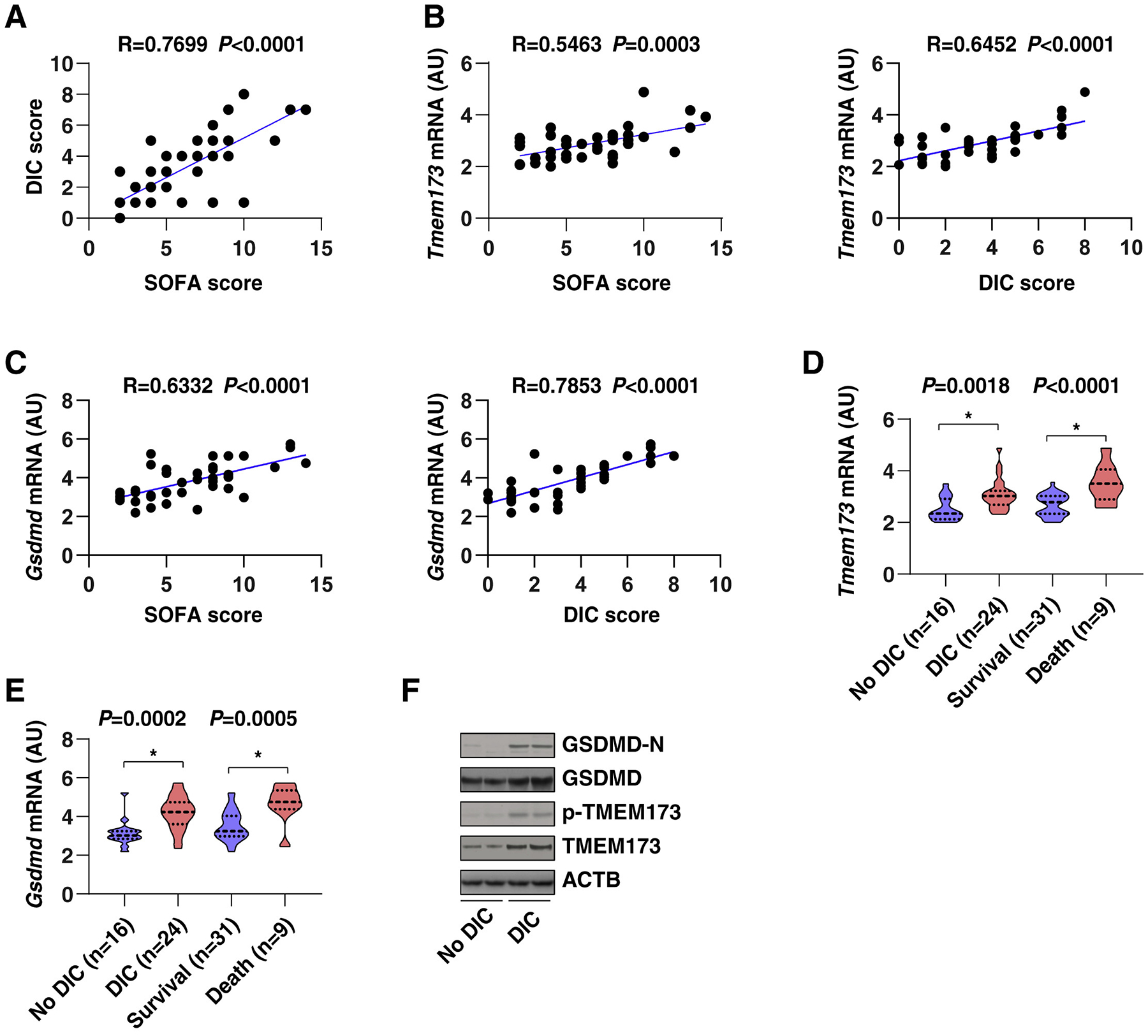
(A–C) The correlation assay between indicated DIC score (A), SOFA score (A), and TMEM173 (B) and GSDMD (C) expression in PBMC from sepsis patients (Pearson rank correlation test).
(D and E) The mRNA level of Tmem173 (D) and Gsdmd (E) in PBMC of indicated sepsis patients (data are presented in a violin plot, t test).
(F) The protein levels of p-TMEM173, TMEM173, GSDMD-N, and GSDMD were upregulated in the DIC group in comparison with non-DIC groups. See also Table S1 and Figure S7.
Discussion
Innate immunity is the first line of defense against infection, but its excessive activation can result in tissue injury and host lethality (Akira et al., 2006). TMEM173 has previously been shown to regulate inflammation and infection in health and disease (Barber, 2015, Motwani et al., 2019). However, the evidence for TMEM173 in the regulation of coagulation activation during lethal infection had not been established. In this study, we revealed a distinct pathway by which myeloid TMEM173 drives F3 release and blood coagulation via calcium-dependent GSDMD activation in bacterial sepsis. Moreover, the expression of TMEM173 and GSDMD in PBMC correlated with the severity of DIC and inflammation in patients with sepsis. Identifying coagulation abnormalities as a critical effector of TMEM173-mediated septic death could open an area of research in immunocoagulation.
TMEM173 was originally identified as a cytosolic nucleic-acid sensor that controls viral infection via the induction of a type I IFN response (Ishikawa and Barber, 2008, Sun et al., 2009, Zhong et al., 2008). This process requires cGAS, a DNA-binding protein, to synthesize the second messenger cGAMP that then actives TMEM173 (Sun et al., 2013). Unlike virus DNA, TMEM173 can directly recognize CDNs from invading bacteria (Marinho et al., 2017). Finally, TMEM173 can activate TBK1 and IRF3, resulting in the enhanced expression of type I IFNs and proinflammatory cytokines (e.g., TNF and IL6) (Liu et al., 2015, Shang et al., 2019, Tanaka and Chen, 2012). We discovered an alternative TMEM173 pathway that leads to F3 release in macrophages and monocytes and contributes to coagulation activation in sepsis. This pathway does not rely on the classical components of the TMEM173 pathway, including cGAS, TBK1, IRF3, and type I IFNs. Thus, downstream of TMEM173, both classical and alternative pathways, are involved in the pathologeny of sepsis and septic shock.
Our study highlights the impact of calcium fluxes on the regulation of TMEM173-mediated coagulation activation. Calcium is an intracellular second messenger, and the ER is the largest calcium storage organelle in eukaryotic cells (Schwarz and Blower, 2016). Production of the classical TMEM173-mediated type I IFNs requires TMEM173 trafficking to the ERGIC, which can be regulated by certain proteins, such as ZDHHC1 and STIM1, in a calcium-independent manner (Dobbs et al., 2015, Gui et al., 2019, Mukai et al., 2016, Srikanth et al., 2019, Zhou et al., 2014). However, these TMEM173 trafficking pathways could be dispensable for TMEM173-mediated F3 release. We demonstrated that the activation of ER-associated TMEM173 promotes ER calcium release that is responsible for F3 release in macrophages and monocytes. Mechanisticcally, TMEM173 promotes ER calcium release via direct binding to ITPR1, a predominant calcium channel on the ER membrane. In contrast, ATP2A2-mediated ER calcium uptake can reverse TMEM173-mediated F3 release (Figure 7G). These findings provide a molecular mechanism for the prolonged ER stress susceptibility associated with intravascular coagulation and septic death in mice.
The molecular mechanism of immunocoagulation has recently been uncovered in a study of inflammasome activation (Wu et al., 2019a, Yang et al., 2019), an event occurring in innate immune responses to many pathogenic infections and tissue damage (Broz and Dixit, 2016, Guo et al., 2015). In particular, CASP1- and/or CASP11-activated GSDMD drives pyroptosis with F3 release, leading to blood coagulation and organismal death in endotoxemia (Wu et al., 2019a, Yang et al., 2019). This process is independent of inflammasome-mediated production of IL1B and IL18 but requires phosphatidylserine exposure (Wu et al., 2019a, Yang et al., 2019), a feature of caspase-mediated apoptosis (Fadok et al., 1998). Additionally, the coagulation protease thrombin can cleave pro-IL1A to activate a subsequent inflammatory cascade, linking the key effector of coagulation to inflammation (Burzynski et al., 2019). Indeed, the levels of proinflammatory mediators, including IL1B, IL1A, TNF, HMGB1, and IL6, are elevated in septic patients with DIC, supporting the notion that DIC is associated with the inflammatory and infectious processes.
Regulated cell death exhibits various molecular and genetic characteristics (Tang et al., 2019) involved in the regulation of innate immunity and infection (Jorgensen et al., 2017). In addition to its effect on inflammation and immunity, TMEM173 is a multifunctional regulator of various types of regulated cell death, including apoptosis (Gulen et al., 2017, Larkin et al., 2017, Wu et al., 2019b), necroptosis (Brault et al., 2018, Sarhan et al., 2019), pyroptosis (Gaidt et al., 2017), NETosis (Lood et al., 2016), and autophagy (Gui et al., 2019, Liu et al., 2019), although distinct signaling pathways could be divergently involved in these effects, depending on varying context. Thus, it is crucial to understand the precise rules that govern the molecular interplay between TMEM173-mediated immune and death signals in diseases.
Our present study demonstrates that TMEM173-mediated ER calcium release promotes cleavage of GSDMD (to generate GSDMD-N) by CASP1/CASP11 or CASP8 in response to E. coli or S. pneumoniae, respectively. These findings are consistent with previous studies showing that E. coli and S. pneumoniae can activate CASP1/CASP11 and CASP8, respectively (Goddard et al., 2019, Kayagaki et al., 2011, Schmeck et al., 2004). The activation of GSDMD-N triggers pyroptosis and requires lipid peroxidation-mediated PLCG1 activation and subsequent calcium influx (Kang et al., 2018). As a negative feedback mechanism resulting from calcium influx, the activation of endosomal sorting complexes required for transport (ESCRT)-dependent membrane repair could limit oxidative injury of the plasma membrane and promote cell survival (Rühl et al., 2018). In addition, GSDMD-mediated potassium efflux can limit TMEM173-mediated type I IFN production in response to bacterial dsDNA (Banerjee et al., 2018), further supporting the notion that electrolyte abnormalities can cause inflammation and immune dysfunction in critically ill patients (Besen et al., 2015). We found that CLP-, E. coli-, or S. pneumoniae-induced coagulation activation and host death were blocked in GSDMD cleavage-site mutant mice or by the administration of PLCG1 inhibitor or anti-F3 antibody, supporting that GSDMD-N-mediated F3 release is involved in lethal DIC.
In summary, our study identifies a previously unknown myeloid TMEM173-driven coagulation activation pathway associated with calcium-mediated GSDMD activation but not with the type I IFN response. We demonstrate that genetic or pharmacological inhibition of the TMEM173-GSDMD pathway can correct DIC and improve animal survival in three relevant models of bacterial sepsis (Gram-negative, Gram-positive, and mixed). Given the complex and heterogeneous nature of human sepsis, the broad significance of our findings needs to be further explored in further preclinical models of infection-driven sepsis, as well as in clinical trials.
Lead Contact and Materials Availability
This study did not generate new or unique reagents. Further information and requests for reagents may be obtained from the Lead Contact, Daolin Tang (Email: daolin.tang@utsouthwestern.edu).
Reagents are as described in Key Resources Table.
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-GSDMD-N antibody | Cell Signaling Technology | 36425; RRID: AB_2799099 |
| Anti-GSDMD antibody | Cell Signaling Technology | 96458; RRID: N/A |
| Anti-TMEM173 antibody | Cell Signaling Technology | 13647; AB_2732796 |
| Anti-phospho-TMEM173 antibody | Cell Signaling Technology | 19781; RRID: AB_2737062 |
| Anti-ITPR1 antibody | Abcam | ab5804; RRID: AB_305124 |
| Anti-ACTB antibody | Cell Signaling Technology | 3700; RRID: AB_2242334 |
| Anti-γ-H2AX antibody | Cell Signaling Technology | 9718; RRID: AB_2118009 |
| Anti-cleaved CASP1 antibody | Cell Signaling Technology | 4199; RRID: AB_1903916 |
| Anti-CASP1 antibody | Cell Signaling Technology | 2225; RRID: AB_2243894 |
| Anti-CASP11 antibody | Cell Signaling Technology | 14340; RRID: AB_2728693 |
| Anti-cleaved CASP8 antibody | Cell Signaling Technology | 9496; RRID: AB_561381 |
| Anti-CASP8 antibody | Cell Signaling Technology | 9746; RRID: AB_2275120 |
| Anti-F3 antibody (monoclonal rat IgG2c; clone #355220) | R&D systems | MAB3178; RRID: N/A |
| Anti-rat IgG2c antibody (clone MRG2c-67) | BioLegend | 407602 RRID: AB_345342 |
| HRP-linked anti-mouse IgG secondary antibody | Cell Signaling Technology | 7076; RRID: AB_330924 |
| HRP-linked anti-rabbit IgG secondary antibody | Cell Signaling Technology | 7074; RRID: AB_2099233 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| TUDCA | Sigma-Aldrich | 580549 |
| 2-APB | Sigma-Aldrich | 100065 |
| 4PBA | Santa Cruz Biotechnology | 1821-12-1 |
| Primaxin | Merck | 0006-3514-58 |
| U73122 | Sigma-Aldrich | 662035 |
| BAPTA-AM | Sigma-Aldrich | 196419 |
| Z-IETD-FMK | Selleck Chemicals | S7314 |
| Thapsigargin | Sigma-Aldrich | T9033 |
| Tunicamycin | Sigma-Aldrich | T7765 |
| Cell lysis buffer (10X) | Cell Signaling Technology | 9803 |
| SDS sample buffer (2X) | Thermo Fisher Scientific | LC2676 |
| SuperSignal West Pico Chemiluminescent Substrate | Thermo Fisher Scientific | 34580 |
| SuperSignal West Femto Maximum Sensitivity Substrate | Thermo Fisher Scientific | 34095 |
| RIPA buffer | Cell Signaling Technology | 9806 |
| Phosphatase inhibitor cocktail | Sigma-Aldrich | P0044 |
| Protease inhibitor cocktail | Promega | G6521 |
| Nutrient agar | Becton Dickinson | 213000 |
| Brain heart infusion agar | Becton Dickinson | 211065 |
| Dulbecco’s Modified Eagle’s Medium | Thermo Fisher Scientific | 11995073 |
| RPMI 1640 | Thermo Fisher Scientific | 11875119 |
| Fetal bovine serum | Millipore | TMS-013-B |
| 1% penicillin and streptomycin | Thermo Fisher Scientific | 15070-063 |
| SsoFast EvaGreen Supermix | Bio-Rad | 172-5204 |
| TBST | Cell Signaling Technology | 9997 |
| 5% nonfat dry milk | Cell Signaling Technology | 9999 |
| Protein A agarose beads | Cell Signaling Technology | 9863 |
| Critical Commercial Assays | ||
| Human IFNβ ELISA kit | R&D systems | DIFNB0 |
| Mouse IFNβ ELISA kit | R&D systems | 42400-1 |
| Human F3 ELISA kit | R&D systems | DCF300 |
| Mouse F3 ELISA kit | Abcam | ab214091 |
| Human fibrin ELISA kit | MyBioSource | MBS265263 |
| Mouse fibrin ELISA kit | MyBioSource | MBS706338 |
| Human D-dimer ELISA kit | Abcam | ab196269 |
| Mouse D-dimer ELISA kit | MyBioSource | MBS723281 |
| Human TNF ELISA kit | R&D Systems | DTA00D |
| Human IL1A ELISA kit | R&D Systems | DLA50 |
| Human IL1B ELISA kit | R&D Systems | DLB50 |
| Human IL6 ELISA kit | R&D Systems | D6050 |
| Human HMGB1 ELISA kit | IBL International | ST51011 |
| iScript cDNA Synthesis Kit | Bio-Rad | 1708890 |
| BCA assay kit | Cell Signaling Technology | 7780 |
| Rneasy Plus Kit | QIAGEN | 74134 |
| Fura-2 Calcium Flux Assay Kit | Abcam | ab176766 |
| EasySep Mouse Monocyte Isolation Kit | STEMCELL Technologies | 19861 |
| Experimental Models: Cell Lines | ||
| THP1 | ATCC | TIB-202 |
| HPBM | STEMCELL Technologies | 70042 |
| Tmem173−/− THP1 | InvivoGen | thpd-kostg |
| Tbk1−/− THP1 | InvivoGen | thpd-kotbk |
| Irf3−/− THP1 | InvivoGen | thpd-koirf3 |
| TMEM173-V155M THP1 | InvivoGen | thpd-m155 |
| TMEM173-N154S THP1 | InvivoGen | thpd-s154 |
| Experimental Models: Organisms/Strains | ||
| Tmem173−/− mice | The Jackson Laboratory | 025805 |
| Tmem173flox/flox mice | The Jackson Laboratory | 031670 |
| Lyz2-Cre mice | The Jackson Laboratory | 004781 |
| Lck-Cre mice | The Jackson Laboratory | 003802 |
| GsdmdI105N/I105N mice | Dr. Vishva M. Dixit | N/A |
| cGAS−/− mice | The Jackson Laboratory | 026554 |
| Irf3−/− | RIKEN BioResource Research Center | RBRC00858 |
| Ifnar1−/− | The Jackson Laboratory | 028288 |
| C57BL/6 wild-type mice | The Jackson Laboratory | 000664 |
| E. coli (Serovar O1:K1:H7) | ATCC | 11775 |
| S. pneumoniae (CIP 104225) | ATCC | 6303 |
| Oligonucleotides | ||
| Mouse Zdhhc1 siRNA | Dharmacon | L-056132-01-0005 |
| Mouse Stim1 siRNA | Dharmacon | L-062376-00-0005 |
| Mouse Atp2a1 siRNA | Dharmacon | L-064501-00-0005 |
| Mouse Atp2a2 siRNA | Dharmacon | L-040968-00-0005 |
| Mouse Atp2a3 siRNA | Dharmacon | L-041023-00-0005 |
| Human Zdhhc1 siRNA | Dharmacon | L-014193-01-0005 |
| Human Stim1 siRNA | Dharmacon | L-011785-00-0005 |
| Human Atp2a1 siRNA | Dharmacon | L-006113-00-0005 |
| Human Atp2a2 siRNA | Dharmacon | L-004082-00-0005 |
| Human Atp2a3 siRNA | Dharmacon | L-006114-00-0005 |
| Human Tmem173 siRNA | Dharmacon | L-024333-00-0005 |
| Human Tbk1 siRNA | Dharmacon | L-003788-00-0005 |
| Human Irf3 siRNA | Dharmacon | L-006875-00-0005 |
| See Table S2 for primers used for qPCR | This paper | Table S2 |
| Gsdmd-N cDNA | Dr. Feng Shao | Shi et al., 2015 |
| Gsdmd-C cDNA | Dr. Feng Shao | Shi et al., 2015 |
| Gsdmd-D275A | Dr. Feng Shao | Shi et al., 2015 |
| Atp2a2 cDNA | OriGene | RC207626 |
| Software and Algorithms | ||
| Image Lab software | Bio-Rad | http://www.bio-rad.com/en-us/product/image-lab-software?ID=KRE6P5E8Z |
| CFX Manager software | Bio-Rad | http://www.bio-rad.com/en-us/sku/1845000-cfx-manager-software?ID=1845000 |
| Prism 8 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
Experimental Model and Subject Details
Cell Culture
A THP1 (TIB-202) cell line was obtained from the American Type Culture Collection (ATCC). The Tmem173−/− (thpd-kostg), Tbk1−/− (thpd-kotbk), Irf3−/− (thpd-koirf3), TMEM173-V155M (thpd-m155), and TMEM173-N154S (thpd-s154) cell lines were obtained from InvivoGen. Primary BMDMs from indicated mice were obtained using 30% L929-cell conditioned medium as a source of CSF2/granulocyte/macrophage colony stimulating factor (Weischenfeldt and Porse, 2008). Primary HPBMs were obtained from STEMCELL Technologies (70042). Tbk1−/− BMDMs were generated by CRISPR/Cas9 technology (KM Bioscience). Monocytes were isolated from whole blood using an EasySep Mouse Monocyte Isolation Kit from STEMCELL Technologies (19861). All cells used were authenticated using STR profiling, and mycoplasma testing was negative.
These cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM; 11995073, Thermo Fisher Scientific) or RPMI 1640 (11875119, Thermo Fisher Scientific) supplemented with 10% heat-inactivated fetal bovine serum (TMS-013-B, Millipore) and 1% penicillin and streptomycin (15070–063, Thermo Fisher Scientific) at 37°C, 95% humidity, and 5% CO2.
Bacterial Strains and Culturing Conditions
E. coli (Serovar O1:K1:H7, 11775) or S. pneumoniae (CIP 104225, 6303) were obtained from ATCC. E. coli was grown in nutrient agar (213000, Becton Dickinson) at 37°C under aerobic conditions. S. pneumoniae was grown in brain heart infusion agar (211065, Becton Dickinson) at 37°C under 5% CO2 conditions. Indicated cells were infected by 25 MOI E. coli or S. pneumoniae. There were no antibiotics in the cell culture medium used for bacterial infection.
Animal Model of Sepsis
WT (000664), Tmem173−/− (025805), cGAS−/− (026554), Ifnar1−/− (028288), Tmem173flox/flox (031670), Lyz2Cre (004781), and LckCre (003802) mice were obtained from the Jackson Laboratory. Irf3−/− mice were obtained from RIKEN BioResource Research Center (RBRC00858). Myeloid cell- (Tmem173Mye−/−) or T cell (Tmem173T−/−) specific–Tmem173-deficient mice were generated by crossing the Tmem173flox/flox mice with Lyz2Cre or LckCre mice, respectively. GsdmdI105N/I105N mice were a gift from Dr. Vishva M. Dixit (Genentech). All mice used in this study were on a C57BL/6J background. Mice were housed with their littermates in groups of 4 or 5 animals per cage and kept on a regular 12 h light and dark cycle (7:00–19:00 light period). Food and water were available ad libitum. Experiments were carried out under pathogen-free conditions with randomly chosen littermates of the same sex, matched by age and body weight. The health status of mouse lines was routinely checked by veterinary staff. We conducted all animal care and experimentation in accordance with the Association for Assessment and Accreditation of Laboratory Animal Care guidelines (http://www.aaalac.org) and with approval from institutional animal care and use committees.
Poly-microbial sepsis model (Chen et al., 2019a, Chen et al., 2019b, Deng et al., 2018b, Kang et al., 2018): Sepsis was induced in male or female C57BL/6J mice (8 to 10 weeks old, 22 to 26 g weight, female or male [1:1]) using a surgical procedure termed CLP (Rittirsch et al., 2009). Briefly, anesthesia was induced with ketamine (80–100 mg/kg/i.p.) and xylazine (10–12.5 mg/kg/i.p.). A small midline abdominal incision was made and the cecum was exteriorized and ligated with 4–0 silk immediately distal to the ileocecal valve without causing intestinal obstruction. The cecum was then punctured once with a 22-gauge needle. The abdomen was closed in two layers and mice were injected subcutaneously with 1 mL Ringer’s solution, including analgesia (0.05 mg/kg buprenorphine). Control IgG, anti-F3 mAbs (10 mg/kg), TUDCA (200 mg/kg), 2-APB (20 mg/kg), U73122 (30 mg/kg) or vehicle were repeatedly administered intraperitoneally to mice at 2, 24, 48, and 72 h after CLP. For a low-grade CLP model, mice were given Primaxin (25 mg/kg; Merck), a broad-spectrum carbapenem antibiotic combination, beginning 2 h after CLP and continuing every 12 h for the first 3 days (Iskander et al., 2016).
Single-bacterial sepsis model: Male or female C57BL/6J mice (8 to 10 weeks old, 22 to 26 g weight, female or male [1:1]) were intraperitoneally given a single dose of E. coli (Serovar O1:K1:H7, 1 × 109 CFU; ATCC) or S. pneumoniae (CIP 104225, 1 × 109 CFU; ATCC). Control IgG, anti-F3 mAbs (10 mg/kg), TUDCA (200 mg/kg), 2-APB (20 mg/kg), U73122 (30 mg/kg) or vehicle were repeatedly administered intraperitoneally to mice at 2, 24, 48, and 72 h after bacterial infection.
Survival was observed for up to 7 days. Blood was collected from anesthetized mice by cardiac puncture using heparinized syringes. Plasma was further obtained from anticoagulated whole blood after removing the blood cells by a centrifugation (2000 g × 15 min) at 4°C. Tissues were cut into pieces and frozen in liquid nitrogen before storing at −80°C until protein extraction, and were homogenized at 4°C in ice-cold RIPA buffer (9806, Cell Signaling Technology) with phosphatase inhibitor cocktail (P0044, Sigma-Aldrich).
Patient Samples
PBMCs and plasma samples from patients with bacterial sepsis were collected from Daping Hospital of the Third Military Medical University at the time of admission to ICU before initiation of treatment. The collection of samples was approved by the institutional review board. Sepsis was established according to the Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3) (Singer et al., 2016), which[REMOVED HYPERLINK FIELD] defines sepsis as life-threatening organ dysfunction caused by a dysregulated host response to infection. Organ dysfunction can be identified as an acute change in total SOFA score of ≥ 2 points due to the infection (Singer et al., 2016). Patients were excluded from the study if they had received a blood transfusion within the past 4 months, a platelet transfusion within the past month, or anticoagulant therapy with the past month. Patients were also excluded if they had a preexisting platelet disorder, such as idiopathic thrombocytopenic purpura, chronic myelogenous leukemia, multiple myeloma, primary myelofibrosis, polycythemia vera, primary thrombocythemia, and thrombotic thrombocytopenic purpura.
Basic demographic and clinical data, including age, sex, infection site, types of infection, SOFA score, DIC score, coagulation markers, length of hospital and ICU stay, and 28-day mortalities were retrieved from the registry (Table S1). The SOFA grades the function of six organ systems on a scale of 0 to 4 depending on the degree of dysfunction using objective measurements (Singer et al., 2016). The maximum SOFA scores were the highest (worst) scores within 24 h of emergency department arrival. The DIC scores were evaluated using the scoring system from the International Society on Thrombosis and Haemostasis (Taylor et al., 2001).
Methods Details
Biochemical and Coagulation Assay
Commercially available ELISA kits were used to measure the concentrations or activity of IFNβ (DIFNB0 or 42400–1, R&D Systems), F3 (DCF300, R&D Systems; ab214091, Abcam), fibrin (MBS265263 or MBS706338, MyBioSource), D-dimer (ab196269, Abcam; MBS723281, MyBioSource), TNF (DTA00D, R&D Systems), IL1A (DLA50, R&D Systems), IL1B (DLB50, R&D Systems), IL6 (D6050, R&D Systems), and HMGB1 (ST51011, IBL International) in the indicated samples. Measurement of GPT/ALT and BUN in the plasma was performed using an IDEXX Catalyst Dx Chemistry Analyzer. PT, APTT, and fibrinogen were measured in an automated coagulometer (Sysmex CA-7000). Platelet count was measured using an IDEXX ProCyte Dx Hematology Analyzer.
RNAi
TARGETplus SMART pool small interfering RNAs (siRNAs) against the indicated genes as described in Key Resources Table were purchased from Dharmacon. This pool was a mixture of four siRNAs provided as a single reagent. The Neon Electroporation System from Invitrogen was used to deliver siRNAs into cells, according to the manufacturer’s instructions. Transfected cells were recovered in complete medium. The medium was replaced at 3 h post electroporation. The cells were cultured for 48 h before further examination.
Q-PCR
Total RNA was extracted using an RNeasy Plus Kit (74134, QIAGEN) according to the manufacturer’s instructions. First-strand cDNA was synthesized from 1 μg of RNA using the iScript cDNA Synthesis kit (1708890, Bio-Rad). Briefly, 20 μL reactions were prepared by combining 4 μl of iScript Select reaction mix, 2 μl of gene-specific enhancer solution, 1 μl of reverse transcriptase, 1 μl of gene-specific assay pool (20 ×, 2 μM), and 12 μl of RNA diluted in Rnase-free water. Quantitative real-time PCR was carried out using synthesized cDNA, primers as described in Key Resources Table, and SsoFast EvaGreen Supermix (172–5204, Bio-Rad). The data were normalized to Rna18s and the fold change was calculated via the 2-ΔΔCt method (Deng et al., 2018b). The relative concentrations of mRNA were expressed in arbitrary units based on the untreated group, which was assigned a value of 1.
Western Blot Analysis
Cells were lysed in Cell Lysis Buffer (9803, Cell Signaling Technology) with protease inhibitor cocktail (G6521, Promega) and phosphatase inhibitor cocktail (P0044, Sigma-Aldrich). Cleared lysates were resolved by SDS-PAGE (3450124, Bio-Rad) and then transferred onto PVDF membranes (1704273, Bio-Rad). The membranes were blocked with Tris-buffered saline Tween 20 (TBST; 9997, Cell Signaling Technology) containing 5% nonfat dry milk (9999, Cell Signaling Technology) for 1 h at room temperature and then incubated with the indicated primary antibodies (1:1000) overnight at 4°C. After being washed with TBST, the membranes were incubated with an HRP-linked anti-mouse IgG secondary antibody (1:1000; 7076, Cell Signaling Technology) or HRP-linked anti-rabbit IgG secondary antibody (1:1000; 7074, Cell Signaling Technology) for 1 h at room temperature. The membranes were washed three times in TBST and then visualized and analyzed with a ChemiDoc Touch Imaging System (1708370, Bio-Rad).
Immunoprecipitation Analysis
Cells were lysed at 4°C in ice-cold RIPA buffer (9806, Cell Signaling Technology) with protease inhibitor cocktail (G6521, Promega), and cell lysates were cleared by a brief centrifugation (12000 g, 10 min). Concentrations of proteins in the supernatant were determined by a BCA Protein Assay Kit (7780, Cell Signaling Technology). Prior to immunoprecipitation, samples containing equal amounts of proteins were pre-cleared with protein A agarose beads (9863, Cell Signaling Technology) at 4°C for 3 h, and subsequently incubated with various irrelevant IgG or specific antibodies (5 μg/mL) in the presence of protein A agarose beads for 2 h or overnight at 4°C with gentle shaking. Following incubation, protein A agarose beads were washed extensively with phosphate-buffered saline and proteins were eluted by boiling in 2 × sodium dodecyl sulfate sample buffer (LC2676, Thermo Fisher Scientific) before sodium dodecyl sulfate polyacrylamide gel electrophoresis.
Measurement of Intracellular Calcium
The cytosolic calcium signal was assayed using a Fura-2 Calcium Flux Assay Kit (ab176766, Abcam) according to the manufacturer’s instructions. Cytosolic calcium increases were presented as the ratio of emitted fluorescence (510 nm) after excitation at 340 and 380 nm, relative to the ratio measured prior to cell stimulation. The relative concentrations of cytosolic calcium were normalized to cell numbers and expressed in arbitrary units based on the control group, which was assigned a value of 1.
Quantification and Statistical analysis
Data are presented as mean ± SD of three independent experiments except where otherwise indicated. All data met the assumptions of the tests (e.g., normal distribution). Unpaired Student’s t tests were used to compare the means of two groups. One-way analysis of variance (ANOVA) was used for comparison between the different groups. When the ANOVA was significant, post hoc testing of differences between groups was performed using the least significant difference test. The correlation analysis was performed with the Pearson rank correlation test. The Kaplan-Meier method was used to compare differences in mortality rates between groups. A two-tailed p value of < 0.05 was considered statistically significant. The exact value of n within the figures is indicated in the figure legends. We did not exclude samples or animals. No statistical methods were used to predetermine sample sizes, but our sample sizes are similar to those generally employed in the field.
Data and Code Availability
This study did not generate any unique datasets or code.
Supplementary Material
Acknowledgments
We thank Dave Primm (Department of Surgery, University of Texas Southwestern Medical Center) for his critical reading of the manuscript. H.W. is supported by grants from the US National Institutes of Health (R01GM063075 and R01AT005076). T.B. is supported by a grant from the US National Institutes of Health (R35GM127027). G.K. is supported by the Ligue contre le Cancer (équipe labelisée); Agence National de la Recherche (ANR) - Projets blancs; ANR under the frame of E-Rare-2, the ERA-Net for Research on Rare Diseases; Association pour la recherché sur le cancer (ARC); Cancéropôle Ile-de-France; Chancelerie des universités de Paris (Legs Poix), Fondation pour la Recherche Médicale (FRM); a donation by Elior; European Research Area Network on Cardiovascular Diseases (ERA-CVD, MINOTAUR); Gustave Roussy Odyssea, the European Union Horizon 2020 Project Oncobiome; Fondation Carrefour; High-end Foreign Expert Program in China (GDW20171100085 and GDW20181100051); Institut National du Cancer (INCa); Inserm (HTE); Institut Universitaire de France; LeDucq Foundation; the LabEx Immuno-Oncology; the RHU Torino Lumière; the Seerave Foundation; the SIRIC Stratified Oncology Cell DNA Repair and Tumor Immune Elimination (SOCRATE); and the SIRIC Cancer Research and Personalized Medicine (CARPEM). J.L. is supported by grants from the National Natural Science Foundation of China (81830048). J.J. is supported by grants from the National Natural Science Foundation of China (81530063). L.Z. is supported by the Excellent Youth Grant of State Key Laboratory of Trauma, Burns, and Combined Injury of China (SKLYQ201901).
Footnotes
Declaration of interests
The authors declare no conflicts of interest or financial interests.
REFERENCES
- Ablasser A, and Chen ZJ (2019). cGAS in action: Expanding roles in immunity and inflammation. Science 363, 363. [DOI] [PubMed] [Google Scholar]
- Aglietti RA, Estevez A, Gupta A, Ramirez MG, Liu PS, Kayagaki N, Ciferri C, Dixit VM, and Dueber EC (2016). GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc. Natl. Acad. Sci. USA 113, 7858–7863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akira S, Uematsu S, and Takeuchi O (2006). Pathogen recognition and innate immunity. Cell 124, 783–801. [DOI] [PubMed] [Google Scholar]
- Banerjee I, Behl B, Mendonca M, Shrivastava G, Russo AJ, Menoret A, Ghosh A, Vella AT, Vanaja SK, Sarkar SN, et al. (2018). Gasdermin D Restrains Type I Interferon Response to Cytosolic DNA by Disrupting Ionic Homeostasis. Immunity 49, 413–426.e5, e415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber GN (2015). STING: infection, inflammation and cancer. Nat. Rev. Immunol 15, 760–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besen BA, Gobatto AL, Melro LM, Maciel AT, and Park M (2015). Fluid and electrolyte overload in critically ill patients: An overview. World J. Crit. Care Med 4, 116–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettigole SE, and Glimcher LH (2015). Endoplasmic reticulum stress in immunity. Annu. Rev. Immunol 33, 107–138. [DOI] [PubMed] [Google Scholar]
- Bianchi ME (2007). DAMPs, PAMPs and alarmins: all we need to know about danger. J. Leukoc. Biol 81, 1–5. [DOI] [PubMed] [Google Scholar]
- Bolivar S, Anfossi R, Humeres C, Vivar R, Boza P, Munoz C, Pardo-Jimenez V, Olivares-Silva F, and Diaz-Araya G (2018). IFN-b Plays Both Pro- and Anti-inflammatory Roles in the Rat Cardiac Fibroblast Through Differential STAT Protein Activation. Front. Pharmacol 9, 1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brault M, Olsen TM, Martinez J, Stetson DB, and Oberst A (2018). Intracellular Nucleic Acid Sensing Triggers Necroptosis through Synergistic Type I IFN and TNF Signaling. J. Immunol 200, 2748–2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broz P, and Dixit VM (2016). Inflammasomes: mechanism of assembly, regulation and signalling. Nat. Rev. Immunol 16, 407–420. [DOI] [PubMed] [Google Scholar]
- Burzynski LC, Humphry M, Pyrillou K, Wiggins KA, Chan JNE, Figg N, Kitt LL, Summers C, Tatham KC, Martin PB, et al. (2019). The Coagulation and Immune Systems Are Directly Linked through the Activation of Interleukin-1a by Thrombin. Immunity 50, 1033–1042.e6, e1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavaillon JM, and Adib-Conquy M (2005). Monocytes/macrophages and sepsis. Crit. Care Med 33 (12, Suppl), S506–S509. [DOI] [PubMed] [Google Scholar]
- Chen X, He WT, Hu L, Li J, Fang Y, Wang X, Xu X, Wang Z, Huang K, and Han J (2016). Pyroptosis is driven by non-selective gasdermin-D pore and its morphology is different from MLKL channel-mediated necroptosis. Cell Res. 26, 1007–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Zeng L, Zhu S, Liu J, Zeh HJ, Kroemer G, Wang H, Billiar TR, Jiang J, Tang D, and Kang R (2019a). cAMP metabolism controls caspase-11 inflammasome activation and pyroptosis in sepsis. Sci. Adv 5, v5562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Zhu S, Zeng L, Wang Q, Sheng Y, Zhou B, Tang D, and Kang R (2019b). AGER-Mediated Lipid Peroxidation Drives Caspase-11 Inflammasome Activation in Sepsis. Front. Immunol 10, 1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa Franco MM, Marim F, Guimaraes ES, Assis NRG, Cerqueira DM, Alves-Silva J, Harms J, Splitter G, Smith J, Kanneganti TD, et al. (2018). Brucella abortus Triggers a cGAS-Independent STING Pathway To Induce Host Protection That Involves Guanylate-Binding Proteins and Inflammasome Activation. J. Immunol 200, 607–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejager L, Vandevyver S, Ballegeer M, Van Wonterghem E, An LL, Riggs J, Kolbeck R, and Libert C (2014). Pharmacological inhibition of type I interferon signaling protects mice against lethal sepsis. J. Infect. Dis 209, 960–970. [DOI] [PubMed] [Google Scholar]
- Deng M, Tang Y, Li W, Wang X, Zhang R, Zhang X, Zhao X, Liu J, Tang C, Liu Z, et al. (2018a). The Endotoxin Delivery Protein HMGB1 Mediates Caspase-11-Dependent Lethality in Sepsis. Immunity 49, 740–753.e7, e747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng W, Zhu S, Zeng L, Liu J, Kang R, Yang M, Cao L, Wang H, Billiar TR, Jiang J, et al. (2018b). The Circadian Clock Controls Immune Checkpoint Pathway in Sepsis. Cell Rep. 24, 366–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, Sun H, Wang DC, and Shao F (2016). Pore-forming activity and structural autoinhibition of the gas- dermin family. Nature 535, 111–116. [DOI] [PubMed] [Google Scholar]
- Dobbs N, Burnaevskiy N, Chen D, Gonugunta VK, Alto NM, and Yan N (2015). STING Activation by Translocation from the ER Is Associated with Infection and Autoinflammatory Disease. Cell Host Microbe 18, 157–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esmon CT (2005). The interactions between inflammation and coagulation. Br. J. Haematol 131, 417–430. [DOI] [PubMed] [Google Scholar]
- Fadok VA, Bratton DL, Frasch SC, Warner ML, and Henson PM (1998). The role of phosphatidylserine in recognition of apoptotic cells by phagocytes. Cell Death Differ. 5, 551–562. [DOI] [PubMed] [Google Scholar]
- Gaidt MM, Ebert TS, Chauhan D, Ramshorn K, Pinci F, Zuber S, O’Duill F, Schmid-Burgk JL, Hoss F, Buhmann R, et al. (2017). The DNA Inflammasome in Human Myeloid Cells Is Initiated by a STING-Cell Death Program Upstream of NLRP3. Cell 171, 1110–1124.e18, e1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallucci S, and Matzinger P (2001). Danger signals: SOS to the immune system. Curr. Opin. Immunol 13, 114–119. [DOI] [PubMed] [Google Scholar]
- Gando S, Levi M, and Toh CH (2016). Disseminated intravascular coagulation. Nat. Rev. Dis. Primers 2, 16037. [DOI] [PubMed] [Google Scholar]
- Ge W, Hu Q, Fang X, Liu J, Xu J, Hu J, Liu X, Ling Q, Wang Y, Li H, et al. (2019). LDK378 improves micro- and macro-circulation via alleviating STING-mediated inflammatory injury in a Sepsis rat model induced by Cecal ligation and puncture. J. Inflamm. (Lond.) 16, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goddard PJ, Sanchez-Garrido J, Slater SL, Kalyan M, Ruano-Gallego D, Marches O, Fernandez LA, Frankel G, and Shenoy AR (2019). Enteropathogenic Escherichia coli Stimulates Effector-Driven Rapid Caspase-4 Activation in Human Macrophages. Cell Rep. 27, 1008–1017.e6, e1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grover SP, and Mackman N (2018). Tissue Factor: An Essential Mediator of Hemostasis and Trigger of Thrombosis. Arterioscler. Thromb. Vasc. Biol 38, 709–725. [DOI] [PubMed] [Google Scholar]
- Gui X, Yang H, Li T, Tan X, Shi P, Li M, Du F, and Chen ZJ (2019). Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature 567, 262–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulen MF, Koch U, Haag SM, Schuler F, Apetoh L, Villunger A, Radtke F, and Ablasser A (2017). Signalling strength determines proapoptotic functions of STING. Nat. Commun 8, 427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Callaway JB, and Ting JP (2015). Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat. Med 21, 677–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, Yang ZH, Zhong CQ, and Han J (2015). Gasdermin D is an executor of pyroptosis and required for interleukin-1b secretion. Cell Res. 25, 1285–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heipertz EL, Harper J, and Walker WE (2017). STING and TRIF Contribute to Mouse Sepsis, Depending on Severity of the Disease Model. Shock 47, 621–631. [DOI] [PubMed] [Google Scholar]
- Holm CK, Rahbek SH, Gad HH, Bak RO, Jakobsen MR, Jiang Z, Hansen AL, Jensen SK, Sun C, Thomsen MK, et al. (2016). Influenza A virus targets a cGAS-independent STING pathway that controls enveloped RNA viruses. Nat. Commun 7, 10680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotchkiss RS, Moldawer LL, Opal SM, Reinhart K, Turnbull IR, and Vincent JL (2016). Sepsis and septic shock. Nat. Rev. Dis. Primers 2, 16045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Q, Ren H, Li G, Wang D, Zhou Q, Wu J, Zheng J, Huang J, Slade DA, Wu X, and Ren J (2019). STING-mediated intestinal barrier dysfunction contributes to lethal sepsis. EBioMedicine 41, 497–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, and Barber GN (2008). STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455, 674–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iskander KN, Vaickus M, Duffy ER, and Remick DG (2016). Shorter Duration of Post-Operative Antibiotics for Cecal Ligation and Puncture Does Not Increase Inflammation or Mortality. PLoS One 11, e0163005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen I, Rayamajhi M, and Miao EA (2017). Programmed cell death as a defence against infection. Nat. Rev. Immunol 17, 151–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kambara H, Liu F, Zhang X, Liu P, Bajrami B, Teng Y, Zhao L, Zhou S, Yu H, Zhou W, et al. (2018). Gasdermin D Exerts Anti-inflammatory Effects by Promoting Neutrophil Death. Cell Rep. 22, 2924–2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang R, Zeng L, Zhu S, Xie Y, Liu J, Wen Q, Cao L, Xie M, Ran Q, Kroemer G, et al. (2018). Lipid Peroxidation Drives Gasdermin D-Mediated Pyroptosis in Lethal Polymicrobial Sepsis. Cell Host Microbe 24, 97–108.e4, e104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S, et al. (2011). Non-canonical inflammasome activation targets caspase-11. Nature 479, 117–121. [DOI] [PubMed] [Google Scholar]
- Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose-Girma M, Phung QT, et al. (2015). Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526, 666–671. [DOI] [PubMed] [Google Scholar]
- Khan MM, Yang WL, and Wang P (2015). Endoplasmic Reticulum Stress in Sepsis. Shock 44, 294–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin B, Ilyukha V, Sorokin M, Buzdin A, Vannier E, and Poltorak A (2017). Cutting Edge: Activation of STING in T Cells Induces Type I IFN Responses and Cell Death. J. Immunol 199, 397–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levi M, and van der Poll T (2010). Inflammation and coagulation. Crit. Care Med 38 (2, Suppl), S26–S34. [DOI] [PubMed] [Google Scholar]
- Levi M, van der Poll T, and ten Cate H (2006). Tissue factor in infection and severe inflammation. Semin. Thromb. Hemost 32, 33–39. [DOI] [PubMed] [Google Scholar]
- Lewis AJ, Seymour CW, and Rosengart MR (2016). Current Murine Models of Sepsis. Surg. Infect. (Larchmt.) 17, 385–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, and Chen ZJ (2018). The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J. Exp. Med 215, 1287–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Zhou H, Wu H, Wu Q, Duan M, Deng W, and Tang Q (2019). STING-IRF3 contributes to lipopolysaccharide-induced cardiac dysfunction, inflammation, apoptosis and pyroptosis by activating NLRP3. Redox Biol. 24, 101215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Sanchez GAM, Tenbrock K, Wittkowski H, Jones OY, Kuehn HS, et al. (2014). Activated STING in a vascular and pulmonary syndrome. N. Engl. J. Med 371, 507–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Cai X, Wu J, Cong Q, Chen X, Li T, Du F, Ren J, Wu YT, Grishin NV, and Chen ZJ (2015). Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 347, aaa2630. [DOI] [PubMed] [Google Scholar]
- Liu D, Wu H, Wang C, Li Y, Tian H, Siraj S, Sehgal SA, Wang X, Wang J, Shang Y, et al. (2019). STING directly activates autophagy to tune the innate immune response. Cell Death Differ. 26, 1735–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, Malech HL, Ledbetter JA, Elkon KB, and Kaplan MJ (2016). Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat. Med 22, 146–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinho FV, Benmerzoug S, Oliveira SC, Ryffel B, and Quesniaux VFJ (2017). The Emerging Roles of STING in Bacterial Infections. Trends Microbiol. 25, 906–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matzinger P (1994). Tolerance, danger, and the extended family. Annu. Rev. Immunol 12, 991–1045. [DOI] [PubMed] [Google Scholar]
- Motwani M, Pesiridis S, and Fitzgerald KA (2019). DNA sensing by the cGAS-STING pathway in health and disease. Nat. Rev. Genet 20, 657–674. [DOI] [PubMed] [Google Scholar]
- Movert E, Lienard J, Valfridsson C, Nordstrom T, Johansson-Lindbom B, and Carlsson F (2018). Streptococcal M protein promotes IL-10 production by cGAS-independent activation of the STING signaling pathway. PLoS Pathog. 14, e1006969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukai K, Konno H, Akiba T, Uemura T, Waguri S, Kobayashi T, Barber GN, Arai H, and Taguchi T (2016). Activation of STING requires palmitoylation at the Golgi. Nat. Commun 7, 11932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orning P, Weng D, Starheim K, Ratner D, Best Z, Lee B, Brooks A, Xia S, Wu H, Kelliher MA, et al. (2018). Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death. Science 362, 1064–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterud B (1998). Tissue factor expression by monocytes: regulation and pathophysiological roles. Blood Coagul. Fibrinolysis 9 (Suppl 1), S9–S14. [PubMed] [Google Scholar]
- Patel P, Walborn A, Rondina M, Fareed J, and Hoppensteadt D (2019). Markers of Inflammation and Infection in Sepsis and Disseminated Intravascular Coagulation. Clin. Appl. Thromb. Hemost 25, 1076029619843338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawlinski R, and Mackman N (2010). Cellular sources of tissue factor in endotoxemia and sepsis. Thromb. Res 125 (Suppl 1), S70–S73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Periasamy M, and Kalyanasundaram A (2007). SERCA pump isoforms: their role in calcium transport and disease. Muscle Nerve 35, 430–442. [DOI] [PubMed] [Google Scholar]
- Prole DL, and Taylor CW (2019). Structure and Function of IP3 Receptors. Cold Spring Harb. Perspect. Biol 11, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhart K, Daniels R, Kissoon N, Machado FR, Schachter RD, and Finfer S (2017). Recognizing Sepsis as a Global Health Priority - A WHO Resolution. N. Engl. J. Med 377, 414–417. [DOI] [PubMed] [Google Scholar]
- Rhee C, Dantes R, Epstein L, Murphy DJ, Seymour CW, Iwashyna TJ, Kadri SS, Angus DC, Danner RL, Fiore AE, et al. ; CDC Prevention Epicenter Program (2017). Incidence and Trends of Sepsis in US Hospitals Using Clinical vs Claims Data, 2009–2014. JAMA 318, 1241–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rittirsch D, Huber-Lang MS, Flierl MA, and Ward PA (2009). Immunodesign of experimental sepsis by cecal ligation and puncture. Nat. Protoc 4, 31–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothmeier AS, Marchese P, Petrich BG, Furlan-Freguia C, Ginsberg MH, Ruggeri ZM, and Ruf W (2015). Caspase-1-mediated pathway promotes generation of thromboinflammatory microparticles. J. Clin. Invest 125, 1471–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruhl S, Shkarina K, Demarco B, Heilig R, Santos JC, and Broz P (2018). ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science 362, 956–960. [DOI] [PubMed] [Google Scholar]
- Samuels JM, Moore HB, and Moore EE (2018). Coagulopathy in Severe Sepsis: Interconnectivity of Coagulation and the Immune System. Surg. Infect. (Larchmt.) 19, 208–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarhan J, Liu BC, Muendlein HI, Li P, Nilson R, Tang AY, Rongvaux A, Bunnell SC, Shao F, Green DR, and Poltorak A (2018). Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. Proc. Natl. Acad. Sci. USA 115, E10888–E10897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarhan J, Liu BC, Muendlein HI, Weindel CG, Smirnova I, Tang AY, Ilyukha V, Sorokin M, Buzdin A, Fitzgerald KA, and Poltorak A (2019). Constitutive interferon signaling maintains critical threshold of MLKL expression to license necroptosis. Cell Death Differ. 26, 332–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmeck B, Gross R, N’Guessan PD, Hocke AC, Hammerschmidt S, Mitchell TJ, Rosseau S, Suttorp N, and Hippenstiel S (2004). Streptococcus pneumoniae-induced caspase 6-dependent apoptosis in lung epithelium. Infect. Immun 72, 4940–4947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz DS, and Blower MD (2016). The endoplasmic reticulum: structure, function and response to cellular signaling. Cell. Mol. Life Sci 73, 79–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang G, Zhang C, Chen ZJ, Bai XC, and Zhang X (2019). Cryo-EM structures of STING reveal its mechanism of activation by cyclic GMP-AMP. Nature 567, 389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, and Shao F (2015). Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665. [DOI] [PubMed] [Google Scholar]
- Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche JD, Coopersmith CM, et al. (2016). The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 315, 801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J, Hu D, He C, Wang T, Liu X, Ma L, Lin Z, and Chen Z (2013). Novel biomarkers for early prediction of sepsis-induced disseminated intravascular coagulation in a mouse cecal ligation and puncture model. J. Inflamm. (Lond.) 10, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soustek MS, Balsa E, Barrow JJ, Jedrychowski M, Vogel R, Jan Smeitink, Gygi SP, and Puigserver P (2018). Inhibition of the ER stress IRE1a inflammatory pathway protects against cell death in mitochondrial complex I mutant cells. Cell Death Dis. 9, 658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srikanth S, Woo JS, Wu B, El-Sherbiny YM, Leung J, Chupradit K, Rice L, Seo GJ, Calmettes G, Ramakrishna C, et al. (2019). The Ca2+ sensor STIM1 regulates the type I interferon response by retaining the signaling adaptor STING at the endoplasmic reticulum. Nat. Immunol 20, 152–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W, Li Y, Chen L, Chen H, You F, Zhou X, Zhou Y, Zhai Z, Chen D, and Jiang Z (2009). ERIS, an endoplasmic reticulum IFN stimulator, activates innate immune signaling through dimerization. Proc. Natl. Acad. Sci. USA 106, 8653–8658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Wu J, Du F, Chen X, and Chen ZJ (2013). Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suschak JJ, Wang S, Fitzgerald KA, and Lu S (2016). A cGAS-Independent STING/IRF7 Pathway Mediates the Immunogenicity of DNA Vaccines. J. Immunol 196, 310–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taabazuing CY, Okondo MC, and Bachovchin DA (2017). Pyroptosis and Apoptosis Pathways Engage in Bidirectional Crosstalk in Monocytes and Macrophages. Cell Chem. Biol 24, 507–514.e4, e504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, and Chen ZJ (2012). STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci. Signal 5, ra20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang D, Kang R, Coyne CB, Zeh HJ, and Lotze MT (2012). PAMPs and DAMPs: signal 0s that spur autophagy and immunity. Immunol. Rev 249, 158–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang D, Kang R, Berghe TV, Vandenabeele P, and Kroemer G (2019). The molecular machinery of regulated cell death. Cell Res. 29, 347–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor FB Jr., Toh CH, Hoots WK, Wada H, and Levi M; Scientific Subcommittee on Disseminated Intravascular Coagulation (DIC) of the International Society on Thrombosis and Haemostasis (ISTH) (2001). Towards definition, clinical and laboratory criteria, and a scoring system for disseminated intravascular coagulation. Thromb. Haemost 86, 1327–1330. [PubMed] [Google Scholar]
- Tilg H, and Peschel C (1996). Interferon-alpha and its effects on the cytokine cascade: a pro- and anti-inflammatory cytokine. Leuk. Lymphoma 23, 55–60. [DOI] [PubMed] [Google Scholar]
- Todd DJ, Lee AH, and Glimcher LH (2008). The endoplasmic reticulum stress response in immunity and autoimmunity. Nat. Rev. Immunol 8, 663–674. [DOI] [PubMed] [Google Scholar]
- van Dam-Mieras MC, Muller AD, van Deijk WA, and Hemker HC (1985). Clotting factors secreted by monocytes and macrophages: analytical considerations. Thromb. Res 37, 9–19. [DOI] [PubMed] [Google Scholar]
- Weischenfeldt J, and Porse B (2008). Bone Marrow-Derived Macrophages (BMM): Isolation and Applications. CSH Protoc 2008, t5080. [DOI] [PubMed] [Google Scholar]
- Wu C, Lu W, Zhang Y, Zhang G, Shi X, Hisada Y, Grover SP, Zhang X, Li L, Xiang B, et al. (2019a). Inflammasome Activation Triggers Blood Clotting and Host Death through Pyroptosis. Immunity 50, 1401–1411.e4, e1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Chen YJ, Dobbs N, Sakai T, Liou J, Miner JJ, and Yan N (2019b). STING-mediated disruption of calcium homeostasis chronically activates ER stress and primes T cell death. J. Exp. Med 216, 867–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Cheng X, Tang Y, Qiu X, Wang Y, Kang H, Wu J, Wang Z, Liu Y, Chen F, et al. (2019). Bacterial Endotoxin Activates the Coagulation Cascade through Gasdermin D-Dependent Phosphatidylserine Exposure. Immunity 51, 983–996.e6, e986. [DOI] [PubMed] [Google Scholar]
- Zelaya H, Rothmeier AS, and Ruf W (2018). Tissue factor at the crossroad of coagulation and cell signaling. J. Thromb. Haemost 16, 1941–1952. [DOI] [PubMed] [Google Scholar]
- Zeng L, Kang R, Zhu S, Wang X, Cao L, Wang H, Billiar TR, Jiang J, and Tang D (2017). ALK is a therapeutic target for lethal sepsis. Sci. Transl. Med 9, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Žgur-Bertok D (2013). DNA damage repair and bacterial pathogens. PLoS Pathog. 9, e1003711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K (2010). Integration of ER stress, oxidative stress and the inflammatory response in health and disease. Int. J. Clin. Exp. Med 3, 33–40. [PMC free article] [PubMed] [Google Scholar]
- Zhang SL, Yu Y, Roos J, Kozak JA, Deerinck TJ, Ellisman MH, Stauderman KA, and Cahalan MD (2005). STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature 437, 902–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong B, Yang Y, Li S, Wang YY, Li Y, Diao F, Lei C, He X, Zhang L, Tien P, and Shu HB (2008). The adaptor protein MITA links virus-sensing re-ceptors to IRF3 transcription factor activation. Immunity 29, 538–550. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Lin H, Wang S, Wang S, Ran Y, Liu Y, Ye W, Xiong X, Zhong B, Shu HB, and Wang YY (2014). The ER-associated protein ZDHHC1 is a positive regulator of DNA virus-triggered, MITA/STING-dependent innate immune signaling. Cell Host Microbe 16, 450–461. [DOI] [PubMed] [Google Scholar]
- Zierhut C, Yamaguchi N, Paredes M, Luo JD, Carroll T, and Funabiki H (2019). The Cytoplasmic DNA Sensor cGAS Promotes Mitotic Cell Death. Cell 178, 302–315.e23, e323. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique datasets or code.