

The Cardiac Safety Research Consortium (CSRC; www.cardiac-safety.org) held a Think Tank on “Detection, Assessment, and Risk Mitigation of Cardiac Safety Signals in Oncology Drug Development” on October 24–25, 2017, at the Food and Drug Administration (FDA) headquarters in Silver Spring, MD.1 The CSRC, a public private partnership, was formed in 2005 as a Critical Path Program and formalized in 2006 under a Memorandum of Understanding between the US FDA and Duke University.2,3
This is a promising period in oncology research, with more than 1,100 medicines and vaccines for cancer in development by American companies alone as of May 2018.4 Oncology accounts for 40% of the global clinical pipeline, with some 1,600 products.5 Over the past 5 years, some 63 cancer drugs have been launched globally.6 Mortality rates in cancer patients have fallen substantially over the last 20–30 years, and the cancer survivor population is aging.7 However, the toxicity of cancer therapies is significant,8,9 with rates of cardiovascular (CV) adverse effects reported to be more than 30%, and CV causes are estimated to be the second most common cause of morbidity and mortality in cancer survivors.10,11
The CV sequelae of cancer therapies are diverse for radiation, traditional chemotherapies, targeted therapies, and other new cancer therapies. These include toxic effects such as hypertension, venous and arterial thromboembolic events, peripheral artery disease, pulmonary hypertension, vasospasm, proteinuria, accelerated atherosclerosis, and metabolic derangements. There are also cardiac toxic effects such as decline in left ventricular ejection fraction (LVEF), congestive heart failure, arrhythmia, myocarditis, pericardial disease, and pericardial effusion.12
Therapies traditionally linked with CV adverse effects include anthracyclines, monoclonal antibodies targeting the human epidermal growth factor receptor (HER2) pathway, and small molecule tyrosine kinase inhibitors, especially the vascular endothelial growth factor signaling pathway inhibitors, which can cause cardiomyopathy.13 In addition, other signaling pathways in cancer cells targeted by novel cancer therapies may also play a role in CV homeostasis. As a result, several of these agents are associated with CV toxic effects ranging from myocardial and vascular dysfunction to arrhythmias.12 Recently, vascular and metabolic complications from novel therapies have emerged as critical considerations in cancer patients.14 Finally, cancer immunotherapies which harness the immune system in the fight against cancer can cause myocarditis, pericarditis, and vasculitis.15–17
Further complicating the clinical picture is the fact that current treatment regimens often include multiple agents with adverse cardiac effects that could be additive or synergistic and transient or irreversible.18 In addition, patients with cancer may also have underlying CV disease, which may be exacerbated by the stress of tumor growth or treatment.18
CV toxicities that are identified with new cancer therapies must be juxtaposed against the prognosis of cancer, the availability of existing therapies in the same class (for the same cancer), and the net benefit of therapy. The threshold for toxicity may be different for a first-in-class therapy for a cancer type that has a poor prognosis and has few existing treatment possibilities versus a later-generation drug for a cancer with multiple existing therapies and a generally good prognosis.
These considerations underline the importance of the expanding discipline of cardio-oncology, which has been the subject of a number of recent professional society statements and guidelines19 and is a focus of attention at bodies including the American College of Cardiology,20 American Society of Clinical Oncology,21 European Society for Medical Oncology,22 European Society of Cardiology,23 National Cancer Center Network, and Canadian Cardiovascular Society.24
The role of preclinical studies in cardiac safety
In response to the growing awareness of potential cardiovascular liabilities of oncologic drugs, preclinical cardiac safety studies are increasingly being used for the early detection (and avoidance) of cardiotoxicity of evolving oncologic drugs. In addition, preclinical models may be useful to dissect mechanisms of toxicities for existing cancer drugs.25 Phenotypic screens are used in early drug discovery efforts to identify and avoid potential safety hazards, as it is often difficult to determine the (multiple) mechanisms involved in various cardiotoxi-cities (which can include electrical, contractile, structural effects). Earliest screening efforts might involve in vitro studies (for higher throughput), with later screening efforts involving in vivo safety pharmacology (focusing on acute functional assessments) and toxicology studies focusing on longer-term effects with morphological readouts. In general, in vitro studies report on acute effects in cellular (myocyte) or ex vivo (isolated tissues or Langendorff hearts) models. In contrast, in vivo studies focus on longer-term effects in animal models. The later studies provide the advantage of evaluating cardiac liabilities in the setting of the entire CV system using end points translatable to clinic (eg, ventricular imaging and ejection fraction measures) but with the disadvantage of limited screening opportunities due to time, cost, and compound requirements. In addition, preclinical models may be useful to dissect mechanisms of toxicities for existing cancer drugs.26
In vitro and in vivo studies serve different roles in different phases of drug discovery (the former generally useful for hazard identification, the later more aligned with risk assessment), and both are essential to provide a more comprehensive assessment of potential cardiac safety liabilities. Preclinical models can also fulfill the secondary role of assessing mitigation of CV effects with combinational drugs or the safety of promising synergistic drug regimens. Identifying the most serious and prevalent clinical cardiac toxicities is critical to guide the development, validation, and use of preclinical studies as translational tools. This is particularly difficult for effects that are not easily detected clinically or may be irreversible situations where preclinical studies could be most valuable. There is clearly a need for collaborations between nonclinical and clinical experts to set relevant end points and identify appropriate biomarkers to guide preclinical studies.
Studies using human induced pluripotent stem-cell derived cardiomyocytes (hiPSC-CMs) are being used to assess cardiotoxicity in vitro, with the presumed advan tage of using human-derived models as test systems. Commercially available cardiomyocytes have spurred growth in this area, with preparations varying in complexity (from single myocytes to 2D cultures, spheroids, organoids, and engineered heart tissues) and varying in functional and morphological characteristics27,28 As different levels of myocyte “maturity” (ie, resemblance to adult native myocytes in form and function) tend to increase with the level of complexity, it is essential to demonstrate assay sensitivity for hiPSCCM preparations under study. Functional end points typically evaluated include field potential waveforms (multielectrode array platforms) to assess electrophysio-logic effects (delayed repolarization and conduction, altered spontaneous beating) and various measures of contractility (sarcomere length changes, edge tracking, various motion detection schemes), with measures of soluble (cardiac troponins) and morphological biomarkers possible.
Cost-effective and scalable procedures to produce hiPSCs with N90% purity are now achievable using a chemically defined process,29 enabling tests with “personalized” cell lines. These cells have a promise in genomic prediction of chemotherapy-induced cardiotoxicity and have been shown to recapitulate individual patients’ predilection to doxorubicin-induced cardiotoxicity (DIC). hiPSC-CMs from breast cancer patients who suffered clinical DIC have been found to be consistently more sensitive to doxorubicin toxicity compared to hiPSC-CMs from patients who did not experience DIC, with a 19-fold difference in sensitivity between cells from the 2 categories of patients. This indicates that iPSC-CMs may be a suitable platform for identifying and verifying the genetic basis and molecular mechanisms of DIC.30
Work is also under way to determine the genetic basis for this type of sensitivity, with a study of anthracycline-induced cardiotoxicity in 280 childhood cancer survivors finding that a particular variant (rs2229774, p.Ser427Leu) in RARG was highly associated with anthracycline-induced cardiotoxicity, showing a 4-fold increase in sensitivity.31 Correction of the rs2229774 variant was found to reverse doxorubicin hypersensitivity in the patients’ iPSCs, providing new insight into the pathophysiology of this severe adverse drug effect.
Oncologic drugs may affect cardiac function due largely to unintended overlapping on-target or off-target effects between cardiac and other tissues. The lack of selectivity of tyrosine kinase inhibitors, along with the distribution of multiple kinases across organs (including the heart), provides one example where it may be difficult to separate antitumor activity from cardiac toxicity. Minimizing and predicting potential tyrosine kinase–induced cardiotoxicity remain important challenges for drug developers and regulatory authorities and highlight the need for developing and implementing robust preclinical models that predict adverse clinical effects.
Early expectations of minimizing cardiac dysfunction with targeted therapeutics (including those against tumor-specific targets such as HER2) may be less successful due to less well-appreciated (or sometimes unknown) overlapping signaling pathways in cardiac and cancerous tissues revealed with drugs and drug combinations. Such pathways could be distinguished by identifying differences in the genomes/proteomes of normal and tumor tissues and obtaining cancer-specific gene-protein expression. The doses required for efficacy could be determined using animal models of disease or by developing 3-dimensional microfluidic organ systems, or tissue chips, which represent an integrated in vitro model of perfused tumor and cardiac tissues, with potential utility in screening. In the future, it may be possible to develop safer oncologic drugs by either identifying cancer-only targets or differentiating exposures required for efficacy from those that cause direct cardiac toxicity.
Overall, a multipronged preclinical approach—including in vitro, in vivo, and in silico approaches—is best suited to identify potential direct cardiotoxic effects of novel oncologic drugs, provide mechanistic insights, define who is at risk of toxicity, explore mechanisms, and inform on preventive and treatment strategies.32
Cardiac safety signals should not impede development of effective drugs
The unique features of early phase oncology studies provide a reason to validate emerging assays and biomarkers in patients to help characterize cardiac risk earlier and more precisely. The potential benefit-risk must always be evaluated; early identification of CV safety signals during drug development should be balanced with the potential benefit. For instance, if a drug being studied in phase 1 showed excellent responses in tumors but causes frequent serious or life-threatening CV toxicities, it should not be developed further. Precise characterization of cardiac safety signals may enable implementation of appropriate cardiac surveillance strategies or, if CV toxicity is dose related, use of lower/safer doses during early-phase clinical trials.
When possible, the CV safety profile of all anticancer drugs should be characterized, including, when appropriate, full characterization of electrophysiology (electrocardiogram/QT), left ventricular (LV) function (imaging/biomarker (BM)) and hemodynamic blood pressure (BP) effects. Depending on the population (metastatic vs curable) and the drug, some battery of cardiac tests may be recommended for each protocol and should depend on the prior knowledge about the drug/drug class. If possible, these assessments should be integrated into early-phase studies, without disrupting or impeding conduct, interpretation, or analysis of trial results. Potential CV safety of investigational anticancer drugs should be defined early in their development, ideally prior to exposing large populations in late phase clinical trials. Wherever possible, CV safety assessments should be integrated into routine early-phase oncology clinical trials with minimal disruption to study design, conduct, interpretation, and analysis.
A particular challenge is to put CV safety findings into context in early-stage oncology trials, which often involve small numbers of participants and may have a single-arm design, making it hard to determine whether the effect is due to the drug or not. Additional testing in later-phase oncology trials with a comparator arm can be informative. Additional challenges include the fact that early-phase studies, including first in human, are conducted in cancer patients, some of whom may be at higher than normal risk for CV disease. Moreover, patients may have already been exposed to therapies that are associated with CV complications. An additional challenge occurs due to requirement to perform dose escalation in early-stage trials often starting at subtherapeutic doses, making interpretation of both safety and efficacy in these patients problematic.
Clinically meaningful end points: Lessons from trastuzumab
It is clearly important to monitor safety signals that are clinically meaningful. For example, in early phase III clinical trials in patients with metastatic HER2-positive breast cancer, trastuzumab was associated with significant cardiac dysfunction and symptomatic heart failure in almost 25% of the patients.33,34 As a result, in subsequent adjuvant studies, routine imaging was required for patients receiving trastuzumab at baseline and every 3 months during 1 year of treatment. In this setting, trastuzumab became rather safe, and incidence of symptomatic heart failure in adjuvant trials was consistently less than 3%. The most common abnormality detected during routine imaging is a significant decline in LVEF; however, the association between an asymptomatic LVEF decline and the risk of subsequent clinical heart failure has not been fully elucidated in this population. Given the inherent variability of LVEF measurement, there is also a risk that patients may be wrongly identified as having cardiotoxicity, which may compromise delivery of curative therapy. Overall, the current available evidence is insufficient to support a specific schedule of cardiac imaging during trastuzumab-based treatment, and further investigation is needed to determine whether routine cardiac monitoring results in improved CV outcomes.35 A more promising approach might be to use existing tools to identify those at highest risk and only apply stringent cardiac monitoring strategies to these individuals. Until such data become available, it is reassuring to know that the rate of symptomatic HF is low based on clinical trial data when routine cardiac monitoring is performed. The goal should be to carry out the right testing and monitoring in the right patient at the right time. In clinical practice, cardiovascular risk factors alone have not been able to predict which patients will develop trastuzumab cardiotoxicity, and the variability in cardiac adverse effects from trastuzumab suggests that genetics may play a role in a patient’s individual risk for cardiotoxicity.
To advance understanding of the optimal management of patients with trastuzumab toxicity, the recently completed SAFE-HEaRt study evaluated cardiac safety of HER2 targeted therapy in patients with HER2-positive breast cancer and mildly reduced LVEF (≥40% no symptoms of heart failure).36 This investigation tested the hypothesis that initiation or continuation of trastuzumab may be safe in patients with mildly reduced LVEF if they concomitantly receive optimal cardiac therapy including β-blockers and angiotensin-converting enzyme inhibitors or angiotensin receptor blockers. Future areas of study may incorporate newer techniques using hiPSC-CMs to predict the cardiotoxic response to trastuzumab.
The role of circulating biomarkers
Biomarkers–which are indicators of normal biological processes, pathogenic processes, or responses to an exposure or intervention–include molecular, histologic, radiographic, and physiologic characteristics. The cardiac biomarkers troponin, brain natriuretic peptide, and myeloperoxidase have potential in detecting subclinical cardiotoxicity during cancer treatment.37 Biomarkers may not provide insight into the mechanism of drug toxicity, however.
Biomarkers can be used at baseline to define at-risk subgroups and help determine optimum cardiac treatment. Biomarkers are often part of comprehensive assessment of patients, in particular if the risk of cardiomyopathy and heart failure is high. The patients with preexisting CV comorbidities could be treated prior to initiation of cancer therapy, avoiding the inappropriate attribution of symptoms to drug toxicity.
Serial biomarkers have potential to define risk and track response to therapy, although the ultimate measure of effectiveness is the clinical outcome. Tracking cardiac biomarkers could identify patients at risk of oxidative stress, providing an early safety signal to facilitate early-stage trials.
To date, cardiac biomarkers have not been deployed optimally, with confusion remaining in areas including appropriate cutoffs and calculation of the reference change interval. Abnormal biomarkers may indicate a need for close surveillance but, based on current knowledge, are unlikely to provide sufficient data to mandate treatment.
In the future, the goal is to reduce morbidity in cancer patients by early risk factor modification, serial monitoring with imaging and/or biomarkers, cardioprotective medical therapy, and optimal medical therapy for cardiotoxicity when it occurs.1
Imaging end points for various outcomes
Cancer therapies can affect the CV system in multiple ways, with appropriate imaging end points reflecting the outcome of interest (Figures 1 and 2). These can be visualized using multiple CV imaging modalities, including echocardiography, positron emission tomography– computed tomography scanning, and cardiac magnetic resonance imaging.
Figure 1.
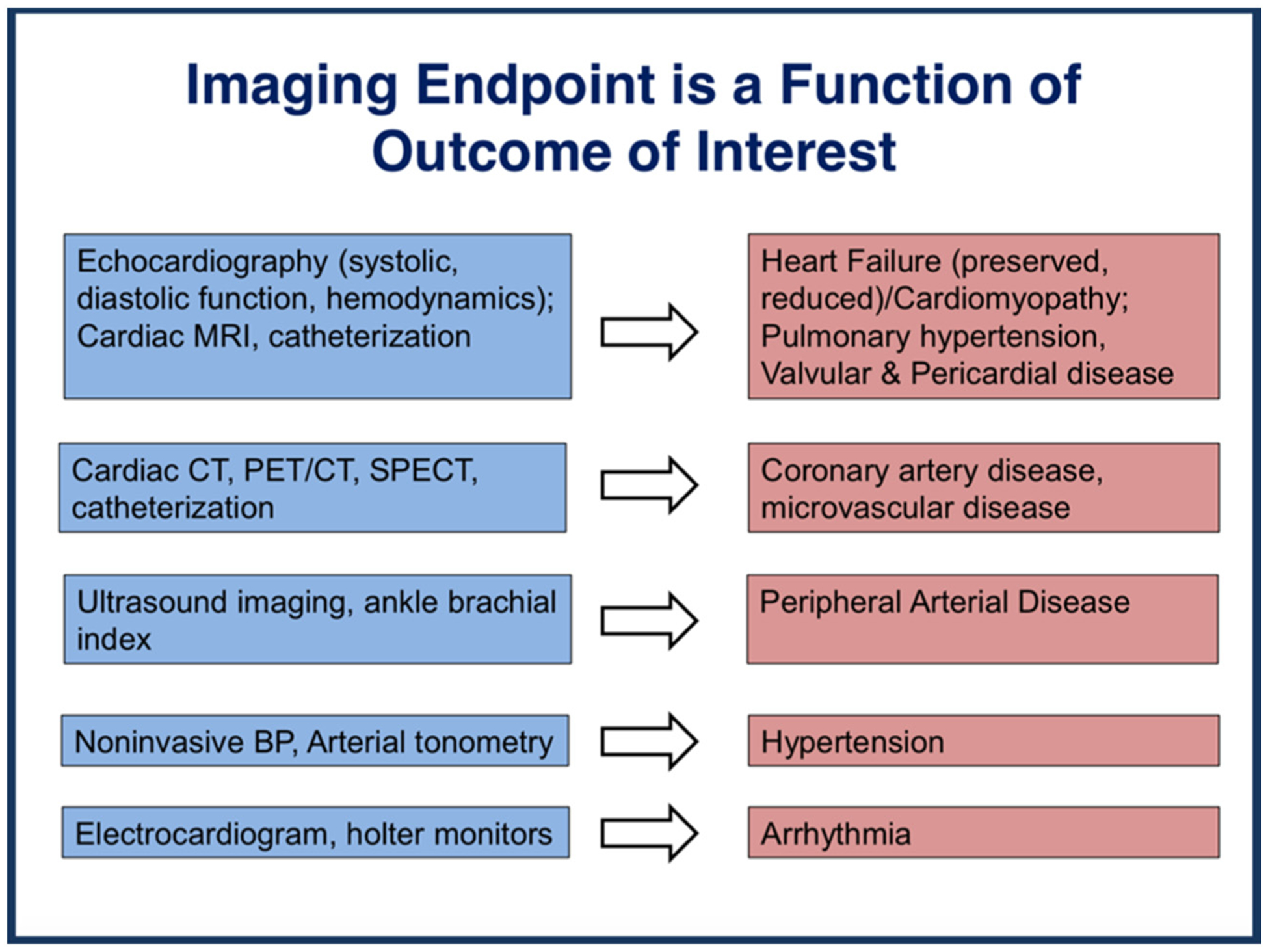
Imaging end point depends on the outcome of interest.
Figure 2.
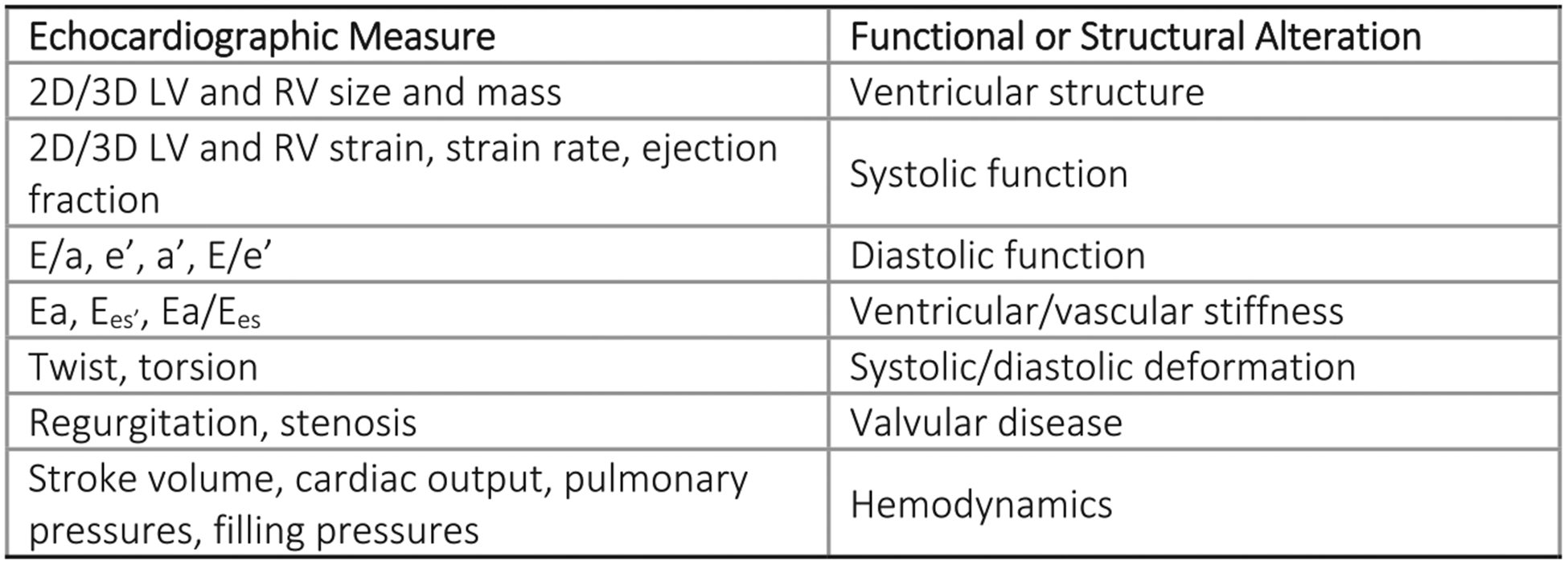
Quantitative echocardiography provides detailed phenotypic data.
Positron emission tomography or single-photon emission computed tomography radiotracers are tools to evaluate myocardial perfusion, cardiac function, and coronary vasculature. Outcome measures include perfusion defects, myocardial blood flow, coronary flow reserve, calcium score, and LVEF; additional measures are markers of inflammation, cell death, and metabolism.
Cardiac magnetic resonance imaging can characterize with high reproducibility cardiac size and function (LVEF, volumes, mass, strain). It is also possible to gain unique qualitative and quantitative insight into myocardial tissue through T1/T2 mapping, extracellular volume index, and delayed enhancement, enabling assessment of edema, inflammation, and fibrosis.
The core laboratory’s role in optimizing data collection and collaboration
The CV imaging core laboratory’s role is to ensure robust and consistent data collection. This includes involvement with protocol development; definition of CV imaging end points and choice of technique (echo, cardiac magnetic resonance); standardized image acquisition; protocol-based site instruction and training; independent, centralized, and standardized analysis and quality control; and data review and interpretation. The core laboratory provides dedicated personnel training, such as webcasts, instructional videos, and face-to-face meetings.38,39 Core laboratory–established data quality and standards enable a rigorous image analysis plan to be developed, with a standard method for image transfer, a secure environment for data storage, and tailored case report forms.
Overall, there are sufficient data to suggest that biomarkers can be useful. However, consensus is needed on the types of testing—such as echo parameters and biomarkers—that should be used routinely so that results can be compared across institutions. The clinical significance of changes in biomarkers in the absence of symptoms needs to be better understood.
When possible, CV assessments should be integrated into early-phase studies, without disrupting or impeding conduct, interpretation, or analysis of trial results. Current limitations of CV safety assessment include multiple cardiotoxic exposures, small numbers, open-label nonrandomized design, selection bias, and variable end point definitions.
Regulatory considerations: Trials should include patients with cardiac risk factors
A unique aspect of oncology drug development is the fact that because registration trials involve relatively small numbers of patients, they poorly predict the post-approval experience—either exaggerating or underestimating CV toxicity. Inclusion of patients with severe preexisting CV disease may translate to significant short-term risk of CV-related morbidity and mortality, potentially confounding the results of cancer trials. As a result, most trials aim to exclude patients with severe CV disease, including acute myocardial infarction, heart failure, stroke, or severe valvular heart disease (ie, severe symptomatic aortic stenosis or mitral regurgitation).
However, the frequency of concurrent cardiovascular disease (CVD) in adults with cancer is relatively high due in part to the increased incidence of both conditions with advancing age and to the higher incidence of cancers in patients with known risk factors for CVD such as obesity or premalignant hematopoietic stem cell mutations. For this reason, a strong argument can be made that clinical trials in cancer should include patients with CVD. Evaluating both disease states concurrently would increase the ability to identify cardiac toxicities of anticancer drugs early in clinical development and help provide guidance to clinicians who treat patients in “real-world” rather than a clinical trial setting.
Several important issues have mitigated against inclusion of patients with known cardiac disease into clinical trials of oncologic agents:
Attribution of causality of an adverse event (AE) to an experimental agent rather than to comorbid conditions is based on clinical judgment. Patients with preexisting CV comorbidities or risk factors make it more difficult for investigators to decide about the causality of the CV AEs.
Most early-stage trials of new anticancer therapeutics enroll patients with late-stage cancer who have exhausted all approved regimens and who have relatively short survival expectations. Deaths on study are to be expected. However, even in patients with objective evidence of progressive disease, the proximate causes of death may be ascribed to failure of an organ system such as the CV system. This is more likely to occur in patients with intrinsic cardiac disease at the time of study enrollment. For example, a patient with preexisting LV dysfunction or coronary artery disease is more likely to develop congestive heart failure or serious arrhythmias during a serious infectious episode than is a patient with a more normal heart at baseline. These events are included in the safety profile of the new agent and may be interpreted to be associated with drug-induced CV toxicity.
Many registration trials follow patients for survival; therefore, trial durations are often not short. However, the main issue is that majority of cancer patients receive a therapy for a short duration and, subsequent to their disease progression, they receive multiple subsequent therapies. Therefore, understanding and detection of late CV toxicities in patients with metastatic cancer are challenging.
Likewise, because exposure to the test agent is often relatively short, serious CV AEs that occur during these studies may delay or even prevent regulatory approval because of concerns of cardiotoxicity. In single-arm trials, as there is no comparator group to serve as a control for the rate of AEs, it is important from the sponsor’s perspective to eliminate as many background events as possible through careful patient screening.
Large trials are necessary to reliably determine the actual rate (compared to background) for CV toxicities. For instance, in CV trials investigating major adverse cardiac event (MACE) in the nondiabetic population, hundreds of patients would be needed to demonstrate noninferiority given the baseline incidence of MACE events in this population.40
Therefore, to advance potentially promising new anticancer therapeutics through drug development and testing, early-stage trials typically have included only patients with minimal preexisting cardiac disease. Early-stage trials should, however, incorporate sensitive screening techniques for cardiac toxicity to rapidly build an adequate safety data base that would allow for evaluation of inclusion of patients at higher CV risk who often are more representative of the therapeutic population. Stratification by CV risk is generally impractical due to the practical limitations of clinical relatively imprecise cardiac biomarkers. A potential solution to assess a compounds effect on a high-risk CV population would be to incorporate a separate substudy of patients with higher CV risk within the framework of a larger phase 3 trial. This approach would provide important insight into the safety and efficacy of a new drug among patients that would likely be more representative of a real-world cancer population.
Looking ahead, it would be ideal if clinical trial eligibility criteria only excluded patients with the highest level of CV risk. This would lead to greater generalizability of study results, making it easier to recruit patients, albeit at the risk of introducing confounding. The alternative is continued use of narrower eligibility criteria, with the pitfall of poor generalizability, longer accrual time, study duration and cost, and limited insight into the effects of CV risk on cancer outcomes. To expand eligibility, a 1-year cutoff could be used, after which mortality due to the CV disease typically plateaus.41,42 Patients who have not had an acute event for 1 year are likely to have a reasonable level of risk for inclusion in clinical studies.
Need for early cardiologist involvement in trial design
There is a pressing need for early involvement of cardiology investigators to ensure appropriate trial design and definition of AE adjudication parameters as part of a broad effort to increase collaboration among clinicians, researchers, sponsors, and regulators to improve cardiac safety in oncology clinical trials. Cardiologists could have valuable input at the initial stages of clinical trial design and throughout the trial to ensure that any early signals of cardiotoxicity are detected and evaluated in close to real time. These could then be shared rapidly with clinicians and regulatory authorities, with timely modification of the protocol if needed. Dedicated adjudication of CV events is critical, and here too, the cardiology community should have a central role in developing standardized criteria and definitions for adjudication. Serious cardiac AEs in particular should be adjudicated as to whether they are likely to have been primary events that might be attributable to the drug or to be due to the patient’s underlying state of health.
Overall benefits of AE collection in clinical trials include the ability to identify events that affect patients; notify investigators, patients, regulators and others; inform the conduct of the trial and risk management; and improve understanding of treatment safety.43
In global trials, many AEs are not detected, and important ADRs are often missed. Information is often incomplete and of limited value, and definitions, data collection, and analysis methods are inconsistent.44 General challenges involved in the collection of AE data include incomplete patient history, concomitant or prior treatments, and comorbidities. Monitoring AEs is complex and labor intensive, and there may be multiple AEs in a single patient. In addition, there is heterogeneity in adjudication. The National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE) are complicated, with the result that reproducible, systematic AE capture is difficult at best.45,46 Key factors contributing to AE underreporting include the lack of a standardized process, lack of training and education, and lack of integrated health information technologies.47
Innovative strategies are needed to obtain high-quality data, related to both exposures and outcomes. A National Cancer Institute–funded Eastern Cooperative Oncology Group and American College of Radiology Imaging Network Cardiotoxicity Working Group has been created to meet these needs, including development of a common case report form for cardio-oncology clinical trials, harmonization of AE reporting of cardiac events by Common Terminology Criteria for Adverse Events, building imaging banks and biobanks of data, and enhancing patient education (www.cardiosmart.org).
As part of an effort to increase resource sharing among academia, sponsors, and regulatory agencies to facilitate AE reporting, an FDA pilot program is under way for providing premarket, real-time safety data. A joint effort between the FDA Office of Hematology and Oncology Products and the Office of Surveillance and Epidemiology, the project has been piloted since 2016 by 4 pharmaceutical companies (Merck, AstraZeneca, Novartis, and Genentech). This uses a different format of the FDA Adverse Event Reporting System from the post-marketing database. The same set of reviewer tools can to be used for pre- and post-marketing surveillance work, and the system could potentially be set up to permit access by FDA and the investigators of a particular development program.
Future patient-focused considerations for AE reporting include the need for standardized reporting systems, ongoing institution-based surveillance, automated surveillance, medical record abstraction to determine AE reports, utilization of external data sources, active surveillance rather than voluntary reporting, training and education, integration of cardiology and cardio-oncology, and the inclusion of patient self-reporting.45,48,49 Registries could potentially be leveraged to collect AE data, with artificial intelligence used over the longer term to sort events.
Future directions in cardio-oncology
Looking ahead, increasing cancer-related survival will drive a need for therapies with an improved long-term cardiac safety profile or for improved strategies for early detection, management, and/or prevention of CV-related AEs. When such events are identified, it will be important to comprehensively phenotype the AE and investigate the underlying mechanism. Patient-specific iPSC-CMs may yield valuable insights into mechanism of cardiotoxicity and inform strategies for prevention and/or treatment.
Opportunities for improvement include the need for a better understanding of cardiotoxicity and repair at the cellular and molecular levels to identify patients at risk of cardiotoxicity, to decrease the risk of cardiotoxicity, and to detect and characterize cardiotoxicity better and at an earlier stage. There is also a need for solid evidence addressing optimal monitoring intervals and for close monitoring of CV safety after drug approval.
Cardio-oncology study designs need to evolve to include careful assessment of both cancer and CV measures, over the short and long term, and to build a full understanding of the clinical impact of changes in sensitive measures of cardiac function.50 As cancer treatments continue to change rapidly, an ongoing, collaborative dialogue between basic, translational, and clinical scientists, and oncologists and cardiologists is essential. Such collaborative efforts are paramount to most efficiently share resources and information toward improving care and minimizing risk of cardiotoxicity. Web-based platforms should be developed to increase awareness of cardiotoxicity among providers and patients and to improve data collection of CV events related to oncology clinical trials.
Finally, clinical trials should be as inclusive as possible, including patients with CV risk factors.51 Enrollment of higher-risk participants is feasible but requires buy-in from oncologists, global regulators, and industry partners and close participation of a cardio-oncologist. Conduct of substudies that include higher-risk patients should be considered, including parallel versus serial trial design. The sponsor’s responsibility to maximize the likelihood of successful trial must be balanced with the need for results to be generalizable to a real-world population—helping to ensure both optimum safety and efficacy at the level of the individual patient.
Acknowledgments
This white paper, drafted by members of the Cardiac Safety Research Consortium (CSRC), examines key issues related to detection, assessment, and risk mitigation of cardiac safety signals in oncology drug development. This was the subject of a CSRC Think Tank held on October 24-25, 2017, at the United States Food and Drug Administration headquarters in Silver Spring, MD. The goal of the meeting was to recommend best practices in detection, assessment, and mitigation of these safety signals. This paper summarizes discussions at the Think Tank, focusing on the current state of knowledge and future directions for cardiooncology—the intersection of cardiovascular disease and oncology—to help reduce cardiovascular morbidity and mortality in cancer survivors.
References
- 1.Larsen CM, Mulvagh SL. Cardio-oncology: what you need to know now for clinical practice and echocardiography. Echo Res Pract 2017;4(1):R33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Finkle J, Bloomfield D, Uhl K, et al. New precompetitive paradigms: focus on cardiac safety. Am Heart J 2009;157(5):825–6http://cardiac-safety.org/wp-content/uploads/2014/11/Finkle-New.pdf. [DOI] [PubMed] [Google Scholar]
- 3.Turner JR, Kowey PR. Rodriguez I, Cabell CH, Gintant G, Green CL, Kunz BL, Mortara J, Sager PT, Stockbridge N, Wright TJ, Finkle J, Krucoff MW; Cardiac Safety Research Consortium. The Cardiac Safety Research Consortium enters its second decade: an invitation to participate. Am Heart J. 2016. July;177:96–101, 10.1016/j.ahj.2016.04.009.Epub 2016 Apr 23http://www.cardiac-safety.org/wp-content/uploads/2016/06/The-Cardiac-Safety-Research-Consortium-enters-its-second-decade.pdf.pdf. [DOI] [PubMed] [Google Scholar]
- 4.PhRMA. List of. Medicines in development for cancer. May 2018;30: 2018.https://www.phrma.org/report/list-of-2018-medicines-in-development-for-cancer. [Google Scholar]
- 5.Albrecht B, Andersen S, Chauhan K, et al. Pursuing breakthroughs in cancer-drug development. Article; January 2018 https://www.mckinsey.com/industries/pharmaceuticals-and-medical-products/our-insights/pursuing-breakthroughs-in-cancer-drug-development.
- 6.IQVIA Institute Report. Global Oncology Trends. Innovation, expansion and disruption. May 2018;24:2018.https://www.iqvia.com/institute/reports/global-oncology-trends-2018. [Google Scholar]
- 7.de Moor JS, Mariotto AB, Parry C, et al. Cancer survivors in the United States: prevalence across the survivorship trajectory and implications for care. Cancer Epidemiol Biomark Prev 2013;22(4):561–70https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3654837/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barac A, Murtagh G, Carver JR, et al. Cardiovascular health of patients with cancer and cancer survivors: a roadmap to the next level. J Am Coll Cardiol 2015;65:2739–46https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4484773/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Siegel R, DeSantis C, Virgo K, et al. Cancer treatment and survivorship statistics, 2012. CA Cancer J Clin 2012 Jul-Aug;62(4):220–41, 10.3322/caac.21149.Epub 2012 Jun 14https://onlinelibrary.wiley.com/doi/epdf/10.3322/caac.21149. [DOI] [PubMed] [Google Scholar]
- 10.Fradley MG, Brown AC, Shields B, et al. Developing a comprehensive cardio-oncology program at a cancer institute: the Moffitt Cancer Center Experience. Oncol Rev 2017;11(340)https://www.ncbi.nlm.nih.gov/pubmed/28781723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Daher IN, Daigle TR, Bhatia N, et al. The prevention of cardiovascular disease in cancer survivors. Tex Heart Inst J 2012;39:190–8https://www.ncbi.nlm.nih.gov/pubmed/22740730. [PMC free article] [PubMed] [Google Scholar]
- 12.Moslehi JJ. Cardiovascular toxic effects of targeted cancer therapies. N Engl J Med 2016. October 13;375(15):1457–67https://www.nejm.org/doi/full/10.1056/NEJMra1100265?url_ver=Z39.88-2003&rfr_id=ori:rid:crossref.org&rfr_dat=cr_pub%3dpubmed. [DOI] [PubMed] [Google Scholar]
- 13.Kenigsberg B, Campia U, Barac A. Cardiovascular side effects of cancer treatments. Clinical Pharmacist, September 2016, Vol 8, No 9, online | DOI: 10.1211/CP.2016.20201651. https://www.pharmaceutical-journal.com/research/review-article/cardiovascular-side-effects-of-cancer-treatments/20201651.article [DOI] [Google Scholar]
- 14.Li W, Croce K, et al. Vascular and metabolic implications of noverl trageted caner therapies: focus on kinase inhibitors. J Am Coll Cardiol 2015. September 8;66(10):1160–78, 10.1016/j.jacc.2015.07.025. https://www.ncbi.nlm.nih.gov/pubmed/?term=26337996. [DOI] [PubMed] [Google Scholar]
- 15.Salem JE, Manouchehri A, et al. Cardiovascular toxicities associated with immune checkpoint inhibitors: an observational, retrospective. pharmacovigilance study Lancet Oncol 2018. December;19(12):1579–89, 10.1016/S1470-2045(18)30608-9. Epub 2018 Nov 12https://www.thelancet.com/journals/lanonc/article/PIIS1470-2045(18)30608-9/fulltext. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moslehi J Salem JE et al. Increased reporting of fatal immune checkpoint inhibitor associated myocarditis. Lancet 2018. March 10;391(10124):933, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johnson DB, Balko JM, et al. Fulminant myocarditis with combination immune checkpoint blockade NEJM 2016. November 3: 375(18). https://www.ncbi.nlm.nih.gov/pubmed/?term=27806233. 1749–1755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ewer MS, Ewer SM. Cardiotoxicity of anticancer treatments. Nat Rev Cardiol. 2015. September;12(9):547–58. doi: 10.1038/nrcardio.2015.65. Epub 2015 May 12 https://www.ncbi.nlm.nih.gov/pubmed/25962976 [DOI] [PubMed] [Google Scholar]
- 19.Tan C, Denlinger C. Cardiovascular toxicity in cancer survivors: current guidelines and future directions. June 29, 2018. ACC website. https://www.acc.org/latest-in-cardiology/articles/2018/06/29/12/57/cv-toxicity-in-cancer-survivors [Google Scholar]
- 20.American College of Cardiology website: https://www.acc.org/clinical-topics/cardio-oncology#sort=%40fcommonsortdate90022%20descending, https://www.acc.org/membership/sections-and-councils/cardio-oncology/about-us/message-from-the-chair and https://www.acc.org/membership/sections-and-councils/cardiooncology
- 21.American Society of Clinical Oncology. Prevention and monitoring of cardiac dysfunction in survivors of adult cancers: ASCO clinical practice guideline. J Clin Oncol. 2016. December 5:JCO2016705400. [Epub ahead of print] https://www.acc.org/~/media/Non-Clinical/Files-PDFs-Excel-MS-Word-etc/Meetings/2017/Course%20PDFs/Cardio%20Oncology/Updated%20Post%20course/Fri%2011%2000%20am%20Guidelines%20Armenian.pdf [DOI] [PubMed] [Google Scholar]
- 22.Curigliano G, Cardinale D, Suter T, Plataniotis G, de Azambuja E, Sandri MT, Criscitiello C, Goldhirsch A, Cipolla C, Roila F; ESMO Guidelines Working Group. Cardiovascular toxicity induced by chemotherapy, targeted agents and radiotherapy: ESMO clinical practice guidelines. Ann Oncol. 2012. October;23 Suppl 7:vii155–66. https://www.esmo.org/Guidelines/Supportive-and-Palliative-Care/Cardiovascular-Toxicity-Induced-Chemotherapy-Targeted-Agents-and-Radiotherapy [DOI] [PubMed] [Google Scholar]
- 23.Zamorano JL, Lancellotti P, Muñoz DR, et al. 2016 ESC position paper on cancer treatments and cardiovascular toxicity developed under the auspices of the ESC Committee for Practice Guidelines. Eur Heart J 2016; 37(36):2768–2801. https://www.esmo.org/Oncology-News/ESC-Position-Paper-on-Anticancer-Treatments-and-Cardiovascular-Toxicity [DOI] [PubMed] [Google Scholar]
- 24.Virani SA, et al. Canadian Cardiovascular Society guidelines for evaluation and management of cardiovascular complications of cancer therapy. Canadian Journal of Cardiology, Volume 32, Issue 7, 831–841. https://www.onlinecjc.ca/article/S0828-282X(16)30004-6/abstract [DOI] [PubMed] [Google Scholar]
- 25.Sharma A, Burridge PW, et al. High-throughput screening of tyrosine kinase inhibitor cardiotoxicity with human induced pluripotent stem cells. Sci Transl Med 2017. February 15;9(377), 10.1126/scitranslmed.aaf2584 Sheng CC, Amiri-Kordestani L, et al. 21st Century Cardio-Oncology: Identifying Cardiac Safety Signals in the Era of Personalized Medicine. JACC Basic Transl Sci. 2016 Aug;1(5):386–398. doi: . https://www.ncbi.nlm.nih.gov/pubmed/?term=28713868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maillet A, Tan K, Chai X, et al. Modeling doxorubicin-induced cardiotoxicity in human pluripotent stem cell derived-cardiomyocytes. Sci Rep 2016. May 4;6, 25333, 10.1038/srep25333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hortigon-Vinagre MP, et al. The use of ratiometric fluorescence measurements of the voltage sensitive dye Di-4-ANEPPS to examine action potential characteristics and drug effects on human induced pluripotent stem cell-derived cardiomyocytes. Toxicol Sci 2016. December;154(2):320–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pointon A, Pilling J, et al. From the cover: high-throughput imaging of cardiac microtissues for the assessment of cardiac contraction during drug discovery. Toxicol Sci 2017. February;155(2):444–57, 10.1093/toxsci/kfw227. [DOI] [PubMed] [Google Scholar]
- 29.Burridge PW, Matsa E, Shukla P, et al. Chemically defined generation of human cardiomyocytes. Nat Methods 2014;11(8):855–60https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4169698/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burridge PW, Li YF, Matsa E, et al. Human induced pluripotent stem cell-derived cardiomyocytes recapitulate the predilection of breast cancer patients to doxorubicin-induced cardiotoxicity. Nat Med 2016;22(5): 547–56https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5086256/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aminkeng F, Bhavsar AP, Visscher H, et al. A coding variant in RARG confers susceptibility to anthracycline-induced cardiotoxicity in childhood cancer. Nat Genet 2015;47(9):1079–84https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4552570/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Op Cit 28.
- 33.Cardiotoxicity Kondapalli L An unexpected consequence of HER2-targeted therapies. American College of Cardiology Expert Analysis, June 2016;7https://www.acc.org/latest-in-cardiology/articles/2016/06/06/09/32/cardiotoxicity. [Google Scholar]
- 34.Kenigsberg B, Wellstein A, Barac A. Left ventricular dysfunction in cancer treatment: is it relevant? JACC Heart Fail 2018. February;6(2): 87–95, 10.1016/j.jchf.2017.08.024. Epub 2017 Dec 1. Review. https://www.ncbi.nlm.nih.gov/pubmed/29413379. [DOI] [PubMed] [Google Scholar]
- 35.Dang CT, Yu AF, Jones LW, et al. Cardiac surveillance guidelines for trastuzumab-containing therapy in early-stage breast cancer: getting to the heart of the matter. J Clin Oncol 2016;34(10):1030–3https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5070558/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lynce F, Barac A, et al. SAFE-HEaRt: rationale and design of a pilot study investigating cardiac safety of HER2 targeted therapy in patients with HER2-positive breast cancer and reduced left ventricular function. Oncologist 2017. May;22(5):518–25, 10.1634/theoncologist.2016-0412. Epub 2017 Mar 17. https://www.ncbi.nlm.nih.gov/pubmed/?term=28314836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tan LL, Lyon AR. Role of biomarkers in prediction of cardiotoxicity during cancer treatment. Curr Treat Options Cardiovasc Med. 2018;20(7):55 Published 2018 Jun 19. doi: 10.1007/s11936-018-0641-z. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6008350/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Douglas PS, et al. Implementation of echocardiography core laboratory best practices: a case study of the PARTNER I Trial. Journal of the American Society of Echocardiography, Volume 26, Issue 4, 348–358.e3. https://www.onlinejase.com/article/S0894-7317(13)00045-X/fulltext [DOI] [PubMed] [Google Scholar]
- 39.Gottdiener JS, et al. American Society of Echocardiography recommendations for use of echocardiography in clinical trials. Journal of the American Society of Echocardiography, Volume 17, Issue 10, 1086–1119. https://www.onlinejase.com/article/S0894-7317(04)00675-3/fulltext [DOI] [PubMed] [Google Scholar]
- 40.Lee Yong-ho, Hong Namki, Chan Joo Lee Sung Ha Park, Lee Byung-Wan. Cha Bong-Soo & Kang Eun Seok. Differential association of ezetimibe-simvastatin combination with major adverse cardiovascular events in patients with or without diabetes: a retrospective propensity score-matched cohort study Scientific Reportsvolume 8. Article number 2018:11925https://www.nature.com/articles/s41598-018-30409-6#Abs1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Levy D, Kenchaiah S, Larson MG, et al. Long-term trends in the incidence of and survival with heart failure. N Engl J Med 2002. October 31;347(18):1397–402https://www.nejm.org/doi/full/10.1056/NEJMoa020265. [DOI] [PubMed] [Google Scholar]
- 42.Smolina K1, Wright FL, Rayner M, Goldacre MJ. Long-term survival and recurrence after acute myocardial infarction in England, 2004 to 2010. Circ Cardiovasc Qual Outcomes. 2012. July 1;5(4):532–40. doi: 10.1161/CIRCOUTCOMES.111.964700. Epub 2012 Jun 26. https://www.ncbi.nlm.nih.gov/pubmed/22740013 [DOI] [PubMed] [Google Scholar]
- 43.Johnson DE. Fusion of nonclinical and clinical data to predict human drug safety. Expert Rev Clin Pharmacol 2013;6(2):185–95, 10.1586/ecp.13.3. https://www.tandfonline.com/doi/full/10.1586/ecp.13.3. [DOI] [PubMed] [Google Scholar]
- 44.Institute of Medicine (US) Committee on Data Standards for Patient Safety. Aspden P, Corrigan JM, Wolcott J, et al. , editors. Washington (DC): National Academies Press (US); 2004. Stroke 2004;35: 533-7https://www.ncbi.nlm.nih.gov/pubmed/25009854. [PubMed] [Google Scholar]
- 45.Miller TP, Li Y, Kavcic M, et al. Accuracy of adverse event ascertainment in clinical trials for pediatric acute myeloid leukemia. J Clin Oncol 2016;34(13):1537–43http://ascopubs.org/doi/abs/10.1200/JCO.2015.65.5860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Seltzer JH, Turner JR, Geiger MJ, et al. Centralized adjudication of cardiovascular end points in cardiovascular and noncardiovascular pharmacologic trials: a report from the Cardiac Safety Research Consortium. Am Heart J 2015. February;169(2):197–204, 10.1016/j.ahj.2014.11.003. Epub 2014 Nov 10. https://www.ncbi.nlm.nih.gov/pubmed/25641528. [DOI] [PubMed] [Google Scholar]
- 47.Stergiopoulos S, Brown CA, Felix T, et al. A survey of adverse event reporting practices among US healthcare professionals. Drug Saf 2016;39(11):1117–27https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5045838/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hurtado-de-Mendoza D, Loaiza-Bonilla A, Bonilla-Reyes PA, et al. (May 18, 2017). Cardio-oncology: cancer therapy-related cardiovascular complications in a molecular targeted era: new concepts and perspectives. Cureus 9(5): e1258 DOI 10.7759/cureus.1258 https://assets.cureus.com/uploads/review_article/pdf/5342/1498143949-20170622-3625-1j9whn2.pdf [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schmidinger M1, Zielinski CC, Vogl UM, Bojic A, Bojic M, Schukro C, Ruhsam M, Hejna M, Schmidinger H. Cardiac toxicity of sunitinib and sorafenib in patients with metastatic renal cell carcinoma. J Clin Oncol. 2008. November 10;26(32):5204–12. doi: 10.1200/JCO.2007.15.6331. Epub 2008 Oct 6. [DOI] [PubMed] [Google Scholar]
- 50.Minasian L, Dimond E, Shelburne N, et al. Evolving cardio-oncology study designs. Global Cardio-Oncology Summit, January 2017;1. [Google Scholar]
- 51.Lichtman, et al. Modernizing clinical trial eligibility criteria: recommendations of the american society of clinical oncology—friends of cancer research organ dysfunction, prior or concurrent malignancy, and comorbidities working group. JCO November 20 2017;35(33): 3753–9. [DOI] [PubMed] [Google Scholar]