

Abstract
Objective:
To determine the effects of empagliflozin on glycerol-derived hepatic gluconeogenesis in obese adults without type 2 diabetes mellitus (T2DM) using oral 13C-labeled glycerol.
Methods:
We performed a randomized, double-blind, placebo-controlled trial in participants with MRI assessment of body fat and measurement of glycerol-derived 13C enrichment in plasma glucose by NMR spectroscopy following ingestion of [U-13C3]-glycerol. Participants were randomized to oral empagliflozin 10 mg once daily or placebo for 3 months. Glycerol-derived 13C enrichment studies were repeated and treatment-differences in mean percent 13C glycerol enrichment in glucose were compared using mixed linear models.
Results:
35 participants completed the study. Empagliflozin increased glycerol-derived 13C enrichment between baseline and follow-up by 6.5% (p=0.005), consistent with less glycerol from visceral adipose tissue (VAT). No difference was found with placebo. Glycerol-derived 13C enrichment was lower in high VAT compared with low VAT participants by 12.6% (p=0.04) but there was no heterogeneity of the treatment effect by baseline VAT. Glycerol-derived 13C enrichment was inversely correlated with VAT but not with weight loss.
Conclusions:
VAT is associated with endogenous glycerol-derived hepatic gluconeogenesis and empagliflozin reduces endogenous glycerol-gluconeogenesis in obese adults without T2DM. These findings suggest a mechanism by which SGLT2 inhibitors may prevent T2DM in obesity.
Introduction
Approximately 40% of adults in the United States are living with obesity (1). Excess fat stored as visceral adipose tissue (VAT) promotes the highest risk for type 2 diabetes mellitus (T2DM) and cardiovascular disease (CVD) in obesity beyond the body mass index (BMI) (2, 3). Excess VAT may alter glucose homeostasis by contributing excess glycerol from overactive lipolysis to stimulate hepatic gluconeogenesis (4, 5). The persistent turnover of mesenteric triglycerides in spite of hyperinsulinemia as seen with visceral obesity delivers glycerol and fatty acids directly into the portal circulation, providing both a gluconeogenic substrate and energy for gluconeogenesis in the liver (6). We previously demonstrated that viscerally obese adults without T2DM had lower enrichment of orally ingested, biologically-labeled (13C) glycerol via hepatic gluconeogenesis compared with BMI-matched, low visceral fat individuals (7), reflecting a dilutional effect from excess unlabeled endogenous glycerol substrates from VAT contributing to gluconeogenesis.
Empagliflozin is a potent and selective sodium glucose co-transporter 2 (SGLT2) inhibitor used for the treatment of T2DM. Empagliflozin has been shown to improve glycemic control, reduce body weight, and improve multiple indices of visceral adiposity in patients with T2DM (8, 9). Furthermore, empagliflozin significantly reduced CVD events in patients with T2DM and established CVD (10).
However, the effects of empagliflozin on VAT function and its relationship to glycerol-derived hepatic gluconeogenesis in viscerally obese adults without T2DM are unknown. Therefore, we tested the hypothesis that empagliflozin would increase exogenous glycerol-derived 13C enrichment in blood glucose by reducing endogenous glycerol-derived hepatic gluconeogenesis from VAT in adults with obesity without T2DM. We also tested the heterogeneity of the treatment effect by amount of VAT to determine if the effect of empagliflozin would be greater among those with higher baseline VAT.
Research Design and Methods
Study population
Healthy individuals with obesity were recruited from the community to participate in the study. For inclusion, participants were required to be age ≥18 years, obese (defined as a BMI ≥30 kg/m2), free from T2DM (both by self-reported medical history and glycosylated hemoglobin (hemoglobin A1c) measurement <6.5% at enrollment), and be able to undergo a neck-to-knee magnetic resonance imaging (MRI) scan for body fat assessment. Participants were excluded if they were pregnant (by urine pregnancy test at the time of enrollment) or breastfeeding, incarcerated, non-obese at the time of enrollment, had donated blood within 6 weeks of enrollment, had a self-reported history of dyslipidemia or chronic liver disease, or a prior allergy or adverse reaction to empagliflozin. Participants completed four visits over a span of ~3 months. At the baseline and completion visits, participants underwent phlebotomy for laboratory and NMR studies, as described below. Body composition assessments were performed at baseline only. Two intervening visits between baseline and completion were also conducted approximately 1 month apart to assess study medication adherence and to evaluate for any adverse effects. All participants provided written informed consent, and the protocol was approved by the Institutional Review Board of the University of Texas Southwestern Medical Center. The trial is registered on ClinicalTrials.gov (ClinicalTrials.gov Identifier: NCT02833415).
Study Procedures
A study timeline is provided in Figure S1. Participants arrived in the morning after an overnight fast. Age, sex, and race/ethnicity were self-reported. Weight, percent body fat, and percent lean mass were measured using an Omron Body Composition Monitor with Scale (model HBF-514C, Omron Healthcare, Lake Forest, IL). Height was measured using a standard scale and BMI was calculated as weight in kilograms divided by height in meters squared. Waist circumference was measured 1 cm above the iliac crest and hip circumference at the widest circumference of the buttocks at the area of the greater trochanters. Blood pressure was measured using an Omron 5 series upper arm blood pressure monitor. A peripheral intravenous catheter was inserted and blood was drawn for analysis of plasma insulin, hemoglobin A1c, triglycerides, adiponectin, C-reactive protein, alanine aminotransferase, norepinephrine, and non-esterified free fatty acids. Samples were analyzed by Quest Diagnostics (Irving, TX or San Juan Capistrano, CA) using standard assays. The homeostasis model assessment of insulin resistance index (HOMA-IR) was calculated by fasting insulin (μIU/ml)×fasting glucose (mg/dL)/405. Plasma free glycerol was measured using an enzymatic assay (Sigma-Aldrich, St. Louis, MO). Blood glucose was analyzed using an YSI 2300 Biochemistry Analyzer (Xylem Inc., Yellow Springs, OH). Plasma glucagon was measured using an ELISA assay (Mercodia, Winston-Salem, NC).
[U-13C3] glycerol (Cambridge Isotope Laboratories, Tewksbury, MA) was prepared in ½ cup of filtered water using the following formula: Weight (kg) × 0.05 g/kg × 1 ml/1.302 g = [U-13C3] glycerol (ml). Participants drank the glycerol followed by 1 cup of filtered water to rinse the bottle. Blood samples were subsequently drawn at +15, +30, +60, +90, +120, +150, and +180 minutes for analysis. For each sample, 0.5 ml of blood was used for blood glucose analysis and 40 ml of each sample was immediately centrifuged and stored on ice to be used for nuclear magnetic resonance (NMR) analysis.
NMR analysis procedures were performed as previously described (7). Briefly, plasma glucose was converted to monoacetone glucose (MAG) for NMR spectral analysis. All NMR spectra were acquired using a Varian INOVA 14.1 T spectrometer (Agilent, Santa Clara, CA) equipped with a 3-mm broadband probe with the observe coil tuned to 13C (150 MHz). Spectra were averaged over ~7000–23,000 scans requiring ~6–20 hours. All NMR spectra were analyzed using the ACD/Labs PC-based NMR spectral analysis program (Advanced Chemistry Development, Inc., Toronto, Canada). The fraction of plasma glucose derived from [U-13C3] glycerol was determined by measuring the total area of each distinct multiplet referenced to the natural abundance 13C signals from methyl groups of MAG (11). Three pathways in glycerol-gluconeogenesis were interrogated by determining the fraction of glycerol-derived 13C enrichment in blood glucose using NMR spectroscopic quantification of 13C-labeled glucose isotopomers: direct hepatic gluconeogenesis, the pentose phosphate pathway (PPP), and the tricarboxylic acid cycle (TCA).
Body composition measurements
Participants underwent MRI scanning on a Philips Achieva 3-T MRI scanner (Philips Healthcare, Amsterdam, Netherlands) using a 6-minute dual-echo Dixon Vibe protocol, providing a water and fat separated volumetric data set covering neck to knees, and a single-slice multiecho Dixon acquisition for proton density fat fraction (PDFF) assessment in the liver. Images of the liver were acquired using a 16-channel SENSE XL Torso coil and images from the rest of the body were acquired using the body coil. For body composition, acquired image data were analyzed for VAT, abdominal subcutaneous adipose tissue (ASAT), thigh muscle volume, and liver PDFF. Briefly, the image analysis consisted of (1) image calibration, (2) fusion of image stacks, (3) image segmentation, and (4) quantification of fat and muscle volumes and included manual quality control by an analysis engineer (12, 13, 14, 15). Body composition analyses were performed using AMRA Profiler Research (AMRA Medical AB, Linköping, Sweden).
Treatment Assignment
After baseline study procedures were completed, participants were randomly assigned to oral empagliflozin 10 mg once daily (Boehringer Ingelheim, Germany) or matching placebo to be taken for the duration of the study. Randomization was performed by a third-party using sealed opaque envelopes and all participants and study personnel were blinded to the treatment assignment until the completion of all study procedures. Study drug and placebo were obtained from a third party pharmacy (Pharmacy Solutions, Inc., Arlington, TX). Drug adherence and adverse events were collected at interval visits through completion. Study participants, investigators, and outcome assessors were blinded to the treatment assignment.
Statistical analysis
For the primary analysis, participants were analyzed within-groups with paired (baseline to follow-up) testing using a modified intention-to-treat analysis stratified by empagliflozin or placebo treatment assignment. For inclusion in the primary analysis, all participants were required to have at least one baseline and follow-up 13C enrichment measurement. In secondary analyses, participants were stratified by VAT group (high: >median vs. low: ≤median value of the study cohort). Pairwise characteristics were compared using Wilcoxon rank sum or signed rank tests as appropriate. The mean ± standard error percent glycerol-derived 13C enrichment in blood glucose versus time for each group was plotted and the area under the enrichment curve was calculated using the trapezoidal rule in all groups. Mixed linear models were used to compare excess glycerol-derived 13C enrichment (%) in blood glucose between groups over all time points at each visit and between baseline and follow-up visits (primary outcome).
There were several secondary outcomes. First we assessed changes in enrichment of [1,2-13C2] glucose (μmol/L) in blood glucose over time between groups as a measure of the portion of [U-13C3] glycerol metabolism through the PPP. Second, we assessed changes in the ratio (%) of [5,6-13C2]/[4,5,6-13C3] in glucose as a measure of the portion of [U-13C3] glycerol metabolism through the TCA cycle compared with direct gluconeogenesis from glycerol (TCA cycle/mitochondria). Third, we assessed baseline differences in glycerol-derived 13C enrichment in blood glucose between the high VAT and low VAT groups. Scatterplots and Spearman correlation coefficients were also examined for the relationships between baseline fat depots and peak glycerol-derived 13C enrichment in glucose, as well as change in peak glycerol-derived 13C enrichment versus change in body weight. For all statistical testing, a 2-sided p-value <0.05 was considered statistically significant. The study was designed to achieve 84% power at a two-sided alpha level of 0.05 to detect at least a 2% difference in glycerol-derived 13C enrichment in glucose within each treatment group (requiring at least 8 paired measurements in each arm) factoring in an estimated dropout rate of 20%. All statistical analyses were performed using SAS version 9.4 software (SAS Corporation, Cary, NC).
Results
A total of 40 patients underwent randomization; 35 received study drug, completed follow-up, and were included in the final analysis (18 participants randomized to the empagliflozin group and 17 to the placebo group, CONSORT flow diagram Figure 1). Median follow-up time was 12.4 (interquartile range 12.0–13.4) weeks. Mean drug compliance (defined as proportion of all drug taken/dispensed across all visits) was 93.5%. There was one adverse event (urinary tract infection) that was appropriately treated and resolved.
Figure 1. CONSORT Study selection diagram.
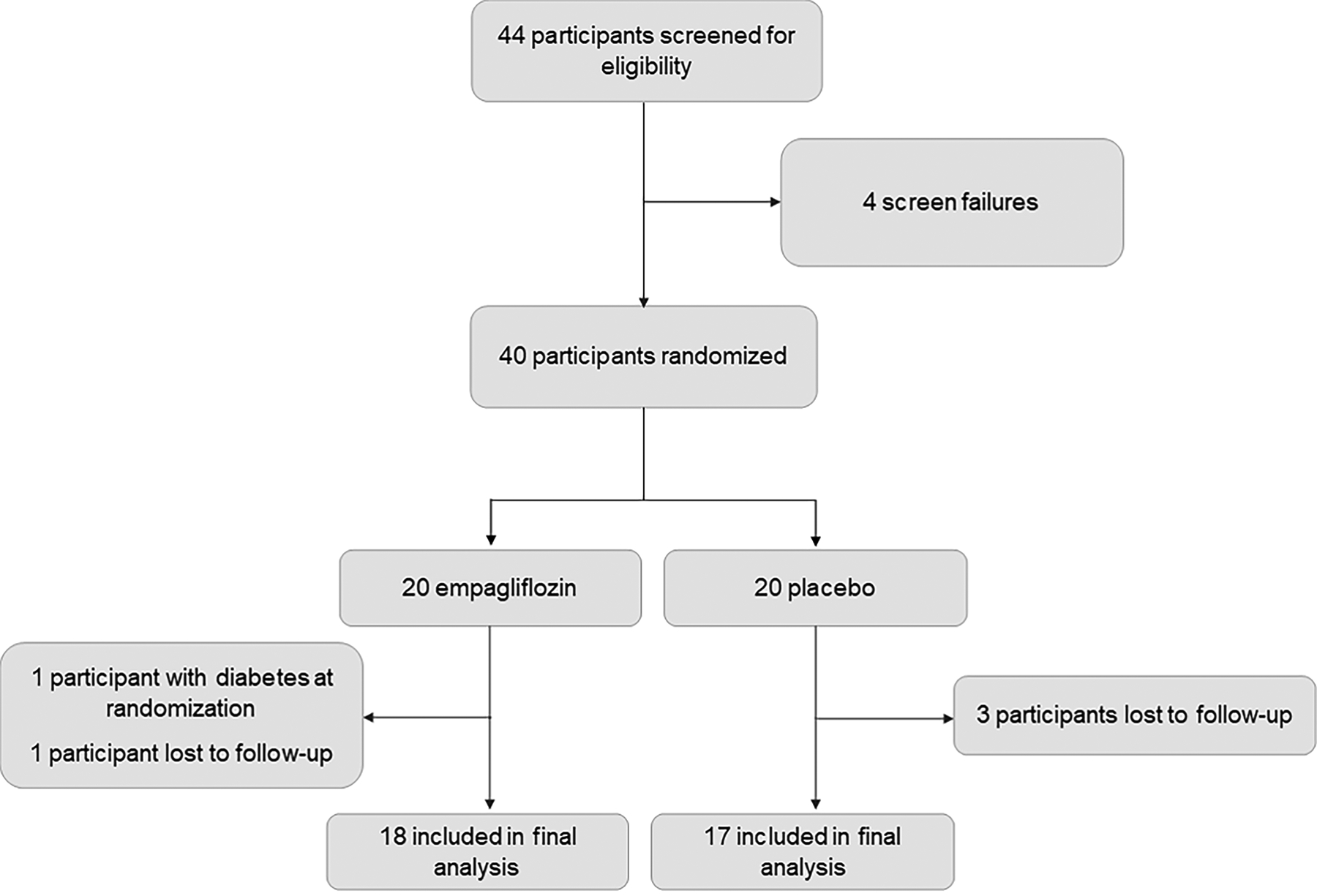
Healthy obese adults without type 2 diabetes were screened for eligibility in the trial and 40 were randomized to empagliflozin/matching placebo. Four participants did not return for follow-up procedures and 1 participant was excluded immediately after randomization due to diagnosis of type 2 diabetes. 35 participants (18 empagliflozin/17 matching placebo) had full data available for the modified intention-to-treat analysis.
Baseline characteristics of the study population overall and stratified by treatment assignment are shown in Table 1. The study cohort was 63% female and 34% black with a median age of 53 years and median BMI of 35 kg/m2. Baseline demographic and clinical characteristics were well balanced between the empagliflozin and placebo groups, although placebo treated participants had a modestly higher waist circumference (43.7 vs. 40.0, p=0.04).
Table 1.
Baseline characteristics of the study population overall and stratified by treatment assignment
| Characteristic | Overall (N=35) | Empagliflozin (N=18) | Placebo (N=17) | P-value |
|---|---|---|---|---|
| Age, years | 53 (46–59) | 50.5 (43–56) | 54.0 (48–59.0) | 0.40 |
| Female | 22 (62.9) | 11 (61.1) | 11 (64.7) | 0.83 |
| Race/ethnicity | 0.50 | |||
| Black | 12 (34.3) | 5 (27.8) | 7 (41.2) | |
| White | 20 (57.1) | 11 (61.1) | 9 (52.9) | |
| Other | 3 (8.6) | 2 (11.1) | 1 (5.9) | |
| Systolic blood pressure (mmHg) | 130 (120–139) | 126 (117–138) | 132 (127–141) | 0.18 |
| Fasting blood glucose (mg/dL) | 96 (91–101) | 96 (90–99) | 96 (92–101) | 0.60 |
| Insulin (μIU/mL) | 13.0 (6.4–17.0) | 10.1 (6.3–13.9) | 15.6 (6.4–24.2) | 0.13 |
| Hemoglobin A1c (%) | 5.8 (5.4–6.0) | 5.6 (5.4–5.9) | 5.9 (5.6–6.1) | 0.09 |
| Triglycerides (mg/dL) | 92 (75–163) | 93 (73–147) | 91 (80–164) | 0.72 |
| Adiponectin (mcg/mL) | 6.0 (4.0–9.0) | 6.0 (5.0–8.0) | 5.0 (4.0–9.0) | 0.59 |
| C-reactive protein (mg/dL) | 0.34 (0.16–0.52) | 0.35 (0.21–0.46) | 0.31 (0.10–0.68) | 0.82 |
| Alanine aminotransferase (ALT, U/L) | 21 (13–32) | 21 (12–34) | 21 (15–29) | 0.91 |
| Norepinephrine (pg/mL) | 346 (253–490) | 353 (297–424) | 339 (242–563) | 0.69 |
| Nonesterified free fatty acids (mmol/L) | 0.65 (0.48–0.81) | 0.62 (0.46–0.74) | 0.66 (0.52–0.86) | 0.20 |
| Plasma free glycerol (μg/μL) | 0.01 (0.01–0.02) | 0.02 (0.01–0.02) | 0.01 (0.01–0.02) | 0.66 |
| Plasma glucagon (pmol/L) | 1.99 (0.25–5.14) | 1.71 (0.25–2.59) | 2.44 (0.25–5.69) | 0.27 |
| HOMA-IR | 3.2 (1.4–4.0) | 2.6 (1.4–3.3) | 3.8 (1.4–5.9) | 0.12 |
| Weight (lbs) | 226 (204–245) | 224 (195–239) | 227 (207–249) | 0.59 |
| Body mass index (kg/m2) | 35 (33–40) | 37 (33–39) | 35 (33–40) | 0.96 |
| Waist circumference (in) | 43 (39–46) | 40 (37–44) | 44 (42–48) | 0.04 |
| Total body fat (%) | 45 (36–50) | 46 (34–49) | 45 (38–53) | 0.35 |
| Total body lean mass (%) | 25 (21–29) | 24 (22–30) | 25 (21–28) | 0.36 |
| Visceral fat (kg) | 5.4 (3.9–7.2) | 5.4 (4.0–7.3) | 4.3 (3.8–6.7) | 0.81 |
| High visceral fat, n(%)* | 17 (51.5) | 10 (62.5) | 7 (41.2) | 0.30 |
| Abdominal subcutaneous fat (kg) | 12.9 (11.4–16.4) | 12.5 (9.3–15.0) | 13.7 (12.4–17.3) | 0.14 |
| Liver fat (%) | 6.5 (5.0–13.6) | 8.5 (5.3–15.3) | 6.0 (5.0–11.6) | 0.42 |
| Total thigh muscle (kg) | 11.1 (9.2–13.5) | 10.7 (9.6–13.5) | 11.5 (9.1–14.1) | 0.98 |
Data are median (interquartile range, IQR) or proportion (%) as appropriate.
High visceral fat defined as greater than the sex-specific median population value
After 12 weeks of treatment, empagliflozin-treated participants showed a trend toward lower body weight and a significant decrease in Hb A1c and systolic blood pressure from baseline that was not seen among the placebo-treated group (Figure 2). There was no significant increase in glucagon levels with empagliflozin (Figure 2) and no significant treatment-related differences were found on fasting plasma glucose (Figure S2), serum insulin, triglycerides, free fatty acids, plasma free glycerol, adiponectin, alanine aminotransferase, norepinephrine, or HOMA-IR levels.
Figure 2. Effects of empagliflozin treatment on weight, glycosylated hemoglobin, systolic blood pressure, and glucagon.
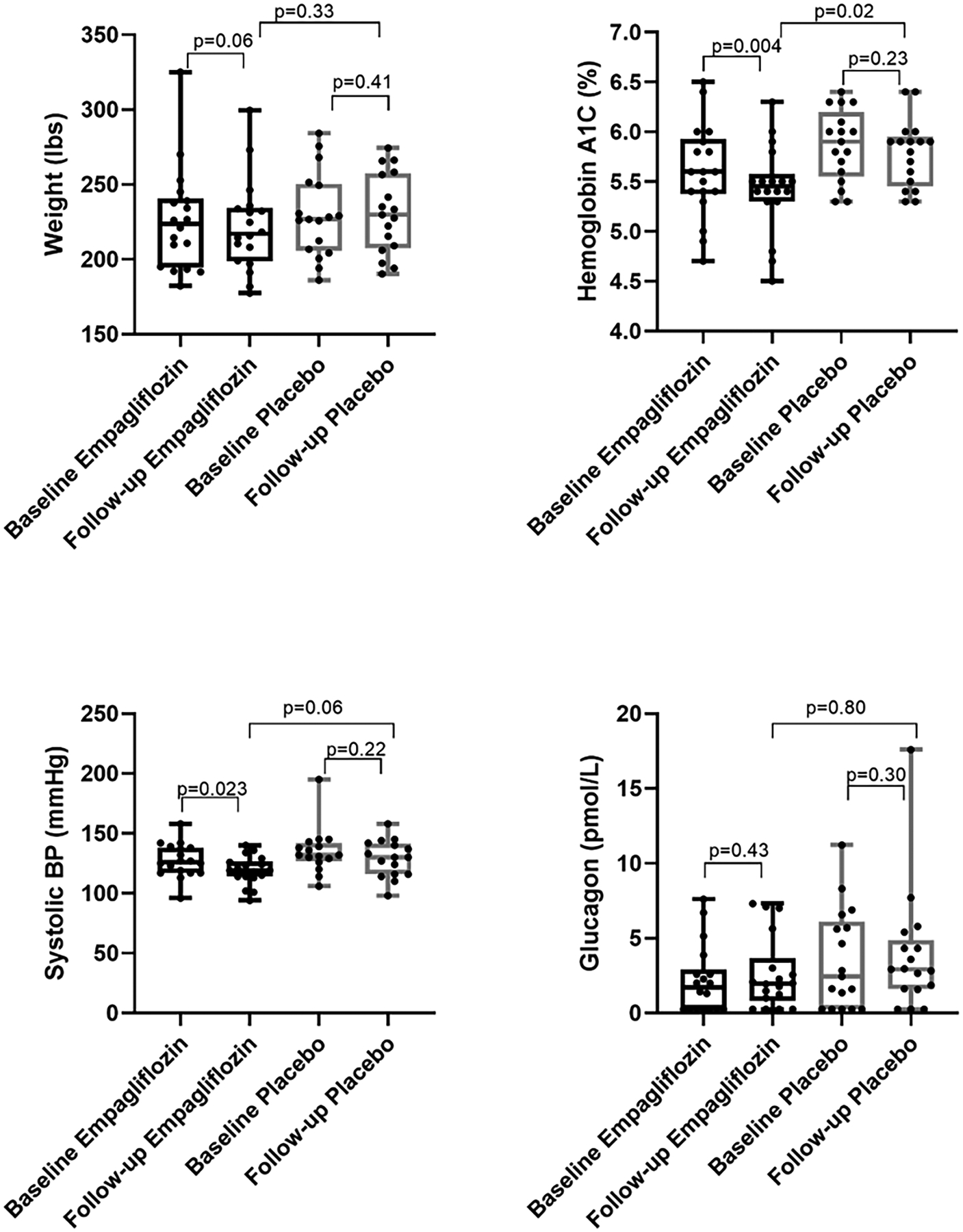
Empagliflozin significantly reduced hemoglobin A1c and systolic blood pressure with a non-significant trend toward greater body weight loss. There was no significant treatment-effect on plasma glucagon levels.
Effect of empagliflozin on glycerol-derived gluconeogenesis
After 12 weeks of treatment, there was a statistically significant 6.5% increase in the area under the curve for glycerol-derived 13C enrichment in glucose between baseline and follow up in the empagliflozin-treated group (p=0.005, Figure 3) while no difference in glycerol-derived 13C enrichment in glucose was seen among those treated with placebo (Figure 3). When assessed using relative percent change by treatment assignment, the empagliflozin-treated group had a 17.8% mean relative increase in glycerol-derived 13C enrichment in glucose at 60 minutes whereas the mean relative change in the placebo-treated group was negligible (0.46%) at 60 minutes (p=0.03, Figure S3). There was no statistical interaction by VAT status on the relationship between empagliflozin treatment and glycerol-derived 13C enrichment in glucose. However, when using relative percent change, there was a trend toward a 17.1% mean relative increase in glycerol-derived 13C enrichment in glucose at 60 minutes in the high VAT group compared with a mean 0.55% relative increase seen in the low VAT group at 60 minutes (p=0.07).
Figure 3. Effects of empagliflozin treatment on excess 13C enrichment in glucose.
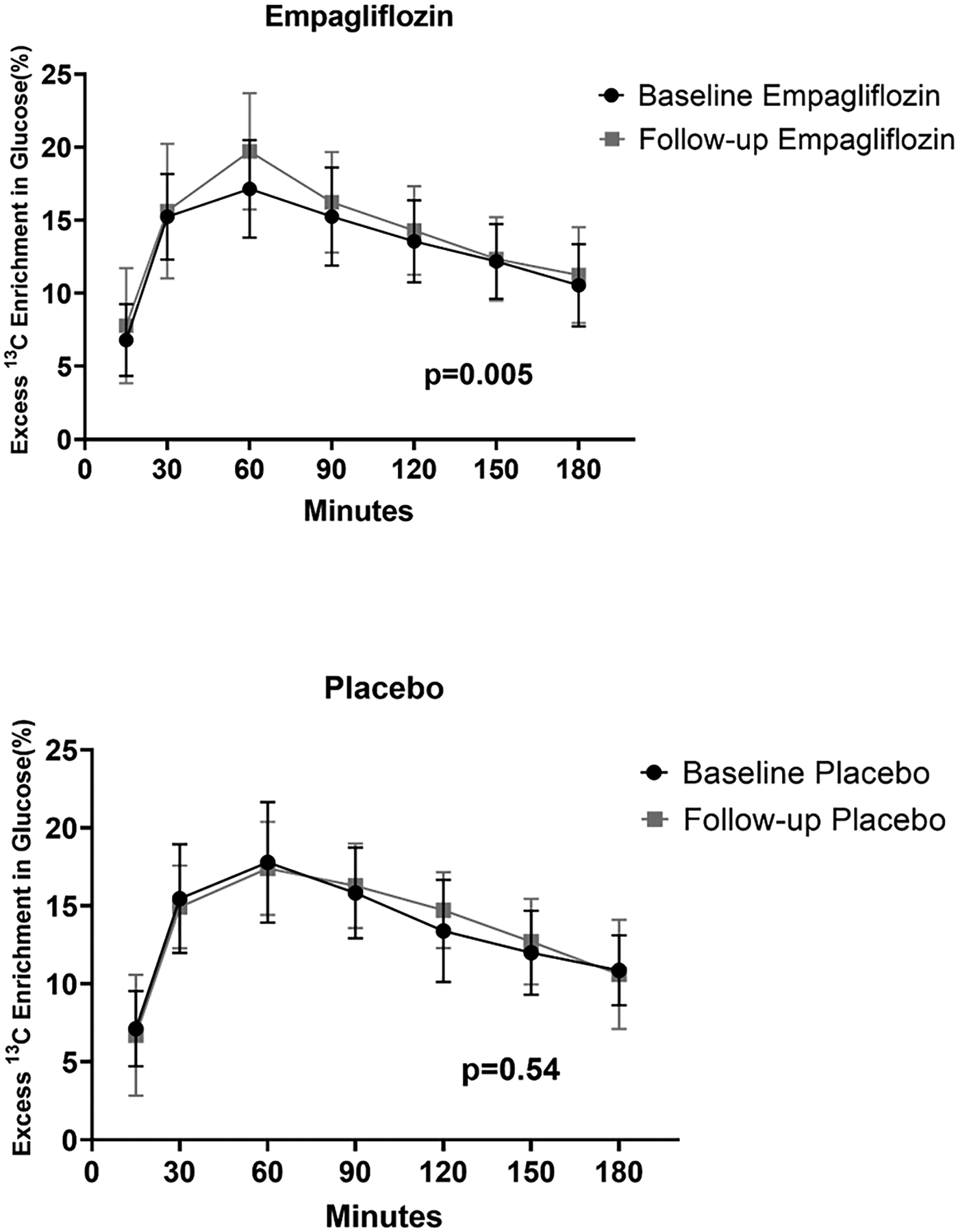
Contribution of [U-13C3] glycerol to glucose production via hepatic gluconeogenesis in participants treated with empagliflozin (top panel) and matching placebo (bottom panel). There was a statistically significant 6.5% increase in the area under the curve for 13C enrichment in glucose in the empagliflozin treated group and no treatment difference with placebo. Baseline to follow-up comparisons were made using mixed linear models to account for all time points at each visit.
No significant difference was seen in the amount of [1,2-13C2] glucose representing PPP activity between baseline and follow up among those randomized to empagliflozin or placebo (Figure S4). The proportion of [U-13C3] glycerol metabolism through the TCA cycle was similar at baseline and follow up in the empagliflozin arm but decreased among those treated with placebo (Figure S5).
Impact of weight and body fat distribution on glycerol-derived gluconeogenesis
The baseline characteristics of the study population stratified by VAT level (defined as > or ≤ the sex-specific median population value) are shown in Table S1. Participants in the high VAT group had statistically significant higher levels of insulin, triglycerides, C-reactive protein, HOMA-IR and liver fat. There was a 12.6% lower area under the curve for glycerol-derived 13C enrichment in glucose in the high VAT group compared to low VAT group at baseline (p= 0.04, Figure 4). Higher VAT was correlated with lower peak glycerol-derived 13C enrichment in glucose (ρ= −0.34, p=0.05), whereas higher abdominal subcutaneous adipose tissue was correlated with higher peak glycerol-derived 13C enrichment in glucose (ρ= 0.43, p=0.01). No correlation between liver fat level and glycerol-derived 13C enrichment in glucose was seen (ρ= −0.05, p=0.77) (Figure S6). There was no significant correlation between change in body weight and change in peak glycerol-derived 13C enrichment in glucose overall (ρ= −0.12, p=0.49) (Figure 5), or in groups stratified by treatment assignment (ρ= −0.04, p=0.87 for empagliflozin, ρ= −0.03, p=0.90 for placebo).
Figure 4. Excess 13C enrichment in glucose stratified by visceral adipose tissue level.
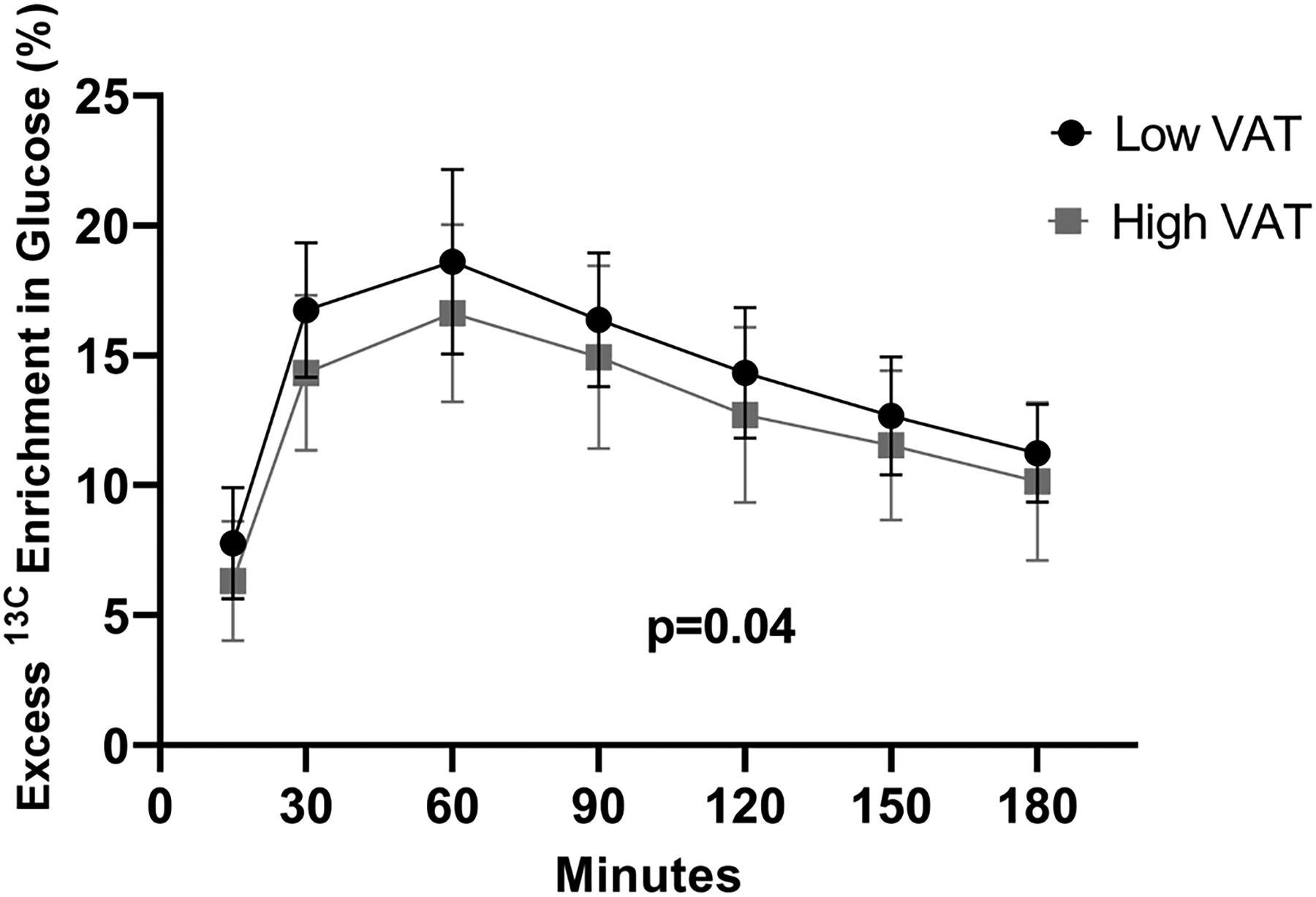
Contribution of [U-13C3] glycerol to glucose production via hepatic gluconeogenesis in participants with high VAT (squares) and low VAT (circles) at baseline. There was a statistically significant 12.6% lower area under the curve for 13C enrichment in glucose in the high VAT group compared to low VAT group. High/low defined as > or ≤ the sex-specific median population value. VAT=visceral adipose tissue.
Figure 5. Change in peak 13C enrichment in glucose by change in body weight.
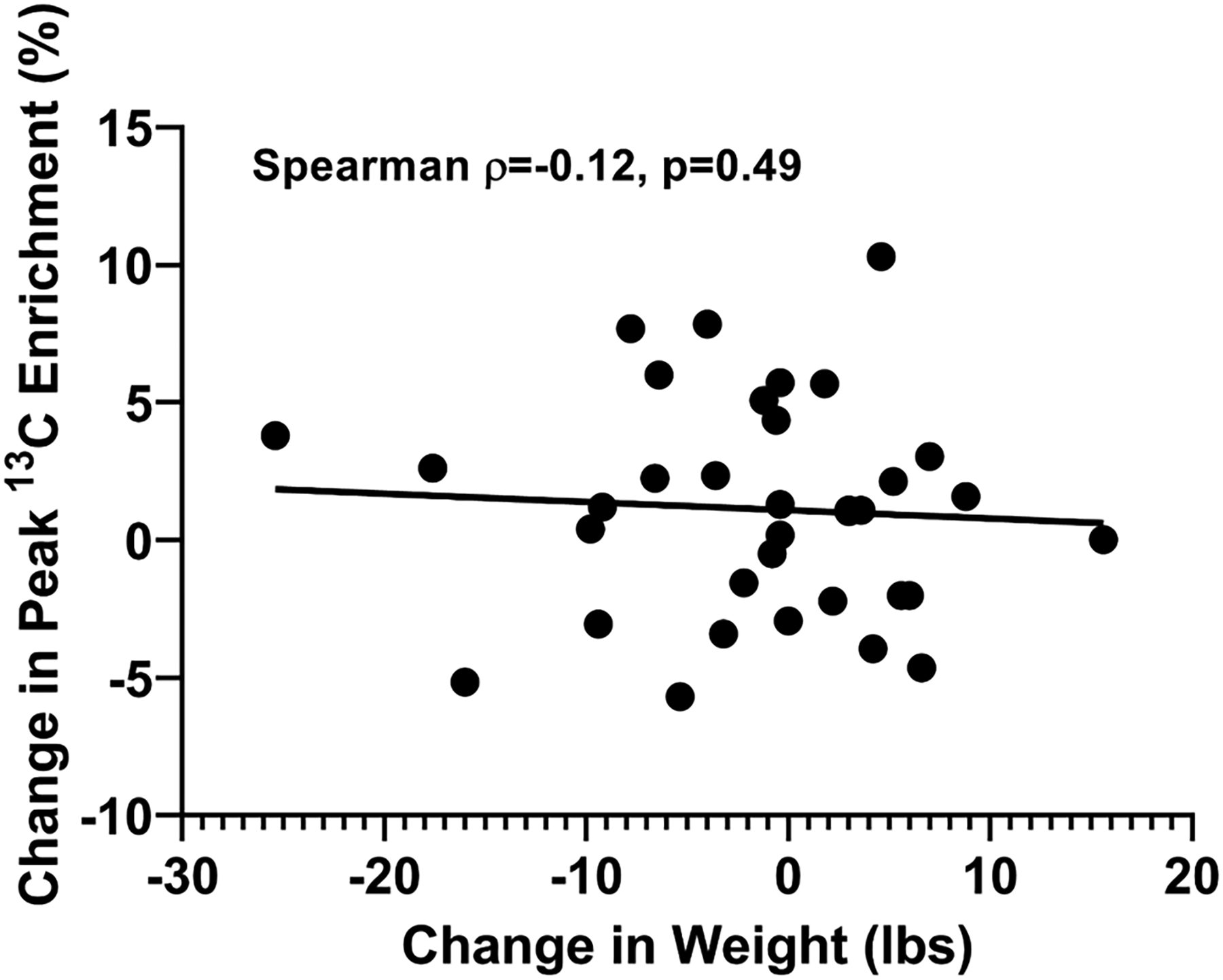
Change in body weight (lbs) was not significantly correlated with change in peak 13C enrichment in blood glucose (Spearman ρ= −0.12, p=0.49).
Discussion:
In this randomized, placebo-controlled clinical trial, we tested and proved the hypothesis that empagliflozin would increase exogenous glycerol-derived 13C enrichment in blood glucose by likely reducing endogenous glycerol-derived hepatic gluconeogenesis from VAT in adults with obesity without T2DM. We did not observe an effect of empagliflozin on glycerol metabolism via the PPP or TCA pathways, although contribution of these pathways to glycerol-gluconeogenesis is relatively minor (7). We also confirmed our prior observation that individuals with high VAT levels had lower glycerol-derived 13C enrichment in glucose compared with those with low VAT. However, we did not see an effect modification of empagliflozin on glycerol-derived 13C enrichment in glucose by VAT level. Overall, our findings support the established hypothesis that overactive lipolysis in VAT chronically delivers excess glycerol to the liver leading to higher risk of hyperglycemia and T2DM. Furthermore, these data are the first to our knowledge to demonstrate that empagliflozin inhibits this pathway and increases exogenous glycerol-derived 13C enrichment in glucose likely reflecting lower glycerol-derived gluconeogenesis from VAT. Notably, changes in glycerol-derived 13C enrichment in glucose were not related to generalized weight loss, suggesting that empagliflozin may act through a VAT-specific mechanism.
The effects of empagliflozin on other biomarkers in our study (e.g. hemoglobin A1c, and systolic blood pressure) are consistent with its known biological effects and support its efficacy and general safety even among non-diabetic obese adults. Notably, empagliflozin reduced HbA1c but did not affect fasting plasma glucose concentration, possibly due to the drug’s effects on mitigating postprandial glucose excursions (16). It is unlikely that our findings are due to treatment-dependent effects on the circulating glucagon or glycerol pools with increased utilization of exogenous glycerol, as there was no differences in plasma glucagon or plasma free glycerol between empagliflozin and placebo-treated groups. Moreover, modeling of the [U-13C3] glycerol flux using mean values for administered [U-13C3] glycerol, body weight, volume of distribution of glucose, and glycerol-derived 13C enrichment in blood glucose in our study, suggests that almost all of the administered [U-13C3] glycerol is accounted for in the experimental system and loss or gain of [U-13C3] glycerol to/from additional sources is unlikely (Table S2). Furthermore, it is unlikely that our results are explained by greater intestinal uptake of the exogenous glycerol after empagliflozin treatment since 1) intestinal uptake depends on local inhibitory compounds that are not known to differ in obesogenic states (17), and 2) there are no SGLT-2 receptors (only SGLT1) present in the intestinal epithelium (18). Although SGLT-2 inhibition may indeed increase endogenous glucose production rates, it is unlikely that our findings are due to increased gluconeogenesis from all substrates since blood glucose levels were similar between groups throughout the experiment.
The exact mechanism responsible for the effects of empagliflozin on glycerol-derived hepatic gluconeogenesis is not completely elucidated. Our results suggest that empagliflozin treatment may decrease visceral adipose tissue thereby reducing its contribution of cold (endogenous) glycerol and/or cold phosphoenolpyruvate carboxykinase-derived substrates into this pathway (Figure 6). Although general weight loss could theoretically result in less lipolytic turnover in fat depots other than VAT, we did not see a significant weight loss with empagliflozin nor did we observe a relationship between decreasing weight and increasing glycerol-derived 13C enrichment in glucose. Furthermore, empagliflozin effects are more likely to be mediated through VAT, as there was no relationship between baseline glycerol-derived 13C enrichment in glucose and liver fat and there was a positive (rather than inverse) relationship between glycerol-derived 13C enrichment in glucose and abdominal subcutaneous fat. A study including 3,300 patients from 5 different clinical trials of empagliflozin versus placebo showed a reduction in multiple indices of visceral adiposity with empagliflozin (9), building compelling evidence for empagliflozin’s potential to directly reduce VAT. Furthermore, empagliflozin has been shown to directly affect the visceral adipocyte in preclinical models of obesity with treatment resulting in less visceral adipocyte hypertrophy, less oxidative stress (19), and less crown-like structures (a hallmark of inflammation in adipose) (20). However, we are unable to confirm this hypothesis from our current study since MRI measurements were not repeated post-treatment. Furthermore, although our findings are consistent with the hypothesis that excess VAT suppressed the incorporation of [U-13C3] glycerol into this gluconeogenic pathway by contributing excessive amounts of endogenous adipose-derived glycerol to the liver, we cannot rule out the possibility that the relative incorporation of other unlabeled gluconeogenic substrates into glucose was reduced compared to glycerol.
Figure 6. Hypothesized effect of empagliflozin on glycerol-derived hepatic gluconeogenesis.
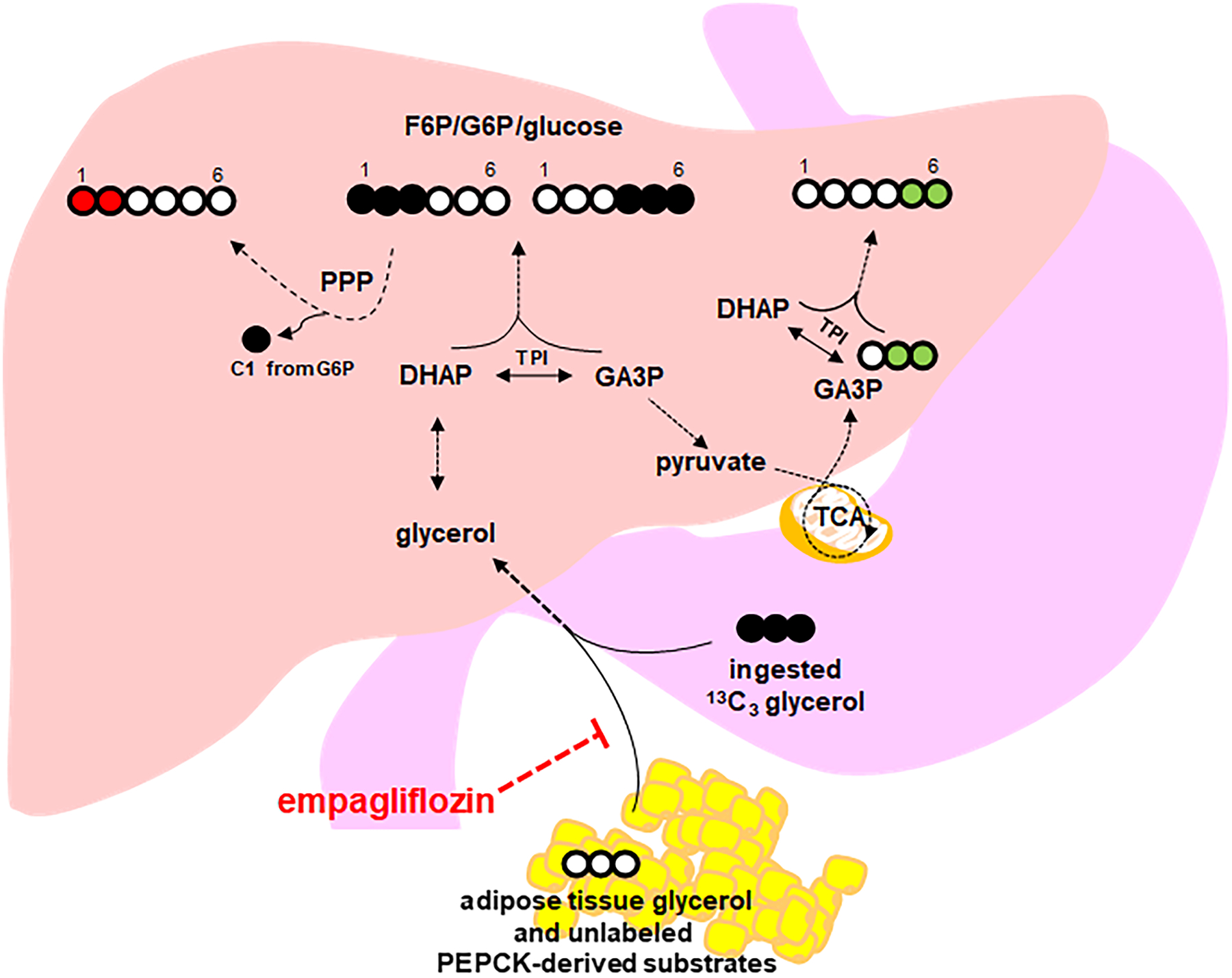
Glycerol-gluconeogenesis is directly interrogated by determining the fraction of 13C3 enrichment in blood glucose using NMR spectroscopic quantification of 13C–labeled glucose isotopomers. Total 13C enrichment in plasma glucose is measured by the sum of all glucose isotopomers with excess 13C. Empagliflozin treatment may decrease visceral adipose tissue thereby reducing its contribution of cold (endogenous) glycerol and/or cold PEPCK-derived substrates into this pathway (red hashed flat arrow). Additional information about specific pathways is derived from specific glucose isotopomers. Initially, glycerol is phosphorylated in the liver by glycerol kinase and is converted to DHAP and GA3P. Direct production of glucose via gluconeogenesis occurs by condensation of DHAP and GA3P to form carbons 1–3 and 4–6 of glucose, respectively. Therefore, quantification of [1,2,3-13C3] glucose and of [4,5,6-13C3] glucose reflects primarily direct hepatic gluconeogenesis from ingested [U-13C3] glycerol (black filled circles). Passage of [1,2,3-13C3] G6P through the PPP produces [1,2-13C2] F6P and subsequently [1,2-13C2] glucose so that the quantification of [1,2-13C2] glucose reflects PPP activity (red filled circles). Metabolism of [U-13C3] glycerol to [U-13C3] pyruvate followed by either decarboxylation to [U-13C2] acetyl-CoA or carboxylation to [1,2,3-13C3] oxaloacetate allows rearrangement of 13C in the TCA cycle to form PEP. Two possible PEP isotopomers through glycerol metabolism in the TCA cycle are [1,2-13C2]- and [2,3-13C2] PEP. Conversion to PEP and metabolism to glucose will produce [4,5-13C2]- or [5,6-13C2] glucose (green filled circles). Since the PPP does not disrupt carbon–carbon bonds in carbons 4–6 of glucose, the presence of [4,5-13C2]- or [5,6-13C2] glucose indicates metabolism of [U-13C3] glycerol through the TCA cycle prior to gluconeogenesis. DHAP = dihydroxyacetone phosphate; GA3P = glyceraldehyde 3-phosphate; G6P = glucose 6-phosphate; F6P = fructose 6-phosphate; NMR = nuclear magnetic resonance; PEP = phosphoenolpyruvate; PEPCK = PEP carboxykinase; PPP = pentose phosphate pathway, TCA = tricarboxylic acid; TPI = triose phosphate isomerase.
Empagliflozin can increase endogenous glucagon after a single dose in persons with T2DM (21). The mechanism by which this occurs has not been fully established (22), and it is unclear to what degree a similar response occurs in those without T2DM. Indeed, we saw a small, but non-significant increase in glucagon in the empagliflozin-treated group. Nonetheless, we would expect that an increase in glucagon and endogenous glucose production with empagliflozin would lead to a decrease in glycerol-derived 13C enrichment in glucose from increased lipolysis and gluconeogenesis. Paradoxically, we found a significant increase in glycerol-derived 13C enrichment in glucose, suggesting the lack of a significant glucagon effect on this pathway.
While this study confirms findings previously described by our group that participants with high VAT have lower glycerol-derived 13C enrichment in glucose when compared to those with low VAT, the magnitude of difference between the high and low VAT groups in the current study was more modest. This finding can likely be explained by the fact that the low VAT group in the current study is more viscerally obese compared with the prior study,(7) with mean VAT 3.8 kg vs. 1.6 kg. This might also potentially explain why we did not observe any effect modification of empagliflozin on glycerol-derived 13C enrichment in glucose by VAT level. Future studies investigating effects of empagliflozin on VAT should aim to enroll participants with greater variation in VAT to better elucidate the biological effects of the drug on visceral obesity.
Strengths of our study include: i) the use of a simple, non-radioactive, in vivo metabolic tracer technique to directly measure the differential enrichment of labeled glycerol vs. adipose-derived glycerol in response to treatment; ii) employing a randomized, placebo-controlled trial design to elucidate the effects of empagliflozin on glycerol-derived hepatic gluconeogenesis; iii) the use of MRI to accurately quantify body fat distribution; and iv) including only participants without T2DM to assess the biological mechanism of this system prior to the onset of T2DM. Several limitations also merit comment. First, the low VAT group had more relative visceral obesity compared with our prior study, making it more challenging to differentiate high/low VAT differences. Second, MRI measurements were not repeated post-treatment so we are unable to directly assess changes in adipose depots after empagliflozin treatment. Third, we did not collect data on physical activity or cardiorespiratory fitness, both strong physiological components of obesity and T2DM outcomes (23, 24), so we are unable to assess their impact on the effects of empagliflozin on glycerol-derived hepatic gluconeogenesis. Fourth, the sample size was modest and a complete physiological model will require larger studies to confirm and extend these findings; however, our study was powered appropriately for the primary outcome and thus our sample size is valid for statistical comparison. Lastly, since the half-life of oral glycerol is on the order of minutes and is likely rapidly utilized by the liver (25), the early part of the 13C3 glycerol enrichment curves (up to ~90 minutes) are most informative for this study. This is further supported by the observation that the 13C3 enrichment curve was seen to plateau after ~90 minutes in both groups; indeed, we saw a similar pattern in a prior study (7). Future studies using this methodology could further simplify the design (using less time and less blood draws for participants) and reduce the number of analyses.
In conclusion, in this first-in-human randomized, double-blind, placebo-controlled clinical trial, we used a simple stable isotope technique to elucidate the in vivo mechanisms underlying the relationship between visceral obesity and glycerol-derived hepatic gluconeogenesis and the inhibitory effects of the SGLT2 inhibitor, empagliflozin, on this pathway. Our findings implicate visceral fat in the pathogenesis of hyperglycemia and T2DM via glycerol-gluconeogenesis and suggest a beneficial role of empagliflozin for the treatment of viscerally obese adults at risk for T2DM.
Supplementary Material
Study Importance Questions.
What is already known about this subject?
Empagliflozin reduces body weight, and indices of visceral adiposity.
Visceral adiposity-related increases in endogenous glycerol-derived hepatic gluconeogenesis can be interrogated using a simple oral 13C labeling method.
What are the new findings in your manuscript?
Empagliflozin increases exogenous glycerol-derived 13C enrichment in blood glucose likely by reducing endogenous glycerol-derived hepatic gluconeogenesis from VAT in adults with obesity without type 2 diabetes mellitus.
Changes in glycerol-derived 13C enrichment in glucose were not related to generalized weight loss, suggesting that empagliflozin may act through a visceral fat-specific mechanism.
How might your results change the direction of research or focus of clinical practice?
Our findings implicate visceral fat in the pathogenesis of hyperglycemia and type 2 diabetes mellitus via glycerol-gluconeogenesis and suggest a beneficial role of empagliflozin for the treatment of viscerally obese adults at risk for T2DM.
Acknowledgements
We thank Lucy Christie, Kelley Derner, Jeannie Baxter, Janet Jerrow, Carol Parcel, Maida Tai, Bienka Milton, Rebecca Murphy, Benjamin Bleiberg, and Wesley Smith for technical support.
Funding Sources: This work was supported by the National Institutes of Health (grants K23DK106520 to Dr. Neeland, P41EB015908 to Dr. Malloy, and R01DK099289 to Dr. Jin), CTSA NIH Grant UL1TR001105 to UT Southwestern, and by the Dedman Family Scholarship in Clinical Care from UT Southwestern to Dr. Neeland.
Footnotes
ClinicalTrials.gov Identifier: NCT02833415
Relationship with Industry/Conflict of Interest: Dr. Neeland has previously received honoraria, consulting, and speaker’s bureau fees, and travel support from Boehringer-Ingelheim/Lilly Alliance, a research grant from Novo Nordisk, and has been a member of the scientific advisory board of AMRA Medical. All other authors have no relationship with industry/conflicts of interest to report.
Data and Resource Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author upon reasonable request. No applicable resources were generated or analyzed during the current study.
Dr. Neeland (guarantor) takes full responsibility for the work as a whole, including study design, access to data, and the decision to submit and publish the manuscript.
References
- 1.Benjamin EJ, Muntner P, Alonso A, Bittencourt MS, Callaway CW, Carson AP, et al. Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation 2019;139: e56–e66. [DOI] [PubMed] [Google Scholar]
- 2.Neeland IJ, Poirier P, Despres JP. Cardiovascular and Metabolic Heterogeneity of Obesity: Clinical Challenges and Implications for Management. Circulation 2018;137: 1391–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lavie CJ, Laddu D, Arena R, Ortega FB, Alpert MA, Kushner RF. Healthy Weight and Obesity Prevention: JACC Health Promotion Series. J Am Coll Cardiol 2018;72: 1506–1531. [DOI] [PubMed] [Google Scholar]
- 4.Puhakainen I, Koivisto VA, Yki-Jarvinen H. Lipolysis and gluconeogenesis from glycerol are increased in patients with noninsulin-dependent diabetes mellitus. J Clin Endocrinol Metab 1992;75: 789–794. [DOI] [PubMed] [Google Scholar]
- 5.Gaidhu MP, Anthony NM, Patel P, Hawke TJ, Ceddia RB. Dysregulation of lipolysis and lipid metabolism in visceral and subcutaneous adipocytes by high-fat diet: role of ATGL, HSL, and AMPK. Am J Physiol Cell Physiol 2010;298: C961–971. [DOI] [PubMed] [Google Scholar]
- 6.Rui L Energy metabolism in the liver. Compr Physiol 2014;4: 177–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Neeland IJ, Hughes C, Ayers CR, Malloy CR, Jin ES. Effects of visceral adiposity on glycerol pathways in gluconeogenesis. Metabolism 2017;67: 80–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ridderstrale M, Andersen KR, Zeller C, Kim G, Woerle HJ, Broedl UC. Comparison of empagliflozin and glimepiride as add-on to metformin in patients with type 2 diabetes: a 104-week randomised, active-controlled, double-blind, phase 3 trial. Lancet Diabetes Endocrinol 2014;2: 691–700. [DOI] [PubMed] [Google Scholar]
- 9.Neeland IJ, McGuire DK, Chilton R, Crowe S, Lund SS, Woerle HJ, et al. Empagliflozin reduces body weight and indices of adipose distribution in patients with type 2 diabetes mellitus. Diab Vasc Dis Res 2016;13: 119–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N Engl J Med 2015;373: 2117–2128. [DOI] [PubMed] [Google Scholar]
- 11.Jin ES, Jones JG, Merritt M, Burgess SC, Malloy CR, Sherry AD. Glucose production, gluconeogenesis, and hepatic tricarboxylic acid cycle fluxes measured by nuclear magnetic resonance analysis of a single glucose derivative. Anal Biochem 2004;327: 149–155. [DOI] [PubMed] [Google Scholar]
- 12.West J, Dahlqvist Leinhard O, Romu T, Collins R, Garratt S, Bell JD, et al. Feasibility of MR-Based Body Composition Analysis in Large Scale Population Studies. PLoS One 2016;11: e0163332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Borga M, Thomas EL, Romu T, Rosander J, Fitzpatrick J, Dahlqvist Leinhard O, et al. Validation of a fast method for quantification of intra-abdominal and subcutaneous adipose tissue for large-scale human studies. NMR Biomed 2015;28: 1747–1753. [DOI] [PubMed] [Google Scholar]
- 14.Karlsson A, Rosander J, Romu T, Tallberg J, Gronqvist A, Borga M, et al. Automatic and quantitative assessment of regional muscle volume by multi-atlas segmentation using whole-body water-fat MRI. J Magn Reson Imaging 2015;41: 1558–1569. [DOI] [PubMed] [Google Scholar]
- 15.West J, Romu T, Thorell S, Lindblom H, Berin E, Holm AS, et al. Precision of MRI-based body composition measurements of postmenopausal women. PLoS One 2018;13: e0192495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nishimura R, Tanaka Y, Koiwai K, Inoue K, Hach T, Salsali A, et al. Effect of empagliflozin monotherapy on postprandial glucose and 24-hour glucose variability in Japanese patients with type 2 diabetes mellitus: a randomized, double-blind, placebo-controlled, 4-week study. Cardiovasc Diabetol 2015;14: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kato T, Hayashi Y, Inoue K, Yuasa H. Glycerol absorption by Na+-dependent carrier-mediated transport in the closed loop of the rat small intestine. Biol Pharm Bull 2005;28: 553–555. [DOI] [PubMed] [Google Scholar]
- 18.Hinnen D The role of the kidney in hyperglycemia: a new therapeutic target in type 2 diabetes mellitus. J Cardiovasc Nurs 2013;28: 157–165. [DOI] [PubMed] [Google Scholar]
- 19.Kusaka H, Koibuchi N, Hasegawa Y, Ogawa H, Kim-Mitsuyama S. Empagliflozin lessened cardiac injury and reduced visceral adipocyte hypertrophy in prediabetic rats with metabolic syndrome. Cardiovasc Diabetol 2016;15: 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Han JH, Oh TJ, Lee G, Maeng HJ, Lee DH, Kim KM, et al. The beneficial effects of empagliflozin, an SGLT2 inhibitor, on atherosclerosis in ApoE (−/−) mice fed a western diet. Diabetologia 2017;60: 364–376. [DOI] [PubMed] [Google Scholar]
- 21.Ferrannini E, Muscelli E, Frascerra S, Baldi S, Mari A, Heise T, et al. Metabolic response to sodium-glucose cotransporter 2 inhibition in type 2 diabetic patients. J Clin Invest 2014;124: 499–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haedersdal S, Lund A, Knop FK, Vilsboll T. The Role of Glucagon in the Pathophysiology and Treatment of Type 2 Diabetes. Mayo Clin Proc 2018;93: 217–239. [DOI] [PubMed] [Google Scholar]
- 23.Fletcher GF, Landolfo C, Niebauer J, Ozemek C, Arena R, Lavie CJ. Promoting Physical Activity and Exercise: JACC Health Promotion Series. J Am Coll Cardiol 2018;72: 1622–1639. [DOI] [PubMed] [Google Scholar]
- 24.Lavie CJ, Ozemek C, Carbone S, Katzmarzyk PT, Blair SN. Sedentary Behavior, Exercise, and Cardiovascular Health. Circ Res 2019;124: 799–815. [DOI] [PubMed] [Google Scholar]
- 25.Beylot M, Martin C, Beaufrere B, Riou JP, Mornex R. Determination of steady state and nonsteady-state glycerol kinetics in humans using deuterium-labeled tracer. J Lipid Res 1987;28: 414–422. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.