

Abstract
Anti-cancer drugs targeting the DNA damage response (DDR) exploit genetic or functional defects in this pathway through synthetic lethal mechanisms. For example, defects in homologous recombination (HR) repair arise in cancer cells through inherited or acquired mutations in BRCA1, BRCA2, or other genes in the Fanconi Anemia/BRCA pathway, and these tumors have been shown to be particularly sensitive to inhibitors of the base excision repair (BER) protein poly (ADP-ribose) polymerase (PARP). Recent work has identified additional genomic and functional assays of DNA repair that provide new predictive and pharmacodynamic biomarkers for these targeted therapies. Here, we examine the development of selective agents targeting DNA repair, including PARP inhibitors, inhibitors of the DNA damage kinases ATR, CHK1, WEE1, and ATM, as well as inhibitors of classical non-homologous end-joining (cNHEJ) and alternative end-joining (Alt-EJ). We also review the biomarkers that guide the use of these agents and current clinical trials with these therapies.
Keywords: PARP inhibitor, DNA repair, Cell cycle kinases, Polymerase theta
eTOC Blurb
In recent years, there has been substantial progress in our understanding of DNA damage response (DDR) pathways which has led to the development of a multitude of DDR inhibitors. This review focuses on therapeutic strategies of utilizing DDR inhibitors and biomarkers that can guide their clinical development.
INTRODUCTION
The maintenance of genomic stability requires a protective cellular response to DNA damaging agents. Genomic instability, which is a hallmark of cancer cells, results from a defect in this DNA Damage Response (DDR). The DDR pathway is an intricately regulated system designed to protect cells against acquired changes to the genome and has evolved to safeguard against intrinsic and extrinsic DNA damage. This pathway encompasses proteins that detect DNA damage, function in DNA repair pathways, and regulate the cell cycle. Oncologists have been therapeutically targeting the DDR pathway for decades with cytotoxic agents. Not surprisingly, many of the mechanisms of sensitivity and resistance to cytotoxic chemotherapy are governed by the DDR pathway. In recent years, new anti-cancer drugs have been generated that inhibit the regulatory pathways governing the DDR response (Table 1). This review will summarize progress made in targeting the DDR pathway and highlight efforts to rationally combine these drugs utilizing biomarkers that predict their sensitivity and resistance.
Table 1.
DDR Inhibitors in Past or Present Clinical Evaluation
| PARP Inhibitors | CHK1 Inhibitor | ATR Inhibitors |
|---|---|---|
| Niraparib (MK-4827)1 | GDC-0575 | Ceralasertib (AZD6738) |
| Olaparib (AZD-2281)1 | LY3300054 | BAY1895344 |
| Talazoparib (BMN-673) 1 | MK-8776 (SCH-900776) | Berzosertib (M6620, VX-970) |
| Rucaparib (AG-014699)1 | Prexasertib (LY2606368) | M4344 (VX-803) |
| Pamiparib (BGB-290) | SRA-737 (CCT245737) | |
| Veliparib (ABT-888) | AZD77622 | |
| CEP-97222 | ||
| E7016 (GPI-21016)2 | ||
| INO-10012 | ||
| DNA-PK Inhibitors | WEE1 Inhibitors | ATM Inhibitors |
| AZD7648 | Adavosertib (AZD1775) | AZD0156 |
| CC-115 | M3541 | |
| M9831 (VX-984) | ||
| Nedisertib (M3814) |
FDA approved
No longer in clinical trials
Molecular Mechanisms of DNA Damage Induced by Cytotoxic Chemotherapy and Radiation
The anti-cancer efficacy of cytotoxic chemotherapy and radiation is derived from their ability to damage DNA. Analysis of the molecular mechanism of how these treatments damage DNA, and are subsequently repaired, has increased our understanding of the DDR pathway and has exposed potential weakness that can be exploited with molecularly targeted therapies. For example, DNA damage caused by the alkylating chemotherapy temozolomide (TMZ), which is commonly used in glioblastoma and pancreatic neuroendocrine tumors, is repaired by MGMT (methylguanine methyltransferase). This finding provides a rationale for generating pharmacological inhibitors of MGMT that may further sensitize tumors to TMZ. To date, the clinical utilization of MGMT inhibitors, such as O6-benzylguanine, has been hampered by significant myelosuppression (Quinn et al., 2009).
Cisplatin mainly exerts damage on DNA by forming intrastrand crosslinks, with the formation of guanine-platinum-guanine and guanine-platinum-adenine adducts (Rocha et al., 2018). While approximately 90% of the crosslinks formed by cisplatin are intrastrand, rarely cisplatin can also generate guanine-platinum-guanine interstrand crosslinks (Rocha et al., 2018). Intrastrand DNA crosslinks are repaired by nucleotide excision repair (NER), a mechanism of single-strand DNA repair which is deficient in patients with xeroderma pigmentosum (Scharer, 2013). Consistent with this, deleterious mutations in an NER pathway helicase, ERRC2, are predictive of cisplatin sensitivity in bladder cancer patients (Van Allen et al., 2014). Repair of interstrand crosslinks, which can also be generated by mitomycin-C, requires both NER and homologous recombination (HR) (Damia and Broggini, 2019; Rocha et al., 2018).
Radiation is a particularly effective anti-cancer therapy because it can sever covalent bonds in the DNA phosphodiester backbone and generate double-strand DNA breaks (Toulany, 2019). Bleomycin, a chemotherapy used in the treatment of testicular cancer and Hodgkin’s lymphoma, forms a complex with Fe2+ and O2 and can thereby cleave DNA and generate double-strand DNA breaks (Chen et al., 2008). Topoisomerase I and II enzymes reduce DNA supercoiling by transiently causing single or double-strand DNA breaks, respectively, to allow DNA unwinding. Topoisomerase I inhibitors, such as irinotecan and topotecan, stabilize the topoisomerase I-DNA cleavage complex and prevents the closure of the single-strand DNA break (Pommier et al., 2010). Likewise topoisomerase II inhibitors, like etoposide and doxorubicin, trap the topoisomerase II-DNA cleavage complex and thereby generate a double-strand DNA break (Pommier et al., 2010).
Regulation of Double-Strand Break Repair
The two canonical pathways of double-strand DNA break repair are homologous recombination (HR) and classical non-homologous end joining (cNHEJ). Homologous recombination is preferred because of its high fidelity in repairing the genome. In contrast, cNHEJ utilizes non-specific ligation to correct DNA breaks, resulting in error-prone repair (Chang et al., 2017). The process of V(D)J recombination in immune cells advantageously harnesses the high percentage of errors introduced by cNHEJ to generate a diverse array of antibodies and T cell receptors. cNHEJ leads to characteristic DNA damage lesions, such as large deletions, resulting in loss of heterozygosity (LOH) and telomeric allelic imbalance. This error pattern can generate a highly recognizable genomic scar in HR-deficient cells, thus yielding an opportunity for therapeutic biomarker development (Table 2). Mechanistically, a key step in cNHEJ is the binding of KU70 and KU80 to the DNA (Chang et al., 2017). This event activates DNA-PK which, in turn, activates a multi-protein complex of XRCC4, Artemis, and DNA Ligase. The importance of DNA-PK, a PI3K-related protein kinase, has driven the development of several DNA-PK inhibitors, including nedisertib (M3814), AZD7648, and M9831 (VX-984) (Blackford and Jackson, 2017) (Table 1).
Table 2:
Predictive and Pharmacodynamic Biomarkers of DNA Repair Inhibitors
| Drug | Target | Predictive Marker | Pharmacodynamic Marker1 |
|---|---|---|---|
| PARPi | HR deficiency | HR gene mutation | ↑γH2AX |
| HRD Score (LOH), Signature 3 | ↓PARylation | ||
| Unstable replication fork2 | ↑Rad51 (in HR proficient cells)3 | ||
| ↑Polθ | |||
| Platinum sensitivity | |||
| Absent RAD51 foci | |||
| ↓Monoubiquitination of FANCD2 | |||
| DNA-PKi | cNHEJ | Loss of Polθ or FEN1 | |
| ATRi | RS4 | ATM deficiency | ↓pCHK1 |
| RS promoting genomic changes4 | ↑γH2AX | ||
| Unstable replication fork | ↑Unstable replication fork | ||
| ↑pRPA, ↑pKAP1 | ↑pKAP1 | ||
| RNase H2deficiency | |||
| CHK1i | RS | p53 deficiency | ↑γH2AX |
| RS promoting genomic changes4 | ↑pCHK1 | ||
| Unstable replication fork | ↑Unstable replication fork | ||
| ↑pRPA, ↑pKAP1 | ↑pRPA, ↑pKAP1 | ||
| WEE1i | RS | p53 deficiency | ↑pCHK1 |
| RS promoting genomic changes4 | ↑γH2AX | ||
| Unstable replication fork | ↑Unstable replication fork | ||
| ↑pRPA, ↑pKAP1 | ↑pRPA | ||
| ↓pCDK1 (pCDC2) | |||
| Polθi | Alt-EJ | HR deficieny5 | ↑Rad51 |
| ↑Polθ | ↑γH2AX | ||
| ↓cNHEJ | ↑Rad51 (in HR proficient cells) | ||
Pharmacodynamic markers are a metric of target and/or pathway engagement
Unstable replications forks are evaluated in the laboratory with DNA fiber assays
RAD51 foci increase in HR proficient cells. RAD51 foci are absent in HR deficient cells
Replicative stress (RS) promoting genomic changes include CCNE1 and MYC amplification and FBXW7 mutations
Polθ shares the same predictive biomarkers as PARP inhibitors.
Unlike cNHEJ, HR uses a template from a sister chromatid to achieve accurate DNA repair. While cNHEJ can occur at any time in the cell cycle, HR occurs only in the S and G2 phases. A trio of three proteins, MRE11–RAD50–NBS1 (also called the MRN complex), initiates the pathway by binding to double strand DNA breaks (Ranjha et al., 2018). The MRN complex, in concert with the CtIP endonuclease and BRCA1, mediates DNA end resection. In addition, the MRN complex activates ATM, which leads to the activation of BRCA1, BRCA2, and PALB2. Following this event, RAD51 loads at the sites of DNA damage and results in the formation of a nucleoprotein filament that then invades the sister chromatid to find homologous DNA sequences that serve as a template for the synthesis of new DNA. Inhibitors of ATM and RAD51 are currently under development.
The HR pathway is deficient in Fanconi Anemia (FA), a rare genetic disease leading to bone marrow failure and increased cancer risk. Tumor cells with a deficiency in the Fanconi Anemia/BRCA pathway are hypersensitive to chemotherapies that cause DNA interstrand crosslinks, such as mitomycin-C and cisplatin, and, in some cases, to PARP inhibitors(Table 2). Cells from FA patients also demonstrate chromosomal instability. Indeed, the BRCA2 breast cancer susceptibility gene was previously mapped to the FANCD1 complementation group (Howlett et al., 2002), thereby establishing the so-called Fanconi Anemia/BRCA pathway.
PARP inhibitors capitalize on the HR deficiency of many cancers in part by inhibiting other DNA repair pathways such as BER. While PARP inhibitors target tumors with intrinsic HR deficiency, additional therapies are under development to inhibit HR in tumors. For example, a first-in-class RAD51 inhibitor, CYT01B, can block HR and has demonstrated preclinical activity in cancer cells expressing activation-induced cytidine deaminase (AID), a protein promoting double-strand DNA breaks (Maclay et al., 2018). Importantly, CYT01B synergizes with PARP and ATR inhibitors in model systems (Maclay et al., 2018).
Unlike cNHEJ, HR requires the generation of a single-strand DNA 3’ overhang at the end of a double-strand DNA break in a process known as end resection. The tumor suppressor p53-binding protein 1 (53BP1) and the shieldin complex, made up of the SHLD1, SHLD2, SHLD3, and REV7 proteins, blocks end resection (Noordermeer et al., 2018). By preventing formation of a single-strand DNA 3’ overhang, these proteins promote cNHEJ. Importantly, regulation of this complex plays a key role in determining whether double-strand DNA breaks are repaired by HR or cNHEJ. BRCA1 and 53BP1 play opposing roles in determining whether HR or cNHEJ is utilized (Escribano-Diaz et al., 2013). BRCA1 stimulates the MRN complex to activate CtIP-mediated 5’ to 3’ end resection to promote HR (Makharashvili and Paull, 2015). Notably, in S phase, BRCA1 activation also leads to decreased amounts of 53BP1 on chromatin (Chapman et al., 2012; Escribano-Diaz et al., 2013; Isono et al., 2017). However, in G1, 53BP1 and its effector protein RIF1 prevent end resection by promoting the assembly of the shieldin complex and activating NHEJ (Chapman et al., 2012; Escribano-Diaz et al., 2013).
TRIP13 is an ATPase that inactivates the shieldin complex and activates the 5’ to 3’ resection of double-strand breaks, thereby promoting HR repair (Clairmont et al., 2020). Importantly, TRIP13 is amplified in many BRCAI-deficient tumors, and its upregulation may contribute to the intrinsic PARP inhibitor resistance of many of these tumors. Accordingly, a small molecule inhibitor of the ATPase domain of TRIP13 may provide a useful means of stabilizing the shieldin complex, promoting cNHEJ, blocking HR, and overcoming intrinsic PARP inhibitor resistance. Early studies suggest that development of TRIP13 inhibitors may be useful not only in the treatment of BRCAI-deficient tumor cells with intrinsic PARP inhibitor resistance, but also in tumors with acquired PARP inhibitor resistance (Clairmont et al., 2020).
The elucidation of a third mechanism of DSB repair has identified important new DDR drug targets. This mechanism, called alternative nonhomologous end-joining (Alt-EJ) or MMEJ, requires “microhomology” (i.e., homologous DNA sequences approximately 2-20 base pairs in length) at the DNA break points (Bennardo et al., 2008; Ranjha et al., 2018). Importantly, Alt-EJ is dependent on PARP1. Following binding of the double-strand break by the MRN complex and PARP1, Polymeraseθ (Polθ) mediates the repair of the break. While the alt-EJ repair system is used rarely in HR proficient tumors, HR deficiency results in increased dependency on alt-EJ (Ceccaldi et al., 2015). The dependence of alt-EJ on Polθ suggests that Polθ inhibitors may be effective for HR-deficient tumors.
The final type of DSB repair is termed single-strand annealing. Similar to Alt-EJ, single strand-annealing requires homologous DNA sites to catalyze the repair of the double-strand break (Bhargava et al., 2016). However, unlike Alt-EJ, single-strand annealing can take place over long stretches of DNA and lead to large deletions which can cause intrachromosomal translocations. Mechanistically, single-strand annealing is inhibited by the RAD51 (Stark et al., 2004). Unlike Alt-EJ, which requires PARP and Polθ, single-strand annealing requires RAD52 to marshal the annealing of the homologous stretches of single-strand DNA (Bennardo et al., 2008; Grimme et al., 2010).
Mechanism of Action of PARP Inhibitors
Two seminal papers introduced the concept that PARP inhibitors have synthetic lethal interaction with HR-deficient tumors (Bryant et al., 2005; Farmer et al., 2005) (Figure 1). PARP1 binding to single-strand DNA breaks generated in BER forms a foundation of the synthetic lethal interaction with HR deficiency (De Vos et al., 2012; Strom et al., 2011). When BER cannot repair single-strand DNA breaks, the single-strand breaks ultimately become double-strand DNA breaks. While HR-proficient cells can rely on the HR repair to correct double-strand DNA breaks, HR-deficient cells are forced to rely on cNHEJ (McCabe et al., 2006). However, the error-prone nature of cNHEJ ultimately leads to genomic instability and cell death (Patel et al., 2011).
Figure 1: Mechanisms of PARP Inhibitor and POLϴ Inhibitor Cytotoxicity.
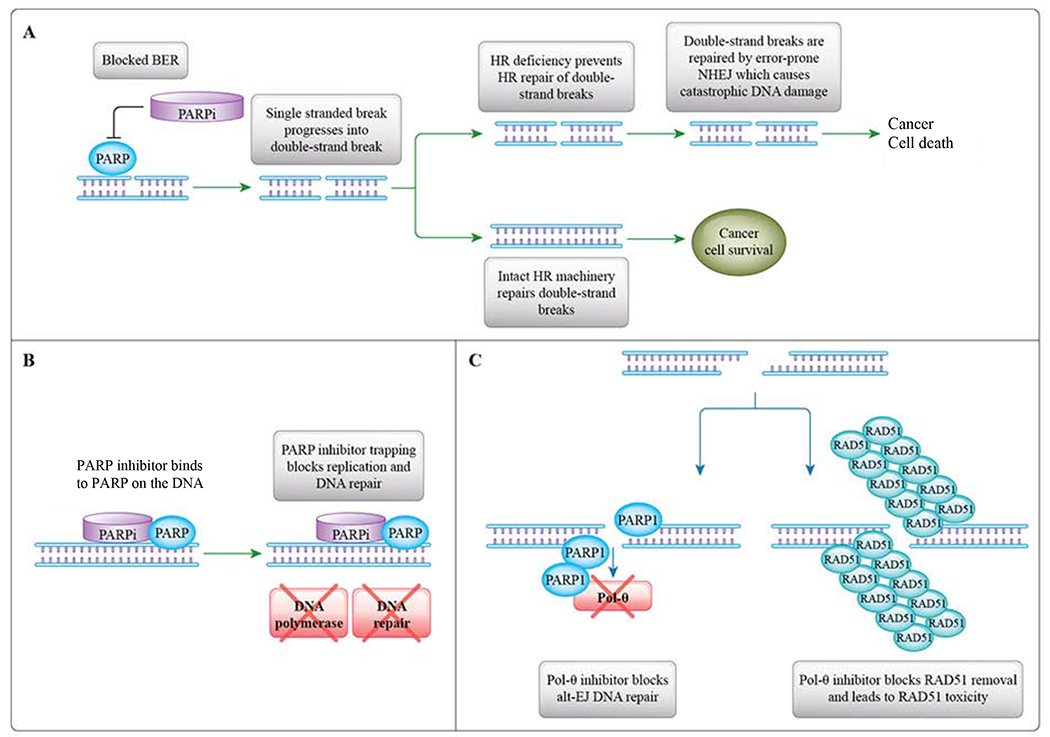
A. PARP inhibition blocks BER single-strand DNA repair. The inability to repair single-strand DNA breaks leads to the accumulation of double-strand DNA breaks. In HR-deficient cancers, cells are dependent on error-prone cNHEJ to repair double strand DNA breaks. The large number of genomic errors introduced by cNHEJ leads to catastrophic DNA damage. B. PARP inhibitor trapping blocks DNA replication and DNA repair. C. Polϴ inhibition blocks the Alt-EJ pathway from repairing double-stand DNA breaks. PARP and POLθ are known to cooperate in Alt-EJ. Inhibition of POLθ, and to a lesser extent, inhibition of PARP, results in the loss of Alt-EJ and the accumulation of toxic RAD51 foci. This mechanism provides a rationale for combining a PARP inhibitor with a POLθ inhibitor for cancer treatment. The combination may be synergistic since it leads to toxic RAD51 accumulation and cell death.
Preclinical and clinical studies of PARP inhibitors soon revealed additional mechanisms of PARP inhibitor activity. PARP inhibitors have been shown to “trap” the PARP enzyme at the site of DNA damage, therefore preventing essential cellular processes such as DNA repair and transcription. Furthermore, the trapped PARP-DNA complex is particularly lethal in HR-deficient cells. Multiple studies have demonstrated differential potency of different PARP inhibitors. In HR-deficient cell lines, the IC50 of talazoparib (5 nM) was significantly lower than that of olaparib (259 nM), rucaparib (609 nM), and veliparib (>10,000 nM) (Shen et al., 2013). One explanation for the increased potency of talazoparib is its strong PARP trapping activity, approximately 100-fold greater than that of other PARP inhibitors (Murai et al., 2014a). Similarly, the relatively weak PARP trapping of veliparib may account for its weaker anti-cancer activity, compared to other PARP inhibitors (Murai et al., 2012).
Clinical trials of PARP inhibitors have led to their approval by the United States Food and Drug Administration (FDA) in several indications, including ovarian, breast, and pancreatic cancer (Table 1)(Geenen et al., 2018; Golan et al., 2019; Konstantinopoulos and Matulonis, 2018). The anti-cancer efficacy and favorable side effect profile of PARP inhibitors has led to their rapid adoption into clinical practice. While myelosuppression can be a limiting toxicity, these orally administered drugs are generally well tolerated, with most patients reporting minimal symptomatic PARP-inhibitor induced toxicities.
Inhibition of Polθ-dependent alternative end-joining
In recent years, there has been growing appreciation for the role of the alt-EJ system in double-strand DNA repair (Bennardo et al., 2008; Wood and Doublié, 2016). Mice deficient in polymerase Polθ are viable and exhibit a mild phenotype (Shima et al., 2004). However, the addition of an HR-inactivating mutation in mice deficient in polymerase Polθ is embryonically lethal (Ceccaldi et al., 2015). Furthermore, shRNA depletion of Polθ, combined with pharmacological PARP inhibition, is cytotoxic in in vitro and in vivo models of HR-deficient ovarian cancer (Ceccaldi et al., 2015).
Polθ plays two critical roles in DNA double-strand repair (Black et al., 2016; Chan et al., 2010). The polymerase domain of Polθ functions in the alt-EJ pathway. In addition, a separate helicase domain of Polθ at the N-terminus has ATPase activity and has nearby RAD51 binding sites. The RAD51-binding activity of Polθ removes the RAD51 molecules that bind to DNA during HR-repair (Ceccaldi et al., 2015; Mateos-Gomez et al., 2015). The removal of RAD51 is crucial since RAD51 accumulation can lead to cellular toxicity. Polθ inhibition may prevent removal of RAD51 foci and thus lead to toxicity in cells with HR alterations, thus providing a mechanism of action of Pole inhibitors (Ceccaldi et al., 2015). Polθ inhibition may be efficacious in both HR-deficient cells through inhibition of the alt-EJ pathway, as well as in cells with acquired PARP inhibitor resistance through disruption of HR restoration.
The potential synthetic lethality of Polθ inhibitors in HR-deficient tumors has created great interest in developing clinical grade Polθ inhibitors (Figure 1C). Whether Polθ inhibitors should be designed which target the C-terminal polymerase domain or the N-terminal ATPase domain remains an unresolved question. Recent evidence indicates that inhibiting the ATPase of Pole results in the accumulation of toxic RAD51 complexes at the sites of abortive HR repair, further suggesting a synthetic lethality mechanism resulting from the knockout of both HR and Pole (Ceccaldi et al., 2015).
Mechanisms of Resistance in HR-deficient Tumors
Cancer cells can develop acquired resistance to PARP inhibition through several mechanisms (Table 3). Similar to other systemic therapies, one mechanism of acquired resistance is through increasing the efflux of the PARP inhibitors out of the cell by upregulating drug-efflux transporters such as the multidrug resistance pumps (MDRs) (Rottenberg et al., 2008). Alterations in the function of the PARP1 protein, by PARP1 mutations which reduce PARP1 binding affinity to DNA or disrupt PARylation, via disruption of PAR removal, also lead to acquired PARP inhibitor resistance (Gogola et al., 2018; Pettitt et al., 2018).
Table 3.
Mechanisms of PARP Inhibitor Resistance
| Mutation | Mechanism | References |
|---|---|---|
| Acquired BRCA1/2 Gene Mutation | Somatic Reversion | (Sakai et al., 2008) |
| Decrease in: | Inactivation of SHLD complex and restoration of HR | (Dev et al., 2018; Jaspers et al., 2013; Nacson et al., 2018; Tomida et al., 2018; Xu et al., 2015) |
| 53BP1 | ||
| RIF1 | ||
| REV7 | ||
| SHLD1, 2, 3 | ||
| Decrease in: | Prevention of fork degradation | (Ray Chaudhuri et al., 2016; Rondinelli et al., 2017a) |
| PTIP | ||
| EZH2 | ||
| Amplification TRIP13 | Removal of SHLD complex and restores HR | (Clairmont et al., 2020) |
| PARP1 loss | Loss of target | (Pettitt et al., 2018) |
| Increased P-glycoprotein | Increased drug efflux | (Henneman et al., 2015) |
| Decreased SLFN11 | Fork stabilization | (Lok et al., 2017) |
| Decreased PARG | Increased PAR chains | (Gogola et al., 2018) |
| Increased TIRR | Blocks 53BP1 binding | (Drane et al., 2017) |
| Decreased DNLL1 | Increased DSB end resection | (He et al., 2018) |
Cancer cells may also develop acquired resistance to PARP inhibitors by either restoring HR repair or by re-establishing replication fork stability (Konstantinopoulos et al., 2015; Ray Chaudhuri et al., 2016; Rondinelli et al., 2017b; Yazinski et al., 2017). Additionally, recent work has shown that these mechanisms may occur together (Yazinski et al., 2017). Reversion mutations in HR genes that restore function of the HR proteins have been observed in samples from patients who have developed acquired resistance to PARP inhibitors. Alternatively, increased expression of hypomorphic mutant proteins capable of RAD51 loading may also restore HR (Johnson et al., 2013).
Notably, HR deficiency has been shown to sensitize cancer cells to platinum-based chemotherapy, and several of the mechanisms that confer resistance to PARP inhibition have also been demonstrated in patients with acquired platinum chemotherapy resistance (Pishvaian et al., 2017; Sakai et al., 2008; Weigelt et al., 2017). Conversely, mechanisms of PARP inhibitor resistance and cisplatin resistance do not always track together (Johnson et al., 2016). For example, REV7 depletion will cause PARP inhibitor resistance by promoting DNA end resection but will also create an acquired vulnerability to platinum based on its role in translesion synthesis in the Fanconi Anemia pathway (Niimi et al., 2014). Moreover, in ovarian cancer, responses to platinum-based chemotherapy have been observed after acquired PARP inhibitor resistance has manifested (Ang et al., 2013).
Predictive and Pharmacodynamic Biomarkers for DDR Inhibitor Drug Development
While PARP inhibitors have shown efficacy in HR-deficient breast and ovarian cancer, there is great need to discover additional biomarkers that can identify tumors susceptible to PARP inhibitors as well as other DDR inhibitors (Table 2). PARP inhibitors are well known to have activity in BRCA1 and BRCA2-mutated tumors; however, PARP inhibitors also demonstrate activity in tumors with other HR mutations such as PALB2, RAD51C, and RAD51D (Chandran and Kennedy, 2018). In addition to mutational analysis, an analysis of BRCA1 methylation demonstrated that homozygous BRCA1 promoter methylation is predictive of response to rucaparib in a cohort of 21 ovarian cancer patients on the ARIEL2 trial (Kondrashova et al., 2018; Swisher et al., 2017).
A genomic mutational signature, termed signature 3, is associated with homologous recombination (HR) repair deficiency (Alexandrov et al., 2013; Polak et al., 2017). While signature 3 shows strong concordance with known pathogenic biallelic mutations in HR genes, many patients display a mutational signature of HR deficiency but do not demonstrate a clear mutation in BRCA1, BRCA2, or PALB2 (Polak et al., 2017), suggesting that additional alterations in the HR pathway can also underlie such a mutational signature. The requirement for whole-genome or whole-exome sequencing for identification of signature 3 in tumor samples has limited its widespread clinical utilization as biomarker; however, computational tools have recently been developed that allow detection of signature 3 using clinical-grade targeted sequencing panels (Gulhan et al., 2019). With this methodology, signature 3 was a powerful predictor of response to a niraparib/pembrolizumab combination in a molecularly unselected population of platinum-resistant ovarian cancer patients (Farkkila et al., 2020).
The possibility of acquired resistance to PARP inhibitors through genetic or non-genetic mechanisms highlights the need for functional biomarkers that can assess HR proficiency. For instance, an immunofluorescence-based RAD51 assay can interrogate HR proficiency in preclinical and clinical samples (Castroviejo-Bermejo et al., 2018; Hill et al., 2018; Mukhopadhyay et al., 2010; Naipal et al., 2014). Since the formation of RAD51 foci at sites of double-strand DNA damage is a critical step in the HR pathway, this assay differentiates between HR-proficient cancers, which form RAD51 foci, and HR-deficient cancers which are unable to form RAD51 foci. One methodology for this assay utilizes irradiated live tumor cells, and this was shown to be predictive of HR-deficient human breast cancer (Naipal et al., 2014). However, since live tumor cells are often not available in clinical specimens, there also has been efforts to perform the RAD51 assay on formalin fixed samples. Using formalin fixed samples, several groups have been able to demonstrate that the RAD51 assay can identify HR-deficient tumor cells that were appropriately PARP inhibitor sensitive (Castroviejo-Bermejo et al., 2018; Hill et al., 2018; Mukhopadhyay et al., 2010).
Platinum sensitivity is increasingly utilized as a biomarker for PARP inhibitor sensitivity in clinical trials (Table 2). The initial correlation of platinum sensitivity with PARP inhibitor sensitivity was observed in ovarian cancer. One study found a marked difference in olaparib sensitivity in platinum-sensitive and -resistant ovarian cancer patients (Fong et al., 2010). In pancreatic cancer, a similar trend has been observed. In a phase 2 trial of BRCA-deficient pancreatic cancer patients, none of the responding patients had previously progressed on platinum-based chemotherapy (Shroff et al., 2018). Building on this observation, the phase 3 POLO trial evaluated the efficacy of olaparib maintenance therapy following platinum-based chemotherapy in pancreatic cancer patients with BRCA1 and BRCA2 germline mutations. This study was limited to patients who were platinum-sensitive, defined as not having disease progression over at least 16 weeks of platinum-containing chemotherapy. Compared to the patients randomized to placebo, the patients treated with maintenance olaparib had a 3.6-month increase in progression-free survival (PFS) (Golan et al., 2019). This study led to the FDA approval of olaparib maintenance therapy in pancreatic cancer patients with BRCA1 and BRCA2 germline mutations.
There has been great interest in developing integrative genomic assays that predict HR deficiency (Lord and Ashworth, 2016). Myriad Genetics developed a homologous recombination deficiency (HRD) assay that identifies HR-deficient tumors based on characteristic genomic findings of these tumors, including Loss of Heterozygosity (LOH), Telomeric Allelic Imbalance (TAI), and Large-scale State Transitions (LST). Another genomic assay, the Foundation Medicine T5 NGS assay, assesses the mutational status of 30 HR genes and also calculates the percentage of genomic LOH (Frampton et al., 2013). Both the Myriad HRD assay and the Foundation Medicine LOH assay have been validated by their ability to predict PARP inhibitor sensitivity in large phase 3 clinical trials in ovarian cancer of niraparib and rucaparib, respectively (Coleman et al., 2017; González-Martín et al., 2019; Swisher et al., 2017). However, while both the Myriad HRD assay and Foundation Medicine LOH assay are predictive of PARP inhibitor sensitivity, they failed to identify all of the platinum-sensitive patients who benefitted from PARP inhibition (Coleman et al., 2017; González-Martín et al., 2019; Swisher et al., 2017). This finding highlights the value of using platinum sensitivity as a surrogate biomarker for PARP inhibitor responsiveness in ovarian cancer.
Schlafen 11 (SLFN11) has emerged as a biomarker of PARP inhibitor sensitivity and resistance. In screens of cancer cell lines for genes predictive of cytotoxic chemotherapy sensitivity, a low level of SLNF11 was a biomarker of resistance to DNA damaging cytotoxic chemotherapies, including topoisomerase inhibitors, platinum chemotherapies, and alkylating agents (Barretina et al., 2012; Zoppoli et al., 2012). Subsequent studies demonstrated that low levels of SLFN11 are also associated with resistance to PARP inhibitors (Murai et al., 2016). While the function of SLFN11 is unknown, a recent study demonstrated that SLFN11 decreases protein synthesis of DDR proteins, such as ATR and ATM (Li et al., 2018). Consistent with this observation, PARP inhibitor resistance caused by low SNLF11 levels, can be overcome by combined inhibition of ATR and PARP (Murai et al., 2016). This has led to a hypothesis that SLNF11 promotes apoptosis in cells under extreme replicative stress, such as that conferred by viral infection or some cancers (Murai et al., 2019).
In addition to predictive biomarkers, the process of DDR drug development also requires pharmacodynamic (PD) biomarkers, which establish that the DDR inhibitor is adequately hitting the target (Table 2). Given their role in repairing DNA damage, increased γH2AX is a universal PD biomarker shared among all DDR repair inhibitors. In addition, PARP or POLθ inhibition results in increased levels of RAD51. A combination of a PARP inhibitor plus a POLθ inhibitor further increases RAD51 foci to toxic levels, further establishing the mechanism of action of POL0 inhibitors (Ceccaldi et al., 2015).
Recent work has demonstrated that mutations in the metabolic enzymes isocitrate dehydrogenase 1 and 2 (IDH1 and IDH2) increase the sensitivity of tumors to PARP inhibition (Molenaar et al., 2018; Sulkowski et al., 2017). A possible mechanism for this is that 2-Hydroxyglutarate (2HG), an oncometabolite produced by mutated IDH1 and IDH2, suppresses HR (Sulkowski et al., 2017). A clinical trial testing whether PARP inhibitor monotherapy has efficacy in IDH1 and IDH2 mutated tumors is currently underway (NCT03212274).
ATR, CHK1, and WEE1 inhibition in HR-deficient tumors
DNA damage-induced checkpoints facilitate the coordination between the DNA damage response and cell cycle control and allow ample time for repair and prevention of the permanent DNA damage produced by replication and mitosis. Two of the PI3K-related protein kinases (PIKKs), ataxia-telangiectasia mutated (ATM) and ataxia-telangiectasia and Rad3-related (ATR), occupy a central role in signaling DNA damage to cell cycle checkpoints and DNA repair pathways (Curtin, 2012). Both checkpoint proteins play a critical role in maintaining the integrity of DNA. ATR homozygous knockout mice are embryonically lethal, while heterozygous ATR knockout mice (ATR−/+) are viable but have increased cancer incidence (Brown and Baltimore, 2000). Conversely, while mice with a homozygous deficiency in ATM are not embryonically lethal, they do demonstrate a markedly increased sensitivity to double-strand DNA breaks caused by y-irradiation (Xu and Baltimore, 1996). In response to y-irradiation, primary cells from ATM-deficient mice did not undergo cell cycle arrest and fail to activate p53 (Xu and Baltimore, 1996).
ATM is primarily activated by double-strand DNA breaks. Activation of ATM leads to initiation of the HR-pathway machinery through the activation of BRCA1, BRCA2, and PALB2 and development of RAD51 foci. In addition to activating HR, ATM also activates CHK2 which in turn activates p53. While ATM is activated by double-strand DNA breaks, ATR is primarily activated by stalling or collapse of DNA replication forks, leading to the accumulation of ssDNA and replicative stress. When a replication fork becomes unstable, because of factors such as DNA damage or a lack of deoxyribonucleotide triphosphates (dNTPs), ssDNA becomes coated with phosphorylated-RPA (pRPA) (Dobbelstein and Sorensen, 2015). The presence of pRPA leads to the activation of the regulatory kinases ATR and its downstream effector CHK1, which govern the cellular response to replicative stress. In addition, activation of ATR/CHK1 leads to the activation of WEE1 and inhibition of CDC25A and CDC25B (Otto and Sicinski, 2017). In this way, activation of the ATR/CHK1/WEE1 pathway compensates for replication stress, resulting in prolongation of the S/G2 phase of the cell cycle (Dobbelstein and Sorensen, 2015).
The critical DDR functions of the ATM and ATR pathways create an opportunity for synthetic lethality. Preclinical investigation has demonstrated that in vitro models of ATM-mutated gastric cancer and chronic lymphocytic leukemia are very sensitive ATR inhibition (Kwok et al., 2016; Min et al., 2017). Accordingly, a phase 1 trial of the ATR inhibitor BAY1895344 showed partial responses in patients whose tumors lacked evidence of ATM protein expression (De Bono et al., 2019). Evaluation of ATM deficiency using immunohistochemistry enables the most comprehensive assessment of ATM expression at the protein level; however, genomic analysis to identify ATM mutations is a robust alternative. Moreover, improved methodologies are needed to identify and distinguish ATM biallelic or dominant-negative mutations.
Acquired resistance to PARP inhibitors remains an unsolved clinical problem. Since the primary mechanisms of PARP inhibitor resistance are 1) the restoration of HR repair and 2) the stabilization of the replication fork, new pharmacological strategies are needed to reverse these processes and to restore PARP inhibitor sensitivity. Inhibition of the ATR-CHK1-WEE1 signaling cascade is likely to prove useful in the treatment of PARP inhibitor resistance (Figure 2). PARP inhibitor treatment results in replicative stress and activation of the ATR/CHK1 pathway (Kim et al., 2017). PARP inhibitor-resistant tumors are hyper-dependent on the ATR-CHK1-WEE1 pathway, and subsequent inhibition of this regulatory pathway may restore PARP inhibitor sensitivity.
Figure 2: The ATR/CHK1/WEE1 pathway compensates for the replicative stress induced by gemcitabine.
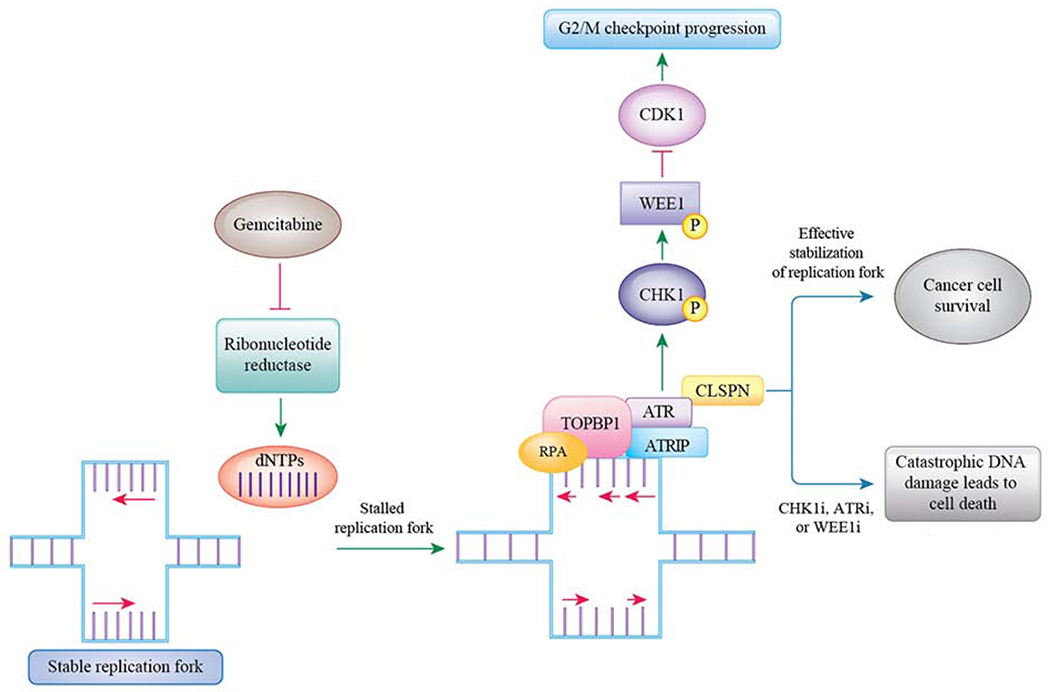
Gemcitabine causes replicative stress by irreversibly inhibiting ribonucleotide reductase and thereby decreasing dNTP concentration. Decreased dNTP concentration causes stalled replication forks. In response to this replicative stress, the ATR/CHK1/WEE1 pathway is activated and this leads to stabilization of the replicative forks. However, inhibitors of the ATR/CHK1/WEE1 pathway block this compensatory pathway. This leads to persistence of unstable replication forks and ultimately causes genomic catastrophe leading to cancer cell death.
Indeed, ATR inhibition can re-sensitize BRCA1-mutated ovarian cancer models to PARP inhibition by reversal of HR and through fork destabilization and blockage of BRCA1-independent RAD51 loading (Yazinski et al., 2017). Similarly, the CHK1 inhibitor prexasertib (LY2606368) overcomes PARP inhibitor resistance in ovarian cancer preclinical models (Parmar et al., 2019). Mechanistically, prexasertib induced cytotoxicity by converting stable replication forks to unstable replications forks (Parmar et al., 2019). Additionally, CHK1 phosphorylates RAD51, an event required for RAD51 nucleofilament formation. CHK1 inhibition therefore can reduce HR and destabilize replication forks (Sorensen et al., 2005).
Early treatment of a tumor with a PARP inhibitor and an ATR-CHK1-WEE1 pathway inhibitor may result in even stronger synergy. For instance, the concurrent exposure to an ATR inhibitor may prevent the acquisition of PARP inhibitor resistance in the first place. Additionally, because tumors are often polyclonal, some clones may be more sensitive to a PARP inhibitor while others are more sensitive to an ATR inhibitor, providing an additional incentive for concurrent upfront treatment with both drugs. Consistent with these findings, ATR or CHK1 inhibitors can synergistically increase cancer cell apoptosis in PDX models of BRCA2-deficient ovarian cancer (Kim et al., 2017). Interestingly, combined ATR and PARP inhibition leads to cytotoxicity by causing premature entry into mitosis (Schoonen et al., 2019).
Inhibition of the ATR-CHK1-WEE1 pathway is complicated by induction of myelosuppression. In phase 1 trials, myelosuppression was the dose limiting toxicity of the ATR inhibitor BAY1895344, the CHK1 inhibitors GDC-0575, and the WEE1 inhibitor adavosertib (De Bono et al., 2019; Do et al., 2015; Italiano et al., 2018; Plummer et al., 2019). A prime example of this challenge was the clinical development of the CHK1i prexasertib. After demonstrating initial encouraging results in ovarian and advanced squamous cell cancers, later phase trials faltered in part because of the treatment limitations of myelosuppression (Lee et al., 2018). Next generation CHK1 inhibitors, such as SRA737 (CCT245737), may mitigate the myelosuppression of other CHK1 inhibitors by decreasing simultaneous inhibition of CHK2 (Walton et al., 2016).
Therapeutic Strategies Designed to Increase Replicative Stress
Although some cancers have an inherent degree of endogenous replicative stress, agents that further increase replicative stress provide a novel treatment strategy. Topoisomerase I and II inhibitors generate replicative stress by preventing the repair of topoisomerase-generated DNA breaks and trapping the topoisomerase enzyme on DNA (Canela et al., 2019; Ubhi and Brown, 2019). This trapping blocks replication forks from proceeding. Furthermore, under normal circumstances, the PARP protein binds to topoisomerase I generated single-strand DNA breaks to stimulate the recruitment of DNA repair proteins such as tyrosyl-DNA phosphodiesterase 1 (TDP1) and XRCC1 (Das et al., 2014; Zhang et al., 2011). Not surprisingly, preclinical data has suggested that topoisomerase I inhibitors and PARP inhibitors are synergistic. Accordingly, preliminary signs of anti-cancer activity were observed in a phase 1 trial of the topoisomerase I inhibitor irinotecan and the PARP inhibitor veliparib (LoRusso et al., 2016; Zhang et al., 2011). Similarly, when compared to irinotecan monotherapy, combining irinotecan and ATR inhibition increases DNA damage and cancer cell death in in vitro and in vivo models (Josse et al., 2014). A phase 1 trial combining topotecan and the M6620 ATR inhibitor demonstrated preliminary signs of clinical benefit in several platinum-resistant small cell lung cancer patients (Thomas et al., 2018). Pharmacodynamic studies on this trial suggested that the topotecan/ATR inhibitor combination can increase the number of double-strand DNA breaks (Thomas et al., 2018).
Another widely recognized means of increasing replicative stress is to create conditions that decrease the cellular concentration of dNTPs, by using drugs such as hydroxyurea and gemcitabine (Ubhi and Brown, 2019). Depriving growing replication forks of dNTPs deprives DNA of its necessary building blocks, resulting in substantial replicative stress. Gemcitabine, a pyrimidine nucleoside analog commonly used for pancreatic and ovarian cancer, exerts its cytotoxicity through incorporation of its product dFdCTP into DNA, thus resulting in replication termination (Brunton et al., 2011). Unlike the pyrimidine analog cytarabine, gemcitabine also targets cancer cells by irreversibly inhibiting ribonucleotide reductase (Brunton et al., 2011; Moore and Cohen, 1967). Purine nucleotide analogs, such as clofarabine, fludarabine, and cladribine, are reversible inhibitors of ribonucleotide reductase (Tsesmetzis et al., 2018). The inhibition of ribonucleotide reductase, an enzyme that generates dNTPs, results in decreased dNTP concentrations and creates replicative stress (Ubhi and Brown, 2019). Pretreatment of tumor cells with gemcitabine stalls replication forks and activates the ATR-CHK1-WEE1 compensatory pathway, thereby stabilizing these stalled forks (Fordham et al., 2018; Koh et al., 2018). Accordingly, sequential treatment with gemcitabine, followed by an inhibitor of the ATR-CHK1-WEE1 pathway, provides a potent anticancer drug combination.
Clinical trials of CHK1 inhibitors have entailed a strategy of combining gemcitabine with CHK1 inhibition (Table 4)(Xiao et al., 2013). Like most therapies that increase replicative stress, a challenge is that these regimens could be too myelosuppressive. However, the high degree of synergy between gemcitabine and CHK1 inhibitors suggests that CHK1 inhibitors may be combined with substantially lower doses of gemcitabine than are typically used clinically. Through this strategy, low-dose gemcitabine generates replicative stress, creates a state dependent on the ATR-CHK1-WEE1 pathway, and primes cancer cells for the use of these kinase inhibitors. An additional advantage of this strategy is that low doses of gemcitabine avoid the myelosuppression that are induced by standard clinical doses. Recent phase 1 studies demonstrate that low doses of gemcitabine at 250 mg/m2 on days 1, 8, and 15 of a 28-day cycle are able to be safely administered with the CHK1 inhibitor SRA737 (Banerji et al., 2019). Despite the low dose of gemcitabine, confirmed partial responses were observed in anogenital and ovarian cancers (Banerji et al., 2019). Notably, partial responses were not observed in a separate trial testing single-agent SRA737 (Plummer et al., 2019).
Table 4.
Selected Clinical Trials Combinations Involving ATM, ATR, CHK1, DNA-PK, and WEE1 Inhibitors
| Combination | Trial Number | Phase | Tumor Type |
|---|---|---|---|
| ATMi/Chemotherapy | |||
| AZD0156/FOLFIRI | NCT02588105 | 1 | Solid Tumor |
| ATMi/PARPi | |||
| AZD0156/Olaparib | NCT02588105 | 1 | Solid Tumor |
| ATRi/PARPi | |||
| Olaparib/Ceralasertib | NCT03462342 | 2 | Ovarian Cancer |
| Niraparib/M4344 | NCT04149145 | 1 | Ovarian Cancer |
| ATRi/Chemotherapy | |||
| Ceralasertib/Gemcitabine | NCT03669601 | 1 | Solid Tumors, Ovarian Cancer |
| ATRi/PD1i | |||
| BAY1895344/Pembrolizumab | NCT04095273 | 1 | Solid Tumor |
| CHK1i/PARPi | |||
| Prexasertib/Olaparib | NCT03057145 | 1 | Solid Tumor |
| CHK1i/Chemotherapy | |||
| LY2880070/Gemcitabine | NCT02632448 | 1 | Solid Tumor |
| CHK1i/PD1i | |||
| Prexasertib/LY3300054 | NCT03495323 | 1 | Solid Tumor |
| DNA-PKi/PD1i | |||
| M3814/Avelumab | NCT03724890 | 1 | Solid Tumor |
| DNA-PKi/Radiation+/−Chemotherapy | |||
| M3814/Capecitabine/Radiation | NCT03770689 | 1 | Solid Tumor |
| WEE1i/PARPi | |||
| Olaparib/AZD1775 | NCT04197713 | 1 | Solid Tumors |
| WEE1i/Chemotherapy | |||
| AZD1775/Cisplatin | NCT03012477 | 2 | Breast Cancer |
| WEE1i/PD1i | |||
| AZD1775/Durvalumab | NCT02617277 | 1 | Solid Tumors |
ATR inhibitors can also counteract replicative stress, suggesting that combining ATR inhibition with gemcitabine may also be an effective treatment strategy. In response to replicative stress, ATR functions to stabilize replication forks and increases RRM2 levels, thereby ensuring adequate levels of dNTPs (Buisson et al., 2015). Hence, ATR inhibition can prevent compensatory increases in RRM2 after its suppression by gemcitabine. The therapeutic effectiveness of combining gemcitabine with ATR inhibition has been shown in preclinical models of multiple tumor types (Fordham et al., 2018; Hall et al., 2014). In a panel of pancreatic cancer cells, the ATR inhibitor ceralasertib (AZD6738) prevented gemcitabine-induced CHK1 phosphorylation, intra-S-phase checkpoint activation, and compensatory RRM2 re-accumulation (Wallez et al., 2018). The combination of gemcitabine and ceralasertib had significant anti-tumor activity in the K8484 pancreatic cancer cell line murine allograft model (Wallez et al., 2018). By blocking the ability of the cancer cell to compensate for gemcitabine-induced replicative stress, ATR inhibitor treatment markedly increases biomarkers of replicative stress (pRPA) and DNA damage (γH2AX).
A randomized phase 2 clinical trial of the ATR inhibitor M6620 (VX-970) combined with gemcitabine, compared with single agent gemcitabine in platinum-resistant ovarian cancer was recently completed (Konstantinopoulos et al., 2020). Combination therapy was well tolerated. Compared to single agent gemcitabine, the gemcitabine/M6620 combination had a statistically-significant 8 week increase in progression-free survival (PFS) [HR 0.57, p = 0.044). A confirmatory phase 3 randomized study is being planned (Konstantinopoulos et al., 2020).
A key clinical question is the differential activity of ATR, CHK1, and WEE1 inhibitors. While the precise mechanistic differences among these DDR drugs is under investigation, some clear distinctions are emerging. First, since ATR signals at the beginning of the DDR damage pathway, alternative pathways to activate CHK1 exist; thus, ATR inhibition appears less potent as a monotherapy but may be more suitable as a partner in combinatorial regimens (Buisson et al., 2015; Young et al., 2019).
Furthermore, in addition to ATR activating CHK1, DNA-PK has also been shown to activate CHK1 (Buisson et al., 2015). CHK1’s two upstream signaling inputs may distinguish the anti-tumor activity of ATR and CHK1 inhibitors. The WEE1 kinase is furthest downstream in the pathway, Intriguingly a study in diffuse large B cell lymphoma suggested that it had increased monotherapy potency compared to single agent ATR inhibition (Young et al., 2019). Further differences between WEE1 inhibition and ATR/CHK1 inhibition include the stage of the cell cycle where they exert their effect. ATR/CHK1 inhibitors interfere with the compensatory mechanisms which regulate S phase, while WEE1 inhibitors release cells into mitosis prematurely (Koh et al., 2018; Young et al., 2019). Finally, while the major toxicity of ATR, CHK1, and WEE1 inhibitors has been myelosuppression, a first generation CHK1 inhibitor, AZD7765, also caused cardiotoxicity, leading to the discontinuation of its development (Sausville et al., 2014).
Given the potential importance of replicative stress in drug efficacy, it is critical to develop biomarkers that can assess replication stress. pRPA and pKAP1, which are both downstream substrates of ATR, have been used as immunohistochemical markers of replicative stress (Hill et al., 2018; Zellweger et al., 2015). A more rigorous approach of evaluating replicative stress is the assessment of replication fork stability using a DNA fiber assay. DNA fiber assays assess replication fork stability by consecutively pulsing live cells with two labeled thymidine analogs, typically 5-iodo-2’-deoxyuridine (IdU) and 5-chloro-2’-deoxyuridine (CldU) (Hill et al., 2018; Quinet et al., 2017). During this assay, the cells are treated with a fork stalling drug, such as hydroxyurea, either after or between the thymidine analog pulses. If incorporation of the two labeled thymidine analogs is equivalent, this indicates the replication fork was stable. However, if there is less incorporation of the second thymidine analog, this result suggests the replication forks containing the second thymidine analog were degraded and hence indicates replication fork instability. The predictive potential of this approach was shown in a recent study of ovarian cancer organoids demonstrating that unstable replication forks correlate with sensitivity to CHK1i and ATRi (Hill et al., 2018). A major shortcoming of DNA fiber assays, especially in clinical settings, is the need for live tumor cells. A more general strategy for assessing whether an experimental drug or drug combination increases replicative stress is to utilize pre-treatment and on-treatment tumor biopsies. Immunohistochemical analysis of these biopsies can assess whether there are increasing levels of pRPA and pKAP1, while organoid cultures generated from the biopsies can be used to perform DNA fiber assays to explore if there is an increase in replication fork instability.
Combination of PARP inhibitors and Cytotoxic Agents
Cytotoxic chemotherapy agents that generate DNA damage may be effective combination partners for PARP inhibitors. Given the mechanism of action of topoisomerase inhibitors and TMZ, combinations of these drugs with PARP inhibitors have been extensively investigated. Topoisomerase I enzymes generate a topoisomerase I cleavage complex which is stabilized by the PARP1 protein (Thomas and Pommier, 2019). This mechanism provides a rationale for a combination of a PARP inhibitor plus a topoisomerase I inhibitor. Similarly, TMZ-induced DNA damage is typically repaired by BER, and the PARP1 protein typically binds to the single-strand DNA generated by this reaction (Murai et al., 2014b). This mechanism suggests that TMZ and PARP inhibitors can also be synergistically combined.
Given the potential synergy of cytotoxic chemotherapy and PARP inhibitors, multiple clinical trials have been conducted exploring these combinations. However, when PARP inhibitors are combined with chemotherapy, achieving full dose chemotherapy has been challenging becaus0e of overlapping myelosuppression (Rajan et al., 2012). While the PARP inhibitor and cytotoxic chemotherapy combination provides enhanced tumoricidal activity, it also risks causing increased normal tissue toxicity. Therefore, the utility of a DDR inhibitor combined with cytotoxic chemotherapy requires establishment of a therapeutic index. If the DDR agent has the same activity in normal tissue and cancer tissue, then no advantage can be achieved. In general, cancer cells exhibit a greater baseline deficiency in the DDR and accumulate more damage than normal cells (Mahamud et al., 2017), and these differences can be leveraged to gain a therapeutic index. Accordingly, profiling DDR defects in specific cancers may guide the combined use of a DDR targeted agent with a cytotoxic agent.
Given the difficulty with myelosuppression, recent trials have explored different dosing strategies. Farago and colleagues found that limiting the administration of olaparib and TMZ 75 mg/m2 daily to the first 7 days of each 21-day cycle was feasible and safe (Farago et al., 2019). Encouragingly, in platinum-sensitive small cell lung cancer patients, the objective response rate to this combination was 47% and the median progression-free survival was 4.5 months. Another strategy being explored involves combining low dose TMZ with PARP inhibition (Stradella et al., 2019).
Combination of DDR Inhibitors with Immunotherapy
A major limitation of DDR inhibitors is the development of acquired resistance. To increase the durability of responses, there is substantial interest in combining DDR inhibitors with immunotherapy agents. The success of PD1 inhibition in cancers with mismatch repair deficiency is proof of principle that neoantigens generated by faulty DNA repair systems can drive an immune response (Le et al., 2017). However, unlike mismatch repair deficient tumors, many tumors do not have a sufficient neoantigen load to drive the immune response. While neoantigen generation by inhibiting DNA repair proteins may be insufficient to drive an immune response, other interactions between DNA repair proteins and the immunoregulators have the potential to catalyze an anti-tumor immune response (Mouw and D’Andrea, 2018). Double-strand DNA breaks can stimulate PD-L1 upregulation, through ATM/ATR/CHK1 signaling, in cancer cells (Sato et al., 2017). Similarly, PARP inhibitor treatment, perhaps through the introduction of DNA damage, also increases PD-L1 expression in breast cancer cells (Jiao et al., 2017). While DNA damage can upregulate PD-L1, the presence of cytoplasmic DNA also stimulates the cyclic GMP–AMP synthase (cGAS)/stimulator of interferon genes (STING) pathway. The cGAS/STING pathway is a component of the innate immune system, a system which evolved to prevent the introduction of viral DNA into the cell. Its activation causes proinflammatory cytokine secretion, such as type 1 interferon (Kwon and Bakhoum, 2019). In an in vivo model of BRCA1-deficient triple-negative breast cancer, PARP inhibitor mediated DNA damage and activated the cGAS/STING pathway, leading to intratumoral CD8 T cell infiltration (Pantelidou et al., 2019).
Multiple clinical trials are underway that are combining DDR inhibitors with PD1 inhibitors. Among DDR inhibitor/immunotherapy combination trials, PARP inhibitors and PD1 inhibitors are the furthest in clinical development. Multiple single arm trials have shown, at best, modest anti-tumor activity. Randomized trials are needed to confirm whether the efficacy of these combinations is superior to PD1 monotherapy (Konstantinopoulos et al., 2019; Vinayak et al., 2019). Given the complexity of interpreting the efficacy of the DDR inhibitor/immunotherapy combination trials, biomarkers for predicting response are being developed. In addition to signature 3 as a biomarker of response in the aforementioned trial of combined niraparib and pembrolizumab in platinum-resistant ovarian cancer, a nanostring-based assay examining interferon gene expression was also predictive of response (Farkkila et al., 2020). Finally, immunocompetent models will be required to preclinically assess combinations of DDR inhibitors with immunotherapies, including appropriate genetically-engineered mouse models, syngeneic models, or humanized mice harboring human cell line xenografts.
CONCLUSIONS
The success of PARP inhibitors in HR-deficient ovarian, breast, and pancreatic cancers highlights the potential of DDR inhibitors. Recently, several other DDR inhibitors, including those targeting ATM, ATR, CHK1, DNA-PK and WEE1 have entered into clinical trials. While many of trials with these agents have been monotherapy trials, there is an increasing number of clinical trials utilizing combinations of these agents.
While there is a temptation to combine drugs in trials because they are available, it is essential that a strong preclinical background provide the rationale of any proposed clinical study. Given the molecular heterogeneity of cancer patients and the risk of mounting toxicities from DDR inhibitor combinations, great care is required to define the optimal clinical setting, dose intensity, and schedule of these agents. Predictive biomarkers, which have been rigorously validated preclinically, can be utilized in DDR inhibitor clinical trial design to define the most suitable patient population. Furthermore, to ensure that clinical studies generate useful mechanistic observations, clinical trials of DDR combinations should incorporate pharmacodynamic biomarkers that can molecularly interrogate whether a drug hits the desired target or pathway. In addition, molecular understanding of the mechanisms of resistance to DDR inhibitors will spur development of strategies that can delay, or reverse, acquired resistance to these agents and optimize clinical benefit from DDR-directed therapies.
Figure 3: Hyperactivation of the ATR/CHK1/WEE1 pathway leads to acquired resistance to PARP inhibitors.
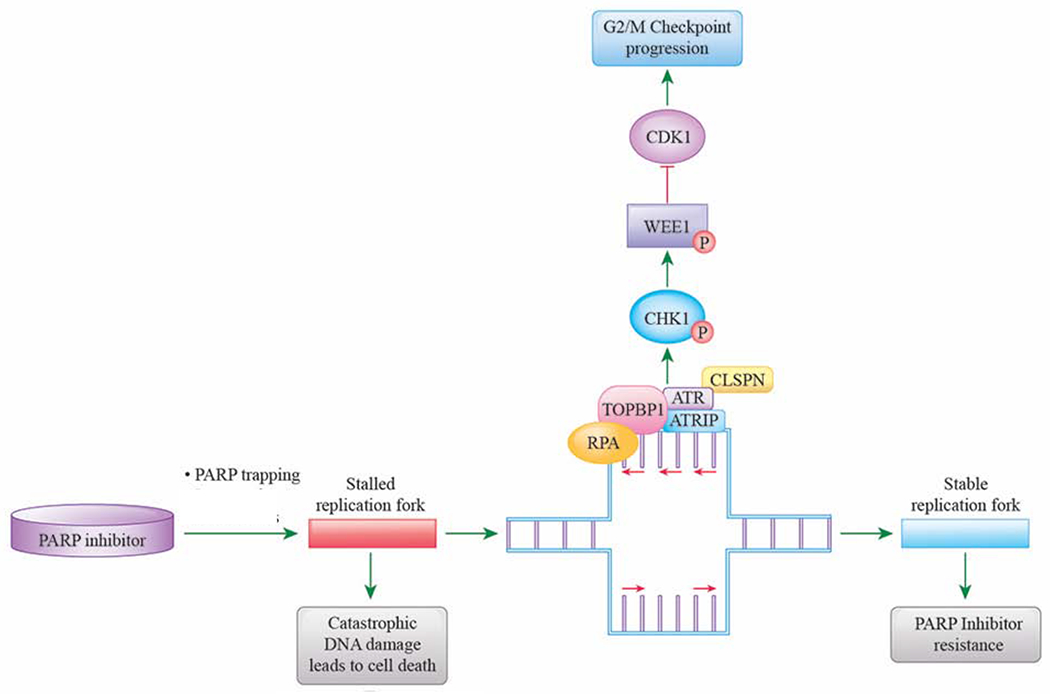
PARP inhibitors, through inhibition of single-strand DNA repair and PARP inhibitor trapping, causes stalled replication forks and ultimately cell death in HR-deficient cancers. Cancer cells can develop resistance to PARP inhibitors by hyperactivation of the compensatory ATR/CHK1/WEE1 pathway, leading to stabilization of replication forks.
Acknowledgements
The work of J.M.C. is supported by the Dana-Farber Cancer Institute Hale Center for Pancreatic Cancer Research, Stand Up To Cancer, and the Lustgarten Foundation. A.J.A. is supported by the Lustgarten Foundation, the Hale Center for Pancreatic Cancer Research, the Doris Duke Charitable Foundation, the Pancreatic Cancer Action Network, NIH-NCI K08 CA218420-02, and U01 CA224146. G.I.S. and A.D.D. receive support from P50 CA127003. A.D.D. is also supported by Stand Up To Cancer and the Lustgarten Foundation.
Authors’ Disclosures of Potential Conflicts of Interest
J.M. Cleary received research funding from Merck, AstraZeneca, and Tesaro, has served as a consultant to Bristol Myers Squib, and received travel funding from Bristol Myers Squib and Roche.
A.J. Aguirre has consulted for Oncorus, Inc., Arrakis Therapeutics, and Merck & Co., Inc, and has research funding from Mirati Therapeutics and Deerfield, Inc. that are unrelated to this project.
G.I. Shapiro has received research funding from Eli Lilly, Merck KGaA/EMD-Serono, Merck, and Sierra Oncology. He has served on advisory boards for Pfizer, Eli Lilly, G1 Therapeutics, Roche, Merck KGaA/EMD-Serono, Sierra Oncology, Bicycle Therapeutics, Fusion Pharmaceuticals, Cybrexa Therapeutics, Astex, Almac, Ipsen, Bayer, Angiex, Daiichi Sankyo, Seattle Genetics, Boehringer Ingelheim, ImmunoMet, Asana, Artios, Atrin and Concarlo Holdings. In addition, he holds a patent entitled, “Dosage regimen for sapacitabine and seliciclib,” also issued to Cyclacel Pharmaceuticals, and a pending patent, entitled, “Compositions and Methods for Predicting Response and Resistance to CDK4/6 Inhibition,” together with Liam Cornell.
A.D. D’Andrea is a consultant/advisory board member for Lilly Oncology, Merck-EMD Serono, Intellia Therapeutics, Sierra Oncology, Cyteir Therapeutics, Third Rock Ventures, AstraZeneca, Ideaya Inc., Cedilla Therapeutics Inc., a stockholder in Ideaya Inc., Cedilla Therapeutics Inc., and Cyteir, and reports receiving commercial research grants from Lilly Oncology and Merck-EMD Serono.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale AL, et al. (2013). Signatures of mutational processes in human cancer. Nature 500, 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ang JE, Gourley C, Powell CB, High H, Shapira-Frommer R, Castonguay V, De Greve J, Atkinson T, Yap TA, Sandhu S, et al. (2013). Efficacy of chemotherapy in BRCA1/2 mutation carrier ovarian cancer in the setting of PARP inhibitor resistance: a multi-institutional study. Clinical cancer research : an official journal of the American Association for Cancer Research 19, 5485–5493. [DOI] [PubMed] [Google Scholar]
- Banerji U, Plummer ER, Moreno V, Ang JE, Quinton A, Drew Y, Hernandez T, Roda D, Carter L, Navarro A, et al. (2019). A phase I/II first-in-human trial of oral SRA737 (a Chk1 inhibitor) given in combination with low-dose gemcitabine in subjects with advanced cancer. Journal of Clinical Oncology 37, 3095–3095. [Google Scholar]
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehár J, Kryukov GV, Sonkin D, et al. (2012). The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennardo N, Cheng A, Huang N, and Stark JM (2008). Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS genetics 4, e1000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhargava R, Onyango DO, and Stark JM (2016). Regulation of Single-Strand Annealing and its Role in Genome Maintenance. Trends Genet 32, 566–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black SJ, Kashkina E, Kent T, and Pomerantz RT (2016). DNA Polymerase θ: A Unique Multifunctional End-Joining Machine. Genes 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackford AN, and Jackson SP (2017). ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Molecular cell 66, 801–817. [DOI] [PubMed] [Google Scholar]
- Brown EJ, and Baltimore D (2000). ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes & development 14, 397–402. [PMC free article] [PubMed] [Google Scholar]
- Brunton L, Chabner B, and Knollman B (2011). Goodman and Gilman’s Phamracological Basis of Therapeutics. (McGraw-Hill Companies, Inc.). [Google Scholar]
- Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, and Helleday T (2005). Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434, 913–917. [DOI] [PubMed] [Google Scholar]
- Buisson R, Boisvert JL, Benes CH, and Zou L (2015). Distinct but Concerted Roles of ATR, DNA-PK, and Chk1 in Countering Replication Stress during S Phase. Molecular cell 59, 1011–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canela A, Maman Y, Huang SN, Wutz G, Tang W, Zagnoli-Vieira G, Callen E, Wong N, Day A, Peters JM, et al. (2019). Topoisomerase II-Induced Chromosome Breakage and Translocation Is Determined by Chromosome Architecture and Transcriptional Activity. Molecular cell 75, 252–266. e258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castroviejo-Bermejo M, Cruz C, Llop-Guevara A, Gutierrez-Enriquez S, Ducy M, Ibrahim YH, Gris-Oliver A, Pellegrino B, Bruna A, Guzman M, et al. (2018). A RAD51 assay feasible in routine tumor samples calls PARP inhibitor response beyond BRCA mutation. EMBO molecular medicine 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccaldi R, Liu JC, Amunugama R, Hajdu I, Primack B, Petalcorin MI, O’Connor KW, Konstantinopoulos PA, Elledge SJ, Boulton SJ, et al. (2015). Homologous-recombination-deficient tumours are dependent on Poltheta-mediated repair. Nature 518, 258–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan SH, Yu AM, and McVey M (2010). Dual roles for DNA polymerase theta in alternative end-joining repair of double-strand breaks in Drosophila. PLoS genetics 6, e1001005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandran EA, and Kennedy I (2018). Significant Tumor Response to the Poly (ADP-ribose) Polymerase Inhibitor Olaparib in Heavily Pretreated Patient With Ovarian Carcinosarcoma Harboring a Germline RAD51D Mutation. 1–4. [DOI] [PubMed]
- Chang HHY, Pannunzio NR, Adachi N, and Lieber MR (2017). Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nature Reviews Molecular Cell Biology 18, 495–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman JR, Sossick AJ, Boulton SJ, and Jackson SP (2012). BRCA1-associated exclusion of 53BP1 from DNA damage sites underlies temporal control of DNA repair. Journal of cell science 125, 3529–3534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Ghorai MK, Kenney G, and Stubbe J (2008). Mechanistic studies on bleomycin-mediated DNA damage: multiple binding modes can result in double-stranded DNA cleavage. Nucleic acids research 36, 3781–3790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clairmont CS, Sarangi P, Ponnienselvan K, Galli LD, Csete I, Moreau L, Adelmant G, Chowdhury D, Marto JA, and D’Andrea AD (2020). TRIP13 regulates DNA repair pathway choice through REV7 conformational change. Nature cell biology 22, 87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman RL, Oza AM, Lorusso D, Aghajanian C, Oaknin A, Dean A, Colombo N, Weberpals JI, Clamp A, Scambia G, et al. (2017). Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 390, 1949–1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtin NJ (2012). DNA repair dysregulation from cancer driver to therapeutic target. Nature reviews. Cancer 12, 801–817. [DOI] [PubMed] [Google Scholar]
- Damia G, and Broggini M (2019). Platinum Resistance in Ovarian Cancer: Role of DNA Repair. Cancers (Basel) 11, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das BB, Huang S.-y.N., Murai J, Rehman I, Amé J-C, Sengupta S, Das SK, Majumdar P, Zhang H, Biard D, et al. (2014). PARP1-TDP1 coupling for the repair of topoisomerase I-induced DNA damage. Nucleic acids research 42, 4435–4449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bono JS, Tan DSP, Caldwell R, Terbuch A, Goh BC, Heong V, Haris NM, Bashir S, Hong DS, Meric-Bernstam F, et al. (2019). First-in-human trial of the oral ataxia telangiectasia and Rad3-related (ATR) inhibitor BAY 1895344 in patients (pts) with advanced solid tumors. Journal of Clinical Oncology 37, 3007–3007. [Google Scholar]
- De Vos M, Schreiber V, and Dantzer F (2012). The diverse roles and clinical relevance of PARPs in DNA damage repair: current state of the art. Biochemical pharmacology 84, 137–146. [DOI] [PubMed] [Google Scholar]
- Dev H, Chiang TW, Lescale C, de Krijger I, Martin AG, Pilger D, Coates J, Sczaniecka-Clift M, Wei W, Ostermaier M, et al. (2018). Shieldin complex promotes DNA end-joining and counters homologous recombination in BRCA1-null cells. Nature cell biology 20, 954–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do K, Wilsker D, Ji J, Zlott J, Freshwater T, Kinders RJ, Collins J, Chen AP, Doroshow JH, and Kummar S (2015). Phase I Study of Single-Agent AZD1775 (MK-1775), a Wee1 Kinase Inhibitor, in Patients With Refractory Solid Tumors. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 33, 3409–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobbelstein M, and Sorensen CS (2015). Exploiting replicative stress to treat cancer. Nature reviews. Drug discovery 14, 405–423. [DOI] [PubMed] [Google Scholar]
- Drane P, Brault ME, Cui G, Meghani K, Chaubey S, Detappe A, Parnandi N, He Y, Zheng XF, Botuyan MV, et al. (2017). TIRR regulates 53BP1 by masking its histone methyl-lysine binding function. Nature 543, 211–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escribano-Diaz C, Orthwein A, Fradet-Turcotte A, Xing M, Young JT, Tkac J, Cook MA, Rosebrock AP, Munro M, Canny MD, et al. (2013). A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Molecular cell 49, 872–883. [DOI] [PubMed] [Google Scholar]
- Farago AF, Yeap BY, Stanzione M, Hung YP, Heist RS, Marcoux JP, Zhong J, Rangachari D, Barbie DA, Phat S, et al. (2019). Combination Olaparib and Temozolomide in Relapsed Small-Cell Lung Cancer. Cancer discovery 9, 1372–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farkkila A, Gulhan DC, Casado J, Jacobson CA, Nguyen H, Kochupurakkal B, Maliga Z, Yapp C, Chen YA, Schapiro D, et al. (2020). Immunogenomic profiling determines responses to combined PARP and PD-1 inhibition in ovarian cancer. Nature communications 11, 1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer H, McCabe N, Lord CJ, Tutt ANJ, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, et al. (2005). Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434, 917–921. [DOI] [PubMed] [Google Scholar]
- Fong PC, Yap TA, Boss DS, Carden CP, Mergui-Roelvink M, Gourley C, De Greve J, Lubinski J, Shanley S, Messiou C, et al. (2010). Poly(ADP)-ribose polymerase inhibition: frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 28, 2512–2519. [DOI] [PubMed] [Google Scholar]
- Fordham SE, Blair HJ, Elstob CJ, Plummer R, Drew Y, Curtin NJ, Heidenreich O, Pal D, Jamieson D, Park C, et al. (2018). Inhibition of ATR acutely sensitizes acute myeloid leukemia cells to nucleoside analogs that target ribonucleotide reductase. Blood advances 2, 1157–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frampton GM, Fichtenholtz A, Otto GA, Wang K, Downing SR, He J, Schnall-Levin M, White J, Sanford EM, An P, et al. (2013). Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nature biotechnology 31, 1023–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geenen JJJ, Linn SC, Beijnen JH, and Schellens JHM (2018). PARP Inhibitors in the Treatment of Triple-Negative Breast Cancer. Clinical pharmacokinetics 57, 427–437. [DOI] [PubMed] [Google Scholar]
- Gogola E, Duarte AA, de Ruiter JR, Wiegant WW, Schmid JA, de Bruijn R, James DI, Guerrero Llobet S, Vis DJ, Annunziato S, et al. (2018). Selective Loss of PARG Restores PARylation and Counteracts PARP Inhibitor-Mediated Synthetic Lethality. Cancer cell 33, 1078–1093. e1012. [DOI] [PubMed] [Google Scholar]
- Golan T, Hammel P, Reni M, Van Cutsem E, Macarulla T, Hall MJ, Park J-O, Hochhauser D, Arnold D, Oh D-Y, et al. (2019). Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. 381, 317–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Martín A, Pothuri B, Vergote I, DePont Christensen R, Graybill W, Mirza MR, McCormick C, Lorusso D, Hoskins P, Freyer G, et al. (2019). Niraparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. 381, 2391–2402. [DOI] [PubMed] [Google Scholar]
- Grimme JM, Honda M, Wright R, Okuno Y, Rothenberg E, Mazin AV, Ha T, and Spies M (2010). Human Rad52 binds and wraps single-stranded DNA and mediates annealing via two hRad52-ssDNA complexes. Nucleic acids research 38, 2917–2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulhan DC, Lee JJ, Melloni GEM, Cortes-Ciriano I, and Park PJ (2019). Detecting the mutational signature of homologous recombination deficiency in clinical samples. Nature genetics 51, 912–919. [DOI] [PubMed] [Google Scholar]
- Hall AB, Newsome D, Wang Y, Boucher DM, Eustace B, Gu Y, Hare B, Johnson MA, Milton S, Murphy CE, et al. (2014). Potentiation of tumor responses to DNA damaging therapy by the selective ATR inhibitor VX-970. Oncotarget 5, 5674–5685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He YJ, Meghani K, Caron MC, Yang C, Ronato DA, Bian J, Sharma A, Moore J, Niraj J, Detappe A, et al. (2018). DYNLL1 binds to MRE11 to limit DNA end resection in BRCA1-deficient cells. Nature 563, 522–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henneman L, van Miltenburg MH, Michalak EM, Braumuller TM, Jaspers JE, Drenth AP, de Korte-Grimmerink R, Gogola E, Szuhai K, Schlicker A, et al. (2015). Selective resistance to the PARP inhibitor olaparib in a mouse model for BRCA1-deficient metaplastic breast cancer. Proceedings of the National Academy of Sciences of the United States of America 112, 8409–8414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill SJ, Decker B, Roberts EA, Horowitz NS, Muto MG, Worley MJ Jr., Feltmate CM, Nucci MR, Swisher EM, Nguyen H, et al. (2018). Prediction of DNA Repair Inhibitor Response in Short-Term Patient-Derived Ovarian Cancer Organoids. Cancer discovery 8, 1404–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howlett NG, Taniguchi T, Olson S, Cox B, Waisfisz Q, De Die-Smulders C, Persky N, Grompe M, Joenje H, Pals G, et al. (2002). Biallelic inactivation of BRCA2 in Fanconi anemia. Science (New York, N.Y.) 297, 606–609. [DOI] [PubMed] [Google Scholar]
- Isono M, Niimi A, Oike T, Hagiwara Y, Sato H, Sekine R, Yoshida Y, Isobe SY, Obuse C, Nishi R, et al. (2017). BRCA1 Directs the Repair Pathway to Homologous Recombination by Promoting 53BP1 Dephosphorylation. Cell reports 18, 520–532. [DOI] [PubMed] [Google Scholar]
- Italiano A, Infante JR, Shapiro GI, Moore KN, LoRusso PM, Hamilton E, Cousin S, Toulmonde M, Postel-Vinay S, Tolaney S, et al. (2018). Phase I study of the checkpoint kinase 1 inhibitor GDC-0575 in combination with gemcitabine in patients with refractory solid tumors. Annals of oncology : official journal of the European Society for Medical Oncology 29, 1304–1311. [DOI] [PubMed] [Google Scholar]
- Jaspers JE, Kersbergen A, Boon U, Sol W, van Deemter L, Zander SA, Drost R, Wientjens E, Ji J, Aly A, et al. (2013). Loss of 53BP1 causes PARP inhibitor resistance in Brca1-mutated mouse mammary tumors. Cancer discovery 3, 68–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao S, Xia W, Yamaguchi H, Wei Y, Chen M-K, Hsu J-M, Hsu JL, Yu W-H, Du Y, Lee H-H, et al. (2017). PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated Immunosuppression. Clinical Cancer Research 23, 3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson N, Johnson SF, Yao W, Li YC, Choi YE, Bernhardy AJ, Wang Y, Capelletti M, Sarosiek KA, Moreau LA, et al. (2013). Stabilization of mutant BRCA1 protein confers PARP inhibitor and platinum resistance. Proceedings of the National Academy of Sciences of the United States of America 110, 17041–17046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SF, Cruz C, Greifenberg AK, Dust S, Stover DG, Chi D, Primack B, Cao S, Bernhardy AJ, Coulson R, et al. (2016). CDK12 Inhibition Reverses De Novo and Acquired PARP Inhibitor Resistance in BrCa Wild-Type and Mutated Models of Triple-Negative Breast Cancer. Cell reports 17, 2367–2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josse R, Martin SE, Guha R, Ormanoglu P, Pfister TD, Reaper PM, Barnes CS, Jones J, Charlton P, Pollard JR, et al. (2014). ATR inhibitors VE-821 and VX-970 sensitize cancer cells to topoisomerase i inhibitors by disabling DNA replication initiation and fork elongation responses. Cancer Res 74, 6968–6979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, George E, Ragland RL, Rafail S, Zhang R, Krepler C, Morgan MA, Herlyn M, Brown EJ, and Simpkins F (2017). Targeting the ATR/CHK1 Axis with PARP Inhibition Results in Tumor Regression in <em>BRCA</em>-Mutant Ovarian Cancer Models. Clinical Cancer Research 23, 3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh S-B, Wallez Y, Dunlop CR, Bernaldo de Quirós Fernández S, Bapiro TE, Richards FM, and Jodrell DI (2018). Mechanistic Distinctions between CHK1 and WEE1 Inhibition Guide the Scheduling of Triple Therapy with Gemcitabine. 78, 3054–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondrashova O, Topp M, Nesic K, Lieschke E, Ho G-Y, Harrell MI, Zapparoli GV, Hadley A, Holian R, Boehm E, et al. (2018). Methylation of all BRCA1 copies predicts response to the PARP inhibitor rucaparib in ovarian carcinoma. Nature communications 9, 3970–3970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstantinopoulos PA, Ceccaldi R, Shapiro GI, and D’Andrea AD (2015). Homologous Recombination Deficiency: Exploiting the Fundamental Vulnerability of Ovarian Cancer. Cancer discovery 5, 1137–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstantinopoulos PA, Cheng S-C, Hendrickson AEW, Penson RT, Schumer ST, Doyle LA, Lee EK, Kohn EC, Duska LR, Crispens MA, et al. (2020). Randomized Phase 2 Study of the ATR inhibitor Berzosertib in Combination with Gemcitabine versus Gemcitabine alone in Platinum Resistant High Grade Serous Ovarian Cancer (HGSOC). Lancet Oncology In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstantinopoulos PA, and Matulonis UA (2018). PARP Inhibitors in Ovarian Cancer: A Trailblazing and Transformative Journey. Clinical cancer research : an official journal of the American Association for Cancer Research 24, 4062–4065. [DOI] [PubMed] [Google Scholar]
- Konstantinopoulos PA, Waggoner S, Vidal GA, Mita M, Moroney JW, Holloway R, Van Le L, Sachdev JC, Chapman-Davis E, Colon-Otero G, et al. (2019). Single-Arm Phases 1 and 2 Trial of Niraparib in Combination With Pembrolizumab in Patients With Recurrent Platinum-Resistant Ovarian Carcinoma. JAMA oncology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwok M, Davies N, Agathanggelou A, Smith E, Oldreive C, Petermann E, Stewart G, Brown J, Lau A, Pratt G, et al. (2016). ATR inhibition induces synthetic lethality and overcomes chemoresistance in TP53- or ATM-defective chronic lymphocytic leukemia cells. Blood 127, 582–595. [DOI] [PubMed] [Google Scholar]
- Kwon J, and Bakhoum SF (2019). The Cytosolic DNA-Sensing cGAS-STING Pathway in Cancer. Cancer discovery. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, Lu S, Kemberling H, Wilt C, Luber BS, et al. (2017). Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science (New York, N.Y.) 357, 409–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JM, Nair J, Zimmer A, Lipkowitz S, Annunziata CM, Merino MJ, Swisher EM, Harrell MI, Trepel JB, Lee MJ, et al. (2018). Prexasertib, a cell cycle checkpoint kinase 1 and 2 inhibitor, in BRCA wild-type recurrent high-grade serous ovarian cancer: a first-in-class proof-of-concept phase 2 study. The Lancet. Oncology 19, 207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Kao E, Malone D, Gao X, Wang JYJ, and David M (2018). DNA damage-induced cell death relies on SLFN11-dependent cleavage of distinct type II tRNAs. Nature structural & molecular biology 25, 1047–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lok BH, Gardner EE, Schneeberger VE, Ni A, Desmeules P, Rekhtman N, de Stanchina E, Teicher BA, Riaz N, Powell SN, et al. (2017). PARP Inhibitor Activity Correlates with SLFN11 Expression and Demonstrates Synergy with Temozolomide in Small Cell Lung Cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 23, 523–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord CJ, and Ashworth A (2016). BRCAness revisited. Nature reviews. Cancer 16, 110–120. [DOI] [PubMed] [Google Scholar]
- LoRusso PM, Li J, Burger A, Heilbrun LK, Sausville EA, Boerner SA, Smith D, Pilat MJ, Zhang J, Tolaney SM, et al. (2016). Phase I Safety, Pharmacokinetic, and Pharmacodynamic Study of the Poly(ADP-ribose) Polymerase (PARP) Inhibitor Veliparib (ABT-888) in Combination with Irinotecan in Patients with Advanced Solid Tumors. Clinical cancer research : an official journal of the American Association for Cancer Research 22, 3227–3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maclay T, Vacca J, McComas C, Castro A, Day M, and Mills K (2018). CYT01B, a Novel RAD51 Inhibitor, Act Synergistically with Both Targeted and Chemotherapeutic Anti-Cancer Agents. Blood 132, 3963–3963. [Google Scholar]
- Mahamud O, So J, Chua MLK, and Bristow RG (2017). Targeting DNA repair for precision radiotherapy: Balancing the therapeutic ratio. Current problems in cancer 41, 265–272. [DOI] [PubMed] [Google Scholar]
- Makharashvili N, and Paull TT (2015). CtIP: A DNA damage response protein at the intersection of DNA metabolism. DNA repair 32, 75–81. [DOI] [PubMed] [Google Scholar]
- Mateos-Gomez PA, Gong F, Nair N, Miller KM, Lazzerini-Denchi E, and Sfeir A (2015). Mammalian polymerase θ promotes alternative NHEJ and suppresses recombination. Nature 518, 254–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCabe N, Turner NC, Lord CJ, Kluzek K, Bialkowska A, Swift S, Giavara S, O’Connor MJ, Tutt AN, Zdzienicka MZ, et al. (2006). Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res 66, 8109–8115. [DOI] [PubMed] [Google Scholar]
- Min A, Im SA, Jang H, Kim S, Lee M, Kim DK, Yang Y, Kim HJ, Lee KH, Kim JW, et al. (2017). AZD6738, A Novel Oral Inhibitor of ATR, Induces Synthetic Lethality with ATM Deficiency in Gastric Cancer Cells. Mol Cancer Ther 16, 566–577. [DOI] [PubMed] [Google Scholar]
- Molenaar RJ, Radivoyevitch T, Nagata Y, Khurshed M, Przychodzen B, Makishima H, Xu M, Bleeker FE, Wilmink JW, Carraway HE, et al. (2018). IDH1/2 Mutations Sensitize Acute Myeloid Leukemia to PARP Inhibition and This Is Reversed by IDH1/2-Mutant Inhibitors. Clinical cancer research : an official journal of the American Association for Cancer Research 24, 1705–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore EC, and Cohen SS (1967). Effects of arabinonucleotides on ribonucleotide reduction by an enzyme system from rat tumor. The Journal of biological chemistry 242, 2116–2118. [PubMed] [Google Scholar]
- Mouw KW, and D’Andrea AD (2018). DNA Repair Deficiency and Immunotherapy Response. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 36, 1710–1713. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay A, Elattar A, Cerbinskaite A, Wilkinson SJ, Drew Y, Kyle S, Los G, Hostomsky Z, Edmondson RJ, and Curtin NJ (2010). Development of a functional assay for homologous recombination status in primary cultures of epithelial ovarian tumor and correlation with sensitivity to poly(ADP-ribose) polymerase inhibitors. Clinical cancer research : an official journal of the American Association for Cancer Research 16, 2344–2351. [DOI] [PubMed] [Google Scholar]
- Murai J, Feng Y, Yu GK, Ru Y, Tang SW, Shen Y, and Pommier Y (2016). Resistance to PARP inhibitors by SLFN11 inactivation can be overcome by ATR inhibition. Oncotarget 7, 76534–76550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murai J, Huang S-YN, Renaud A, Zhang Y, Ji J, Takeda S, Morris J, Teicher B, Doroshow JH, and Pommier Y (2014a). Stereospecific PARP Trapping by BMN 673 and Comparison with Olaparib and Rucaparib. Molecular Cancer Therapeutics 13, 433–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murai J, Huang SY, Das BB, Renaud A, Zhang Y, Doroshow JH, Ji J, Takeda S, and Pommier Y (2012). Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res 72, 5588–5599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murai J, Thomas A, Miettinen M, and Pommier Y (2019). Schlafen 11 (SLFN11), a restriction factor for replicative stress induced by DNA-targeting anti-cancer therapies. Pharmacology & therapeutics 201, 94–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murai J, Zhang Y, Morris J, Ji J, Takeda S, Doroshow JH, and Pommier Y (2014b). Rationale for poly(ADP-ribose) polymerase (PARP) inhibitors in combination therapy with camptothecins or temozolomide based on PARP trapping versus catalytic inhibition. The Journal of pharmacology and experimental therapeutics 349, 408–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nacson J, Krais JJ, Bernhardy AJ, Clausen E, Feng W, Wang Y, Nicolas E, Cai KQ, Tricarico R, Hua X, et al. (2018). BRCA1 Mutation-Specific Responses to 53BP1 Loss-Induced Homologous Recombination and PARP Inhibitor Resistance. Cell reports 24, 3513–3527. e3517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naipal KAT, Verkaik NS, Ameziane N, van Deurzen CHM, ter Brugge P, Meijers M, Sieuwerts AM, Martens JW, O’Connor MJ, Vrieling H, et al. (2014). Functional <em>Ex Vivo</em> Assay to Select Homologous Recombination–Deficient Breast Tumors for PARP Inhibitor Treatment. 20, 4816–4826. [DOI] [PubMed] [Google Scholar]
- Niimi K, Murakumo Y, Watanabe N, Kato T, Mii S, Enomoto A, Asai M, Asai N, Yamamoto E, Kajiyama H, et al. (2014). Suppression of REV7 enhances cisplatin sensitivity in ovarian clear cell carcinoma cells. Cancer science 105, 545–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noordermeer SM, Adam S, Setiaputra D, Barazas M, Pettitt SJ, Ling AK, Olivieri M, Alvarez-Quilon A, Moatti N, Zimmermann M, et al. (2018). The shieldin complex mediates 53BP1-dependent DNA repair. Nature 560, 117–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto T, and Sicinski P (2017). Cell cycle proteins as promising targets in cancer therapy. Nature reviews. Cancer 17, 93–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantelidou C, Sonzogni O, De Oliveria Taveira M, Mehta AK, Kothari A, Wang D, Visal T, Li MK, Pinto J, Castrillon JA, et al. (2019). PARP Inhibitor Efficacy Depends on CD8(+) T-cell Recruitment via Intratumoral STING Pathway Activation in BRCA-Deficient Models of Triple-Negative Breast Cancer. Cancer discovery 9, 722–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parmar K, Kochupurakkal BS, Lazaro J-B, Wang ZC, Palakurthi S, Kirschmeier PT, Yang C, Sambel LA, Färkkilä A, Reznichenko E, et al. (2019). The CHK1 Inhibitor Prexasertib Exhibits Monotherapy Activity in High-Grade Serous Ovarian Cancer Models and Sensitizes to PARP Inhibition. Clinical Cancer Research 25, 6127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AG, Sarkaria JN, and Kaufmann SH (2011). Nonhomologous end joining drives poly(ADP-ribose) polymerase (PARP) inhibitor lethality in homologous recombination-deficient cells. Proceedings of the National Academy of Sciences of the United States of America 108, 3406–3411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettitt SJ, Krastev DB, Brandsma I, Drean A, Song F, Aleksandrov R, Harrell MI, Menon M, Brough R, Campbell J, et al. (2018). Genome-wide and high-density CRISPR-Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance. Nature communications 9, 1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pishvaian MJ, Biankin AV, Bailey P, Chang DK, Laheru D, Wolfgang CL, and Brody JR (2017). BRCA2 secondary mutation-mediated resistance to platinum and PARP inhibitor-based therapy in pancreatic cancer. British Journal Of Cancer 116, 1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plummer ER, Kristeleit RS, Cojocaru E, Haris NM, Carter L, Jones RH, Blagden SP, Evans TRJ, Arkenau H-T, Sarker D, et al. (2019). A first-in-human phase I/II trial of SRA737 (a Chk1 Inhibitor) in subjects with advanced cancer. 37, 3094–3094. [Google Scholar]
- Polak P, Kim J, Braunstein LZ, Karlic R, Haradhavala NJ, Tiao G, Rosebrock D, Livitz D, Kubler K, Mouw KW, et al. (2017). A mutational signature reveals alterations underlying deficient homologous recombination repair in breast cancer. Nature genetics 49, 1476–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pommier Y, Leo E, Zhang H, and Marchand C (2010). DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chemistry & biology 17, 421–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinet A, Carvajal-Maldonado D, Lemacon D, and Vindigni A (2017). Chapter Three - DNA Fiber Analysis: Mind the Gap! In Methods in Enzymology, Eichman BF, ed. (Academic Press; ), pp. 55–82. [DOI] [PubMed] [Google Scholar]
- Quinn JA, Jiang SX, Reardon DA, Desjardins A, Vredenburgh JJ, Rich JN, Gururangan S, Friedman AH, Bigner DD, Sampson JH, et al. (2009). Phase II trial of temozolomide plus o6-benzylguanine in adults with recurrent, temozolomide-resistant malignant glioma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 27, 1262–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajan A, Carter CA, Kelly RJ, Gutierrez M, Kummar S, Szabo E, Yancey MA, Ji J, Mannargudi B, Woo S, et al. (2012). A phase I combination study of olaparib with cisplatin and gemcitabine in adults with solid tumors. Clinical cancer research : an official journal of the American Association for Cancer Research 18, 2344–2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranjha L, Howard SM, and Cejka P (2018). Main steps in DNA double-strand break repair: an introduction to homologous recombination and related processes. Chromosoma 127, 187–214. [DOI] [PubMed] [Google Scholar]
- Ray Chaudhuri A, Callen E, Ding X, Gogola E, Duarte AA, Lee JE, Wong N, Lafarga V, Calvo JA, Panzarino NJ, et al. (2016). Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 535, 382–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha CRR, Silva MM, Quinet A, Cabral-Neto JB, and Menck CFM (2018). DNA repair pathways and cisplatin resistance: an intimate relationship. Clinics (Sao Paulo) 73, e478s–e478s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rondinelli B, Gogola E, Yucel H, Duarte AA, van de Ven M, van der Sluijs R, Konstantinopoulos PA, Jonkers J, Ceccaldi R, Rottenberg S, et al. (2017a). EZH2 promotes degradation of stalled replication forks by recruiting MUS81 through histone H3 trimethylation. Nature cell biology 19, 1371–1378. [DOI] [PubMed] [Google Scholar]
- Rondinelli B, Gogola E, Yücel H, Duarte AA, van de Ven M, van der Sluijs R, Konstantinopoulos PA, Jonkers J, Ceccaldi R, Rottenberg S, et al. (2017b). EZH2 promotes degradation of stalled replication forks by recruiting MUS81 through histone H3 trimethylation. Nature cell biology 19, 1371. [DOI] [PubMed] [Google Scholar]
- Rottenberg S, Jaspers JE, Kersbergen A, van der Burg E, Nygren AO, Zander SA, Derksen PW, de Bruin M, Zevenhoven J, Lau A, et al. (2008). High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proceedings of the National Academy of Sciences of the United States of America 105, 17079–17084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai W, Swisher EM, Karlan BY, Agarwal MK, Higgins J, Friedman C, Villegas E, Jacquemont C, Farrugia DJ, Couch FJ, et al. (2008). Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature 451, 1116–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato H, Niimi A, Yasuhara T, Permata TBM, Hagiwara Y, Isono M, Nuryadi E, Sekine R, Oike T, Kakoti S, et al. (2017). DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nature communications 8, 1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sausville E, Lorusso P, Carducci M, Carter J, Quinn MF, Malburg L, Azad N, Cosgrove D, Knight R, Barker P, et al. (2014). Phase I dose-escalation study of AZD7762, a checkpoint kinase inhibitor, in combination with gemcitabine in US patients with advanced solid tumors. Cancer chemotherapy and pharmacology 73, 539–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharer OD (2013). Nucleotide excision repair in eukaryotes. Cold Spring Harbor perspectives in biology 5, a012609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoonen PM, Kok YP, Wierenga E, Bakker B, Foijer F, Spierings DCJ, and van Vugt M (2019). Premature mitotic entry induced by ATR inhibition potentiates olaparib inhibition-mediated genomic instability, inflammatory signaling, and cytotoxicity in BRCA2-deficient cancer cells. Molecular oncology 13, 2422–2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Rehman FL, Feng Y, Boshuizen J, Bajrami I, Elliott R, Wang B, Lord CJ, Post LE, and Ashworth A (2013). BMN 673, a Novel and Highly Potent PARP1/2 Inhibitor for the Treatment of Human Cancers with DNA Repair Deficiency. Clinical Cancer Research 19, 5003–5015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shima N, Munroe RJ, and Schimenti JC (2004). The mouse genomic instability mutation chaos1 is an allele of Polq that exhibits genetic interaction with Atm. Molecular and cellular biology 24, 10381–10389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shroff RT, Hendifar A, McWilliams RR, Geva R, Epelbaum R, Rolfe L, Goble S, Lin KK, Biankin AV, Giordano H, et al. (2018). Rucaparib Monotherapy in Patients With Pancreatic Cancer and a Known Deleterious BRCA Mutation. JCO precision oncology 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen CS, Hansen LT, Dziegielewski J, Syljuasen RG, Lundin C, Bartek J, and Helleday T (2005). The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nature cell biology 7, 195–201. [DOI] [PubMed] [Google Scholar]
- Stark JM, Pierce AJ, Oh J, Pastink A, and Jasin M (2004). Genetic steps of mammalian homologous repair with distinct mutagenic consequences. Molecular and cellular biology 24, 9305–9316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stradella A, Johnson ML, Goel S, Chandana SR, Galsky MD, Calvo E, Moreno V, Park H, Arkenau H, Cervantes A, et al. (2019). Updated results of the PARP1/2 inhibitor pamiparib in combination with low-dose (ld) temozolomide (TMZ) in patients (pts) with locally advanced or metastatic solid tumor. Annals of Oncology 30. [Google Scholar]
- Strom CE, Johansson F, Uhlen M, Szigyarto CA, Erixon K, and Helleday T (2011). Poly (ADP-ribose) polymerase (PARP) is not involved in base excision repair but PARP inhibition traps a single-strand intermediate. Nucleic acids research 39, 3166–3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulkowski PL, Corso CD, Robinson ND, Scanlon SE, Purshouse KR, Bai H, Liu Y, Sundaram RK, Hegan DC, Fons NR, et al. (2017). 2-Hydroxyglutarate produced by neomorphic IDH mutations suppresses homologous recombination and induces PARP inhibitor sensitivity. Science translational medicine 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swisher EM, Lin KK, Oza AM, Scott CL, Giordano H, Sun J, Konecny GE, Coleman RL, Tinker AV, O’Malley DM, et al. (2017). Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): an international, multicentre, open-label, phase 2 trial. The Lancet. Oncology 18, 75–87. [DOI] [PubMed] [Google Scholar]
- Thomas A, and Pommier Y (2019). Targeting Topoisomerase I in the Era of Precision Medicine. Clinical cancer research : an official journal of the American Association for Cancer Research 25, 6581–6589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas A, Redon CE, Sciuto L, Padiernos E, Ji J, Lee MJ, Yuno A, Lee S, Zhang Y, Tran L, et al. (2018). Phase I Study of ATR Inhibitor M6620 in Combination With Topotecan in Patients With Advanced Solid Tumors. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 36, 1594–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomida J, Takata KI, Bhetawal S, Person MD, Chao HP, Tang DG, and Wood RD (2018). FAM35A associates with REV7 and modulates DNA damage responses of normal and BRCA1-defective cells. The EMBO journal 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toulany M (2019). Targeting DNA Double-Strand Break Repair Pathways to Improve Radiotherapy Response. Genes 10, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsesmetzis N, Paulin CBJ, Rudd SG, and Herold N (2018). Nucleobase and Nucleoside Analogues: Resistance and Re-Sensitisation at the Level of Pharmacokinetics, Pharmacodynamics and Metabolism. Cancers (Basel) 10, 240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubhi T, and Brown GW (2019). Exploiting DNA Replication Stress for Cancer Treatment. Cancer Res 79, 1730–1739. [DOI] [PubMed] [Google Scholar]
- Van Allen EM, Mouw KW, Kim P, Iyer G, Wagle N, Al-Ahmadie H, Zhu C, Ostrovnaya I, Kryukov GV, O’Connor KW, et al. (2014). Somatic ERCC2 mutations correlate with cisplatin sensitivity in muscle-invasive urothelial carcinoma. Cancer discovery 4, 1140–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinayak S, Tolaney SM, Schwartzberg L, Mita M, McCann G, Tan AR, Wahner-Hendrickson AE, Forero A, Anders C, Wulf GM, et al. (2019). Open-Label Clinical Trial of Niraparib Combined With Pembrolizumab for Treatment of Advanced or Metastatic Triple-Negative Breast Cancer. JAMA oncology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallez Y, Dunlop CR, Johnson TI, Koh SB, Fornari C, Yates JWT, Bernaldo de Quiros Fernandez S, Lau A, Richards FM, and Jodrell DI (2018). The ATR Inhibitor AZD6738 Synergizes with Gemcitabine In Vitro and In Vivo to Induce Pancreatic Ductal Adenocarcinoma Regression. Mol Cancer Ther 17, 1670–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton MI, Eve PD, Hayes A, Henley AT, Valenti MR, De Haven Brandon AK, Box G, Boxall KJ, Tall M, Swales K, et al. (2016). The clinical development candidate CCT245737 is an orally active CHK1 inhibitor with preclinical activity in RAS mutant NSCLC and Emicro-MYC driven B-cell lymphoma. Oncotarget 7, 2329–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigelt B, Comino-Mendez I, de Bruijn I, Tian L, Meisel JL, Garcia-Murillas I, Fribbens C, Cutts R, Martelotto LG, Ng CKY, et al. (2017). Diverse BRCA1 and BRCA2 Reversion Mutations in Circulating Cell-Free DNA of Therapy-Resistant Breast or Ovarian Cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 23, 6708–6720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood RD, and Doublié S (2016). DNA polymerase θ (POLQ), double-strand break repair, and cancer. DNA repair 44, 22–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y, Ramiscal J, Kowanetz K, Del Nagro C, Malek S, Evangelista M, Blackwood E, Jackson PK, and O’Brien T (2013). Identification of preferred chemotherapeutics for combining with a CHK1 inhibitor. Mol Cancer Ther 12, 2285–2295. [DOI] [PubMed] [Google Scholar]
- Xu G, Chapman JR, Brandsma I, Yuan J, Mistrik M, Bouwman P, Bartkova J, Gogola E, Warmerdam D, Barazas M, et al. (2015). REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature 521, 541–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, and Baltimore D (1996). Dual roles of ATM in the cellular response to radiation and in cell growth control. Genes & development 10, 2401–2410. [DOI] [PubMed] [Google Scholar]
- Yazinski SA, Comaills V, Buisson R, Genois MM, Nguyen HD, Ho CK, Todorova Kwan T, Morris R, Lauffer S, Nussenzweig A, et al. (2017). ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor-resistant BRCA-deficient cancer cells. Genes & development 31, 318–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young LA, O’Connor LO, de Renty C, Veldman-Jones MH, Dorval T, Wilson Z, Jones DR, Lawson D, Odedra R, Maya-Mendoza A, et al. (2019). Differential Activity of ATR and WEE1 Inhibitors in a Highly Sensitive Subpopulation of DLBCL Linked to Replication Stress. Cancer Res 79, 3762–3775. [DOI] [PubMed] [Google Scholar]
- Zellweger R, Dalcher D, Mutreja K, Berti M, Schmid JA, Herrador R, Vindigni A, and Lopes M (2015). Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J Cell Biol 208, 563–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YW, Regairaz M, Seiler JA, Agama KK, Doroshow JH, and Pommier Y (2011). Poly(ADP-ribose) polymerase and XPF-ERCC1 participate in distinct pathways for the repair of topoisomerase I-induced DNA damage in mammalian cells. Nucleic acids research 39, 3607–3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoppoli G, Regairaz M, Leo E, Reinhold WC, Varma S, Ballestrero A, Doroshow JH, and Pommier Y (2012). Putative DNA/RNA helicase Schlafen-11 (SLFN11) sensitizes cancer cells to DNA-damaging agents. Proceedings of the National Academy of Sciences of the United States of America 109, 15030–15035. [DOI] [PMC free article] [PubMed] [Google Scholar]