

Abstract
Ribonucleotide reductases (RNRs) catalyze the de novo conversion of nucleotides to deoxynucleotides in all organisms, controlling their relative ratios and abundance and in doing so play an important role in fidelity of DNA replication and repair. RNR’s central role in nucleic acid metabolism has resulted in five therapeutics that inhibit human RNR. In this review we discuss the structural, dynamic, and mechanistic aspects of RNR activity and regulation, primarily for the human and E. coli class Ia enzymes. The unusual radical-based organic chemistry of nucleotide reduction, the inorganic chemistry of the essential metallo-cofactor biosynthesis/maintenance, the transport of a radical over a long distance and the dynamics of subunit interactions all present distinct entry points toward RNR inhibition relevant for drug discovery. Our current mechanistic understanding of small molecules that target different elements of RNR function are described, including downstream pathways that lead to cell cytotoxicity. We conclude by summarizing novel and emergent RNR targeting motifs for cancer and antibiotic therapeutics.
Keywords: ribonucleotide reductases, structures, mechanisms, therapeutics
Introduction.
The availability of adequate and balanced deoxynucleotide pools is essential for accurate DNA replication and repair and consequently genome stability. Deoxynucleotides are supplied universally in all organisms by a de novo pathway catalyzed by ribonucleotide reductases (RNRs) that convert RNA building blocks to DNA building blocks (1–3). Deoxynucleotides can also be generated in an organism-, environment-, and disease-specific fashion by nucleoside (or nucleotide) salvage pathways (4). Our current understanding of the unique organic (5) and inorganic chemistry (6) of RNRs, have been revealed, in part, by our understanding of clinically used therapeutics that target the universal radical-mediated nucleotide reduction mechanism, and the specific metallo-cofactor biosynthetic and repair pathways. The ensemble of studies led to the first structures of class I RNRs at low resolution (7–10), and more recently, to high resolution structures in trapped active and inhibited states (8, 11, 12). These new studies suggest, in combination with inhibitors of specific signaling pathways downstream of RNR, that the time is right to revisit RNR as a target for antibacterial, antiviral, as well as anticancer agents.
All RNRs catalyze the conversion of nucleoside diphosphates (NDPs) or triphosphates (NTPs) to deoxynucleotides (dNDP or dNTP, Figure 1A). The RNR’s share a common active site architecture located in subunit α that houses 3 essential cysteines (Figure 1B) (13–15). Two cysteines (bottom face) provide the reducing equivalents to make dNDPs and the third cysteine (top face) is transiently oxidized to a thiyl radical (−S•) that initiates NDP reduction (16). Distinct metallo-cofactors catalyze this oxidation (Figure 1C) and they are the main basis for RNR classification (Ia-e, II, III), though a recently discovered non-metallo-cofactor, 2,3-dihydroxyphenylalaninine radical (DOPA•) breaks this paradigm (17–19). This review focuses on the class I RNRs that share a distinct mechanism by which a transient thiyl radical is generated, and whose formation requires a second subunit β that houses the cofactor oxidant (Figure 1C).
Figure 1.
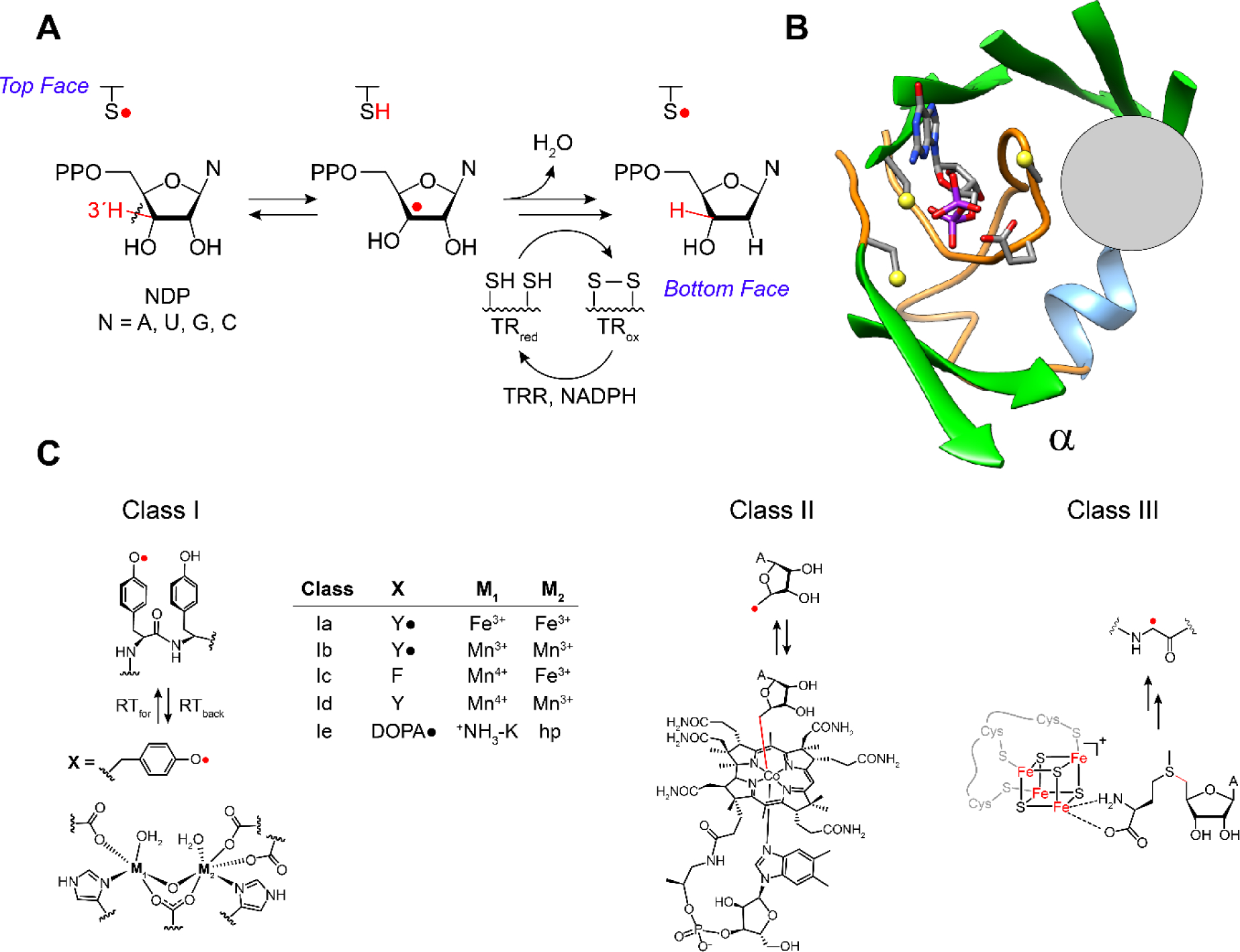
A RNRs catalyze the conversion of nucleoside di- or triphosphates, ND(T)Ps, to deoxynucleoside di- or triphosphate, dND(T)Ps. B The reduction occurs in the active site in subunit α composed of a 10 stranded β barrel with three cysteines and the conserved placement of the oxidant (gray circle, panel B) involved in thiyl radical formation (−S•, top face in A) that initiates NDP reduction. The bottom face thiols in A deliver the reducing equivalents and themselves become oxidized. C The oxidants are distinct among the RNR classes (I, II and III) represented here by a gray circle that is juxtaposed with the thiyl radical loop. Substrate and four essential residues, including the three essential cysteines and E441, are shown as sticks. C The class Ia RNRs use a diferric-tyrosyl radical (Y•) cofactor (M1, M2 = Fe3+) that is located in subunit β (left, bottom) to regenerate a radical species in the active site in subunit α. The oxidation occurs over a distance of ~33 Å by long range radical transfer to first generate a Y• in subunit α (under the gray circle), and second generate -S• on an adjacent cysteine (top face in A). In other class I RNRs (Ib-Ie) the oxidation also occurs by long range radical transfer across α and β, but involves distinct metallo-oxidants (X, M1, M2). In the case of the class II and III RNRs the oxidants, the 5′-deoxyadenosyl radical generated from adenosylcobalamin (class II) and the glycyl radical (class III) generated from S- adenosylmethionine and an FeS cluster, are located adjacent to the cysteine to be oxidized (gray circle). A = adenine base.
Docking model and Radical Transfer (RT) pathway.
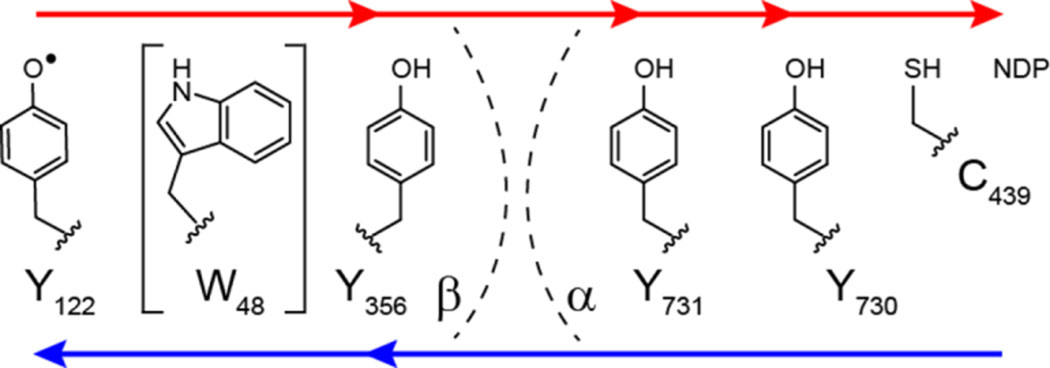
Reichard and coworkers in 1969 discovered the class Ia E. coli RNR and proposed that active enzyme is an α2β2 complex (20, 21). However, it wasn’t until 1994 that Eklund et al. (13) reported the X-ray structure of α2 (Figure 2B), which together with their earlier structure of β2 (Figure 2A) led to a symmetrical “docking model” based on subunit shape complementarity (Figure 2C). This model has guided experimentation until recently. A fascinating feature of the docking model is that the diferric-tyrosyl radical cofactor (Fe3+2-Y122•, Figure 2C) in β is ~ 35 Å away from C439 (E. coli numbering), which is oxidized in the α subunit. A turnover frequency for dNDP production of 2 to 10 s−1, together with the long distance between Y122• and C439, engender a RT pathway (7, 14): Y122•[β] ⇌ [W48[β]] ⇌ Y356[β] to Y731[α] ⇌ Y730[α] ⇌ C439[α] (Figure 3, noting that [W48] involvement has not yet been demonstrated).
Figure 2.
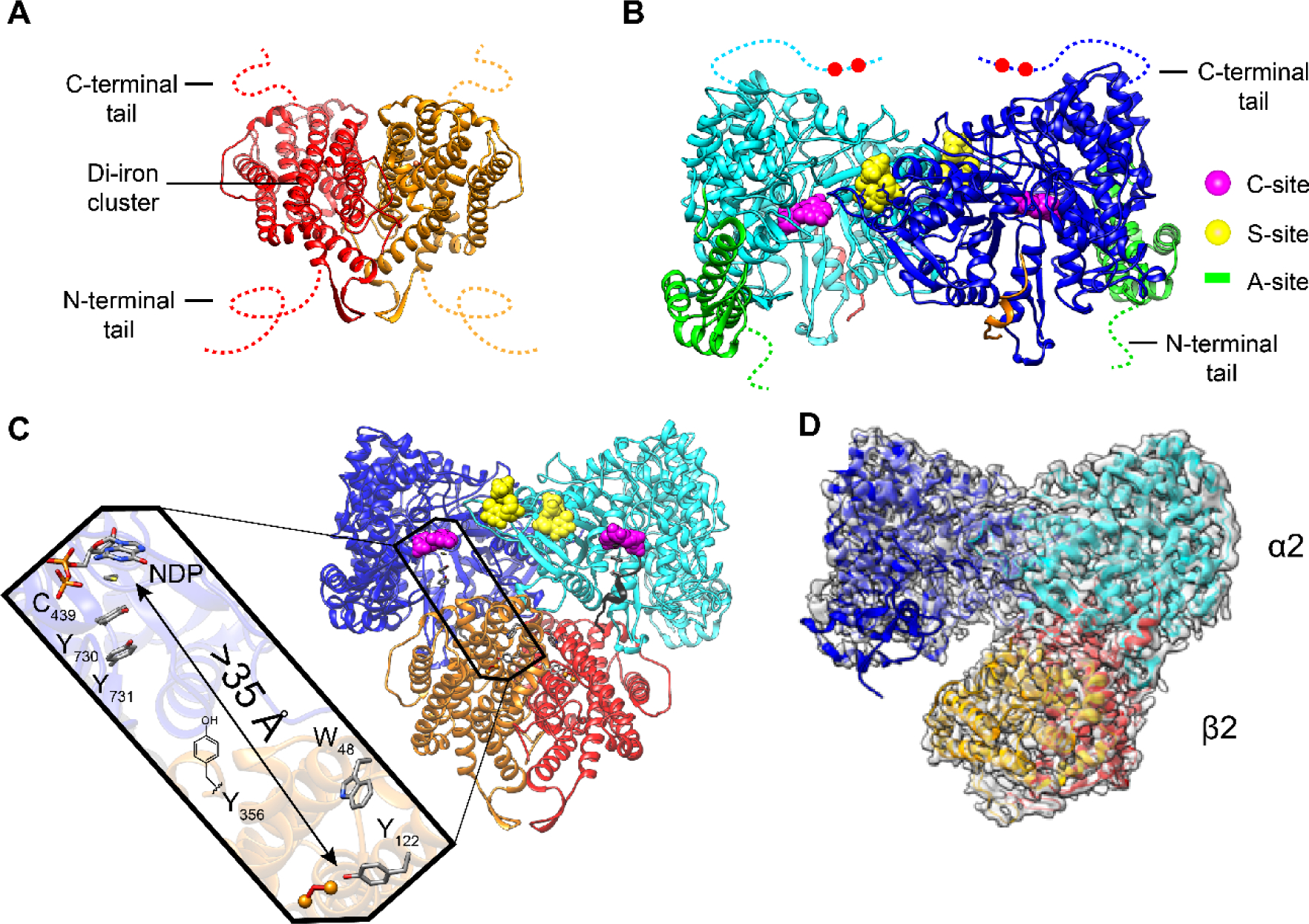
Structural models of class Ia RNR from E. coli. X-ray structures of A β2 (15), B α2 (13), C the Eklund docking model of α2β2 (13), and D a cryo-EM structure of an active α2β2 with two mutations in β2: F3Y122/E52Q (11). A β2, a homodimer in red/orange with disordered C-terminal tail residues (dashed lines 341–375 E. coli). B α2, a homodimer in light and dark blue with disordered C-terminal tail residues (dashed lines 737–761) that houses the two cysteines (red balls) which re-reduce the active site disulfide formed on NDP reduction (Figure 1A). α2 also houses the A-site (activity site or cone domain) that binds ATP (that activates RNR) or dATP (that inactivates RNR) in green; the C-site (catalytic site that binds CDP, UDP, GDP, and ADP) in magenta; the S-site (specificity site that binds the effectors dATP, ATP, TTP dGTP) in yellow. C Docking model of α2β2 with the long-range radical transfer pathway (left) (14). Shown also is a peptide in gray (residues 360–375 of β2) proposed to represent the tail of β2 responsible for α2 binding. D Asymmetric complex formed when F3Y122/E52Q-β2, interacts with α2, GDP, and TTP. 3.6-Å resolution cryo-EM density shown in transparent gray. This structure of the active α2β2 can be compared with the symmetric docking model in C.
Figure 3.
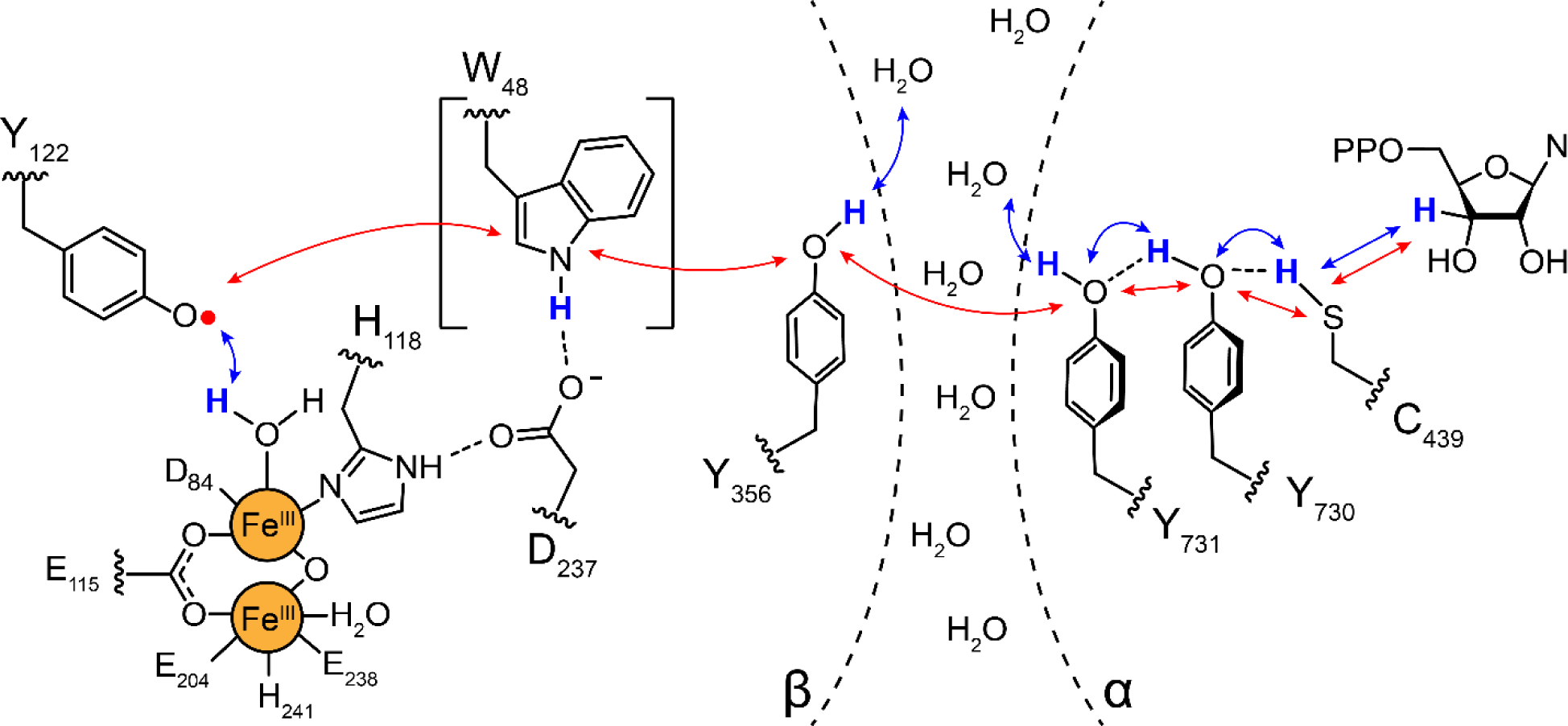
Proposed radical transfer pathway in Ia RNRs (E. coli numbering) within the α2β2 complex. Binding of substrate and allosteric effector (not shown) to α2, triggers radical transfer from the Y122• of the Fe3+2-Y122• cofactor in β, through three transient Y•s (356-β across the subunit interface to 731-α and 730-α). The Y730•- α (under the gray circle in Figure 1B) then oxidizes the active site cysteine to a −S• that initiates NDP reduction (Figure 1A). Subsequent to dNDP formation, the Y122• is regenerated by reverse radical transfer. W48-β is bracketed as its role in the pathway has not been established. Each step in the pathway is proposed to involve distinct proton (H+, blue arrows) coupled electron (e−, red arrows) transfer (PCET) steps indicated by blue and red arrows (7).
Rate limiting physical step(s) mask both the NDP reduction and the RT chemistry. These processes are conformationally gated by proper substrate and effector binding to α2 and its association with β2 (22). The “stable” Y122• in β2 is transiently reduced and re-oxidized on each turnover, and RT through the pathway involves distinct proton-coupled electron transfer (PCET) steps at each pathway residue (Figure 3). The first step in RT is proposed to occur at the metal cofactor in β2, triggered by substrate and effector binding in α2 more than 35–40 Å away. Studies using site-specifically incorporated tyrosine analogs with altered reduction potentials, high field, multifrequency, electron paramagnetic resonance (EPR) methods, structural analysis, and RT photoinitiation in photosensitized RNRs (photo-RNRs) have provided insight into each of the proposed steps (7). Our current understanding of this pathway suggests that the thermodynamic landscape of the radical transfer process (Y122• to C439) is uphill by greater than 200 mV, and that the NDP reduction reaction, which also involves an uphill 3′-hydrogen atom abstraction, is driven to the right by rapid and irreversible loss of water during NDP reduction (Figure 1A). This pathway design avoids buildup of the highly reactive protein radical intermediates such as Y•, which has a reduction potential of 0.96 V vs the normal hydrogen electrode). Reduction of any of the Y• intermediates in the pathway (Figure 3) would inactivate RNR ending in catastrophic consequences for the organism (23, 24). Accordingly, the RT pathway provides a target of opportunity for future drug design.
Evidence for the docking model (Figure 2C) has been provided by trapping pathway radicals using tyrosine analogs with perturbed reduction potentials (Figures 3 and 4A), and active site radicals in α2 using MBIs (Figure 4B) (25). As briefly summarized, pulsed electron-electron double resonance (PELDOR) spectroscopy and negative stain electron microscopy (EM) have allowed spectroscopic analysis of these trapped α2β2 complexes.
Figure 4.
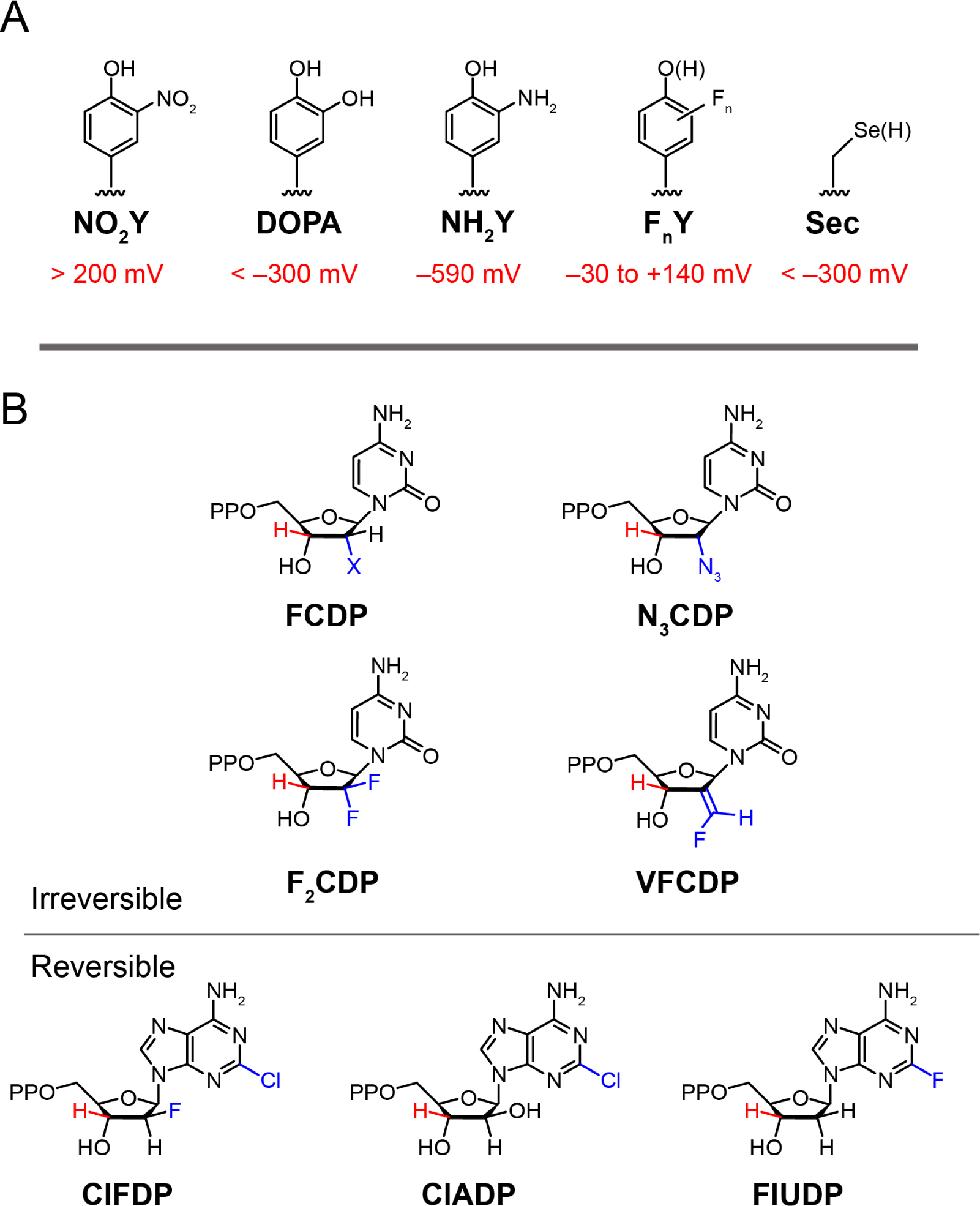
Structures of unnatural amino acids and nucleotide analogs used to study class Ia RNRs. A Unnatural amino acids that have been site-specifically incorporated in place of the tyrosines or cysteine within the radical transfer pathway (Figure 3) and their reduction potentials vs. tyrosine for NO2Y, DOPA, NH2Y, FnYs and vs. cysteine for seleocysteine (Sec) at pH 7 (24). 3-Aminotyrosine (NH2Y) is 590 mv easier to oxidize than Y. Fluorinated Ys (FnY where n = 2 or 3) have allowed for tuning of the reduction potential over 170 mV depending on the number of Fs and their substitution pattern. B Nucleoside 5′-diphosphate are irreversible and reversible inhibitors of RNR. The irreversible inhibitors are mechanism based as the 3′ C-H bond (red) of the inhibitor must be cleaved as with the normal substrate (Figure 1A), before distinct radical chemistry in each case occurs that causes enzyme inactivation.
3-Aminotyrosine (NH2Y)-RNR (Figure 4) and PELDOR analysis: low resolution evidence for the docking model.
NH2Y is easier to oxidize than Y by 590 mV (Figure 4A). When NH2Y replaces a pathway Y in α or β and is incubated with the second subunit, substrate, and specificity effector, it functions as an efficient radical trap forming a 3-aminotyrosyl radical (NH2Y•) (10, 25) that is unable to oxidize the next residue in the pathway. Under these conditions, 0.5 equivalents of total Y122• in β2 is reduced and a stoichiometric amount of NH2Y• is formed in (α/β pair, one side). Because the chemistry in one α/β pair is incomplete, 0.5 equivalents of Y122• resides in the adjacent α/β pair (on the other side), which is unable to carry out chemistry. These results require asymmetry within α2β2 and are described as half-sites reactivity.
In these trapped complexes, PELDOR spectroscopy has been used to measure the distance between the NH2Y• (located at 356-β or 731-α or 730-α) in one α/β pair and the Y122• on the adjacent pair, as shown in Figure 5A. Studies with the MBI N3CDP (Figure 4B), which forms a nitrogen centered radical (N•) covalently bound to a cysteine in the active site of α2, also allows a distance measurement. Additionally, RNR mutants in which Y122• is replaced with 2,3,5-F3-Y122• or NO2Y122• (Figure 4A), which are “hotter” oxidants than Y122•, also generate pathway radicals and demonstrate half-site reactivity (26). Distances measured to date are summarized in Table 1 and they are consistent with the docking model (Figure 2C) (25, 26). An unexpected outcome of these experiments was that when radicals are trapped within the pathway, the α2β2 complex exhibits increased subunit affinity, thus enabling its isolation. A number of these complexes have been examined by negative stain EM and have revealed structures resembling the docking complex (15–30 Å resolution (Figure 5B) (7). Our current model from these studies with perturbants is that for wild type RNR, radical transfer and catalysis occur initially within one α/β pair of an asymmetric α2β2, which triggers rapid chemistry within the second α/β pair. The mechanism of switching remains to be determined.
Figure 5.
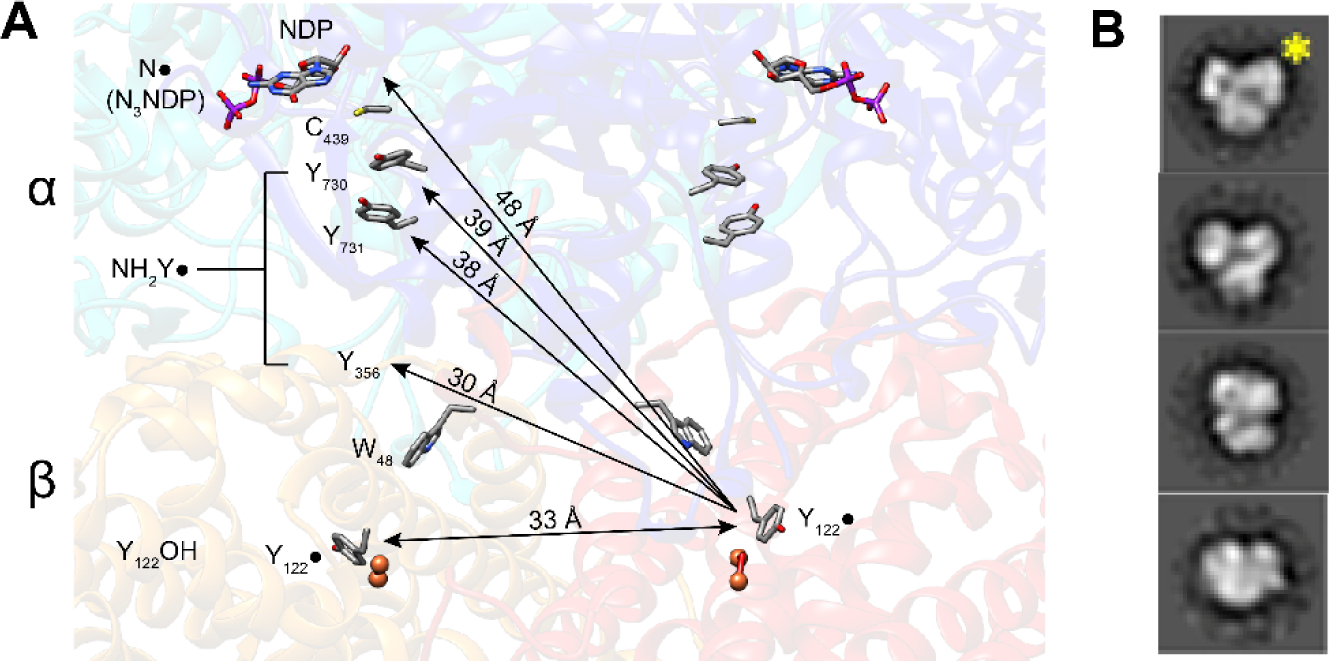
Support for the Eklund docking model (Figure 2C). A PELDOR spectroscopy used to measure distances between Y122• in the unreacted α/β pair (right) and the trapped radicals (NH2Y• or N•) in the reactive α/β pair where Y122 is reduced (YOH, left). B Representative negative stain EM 2D class averages of the structures of the NH2Y730• trapped in α2β2 complex (25). The view with the yellow star resembles the Eklund docking model shown in Figure 2C.
Table 1.
Pathway radicals trapped in E. coli class Ia RNR by site-specifically incorporating unnatural amino acids or reaction with N3UDP; PELDOR distances (< ± 1 Å) are given in last column. ● corresponds to forward RT and ● to reverse RT, as indicated by the direction of the arrows on the pathway shown below the table.
| α | β | Y122 | Y356 | Y731 | Y730 | C439 | NDP | Y122•-X• distance (Å) |
|---|---|---|---|---|---|---|---|---|
| WT | WT | ●/● | - | - | - | - | - | 33 |
| WT | DOPA356 | - | ● | - | - | - | - | 30 |
| WT | NH2Y356 | - | ● | - | - | - | - | 30 |
| Y731F | F3Y122 | - | ● | - | - | - | - | 30 |
| NH2Y731 | WT | - | - | ● | - | - | - | 38 |
| NH2Y731/R411A | WT | - | - | ● | - | - | - | 35 |
| NH2Y730 | WT | - | - | - | ● | - | - | 39 |
| WT | WT | - | - | - | - | - | N3UDP● | 48 |
| WT | F3Y122 | - | ● | - | - | - | - | 30 |
| WT | NO2Y122 | - | ● | - | - | - | - | 30 |
 | ||||||||
Higher resolution structures of RNRs
Inhibited structures in vitro.
dATP is a universal inhibitor of all class Ia RNRs. It binds to the N-terminal domain of α (cone domain, green, Figure 2B). Two independent studies by the Dealwis and Walt groups (2011) (8) and Drennan and Asturias groups (2018) (27) revealed structures of eukaryotic dATP-inhibited states. Fairman et al studying S. cerevisiae RNR observed crystallographically, an α6 hexameric ring structure (6.6 Å). Brignole et al studying human α with CDP, dATP and a small amount of ATP, observed by cryo-EM, a similar, but higher resolution hexameric ring structure (Figure 6B, 3.3Å). Both groups also reported negative stain EM studies (28 and 30 Å) (8, 27) of a dATP inhibited state in the presence of both α and β (1:1). Despite the stoichiometry of the subunits, in both cases, less than 1β2/α6 was observed. Both eukaryotic dATP inhibited states are a trimer of dimers with the cone domains responsible for the dimer interfaces. In addition, with the human RNR, small-angle X-ray scattering data on this state suggested that β2 could not enter the hole in the α6 ring structure, and hence that an active α2β2 is not accessible (28).
Figure 6.
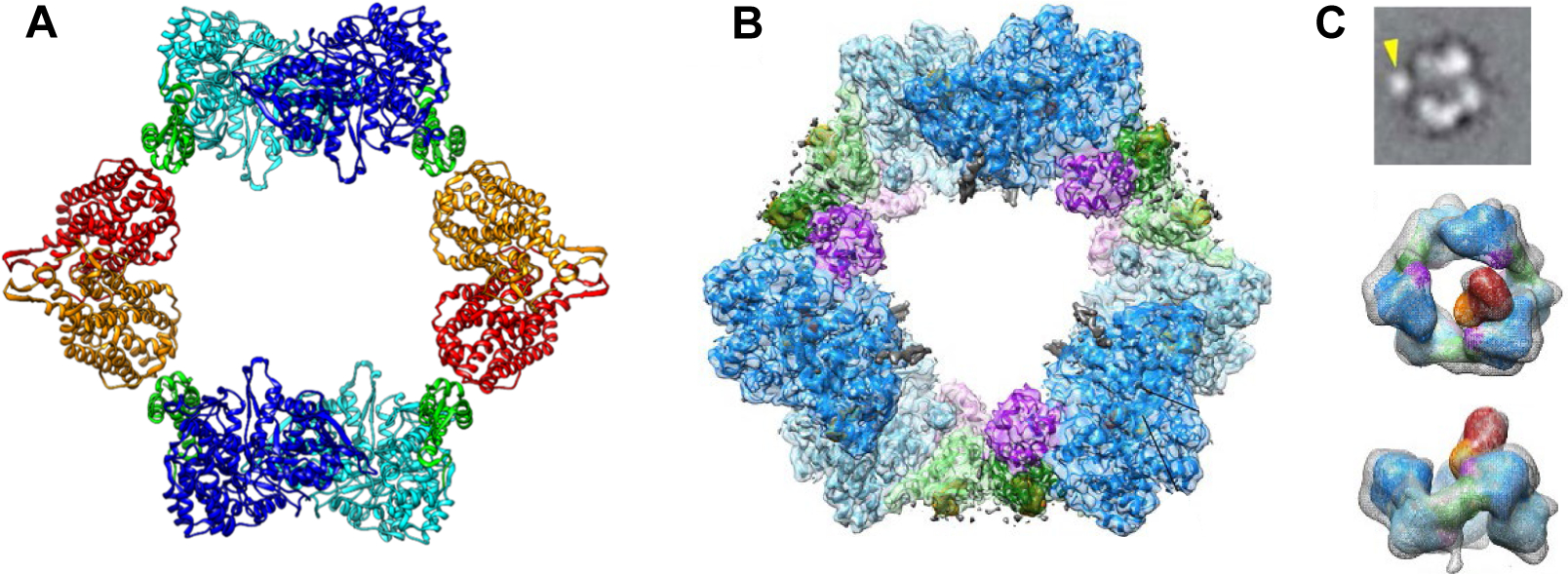
Structures of dATP inhibited states of class Ia RNRs. A X-ray structure of dATP inhibited E. coli class Ia RNR (29) is an α4β4 ring structure with a hole in the middle, composed of alternating α2 (light and dark blue with the cone domains in green) and β2 (orange and red) subunits. Note the importance of the cone domain in the α/β interaction. B Cryo-EM structure of dATP inhibited human class Ia RNR (27) is a hexameric α6 ring with a hole in the middle. A subunits are in light blue and dark blue with cone domain in light and dark green and a three-helix insertion in purple (residues 638–681). Note the importance of the cone domain in the α/α interactions. C) Cryo-EM structure of α/β (1:1)clofarabine triphosphate (ClFTP) inhibited human class Ia RNR (27). Top panel is a representative cryo -EM 2D class average image generated from α, β and clofarabine triphosphate (ClFTP) that shows β (arrow) interacting with α. The middle and bottom panels are two views of the 3D reconstruction of the same data set. The bottom is rotated 90° from the middle image. Only a fraction of the α6 rings in these images have a single and variably positioned β.
The structure of the dATP inhibited E. coli Ia RNR generated from α:β (1:1) in the presence of dATP is distinct from its eukaryotic counterparts. Drennan and collaborators using a variety of biophysical methods, reported an α4β4 ring complex with alternating α2 and β2s and a hole in the center (Figure 6A) (29, 30). In this structure, in contrast with the eukaryotic inhibited state, the cone domain (green) interacts with β2. The most intriguing result is that the distance between Y122•-β2 and C439 in α2 (Figure 2C) has increased from 35 to 60 Å, shutting down radical transfer and consequently nucleotide reduction. Thus, despite the distinct quaternary structures of the dATP-inhibited states, a common mechanism of inhibition emerges that involves inability of β2 to form an active α2β2 state.
Inhibited structures in vivo.
The presence of these inhibited states in cells (Figure 6) are important to establish. Drennan et al. used their structural insight from the E. coli α4β4 complex and site-directed mutagenesis to disrupt the α2-cone domain-β2 interface. Activity assays and negative stain EM analysis of several mutants showed that dATP no longer inhibited RNR and, that no α4β4 was detected (31). This study, in concert with genetic experiments on E. coli using a random mutagenesis protocol, a screen for altered dNTP pools, and genome sequencing, identified RNR with mutations at the same interface (32). The biochemical and genetic studies together suggest that the dATP-inhibited state of E. coli α4β4 occurs in vivo. Studies with clofarabine (ClF) (33) and other nucleoside therapeutic (cladribine (ClA) and fludarabine (FlU)) inhibitors of RNR (Figure 4B) (34) have demonstrated α6 formation in several human cell lines treated with sublethal doses of the nucleosides (35). The distinct inhibitory structures of the Ia RNRs (Figure 6) will be discussed further in the section on mechanism-based and reversible inhibitors as a new strategy to target RNRs.
Toward Active α2β2 structures.
Our studies using fluorinated tyrosine analogs (Figure 4A) combined with bioinformatics and the docking model of α2β2 (Figure 2C) to identify residues within the α/β subunit interface including E52 in β, led us to investigate the E. coli double mutant of β2 (E52Q and F3Y122•) (11). The F3Y122 substitution allowed trapping of the Y356• on the RT pathway (Figure 3) resulted in a tighter subunit affinity, Kd < 0.4 nM as compared with 0.2 μM for the control with E52Q-β2 (36). Although E52Q-β2 when incubated with any substrate and effector resulted in completely inactive RNR, incubation of the double mutant β2 (E52Q and F3Y122•) with α2 (or His6-α2), GDP, and TTP produced 0.5 equivalents of Y356• and 0.5 equivalents of dGDP consistent with half-sites reactivity (36). The resulting α2(E52Q and F3Y122• β2) complex gave rise to a near atomic resolution (3.3 to 5 Å) cryo-EM structure shown in Figure 2D which is asymmetric, consistent with the half-sites reactivity. In line with the biochemistry, the structure of α (left, Figure 2D) has generated a disulfide in the active site that we presume gives rise to the dGDP. With the α/β pair on the right side of the complex in Figure 2D, residues 341–375 in β are visualized for the first time (Figure 2A). In addition, GDP/TTP are apparent and the location of Y356 is finally revealed for the first time as part of the entire radical transfer pathway (Figures 2C and 3) (11). The details of this structure have recently been submitted for publication. Our ability to trap radicals at different residues within the pathway summarized in Table 1 and the increased subunit affinity observed under these conditions, suggests that this approach may lead to additional cryo-EM structures that will provide insight about the dynamics of this amazing machine.
New mechanistic insight about the chemistry of NDP reduction
Model for disulfide re-reduction and conformational gating.
In the E. coli RNR, the rate-limiting step(s) are physical (Figure 7 step B, E and G), involving conformational changes that mask the chemistry of long-range reversible RT and dNDP formation (22, 37). The observed rate constants (kcat) for dNDP formation in the absence of an external reductant (steps A through E), range from 2 to 5–10 s−1 with substrate and substrate/effector, respectively. In the presence of the physiological reductants (in cells or in steady state assays), additional conformational changes become rate limiting (1–2 s−1, step H) and likely involve either α/β subunit dissociation, conformational changes associated with the re-reduction of the active site disulfide by the C-terminal tail of α (steps F, G, H), or both, and are protein concentration dependent. Different αs have distinct cysteine configurations within their C-terminal tails (red balls in the C-terminal tails in Figure 2B) and require organism-specific reductants (e.g. thioredoxin (TR), NrdH, glutaredoxin (Grx), and thioredoxin reductase (TRR) and glutaredoxin reductase) (38–41).
Figure 7.
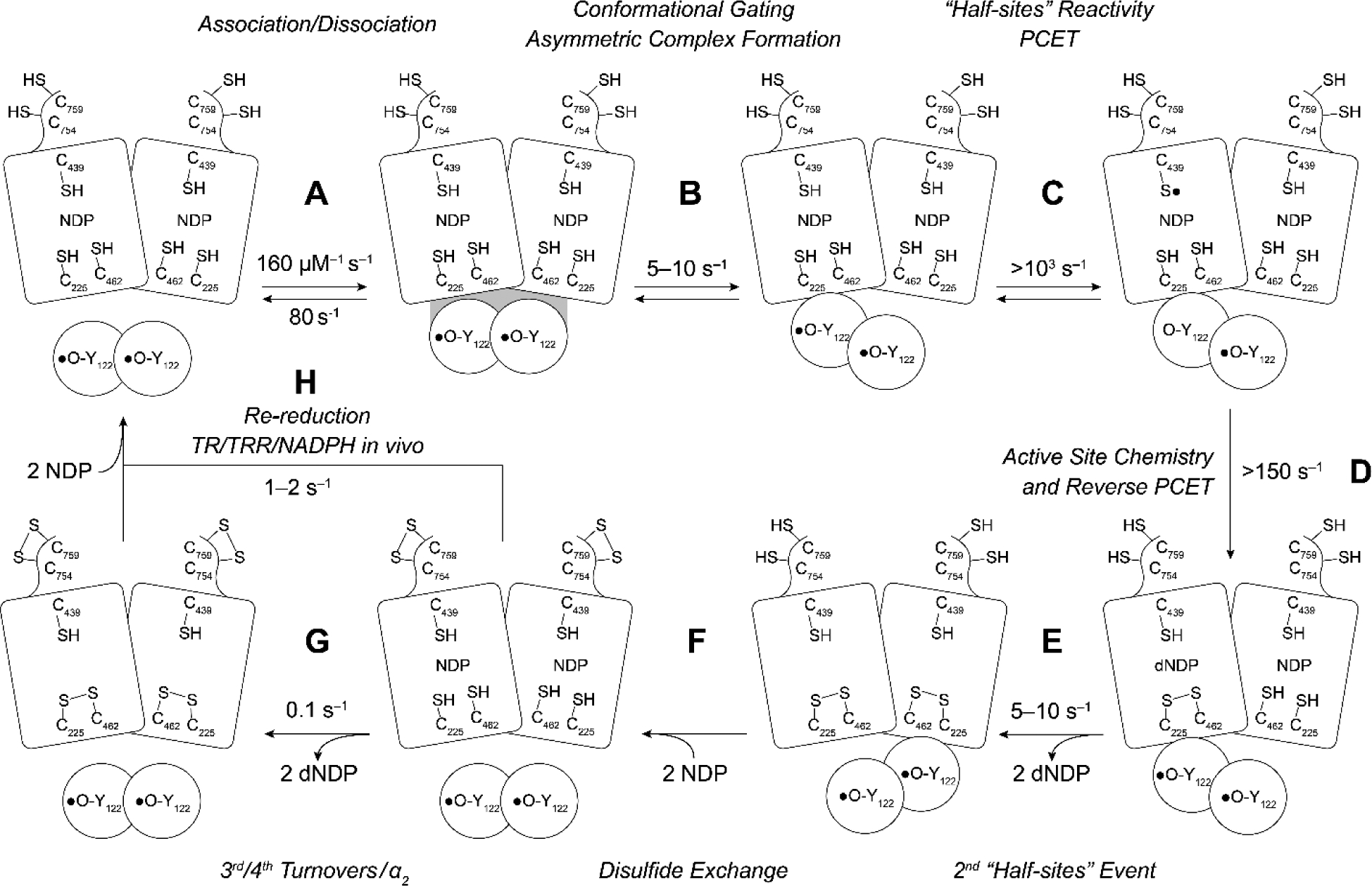
Resetting RNR for single and multiple turnovers. The model assumes a 1:1 α to β ratio, that Y122• is distributed equally between each β, and that the wt-α2β2 complex is asymmetric. In the absence of external reductants, 2 dCDPs are generated at 2 (substrate only) to 5–10 s−1 (substrate and effector) that arise from chemistry at each α of α2 (step E). Steps B and E are rate-limiting and conformationally gated. In an assay in the absence of an external reductant, two additional dCDPs are formed at 0.1 s−1 (step G). In the presence of an external reductant such as thioredoxin (TR) and TR reductase (TRR), under steady state conditions, step H becomes rate-limiting.
To unmask the mechanistic details of dNDP formation, multiple methods have been employed including site-directed mutagenesis, insertion of unnatural amino acids (Figure 4A), photosensitized RNRs (photo-β2), and MBIs (Figure 4B). In subsequent sections, we describe how this information combined with structural studies have provided insight into new therapeutic strategies.
NDP reduction mechanism.
Our current mechanism for nucleotide reduction is shown in Figure 8 (5, 43). The important features are that a −S• (C439, E. coli) initiates the reduction by removal of the NDP 3′-H (step A) (44). E441 facilitates this step by functioning as a base catalyst for 3′-OH deprotonation (1 to 2) (45). This reaction is driven to the right by the rapid, irreversible loss of water catalyzed by C225 (step B). The proposal for the reductive half-reaction (steps C and D) is that the 3′-keto-2′-radical [3] is reduced by PCET to generate the 3′-ketodeoxynucleotide and the three electron, disulfide radical anion [4]. This species then reduces the 3′-ketone by another PCET step (step D) where the proton is supplied by the protonated E441. In the last step, the H-atom abstracted from the 3′ position of NDP is returned to the same position to form dNDP and the C439•, that reoxidizes Y122 in β2 on each turnover.
Figure 8.
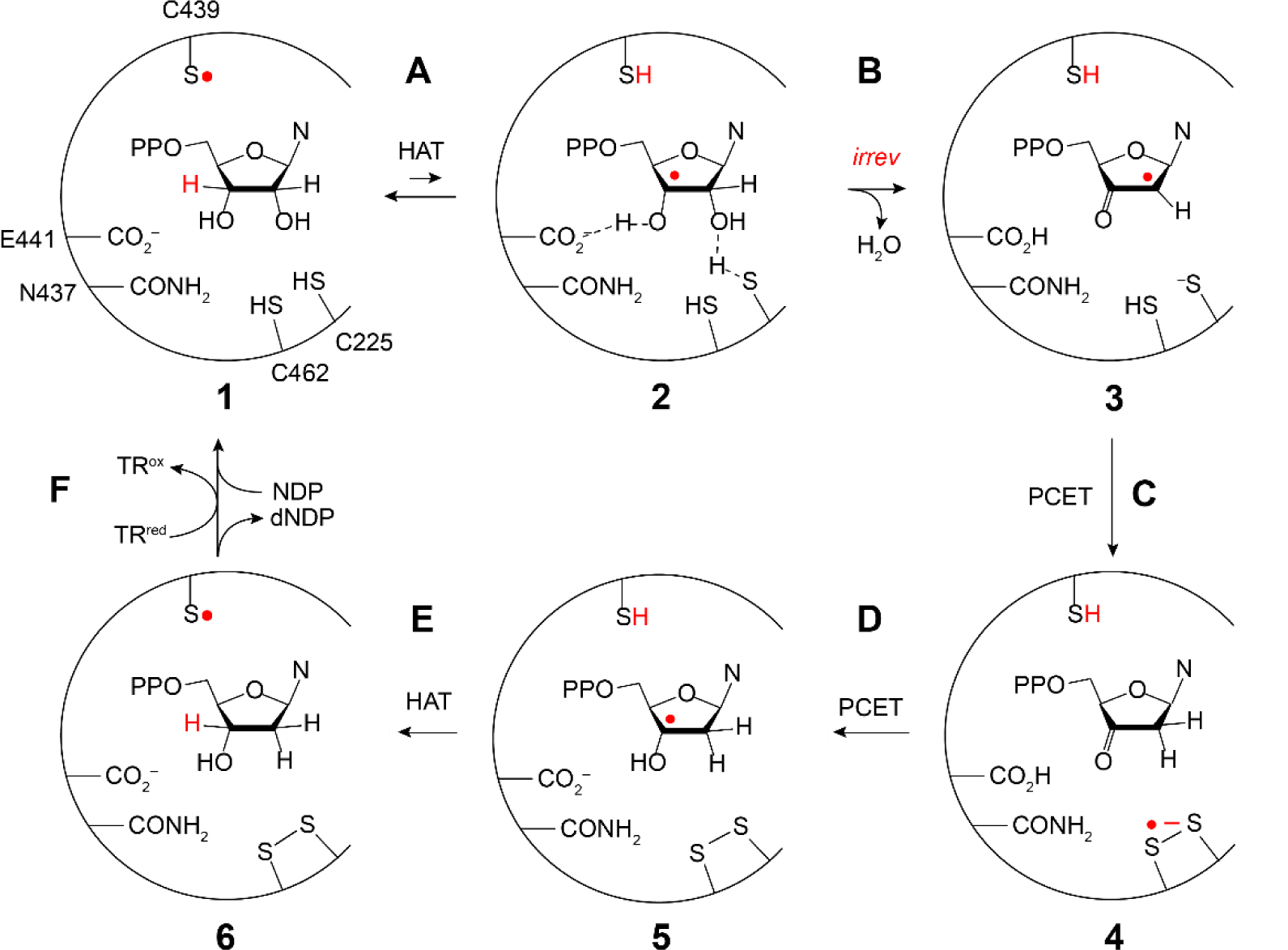
Mechanism of −S• mediated NDP reduction by most RNRs (5). Steps A–F are the proposed steps in the reaction and 1–6 represent the active site participants in each step. Note that steps A, C, D, and E involve either hydrogen atom transfer (HAT) or proton coupled electron transfer (PCET) processes.
Role of multiple thiyl radicals.
The role of the −S• initiator (C439, Figure 8) was previously established based on studies with the class II, adenosylcobalamin-dependent ribonucleotide triphosphate reductase. An exchange coupled −S•-cob(II)alamin species, detected by EPR and UV-vis absorption stopped flow spectroscopies, was shown to be chemically and kinetically competent in deoxynucleotide formation (46, 47). The structural homology and conserved residues in the active site of all RNRs (Figure 1B) have thus been used to infer the universal involvement of −S• in initiating 3′-H atom abstraction (16). Support for −S• involvement in the reductive half-reaction (Figure 8 step C and D) comes from studies with E441Q-α/β/CDP/TTP where a disulfide radical anion was spectroscopically identified due to the absence of a required proton from E441 for the PCET step D (48). Although −S• chemistry has been proposed for many enzymatic reactions, RNR is the only enzymatic system where this intermediate has been detected (46, 47).
PhotoRNRs unmask rate constants for NDP reduction chemistry.
The development of methods to uncouple conformational gating and unmask chemistry has allowed unprecedented insight into the active site chemistry, including thiyl radical mediated hydrogen atom abstraction (Figure 8, step A) (49, 50) and the subsequent rate-limiting 3′-ketodeoxynucleotide reduction (step D, 4 to 5) (23,26). In the former case, we designed a method for photosensitization of RNR (Figure 9A) in which a photooxidant, bromomethylpyridyl rhenium(I) tricarbonyl phenanthroline ([Re]), is covalently attached to a single surface-exposed cysteine in S355C-β2 mutant; the Y122• in β2 is reduced and Y356 in β2 is replaced with a fluorinated tyrosine (FnY356, Figure 4A). This photoβ2 in complex with α2, substrate, and effector, can be rapidly (ns) oxidized to a FnY356•-β2 state upon illumination (Figure 9B). The photochemically generated radical rapidly equilibrates with the radical transfer pathway in α2, ultimately oxidizing C439, initiating cleavage of the 3′ C-H bond of NDP. Comparison of the FnY356• decay, observed by transient absorption spectroscopy, in the presence of 3′-[1(2)H]-CDP established a lower limit for the −S• mediated H-atom abstraction (step A) of 1.3 × 104 s−1 and an isotope effect of ≥7 (51)! Note that the kcat for RNR is 2–10 s−1. The RT chemistry is thus very fast (!) and unmasked for the first time using this method.
Figure 9.
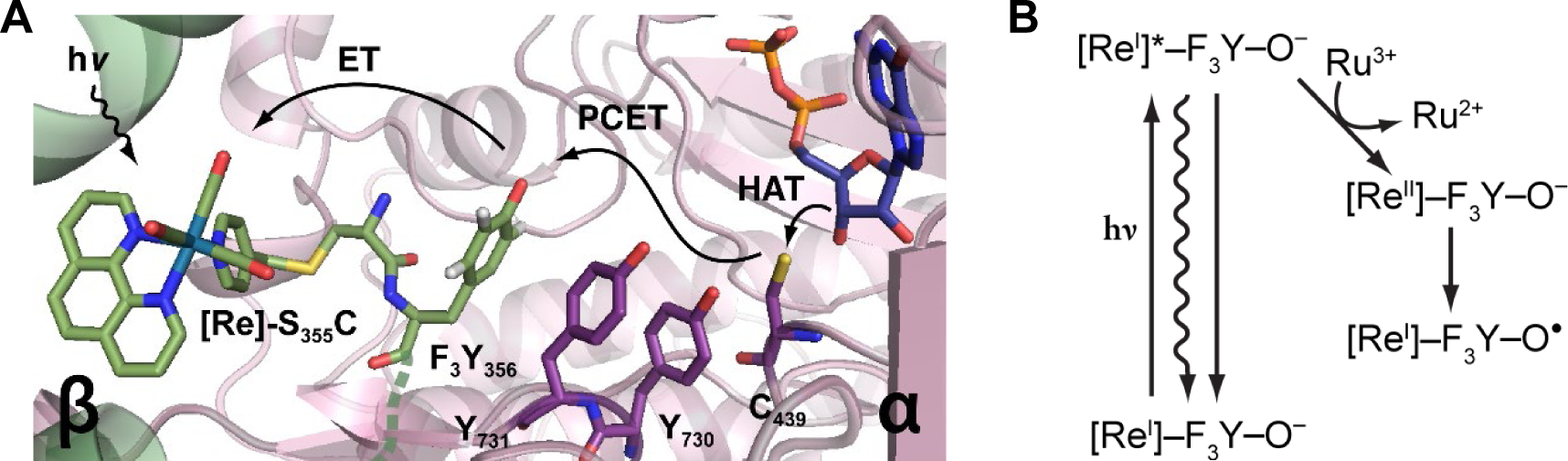
Use of photoRNRs to unmask rates of chemical steps. A Schematic of photoβ2 with rhenium photooxidant [Re] attached covalently to C355 and Y122OH-β2 (that is, Y122• is reduced) complexed with CDP, ATP, and α2 (49). In the case shown, Y356 in β is replaced with the unnatural amino acid 2,3,5-F3Y356. (Figure 4). B Light initiates the reaction and the presence of the flash quencher, Ru(NH3)6Cl3, prevents charge recombination (left) and generates [ReII]-F3Y-O− (right), that rapidly drives 2,3,5-F3Y356 oxidation to the 3,5-F3Y356• that initiates chemistry within α2. Different mechanisms of oxidation shown are electron transfer (ET), PCET, proton coupled electron transfer, and HAT, hydrogen atom transfer.
The subsequent rate-limiting 3′-ketodeoxynucleotide reduction (step D, Figure 8) has been examined by incorporating tyrosine analogs with altered reduction potentials in place of Y122 in β. Use of these “hotter” oxidants drives radical transfer and also uncouples conformational gating. Specifically, F3Y122• and NO2Y122• (vida supra) have higher reduction potentials than the native Y122•(β2) by 80 and >200 mV, respectively (23, 26), as determined from the independent measurements of formal reduction potentials in the small 3-helix bundle protein (24). These β2 mutants have been studied in an effort to observe the slow step(s) within the proposed chemistry, specifically the PCET reduction of the 3′-ketone by the disulfide radical anion (Figure 8, 4 to 5, step D). When NO2Y122•-β2 (or F3Y122•-β2) is mixed with α2/CDP/ATP, dCDP formation occurred at ~150 s−1 (30 s−1). This rate constant is similar to the 50 s−1 measured for dCTP formation; the latter is catalyzed by class II RNR (52) where the active site chemistry is not masked by physical steps and is much faster than the wt turnover of 1–2 s−1.
MBIs and reversible inhibitors to understand mechanism and design of new therapeutics.
The MBIs 2′-halo (X)-2′-deoxyNDPs (XNDP, X = Cl, F; Figures 4B irreversible and Figure 10) have played a pivotal role in our current understanding of the mechanism of nucleotide reduction (53). In 1976 Thelander and Eckstein et al. reported that ClCDP incubated with E. coli RNR resulted in time-dependent release of Cl− and cytosine, and that the α subunit was inactivated (54). These observations provided the impetus for studies using labeled nucleotide analogs, which led to the general model for inhibition shown in Figure 10. As with the NDP substrate, the −S• abstracts the 3′-H (step A) to generate 2. The outcome of the reaction depends on whether and how the loss of X at 2′-C is catalyzed by the enzyme. From 3, the 2′-delocalized radical can be reduced from the top face by H-atom transfer mediated by C439 (red H) or the bottom face (blue H) facilitated by C225. With Cl(F)NDP, a 3′-ketodeoxynucleotide is generated (7) that dissociates from the active site (when X is not protonated). Intermediate 7 can decompose on a minute time scale to nucleic acid base, pyrophosphate (PPi), and a furanone (8) that non-specifically alkylates the α subunit. If the reduction of 3 is by C439, then reverse RT can effectively regenerate the Y122• in β2. However, if reduction occurs from the bottom face, Y122 remains reduced and β2 is inactivated. Thus, α and/or β can be inactivated via distinct mechanisms. With both XNDPs (X = Cl, F), if X is protonated (XH, step B above arrow), then dNDP is formed. The details of RNR inactivation in vitro and in vivo depend on the identities of substrate and effector, leaving group (X), and the reductant. In all cases, α inactivation requires Y122• reduction. These inhibitors inactivate class I, II, and III RNRs by a common mechanism suggesting similar active sites (Figure 1B). The involvement of the α C-terminal cysteines in enzyme inhibition (Figure 2B, red balls) is not well understood as their covalent linkage to 8 is reversible, precluding isolation and characterization of alkylated α.
Figure 10.
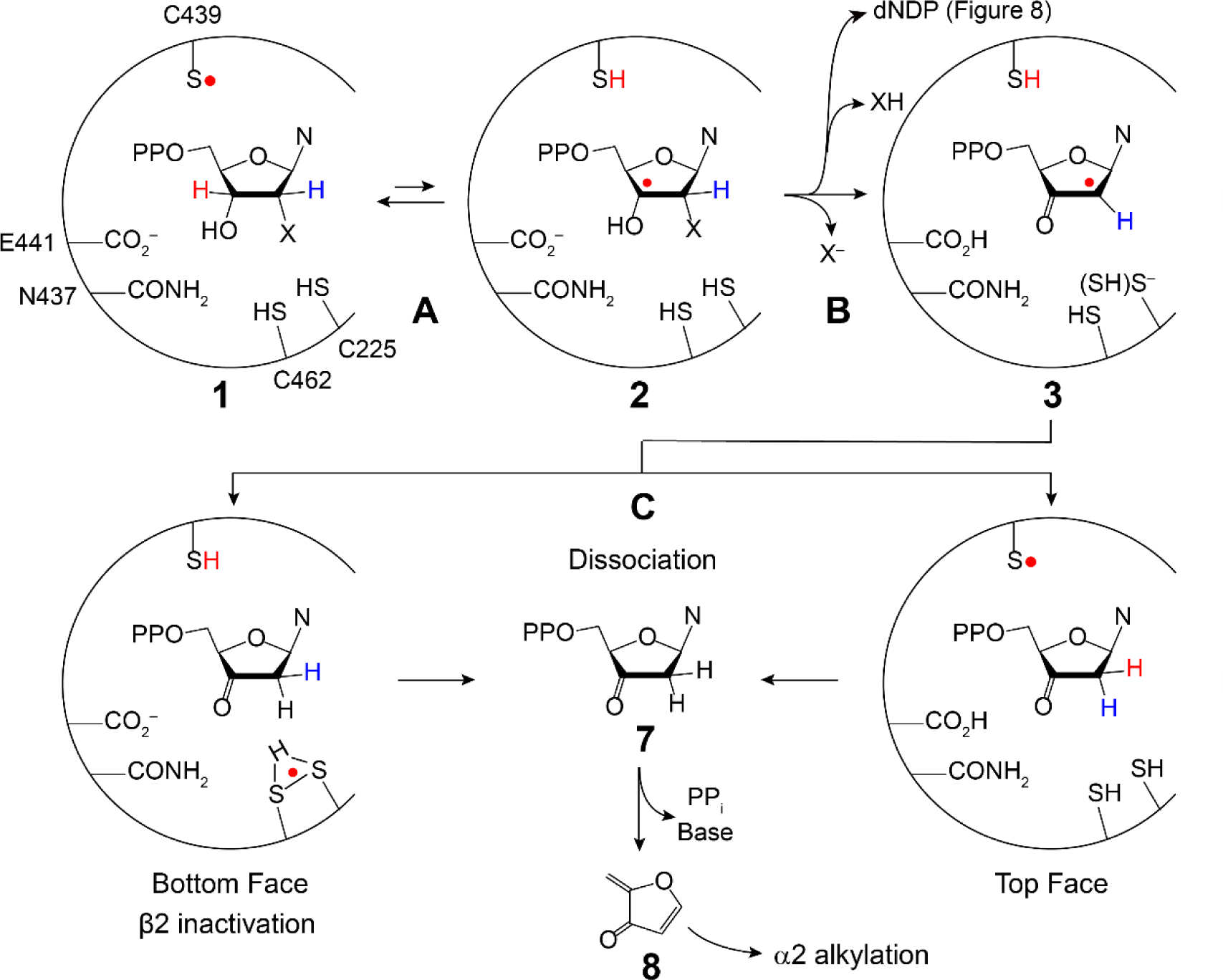
Generic mechanism for 2′ X-dNDP (X = F, Cl, N3, F2 (Figure 4)) mechanism-based inhibition of RNRs with loss of X− in step B (43, 53). 3 is formed with bottom face protonation states of the thiols unknown, which can be reduced from the top face by SH to produce 7 (right) or the bottom face to produce 7 (left) which dissociates from the active site and then decomposes to generate the products (PPi. Base and 8). 8 can alkylate the α subunit. Alternatively, if in conversion of 2 to 3 (step B), XH is eliminated, and the normal product dNDP is formed.
In contrast to XNDPs (X = Cl, F), there are a number of MBIs (X = N3, F2, VF, Figure 4B) (55–57) that share similar chemistry in steps A and B (Figure 10), but then undergo distinct chemistry controlled by X and the active site cavity. Unraveling the mechanism by which N3NDP inactivates all RNRs defined the strategy to study the mechanism of action of the clinically used nucleoside therapeutics, gemcitabine (F2C) and clofarabine (ClF). F2CDP is an irreversible inhibitor (58) and, in contrast to expectations, ClFDP and ClFTP are reversible, non-covalent inhibitors (Figure 4B) (59–61).
2′-Azido-2′-deoxynucleotide (N3NDP).
N3NDPs (N = C (shown in Figure 4B), U or A) are substoichiometric MBIs that were also first reported by Thelander and Eckstein (54). Extensive studies with N3UDP revealed that its incubation with α2β2 resulted in rapid loss of ~90% RNR activity concomitant with loss of only 0.5 equivalents of Y122•. The Y• loss was biphasic with the fast phase accompanied by formation of a nucleotide-based nitrogen centered radical (N•) derived from the N3 moiety that was structurally identified using isotopically labeled N3UDPs and EPR methods (Figure 11) (62, 63). The N• species then slowly decomposes to form nucleoside base (blue N in Figure 11), PPi, and 8. The α/β subunits dissociate, and subsequent to α2β2 complex reformation, more Y• is lost and N• formed. While these observations were perplexing at the time, the many recent examples of “half-site” reactivity (Table 1) and RNR asymmetry (Figure 2D), now place these observations on firm footing.
Figure 11.
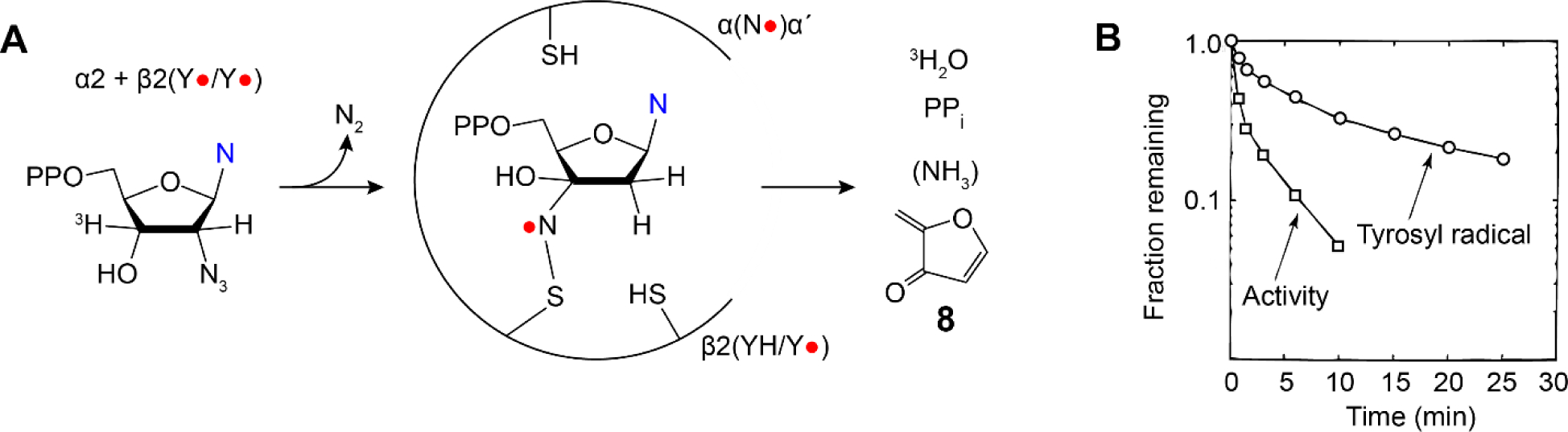
N3NDP (Figure 4B) is a potent inhibitor of all Ia RNRs (62,63). Studies of this inhibitor provided a glimpse of unprecedented chemistry associated with reactive radical species in an active site cavity and the challenges associated with radical structure elucidation. It also provided the first evidence for half-sites reactivity.
In vitro N3CDP inhibits β2 by reduction of the essential Y122• whereas in vivo, the nucleoside analog N3C is not cytotoxic. In cells, N3C is not readily phosphorylated to N3CMP by deoxycytidine kinase, presaging the importance of the specificity of kinases in generation nucleotide (di and triphosphate) therapeutics.
Gemcitabine (F2C) and clofarabine (ClF), clinically used nucleoside therapeutics that inhibit human RNR.
F2C and ClF are used clinically as cancer therapeutics (Figure 4B). F2C targets a broad spectrum of solid tumors (pancreatic, metastatic breast, lung) and hematological cancers. In the clinic, this compound is used in combination with DNA damaging agents such as cis-platin or small molecule inhibitors of signaling pathways that affect the cells response to DNA replication stress (3, 64–66). ClF is limited to hematological cancers (AML, ALL). Both agents inhibit DNA synthesis. RNR is the upstream target of the diphosphate forms of these compounds (F2CDP, ClFDP), whose inhibition alters dNTP pools. Additionally, the triphosphate forms of these compounds (F2CTP, ClFTP) inhibit DNA polymerases by incorporation into DNA. The mechanisms, however, by which these compounds inhibit RNR and DNA synthesis, are distinct.
F2CDP.
F2C was synthesized independently by research groups at Merrill Dow and Lilly (58, 67). Studies from Plunkett and the Lilly group demonstrated that F2C inhibited growth of a variety of tumor cell lines and that the cytotoxicity resulted from inhibition of multiple targets including DNA polymerases and RNR (67–69). Biochemical studies on E. coli and human RNR established that F2CDP is a time-dependent irreversible inhibitor and that inactivation occurs with one equivalent per α2 subunit. Studies using isotopically labeled F2CDPs established that the products of the inactivation were distinct depending on whether they were carried out in the presence or in the absence of reductant (70–72).
In the presence of reductant (TR/TRR or DTT), 2F−, cytosine, PPi and one alkylated α-cysteine (C225) per α2 were identified and no Y122• in β2 was lost. Under these conditions, while only 0.5 equivalents of α is inactivated and β remains active, all enzymatic activity is lost. Analysis of the inhibited reaction mixture by SDS-PAGE with no heating revealed that α migrated as a 60:40 ratio of 80 kDa (endogenous α molecular weight) to 110 kDa (modified α) (Figure 12A and B). In cancer cell lines incubated with F2C, SDS PAGE analysis of cell lysate revealed that the α subunit also migrated in a 60:40 ratio (Figure 12B), similar to the in vitro studies (73). Similar experiments in the absence of reductant, resulted in 50% loss of the β2-Y122• and formation of an equivalent amount of a new, nucleotide-based radical (structure shown in Figure 12C). This radical slowly breaks down to cytosine and PPi. Inhibition was accompanied by loss of 2F−, but the α subunit was not covalently labeled. Thus, both in the presence and absence of reductant, 1 F2CDP/α2 is sufficient for inhibition, although the underlying mechanisms of inactivation are distinct.
Figure 12.
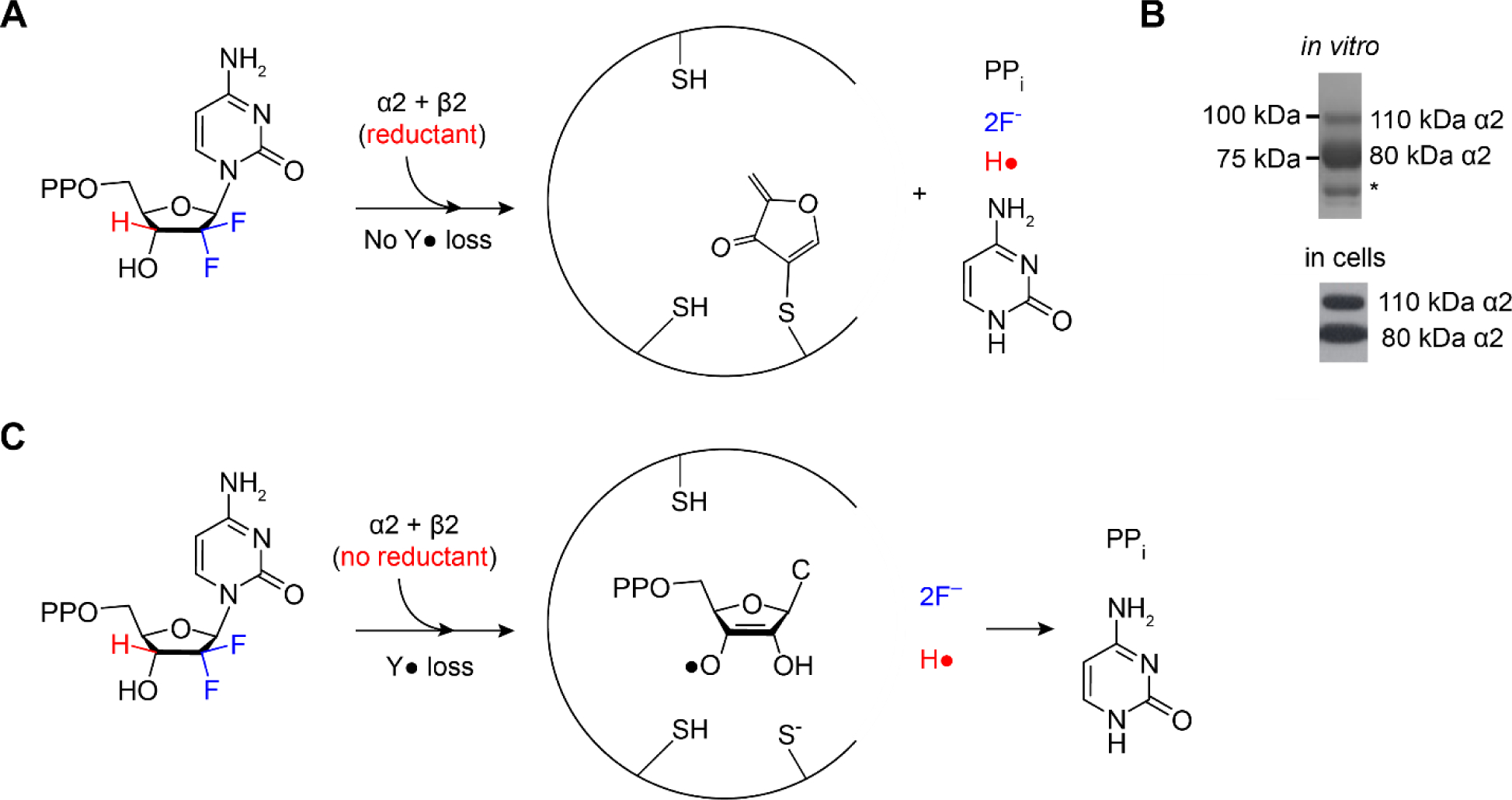
Products of RNR inhibition experiments depend on whether reductant is used. A F2CDP is a MBI of E. coli and human RNRs in the presence of reductant (protein TR or dithiothreitol) in vitro (70–72), B in cell lines (73), and C in the absence of reductant in vitro. A and C show the products produced during RNR inactivation. B shows SDS-PAGE of RNR from studies in vitro and cell lysate without boiling after incubation with F2CDP (in vitro) or F2C (in cells). Wt-α (80 kDa, C), migrates as 110 kDa.
To account for the complete inactivation of RNR with only 0.5 equivalents F2CDP/α, we proposed that the α/β subunit affinity increased and switching to the second α/β pair for additional chemistry is prevented. To test this possibility, inactivated E. coli and human RNR were subjected to size exclusion chromatography (SEC) analysis. The former showed a species consistent with an apparent molecular weight for α2β2 and the latter with α6β6 (74). In the control, in the absence of F2CDP, the subunits separate with β eluting as a dimer and α as a mixture of monomers and dimers consistent with weak subunit interactions. Based on our recent EM analyses of a mixture of α and β (1:1) with ClFTP (Figure 6C and the next section) (27), the size with human-RNR attributed to α6β6 is not possible. Altered molecular weights using SEC analysis can be attributed to unusual, non-globular, shapes (α6β2) or altered quaternary structure(s). Fibril structures for example have been reported with human α and ATP (27) and with Bacillus subtilis class Ib (α/β) RNR (75).
ClFDP and ClFTP (reversible).
Early studies by Plunkett et al. established the toxicity of ClF towards many cell lines (CEM, K562, Hep2). In cell-free systems, ClFTP inhibits RNR and DNA polymerase α and ε (59, 60, 66, 76). The observation that the ClFTP:ClFDP ratio in some cells was 7:1, led to the proposal that ClFTP was a reversible inhibitor of ATP binding to the A-site of α (Figure 2B, cone domain (green)). To better understand how RNR is targeted, kinetic and biochemical studies were undertaken with both ClFDP and ClFTP (61). ClFDP was shown to be a reversible, time-dependent, slow binding, inhibitor to the C-site. The kinetic analysis revealed a two-step binding mechanism with a KI* of 17 nM. ClFTP exhibits reversible, time-independent A-site binding. With ClFTP in 5-fold excess relative to RNR under physiological conditions, RNR activity was rapidly and completely lost with a KI of 40 nM. With sample dilution and follow-up assays, enzyme activity was recovered over 30 min but only to 50% of the initial value. The t1/2 of the human Y• in β2 is 30 min at 37 °C (61) and the α subunit is prone to oxidation, making the kinetic measurements challenging; further studies are required. To determine if the observed inhibition was associated with changes in RNR’s quaternary structure, studies of ClFTP (ClFDP) with α, with and without allosteric effector (dGTP), were each examined by SEC. In the absence of the corresponding nucleotide in the elution buffer, α migrated as α6 in the presence of either ClFTP and ClFDP. This result is distinct from dATP-α6 When dATP is absent from the elution buffer during SEC, the hexamer rapidly reverts to α monomer. Thus, the presence of ClFDP or ClFTP alters α’s quaternary structure such that, even subsequent to ClFD(T)P dissociation, α6 remains trapped in the inactive state! β2 had no effect on the inhibition or migration on SEC analysis. Structures of ClFTP mixed with human α/β (1:1) were examined by cryo-EM and solved to 30-Å resolution (Figure 6C). Less than 1 β2 per 3 α2 was observed and it appeared randomly positioned on the exterior or on top of a hexameric ring structure (Figure 6C) (27). In support of this model, experiments with D57N-α, where the mutation in the cone domain prevents hexamerization of α, revealed that neither ClFD(T)P treated mutant-RNR, nor ClF treated cells with mutant RNR were inhibited. Finally, E. coli RNR, which does not form α6 structures, is not inhibited by ClFDP (61).
The dynamics of quaternary structure interconversions offer an opportunity to inhibit RNRs through unconventional mechanisms. The flexible cone domains (Figure 2, Figure 6A and B) (27, 30) play critical, but distinct roles in these states. Strengthening or weakening the interactions responsible for these quaternary structures with small molecules could alter RNR activity.
To assess the importance of the hexameric state of human-RNR, studies were carried out with His6-α expressed at 3 or 30× endogenous levels in COS cells that were then treated with noncytotoxic levels of ClF for 3 h. Analysis of the 30× material purified by Ni-affinity chromatography revealed the α6 state was present; with 3× endogenous levels crosslinking was required to detect α6. The α6 state from these and other studies is likely the inhibited state inside the cell in the presence of ClFTP and dATP (33).
A recent extension of this strategy to other adenosine analog therapeutics, cladribine (ClA) and fludarabine (FlU) (Figure 4B), was reported (34). In vitro studies of ClADP and ClATP interactions with human-α revealed α6 formation. Further assessment of the hexameric structures and their relationship to cell cytotoxicity is an ongoing challenge. Collectively, the results in cells and in vitro with these adenosine inhibitors suggest a potential new way to target RNR: trapping α in an inhibited state with a small molecule.
Pleiotropic modes of cytotoxicity of F2C and ClF.
With both F2C and ClF, the mechanisms of cytotoxicity require nucleoside uptake and metabolism (64, 65, 77). As noted above, the diphosphates and triphosphates of F2C and ClF inhibit RNR and DNA polymerases, respectively (60, 78), the latter by chain termination. The consequences of DNA inhibition involving both targets are DNA replication stress that manifests as stalled or collapsed DNA replication forks, DNA single or strand breaks, which can lead to cell cycle arrest, DNA repair or programmed cell death (64) (see Figure 13).
Figure 13.
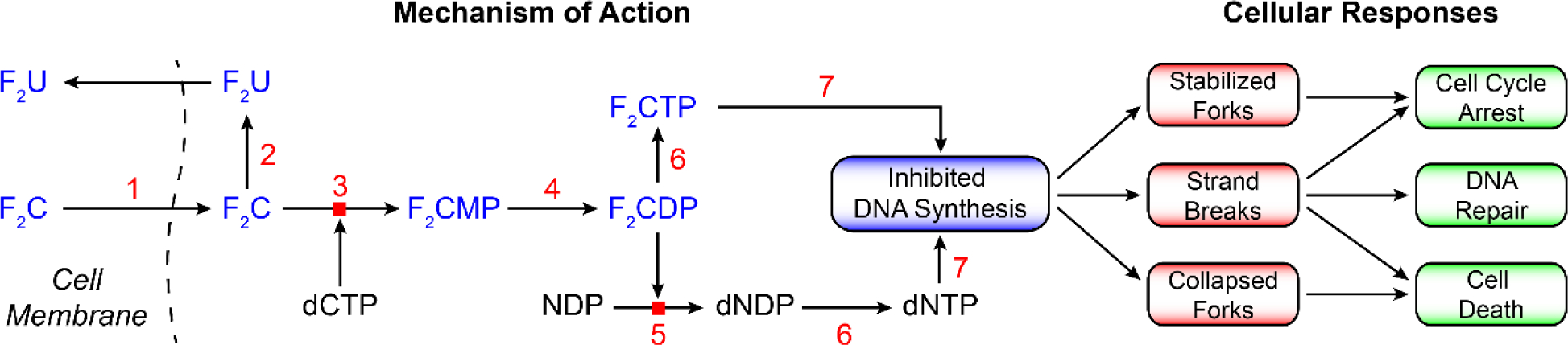
A general scheme for metabolism of nucleosides using F2C as an example (64). F2C and ClF therapeutics require cellular uptake and phosphorylation to the appropriate state recognized by target enzymes. The former is mediated by nucleoside transporters ENT1, ENT2, and CNT [1]. Once inside the cell, both F2C and ClF are phosphorylated to the monophosphate by deoxycytidine kinase [3] and subsequently to the di and triphosphates by cellular kinases [4 and 6]. Deoxycytidine kinase has unusual specificity in that it phosphorylates both pyrimidines and purines. The concentrations of the monophosphates are in general > than the triphosphates ≫ than the diphosphates, are cell type distinct, and influence therapeutic outcomes. [5] is RNR, [7] is DNA polymerase, and [2] is cytidine deaminase.
F2CDP, a potent MBI of RNR, results in lower dNDP and consequently dNTP pools. Reduced dCTP, a feedback inhibitor of deoxycytidine kinase (3, Figure 13), results in enhanced production of F2CMP, leading to elevated levels of F2CTP. The F2CTP is then able to more effectively compete with lowered dNTPs pools to inhibit DNA synthesis. F2C’s broad spectrum of solid tumor inhibition, distinct from other nucleoside therapeutics such as araC, may be associated with the pleiotropic metabolic effects (Figure 13) resulting in its self-potentiation (69).
ClF is also phosphorylated by (3, Figure 13) and subsequently by distinct kinases to afford ClFDP and ClFTP. Its stability (due to F/Cl substitution) is increased relative to other adenosine analogs (ClA, FlU, Figure 4B) by its resistance to metabolism by purine nucleoside phosphorylase and adenosine deaminase. Down-stream consequences of DNA synthesis inhibition by F2C and ClF are actively being pursued. F2C is being investigated in combination with DNA damage response inhibitors of Chk1 (64, 68, 79, 80), with inhibitors of ATR in the same pathway (81), and with DNA repair enzyme inhibitors (65, 82). In addition, F2C is often used in combination with cis-platin that enhances DNA damage and alters the downstream consequences. The ability to monitor the consequences of treatment with combinations of therapeutics using genomics, phospho-transcriptomics and metabolomics, have and will continue to aid in new approaches (65, 83).
Reversible C-site binders lacking phosphoryl groups.
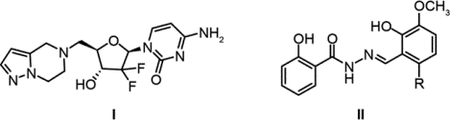
Two compounds (I and II) have recently been reported to inhibit human RNR by binding reversibly to the C-site of α. In contrast to ClFDP and ClADP (Figure 4B), these small molecules lack the diphosphate moiety thought to be essential for substrate recognition. A 5′-substituted amine of F2C (I), for example, is reported to inhibit RNR in vitro and in vivo (85). The unusual diphosphate binding site for NDP in α (no lysine, arginine or Mg2+) suggests that amine substitution might avoid issues associated with cellular uptake and phosphorylation (Figure 13). In a second case, Dealwis et al., reported molecules with a naphthyl salicyl hydrazone scaffold (II) that target the C-site of α and bind reversibly (86, 87). Decorating the scaffold, with an appropriately placed electrophilic moiety such as ClCH2CO-(R″), could result in alkylation of one of the C-site cysteines, analogous to the mechanisms of covalent protein kinase inhibitors (88).
Reversible inhibitors that disrupt α/β subunit affinity.
The C-terminal tails (30 to 35 amino acids) of all β2 subunits are disordered (Figure 2A) (14, 89–93), distinct, and predominantly responsible for subunit affinity (Figure 14). Early studies of Herpes simplex viruses (HSV-1, HSV-2), which encode for their own RNRs, provide an example of this approach (94, 95). Peptidomimetics of their tail were successfully developed that disrupted the α/β subunit interaction in vitro and in a murine ocular model of HSV-1 induced keratitis (96). The new structure of the E. coli α2β2 (Figure 2D) , which reveals for the first time the tail interaction (residues 341 to 375) with the α subunit (11), may suggest new approaches to disruption of this interface.
Figure 14.
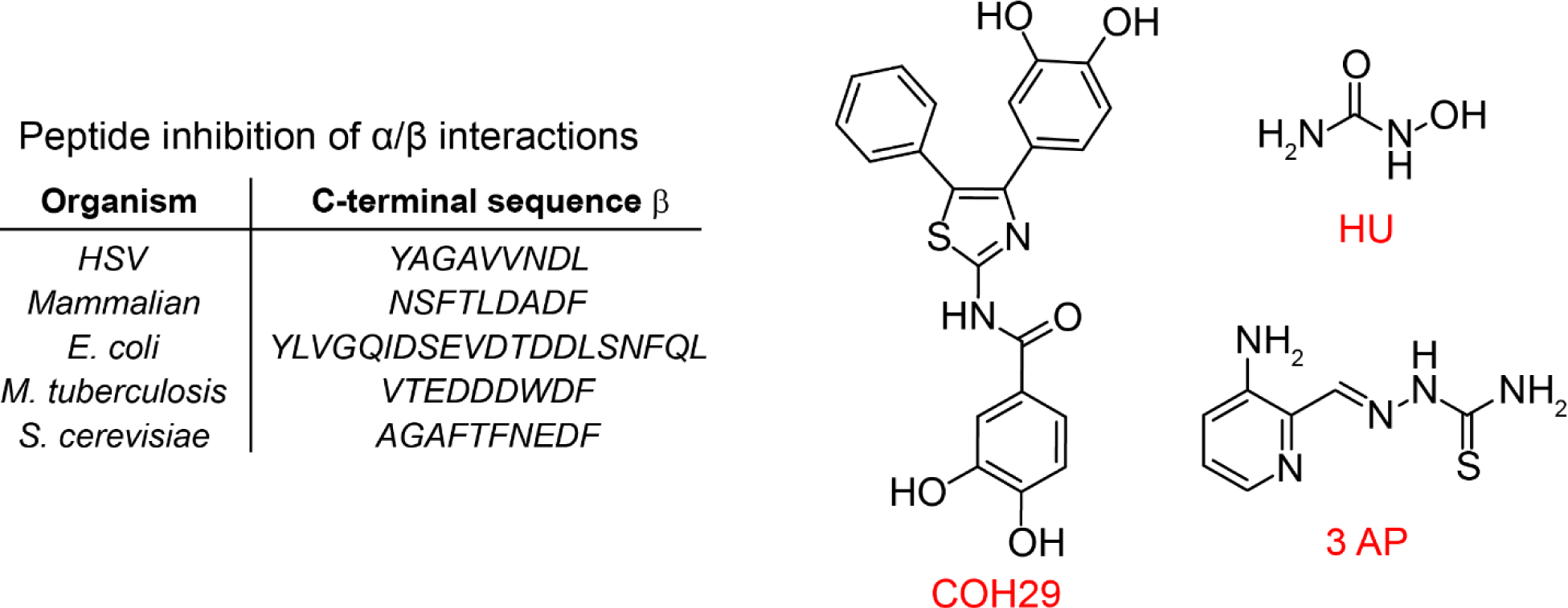
Targeting the α/β interface of active RNR to prevent active complex formation with peptidomimetics and COH29. Targeting formation and repair of Me23+-Y• (Me is Fe)-cofactor of β2 with HU and triapine, 3-AP.
A second example of subunit disruption was reported by Yen’s group (82, 97). They used the structure of human-p53β2 and computer modeling to identify a pocket in each β subunit close to the C-terminal tail but removed from the buried Fe3+2-Y• cluster (Figure 1C) essential for β2 stability. Virtual screening and additional experiments led to the identification of COH29 (Figure 14) that exhibited cytotoxicity to many of the NIH 60 cancer cell lines and caused S-phase cell cycle arrest. COH29 enhanced cytotoxicity of BRAC1 deficient HCC1937 cells. This report provides an example of DNA repair inhibition (98) (in this case genetically) that potentiated the effects of the RNR inhibitor COH29.
Biosynthesis and repair of the essential diferric-Y• cofactor of Ia RNRs: targeting the β2 cofactor.
Whereas ClF and F2C target α and the α/β subunits of RNR, respectively, hydroxyurea (HU) and triapine (3-AP, a thiosemicarbazone) (Figure 14) target reduction of the essential Y• (83, 99, 100) cofactor in β2 and/or interfere with cofactor assembly and/or its repair if the essential Y• gets reduced (Figure 1C). HU is used clinically, predominantly in combination with other therapeutics (65), although recent studies suggest that RNR is not a key target of its cytotoxicity (99, 101, 102). Triapine (3-AP) continues to be examined in clinical trials, but has not yet been approved for clinical use (83, 100). Although the upstream target of both these compounds is RNR, the downstream pathways that lead to cytotoxicity are pleiotropic and distinct in different organisms. Herein we focus on HU and 3-AP inhibition of RNR in vitro and in the early stage of cell culture where cell viability remains high. Even under these conditions, their detailed mechanism(s) of RNR inhibition require further exploration.
Background for metallo-cluster metabolism.
The class Ia RNRs require a Fe3+2-Y• cofactor in β2 to initiate NDP reduction in α2 with activity being directly proportional to the concentration of Y• (Figure 1C and Figure 3). The t1/2 of the Y• in the cluster of different Ia β2s is variable, ranging from 4 days in E. coli at 4° C to 30 min in humans at 37 °C. In addition, recombinant expression of β from different organisms results in variable amounts of active cofactor (0 to 1 Fe3+2-Y•/ β2) (6, 103). In general, therefore, the β2 cofactor must be loaded by self-assembly using Fe2+and O2 with variable outcomes (6, 104). In the past two decades, the importance of biosynthetic pathways has been established for FeS cluster cofactor assembly that, in turn, has been linked to mono- and dinuclear non-heme iron cofactor formation including the RNR cofactor (105). Although much remains to be learned, genetic studies in E. coli and S. cerevisiae, and biochemical studies in vitro on these Ia β2s, have suggested that there are pathways not only for cofactor biosynthesis, but also for its maintenance, and activity regulation (Figure 15). Our general model for Fe3+2-Y• cofactor biosynthesis indicates the requirement for a chaperone protein(s) (106) to alter the apo-β2 conformation for optimized Fe2+ loading, an Fe2+ carrier protein or small molecule that delivers Fe2+ to apo-β2, and a reducing equivalent delivery mechanism required for cluster assembly with O2 as the oxidant (107). Studies in vivo in E. coli (108) and S. cerevisiae (109) reveal that cluster assembly can yield β2 with each β subunit having 2 Fe2+ and 1 Y•, that is, quantitative loading. In vitro however, E. coli β2, loading gives rise to 66% of active cofactor and 33% of inactive diferric cluster with no Y•. In both in vivo and in vitro loading, the activity of RNR per Y• is the same, suggesting identical cofactor structures.
Figure 15.
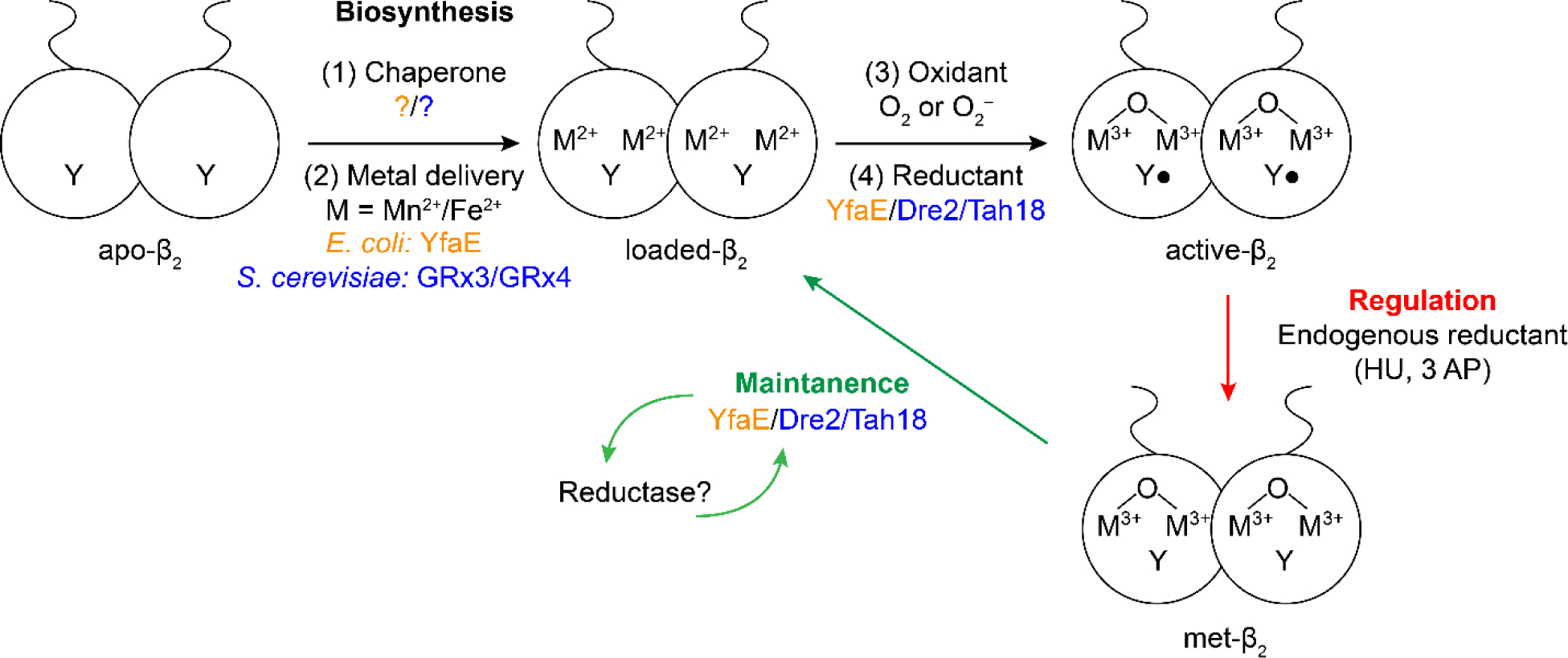
Model for Me23+-Y• (Me is Fe or Mn) cofactor biosynthesis (black), maintenance (green) and regulation (red) with factors identified from E. coli in orange and from S cerevisiae in blue (6). S. cerevisiae counterparts are found in humans. Regulation can occur by endogenous reductants or by therapeutics such as HU and triapine (Figure 14).
Hydroxyurea (HU).
HU (Figure 14) has been studied since the 1960s. Based on EPR analysis of prokaryotic and eukaryotic cells or of purified β2 with a self-assembled Fe3+2-Y•, HU treatment results in reduction of the Y• to YOH. In vitro, the iron cluster of human β2 is also reduced (Fe2+2-YOH), whereas in E. coli it remains in the Fe3+ state (Fe3+2-YOH). HU reduction of β2 alone is slow (0.45 M−1 s−1) and there is no evidence for its binding to either E. coli or human-β2 (107, 110, 111). The chemical mechanism of Y• reduction and the structure of the resulting cluster remain unknown (112). Studies by the Sjöberg lab in vitro have shown that HU mediated loss of RNR activity is potentiated ten-fold by complexation of β2 with α2, substrate and effector (113). This result led to their suggestion that its reduction of Y122• is not direct, but instead might be trapping a “transient” pathway radical at the α/β subunit interface (Figure 3). Studies on the reduction by HU of the Mn4+-Fe3+-β2 cofactor in the C. trachomatis Ic RNR (Figure 1C), an Fe3+2-Y• surrogate, were also interpreted to suggest that HU intercepts a pathway radical at the α/β interface (112). Furthermore, Mn4+-Fe3+-β2 in the presence of α2, CDP, and ATP is reduced by HU to a Mn3+-Fe3+ cluster with half-sites reactivity involving a fast phase and a slow phase, with apparent saturation by HU for the fast phase. These studies support HU binding and targeting of the radical transfer pathway (112, 114). The consequences of the HU reduced cofactor state in E. coli and mammalian cells are still unclear; however, since the proteins identified in S. cerevisiae for β2 cofactor biosynthesis and maintenance are also found in mammalian cells (107), Y• regeneration is one possible fate (maintenance pathway, Figure 15) and requires further investigation.
RNR inhibition by HU blocks DNA replication. Two papers have suggested that cytotoxicity from extended HU exposure of E. coli (100) or S. cerevisiae (102) cells is linked to reactive oxygen species (such as HO•) mediated damage. Vernis et al. showed that HU resistance in S. cerevisiae led to enhanced production of the cytosolic FeS cluster biosynthetic machinery including Dre2/Tah18 (102). We have shown the importance of these two proteins in the assembly of the β2 cofactor in S. cerevisiae (107).
Triapine.
Triapine (3-AP, Figure 14) has been extensively investigated since its introduction in the 1990’s and its cytotoxic effects have inspired the synthesis of many additional thiosemicarbazones. However, studies on these analogs reveal that the mechanism of cytotoxicity changes with structure. The complexity arises from their distinct abilities to bind Fe2+ and Fe3+ (also Cu2+ and Zn2+, not discussed) and the resultant ligand field imposed iron redox chemistry (100). The results reported by different groups (115, 116) in different mammalian cell lines, primarily at late stages of 3-AP treatment have thus made it challenging to compare and evaluate the outcomes between the different studies.
Our recent studies in cell culture in the “early” stages of 3-AP treatment are summarized and provide a framework for thinking about the issues and evaluation of their potential as therapeutics that target β2 (115). Three mammalian cell lines (K565, COS-1 and HU resistant TA3) treated with 3-AP and analyzed by whole cell EPR, revealed loss of the RNR Y• and assays of the corresponding cell lysates revealed loss of RNR activity. Immunoprecipitation of β2 from 55Fe treated and non-treated cells revealed similar iron content. These and additional studies suggested that the Y• loss is the major mode of RNR inhibition with iron loading remaining unchanged. Although the oxidation state of the bound iron is unknown, we know from in vitro studies that the Fe2+ loaded β2 can assemble rapidly into the native Fe3+2-Y• cofactor, consistent with a maintenance pathway (Figure 15, green). Our model is that Fe2+-(3-AP) is the active species involved in β2 inhibition and that in the continued presence of Fe2+/ Fe3+, RNR is susceptible to Fe2+-(3-AP) inhibition by direct Y• reduction. Recent studies by Gräslund et al. using [3H]-3-AP and a docking model of 3-AP to mouse β2, resulted in the proposal of a specific 3-AP binding site (116). However, neither 3-AP or Fe2+-(3-AP) binding to β2 has been observed. In our opinion, the mechanism of action of these compounds requires further study. Finally, our studies at early times subsequent to 3-AP treatment, in contrast with latter stage studies of others, indicate that reactive oxygen species are not responsible for RNR activity loss.
From the above discussion, how 3-AP and HU inhibit RNR and the relationship between their RNR inhibition and cell cytotoxicity still remain a mystery. Although interference with cluster assembly/maintenance might yield effective therapeutics, better understanding of the biology of Fe3+2-Y• pathways is required. The recent discovery, however, of Mn3+2-Y• cofactors in β2 of Ib RNRs (Figure 1C) and the identification of a NrdI-β2 interaction essential for oxidant delivery (O2•−) for active cofactor formation (Figure 15) (6, 117), suggests that disruption of this protein/protein interface could provide proof of principle for targeting cofactor pathways in pathogenic bacteria. The link between the Ia Fe3+2-Y• pathway, iron homeostasis and oxidative stress will make selective targeting difficult. However, for pathogenic organisms with Mn and or Mn/Fe clusters (Figure 1C Ib–Id), interference with cluster assembly, may well provide a new therapeutic target.
Summary points.
The quaternary structures of the class Ia RNR α subunit are nucleotide-dependent and distinct. α structures detected to data are: α2, non-canonical α2 (118), α4, α6, and fibrils (27, 75).
dATP-inhibited RNR structures include α6 (human), α4β4 (E. coli), α4 (P. aeruginosa) (119), and a double helical fibril of canonical and non-canonical α2s (Ib, B. subtilis) (75).
dATP inhibited states appear to interfere with the radical transfer pathway and thiyl radical formation by preventing β2 from achieving a productive α2β2 complex.
ClFDP (ClFTP) bind to human-RNR and form conformationally “stable” α6 state(s), even subsequent to their dissociation.
N3NDP and F2CDP are mechanism-based inhibitors of class Ia RNRs with one inhibitor/α2 in the α2β2 complex, half-sites reactivity; half-sites reactivity.
Incorporation of unnatural amino acids (F3Y122•or NO2Y122•-β or NH2Y356-β, NH2Y 731 or NH2Y 730-α) and incubation with the second subunit, substrate, and effector, trap radicals within the pathway and increase α/β subunit affinity.
The reaction of F3Y122•/ E52Q-β2 with α2, substrate, and effector, results in an asymmetric, active, and kinetically trapped α2β2 complex, whose structure has been determined by cryo-EM.
Future studies.
Continued studies of the structure, dynamics, and biology of RNRs
Trapping of additional α2β2 complexes of RNR with mechanism-based inhibitors, and unnatural amino acid-mutants may provide distinct and higher resolution structures.
The active form of a mutant E. coli Ia RNR has been shown by cryo-EM analysis to be an asymmetric and dynamic α2β2. The relationship of this structure to the wt enzyme and the structure of the human active complex remain to be established.
New ways to target RNRs
Identification of small molecules that can trap human and bacterial RNRs in distinct inhibited quaternary structures.
The discovery of biosynthetic pathways for dimetallo-Y• cluster assembly in Ia and Ib RNRs suggest that targeting the metal center formation, such as disruption of NrdI-NrdF interaction in the assembly of the class Ib, Mn3+2-Y• cofactor, might be possible.
The omics revolution (proteomics, phosphomics, transcriptomics) and a refined understanding of nucleotide metabolism are providing new insight into RNR regulation. This knowledge will lead to combination chemotherapies using RNR inhibitors in conjunction with inhibitors of downstream signaling pathways.
BOX 1 The model (Figure 7) encompasses weak, dynamic, subunit interactions that change with subunit and dNTP concentrations by altering RNR quaternary structure and activity. The assays/issues for the E. coli RNR are described in BioRxiv (42) and are essential for development of in vitro and in vivo high throughput screens for RNR inhibitors. The same issues are likely to be encountered, to different extents, with other Ia and Ib RNRs.
BOX2 Moonlighting function of α. A recent report (35) and review (84) provide support for a “moonlighting” function for α, independent of its ability with β to make dNDPs in the cytosol of the cell. Using a C→S mutant that inactivates formation of −S• in α (Figure 1A), a small amount of α was detected in the nucleus of the cell in an α6 state. Yeast two hybrid experiments with cDNA from HELA cells revealed that α interacts with ZRANB3, a protein that forms a complex with PCNA, the sliding clamp that together with DNA polymerase promotes DNA synthesis in non-stressed cells. Nuclear localized α inhibits the interaction of ZRANB3 with PCNA resulting in inhibition of DNA synthesis. This study potentially provides an explanation for the tumor suppressor activity reported for α (3).
References
- 1.Hofer A, Crona M, Logan DT, Sjöberg BM. 2012. DNA building blocks: keeping control of manufacture. Crit. Rev. Biochem. Mol. Biol 47: 50–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guarino E, Salguero I, Kearsey SE. 2014. Cellular regulation of ribonucleotide reductase in eukaryotes. Semin. Cell Dev. Biol 30: 97–103 [DOI] [PubMed] [Google Scholar]
- 3.Aye Y, Li M, Long MJC, Weiss RS. 2014. Ribonucleotide reductase and cancer: biological mechanisms and targeted therapies. Oncogene 34: 2011–21 [DOI] [PubMed] [Google Scholar]
- 4.Le TM, Poddar S, Capri JR, Abt ER, Kim W, et al. 2017. ATR inhibition facilitates targeting of leukemia dependence on convergent nucleotide biosynthetic pathways. Nat. Commun 8: 241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Licht S, Stubbe J. 1999. Mechanistic investigations of ribonucleotide reductases. Compr. Nat. Prod. Chem 5: 163–203 [Google Scholar]
- 6.Cotruvo JA, Stubbe J. 2011. Class I ribonucleotide reductases: metallocofactor assembly and repair in vitro and in vivo. Annu. Rev. Biochem 80: 733–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Minnihan EC, Nocera DG, Stubbe J. 2013. Reversible, long-range radical transfer in E. coli class Ia ribonucleotide reductase. Acc. Chem. Res 46: 2524–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fairman JW, Wijerathna SR, Ahmad MF, Xu H, Nakano R, et al. 2011. Structural basis for allosteric regulation of human ribonucleotide reductase by nucleotide-induced oligomerization. Nat. Struct. Mol. Biol 18: 316–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bennati M, Weber A, Antonic J, Perlstein DL, Robblee JH, Stubbe J. 2003. Pulsed ELDOR spectroscopy measured the distance between the two tyrosyl radicals in the R2 subunit of the E. coli ribonucleotide reductase. J. Am. Chem. Soc 125: 14988–9 [DOI] [PubMed] [Google Scholar]
- 10.Seyedsayamdost MR, Chan CTY, Mugnaini V, Stubbe J, Bennati M. 2007. PELDOR spectroscopy with DOPA-β2 and NH2Y-α2s: distance measurements between residues involved in the radical propagation pathway of E. coli ribonucleotide reductase. J. Am. Chem. Soc 129: 15748–49 [DOI] [PubMed] [Google Scholar]
- 11.Kang G 2019. submitted. Massachusetts Institute of Technology, Cambridge, Massachusetts [Google Scholar]
- 12.Zimanyi CM, Chen PY, Kang G, Funk MA, Drennan CL. 2016. Molecular basis for allosteric specificity regulation in class Ia ribonucleotide reductase from Escherichia coli. eLIFE 5: e07141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Uhlin U, Eklund H. 1994. Structure of ribonucleotide reductase protein R1. Nature 370: 533–39 [DOI] [PubMed] [Google Scholar]
- 14.Eklund H, Uhlin U, Färnegårdh M, Logan DT, Nordlund P. 2001. Structure and function of the radical enzyme ribonucleotide reductase. Prog. Biophys. Mol. Biol 77: 177–268 [DOI] [PubMed] [Google Scholar]
- 15.Nordlund P, Sjöberg BM, Eklund H. 1990. Three-dimensional structure of the free radical protein of ribonucleotide reductase. Nature 345: 593–8 [DOI] [PubMed] [Google Scholar]
- 16.Stubbe J 1998. Ribonucleotide reductases in the twenty-first century. Proc. Natl. Acad. Sci. U.S.A 95: 2723–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stubbe J, Seyedsayamdost MR. 2019. Discovery of a new class I ribonucleotide reductase with an essential DOPA radical and NO metal as an initiator of long-range radical transfer. Biochemistry 58: 435–37 [DOI] [PubMed] [Google Scholar]
- 18.Srinivas V, Lebrette H, Lundin D, Kutin Y, Sahlin M, et al. 2018. Metal-free ribonucleotide reduction powered by a DOPA radical in Mycoplasma pathogens. Nature 563: 416–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Blaesi EJ, Palowitch GM, Hu K, Kim AJ, Rose HR, et al. 2018. Metal-free class Ie ribonucleotide reductase from pathogens initiates catalysis with a tyrosine-derived dihydroxyphenylalanine radical. Proc. Natl. Acad. Sci. U.S.A 115: 10022–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jordan A, Reichard P. 1998. Ribonucleotide reductases. Annu. Rev. Biochem 67: 71–98 [DOI] [PubMed] [Google Scholar]
- 21.Nordlund P, Reichard P. 2006. Ribonucleotide reductases. Annu. Rev. Biochem 75: 681–706 [DOI] [PubMed] [Google Scholar]
- 22.Ge J, Yu G, Ator MA, Stubbe J. 2003. Pre-steady-state and steady-state kinetic analysis of E. coli class I ribonucleotide reductase. Biochemistry 42: 10071–83 [DOI] [PubMed] [Google Scholar]
- 23.Ravichandran KR, Taguchi AT, Wei Y, Nocera DG, Stubbe J. 2016. A >200 meV uphill thermodynamic landscape for radical transport in E. coli ribonucleotide reductase determined using fluorotyrosine-substituted enzymes. J. Am. Chem. Soc 138: 13706–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ravichandran KR, Zong AB, Taguchi AT, Nocera DG, Stubbe J, Tommos C. 2017. Formal reduction potentials of difluorotyrosine and trifluorotyrosine protein residues: defining the thermodynamics of multistep radical transfer. J. Am. Chem. Soc 139: 2994–3004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Minnihan EC, Ando N, Brignole EJ, Olshansky L, Chittuluru J, et al. 2013. Generation of a stable, aminotyrosyl radical-induced alpha2beta2 complex of Escherichia coli class Ia ribonucleotide reductase. Proc. Natl. Acad. Sci. U.S.A 110: 3835–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yokoyama K, Smith AA, Corzilius B, Griffin RG, Stubbe J. 2011. Equilibration of tyrosyl radicals (Y356•, Y731•, Y730•) in the radical propagation pathway of the Escherichia coli class Ia ribonucleotide reductase. J. Am. Chem. Soc 133: 18420–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brignole EJ, Tsai K-L, Chittuluru J, Li H, Aye Y, et al. 2018. 3.3-Å resolution cryo-EM structure of human ribonucleotide reductase with substrate and allosteric regulators bound. Elife 7: e31502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ando N, Li H, Brignole EJ, Thompson S, McLaughlin MI, et al. 2016. Allosteric inhibition of human ribonucleotide reductase by dATP entails the stabilization of a hexamer. Biochemistry 55: 373–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ando N, Brignole EJ, Zimanyi CM, Funk MA, Yokoyama K, et al. 2011. Structural interconversions modulate activity of Escherichia coli ribonucleotide reductase. Proc. Natl. Acad. Sci. U.S.A 108: 21046–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zimanyi CM, Ando N, Brignole EJ, Asturias FJ, Stubbe J, Drennan CL. 2012. Tangled up in knots: structures of inactivated forms of E. coli class Ia ribonucleotide reductase. Structure 20: 1374–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen PY, Funk MA, Brignole EJ, Drennan CL. 2018. Disruption of an oligomeric interface prevents allosteric inhibition of Escherichia coli class Ia ribonucleotide reductase. J. Biol. Chem 293: 10404–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ahluwalia D, Bienstock RJ, Schaaper RM. 2012. Novel mutator mutants of E. coli nrdAB ribonucleotide reductase: Insight into allosteric regulation and control of mutation rates. DNA Repair 11: 480–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aye Y, Brignole EJ, Long MJ, Chittuluru J, Drennan CL, et al. 2012. Clofarabine targets the large subunit (alpha) of human ribonucleotide reductase in live cells by assembly into persistent hexamers. Chem. Biol 19: 799–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wisitpitthaya S, Zhao Y, Long MJC, Li M, Fletcher EA, et al. 2016. Cladribine and fludarabine nucleotides induce distinct hexamers defining a common mode of reversible RNR inhibition. ACS Chem. Biol 11: 2021–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fu Y, Long MJC, Wisitpitthaya S, Inayat H, Pierpont TM, et al. 2018. Nuclear RNR-alpha antagonizes cell proliferation by directly inhibiting ZRANB3. Nat. Chem. Biol 14: 943–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin Q, Parker MJ, Taguchi AT, Ravichandran K, Kim A, et al. 2017. Glutamate 52-β at the α/β subunit interface of Escherichia coli class Ia ribonucleotide reductase is essential for conformational gating of radical transfer. J. Biol. Chem 292: 9229–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Erickson HK. 2001. Kinetics in the pre-steady state of the formation of cystines in ribonucleoside diphosphate reductase: evidence for an asymmetric complex. Biochemistry 40: 9631–37 [DOI] [PubMed] [Google Scholar]
- 38.Lou M, Liu Q, Ren GP, Zeng JL, Xiang XP, et al. 2017. Physical interaction between human ribonucleotide reductase large subunit and thioredoxin increases colorectal cancer malignancy. J. Biol. Chem 292: 9136–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Makhlynets O, Boal AK, Rhodes DV, Kitten T, Rosenzweig AC, Stubbe J. 2014. Streptococcus sanguinis class Ib ribonucleotide reductase: high activity with both iron and manganese cofactors and structural insights. J. Biol. Chem 289: 6259–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lu J, Holmgren A. 2014. The thioredoxin antioxidant system. Free Rad. Biol. Med 66: 75–87 [DOI] [PubMed] [Google Scholar]
- 41.Parker MJ, Stubbe J. 2014. Bacillus subtilis class Ib ribonucleotide reductase: high activity and dynamic subunit interactions. Biochemistry 53: 766–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ravichandran KR, Olshansky L, Nocera DG, Stubbe J. Subunit interaction dynamics of the E. coli class Ia ribonucleotide reductase. BioRxiv submitted [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stubbe J, van der Donk WA. 1998. Protein radicals in enzyme catalysis. Chem. Rev 98: 705–62 [DOI] [PubMed] [Google Scholar]
- 44.Stubbe J, Ackles D. 1980. On the mechanism of ribonucleoside diphosphate reductase from Escherichia coli. Evidence for 3′-C–H bond cleavage. J. Biol. Chem 255: 8027–30 [PubMed] [Google Scholar]
- 45.Lenz R, Giese B. 1997. Studies on the mechanism of ribonucleotide reductases. J. Am. Chem. Soc 119: 2784–94 [Google Scholar]
- 46.Licht SS, Booker S, Stubbe J. 1999. Studies on the catalysis of carbon-cobalt bond homolysis by ribonucleoside triphosphate reductase: evidence for concerted carbon-cobalt bond homolysis and thiyl radical formation. Biochemistry 38: 1221–33 [DOI] [PubMed] [Google Scholar]
- 47.Licht S, Gerfen GJ, Stubbe J. 1996. Thiyl radicals in ribonucleotide reductases. Science 271: 477–81 [DOI] [PubMed] [Google Scholar]
- 48.Lawrence CC, Bennati M, Obias HV, Bar G, Griffin RG, Stubbe J. 1999. High-field EPR detection of a disulfide radical anion in the reduction of cytidine 5′-diphosphate by the E441Q-R1 mutant of Escherichia coli ribonucleotide reductase. Proc. Natl. Acad. Sci. U.S.A 96: 8979–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pizano AA, Lutterman DA, Holder PG, Teets TS, Stubbe J, Nocera DG. 2011. Photo-ribonucleotide reductase beta2 by selective cysteine labeling with a radical phototrigger. Proc. Natl. Acad. Sci. U.S.A 109: 39–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chang MCY, Yee CS, Stubbe J, Nocera DG. 2004. Turning on ribonucleotide reductase by light-initiated amino acid radical generation. Proc. Natl. Acad. Sci. U.S.A 101: 6882–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Olshansky L, Pizano AA, Wei Y, Stubbe J, Nocera DG. 2014. Kinetics of hydrogen atom abstraction from substrate by an active site thiyl radical in ribonucleotide reductase. J. Am. Chem. Soc 136: 16210–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Licht SS, Lawrence CC, Stubbe J. 1999. Class II ribonucleotide reductases catalyze carbon-cobalt bond reformation on every turnover. J. Am. Chem. Soc 121: 7463–68 [Google Scholar]
- 53.Stubbe J, van der Donk WA. 1995. Ribonucleotide reductases: Radical enzymes with suicidal tendencies. Chem. Biol 2: 793–801 [DOI] [PubMed] [Google Scholar]
- 54.Thelander L, Larsson B, Hobbs J, Eckstein F. 1976. Active site of ribonucleoside diphosphate reductase from Escherichia coli. J. Biol. Chem 251: 1396–405 [PubMed] [Google Scholar]
- 55.van der Donk WA, Yu G, Silva DJ, Stubbe J, McCarthy JR, et al. 1996. Inactivation of Ribonucleotide Reductase by (E)-2′-Fluoromethylene-2′-deoxycytidine 5′-Diphosphate: A Paradigm for Nucleotide Mechanism-Based Inhibitors. Biochemistry 35: 8381–91 [DOI] [PubMed] [Google Scholar]
- 56.Bitonti AJ, Dumont JA, Bush TL, Cashman EA, Cross-Doersen DE, et al. 1994. Regression of human breast tumor xenografts in response to (E)-2′-deoxy-2′-(fluoromethylene)cytidine, an inhibitor of ribonucleoside diphosphate reductase. Cancer Res 54: 1485–90 [PubMed] [Google Scholar]
- 57.Zhou Y, Achanta G, Pelicano H, Gandhi V, Plunkett W, Huang P. 2002. Action of (E)-2′-deoxy-2′-(fluoromethylene)cytidine on DNA metabolism: incorporation, excision, and cellular response. Mol. Pharmacol 61: 222–9 [DOI] [PubMed] [Google Scholar]
- 58.Baker CH, Banzon J, Bollinger JM, Stubbe J, Samano V, et al. 1991. 2′-Deoxy-2′-methylenecytidine and 2′-deoxy-2′,2′-difluorocytidine 5′-diphosphates: potent mechanism-based inhibitors of ribonucleotide reductase. J. Med. Chem 34: 1879–84 [DOI] [PubMed] [Google Scholar]
- 59.Xie KC, Plunkett W. 1996. Deoxynucleotide pool depletion and sustained inhibition of ribonucleotide reductase and DNA synthesis after treatment of human lymphoblastoid cells with 2-chloro-9-(2-deoxy-2-fluoro-beta-D-arabinofuranosyl) adenine. Cancer Res 56: 3030–7 [PubMed] [Google Scholar]
- 60.Xie C, Plunkett W. 1995. Metabolism and actions of 2-chloro-9-(2-deoxy-2-fluoro-β-D-arabinofuranosyl)-adenine in human lymphoblastoid cells. Cancer Res 55: 2847–52 [PubMed] [Google Scholar]
- 61.Aye Y, Stubbe J. 2011. Clofarabine 5′-di and -triphosphates inhibit human ribonucleotide reductase by altering the quaternary structure of its large subunit. Proc. Natl. Acad. Sci. U.S.A 108: 9815–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fritscher J, Artin E, Wnuk S, Bar G, Robblee JH, et al. 2005. Structure of the nitrogencentered radical formed during inactivation of E. coli ribonucleotide reductase by 2′-azido-2′-deoxyuridine-5′-diphosphate: trapping of the 3′-ketonucleotide. J. Am. Chem. Soc 127: 7729–38 [DOI] [PubMed] [Google Scholar]
- 63.Sjöberg BM, Gräslund A, Eckstein F. 1983. A substrate radical intermediate in the reaction between ribonucleotide reductase from Escherichia coli and 2′-azido-2′-deoxynucleoside diphosphates. J. Biol. Chem 258: 8060–7 [PubMed] [Google Scholar]
- 64.Ewald B, Sampath D, Plunkett W. 2008. Nucleoside analogs: molecular mechanisms signaling cell death. Oncogene 27: 6522–37 [DOI] [PubMed] [Google Scholar]
- 65.Murai J 2017. Targeting DNA repair and replication stress in the treatment of ovarian cancer. Int. J. Clin. Oncol 22: 619–28 [DOI] [PubMed] [Google Scholar]
- 66.Bonate PL, Arthaud L, Cantrell WR Jr., Stephenson K, Secrist JA, Weitman S. 2006. Discovery and development of clofarabine: a nucleoside analogue for treating cancer. Nat. Rev. Drug Discov 5: 855–63 [DOI] [PubMed] [Google Scholar]
- 67.Hertel LW, Boder GB, Kroin JS, Rinzel SM, Poore GA, Todd GC, Grindey GB 1990. Evaluation of the antitumor-activity of gemcitabine (2′,2′-difluoro-2′-deoxycytidine). Cancer Res 50: 4417–22 [PubMed] [Google Scholar]
- 68.Ewald B, Sampath D, Plunkett W. 2007. H2AX phosphorylation marks gemcitabine-induced stalled replication forks and their collapse upon S-phase checkpoint abrogation. Mol. Cancer. Ther 6: 1239–48 [DOI] [PubMed] [Google Scholar]
- 69.Plunkett W, Huang P, Searcy CE, Gandhi V. 1996. Gemcitabine: preclinical pharmacology and mechanisms of action. Semin. Oncol 23: 3–15 [PubMed] [Google Scholar]
- 70.van der Donk WA, Yu GX, Pérez L, Sanchez RJ, Stubbe J, Samano V. Robins MJ 1998. Detection of a new substrate-derived radical during inactivation of ribonucleotide reductase from Escherichia coli by gemcitabine 5′-diphosphate. Biochemistry 37: 6419–26 [DOI] [PubMed] [Google Scholar]
- 71.Artin E, Wang J, Lohman GJ, Yokoyama K, Yu G, et al. 2009. Insight into the mechanism of inactivation of ribonucleotide reductase by gemcitabine 5′-diphosphate in the presence or absence of reductant. Biochemistry 48: 11622–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang J, Lohman GJ, Stubbe J. 2009. Mechanism of inactivation of human ribonucleotide reductase with p53R2 by gemcitabine 5′-diphosphate. Biochemistry 48: 11612–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chen Z, Zhou J, Zhang Y, Bepler G. 2011. Modulation of the ribonucleotide reductase M1-gemcitabine interaction in vivo by N-ethylmaleimide. Biochem. Biophys. Res. Commun 413: 383–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang J, Lohman GJ, Stubbe J. 2007. Enhanced subunit interactions with gemcitabine-5′-diphosphate inhibit ribonucleotide reductases. Proc. Natl. Acad. Sci. U.S.A 104: 14324–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Thomas WC, Brooks FP, Burnim AA, Bacik JP, Stubbe J, et al. 2019. Convergent allostery in ribonucleotide reductase. Nat. Commun 10: 2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ghanem H, Jabbour E, Faderl S, Ghandhi V, Plunkett W, Kantarjian H. 2010. Clofarabine in leukemia. Expert. Rev. Hematol 3: 15–22 [DOI] [PubMed] [Google Scholar]
- 77.Wong A, Soo RA, Yong WP, Innocenti F. 2009. Clinical pharmacology and pharmacogenetics of gemcitabine. Drug Metab. Rev 41: 77–88 [DOI] [PubMed] [Google Scholar]
- 78.Xie C, Plunkett W. 1995. Metabolism and actions of 2-chloro-9-(2-deoxy-2-fluoro-beta-D-arabinofuranosyl)-adenine in human lymphoblastoid cells. Cancer Res 55: 2847–52 [PubMed] [Google Scholar]
- 79.Warren NJH, Eastman A. 2019. Inhibition of checkpoint kinase 1 following gemcitabine-mediated S phase arrest results in CDC7- and CDK2-dependent replication catastrophe. J. Biol. Chem 294: 1763–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liu Y, Li Y, Wang X, Liu F, Gao P, et al. 2017. Gemcitabine and chk1 inhibitor AZD7762 synergistically suppress the growth of Lkb1-deficient lung adenocarcinoma. Cancer Res 77: 5068–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fordham SE, Blair HJ, Elstob CJ, Plummer R, Drew Y, et al. 2018. Inhibition of ATR acutely sensitizes acute myeloid leukemia cells to nucleoside analogs that target ribonucleotide reductase. Blood Adv 2: 1157–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chen YR, Tsou B, Hu S, Ma H, Liu X, et al. 2016. Autophagy induction causes a synthetic lethal sensitization to ribonucleotide reductase inhibition in breast cancer cells. Oncotarget 7: 1984–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mannargudi MB, Deb S. 2017. Clinical pharmacology and clinical trials of ribonucleotide reductase inhibitors: is it a viable cancer therapy? J. Cancer Res. Clin. Oncol 143: 1499–529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Long MJC, Van Hall-Beauvais A, Aye Y. 2019. The more the merrier: how homo-oligomerization alters the interactome and function of ribonucleotide reductase. Curr. Opin. Chem. Biol 54:10–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Labroli MA, Dwyer MP, Shen R, Popovici-Muller J, Pu Q, et al. 2014. The identification of novel 5′-amino gemcitabine analogs as potent RRM1 inhibitors. Bioorg. Med. Chem 22: 2303–10 [DOI] [PubMed] [Google Scholar]
- 86.Misko TA, Liu YT, Harris ME, Oleinick NL, Pink J, et al. 2019. Structure-guided design of anti-cancer ribonucleotide reductase inhibitors. J. Enz. Inhib. Med. Chem 34: 438–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ahmad MF, Alam I, Huff SE, Pink J, Flanagan SA, et al. 2017. Potent competitive inhibition of human ribonucleotide reductase by a nonnucleoside small molecule. Proc. Natl. Acad. Sci. U.S.A 114: 8241–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Backus KM, Cao J, Maddox SM. 2019. Opportunities and challenges for the development of covalent chemical immunomodulators. Bioorg. Med. Chem 27: 3421–39 [DOI] [PubMed] [Google Scholar]
- 89.Moss N, Beaulieu P, Duceppe JS, Ferland JM, Gauthier J, et al. 1995. Peptidomimetic inhibitors of herpes simplex virus ribonucleotide reductase: a new class of antiviral agents. J. Med. Chem 38: 3617–23 [DOI] [PubMed] [Google Scholar]
- 90.Climent I, Sjöberg BM, Huang CY. 1991. Carboxyl-terminal peptides as probes for Escherichia coli ribonucleotide reductase subunit interaction: kinetic analysis of inhibition studies. Biochemistry 30: 5164–71 [DOI] [PubMed] [Google Scholar]
- 91.Climent I, Sjöberg BM, Huang CY. 1992. Site-directed mutagenesis and deletion of the carboxyl terminus of Escherichia coli ribonucleotide reductase protein R2 - effects on catalytic activity and subunit interaction. Biochemistry 31: 4801–7 [DOI] [PubMed] [Google Scholar]
- 92.Xu H, Fairman JW, Wijerathna SR, Kreischer NR, LaMacchia J, et al. 2008. The structural basis for peptidomimetic inhibition of eukaryotic ribonucleotide reductase: a conformationally flexible pharmacophore. J. Med. Chem 51: 4653–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cooperman BS, Gao Y, Tan C, Kashlan OB, Kaur J. 2005. Peptide inhibitors of mammalian ribonucleotide reductase. Adv. Enz. Reg 45: 112–25 [DOI] [PubMed] [Google Scholar]
- 94.Cohen EA, Gaudreau P, Brazeau P, Langelier Y. 1986. Specific inhibition of herpesvirus ribonucleotide reductase by a nonapeptide derived from the carboxy terminus of subunit 2. Nature 321: 441–43 [DOI] [PubMed] [Google Scholar]
- 95.Dutia BM, Frame MC, Subak-Sharpe JH, Clark WN, Marsden HS. 1986. Specific inhibition of herpesvirus ribonucleotide reductase by synthetic peptides. Nature 321: 439–41 [DOI] [PubMed] [Google Scholar]
- 96.Liuzzi M, Déziel R, Moss N, Beaulieu P, Bonneau A-M, et al. 1994. A potent peptidomimetic inhibitor of HSV ribonucleotide reductase with antiviral activity in vivo. Nature 372: 695–98 [DOI] [PubMed] [Google Scholar]
- 97.Zhou B, Su L, Hu S, Hu W, Yip ML, et al. 2013. A small-molecule blocking ribonucleotide reductase holoenzyme formation inhibits cancer cell growth and overcomes drug resistance. Cancer Res 73: 6484–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chen MC, Zhou B, Zhang K, Yuan YC, Un F, et al. 2015. The novel ribonucleotide reductase inhibitor COH29 inhibits DNA repair in vitro. Mol. Pharmacol 87: 996–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Singh A, Xu YJ. 2016. The Cell Killing Mechanisms of Hydroxyurea. Genes (Basel) 7: 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Heffeter P, Pape VF, Enyedy EA, Keppler BK, Szakacs G, Kowol CR. 2019. Anticancer thiosemicarbazones: chemical properties, interaction with iron metabolism, and resistance development. Antiox. Redox Sig 30: 1062–82 [DOI] [PubMed] [Google Scholar]
- 101.Davies BW, Kohanski MA, Simmons LA, Winkler JA, Collins JJ, Walker GC. 2009. Hydroxyurea induces hydroxyl radical-mediated cell death in Escherichia coli. Mol. Cell 36: 845–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Huang ME, Facca C, Fatmi Z, Baille D, Benakli S, Vernis L. 2016. DNA replication inhibitor hydroxyurea alters Fe-S centers by producing reactive oxygen species in vivo. Sci. Rep 6: 29361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Stubbe J, Cotruvo JA. 2011. Control of metallation and active cofactor assembly in the class Ia and Ib ribonucleotide reductases: diiron or dimanganese? Curr. Op. Chem. Bio 15: 284–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Atkin CL, Thelander L, Reichard P, Lang G. 1973. Iron and free radical in ribonucleotide reductase. Exchange of iron and Mössbauer spectroscopy of the protein B2 subunit of the Escherichia coli enzyme. J. Biol. Chem 248: 7464–72 [PubMed] [Google Scholar]
- 105.Mühlenhoff U, Molik S, Godoy JR, Uzarska MA, Richter N, et al. 2010. Cytosolic monothiol glutaredoxins function in intracellular iron sensing and trafficking via their bound iron-sulfur cluster. Cell Metab 12: 373–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sluder IT, Nitika, Knighton LE, Truman AW. 2018. The Hsp70 co-chaperone Ydj1/HDJ2 regulates ribonucleotide reductase activity. PLoS Genet 14: e1007462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zhang Y, Li H, Zhang C, An X, Liu L, et al. 2014. Conserved electron donor complex Dre2-Tah18 is required for ribonucleotide reductase metallocofactor assembly and DNA synthesis. Proc. Natl. Acad. Sci. U.S.A 111: E1695–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hristova D, Wu CH, Jiang W, Krebs C, Stubbe J. 2008. Importance of the maintenance pathway in the regulation of the activity of Escherichia coli ribonucleotide reductase. Biochemistry 47: 3989–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ortigosa AD, Hristova D, Perlstein DL, Zhang Z, Huang M, Stubbe J. 2006. Determination of the in vivo stoichiometry of tyrosyl radical per ββ′ in Saccharomyces cerevisiae ribonucleotide reductase. Biochemistry 45: 12282–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lassmann G, Thelander L, Gräslund A. 1992. EPR stopped-flow studies of the reaction of the tyrosyl radical of protein R2 from ribonucleotide reductase with hydroxyurea. Biochem. Biophys. Res. Commun 188: 879–87 [DOI] [PubMed] [Google Scholar]
- 111.Swarts JC, Aquino MA, Han JY, Lam KY, Sykes AG. 1995. Kinetic studies on the reduction of the tyrosyl radical of the R2 subunit of E. coli ribonucleotide reductase. Biochim. Biophys. Acta 1247: 215–24 [DOI] [PubMed] [Google Scholar]
- 112.Jiang W, Xie J, Varano PT, Krebs C, Bollinger JM. 2010. Two distinct mechanisms of inactivation of the class Ic ribonucleotide reductase from Chlamydia trachomatis by hydroxyurea: implications for the protein gating of intersubunit electron transfer. Biochemistry 49: 5340–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Karlsson M, Sahlin M, Sjöberg BM. 1992. Escherichia coli ribonucleotide reductase. Radical susceptibility to hydroxyurea is dependent on the regulatory state of the enzyme. J. Biol. Chem 267: 12622–6 [PubMed] [Google Scholar]
- 114.Bollinger JM Jr., Jiang W, Green MT, Krebs C. 2008. The manganese(IV)/iron(III) cofactor of Chlamydia trachomatis ribonucleotide reductase: structure, assembly, radical initiation, and evolution. Curr. Opin. Struct. Biol 18: 650–7 [DOI] [PubMed] [Google Scholar]
- 115.Aye Y, Long MJ, Stubbe J. 2012. Mechanistic studies of semicarbazone triapine targeting human ribonucleotide reductase in vitro and in mammalian cells: tyrosyl radical quenching not involving reactive oxygen species. J. Biol. Chem 287: 35768–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Popović-Bijelić A, Kowol CR, Lind ME, Luo J, Himo F, et al. 2011. Ribonucleotide reductase inhibition by metal complexes of Triapine (3-aminopyridine-2-carboxaldehyde thiosemicarbazone): a combined experimental and theoretical study. J. Inorg. Biochem 105: 1422–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Boal AK, Cotruvo JA Jr., Stubbe J, Rosenzweig AC. 2010. Structural basis for activation of class Ib ribonucleotide reductase. Science 329: 1526–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Parker MJ, Maggiolo AO, Thomas WC, Kim A, Meisburger SP, et al. 2018. An endogenous dAMP ligand in Bacillus subtilis class Ib RNR promotes assembly of a noncanonical dimer for regulation by dATP. Proc. Natl. Acad. Sci. U.S.A 115: E4594–E603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Johansson R, Jonna Venkateswara R, Kumar R, Nayeri N, Lundin D, et al. 2016. Structural mechanism of allosteric activity regulation in a ribonucleotide reductase with double ATP cones. Structure 24: 906–17 [DOI] [PubMed] [Google Scholar]