

Abstract
Methylation of proteins has considerable impacts on physiological processes including signal transduction, DNA damage repair, transcriptional regulation, gene activation and inhibition of gene expression. However, the traditional proteomics-based approach suffers from limited identification rates of these critical methylation sites on endogenous peptides. In this work, a peptidomics-based workflow was established to discover and characterize the global methylome of endogenous peptides in human cells. The reliability of our strategy was validated by methyl-SILAC labeling, resulting in 83% true-positive identifications in HeLa cell line. We applied this approach to seven cell lines of human, and 700 methylated forms on 646 putative methylation sites were identified in total, with over 61% of the methylation sites being newly identified. This study provides a complementary strategy for traditional proteomics-based approach that enables identification of missing methylation sites and creates a first methylome draft of endogenous peptides of human cell lines, offering a valuable resource for in-depth studies of biological functions of methylated endogenous peptides.
Keywords: Endogenous peptides, Peptide methylation, Mass spectrometry, Peptidomics, Post-translational modification (PTM), Methylome
Graphical Abstract

Introduction
There has been accumulating evidence suggesting that endogenous peptides play important roles in various biological processes [1]. Endogenous peptides are typically derived from precursor proteins or prohormones. By diversified proteolytic processes in endoplasmic reticulum, Golgi apparatus and secretory granules, prohormones are cleaved and further undergo post-translational modifications (PTMs) to generate mature forms of peptides with biological functions. In addition to degradation, short open reading frames (sORF) in the 5′-untranslated region of eukaryotic mRNAs have also been shown to produce detectable polypeptides, termed short open reading frame-encoded peptides (SEP), which have specific cellular localizations and functions [2].
Endogenous peptides that range in size from 2–100 amino acids in length [3], with masses below 10 kDa, are diffusely present in organs and body fluids (blood, urine, tears, sweat, saliva, cerebrospinal fluid, etc.) and participate in cell differentiation, gene expression, neurotransmission regulation and modulation, tumor development as well as immune cell infiltration, reflecting the metabolic level of lesions [4]. In addition, endogenous peptides have been identified as biomarkers for early diagnosis of diseases. For example, high-throughput MALDI-TOF MS-based peptidomic analysis has been conducted to profile the urine peptidome of the patient with major depressive disorder (MDD), leading to identification of 5 endogenous peptides as potential biomarkers [5]. The biological functions of endogenous peptides have been extensively studied. For instance, Pep5, an intercellular peptide derived from G1/S cyclin D2 protein induces cell death in HeLa cell and several other tumor cells and reduces by 50% the volume of the rat C6 glioblastoma [6] in vivo. More than 20 antimicrobial peptides participate in innate immunity, including defensins which act as internal antibiotics and are able to regulate intestinal microorganism [7]. However, the PTMs of endogenous peptides of cells are rarely reported.
In contrast to proteomics, peptidomics studies generally do not use proteolytic enzymes to characterize the primary sequence of the peptide [8]. Moreover, the mobilities of peptides are higher than those of proteins, which makes it very difficult to focus peptides in a gel for gel-based electrophoretic separation. Thus, strategies different from traditional proteomics need to be developed for analysis of endogenous peptides. Centrifugal ultrafiltration with various sizes of molecular weight cut off (MWCO) was commonly used in peptidomics. Multiple MWCOs (100 kDa, 10 kDa, 3 kDa) coupled with RP-HPLC were utilized to characterize the secreted peptidome of human colon tumor (LIM1215, LIM1863) [9]. Another study employed 10 kDa MWCO together with size exclusion chromatography (SEC) to isolate peptides in mouse livers [10]. In addition to membrane-based enrichment method, nanoporous silica materials had also been used to isolate low molecular weight (LMW) proteins and peptides from the serum of nude mice with MDA-MB-231 human breast cancer lung metastasis [11] as well as human plasma [12]. Even though the physicochemical properties of nanoporous materials, like pore size, pore structure and surface affinity, had been optimized to get better enrichment efficiency, the complexity and cost of the experiments were higher than extraction methods based on selective precipitation. Selective precipitation with acid addition or organic solvent to remove the large abundant proteins had been a typical and effective strategy for peptide isolation and purification. Different agents had been reported previously [13, 14], which provided alternative and more straightforward means for peptide enrichment.
Post-translational modification analysis has been an important aspect in proteomics and peptidomics. In addition to phosphorylation [15, 16] and glycosylation [17], methylation also has considerable impact on protein function and underlying physiological processes, including signal transduction, repair of DNA damage, transcriptional regulation, gene activation and inhibition of gene expression. Methylation occurs more frequently on arginine and lysine, compared with other amino acid residues (aspartic acid, asparagine, glutamine, glutamic acid, histidine, cysteine) and N terminus or C terminus of proteins. N-methylation is the most abundant event compared with O-methylation and S-methylation and commonly occurs at arginine (Arg) and lysine (Lys) in six forms [18], including mono-methylated, asymmetric di-methylated and symmetric di-methylated arginine, as well as mono-, di-, and tri-methylated lysine. The global profiling of methylated endogenous peptides in cells is challenging and often complicated due to low abundance and low stoichiometry. Therefore, enrichment of methylated peptides is often needed for methylation identification. Three different enrichment methods were compared by Uhlmann et al. [19], including strong cation exchange (SCX), isoelectric focusing (IEF), and hydrophilic interaction chromatography (HILIC), with the results indicating that HILIC had an excellent capacity to enrich methylated peptides.
Inspired by these prior studies, we exploited the antibody-free enrichment strategy of DOMAIN (De-glycO-assisted MethylAtion site IdeNtification) [20], which was previously developed by our group based on HILIC enrichment, to semi-specifically enrich methylated peptides. In the current work, two highly complementary extraction methods (hot water and acidified methanol) were employed to extract endogenous peptides from human cells. Methylated endogenous peptides were then enriched by DOMAIN technique and detected by liquid chromatography-tandem mass spectrometry (LC-MS/MS) (Fig.1). We further validated the results by screening of methylated endogenous peptides in hM-SILAC (heavy methyl stable isotope labeling by amino acid cell culture) labeled HeLa cells. This work represents the first report on large-scale characterization of methylation of endogenous peptides in cancer cells.
Fig. 1.

Workflow for the analysis of methylated endogenous peptides. Endogenous peptides were extracted by hot water and acidified methanol. Glycosylated peptides were digested with PNGase F to minimize their interference for methylpeptide enrichment on the HILIC column. Methylated peptides were enriched by HILIC-tips and analyzed by LC-MS/MS.
Experimental Procedures
Cell Culture.
HeLa (human cervical cancer cell), SGC7901 (human gastric cancer cell), HepG2 (human hepatocellular cancer cell), A549 (human lung cancer cell), 293T (human renal epithelial cell), were cultured in DMEM medium (Hyclone) supplemented with 10% fetal bovine serum (Gibco), 1% penicillin/streptomycin (Sigma-Aldrich) and incubated at 37 °C in a humidified chamber with 5% CO2. THP1 (human monocytic cell) cells were culture in RPMI 1640 medium (Hyclone) supplemented with 10% fetal bovine serum (Gibco), 1% penicillin/streptomycin (Sigma-Aldrich) and incubated at 37 °C in a humidified chamber with 5% CO2. For SILAC labeling, HeLa cells were grown in SILAC DMEM medium (Thermo Scientific) supplemented with 10% dialyzed FBS (Gibco), 1% penicillin/streptomycin (Sigma-Aldrich), L-lysine (Sigma), L-arginine (Sigma), L-leucine (Sigma), L- and CD3-methionine (Cambridge Isotope Laboratory, Inc.), and were grown at 37 °C in a humidified 5% CO2-containing atmosphere for at least 10 cell doublings. The SILAC label efficiency was measured by mass spectrometry.
Extraction of Endogenous Peptides.
Cells were digested by trypsin (Gibco) and collected into 1.5 mL tubes by centrifugation at 1000 rpm for 5 min. Cells were washed with PBS (Gibco, pH 7.4) for three times before extraction and then divided into two parts. To enhance the efficiency of endogenous peptide extraction, both hot water and acidified methanol were used in the extraction steps.
For water extraction method, as previously described by Fricker et al. [21], cells were resuspended with 1 mL of hot water (80 °C) and incubated in metal bath at 80 °C for 20 min. The tube was then centrifuged at 4 °C, 13000 rpm for 15 min and stored at −80 °C overnight. The sample was thawed the next day and centrifuged at 4 °C, 13000 rpm for 30 min. The supernatant was transferred to a new 1.5 mL tube, evaporated to 750 μL by vacuum centrifuge and cooled on ice. 75 μL of pre-cooled 0.1 M HCl was added in the sample to a final concentration of 10 mM. After incubation on ice for 15 min, the tube was centrifuged at 4 °C, 13000 rpm for 40 min. The supernatant was collected and evaporated for further treatment.
For another method, cells were resuspended with 1 mL of ice-cooled acidified methanol (90% methanol, 1% acetic acid, 9% water, v/v), sonicated two minutes at 30% power and centrifuged at 4 °C, 20000 xg for 10 min. The supernatant was transferred to a new tube while the pellet was resuspended with 400 μL of acidified methanol and another cycle of sonication and centrifugation was repeated as previously described. The supernatants collected from two steps were combined and evaporated in vacuum centrifuge.
Sample Desalting.
Endogenous peptides extracted using hot water and acidified methanol were resuspended with 400 μL of 0.1% formic acid (FA) separately and prepared for desalting. The C18 Solid Phase Extraction (SPE) column (Phenomenex, 30 mg) was washed with 100% acetonitrile (ACN) and 0.1% FA successively for three times to activate the column. The endogenous peptide samples were loaded on the column for three times. After that, the SPE column was washed with 0.1% FA to remove the unbound impurities and salt. Finally, the peptides were eluted by 40% ACN, 0.1% FA from the SPE column and the eluate was collected in an EP tube and dried by SpeedVac.
Deglycosylation.
Following our previously reported protocol [20], deglycosylation prior to methylated peptide enrichment using the HILIC method can effectively increase the enrichment efficiency. Based on this observation, the desalted sample was deglycosylated by PNGase F to reduce the interference of glycosylated peptides for methylated peptide enrichment via HILIC. The dried sample was dissolved in 20 mM HEPES solution and was incubated with PNGaes F (500 units/μL) at 37 °C for 4 h. The digested product was heated in metal bath at 95 °C for 3 min to denature the PNGase F and evaporated in vacuum.
Methylated-Peptide Enrichment by HILIC-tip.
Approximately 100 μg desalted sample was dissolved in 100 μL of binding buffer (80% ACN, 5% FA). After that, 3 mg of ZIC-HILIC (Merck, particle size 10 μm) beads were suspended and incubated with 1 mL of 0.5% FA for 5 min on vertical rotating mixer to activate the HILIC media. After dislodging the 0.5% FA, the HILIC beads were incubated with 1 mL of binding buffer for 5 min. The binding buffer was removed and HILIC beads were resuspended and incubated with sample solution for 2 h. The mixture was then loaded on a T-400 pipet tip, which was prefilled with a C8 disk (EMpore) for five times. The HILIC-tip was washed ten times by binding buffer to eliminate unbound peptides and methylated peptides were eluted by 200 μL of elution buffer (40% ACN, 0.5% FA). 1 μg of eluted peptides from each of the two extraction methods was took out, mixed well and prepared for MS analysis.
LC-MS/MS Analysis.
The endogenous methylated peptides were analyzed using an Easy-nLC1200 nano-HPLC system (Thermo Fisher Scientific) coupled to an Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher Scientific). The sample was resuspended in 0.1% FA and captured on a trap column (3 μm C18 particles, 2 cm×100 μm ID). The loaded peptides were separated on a 30 cm analytical column (1.9 μm C18 particles, 30 cm×150 μm ID) at a 500 μL/min flow rate for 150 min. Aqueous solution with 0.1% FA and 80% ACN with 0.1% FA were utilized as solvents A and B, respectively. More detailed LC parameters were shown in Table S1. The electrospray voltage was 2.1 kV. The mass spectrometer was operated in a data-dependent acquisition mode with a resolution of 120,000 at full scan mode. Survey scan was acquired after accumulating 5e5 ions in Orbitrap for m/z 300–1400, the top 30 most intense ions in each scan were automatically selected for HCD fragmentation with normalized collision energy of 32% and measured in Orbitrap analyzer operating at a resolution of 15,000. The maximum injection time was 80 ms for full scans and 100 ms for MS/MS scans; and dynamic exclusion of previously acquired precursor ions was enabled at 25 s.
Database Search and Data Analysis.
The mass spectrometry raw data files were analyzed using the PEAKS Studio 8.5 (Bioinformatics Solutions Inc. Canada) [22]. All three technical replicates were searched individually against a human protein database downloaded from UniProt encompassing 20180 entries. A de novo process was performed before the database search. The mass tolerance was set to 10 ppm for precursor ions and 0.02 Da for fragment ions. Mono-methylation, di-methylation and tri-methylation were set as variable modifications. For SILAC sample, heavy-mono-methylation, heavy-di-methylation and heavy-tri-methylation were also added. No enzyme was selected and up to 3 variable post-translational modifications were allowed for each peptide. The discovered peptides were filtered by false discover rate (FDR) of 1% and A Score of 20. The iceLogo [23] was used to analyze the sequence characteristics of methylated peptides and ±6 residues around methylated sites were subjected to analysis. The DAVID Bioinformatics [24, 25] database was utilized to conduct GO analysis.
Results and Discussion
Extraction and Enrichment of Methylated Endogenous Peptides.
Cell samples contain a large number of endogenous peptides, which have highly variable abundances, physical and chemical properties. The use of different extraction solvents provides distinct environments for peptide dissolution. Hot water and acidified methanol were two mainly used reagents in the extraction of peptides as previously reported [21, 26–28], we speculated that the combined use of these two methods may improve the extraction coverage. To examine the extraction efficiency, the endogenous peptide samples derived from HeLa cells by two methods were desalted by C18 SPE column and analyzed using LC-MS/MS. As show in Fig.2a, most of endogenous peptides extracted by acidified methanol were diluted and were distributed in the first 40 minutes, while the extracts of hot water had a unique distribution in the last 30 minutes. The retention time (RT) analysis illustrated the benefit of complementary methods for extraction of endogenous peptides.
Fig. 2.
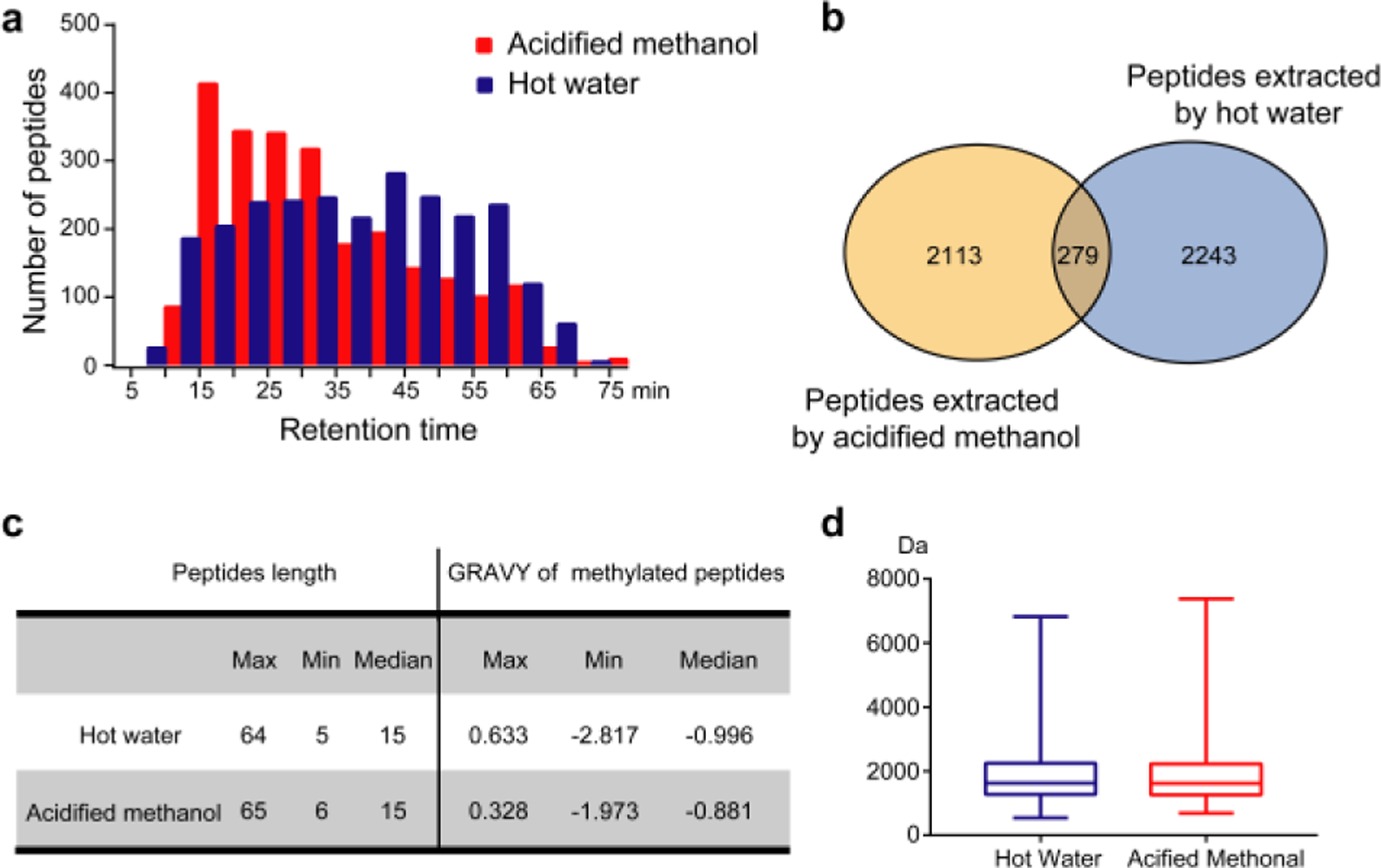
Comparisons of the two extraction methods. (a) The histogram distribution of endogenous peptides extracted by two methods during LC elution. (b) Venn diagram comparing the numbers of endogenous peptides obtained from two extraction methods. (c) Physicochemical properties of endogenous peptides and methylated peptides identified from HeLa cells. (d) Box plot of molecular weights of endogenous peptides extracted by hot water and acidified methanol from HeLa cells.
Next, we evaluated methods at the level of identified peptides. The raw data of the MS result was searched against the human protein database from UniProt using PEAKS (Fig.2b). A total number of 4635 endogenous peptides were identified in the first replicate (an average of 5395 peptides in two replicates), while the co-identified endogenous peptides were 6.4% of the total peptides. Comparing endogenous peptides with identical retention time (Figs. 3b and 3d) that isolated by two reagents mentioned above, we found that some peptides derived from acidified methanol, such as ESRAQLGGPEAAKSDETAAK in Fig. 3a, which show lower intensities of precursor and fragment ions, resulting in poor spectral quality, were accurately identified with high quality tandem MS spectra in samples extracted by hot water. Similar phenomenon was also observed in extracted peptides by hot water (Fig. 3c). Different solvents could be favorable for extracting different kinds of peptides and enhance the signals of peptides in MS detection. These results further support that the combined usage of the two complementary methods can efficiently reduce the omission of endogenous peptides and increase the overall coverage of peptidome identification. Endogenous peptides extracted by two methods span a wide range from 5 to 65 amino acids in length with molecular weights below 10 kDa, and most of these peptides contain around 15 amino acids (Fig. 2c and 2d), which is consistent with the sequence length of endogenous peptides. Furthermore, an average of 174 methylated peptides have been identified in each replicate. The majority of methylated peptides identified in the first replicate are hydrophilic with the median GRAVY score of −0.99 and −0.88 (Fig. 2c), which is in accordance with the previous report [20]. Our results indicate that complementary use of the two extraction methods can dramatically improve the identification rate of endogenous peptides.
Fig. 3.

Mass spectrometric evaluation of the complementary use of hot water and acidified methanol for methylated peptides extraction. (a) The MS/MS fragmentation pattern and (b) the precursor of peptide ESRAQLGGPEAAKSDETAAK obtained by two extraction methods. (c) The MS/MS fragment and (d) the precursor of peptide TQEKNPLPSKETIEQEKQ obtained by two extraction methods.
Evaluation of the Confidence of the Methylated Peptide Identifications.
It had been reported that the side chains of aspartic acid and glutamic acid could be attacked by OH group of methanol which induced in vitro methylation that caused false identification [29]. Although there was no such evidence that methanol or acetic acid interfered with the methylation of K and R, we tested the reliability of our strategy by hM-SILAC labelling. Cells were respectively cultured in medium containing light and heavy (CH3 and CD3) methionine. During the cell metabolism, the methionine was converted to S-adenosyl methionine (AdoMet). The heavy or light AdoMet acted as a methyl donor to incorporate isotopic labeled methyl groups to methylation sites, which generated mass differences of 3 Da, 6 Da and 9 Da respectively on mono-, di, and tri-methylated peptides. In our study, the endogenous peptides were prepared from SILAC labeled cells with 98% substitution rate. Subsequently, the heavy and light labeled methyl peptides were mixed at a ratio of 1:1, followed by further enrichment and LC-MS/MS analysis. If the identified methylated peptide was a true-positive result, it could be metabolically labeled to show a mass shift compared with the unlabeled methyl peptide (Fig. 4a). In contrast, there was no observed peak pair for the false-positive identifications.
Fig. 4.

Bioinformatics analysis of identified methylated peptides in 7 cell lines. (a) GO molecular functions, GO biological processes and GO cellular components enriched in the precursor proteins of methylated peptides. (b) Heatmap of the methylation sites identified in seven cell lines. The adjusted p value <0.05 was taken as the cut off criteria for GO analysis. (c) (d) Motifs enriched around the identified Lys and Arg methylated sites within ± 6 amino acids distance.
A total 104 methylated peptides were detected, and more than 83% of them showed SILAC pairs (Fig. 4b). The mass errors of methylated peptides with SILAC pairs were within 10 ppm (Figs. 4c and 4d), significantly lower than that without SILAC pairs. Nonetheless, the hM-SILAC experiments confirmed that the majority of the identified methylated peptides were resulted from in vivo methylation occurring via S-adenosyl-L-methionine (SAM) –dependent pathways. One of the possible reasons for the false positive identification was the random degradation of proteins during sample preparation. Although caution has been exercised during experimental operation, the degradation of some proteins was still inevitable. However, the high positive rate of methylation identification confirmed the identifications of methylated peptides and also demonstrated reliability of our strategy.
Methylation Sites and Forms Identified in Different Cells.
Since the credibility of our strategy was verified by the hM-SILAC experiment, we applied the approach to enrich and analyze methylated endogenous peptides in seven cell lines including HeLa, A549, HepG2, SGC7901, MCF7, THP1 and 293T to simply profile and characterize the methylation of endogenous peptides in human. The Pearson correlation coefficient values were greater than 0.95 among three technical replicates of each cell lines, indicating high reproducibility of our strategy (Fig. 5a). The methylated peptides were exported by filtering with stringent criteria from the database searching results. Furthermore, manual curation was conducted using criteria: (1) the fragment ions on methylated sites; (2) the major peaks (peaks with the highest abundance) were matched along with at least four consecutive b/y fragment ions; or (3) the minor peaks were matched along with at least six consecutive b/y ion fragments. In total, 700 methylated forms on 646 methylated sites were identified in all seven cell lines, as shown in Fig. 5b. Among them, 435 methylated forms were on the lysine residues, and 265 methylation occurred on the arginine residues. Compared with the identified protein methylation sites which were included in PhosphoSitePlus (http://www.phosphosite.org) [30], 69.7% methylated forms (61.7% of methylated sites) were newly found in our study (Fig. 5c). Our data indicated that a large proportion of methylation modifications were missing in previous reports, and our integrated peptidomics approach can significantly expand the characterization of new methylation sties in methylome. The lists of methylation forms and methylation sites were respectively shown in Table S2 and Table S3.
Fig. 5.

Global view of methylpeptidome of seven cell lines. (a) Pearson correlation coefficient values for identification results of seven cell lines. Each experiment contains three technical replicates. (b) Bar plots showing the identified methylated forms of different cells. (c) Bar plots showing new methylated forms identified in different cells. (d) MS/MS spectra of annotated sites (P68431–28K) containing three forms of lysine methylation, including mono-methylation (upper), di-methylation (middle) and tri-methylation (bottom).
Our approach allowed identification of diverse methylation forms on arginine and lysine. The dataset included 22.5% of methylation sites with multiple forms. For example, mono-, di- and tri-methylated lysine were simultaneously observed on the position 10 of precursor protein P68431 in SGC7901, A549 and HeLa cells (Table S2). The methylated lysine 10 of histone H3 generated a binding site for HP1 protein which was implicated in gene silencing and supra-nucleosomal chromatin structure [31]. In addition, three methylated forms of lysine were found on the position 28 of precursor protein P68431 (human histone H3.1) (Fig. 5d) in all cell lines except for 293T, which were reported to be involved in transcription silencing [32]. Moreover, the acetylation on the same position of precursor protein mentioned above had also been found in a previous study [33], suggesting potential functional switch between transcriptional activation and silencing. Mono- and di-methylated arginine were simultaneously observed on the position 468 of precursor protein Q9UN86, while only mono-methylation form was annotated by a previous study [34]. Previous studies indicated that protein methylation was a dynamically regulated process. Our dataset provided a valuable resource of full complement of methylated peptides with site-specific information, which could help further understand about the regulatory dynamics and the underlying mechanisms of this important PTM in various biological processes.
Bioinformatics Analysis of Identified Methylome.
So far, there are no reports on function of methylated endogenous peptides. In this work, we analyzed the function of the precursor proteins, which may provide insights into the potential biological function of methylated endogenous peptides. Gene ontology annotation analysis was conducted by using bioinformatic tool DAVID [24]. Precursor proteins of methylated peptides were mainly distributed in nucleus, extracellular exosomes, nucleoplasms, ribosomes and intracellular ribonuleoprotein complexes. In terms of the category of biological process, precursor proteins were mostly involved in SRP-dependent co-translational membrane targeting, translational initiation, viral transcription, nuclear-transcribed mRNA catabolic process, mRNA splicing via spliceosome (Fig. 6a). Additionally, the precursor proteins participated in poly(A) RNA binding were more enriched than those in nucleotide binding, RNA binding and structural constituent of ribosome and protein binding.
Fig. 6.

Evaluation of confidence of the methylated peptide identifications. (a) Mass spectrum showing the isotopic distribution of light-labeled and heavy-labeled methylpeptide, RGPPPPPPGR(28.03)GGR(28.03)GGSR(28.03)A. (b) Pie chart showing the percentage of methylated peptides with SILAC pair. (c) Box plot of precursor mass errors of peptides with/without SILAC pairs. (d) Plot of PEAKS peptide score versus precursor mass error.
As shown in Fig.5a, the distribution of endogenous peptides among seven cell lines revealed apparent differences. Since the Pearson correlation coefficient values among three replicates of different cells were high, supporting the reliability and robustness of our method, we considered the distribution characteristics mentioned above were caused by biological differences, rather than applied method or other human interferences. Thus, we clustered the methylation sites co-expressed in seven cell lines as well as those specifically expressed in single cell types. The GO analysis results of precursor proteins from co-expressed methylation sites indicated that the viral transcription and nucleosome assembly were the top two enriched biological processes (Fig. 6b). The precursor proteins of cell-specific methylation sites exhibit diverse biological processes. For instance, the methylation site P68104–36K specifically occurred in MCF7 cell. EEF1A1 was reported upregulated in invasive breast cancer cells, suggesting a role in mediating invasive activity [35]. The methylation on K36 of eEF1A1 protein which was catalyzed by eEF1A-KMT4 was reported to affect few specific codons and in turn regulated the eEF1A1 function [36].
To investigate sequence specificity of the identified methylated peptidome, iceLogo [23] was used to align the sequence motifs. As shown in Fig. 6c, a typical motif RGG was significantly enriched. However, no significantly enriched motifs were found on methylation sites of lysine (Fig. 6d), compared with arginine, suggesting more sequence diversity and possible different regulatory mechanisms. Our results obtained by peptidomics approaches were consistent with previously reported ones by traditional proteomics approaches [19, 20, 37, 38]. With the increasing number of methylation sites to be found and verified in future peptidome studies, the discovery of these motifs provided the possibility for further study on the mechanism and functions of endogenous peptide methylation. More detailed information about motifs on methylated Arg and Lys in each cell line could be found in Fig. S1 and Fig. S2.
Conclusions
In this work, a peptidomics-based approach was established for the global analysis of methylated endogenous peptides in human cells. The combined use of hot water and acidified methanol extraction significantly increased the coverage of resulting endogenous peptides, which enabled in-depth profiling of methylation in cells. The hM-SILAC experiments indicated that the majority of methylpeptides were identified with SILAC pairs. The high positive rate of identification suggested that the extraction methods could reliably identify methylation on arginine and lysine, without the induction of exogenous interference. Our peptidomic study provided the first global view of methylated endogenous peptides in several human cell lines, which serves as a valuable resource for future investigation of functional roles of these methylated endogenous peptides.
Supplementary Material
Acknowlegements
This work is supported by the National Key R&D Program of China (No. 2016YFA0501302 to CJ and No. 2017YFA0505702 to CJ), the National Science Foundation of China (No. 21675006 to CJ) and a fund (BWS17J025 to CJ). LL acknowledges funding support from National Institutes of Health grants RF1AG052324 (to LL), R01 DK071801 (to LL), and U01CA231081 (to LL). We thank the mass spectrometry facility of the National Center for Protein Science-Beijing (PHOENIX Center) for accessing the instruments. We thank Dr. Min Ma, Qianqian Wang, Ting Zhao from the National Center for Protein Science-Beijing (PHOENIX Center) for helpful discussions and insightful suggestions on this project.
Footnotes
Supplemental Information: Additional information available as noted in the text. All mass spectrometry data have been deposited to the iProx with the dataset identifier IPX0001467000. All other data supporting the findings of this study are available from the corresponding authors upon reasonable request.
References
- 1.Adermann K, John H, Standker L, Forssmann WG: Exploiting natural peptide diversity: novel research tools and drug leads. Current opinion in biotechnology. 15, 599–606 (2004) [DOI] [PubMed] [Google Scholar]
- 2.Slavoff SA, Mitchell AJ, Schwaid AG, Cabili MN, Ma J, Levin JZ, Karger AD, Budnik BA, Rinn JL, Saghatelian A: Peptidomic discovery of short open reading frame-encoded peptides in human cells. Nature chemical biology. 9, 59–64 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fricker LD Neuropeptides and other bioactive peptides: from discovery to function. Colloquium Series on Neuropeptides. Morgan & Claypool Life Sciences. 1(2), 1–122 (2012) [Google Scholar]
- 4.Richards AL, Hebert AS, Ulbrich A, Bailey DJ, Coughlin EE, Westphall MS, Coon JJ: One-hour proteome analysis in yeast. Nature protocols. 10, 701–714 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang Y, Chen J, Chen L, Zheng P, Xu HB, Lu J, Zhong J, Lei Y, Zhou C, Ma Q, Li Y, Xie P: Urinary peptidomics identifies potential biomarkers for major depressive disorder. Psychiatry research. 217, 25–33 (2014) [DOI] [PubMed] [Google Scholar]
- 6.de Araujo CB, Russo LC, Castro LM, Forti FL, do Monte ER, Rioli V, Gozzo FC, Colquhoun A, Ferro ES: A novel intracellular peptide derived from g1/s cyclin d2 induces cell death. The Journal of biological chemistry. 289, 16711–16726 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Salzman NH, Hung K, Haribhai D, Chu H, Karlsson-Sjoberg J, Amir E, Teggatz P, Barman M, Hayward M, Eastwood D, Stoel M, Zhou Y, Sodergren E, Weinstock GM, Bevins CL, Williams CB, Bos NA: Enteric defensins are essential regulators of intestinal microbial ecology. Nature immunology. 11, 76–83 (2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schrader M, Schulz-Knappe P: Peptidomics technologies for human body fluids. Trends in biotechnology. 19, S55–60 (2001) [DOI] [PubMed] [Google Scholar]
- 9.Greening DW, Kapp EA, Ji H, Speed TP, Simpson RJ: Colon tumour secretopeptidome: insights into endogenous proteolytic cleavage events in the colon tumour microenvironment. Biochimica et biophysica acta. 1834, 2396–2407 (2013) [DOI] [PubMed] [Google Scholar]
- 10.Hu L, Li X, Jiang X, Zhou H, Jiang X, Kong L, Ye M, Zou H: Comprehensive peptidome analysis of mouse livers by size exclusion chromatography prefractionation and nanoLC-MS/MS identification. Journal of proteome research. 6, 801–808 (2007) [DOI] [PubMed] [Google Scholar]
- 11.Fan J, Deng X, Gallagher JW, Huang H, Huang Y, Wen J, Ferrari M, Shen H, Hu Y: Monitoring the progression of metastatic breast cancer on nanoporous silica chips. Philosophical transactions. Series A, Mathematical, physical, and engineering sciences. 370, 2433–2447 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Terracciano R, Gaspari M, Testa F, Pasqua L, Tagliaferri P, Cheng MM, Nijdam AJ, Petricoin EF, Liotta LA, Cuda G, Ferrari M, Venuta S: Selective binding and enrichment for low-molecular weight biomarker molecules in human plasma after exposure to nanoporous silica particles. Proteomics. 6, 3243–3250 (2006) [DOI] [PubMed] [Google Scholar]
- 13.Chertov O, Biragyn A, Kwak LW, Simpson JT, Boronina T, Hoang VM, Prieto DA, Conrads TP, Veenstra TD, Fisher RJ: Organic solvent extraction of proteins and peptides from serum as an effective sample preparation for detection and identification of biomarkers by mass spectrometry. Proteomics. 4, 1195–1203 (2004) [DOI] [PubMed] [Google Scholar]
- 14.Jiang L, He L, Fountoulakis M: Comparison of protein precipitation methods for sample preparation prior to proteomic analysis. Journal of chromatography. A. 1023, 317–320 (2004) [DOI] [PubMed] [Google Scholar]
- 15.Chen IH, Xue L, Hsu CC, Paez JS, Pan L, Andaluz H, Wendt MK, Iliuk AB, Zhu JK, Tao WA: Phosphoproteins in extracellular vesicles as candidate markers for breast cancer. Proceedings of the National Academy of Sciences of the United States of America. 114, 3175–3180 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schunter AJ, Yue X, Hummon AB: Phosphoproteomics of colon cancer metastasis: comparative mass spectrometric analysis of the isogenic primary and metastatic cell lines SW480 and SW620. Analytical and bioanalytical chemistry. 409, 1749–1763 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu Y, Shah P, Clark DJ, Ao M, Zhang H: Reanalysis of Global Proteomic and Phosphoproteomic Data Identified a Large Number of Glycopeptides. Analytical chemistry. 90, 8065–8071 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Q, Wang K, Ye M: Strategies for large-scale analysis of non-histone protein methylation by LC-MS/MS. The Analyst. 142, 3536–3548 (2017) [DOI] [PubMed] [Google Scholar]
- 19.Uhlmann T, Geoghegan VL, Thomas B, Ridlova G, Trudgian DC, Acuto O: A method for large-scale identification of protein arginine methylation. Molecular & cellular proteomics : MCP. 11, 1489–1499 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ma M, Zhao X, Chen S, Zhao Y, Yang L, Feng Y, Qin W, Li L, Jia C: Strategy Based on Deglycosylation, Multiprotease, and Hydrophilic Interaction Chromatography for Large-Scale Profiling of Protein Methylation. Analytical chemistry. 89, 12909–12917 (2017) [DOI] [PubMed] [Google Scholar]
- 21.Fricker LD, Gelman JS, Castro LM, Gozzo FC, Ferro ES: Peptidomic analysis of HEK293T cells: Effect of the proteasome inhibitor epoxomicin on intracellular peptides. Journal of proteome research. 11, 1981 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang J, Xin L, Shan B, Chen W, Xie M, Yuen D, Zhang W, Zhang Z, Lajoie GA, Ma B: PEAKS DB: de novo sequencing assisted database search for sensitive and accurate peptide identification. Molecular & cellular proteomics : MCP. 11, M111 010587 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Colaert N, Helsens K, Martens L, Vandekerckhove J, Gevaert K: Improved visualization of protein consensus sequences by iceLogo. Nature methods. 6, 786–787 (2009) [DOI] [PubMed] [Google Scholar]
- 24.Huang da W, Sherman BT, Lempicki RA: Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic acids research. 37, 1–13 (2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang da W, Sherman BT, Lempicki RA: Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols. 4, 44–57 (2009) [DOI] [PubMed] [Google Scholar]
- 26.DeLaney K, Buchberger A, Li L: Identification, Quantitation, and Imaging of the Crustacean Peptidome. Methods in molecular biology. 1719, 247–269 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang X, Petruzziello F, Zani F, Fouillen L, Andren PE, Solinas G, Rainer G: High identification rates of endogenous neuropeptides from mouse brain. Journal of proteome research. 11, 2819–2827 (2012) [DOI] [PubMed] [Google Scholar]
- 28.Gelman JS, Sironi J, Castro LM, Ferro ES, Fricker LD: Peptidomic Analysis of Human Cell Lines. Journal of proteome research. 10, 1583–1592 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jung SY, Li Y, Wang Y, Chen Y, Zhao Y, Qin J: Complications in the assignment of 14 and 28 Da mass shift detected by mass spectrometry as in vivo methylation from endogenous proteins. Analytical chemistry. 80, 1721–1729 (2008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V, Skrzypek E: PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic acids research. 43, D512–520 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lachner M, O’Carroll D, Rea S, Mechtler K, Jenuwein T: Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature. 410, 116–120 (2001) [DOI] [PubMed] [Google Scholar]
- 32.Kuzmichev A, Nishioka K, Erdjument-Bromage H, Tempst P, Reinberg D: Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes & development. 16, 2893–2905 (2002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Garcia BA, Hake SB, Diaz RL, Kauer M, Morris SA, Recht J, Shabanowitz J, Mishra N, Strahl BD, Allis CD, Hunt DF: Organismal differences in post-translational modifications in histones H3 and H4. The Journal of biological chemistry. 282, 7641–7655 (2007) [DOI] [PubMed] [Google Scholar]
- 34.Guo A, Gu H, Zhou J, Mulhern D, Wang Y, Lee KA, Yang V, Aguiar M, Kornhauser J, Jia X, Ren J, Beausoleil SA, Silva JC, Vemulapalli V, Bedford MT, Comb MJ: Immunoaffinity enrichment and mass spectrometry analysis of protein methylation. Molecular & cellular proteomics : MCP. 13, 372–387 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhu G, Reynolds L, Crnogorac-Jurcevic T, Gillett CE, Dublin EA, Marshall JF, Barnes D, D’Arrigo C, Van Trappen PO, Lemoine NR, Hart IR: Combination of microdissection and microarray analysis to identify gene expression changes between differentially located tumour cells in breast cancer. Oncogene. 22, 3742–3748 (2003) [DOI] [PubMed] [Google Scholar]
- 36.Jakobsson ME, Malecki J, Nilges BS, Moen A, Leidel SA, Falnes PO: Methylation of human eukaryotic elongation factor alpha (eEF1A) by a member of a novel protein lysine methyltransferase family modulates mRNA translation. Nucleic acids research. 45, 8239–8254 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang K, Dong M, Mao J, Wang Y, Jin Y, Ye M, Zou H: Antibody-Free Approach for the Global Analysis of Protein Methylation. Analytical chemistry. 88, 11319–11327 (2016) [DOI] [PubMed] [Google Scholar]
- 38.Bremang M, Cuomo A, Agresta AM, Stugiewicz M, Spadotto V, Bonaldi T: Mass spectrometry-based identification and characterisation of lysine and arginine methylation in the human proteome. Molecular bioSystems. 9, 2231–2247 (2013) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.